
Histone Deacetylase Inhibitors Restore Cancer Cell Sensitivity towards T Lymphocytes Mediated Cytotoxicity in Pancreatic Cancer

 , , ,
, , ,
Abstract
:Simple Summary
Abstract
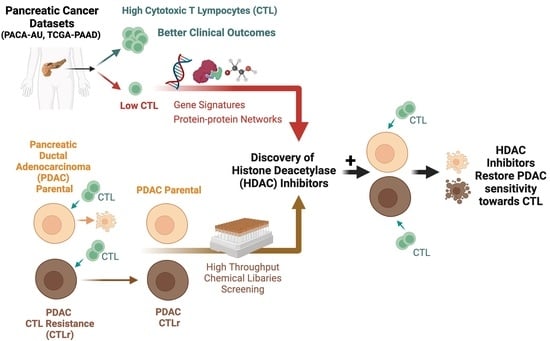
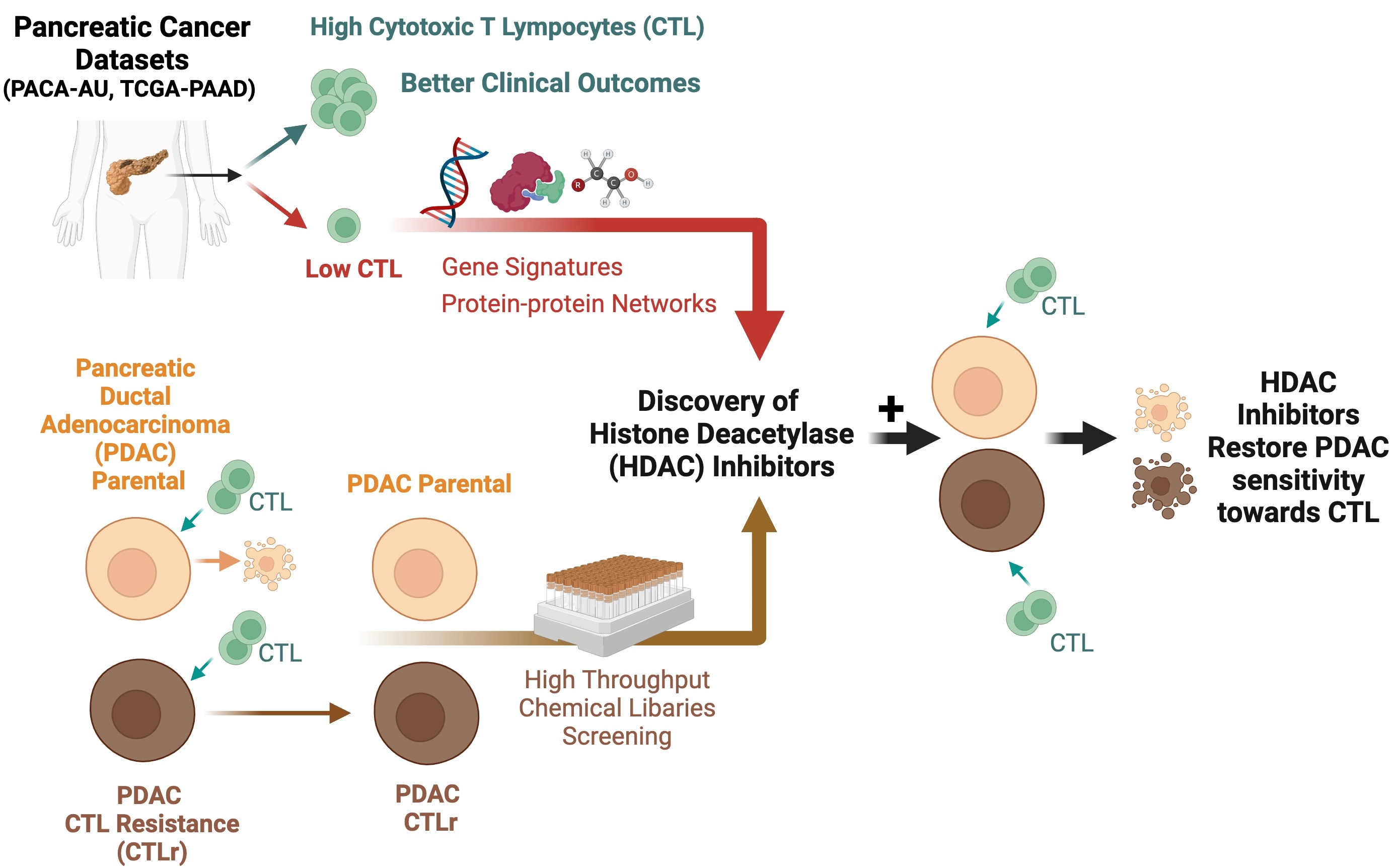
1. Introduction
2. Materials and Methods
2.1. Characterisation of the CTL Activity in PDAC Patients
2.2. Identification of Differentially Expressed Genes Associated with the Low CTL Activity in PDAC Patients
2.3. Determination of Perturbagens That Target PDAC with Low CTL Killing Activity
2.4. Cell Lines and Cell Cultures
2.5. Establishment of PDAC Cells with Acquired Resistance towards CTL-Induced Cytotoxicity
2.6. High-Throughput Chemical Library Screening
2.7. Western Blotting
2.8. HDAC Inhibitor Efficacy Study
3. Results
3.1. High CTL Activity Is Associated with Better Overall Survival and Disease Progression in PDAC Patients
3.2. Identification of Gene Signature Associated with Low CTL Killing Activity in PDAC Patients
3.3. Identification of Perturbagens Targeting PDAC Gene Signature Associated with Low CTL Killing Activity
3.4. Identification of Inhibitors Targeting CTL-Resistant PDAC via High-Throughput Chemical Library Screening
3.5. HDAC Inhibitors Restore CTL Sensitivity towards CTL-Resistant PDAC Cells
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Gentiluomo, M.; Canzian, F.; Nicolini, A.; Gemignani, F.; Landi, S.; Campa, D. Germline genetic variability in pancreatic cancer risk and prognosis. Semin. Cancer Biol. 2022, 79, 105–131. [Google Scholar] [CrossRef] [PubMed]
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Rahib, L.; Smith, B.D.; Aizenberg, R.; Rosenzweig, A.B.; Fleshman, J.M.; Matrisian, L.M. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014, 74, 2913–2921. [Google Scholar] [CrossRef] [Green Version]
- Looi, C.K.; Hii, L.W.; Ngai, S.C.; Leong, C.O.; Mai, C.W. The Role of Ras-Associated Protein 1 (Rap1) in Cancer: Bad Actor or Good Player? Biomedicines 2020, 8, 334. [Google Scholar] [CrossRef]
- Gan, L.L.; Hii, L.W.; Wong, S.F.; Leong, C.O.; Mai, C.W. Molecular Mechanisms and Potential Therapeutic Reversal of Pancreatic Cancer-Induced Immune Evasion. Cancers 2020, 12, 1872. [Google Scholar] [CrossRef]
- Looi, C.K.; Chung, F.F.; Leong, C.O.; Wong, S.F.; Rosli, R.; Mai, C.W. Therapeutic challenges and current immunomodulatory strategies in targeting the immunosuppressive pancreatic tumor microenvironment. J. Exp. Clin. Cancer Res. 2019, 38, 162. [Google Scholar] [CrossRef]
- Chen, D.S.; Mellman, I. Oncology meets immunology: The cancer-immunity cycle. Immunity 2013, 39, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Protti, M.P.; De Monte, L. Immune infiltrates as predictive markers of survival in pancreatic cancer patients. Front. Physiol. 2013, 4, 210. [Google Scholar] [CrossRef] [Green Version]
- Grywalska, E.; Pasiarski, M.; Gozdz, S.; Rolinski, J. Immune-checkpoint inhibitors for combating T-cell dysfunction in cancer. Onco Targets Ther. 2018, 11, 6505–6524. [Google Scholar] [CrossRef] [Green Version]
- McDermott, D.F.; Sosman, J.A.; Sznol, M.; Massard, C.; Gordon, M.S.; Hamid, O.; Powderly, J.D.; Infante, J.R.; Fasso, M.; Wang, Y.V.; et al. Atezolizumab, an Anti-Programmed Death-Ligand 1 Antibody, in Metastatic Renal Cell Carcinoma: Long-Term Safety, Clinical Activity, and Immune Correlates From a Phase Ia Study. J. Clin. Oncol. 2016, 34, 833–842. [Google Scholar] [CrossRef]
- Li, H.B.; Yang, Z.H.; Guo, Q.Q. Immune Checkpoint Inhibition for Pancreatic Ductal Adenocarcinoma: Limitations and Prospects: A Systematic Review. Cell Commun. Signal. 2021, 19, 117. [Google Scholar] [CrossRef]
- Subramanian, I.; Verma, S.; Kumar, S.; Jere, A.; Anamika, K. Multi-omics Data Integration, Interpretation, and Its Application. Bioinform. Biol. Insights 2020, 14, 1177932219899051. [Google Scholar] [CrossRef] [Green Version]
- Hutter, C.; Zenklusen, J.C. The Cancer Genome Atlas: Creating Lasting Value beyond Its Data. Cell 2018, 173, 283–285. [Google Scholar] [CrossRef]
- Gonzalez-Reymundez, A.; de Los Campos, G.; Gutierrez, L.; Lunt, S.Y.; Vazquez, A.I. Prediction of years of life after diagnosis of breast cancer using omics and omic-by-treatment interactions. Eur. J. Hum. Genet. 2017, 25, 538–544. [Google Scholar] [CrossRef] [Green Version]
- Mishra, N.K.; Southekal, S.; Guda, C. Survival Analysis of Multi-Omics Data Identifies Potential Prognostic Markers of Pancreatic Ductal Adenocarcinoma. Front. Genet. 2019, 10, 624. [Google Scholar] [CrossRef] [Green Version]
- Baek, B.; Lee, H. Prediction of survival and recurrence in patients with pancreatic cancer by integrating multi-omics data. Sci. Rep. 2020, 10, 18951. [Google Scholar] [CrossRef]
- Zuo, H.; Chen, L.; Li, N.; Song, Q. Identification of a Ubiquitination-Related Gene Risk Model for Predicting Survival in Patients With Pancreatic Cancer. Front. Genet. 2020, 11, 612196. [Google Scholar] [CrossRef]
- Grunwald, B.T.; Devisme, A.; Andrieux, G.; Vyas, F.; Aliar, K.; McCloskey, C.W.; Macklin, A.; Jang, G.H.; Denroche, R.; Romero, J.M.; et al. Spatially confined sub-tumor microenvironments in pancreatic cancer. Cell 2021, 184, 5577–5592.e5518. [Google Scholar] [CrossRef]
- Li, Y.; Seto, E. HDACs and HDAC Inhibitors in Cancer Development and Therapy. Cold Spring Harb. Perspect. Med. 2016, 3, a026831. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Tamayo, P.; Mootha, V.K.; Mukherjee, S.; Ebert, B.L.; Gillette, M.A.; Paulovich, A.; Pomeroy, S.L.; Golub, T.R.; Lander, E.S.; et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 2005, 102, 15545–15550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hackl, H.; Charoentong, P.; Finotello, F.; Trajanoski, Z. Computational genomics tools for dissecting tumour-immune cell interactions. Nat. Rev. Genet. 2016, 17, 441–458. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef] [PubMed]
- Soo, H.C.; Chung, F.F.; Lim, K.H.; Yap, V.A.; Bradshaw, T.D.; Hii, L.W.; Tan, S.H.; See, S.J.; Tan, Y.F.; Leong, C.O.; et al. Cudraflavone C Induces Tumor-Specific Apoptosis in Colorectal Cancer Cells through Inhibition of the Phosphoinositide 3-Kinase (PI3K)-AKT Pathway. PLoS ONE 2017, 12, e0170551. [Google Scholar] [CrossRef] [Green Version]
- Er, J.L.; Goh, P.N.; Lee, C.Y.; Tan, Y.J.; Hii, L.W.; Mai, C.W.; Chung, F.F.; Leong, C.O. Identification of inhibitors synergizing gemcitabine sensitivity in the squamous subtype of pancreatic ductal adenocarcinoma (PDAC). Apoptosis 2018, 23, 343–355. [Google Scholar] [CrossRef]
- Samart, K.; Tuyishime, P.; Krishnan, A.; Ravi, J. Reconciling multiple connectivity scores for drug repurposing. Brief. Bioinform. 2021, 22, bbab161. [Google Scholar] [CrossRef]
- Song, D.S.S.; Leong, S.W.; Ng, K.W.; Abas, F.; Shaari, K.; Leong, C.O.; Chung, F.F.; Mai, C.W.; Hii, L.W.; Tan, P.J.; et al. Novel 2-Benzoyl-6-(2,3-Dimethoxybenzylidene)-Cyclohexenol Confers Selectivity toward Human MLH1 Defective Cancer Cells through Synthetic Lethality. SLAS Discov. 2019, 24, 548–562. [Google Scholar] [CrossRef]
- Konig, R.; Chiang, C.Y.; Tu, B.P.; Yan, S.F.; DeJesus, P.D.; Romero, A.; Bergauer, T.; Orth, A.; Krueger, U.; Zhou, Y.; et al. A probability-based approach for the analysis of large-scale RNAi screens. Nat. Methods 2007, 4, 847–849. [Google Scholar] [CrossRef]
- Hii, L.W.; Chung, F.F.; Soo, J.S.; Tan, B.S.; Mai, C.W.; Leong, C.O. Histone deacetylase (HDAC) inhibitors and doxorubicin combinations target both breast cancer stem cells and non-stem breast cancer cells simultaneously. Breast Cancer Res. Treat. 2020, 179, 615–629. [Google Scholar] [CrossRef]
- Hontecillas-Prieto, L.; Flores-Campos, R.; Silver, A.; de Alava, E.; Hajji, N.; Garcia-Dominguez, D.J. Synergistic Enhancement of Cancer Therapy Using HDAC Inhibitors: Opportunity for Clinical Trials. Front. Genet. 2020, 11, 578011. [Google Scholar] [CrossRef]
- Zhang, S.; Gong, Z.; Oladimeji, P.O.; Currier, D.G.; Deng, Q.; Liu, M.; Chen, T.; Li, Y. A high-throughput screening identifies histone deacetylase inhibitors as therapeutic agents against medulloblastoma. Exp. Hematol. Oncol. 2019, 8, 30. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissTargetPrediction: Updated data and new features for efficient prediction of protein targets of small molecules. Nucleic Acids Res 2019, 47, W357–W364. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, A.; Narayan, R.; Corsello, S.M.; Peck, D.D.; Natoli, T.E.; Lu, X.; Gould, J.; Davis, J.F.; Tubelli, A.A.; Asiedu, J.K.; et al. A Next Generation Connectivity Map: L1000 Platform and the First 1,000,000 Profiles. Cell 2017, 171, 1437–1452.e1417. [Google Scholar] [CrossRef]
- Giri, B.; Sharma, P.; Jain, T.; Ferrantella, A.; Vaish, U.; Mehra, S.; Garg, B.; Iyer, S.; Sethi, V.; Malchiodi, Z.; et al. Hsp70 modulates immune response in pancreatic cancer through dendritic cells. Oncoimmunology 2021, 10, 1976952. [Google Scholar] [CrossRef]
- Collisson, E.A.; Bailey, P.; Chang, D.K.; Biankin, A.V. Molecular subtypes of pancreatic cancer. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 207–220. [Google Scholar] [CrossRef]
- Yang, X.; Lin, J.; Wang, G.; Xu, D. Targeting Proliferating Tumor-Infiltrating Macrophages Facilitates Spatial Redistribution of CD8(+) T Cells in Pancreatic Cancer. Cancers 2022, 14, 1474. [Google Scholar] [CrossRef]
- Hiraoka, N.; Onozato, K.; Kosuge, T.; Hirohashi, S. Prevalence of FOXP3+ regulatory T cells increases during the progression of pancreatic ductal adenocarcinoma and its premalignant lesions. Clin. Cancer Res. 2006, 12, 5423–5434. [Google Scholar] [CrossRef] [Green Version]
- Kiryu, S.; Ito, Z.; Suka, M.; Bito, T.; Kan, S.; Uchiyama, K.; Saruta, M.; Hata, T.; Takano, Y.; Fujioka, S.; et al. Prognostic value of immune factors in the tumor microenvironment of patients with pancreatic ductal adenocarcinoma. BMC Cancer 2021, 21, 1197. [Google Scholar] [CrossRef]
- Hou, Y.C.; Chao, Y.J.; Hsieh, M.H.; Tung, H.L.; Wang, H.C.; Shan, Y.S. Low CD8(+) T Cell Infiltration and High PD-L1 Expression Are Associated with Level of CD44(+)/CD133(+) Cancer Stem Cells and Predict an Unfavorable Prognosis in Pancreatic Cancer. Cancers 2019, 11, 541. [Google Scholar] [CrossRef] [Green Version]
- Carstens, J.L.; Correa de Sampaio, P.; Yang, D.; Barua, S.; Wang, H.; Rao, A.; Allison, J.P.; LeBleu, V.S.; Kalluri, R. Spatial computation of intratumoral T cells correlates with survival of patients with pancreatic cancer. Nat. Commun. 2017, 8, 15095. [Google Scholar] [CrossRef]
- Karamitopoulou, E. Tumour microenvironment of pancreatic cancer: Immune landscape is dictated by molecular and histopathological features. Br. J. Cancer 2019, 121, 5–14. [Google Scholar] [CrossRef]
- Harlin, H.; Kuna, T.V.; Peterson, A.C.; Meng, Y.; Gajewski, T.F. Tumor progression despite massive influx of activated CD8(+) T cells in a patient with malignant melanoma ascites. Cancer Immunol. Immunother. 2006, 55, 1185–1197. [Google Scholar] [CrossRef]
- Rosenberg, S.A.; Sherry, R.M.; Morton, K.E.; Scharfman, W.J.; Yang, J.C.; Topalian, S.L.; Royal, R.E.; Kammula, U.; Restifo, N.P.; Hughes, M.S.; et al. Tumor progression can occur despite the induction of very high levels of self/tumor antigen-specific CD8+ T cells in patients with melanoma. J. Immunol. 2005, 175, 6169–6176. [Google Scholar] [CrossRef] [Green Version]
- Colombo, M.; Mirandola, L.; Chiriva-Internati, M.; Basile, A.; Locati, M.; Lesma, E.; Chiaramonte, R.; Platonova, N. Cancer Cells Exploit Notch Signaling to Redefine a Supportive Cytokine Milieu. Front. Immunol. 2018, 9, 1823. [Google Scholar] [CrossRef]
- Li, S.; Wang, Z.; Li, X.J. Notch signaling pathway suppresses CD8(+) T cells activity in patients with lung adenocarcinoma. Int. Immunopharmacol. 2018, 63, 129–136. [Google Scholar] [CrossRef]
- Yu, W.; Wang, Y.; Guo, P. Notch signaling pathway dampens tumor-infiltrating CD8(+) T cells activity in patients with colorectal carcinoma. Biomed. Pharmacother. 2018, 97, 535–542. [Google Scholar] [CrossRef]
- Zhou, J.X.; Zhou, L.; Li, Q.J.; Feng, W.; Wang, P.M.; Li, E.F.; Gong, W.J.; Kou, M.W.; Gou, W.T.; Yang, Y.L. Association between high levels of Notch3 expression and high invasion and poor overall survival rates in pancreatic ductal adenocarcinoma. Oncol. Rep. 2016, 36, 2893–2901. [Google Scholar] [CrossRef] [Green Version]
- Mazur, P.K.; Einwachter, H.; Lee, M.; Sipos, B.; Nakhai, H.; Rad, R.; Zimber-Strobl, U.; Strobl, L.J.; Radtke, F.; Kloppel, G.; et al. Notch2 is required for progression of pancreatic intraepithelial neoplasia and development of pancreatic ductal adenocarcinoma. Proc. Natl. Acad. Sci. USA 2010, 107, 13438–13443. [Google Scholar] [CrossRef] [Green Version]
- Qi, Z.; Xu, Z.; Zhang, L.; Zou, Y.; Li, J.; Yan, W.; Li, C.; Liu, N.; Wu, H. Overcoming resistance to immune checkpoint therapy in PTEN-null prostate cancer by intermittent anti-PI3Kalpha/beta/delta treatment. Nat. Commun. 2022, 13, 182. [Google Scholar] [CrossRef]
- Shao, L.; Yu, Q.; Xia, R.; Zhang, J.; Gu, S.; Yu, D.; Zhuang, Z. B7-H3 on breast cancer cell MCF7 inhibits IFN-γ release from tumour-infiltrating T cells. Pathol. Res. Pract. 2021, 224, 153461. [Google Scholar] [CrossRef]
- Wu, Q.; Tian, A.L.; Li, B.; Leduc, M.; Forveille, S.; Hamley, P.; Galloway, W.; Xie, W.; Liu, P.; Zhao, L.; et al. IGF1 receptor inhibition amplifies the effects of cancer drugs by autophagy and immune-dependent mechanisms. J. Immunother. Cancer 2021, 9, e002722. [Google Scholar] [CrossRef] [PubMed]
- Klein, K.; Stoiber, D.; Sexl, V.; Witalisz-Siepracka, A. Untwining Anti-Tumor and Immunosuppressive Effects of JAK Inhibitors-A Strategy for Hematological Malignancies? Cancers 2021, 13, 2611. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; McMillan, M.T.; Zhao, X.; Wang, Z.; Jiang, L.; Karnak, D.; Lima, F.; Parsels, J.D.; Parsels, L.A.; Lawrence, T.S.; et al. DNA-PK inhibition and radiation promote anti-tumoral immunity through RNA Polymerase III in pancreatic cancer. Mol. Cancer Res. 2022, 20, 1137–1150. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Li, X.; Zhao, H.; Liu, M.; Du, J.; Zhang, J.; Yang, X.; Hou, X.; Fang, H. Discovery of DNA-Targeting HDAC Inhibitors with Potent Antitumor Efficacy In Vivo That Trigger Antitumor Immunity. J. Med. Chem. 2022, 65, 3667–3683. [Google Scholar] [CrossRef] [PubMed]
- Powell, R.M.; Peeters, M.J.W.; Rahbech, A.; Aehnlich, P.; Seremet, T.; Thor Straten, P. Small Molecule Inhibitors of MERTK and FLT3 Induce Cell Cycle Arrest in Human CD8(+) T Cells. Vaccines 2021, 9, 1294. [Google Scholar] [CrossRef] [PubMed]
- Booth, L.; Roberts, J.L.; Poklepovic, A.; Dent, P. Prior exposure of pancreatic tumors to [sorafenib + vorinostat] enhances the efficacy of an anti-PD-1 antibody. Cancer Biol. Ther. 2019, 20, 109–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christmas, B.J.; Rafie, C.I.; Hopkins, A.C.; Scott, B.A.; Ma, H.S.; Cruz, K.A.; Woolman, S.; Armstrong, T.D.; Connolly, R.M.; Azad, N.A.; et al. Entinostat Converts Immune-Resistant Breast and Pancreatic Cancers into Checkpoint-Responsive Tumors by Reprogramming Tumor-Infiltrating MDSCs. Cancer Immunol. Res. 2018, 6, 1561–1577. [Google Scholar] [CrossRef] [Green Version]
- Sim, W.; Lim, W.M.; Hii, L.W.; Leong, C.O.; Mai, C.W. Targeting pancreatic cancer immune evasion by inhibiting histone deacetylases. World J. Gastroenterol. 2022, 28, 1934–1945. [Google Scholar] [CrossRef]
- Tang, Z.; Ding, S.; Huang, H.; Luo, P.; Qing, B.; Zhang, S.; Tang, R. HDAC1 triggers the proliferation and migration of breast cancer cells via upregulation of interleukin-8. Biol. Chem. 2017, 398, 1347–1356. [Google Scholar] [CrossRef]
- Zhong, L.; Sun, S.; Yao, S.; Han, X.; Gu, M.; Shi, J. Histone deacetylase 5 promotes the proliferation and invasion of lung cancer cells. Oncol. Rep. 2018, 40, 2224–2232. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, A.; Denkert, C.; Budczies, J.; Buckendahl, A.C.; Darb-Esfahani, S.; Noske, A.; Muller, B.M.; Bahra, M.; Neuhaus, P.; Dietel, M.; et al. High class I HDAC activity and expression are associated with RelA/p65 activation in pancreatic cancer in vitro and in vivo. BMC Cancer 2009, 9, 395. [Google Scholar] [CrossRef] [Green Version]
- Peulen, O.; Gonzalez, A.; Peixoto, P.; Turtoi, A.; Mottet, D.; Delvenne, P.; Castronovo, V. The anti-tumor effect of HDAC inhibition in a human pancreas cancer model is significantly improved by the simultaneous inhibition of cyclooxygenase 2. PLoS ONE 2013, 8, e75102. [Google Scholar] [CrossRef] [Green Version]
- Fenichel, M.P. FDA approves new agent for multiple myeloma. J. Natl. Cancer Inst. 2015, 107, 5–6. [Google Scholar] [CrossRef] [Green Version]
- Sawas, A.; Radeski, D.; O’Connor, O.A. Belinostat in patients with refractory or relapsed peripheral T-cell lymphoma: A perspective review. Ther. Adv. Hematol. 2015, 6, 202–208. [Google Scholar] [CrossRef] [Green Version]
- Rana, Z.; Diermeier, S.; Hanif, M.; Rosengren, R.J. Understanding Failure and Improving Treatment Using HDAC Inhibitors for Prostate Cancer. Biomedicines 2020, 8, 22. [Google Scholar] [CrossRef] [Green Version]
- Mazziotta, C.; Lanzillotti, C.; Gafa, R.; Touze, A.; Durand, M.A.; Martini, F.; Rotondo, J.C. The Role of Histone Post-Translational Modifications in Merkel Cell Carcinoma. Front. Oncol. 2022, 12, 832047. [Google Scholar] [CrossRef]
- Schizas, D.; Mastoraki, A.; Naar, L.; Tsilimigras, D.I.; Katsaros, I.; Fragkiadaki, V.; Karachaliou, G.S.; Arkadopoulos, N.; Liakakos, T.; Moris, D. Histone Deacetylases (HDACs) in Gastric Cancer: An Update of Their Emerging Prognostic and Therapeutic Role. Curr. Med. Chem. 2020, 27, 6099–6111. [Google Scholar] [CrossRef]
- Suraweera, A.; O’Byrne, K.J.; Richard, D.J. Combination Therapy with Histone Deacetylase Inhibitors (HDACi) for the Treatment of Cancer: Achieving the Full Therapeutic Potential of HDACi. Front. Oncol. 2018, 8, 92. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Ortiz, A.; Serrador, J.M. Nitric Oxide Signaling in T Cell-Mediated Immunity. Trends Mol. Med. 2018, 24, 412–427. [Google Scholar] [CrossRef]
- Bonavida, B.; Garban, H. Nitric oxide-mediated sensitization of resistant tumor cells to apoptosis by chemo-immunotherapeutics. Redox Biol. 2015, 6, 486–494. [Google Scholar] [CrossRef] [Green Version]
- Colussi, C.; Mozzetta, C.; Gurtner, A.; Illi, B.; Rosati, J.; Straino, S.; Ragone, G.; Pescatori, M.; Zaccagnini, G.; Antonini, A.; et al. HDAC2 blockade by nitric oxide and histone deacetylase inhibitors reveals a common target in Duchenne muscular dystrophy treatment. Proc. Natl. Acad. Sci. USA 2008, 105, 19183–19187. [Google Scholar] [CrossRef] [Green Version]
- Rossig, L.; Li, H.; Fisslthaler, B.; Urbich, C.; Fleming, I.; Forstermann, U.; Zeiher, A.M.; Dimmeler, S. Inhibitors of histone deacetylation downregulate the expression of endothelial nitric oxide synthase and compromise endothelial cell function in vasorelaxation and angiogenesis. Circ. Res. 2002, 91, 837–844. [Google Scholar] [CrossRef] [Green Version]
- Suliman, B.A.; Xu, D.; Williams, B.R. HDACi: Molecular mechanisms and therapeutic implications in the innate immune system. Immunol. Cell Biol. 2012, 90, 23–32. [Google Scholar] [CrossRef]
- Lewis, E.C.; Blaabjerg, L.; Storling, J.; Ronn, S.G.; Mascagni, P.; Dinarello, C.A.; Mandrup-Poulsen, T. The oral histone deacetylase inhibitor ITF2357 reduces cytokines and protects islet beta cells in vivo and in vitro. Mol. Med. 2011, 17, 369–377. [Google Scholar] [CrossRef]
- Gurbuz, N.; Ashour, A.A.; Alpay, S.N.; Ozpolat, B. Down-regulation of 5-HT1B and 5-HT1D receptors inhibits proliferation, clonogenicity and invasion of human pancreatic cancer cells. PLoS ONE 2014, 9, e110067. [Google Scholar] [CrossRef] [Green Version]
- Vojinovic, J.; Damjanov, N.; D’Urzo, C.; Furlan, A.; Susic, G.; Pasic, S.; Iagaru, N.; Stefan, M.; Dinarello, C.A. Safety and efficacy of an oral histone deacetylase inhibitor in systemic-onset juvenile idiopathic arthritis. Arthritis Rheum. 2011, 63, 1452–1458. [Google Scholar] [CrossRef]
- Rambaldi, A.; Iurlo, A.; Vannucchi, A.M.; Martino, B.; Guarini, A.; Ruggeri, M.; von Bubnoff, N.; De Muro, M.; McMullin, M.F.; Luciani, S.; et al. Long-term safety and efficacy of givinostat in polycythemia vera: 4-year mean follow up of three phase 1/2 studies and a compassionate use program. Blood Cancer J. 2021, 11, 53. [Google Scholar] [CrossRef]
- Furlan, A.; Monzani, V.; Reznikov, L.L.; Leoni, F.; Fossati, G.; Modena, D.; Mascagni, P.; Dinarello, C.A. Pharmacokinetics, safety and inducible cytokine responses during a phase 1 trial of the oral histone deacetylase inhibitor ITF2357 (givinostat). Mol. Med. 2011, 17, 353–362. [Google Scholar] [CrossRef]
- Glauben, R.; Batra, A.; Stroh, T.; Erben, U.; Fedke, I.; Lehr, H.A.; Leoni, F.; Mascagni, P.; Dinarello, C.A.; Zeitz, M.; et al. Histone deacetylases: Novel targets for prevention of colitis-associated cancer in mice. Gut 2008, 57, 613–622. [Google Scholar] [CrossRef]
- Topper, M.J.; Vaz, M.; Chiappinelli, K.B.; DeStefano Shields, C.E.; Niknafs, N.; Yen, R.C.; Wenzel, A.; Hicks, J.; Ballew, M.; Stone, M.; et al. Epigenetic Therapy Ties MYC Depletion to Reversing Immune Evasion and Treating Lung Cancer. Cell 2017, 171, 1284–1300.e1221. [Google Scholar] [CrossRef] [Green Version]
- Vo, D.D.; Prins, R.M.; Begley, J.L.; Donahue, T.R.; Morris, L.F.; Bruhn, K.W.; de la Rocha, P.; Yang, M.Y.; Mok, S.; Garban, H.J.; et al. Enhanced antitumor activity induced by adoptive T-cell transfer and adjunctive use of the histone deacetylase inhibitor LAQ824. Cancer Res. 2009, 69, 8693–8699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woods, D.M.; Woan, K.; Cheng, F.; Wang, H.; Perez-Villarroel, P.; Lee, C.; Lienlaf, M.; Atadja, P.; Seto, E.; Weber, J.; et al. The antimelanoma activity of the histone deacetylase inhibitor panobinostat (LBH589) is mediated by direct tumor cytotoxicity and increased tumor immunogenicity. Melanoma Res. 2013, 23, 341–348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali, A.I.; Wang, M.; von Scheidt, B.; Dominguez, P.M.; Harrison, A.J.; Tantalo, D.G.M.; Kang, J.; Oliver, A.J.; Chan, J.D.; Du, X.; et al. A Histone Deacetylase Inhibitor, Panobinostat, Enhances Chimeric Antigen Receptor T-cell Antitumor Effect Against Pancreatic Cancer. Clin. Cancer Res. 2021, 27, 6222–6234. [Google Scholar] [CrossRef] [PubMed]
- Tay, R.E.; Olawoyin, O.; Cejas, P.; Xie, Y.; Meyer, C.A.; Ito, Y.; Weng, Q.Y.; Fisher, D.E.; Long, H.W.; Brown, M.; et al. Hdac3 is an Epigenetic Inhibitor of the Cytotoxicity Program in CD8 T Cells. J. Exp. Med. 2020, 217, e20191453. [Google Scholar] [CrossRef] [PubMed]
- Manning, J.; Indrova, M.; Lubyova, B.; Pribylova, H.; Bieblova, J.; Hejnar, J.; Simova, J.; Jandlova, T.; Bubenik, J.; Reinis, M. Induction of MHC class I molecule cell surface expression and epigenetic activation of antigen-processing machinery components in a murine model for human papilloma virus 16-associated tumours. Immunology 2008, 123, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Sun, T.; Li, Y.; Yang, W.; Wu, H.; Li, X.; Huang, Y.; Zhou, Y.; Du, Z. Histone deacetylase inhibition up-regulates MHC class I to facilitate cytotoxic T lymphocyte-mediated tumor cell killing in glioma cells. J. Cancer 2019, 10, 5638–5645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gameiro, S.R.; Malamas, A.S.; Tsang, K.Y.; Ferrone, S.; Hodge, J.W. Inhibitors of histone deacetylase 1 reverse the immune evasion phenotype to enhance T-cell mediated lysis of prostate and breast carcinoma cells. Oncotarget 2016, 7, 7390–7402. [Google Scholar] [CrossRef] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rank | Score | Name | Description |
|---|---|---|---|
| 1 | −98.48 | Linsitinib | IGF-1 inhibitor |
| 2 | −98.34 | AZD-8055 | mTOR inhibitor |
| 3 | −98.09 | Palbociclib | CDK inhibitor |
| 4 | −97.89 | Apicidin | HDAC inhibitor |
| 5 | −97.85 | HG-5-113-01 | Protein kinase inhibitor |
| 6 | −97.71 | KU-0063794 | mTOR inhibitor |
| 7 | −97.43 | Selumetinib | MEK inhibitor |
| 8 | −97.37 | HSP90-inhibitor | HSP inhibitor |
| 9 | −97.11 | PI-828 | PI3K inhibitor |
| 10 | −96.93 | PI-103 | mTOR inhibitor |
| 11 | −96.89 | ENMD-2076 | FLT3 inhibitor |
| 12 | −96.44 | PIK-90 | PI3K inhibitor |
| 13 | −96.33 | WYE-354 | mTOR inhibitor |
| 14 | −96.22 | Panobinostat | HDAC inhibitor |
| 15 | −96.16 | Dactolisib | mTOR inhibitor |
| 16 | −95.53 | Scriptaid | HDAC inhibitor |
| 17 | −95.17 | ISOX | HDAC inhibitor |
| 18 | −95.14 | GDC-0941 | PI3K inhibitor |
| 19 | −94.99 | Aminopurvalanol-a | Tyrosine kinase inhibitor |
| 20 | −94.94 | Lestaurtinib | FLT3 inhibitor |
| 21 | −94.73 | GSK-1059615 | PI3K inhibitor |
| 22 | −94.59 | Topotecan | Topoisomerase inhibitor |
| 23 | −94.36 | MK-2206 | AKT inhibitor |
| 24 | −94.24 | Staurosporine | PKC inhibitor |
| 25 | −94.15 | NCH-51 | HDAC inhibitor |
| 26 | −94.14 | PP-1 | SRC inhibitor |
| 27 | −93.94 | PP-30 | RAF inhibitor |
| 28 | −93.9 | Vorinostat | HDAC inhibitor |
| 29 | −93.81 | TG-101348 | FLT3 inhibitor |
| 30 | −93.42 | BMS-536924 | IGF-1 inhibitor |
| 31 | −93.41 | Fostamatinib | SYK inhibitor |
| 32 | −93.13 | OSI-027 | mTOR inhibitor |
| 33 | −93.05 | THM-I-94 | HDAC inhibitor |
| 34 | −92.53 | AKT-inhibitor-1-2 | AKT inhibitor |
| 35 | −92.39 | Camptothecin | Topoisomerase inhibitor |
| 36 | −92.19 | Temsirolimus | mTOR inhibitor |
| 37 | −91.99 | Idarubicin | Topoisomerase inhibitor |
| 38 | −91.6 | VER-155008 | HSP inhibitor |
| 39 | −91.54 | NU-7441 | DNA dependent protein kinase inhibitor |
| 40 | −90.96 | ALW-II-38-3 | Ephrin inhibitor |
| 41 | −90.77 | PHA-793887 | CDK inhibitor |
| 42 | −90.53 | TPCA-1 | IKK inhibitor |
| 43 | −90.49 | KU-0060648 | DNA dependent protein kinase inhibitor |
| 44 | −90.24 | Vemurafenib | RAF inhibitor |
| Rank | Score | Perturbational Class |
|---|---|---|
| 1 | −98.13 | PI3K inhibitor |
| 2 | −98.03 | mTOR inhibitor |
| 3 | −96.04 | IGF-1 inhibitor |
| 4 | −93.68 | JAK inhibitor |
| 5 | −91.78 | DNA dependent protein kinase inhibitor |
| 6 | −91.76 | HDAC inhibitor |
| 7 | −90.11 | FLT3 inhibitor |
| Rank | Target | Hits | Log p-Value (RSA) | |
|---|---|---|---|---|
| Parental SW1990 | CTL-Resistant SW1990 | |||
| 1 | HSP | 10/12 | −9.77 | −10.74 |
| 2 | HDAC | 13/22 | −9.24 | −9.44 |
| 3 | Proteasome | 6/9 | −6.03 | −6.31 |
| 4 | CDK | 9/16 | −9.59 | −5.44 |
| 5 | mTOR | 6/29 | −6.71 | −5.10 |
| 6 | Microtubule associated | 1/8 | −4.56 | −4.96 |
| 7 | Aurora kinase | 7/23 | −2.95 | −4.50 |
| 8 | Bcr-Abl | 3/12 | −3.23 | −4.45 |
| 9 | EGFR | 6/31 | −6.62 | −4.27 |
| 10 | c-Met | 4/18 | −2.01 | −3.46 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Looi, C.-K.; Gan, L.-L.; Sim, W.; Hii, L.-W.; Chung, F.F.-L.; Leong, C.-O.; Lim, W.-M.; Mai, C.-W. Histone Deacetylase Inhibitors Restore Cancer Cell Sensitivity towards T Lymphocytes Mediated Cytotoxicity in Pancreatic Cancer. Cancers 2022, 14, 3709. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14153709
Looi C-K, Gan L-L, Sim W, Hii L-W, Chung FF-L, Leong C-O, Lim W-M, Mai C-W. Histone Deacetylase Inhibitors Restore Cancer Cell Sensitivity towards T Lymphocytes Mediated Cytotoxicity in Pancreatic Cancer. Cancers. 2022; 14(15):3709. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14153709
Chicago/Turabian StyleLooi, Chin-King, Li-Lian Gan, Wynne Sim, Ling-Wei Hii, Felicia Fei-Lei Chung, Chee-Onn Leong, Wei-Meng Lim, and Chun-Wai Mai. 2022. "Histone Deacetylase Inhibitors Restore Cancer Cell Sensitivity towards T Lymphocytes Mediated Cytotoxicity in Pancreatic Cancer" Cancers 14, no. 15: 3709. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14153709