

Targeting KRAS in PDAC: A New Way to Cure It?

1
State Key Laboratory of Electroanalytical Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun 130022, China
2
School of Pharmacy, Changchun University of Chinese Medicine, Changchun 130021, China
3
Department of Chemistry and Physics, Stony Brook University, Stony Brook, NY 11794-3400, USA
*
Authors to whom correspondence should be addressed.
Cancers 2022, 14(20), 4982; https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14204982
Submission received: 25 August 2022
/
Revised: 29 September 2022
/
Accepted: 2 October 2022
/
Published: 11 October 2022
(This article belongs to the Special Issue Molecular Genetics of Pancreatic Cancer and Translational Challenges)
Abstract
:Simple Summary
Pancreatic cancer is one of the most intractable malignant tumors worldwide, and is known for its refractory and poor prognosis. Pancreatic ductal adenocarcinoma (PDAC) is the most common type of pancreatic cancer. KRAS is the most commonly mutated oncogene in PDAC. It has been considered the “untargetable” oncogene for decades until the emergence of G12C inhibitors, which put an end to this dilemma by covalent binding to the switch-II pocket of the G12C mutant protein. However, G12C inhibitors showed remarkable efficacy against non-small-cell lung cancer (NSCLC), while the G12C mutation is rare in PDAC. Based on the successful experience of G12C inhibitors, targeting KRAS G12D/V, which forms the majority of KRAS mutations in PDAC, is gradually being regarded as a potential therapy.
Abstract
Pancreatic cancer is one of the most intractable malignant tumors worldwide, and is known for its refractory nature and poor prognosis. The fatality rate of pancreatic cancer can reach over 90%. In pancreatic ductal carcinoma (PDAC), the most common subtype of pancreatic cancer, KRAS is the most predominant mutated gene (more than 80%). In recent decades, KRAS proteins have maintained the reputation of being “undruggable” due to their special molecular structures and biological characteristics, making therapy targeting downstream genes challenging. Fortunately, the heavy rampart formed by KRAS has been broken down in recent years by the advent of KRASG12C inhibitors; the covalent inhibitors bond to the switch-II pocket of the KRASG12C protein. The KRASG12C inhibitor sotorasib has been received by the FDA for the treatment of patients suffering from KRASG12C-driven cancers. Meanwhile, researchers have paid close attention to the development of inhibitors for other KRAS mutations. Due to the high incidence of PDAC, developing KRASG12D/V inhibitors has become the focus of attention. Here, we review the clinical status of PDAC and recent research progress in targeting KRASG12D/V and discuss the potential applications.
1. Introduction
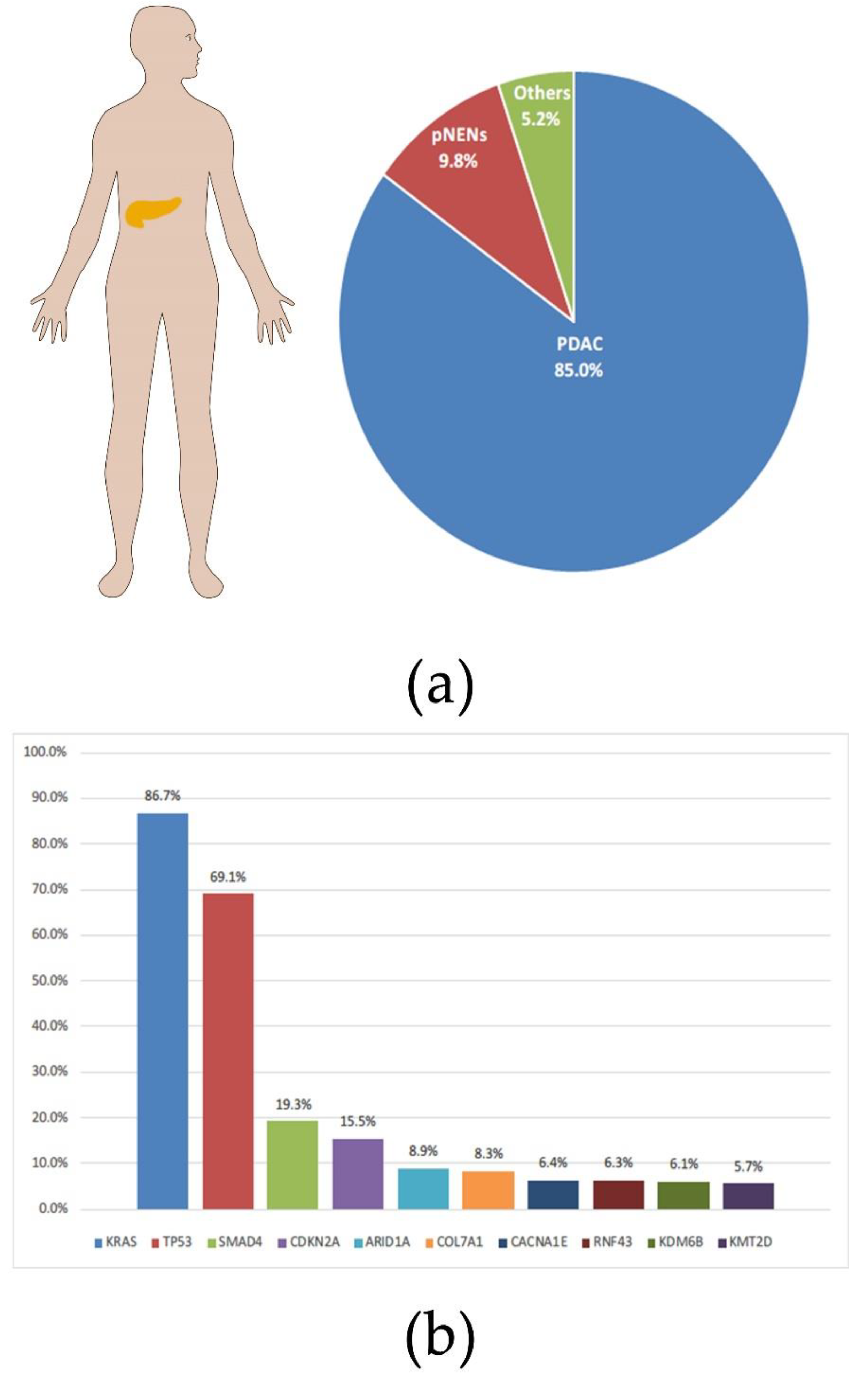
Pancreatic cancer is a malignant tumor with a high incidence and poor prognosis. It was estimated that more than 450,000 people died of pancreatic cancer in 2020, with a case fatality rate of over 90% [1]. To make matters worse, the incidence of pancreatic cancer continues to rise worldwide; according to statistics from the United States, the incidence of pancreatic cancer in both genders has reached the top ten [2]. Pancreatic ductal adenocarcinoma (PDAC) is the main histological subtype of pancreatic cancer, accounting for more than 80% (Figure 1a) [3]. The lack of screening and early metastasis are major reasons for the high mortality rate of pancreatic cancer [4]. The result of a comparative study showed that PDAC had four molecular subtypes correlating with histopathological characteristics, squamous, pancreatic progenitor, immunogenic, and aberrantly differentiated endocrine exocrine (ADEX) [5]. The oncogenic driver plays an important role in the proliferation and metastasis of tumors [6]. Therefore, targeted therapy for oncogenic drivers has considerable prospects in the treatment of PDAC. Most PDACs have been identified to contain the following four driver mutations: the Kirsten rat sarcoma (KRAS), the cyclin dependent kinase inhibitor 2 (CDKN2A), the tumor suppressor protein 53 (TP53), and the Small Mothers Against Decapentaplegic homolog 4 (SMAD4) (Figure 1b) [7]. It is worth noting that the incidence of the KRAS mutation in PDAC reaches an astonishing 86% [8]. There is no doubt that targeted KRAS therapy will be key to improving the poor prognosis of PDAC.
However, due to the lack of binding domains on its surface, multiple and complex downstream pathway branches and other reasons [9], the design of KRAS targeted drugs remained in a state of stagnation for a long time, until Shokat and colleagues discovered a molecular screening method for the KRASG12C mutant (glycine mutated into cysteine) in 2013 [10]. KRAS targeted drug development has just entered a state of rapid development. To date, a number of KRAS-targeted drugs and related pathway drugs have entered clinical and preclinical studies [11]. Unfortunately, all KRAS inhibitors that are clinically approved currently target KRASG12C, while the proportion of KRASG12C mutations in PDAC is extremely low (about 1%) [11,12]. However, since there are many similar structures among the KRAS mutant subtypes, it is possible to develop other KRAS inhibitors based on KRASG12C inhibitors. Researchers have developed a KRASG12D inhibitor named MRTX1133, whose molecular structure is based on MRTX849. MRTX1133 has achieved good results in both in vitro and in vivo models [13]. More recently, Wang’s group used a multidisciplinary approach to identify the “non-signaling open conformation” existing in KRAS–GTP hydrolysis as a potential target for the treatment of KRAS-dependent non-small-cell lung cancer and pancreatic cancer [14,15,16]. Thus, the development of pan-KRAS inhibitors is being pursued extensively for pancreatic cancer therapy due to drug resistance. This review will summarize the current clinical status of PDAC and the prospect of targeting KRAS for PDAC therapy.
2. Clinical Status of PDAC
2.1. Living Conditions of Patients with PDAC
Among all types of malignancies, the incidence of PDAC is at a fairly high level (2.6% ranked 14th in 2020), while the mortality rate is much higher (4.7% ranked 7th in 2020) [1]. In many countries, the trend in the incidence and mortality of PDAC has either remained essentially the same or has increased slightly, which may be due to obesity, diabetes, alcohol, and other factors [17]. The five-year survival of PDAC is not good either, statistically less than 10% [18,19]. This can be increased to 25% if resection is performed during an operable period [20]. However, due to the lack of screening methods, how to detect PDAC in time within the operable period has become a challenging issue. Currently, ultrasound and magnetic resonance imaging (MRI) detection are only applied to people with a family genetic history and related risk gene mutations [21]. Given this status quo, with the improving prognosis of other cancers, PDAC is becoming or has become the primary cause of cancer-related death in many countries [22]. By 2025, pancreatic cancer in Europe is expected to be the third leading cause of death among all cancers [23].
2.2. Clinical Treatment of PDAC
2.2.1. For Patients with Resectable Tumors
At present, surgical resection is still one of the best treatments for PDAC in the clinic, as long as the patient’s tumor is still in the resectable stage [24]. The surgery to remove the tumor varies depending on the exact location of the tumor in the pancreas. Approximately 80% of PDAC occurs at the head of the pancreas. Pancreaticoduodenectomy is commonly used for tumor resection at this location, which is safer and has a better prognosis than traditional open surgery assisted by laparoscopy or robotics [25,26,27,28,29]. For tumors located elsewhere in the pancreas, terminal pancreatectomy is usually performed [30].
In addition to surgery, adjuvant therapy with chemotherapy drugs has also been shown to benefit patients. For patients with resectable or borderline resectable PDAC, postoperative adjuvant chemotherapy for six months with fluorouracil and leucovorin improves median survival (19.7 months in 238 patients vs. 14.0 months in 235 patients, p < 0.001) [31]. The European Study Group for Pancreatic Cancer (ESPAC) conducted a series of trials that determined the benefits of gemcitabine alone and in combination with capecitabine for postoperative adjuvant therapy [32,33,34]. In contrast, the recurrence rate of PDAC after the above-mentioned adjuvant therapy is still very high. According to statistics, 70% of patients will relapse within two years [32,33,35]. FOLFIRINOX, a combination chemotherapy using fluorouracil, irinotecan, folate and oxaliplatin, has been shown to extend disease-free survival significantly compared with gemcitabine chemotherapy (12.8 months vs. 21.6 months), making it a reliable adjunct treatment for patients with PDAC after tumor resection [36]. However, due to the lack of further evidence on the extent of the benefits of adjuvant chemotherapy and on the overall survival of patients with PDAC [37], the use of adjuvant chemotherapy still requires further exploration. A number of novel adjuvant therapies are currently in clinical trials, such as the ESPAC5F trial, the NEOLAP-AIO-PAK0113 trial, and the SWOG 1505 trial [38].
2.2.2. For Patients Who Are Unsuitable for Surgery
Current screening for PDAC targets high-risk populations, and there is no effective screening for the general public due to the relatively low incidence in this population [39]. As a result, some patients have missed the resectable stage by the time PDAC is diagnosed. Coupled with the early onset of metastasis in PDAC, most patients have advanced-stage tumors at the time of diagnosis [40]. For PDAC patients with locally advanced or distant metastases, systemic chemotherapy is currently an effective therapy. The combination of gemcitabine and nab-paclitaxel, as well as FOLFIRINOX, is a commonly used chemotherapy regimen [37,41]. Although a small percentage of patients with advanced PDAC will have their tumors shrink to an operable size after chemotherapy, the vast majority will not be curable. The primary purpose of systemic chemotherapy for PDAC patients with locally advanced or distant metastases is to slow disease progression and prolong life. A retrospective analysis of clinical cases showed that patients who were younger or in better overall physical condition benefited more from FOLFIRINOX, as evidenced by an increase in overall survival [42,43]. Additionally, for patients who are not candidates for combination chemotherapy, gemcitabine monotherapy has improved their survival [44].
Biomarker-targeted therapy is a novel treatment modality, and some progress has been made in the treatment of PDAC. Mutations in BRCA1 and BRCA2 genes are often found in breast cancer patients, and approximately 5% of pancreatic cancer patients also have mutations in these genes [5,43]. Previous trials have shown the positive effects of PARP inhibitors in breast and ovarian cancer patients involving BRCA1 or BRCA2 mutations, with similar findings in pancreatic cancer patients [45,46]. Results from the phase III POLO trial showed that the use of olaparib prolonged progression-free survival in patients with metastatic pancreatic cancer of the germline BRCA mutation compared to the placebo group [47]. In 2019, the FDA approved olaparib, a PARP inhibitor, for the treatment of germline BRCA-mutated metastatic pancreatic adenocarcinoma [48].
Overall, although the clinical therapies for pancreatic cancer are being pursued, the gains have been relatively limited. On the other hand, research into novel therapies, such as targeted therapies and immunotherapies, has the potential to break through present therapeutic dilemmas. It is exciting to note that research on targeted drugs for KRAS, a member of the RAS gene family that has long been considered undruggable, has made tremendous breakthroughs in recent years and has benefited NSCLC patients [49,50,51]. It is clear that PDAC, as a tumor that also contains a high percentage of KRAS mutations, is likely to benefit from this.
3. KRAS Mutations in PDAC
RAS (rat sarcoma virus) genes constitute one of the most commonly mutated gene families in malignant tumors [52]. The RAS gene family includes three genes: KRAS, HRAS and NRAS. KRAS is the most common mutation type of the RAS gene, accounting for 80% of RAS gene-related malignancies. The KRAS gene encodes two splice variants using different exon 4 s, producing KRAS4A and KRAS4B. It has been experimentally demonstrated that both the isoforms are associated with tumor formation [11]. KRAS mutations have mainly been found in lung cancer (32%), PDAC (86%), and colon cancer (41%) [53,54,55]. The most common isoforms of KRAS in PDAC are KRASG12D (45%) and KRASG12V (35%) [56].
3.1. Molecular Mechanism of KRAS Mutations
From the perspective of function, the protein expressed by the KRAS gene is a purine nucleotide binding protein located on the cell membrane and has the activity of GTPase [57]. KRAS protein, as a binary switch of guanosine diphosphate (GDP)/guanosine triphosphate (GTP), controls important signal transduction from activated membrane receptors to intracellular molecules [58]. In the inactive state, KRAS protein binds to GDP [59]. When stimulated by relevant signal molecules (such as epidermal growth factor receptor EGFR), the binding ability of KRAS protein to GDP is weakened. GTP takes the place of GDP to bind to the RAS protein, and the KRAS protein is, therefore, activated to bind with downstream signal molecules as monomers or dimers for signal transduction. Then, with the effect of GTP-activated proteins (GAPs), the GTPase activity of KRAS is significantly increased, and GTP combined with KRAS is hydrolyzed into GDP, restoring KRAS to its inactivated state [60]. However, in tumor cells, KRAS gene mutation leads to the loss of GTPase activity in the KRAS protein, which makes it unable to hydrolyze GTP into GDP after binding with GTP, entering the inactivation state; this finally leads to the continuous activation of the downstream pathway, resulting in malignant proliferation, metastasis and anti-apoptosis of tumor cells [60,61]. Intrinsic GTPase and GTP-GDP exchange efficiency can differ between several mutant types of KRAS. For example, KRASG13 mutation is more sensitive to NF1-GAP-mediated hydrolytic activity, while KRASG12 and KRASQ61 mutations are insensitive to it [62]. Another example is that the KRASG12C mutant type has similar intrinsic GTPase activity to the wild type, whereas other KRAS mutants have lower intrinsic GTPase activity than the wild type. [20]. In fact, the KRASG12C inhibitor was designed with this characteristic in mind [10].
It is also worth mentioning that the oncogenicity and drug resistance of mutant KRAS is related to its dimerization with wild-type KRAS [63]. The exact relationship between them needs to be studied in depth.
3.2. Progress of PDAC with KRAS Mutations
The link between KRAS mutations and PDAC prognosis has been the focus of research, and several recent studies have further illustrated their relationship. Itonaga and colleagues analyzed the personal information of 110PDAC patients who underwent histological diagnosis from 2017 to 2019. All of these patients underwent first-line therapy with gemcitabine and nab-paclitaxel. Patients were analyzed for the presence of KRAS mutations and grouped through the quenching probe method. Then, progression-free survival (PFS) and overall survival (OS) were compared between the two groups. The study showed that patients with wild-type KRAS genes had much longer PFS and OS than patients with KRAS mutations (6.9/5.3 months (p = 0.044) vs. 19.9/11.8 months (p = 0.037), respectively) [64]. In patients with surgically resectable tumors, KRAS gene mutations can also affect their prognosis after undergoing surgery. The analysis of patient data collected from Memorial Sloan Kettering (MSK) showed that patients with KRAS mutations had a worse prognosis after the surgical removal of the tumor [65].
With the development of next-generation sequencing (NGS), it has become possible to measure the mutation frequency of the alleles in tumor samples [66,67]. As PDAC tumors are highly heterogeneous [68], the proportion of malignant cells in tumors may vary greatly from patient to patient. Nauheim and colleagues studied microdissection samples from 144 PDAC patients who had undergone classic pancreaticoduodenectomy (PD) (classic Whipple) or pylorus-preserving PD (PPPD). KRAS mutations were present in 121 patients (84%). Studies show that patients with a high frequency of KRAS mutations (more than or equal to 20%, n = 29) have larger tumors, higher postoperative distal recurrence rates, and shorter disease-free survival after surgery than those with a low frequency of KRAS mutations (less than 20%, n = 29) [69]. Another study found that PDAC patients who received FOLFIRINOX chemotherapy followed by the surgical resection of tumors had new KRAS mutations in their cell-free DNA compared to those before treatment [70]. The relationship between increased KRAS mutations and chemotherapy, as well as the surgical resection of tumors, still warrants further exploration.
Research has progressed on the specific molecular mechanisms by which KRAS gene mutations worsen the prognosis of PDAC patients. It has been shown that KRASG12D, the most predominant KRAS mutant phenotype in PDAC, induces the overexpression of SUMO-activating enzyme subunit 1 (SAE1), which can lead to heterogeneous nuclear ribonucleoprotein A1 (hnRNPA1) being SUMOylated. SUMOylated hnRNPA1 is packaged by extracellular vesicles (EVs) and transported to human lymphatic endothelial cells (HLECs), ultimately promoting lymphatic vessel proliferation and lymph node metastasis [11,61].
4. KRAS Inhibitors for PDAC
4.1. KRASG12C Inhibitors
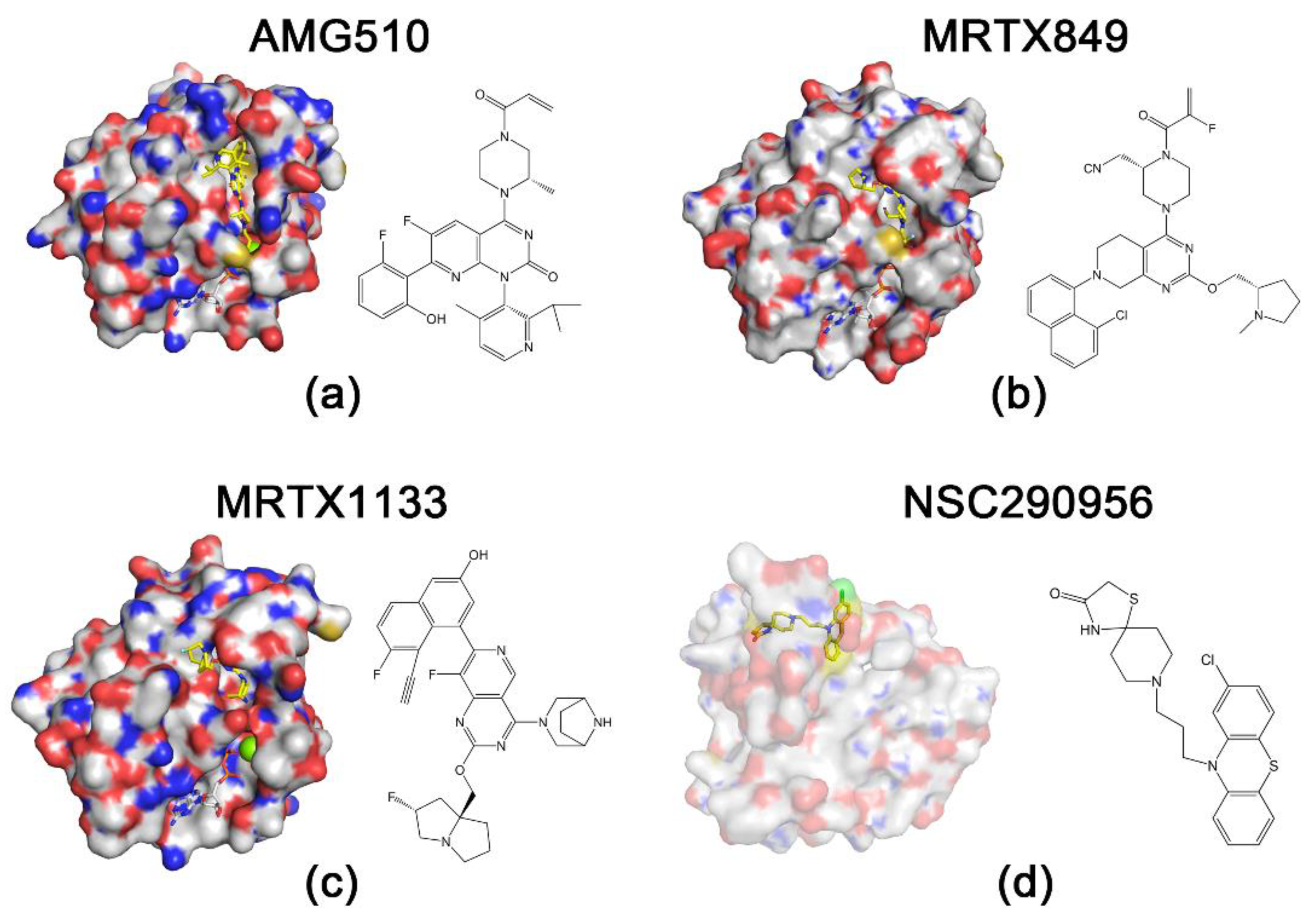
KRASG12C inhibitors have shown excellent results in the treatment of non-small cell lung cancer, and studies on their efficacy for other solid tumors are still advancing [71]. A phase 1 trial (NCT03600883) evaluating the various aspects of sotorasib (AMG510) performance showed that sotorasib has good antitumor activity against solid tumors containing KRASG12C mutations [49] (Figure 2). Another KRASG12C inhibitor, MRTX849, validated its antitumor activity against KRASG12C mutation-containing tumors in a mouse xenograft model [72]. However, none of the KRASG12C inhibitors have been approved by the FDA as a treatment for pancreatic cancer. Although the frequency of KRASG12C mutations in PDAC patients is abnormally high in some regions, for example, more than 60% in Japan [73], the frequency of KRASG12C mutations in PDAC patients worldwide remains quite low, which leads to a limited prospect for the clinical treatment of PDAC using KRASG12C inhibitors [11,74].
4.2. KRASG12D Inhibitors
4.2.1. MRTX1133
While sotorasib has been approved by the FDA for the treatment of KRASG12C mutation-containing NSCLC [51], the development of other KRAS mutation inhibitors has come to a standstill. One of the main reasons hindering the development of KRASG12D inhibitors, which has been mentioned previously, is the low rate of intrinsic GTP hydrolysis in the KRASG12D mutant [60]. KRAS mutations lead to a decrease in intrinsic GTPase activity, which further decreases the rate of GTP hydrolysis and ultimately continues to activate downstream pathways and produce carcinogenesis [61]. The intrinsic hydrolysis rate of the KRASG12C mutation is equivalent to approximately 70% of that of the wild-type KRAS, while the intrinsic hydrolysis rate of the KRASG12D mutation is only less than 30% [60]. This disadvantage poses a challenge for the design of KRASG12D inhibitors. It is also challenging to determine whether the inhibitor has sufficient affinity for 12-aspartate involved in the KRASG12D mutant to avoid binding to wild-type KRAS. In February 2022, Mirati Therapeutics announced a selective non-covalent inhibitor, MRTX1133 of KRASG12D (Figure 2). The structure of MRTX1133 is based on MRTX849, a KRASG12C inhibitor developed by Mirati Therapeutics. The investigators introduced a salt bridge between the inhibitor and 12-aspartate to enhance the reversible affinity for KRASG12D. This strengthened the selectivity of the inhibitor for KRASG12D through a series of modifications to avoid binding to wild-type KRAS. Compared to several KRASG12C inhibitors whose reversible affinity for the target is in the micromolar range [50,75,76], MRTX1133 has a picomolar range of reversible affinity for KRASG12D. Although MTRX1133 binds weakly to KRAS proteins in the GDP state, it also has the ability to bind to KRAS proteins in the GTP state [77]. This will lead to new ideas for combination therapy studies of KRAS inhibitors. In a previous study, MRTX1133 achieved excellent results in a mouse xenograft model of pancreatic cancer, with a 94% reduction in tumor volume at 3 mg/kg BID (IP) compared to the control group [13].
4.2.2. Peptide Nucleic Acids (PNAs)
Peptide Nucleic Acids (PNAs) are synthetic nucleotide analogs whose molecular structures are very similar to those of DNA and RNA [78]. PNAs have good hybridization properties and can specifically bind to complementary DNA or RNA, distinguishing similar sequences even at the level of single base mismatches [79,80]. Meanwhile, PNAs can bind specifically to the mRNA of the target gene and inhibit its translation process [81]. Moreover, PNAs have stable chemical structures and are not easily degraded by nucleases or proteases. Based on the above characteristics, treatment using PNAs has great potential to become a new tool in the fight against malignant tumors. In a recent study, several PNAs were designed for the KRASG12D mutated gene fragment and tested in the human metastatic pancreatic adenocarcinoma cell line AsPC-1 containing the KRASG12D mutation. The results showed that PNAs significantly inhibited tumor cell activity and reduced the expression of the KRASG12D mutated gene [82]. The successful inhibition of the KRASG12D mutant gene by PNAs at the cellular level raises the possibility for subsequent animal experiments.
4.3. Pan-RAS Inhibitors
Compared to specific inhibitors, pan-RAS inhibitors have broader applicability and can provide treatment for patients with different types of KRAS mutations. Additionally, pan-RAS inhibitors can avoid drug resistance caused by the compensatory activation of wild-type KRAS. Although this class of inhibitors suffers from high toxicity and off-target inhibition, it still has great research potential [83]. Several pan-RAS inhibitors have been shown to have good specificity for RAS mutations, and animal models have tolerated these inhibitors to an appreciable degree [84,85].
Nassar et al. revealed that there are three distinct but equally populated conformations in the process of HRAS-GTP hydrolysis and nucleotide exchange, one of which is the “non-signaling open conformation” state [86]. Due to the same hydrolysis process and the structural homology, the state also appears in KRAS [87]. Using nuclear magnetic resonance (NMR) analysis, the researchers uncovered that the HRASG60A-GppNp complex adopts an “open conformation” at the switch 1 region and abolishes the biological activity of HRAS [86,88]. Recent studies have indicated extremely open switch 1 conformations of KRAS [89]. This implies that the “open conformation” may be a convergent point for survival signaling in KRAS-driven cancer, and agents locking this “open conformation” may theoretically block KRAS-dependent signaling. Most recently, Jin Wang’s group used a Specificity Affinity (SPA)-based virtual screening strategy to develop small-molecule inhibitors that stabilize the “open conformation”. This process led to the selection of three hits (NSC290956, NSC48693, and NSC48160) from 2000 compounds by individually docking compounds in the National Cancer Institute diversity compound sets to the “open non-signaling intermediate conformation” of RAS [89]. Of these, NSC290956 (also termed Spiclomazine or APY606) manifested potent efficacy against the proliferation of KRAS-driven pancreatic cancer cell lines CFPAC-1 (KRASG12V), MIA PaCa-2 (KRASG12C), Capan-1 (KRASG12V), SW1990 (KRASG12T) and BxPC-3 (wild-type KRAS) and pancreatic cancer cells but showed much less toxicity towards human normal cells [15,90,91]. NSC48160 inhibited the survival and growth of KRAS-driven pancreatic cancer cells CPFAC-1 (KRASG12V) and BxPC-3 (wild-type KRAS) by using MTT and colony-forming assays [16]. Liu et al. found that NSC48160 selectively induced apoptosis in pancreatic cancer MIA PaCa-2 (KRASG12C) cells as compared to human normal HEK-293 and HL-7702 cells [92]. Liu et al. further found that the inhibitory effects of small-molecule NSC48693 on KRAS-driven cancer cells were greater than NSC48160 for CFPAC-1(KRASG12V), MIA PaCa-2 (KRASG12C) and BxPC-3 (wild-type KRAS) cells [93]. Interestingly, the cytotoxic effect of NSC48693 on the human normal cell line (HL-7702) was lower than that on cancer cell lines (CFPAC-1, MIA PaCa-2 and BxPC-3). Together, this research provides functional insights into the “open conformation” and validates three hits acting as pan-KRAS inhibitors to induce the apoptosis of pancreatic cancer cells.
5. Drug Resistance Mechanisms of KRAS Inhibitors
To date, MRTX1133 has been tested in preclinical studies as a KRASG12D inhibitor but has not entered clinical trials. Therefore, the analysis of the resistance mechanism of KRASG12D inhibitors needs to be carried out on the resistance status of KRASG12C inhibition that has been applied in the clinic (Figure 3). Studies have been conducted to analyze tissue or fluid samples from 43 patients treated with the KRASG12C inhibitor sotorasib [94]. A comparison of pre-treatment and post-treatment samples revealed that 27 patients developed multiple gene mutations after treatment, including KRAS, NRAS, BRAF, EGFR, FGFR2, and MYC. As mutations occurring in a single patient are insufficient to suggest an association with drug resistance, the investigators constructed corresponding cellular models and xenograft models for further exploration. Their studies showed that the induction of KRASG12V, NRASQ61K or MRASQ71R (a small GTPase involved in regulating the dimerization and activation of CRAF, a signaling molecule of the MAPK-ERK pathway downstream of KRAS [95]) mutations in tumor cell lines containing KRASG12C mutations reduced the inhibitory effect of KRASG12C inhibitor sotorasib on downstream signaling and failed to significantly alter the level of endogenous KRAS activation. As PDAC is highly heterogeneous, the further application of KRASG12D inhibitors to PDAC has a high potential to increase the frequency of the mutated genes mentioned above.
MAPK-ERK and PI3K-AKT-mTOR pathways are two important RAS downstream signaling pathways, and their signaling is regulated by RAS proteins [96]. Notably, the PI3K-AKT-mTOR pathway is not only regulated by RAS proteins, but also by various signaling molecules, including PDK-1 and IGF1 [97]. It has been demonstrated that after silencing the KRAS gene in pancreatic cancer cell lines containing KRAS mutations using short hairpin RNAs (shRNAs), some of the cell lines still survive, exhibiting dependence on PI3K [98]. Other studies have identified that YAP1 overexpression regulated by PI3K is a way for tumor cells to evade KRAS inhibition [99,100,101]. However, the relationship between PI3K and tumor cells evading KRAS inhibition still requires further study. Research on the relationship between the MAPK-ERK pathway and drug resistance mechanisms also continues, but previous clinical attempts to target MEK have not yielded encouraging results [102,103,104].
The switch-II pocket is located next to the cys12 residue of the KRASG12C mutant protein, which is also the binding site for a series of KRASG12C inhibitors represented by sotorasib [50,105]. If the structure of this pocket is changed, the inhibitors that target it are likely to be ineffective. In a phase 1 study of MRTX849, a 67-year-old patient with metastatic KRASG12C-mutated NSCLC was found to have tumor shrinkage of approximately 32% after treatment with MRTX849, but the tumor progressed again after four months. A KRAS mutation, KRASY96D, was detected in this patient [106]. After intermolecular interaction simulations, it was found that the amino acid substitution at the Y96 site broke the hydrogen bond between it and sotorasib and MRTX849, leading to the breakdown of the inhibitor. This unique KRAS secondary alteration may be a common weakness of KRASG12C inhibitors currently applied in the clinic.
6. Strategies to Circumvent Drug Resistance
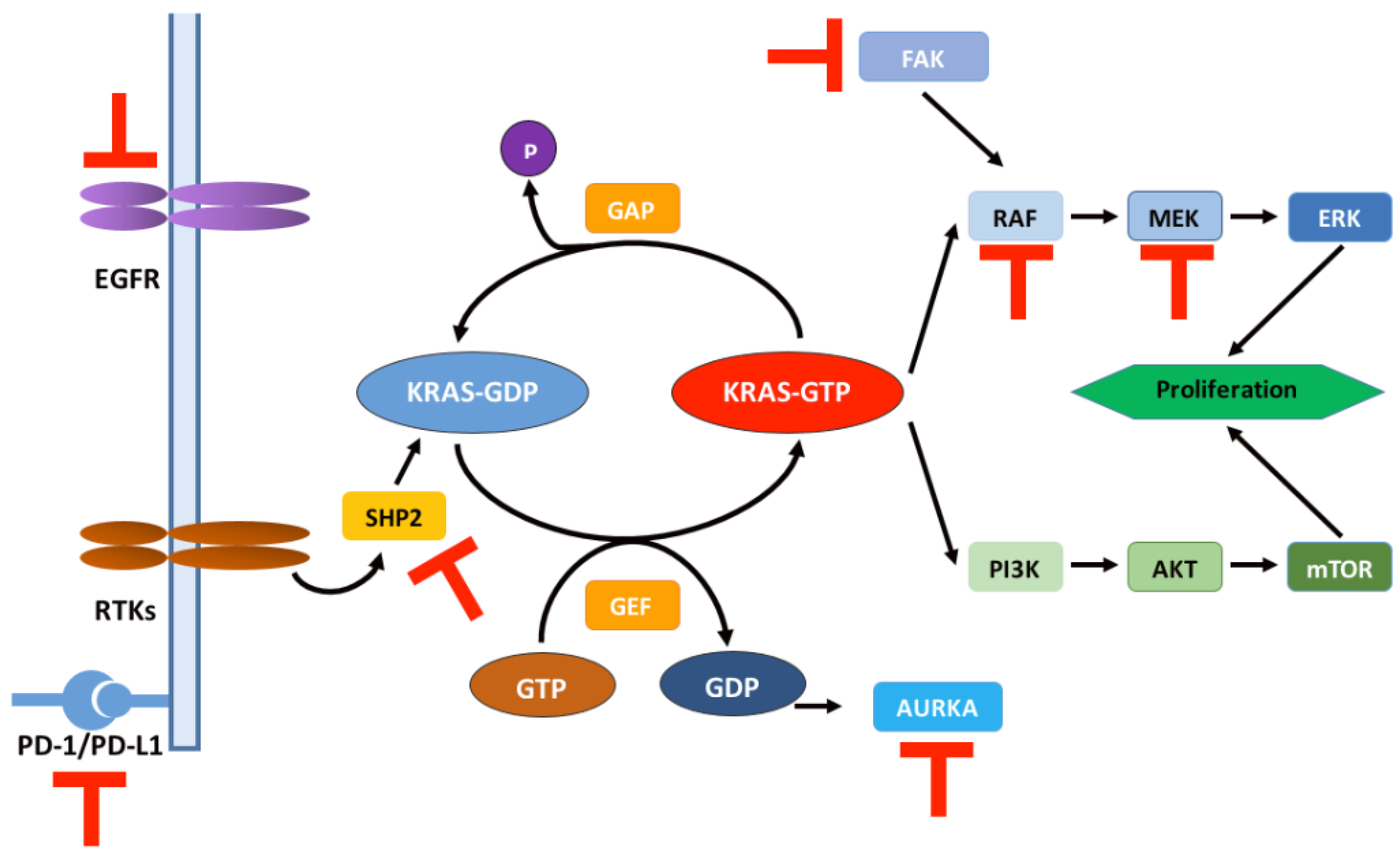
Combination therapy is currently an effective way to overcome drug resistance, and the exploration of combination therapy with KRAS inhibitors is underway. Several clinical trials of combination therapy for KRASG12C inhibitors have been conducted (Table 1). The combination of KRASG12C inhibitors with RAS upstream pathway inhibitors is a hot research topic nowadays, among which the combination with SHP2 inhibitors is the most popular. SHP2 is a non-receptor protein tyrosine phosphatase encoded by the PTPN11 gene, which can be activated by dephosphorylating KRAS proteins through binding to them. KRAS is phosphorylated via Src kinase on a conserved tyrosine at position 32 of the switch I region. This phosphorylation inhibits the binding of the effector Raf while promoting the involvement of GAPs and the hydrolysis of GTP [107]. SHP2 inhibition not only rescued RTK-driven acquired resistance to MEK inhibition but also had inhibitory effects on preclinical tumor models containing mutations in RAS-related pathways [108,109,110]. Moreover, the combination of SHP2 inhibitors with KRASG12C inhibitors has been shown to overcome KRASG12C inhibitor resistance and direct a better tumor microenvironment in PDAC models [111]. Related drug combination clinical trials are also in progress [112].
The combination of KRAS inhibitors with downstream pathway inhibitors is less commonly reported, and the combined targeting of the MAPK-ERK and PI3K-AKT-mTOR pathways was ineffective in pancreatic cancer models. However, this has been changed with the addition of histone deacetylase (HDAC, a kind of epigenetic modifier) to the above combination targeting regimen [113]. In fact, the inhibitory effect of HDAC was dominant, and the combined targeting of the MAPK-ERK and PI3K-AKT-mTOR pathways enhanced this inhibitory effect [114,115,116].
Additionally, the combined targeting of KRASG12C with cell cycle checkpoints or immune checkpoints is promising [50,72,117]; the triple combined inhibition of KRASG12C/SHP2/PD-L1 leads to severe tumor regression in PDAC mouse models [108]. Recent studies have shown that the nuclear export protein exportin 1 (XPO1) relieves tumor cells from resistance to KRASG12C inhibitors [118]. This effect has been demonstrated in a mouse xenograft model. XPO1 has the function of transporting protein cargo from the nucleus to the cell, thus maintaining cellular homeostasis [119]. Additionally, KRAS mutant NSCLC cells are dependent on this mode of transport [120]. This combination therapy may also be applicable to the treatment of patients with PDAC containing the KRASG12C mutation.
7. Conclusions and Prospect
Pancreatic cancer is known for its high mortality rate and short survival period. Although some progress has been made in recent years in terms of early diagnosis, perioperative management and systemic treatment, the prognosis of patients has not improved significantly [30]. As the most common type of pancreatic cancer, PDAC has become the focus of clinical and preclinical studies. Since KRAS is one of the most frequently occurring oncogenic mutations in PDAC, the introduction of its inhibitors has opened a new avenue for the clinical treatment of PDAC. Nevertheless, KRASG12C mutations account for a very small proportion of KRAS mutations in PDAC, and KRASG12C inhibitors currently used in clinical practice have very limited efficacy in PDAC patients. Fortunately, KRASG12D inhibitors have been developed and put into preclinical trials, while the exploration of KRASG12V inhibitors is also in progress [121]. It is believed that in the near future, KRASG12D/V inhibitors will provide a new perspective on curing PDAC.
Author Contributions
Conceptualization, Z.L. and J.W.; writing—original draft preparation, Q.H.; writing—review and editing, Z.L. and J.W.; visualization, Q.H.; supervision, J.W.; project administration, J.W.; funding acquisition, Z.L. All authors have read and agreed to the published version of the manuscript.
Funding
Q.H. and Z.L. were funded by the National Natural Science Foundation of China (81573448 and 21721003), the Ministry of Science and Technology of China (2016YFA0203200 and 2013YQ170585), and the Scientific Instrument Developing Project of the Chinese Academy of Sciences (YJKYYQ20180038).
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
The authors would like to acknowledge the American Association for Cancer Research and its financial and material support in the development of the AACR Project GENIE registry, as well as members of the consortium for their commitment to data sharing. Interpretations are the responsibility of study authors.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the collection and analysis of data, in the writing of the manuscript, or in the decision to publish the results.
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Ferlay, J.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef]
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer Statistics, 2021. CA Cancer J. Clin. 2021, 71, 7–33. [Google Scholar] [CrossRef]
- Haeberle, L.; Esposito, I. Pathology of pancreatic cancer. Transl. Gastroenterol. Hepatol. 2019, 4, 50. [Google Scholar] [CrossRef]
- Chiorean, E.G.; Coveler, A.L. Pancreatic cancer: Optimizing treatment options, new, and emerging targeted therapies. Drug Des. Devel. Ther. 2015, 9, 3529–3545. [Google Scholar] [CrossRef] [Green Version]
- Bailey, P.; Chang, D.K.; Nones, K.; Johns, A.L.; Patch, A.M.; Gingras, M.C.; Miller, D.K.; Christ, A.N.; Bruxner, T.J.; Quinn, M.C.; et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016, 531, 47–52. [Google Scholar] [CrossRef]
- Sharma, S.V.; Bell, D.W.; Settleman, J.; Haber, D.A. Epidermal growth factor receptor mutations in lung cancer. Nat. Rev. Cancer 2007, 7, 169–181. [Google Scholar] [CrossRef]
- Kamisawa, T.; Wood, L.D.; Itoi, T.; Takaori, K. Pancreatic cancer. Lancet 2016, 388, 73–85. [Google Scholar] [CrossRef]
- Consortium, A.P.G. AACR Project GENIE: Powering Precision Medicine through an International Consortium. Cancer Discov. 2017, 7, 818–831. [Google Scholar] [CrossRef] [Green Version]
- Cox, A.D.; Fesik, S.W.; Kimmelman, A.C.; Luo, J.; Der, C.J. Drugging the undruggable RAS: Mission possible? Nat. Rev. Drug Discov. 2014, 13, 828–851. [Google Scholar] [CrossRef] [Green Version]
- Ostrem, J.M.; Peters, U.; Sos, M.L.; Wells, J.A.; Shokat, K.M. K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013, 503, 548–551. [Google Scholar] [CrossRef]
- Moore, A.R.; Rosenberg, S.C.; McCormick, F.; Malek, S. Author Correction: RAS-targeted therapies: Is the undruggable drugged? Nat. Rev. Drug Discov. 2020, 19, 902. [Google Scholar] [CrossRef] [PubMed]
- Turpin, A.; Neuzillet, C.; Colle, E.; Dusetti, N.; Nicolle, R.; Cros, J.; de Mestier, L.; Bachet, J.B.; Hammel, P. Therapeutic advances in metastatic pancreatic cancer: A focus on targeted therapies. Ther. Adv. Med. Oncol. 2022, 14, 17588359221118019. [Google Scholar] [PubMed]
- Wang, X.; Allen, S.; Blake, J.F.; Bowcut, V.; Briere, D.M.; Calinisan, A.; Dahlke, J.R.; Fell, J.B.; Fischer, J.P.; Gunn, R.J.; et al. Identification of MRTX1133, a Noncovalent, Potent, and Selective KRAS(G12D) Inhibitor. J. Med. Chem. 2022, 65, 3123–3133. [Google Scholar] [CrossRef]
- Zhang, J.; Liu, Z.; Zhao, W.; Yin, X.; Zheng, X.; Liu, C.; Wang, J.; Wang, E. Discovery of Small Molecule NSC290956 as a Therapeutic Agent for KRas Mutant Non-Small-Cell Lung Cancer. Front. Pharmacol. 2021, 12, 797821. [Google Scholar] [CrossRef] [PubMed]
- Guo, N.; Liu, Z.; Zhao, W.; Wang, E.; Wang, J. Small Molecule APY606 Displays Extensive Antitumor Activity in Pancreatic Cancer via Impairing Ras-MAPK Signaling. PLoS ONE 2016, 11, e0155874. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Liu, Z.; Zhao, W.; Zheng, X.; Wang, J.; Wang, E. A small-molecule induces apoptosis and suppresses metastasis in pancreatic cancer cells. Eur. J. Pharm. Sci. 2013, 48, 658–667. [Google Scholar] [CrossRef]
- Arnold, M.; Abnet, C.C.; Neale, R.E.; Vignat, J.; Giovannucci, E.L.; McGlynn, K.A.; Bray, F. Global Burden of 5 Major Types of Gastrointestinal Cancer. Gastroenterology 2020, 159, 335–349.e15. [Google Scholar] [CrossRef]
- Chhoda, A.; Lu, L.; Clerkin, B.M.; Risch, H.; Farrell, J.J. Current Approaches to Pancreatic Cancer Screening. Am. J. Pathol. 2019, 189, 22–35. [Google Scholar] [CrossRef] [Green Version]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2019. CA Cancer J. Clin. 2019, 69, 7–34. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.; Jansen, L.; Balavarca, Y.; Babaei, M.; van der Geest, L.; Lemmens, V.; Van Eycken, L.; De Schutter, H.; Johannesen, T.B.; Primic-Zakelj, M.; et al. Stratified survival of resected and overall pancreatic cancer patients in Europe and the USA in the early twenty-first century: A large, international population-based study. BMC Med. 2018, 16, 125. [Google Scholar] [CrossRef]
- Kleeff, J.; Korc, M.; Apte, M.; La Vecchia, C.; Johnson, C.D.; Biankin, A.V.; Neale, R.E.; Tempero, M.; Tuveson, D.A.; Hruban, R.H.; et al. Pancreatic cancer. Nat. Rev. Dis. Prim. 2016, 2, 16022. [Google Scholar] [CrossRef] [PubMed]
- Pourshams, A.; Sepanlou, S.G.; Ikuta, K.S.; Bisignano, C.; Safiri, S.; Roshandel, G.; Sharif, M.; Khatibian, M.; Fitzmaurice, C.; Nixon, M.R.; et al. The global, regional, and national burden of pancreatic cancer and its attributable risk factors in 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol. Hepatol. 2019, 4, 934–947. [Google Scholar] [CrossRef] [Green Version]
- Ferlay, J.; Partensky, C.; Bray, F. More deaths from pancreatic cancer than breast cancer in the EU by 2017. Acta Oncol. 2016, 55, 1158–1160. [Google Scholar] [CrossRef] [PubMed]
- Millikan, K.W.; Deziel, D.J.; Silverstein, J.C.; Kanjo, T.M.; Christein, J.D.; Doolas, A.; Prinz, R.A. Prognostic factors associated with resectable adenocarcinoma of the head of the pancreas. Am. Surg. 1999, 65, 618–623. [Google Scholar] [PubMed]
- Slack, J. Developmental biology of the pancreas. Development 1995, 121, 1569–1580. [Google Scholar] [CrossRef] [PubMed]
- Kendrick, M.L.; Cusati, D. Total laparoscopic pancreaticoduodenectomy: Feasibility and outcome in an early experience. Arch. Surg. 2010, 145, 19–23. [Google Scholar] [CrossRef] [Green Version]
- Palanivelu, C.; Jani, K.; Senthilnathan, P.; Parthasarathi, R.; Rajapandian, S.; Madhankumar, M.V. Laparoscopic pancreaticoduodenectomy: Technique and outcomes. J. Am. Coll. Surg. 2007, 205, 222–230. [Google Scholar] [CrossRef]
- Zureikat, A.H.; Breaux, J.A.; Steel, J.L.; Hughes, S.J. Can laparoscopic pancreaticoduodenectomy be safely implemented? J. Gastrointest. Surg. 2011, 15, 1151–1157. [Google Scholar] [CrossRef]
- Croome, K.P.; Farnell, M.B.; Que, F.G.; Reid-Lombardo, K.M.; Truty, M.J.; Nagorney, D.M.; Kendrick, M.L. Total laparoscopic pancreaticoduodenectomy for pancreatic ductal adenocarcinoma: Oncologic advantages over open approaches? Ann. Surg. 2014, 260, 633–638, discussion 638–640. [Google Scholar] [CrossRef]
- Mizrahi, J.D.; Surana, R.; Valle, J.W.; Shroff, R.T. Pancreatic cancer. Lancet 2020, 395, 2008–2020. [Google Scholar] [CrossRef]
- Neoptolemos, J.P.; Dunn, J.A.; Stocken, D.D.; Almond, J.; Link, K.; Beger, H.; Bassi, C.; Falconi, M.; Pederzoli, P.; Dervenis, C.; et al. Adjuvant chemoradiotherapy and chemotherapy in resectable pancreatic cancer: A randomised controlled trial. Lancet 2001, 358, 1576–1585. [Google Scholar] [CrossRef]
- Oettle, H.; Post, S.; Neuhaus, P.; Gellert, K.; Langrehr, J.; Ridwelski, K.; Schramm, H.; Fahlke, J.; Zuelke, C.; Burkart, C.; et al. Adjuvant chemotherapy with gemcitabine vs. observation in patients undergoing curative-intent resection of pancreatic cancer: A randomized controlled trial. JAMA 2007, 297, 267–277. [Google Scholar] [CrossRef] [PubMed]
- Neoptolemos, J.P.; Stocken, D.D.; Bassi, C.; Ghaneh, P.; Cunningham, D.; Goldstein, D.; Padbury, R.; Moore, M.J.; Gallinger, S.; Mariette, C.; et al. Adjuvant chemotherapy with fluorouracil plus folinic acid vs. gemcitabine following pancreatic cancer resection: A randomized controlled trial. JAMA 2010, 304, 1073–1081. [Google Scholar] [CrossRef]
- Neoptolemos, J.P.; Palmer, D.H.; Ghaneh, P.; Psarelli, E.E.; Valle, J.W.; Halloran, C.M.; Faluyi, O.; O’Reilly, D.A.; Cunningham, D.; Wadsley, J.; et al. Comparison of adjuvant gemcitabine and capecitabine with gemcitabine monotherapy in patients with resected pancreatic cancer (ESPAC-4): A multicentre, open-label, randomised, phase 3 trial. Lancet 2017, 389, 1011–1024. [Google Scholar] [CrossRef] [Green Version]
- Sinn, M.; Bahra, M.; Liersch, T.; Gellert, K.; Messmann, H.; Bechstein, W.; Waldschmidt, D.; Jacobasch, L.; Wilhelm, M.; Rau, B.M.; et al. CONKO-005: Adjuvant Chemotherapy With Gemcitabine Plus Erlotinib Versus Gemcitabine Alone in Patients After R0 Resection of Pancreatic Cancer: A Multicenter Randomized Phase III Trial. J. Clin. Oncol. 2017, 35, 3330–3337. [Google Scholar] [CrossRef] [PubMed]
- Conroy, T.; Hammel, P.; Hebbar, M.; Ben Abdelghani, M.; Wei, A.C.; Raoul, J.L.; Chone, L.; Francois, E.; Artru, P.; Biagi, J.J.; et al. FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N. Engl. J. Med. 2018, 379, 2395–2406. [Google Scholar] [CrossRef]
- Philip, P.A.; Lacy, J.; Portales, F.; Sobrero, A.; Pazo-Cid, R.; Manzano Mozo, J.L.; Kim, E.J.; Dowden, S.; Zakari, A.; Borg, C.; et al. Nab-paclitaxel plus gemcitabine in patients with locally advanced pancreatic cancer (LAPACT): A multicentre, open-label phase 2 study. Lancet Gastroenterol. Hepatol. 2020, 5, 285–294. [Google Scholar] [CrossRef]
- Lo, W.; Zureikat, A. Neoadjuvant therapy in pancreatic cancer: A review and update on recent trials. Curr. Opin. Gastroenterol. 2022, 38, 521–531. [Google Scholar] [CrossRef]
- Screening for testicular cancer:, U.S.Preventive Services Task Force reaffirmation recommendation statement. Ann. Intern. Med. 2011, 154, 483–486. [CrossRef] [Green Version]
- Zhao, Y.; Wang, Y.; Chen, W.; Bai, S.; Peng, W.; Zheng, M.; Yang, Y.; Cheng, B.; Luan, Z. Targeted intervention of eIF4A1 inhibits EMT and metastasis of pancreatic cancer cells via c-MYC/miR-9 signaling. Cancer Cell Int. 2021, 21, 670. [Google Scholar] [CrossRef]
- Marthey, L.; Sa-Cunha, A.; Blanc, J.F.; Gauthier, M.; Cueff, A.; Francois, E.; Trouilloud, I.; Malka, D.; Bachet, J.B.; Coriat, R.; et al. FOLFIRINOX for locally advanced pancreatic adenocarcinoma: Results of an AGEO multicenter prospective observational cohort. Ann. Surg. Oncol. 2015, 22, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Chan, K.K.W.; Guo, H.; Cheng, S.; Beca, J.M.; Redmond-Misner, R.; Isaranuwatchai, W.; Qiao, L.; Earle, C.; Berry, S.R.; Biagi, J.J.; et al. Real-world outcomes of FOLFIRINOX vs. gemcitabine and nab-paclitaxel in advanced pancreatic cancer: A population-based propensity score-weighted analysis. Cancer Med. 2020, 9, 160–169. [Google Scholar] [CrossRef] [PubMed]
- Holter, S.; Borgida, A.; Dodd, A.; Grant, R.; Semotiuk, K.; Hedley, D.; Dhani, N.; Narod, S.; Akbari, M.; Moore, M.; et al. Germline BRCA Mutations in a Large Clinic-Based Cohort of Patients With Pancreatic Adenocarcinoma. J. Clin. Oncol. 2015, 33, 3124–3129. [Google Scholar] [CrossRef]
- Wang, Y.; Camateros, P.; Cheung, W.Y. A Real-World Comparison of FOLFIRINOX, Gemcitabine Plus nab-Paclitaxel, and Gemcitabine in Advanced Pancreatic Cancers. J. Gastrointest. Cancer 2019, 50, 62–68. [Google Scholar] [CrossRef] [PubMed]
- Shroff, R.T.; Hendifar, A.; McWilliams, R.R.; Geva, R.; Epelbaum, R.; Rolfe, L.; Goble, S.; Lin, K.K.; Biankin, A.V.; Giordano, H.; et al. Rucaparib Monotherapy in Patients With Pancreatic Cancer and a Known Deleterious BRCA Mutation. JCO Precis. Oncol. 2018, 2018, PO.17.00316. [Google Scholar] [CrossRef]
- De, P.; Sun, Y.; Carlson, J.H.; Friedman, L.S.; Leyland-Jones, B.R.; Dey, N. Doubling down on the PI3K-AKT-mTOR pathway enhances the antitumor efficacy of PARP inhibitor in triple negative breast cancer model beyond BRCA-ness. Neoplasia 2014, 16, 43–72. [Google Scholar] [CrossRef] [Green Version]
- Golan, T.; Hammel, P.; Reni, M.; Van Cutsem, E.; Macarulla, T.; Hall, M.J.; Park, J.O.; Hochhauser, D.; Arnold, D.; Oh, D.Y.; et al. Maintenance Olaparib for Germline BRCA-Mutated Metastatic Pancreatic Cancer. N. Engl. J. Med. 2019, 381, 317–327. [Google Scholar] [CrossRef]
- US FDA. FDA Approves Olaparib for gBRCAm Metastatic Pancreatic Adenocarcinoma. Available online: https://www.fda.gov/drugs/resources-information-approved-drugs/fda-approves-olaparib-gbrcam-metastatic-pancreatic-adenocarcinoma (accessed on 30 December 2019).
- Fakih, M.; O’Neil, B.; Price, T.J.; Falchook, G.S.; Desai, J.; Kuo, J.; Govindan, R.; Rasmussen, E.; Morrow, P.K.H.; Ngang, J.; et al. Phase 1 study evaluating the safety, tolerability, pharmacokinetics (PK), and efficacy of AMG 510, a novel small molecule KRAS(G12c) inhibitor, in advanced solid tumors. Meeting Abstract. J. Clin. Oncol. 2019, 37, 3003. [Google Scholar] [CrossRef]
- Canon, J.; Rex, K.; Saiki, A.Y.; Mohr, C.; Cooke, K.; Bagal, D.; Gaida, K.; Holt, T.; Knutson, C.G.; Koppada, N.; et al. The clinical KRAS(G12C) inhibitor AMG 510 drives anti-tumour immunity. Nature 2019, 575, 217–223. [Google Scholar] [CrossRef]
- Nakajima, E.C.; Drezner, N.; Li, X.; Mishra-Kalyani, P.S.; Liu, Y.; Zhao, H.; Bi, Y.; Liu, J.; Rahman, A.; Wearne, E.; et al. FDA Approval Summary: Sotorasib for KRAS G12C-Mutated Metastatic NSCLC. Clin. Cancer Res. 2022, 28, 1482–1486. [Google Scholar] [CrossRef]
- Soh, J.; Okumura, N.; Lockwood, W.W.; Yamamoto, H.; Shigematsu, H.; Zhang, W.; Chari, R.; Shames, D.S.; Tang, X.; MacAulay, C.; et al. Oncogene mutations, copy number gains and mutant allele specific imbalance (MASI) frequently occur together in tumor cells. PLoS ONE 2009, 4, e7464. [Google Scholar] [CrossRef] [PubMed]
- The Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [Green Version]
- The Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature 2014, 511, 543–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Cancer Genome Atlas Research Network. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell. 2017, 32, 185–203.e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takai, Y.; Sasaki, T.; Matozaki, T. Small GTP-binding proteins. Physiol. Rev. 2001, 81, 153–208. [Google Scholar] [CrossRef]
- Drugan, J.K.; Rogers-Graham, K.; Gilmer, T.; Campbell, S.; Clark, G.J. The Ras/p120 GTPase-activating protein (GAP) interaction is regulated by the p120 GAP pleckstrin homology domain. J. Biol. Chem. 2000, 275, 35021–35027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bos, J.L.; Rehmann, H.; Wittinghofer, A. GEFs and GAPs: Critical elements in the control of small G proteins. Cell 2007, 129, 865–877. [Google Scholar] [CrossRef] [Green Version]
- Pamonsinlapatham, P.; Hadj-Slimane, R.; Lepelletier, Y.; Allain, B.; Toccafondi, M.; Garbay, C.; Raynaud, F. p120-Ras GTPase activating protein (RasGAP): A multi-interacting protein in downstream signaling. Biochimie 2009, 91, 320–328. [Google Scholar] [CrossRef] [PubMed]
- Hunter, J.C.; Manandhar, A.; Carrasco, M.A.; Gurbani, D.; Gondi, S.; Westover, K.D. Biochemical and Structural Analysis of Common Cancer-Associated KRAS Mutations. Mol. Cancer Res. 2015, 13, 1325–1335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostrem, J.M.; Shokat, K.M. Direct small-molecule inhibitors of KRAS: From structural insights to mechanism-based design. Nat. Rev. Drug Discov. 2016, 15, 771–785. [Google Scholar] [CrossRef]
- Rabara, D.; Tran, T.H.; Dharmaiah, S.; Stephens, R.M.; McCormick, F.; Simanshu, D.K.; Holderfield, M. KRAS G13D sensitivity to neurofibromin-mediated GTP hydrolysis. Proc. Natl. Acad. Sci. USA 2019, 116, 22122–22131. [Google Scholar] [CrossRef]
- Ambrogio, C.; Kohler, J.; Zhou, Z.W.; Wang, H.; Paranal, R.; Li, J.; Capelletti, M.; Caffarra, C.; Li, S.; Lv, Q.; et al. KRAS Dimerization Impacts MEK Inhibitor Sensitivity and Oncogenic Activity of Mutant KRAS. Cell 2018, 172, 857–868.e15. [Google Scholar] [CrossRef] [PubMed]
- Itonaga, M.; Ashida, R.; Murata, S.I.; Yamashita, Y.; Hatamaru, K.; Tamura, T.; Kawaji, Y.; Kayama, Y.; Emori, T.; Kawai, M.; et al. Kras Gene Analysis Using Liquid-Based Cytology Specimens Predicts Therapeutic Responses and Prognosis in Patients with Pancreatic Cancer. Cancers 2022, 14, 551. [Google Scholar] [CrossRef]
- McIntyre, C.A.; Lawrence, S.A.; Richards, A.L.; Chou, J.F.; Wong, W.; Capanu, M.; Berger, M.F.; Donoghue, M.T.A.; Yu, K.H.; Varghese, A.M.; et al. Alterations in driver genes are predictive of survival in patients with resected pancreatic ductal adenocarcinoma. Cancer 2020, 126, 3939–3949. [Google Scholar] [CrossRef]
- Dong, L.; Wang, S.; Fu, B.; Wang, J. Evaluation of droplet digital PCR and next generation sequencing for characterizing DNA reference material for KRAS mutation detection. Sci. Rep. 2018, 8, 9650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, M.T.; Mosier, S.L.; Thiess, M.; Beierl, K.F.; Debeljak, M.; Tseng, L.H.; Chen, G.; Yegnasubramanian, S.; Ho, H.; Cope, L.; et al. Clinical validation of KRAS, BRAF, and EGFR mutation detection using next-generation sequencing. Am. J. Clin. Pathol. 2014, 141, 856–866. [Google Scholar] [CrossRef] [Green Version]
- Haque, M.R.; Wessel, C.R.; Leary, D.D.; Wang, C.; Bhushan, A.; Bishehsari, F. Patient-derived pancreatic cancer-on-a-chip recapitulates the tumor microenvironment. Microsyst. Nanoeng. 2022, 8, 36. [Google Scholar] [CrossRef] [PubMed]
- Nauheim, D.; Moskal, D.; Renslo, B.; Chadwick, M.; Jiang, W.; Yeo, C.J.; Nevler, A.; Bowne, W.; Lavu, H. KRAS mutation allele frequency threshold alters prognosis in right-sided resected pancreatic cancer. J. Surg. Oncol. 2022, 126, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Dopico, P.J.; Le, M.N.; Burgess, B.; Yang, Z.; Zhao, Y.; Wang, Y.; George, T.J.; Fan, Z.H. Longitudinal Study of Circulating Biomarkers in Patients with Resectable Pancreatic Ductal Adenocarcinoma. Biosensors 2022, 12, 206. [Google Scholar] [CrossRef]
- Li, Z.; Zhuang, H.; Chen, X.; Zhang, Y.; Ma, Z.; Wang, S.; Yan, Q.; Zhou, Z.; Huang, S.; Zhang, C.; et al. Identification of MBOAT2 as an Unfavorable Biomarker Correlated with KRAS Activation and Reduced CD8(+) T-Cell Infiltration in Pancreatic Cancer. J. Oncol. 2022, 2022, 4269733. [Google Scholar] [CrossRef]
- Hallin, J.; Engstrom, L.D.; Hargis, L.; Calinisan, A.; Aranda, R.; Briere, D.M.; Sudhakar, N.; Bowcut, V.; Baer, B.R.; Ballard, J.A.; et al. The KRAS(G12C) Inhibitor MRTX849 Provides Insight toward Therapeutic Susceptibility of KRAS-Mutant Cancers in Mouse Models and Patients. Cancer Discov. 2020, 10, 54–71. [Google Scholar] [CrossRef] [Green Version]
- Brauswetter, D.; Gurbi, B.; Varga, A.; Varkondi, E.; Schwab, R.; Banhegyi, G.; Fabian, O.; Keri, G.; Valyi-Nagy, I.; Petak, I. Molecular subtype specific efficacy of MEK inhibitors in pancreatic cancers. PLoS ONE 2017, 12, e0185687. [Google Scholar] [CrossRef]
- Zhou, L.; Baba, Y.; Kitano, Y.; Miyake, K.; Zhang, X.; Yamamura, K.; Kosumi, K.; Kaida, T.; Arima, K.; Taki, K.; et al. KRAS, BRAF, and PIK3CA mutations, and patient prognosis in 126 pancreatic cancers: Pyrosequencing technology and literature review. Med. Oncol. 2016, 33, 32. [Google Scholar] [CrossRef] [PubMed]
- Fell, J.B.; Fischer, J.P.; Baer, B.R.; Blake, J.F.; Bouhana, K.; Briere, D.M.; Brown, K.D.; Burgess, L.E.; Burns, A.C.; Burkard, M.R.; et al. Identification of the Clinical Development Candidate MRTX849, a Covalent KRAS(G12C) Inhibitor for the Treatment of Cancer. J. Med. Chem. 2020, 63, 6679–6693. [Google Scholar] [CrossRef] [Green Version]
- Hansen, R.; Peters, U.; Babbar, A.; Chen, Y.; Feng, J.; Janes, M.R.; Li, L.S.; Ren, P.; Liu, Y.; Zarrinkar, P.P. The reactivity-driven biochemical mechanism of covalent KRAS(G12C) inhibitors. Nat. Struct. Mol. Biol. 2018, 25, 454–462. [Google Scholar] [CrossRef] [PubMed]
- Vasta, J.D.; Peacock, D.M.; Zheng, Q.; Walker, J.A.; Zhang, Z.; Zimprich, C.A.; Thomas, M.R.; Beck, M.T.; Binkowski, B.F.; Corona, C.R.; et al. KRAS is vulnerable to reversible switch-II pocket engagement in cells. Nat. Chem. Biol. 2022, 18, 596–604. [Google Scholar] [CrossRef] [PubMed]
- Pellestor, F.; Paulasova, P. The peptide nucleic acids (PNAs), powerful tools for molecular genetics and cytogenetics. Eur. J. Hum. Genet. 2004, 12, 694–700. [Google Scholar] [CrossRef]
- Dean, D.A. Peptide nucleic acids: Versatile tools for gene therapy strategies. Adv. Drug Deliv. Rev. 2000, 44, 81–95. [Google Scholar] [CrossRef] [Green Version]
- De Cola, C.; Manicardi, A.; Corradini, R.; Izzo, I.; De Riccardis, F. Carboxyalkyl peptoid PNAs: Synthesis and hybridization properties. Article. Tetrahedron 2012, 68, 499–506. [Google Scholar] [CrossRef]
- Chiarantini, L.; Cerasi, A.; Fraternale, A.; Millo, E.; Benatti, U.; Sparnacci, K.; Laus, M.; Ballestri, M.; Tondelli, L. Comparison of novel delivery systems for antisense peptide nucleic acids. J. Control. Release 2005, 109, 24–36. [Google Scholar] [CrossRef]
- Shai, A.; Galouk, E.; Miari, R.; Tareef, H.; Sammar, M.; Zeidan, M.; Rayan, A.; Falah, M. Inhibiting mutant KRAS G12D gene expression using novel peptide nucleic acid-based antisense: A potential new drug candidate for pancreatic cancer. Oncol. Lett. 2022, 23, 130. [Google Scholar] [CrossRef] [PubMed]
- Coley, A.B.; Ward, A.; Keeton, A.B.; Chen, X.; Maxuitenko, Y.; Prakash, A.; Li, F.; Foote, J.B.; Buchsbaum, D.J.; Piazza, G.A. Pan-RAS inhibitors: Hitting multiple RAS isozymes with one stone. Adv. Cancer Res. 2022, 153, 131–168. [Google Scholar] [PubMed]
- Keeton, A.B.; Ward, A.; Chen, X.; Valiyaveettil, J.; Zhu, B.; Ramirez-Alcantara, V. Abstract 2707: A novel RAS inhibitor, MCI-062, inhibits colon tumor growth in vivo and activates antitumor immunity. Cancer Res. 2019, 79, 2707. [Google Scholar] [CrossRef]
- Welsch, M.E.; Kaplan, A.; Chambers, J.M.; Stokes, M.E.; Bos, P.H.; Zask, A.; Zhang, Y.; Sanchez-Martin, M.; Badgley, M.A.; Huang, C.S.; et al. Multivalent Small-Molecule Pan-RAS Inhibitors. Cell 2017, 168, 878–889.e29. [Google Scholar] [CrossRef] [Green Version]
- Rognan, D. Rational design of protein–protein interaction inhibitors. Med. Chem. Commun. 2015, 6, 51–60. [Google Scholar] [CrossRef]
- Khan, I.; Rhett, J.M.; O’Bryan, J.P. Therapeutic targeting of RAS: New hope for drugging the “undruggable”. Biochim. Biophys. Acta. Mol. Cell Res. 2020, 1867, 118570. [Google Scholar] [CrossRef]
- Gorgulla, C.; Boeszoermenyi, A.; Wang, Z.F.; Fischer, P.D.; Coote, P.W.; Padmanabha Das, K.M.; Malets, Y.S.; Radchenko, D.S.; Moroz, Y.S.; Scott, D.A.; et al. An open-source drug discovery platform enables ultra-large virtual screens. Nature 2020, 580, 663–668. [Google Scholar] [CrossRef]
- Zheng, X.; Liu, Z.; Li, D.; Wang, E.; Wang, J. Rational drug design: The search for Ras protein hydrolysis intermediate conformation inhibitors with both affinity and specificity. Curr. Pharm. Des. 2013, 19, 2246–2258. [Google Scholar] [CrossRef]
- Guo, X.; Zhao, W.; Liu, Z.; Wang, J. Spiclomazine displays a preferential anti-tumor activity in mutant KRas-driven pancreatic cancer. Oncotarget 2018, 9, 6938–6951. [Google Scholar] [CrossRef] [Green Version]
- Zhao, W.; Li, D.; Liu, Z.; Zheng, X.; Wang, J.; Wang, E. Spiclomazine induces apoptosis associated with the suppression of cell viability, migration and invasion in pancreatic carcinoma cells. PLoS ONE 2013, 8, e66362. [Google Scholar] [CrossRef]
- Liu, Z.; Li, D.; Zheng, X.; Wang, E.; Wang, J. Selective induction of apoptosis: Promising therapy in pancreatic cancer. Curr. Pharm. Des. 2013, 19, 2259–2268. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Li, D.; Zhao, W.; Zheng, X.; Wang, J.; Wang, E. A potent lead induces apoptosis in pancreatic cancer cells. PLoS ONE 2012, 7, e37841. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Murciano-Goroff, Y.R.; Xue, J.Y.; Ang, A.; Lucas, J.; Mai, T.T.; Da Cruz Paula, A.F.; Saiki, A.Y.; Mohn, D.; Achanta, P.; et al. Diverse alterations associated with resistance to KRAS(G12C) inhibition. Nature 2021, 599, 679–683. [Google Scholar] [CrossRef]
- Boned Del Rio, I.; Young, L.C.; Sari, S.; Jones, G.G.; Ringham-Terry, B.; Hartig, N.; Rejnowicz, E.; Lei, W.; Bhamra, A.; Surinova, S.; et al. SHOC2 complex-driven RAF dimerization selectively contributes to ERK pathway dynamics. Proc. Natl. Acad. Sci. USA 2019, 116, 13330–13339. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malumbres, M.; Barbacid, M. RAS oncogenes: The first 30 years. Nat. Rev. Cancer 2003, 3, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Franke, T.F. PI3K/Akt: Getting it right matters. Oncogene 2008, 27, 6473–6488. [Google Scholar] [CrossRef] [Green Version]
- Muzumdar, M.D.; Chen, P.Y.; Dorans, K.J.; Chung, K.M.; Bhutkar, A.; Hong, E.; Noll, E.M.; Sprick, M.R.; Trumpp, A.; Jacks, T. Survival of pancreatic cancer cells lacking KRAS function. Nat. Commun. 2017, 8, 1090. [Google Scholar] [CrossRef] [Green Version]
- Kapoor, A.; Yao, W.; Ying, H.; Hua, S.; Liewen, A.; Wang, Q.; Zhong, Y.; Wu, C.J.; Sadanandam, A.; Hu, B.; et al. Yap1 Activation Enables Bypass of Oncogenic Kras Addiction in Pancreatic Cancer. Cell 2019, 179, 1239. [Google Scholar] [CrossRef] [Green Version]
- Shao, D.D.; Xue, W.; Krall, E.B.; Bhutkar, A.; Piccioni, F.; Wang, X.; Schinzel, A.C.; Sood, S.; Rosenbluh, J.; Kim, J.W.; et al. KRAS and YAP1 converge to regulate EMT and tumor survival. Cell 2014, 158, 171–184. [Google Scholar] [CrossRef] [Green Version]
- Yu, C.; Zhang, M.; Song, J.; Zheng, X.; Xu, G.; Bao, Y.; Lan, J.; Luo, D.; Hu, J.; Li, J.J.; et al. Integrin-Src-YAP1 signaling mediates the melanoma acquired resistance to MAPK and PI3K/mTOR dual targeted therapy. Mol. Biomed. 2020, 1, 12. [Google Scholar] [CrossRef]
- Infante, J.R.; Fecher, L.A.; Falchook, G.S.; Nallapareddy, S.; Gordon, M.S.; Becerra, C.; DeMarini, D.J.; Cox, D.S.; Xu, Y.; Morris, S.R.; et al. Safety, pharmacokinetic, pharmacodynamic, and efficacy data for the oral MEK inhibitor trametinib: A phase 1 dose-escalation trial. Lancet Oncol. 2012, 13, 773–781. [Google Scholar] [CrossRef]
- Jänne, P.A.; van den Heuvel, M.M.; Barlesi, F.; Cobo, M.; Mazieres, J.; Crinò, L.; Orlov, S.; Blackhall, F.; Wolf, J.; Garrido, P.; et al. Selumetinib Plus Docetaxel Compared With Docetaxel Alone and Progression-Free Survival in Patients With KRAS-Mutant Advanced Non-Small Cell Lung Cancer: The SELECT-1 Randomized Clinical Trial. JAMA 2017, 317, 1844–1853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blumenschein, G.R., Jr.; Smit, E.F.; Planchard, D.; Kim, D.W.; Cadranel, J.; De Pas, T.; Dunphy, F.; Udud, K.; Ahn, M.J.; Hanna, N.H.; et al. A randomized phase II study of the MEK1/MEK2 inhibitor trametinib (GSK1120212) compared with docetaxel in KRAS-mutant advanced non-small-cell lung cancer (NSCLC)†. Ann. Oncol. 2015, 26, 894–901. [Google Scholar] [CrossRef] [PubMed]
- Papadopoulos, K.P.; Ou, S.H.I.; Johnson, M.L.; Christensen, J.; Velastegui, K.; Potvin, D.; Faltaos, D.; Chao, R.C. A phase I/II multiple expansion cohort trial of MRTX849 in patients with advanced solid tumors with KRAS G12C mutation. Meeting Abstract. J. Clin. Oncol. 2019, 37, 1. [Google Scholar] [CrossRef]
- Tanaka, N.; Lin, J.J.; Li, C.; Ryan, M.B.; Zhang, J.; Kiedrowski, L.A.; Michel, A.G.; Syed, M.U.; Fella, K.A.; Sakhi, M.; et al. Clinical Acquired Resistance to KRAS(G12C) Inhibition through a Novel KRAS Switch-II Pocket Mutation and Polyclonal Alterations Converging on RAS-MAPK Reactivation. Cancer Discov. 2021, 11, 1913–1922. [Google Scholar] [CrossRef]
- Bunda, S.; Burrell, K.; Heir, P.; Zeng, L.; Alamsahebpour, A.; Kano, Y.; Raught, B.; Zhang, Z.Y.; Zadeh, G.; Ohh, M. Inhibition of SHP2-mediated dephosphorylation of Ras suppresses oncogenesis. Nat. Commun. 2015, 6, 8859. [Google Scholar] [CrossRef] [Green Version]
- Fedele, C.; Ran, H.; Diskin, B.; Wei, W.; Jen, J.; Geer, M.J.; Araki, K.; Ozerdem, U.; Simeone, D.M.; Miller, G.; et al. SHP2 Inhibition Prevents Adaptive Resistance to MEK Inhibitors in Multiple Cancer Models. Cancer Discov. 2018, 8, 1237–1249. [Google Scholar] [CrossRef] [Green Version]
- Lu, H.; Liu, C.; Velazquez, R.; Wang, H.; Dunkl, L.M.; Kazic-Legueux, M.; Haberkorn, A.; Billy, E.; Manchado, E.; Brachmann, S.M.; et al. SHP2 Inhibition Overcomes RTK-Mediated Pathway Reactivation in KRAS-Mutant Tumors Treated with MEK Inhibitors. Mol. Cancer Ther. 2019, 18, 1323–1334. [Google Scholar] [CrossRef] [Green Version]
- Nichols, R.J.; Haderk, F.; Stahlhut, C.; Schulze, C.J.; Hemmati, G.; Wildes, D.; Tzitzilonis, C.; Mordec, K.; Marquez, A.; Romero, J.; et al. RAS nucleotide cycling underlies the SHP2 phosphatase dependence of mutant BRAF-, NF1- and RAS-driven cancers. Nat. Cell. Biol. 2018, 20, 1064–1073. [Google Scholar] [CrossRef]
- Fedele, C.; Li, S.; Teng, K.W.; Foster, C.J.R.; Peng, D.; Ran, H.; Mita, P.; Geer, M.J.; Hattori, T.; Koide, A.; et al. SHP2 inhibition diminishes KRASG12C cycling and promotes tumor microenvironment remodeling. J. Exp. Med. 2021, 218, e20201414. [Google Scholar] [CrossRef]
- Hata, A.N.; Shaw, A.T. Resistance looms for KRAS(G12C) inhibitors. Nat. Med. 2020, 26, 169–170. [Google Scholar] [CrossRef] [PubMed]
- Ischenko, I.; Petrenko, O.; Hayman, M.J. A MEK/PI3K/HDAC inhibitor combination therapy for KRAS mutant pancreatic cancer cells. Oncotarget 2015, 6, 15814–15827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jane, E.P.; Premkumar, D.R.; Addo-Yobo, S.O.; Pollack, I.F. Abrogation of mitogen-activated protein kinase and Akt signaling by vandetanib synergistically potentiates histone deacetylase inhibitor-induced apoptosis in human glioma cells. J. Pharmacol. Exp. Ther. 2009, 331, 327–337. [Google Scholar] [CrossRef]
- Ozaki, K.; Kosugi, M.; Baba, N.; Fujio, K.; Sakamoto, T.; Kimura, S.; Tanimura, S.; Kohno, M. Blockade of the ERK or PI3K-Akt signaling pathway enhances the cytotoxicity of histone deacetylase inhibitors in tumor cells resistant to gefitinib or imatinib. Biochem. Biophys. Res. Commun. 2010, 391, 1610–1615. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, A.R.; Robey, R.W.; Luchenko, V.L.; Zhan, Z.; Piekarz, R.L.; Gillet, J.P.; Kossenkov, A.V.; Wilkerson, J.; Showe, L.C.; Gottesman, M.M.; et al. MAPK pathway activation leads to Bim loss and histone deacetylase inhibitor resistance: Rationale to combine romidepsin with an MEK inhibitor. Blood 2013, 121, 4115–4125. [Google Scholar] [CrossRef] [Green Version]
- Lou, K.; Steri, V.; Ge, A.Y.; Hwang, Y.C.; Yogodzinski, C.H.; Shkedi, A.R.; Choi, A.L.M.; Mitchell, D.C.; Swaney, D.L.; Hann, B.; et al. KRAS(G12C) inhibition produces a driver-limited state revealing collateral dependencies. Sci. Signal. 2019, 12, eaaw9450. [Google Scholar] [CrossRef] [PubMed]
- Khan, H.Y.; Nagasaka, M.; Li, Y.; Aboukameel, A.; Uddin, M.H.; Sexton, R.; Bannoura, S.; Mzannar, Y.; Al-Hallak, M.N.; Kim, S.; et al. Inhibitor of the Nuclear Transport Protein XPO1 Enhances the Anticancer Efficacy of KRAS G12C Inhibitors in Preclinical Models of KRAS G12C-Mutant Cancers. Cancer Res. Commun. 2022, 2, 342–352. [Google Scholar] [CrossRef] [PubMed]
- Azmi, A.S.; Uddin, M.H.; Mohammad, R.M. The nuclear export protein XPO1-from biology to targeted therapy. Nat. Rev. Clin. Oncol. 2021, 18, 152–169. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; McMillan, E.; Kim, H.S.; Venkateswaran, N.; Makkar, G.; Rodriguez-Canales, J.; Villalobos, P.; Neggers, J.E.; Mendiratta, S.; Wei, S.; et al. XPO1-dependent nuclear export is a druggable vulnerability in KRAS-mutant lung cancer. Nature 2016, 538, 114–117. [Google Scholar] [CrossRef] [Green Version]
- Koltun, E.; Cregg, J.; Rice, M.A.; Whalen, D.M.; Freilich, R.; Jiang, J.J.; Hansen, R.; Bermingham, A.; Knox, J.E.; Dinglasan, J.; et al. First-in-class, orally bioavailable KRAS(G12V)(ON) tri-complex inhibitors, as single agents and in combinations, drive profound anti-tumor activity in preclinical models of KRAS(G12V) mutant cancers. Meeting Abstract. Cancer Res. 2021, 81, 2. [Google Scholar] [CrossRef]
Figure 1.
Percentage of pancreatic cancer by type and the probability of related mutations. (a) The proportion of main subtypes in pancreatic cancer. (b) Frequencies of mutations in individual genes contained in PDAC. All charts were constructed based on data from AACR Project GENIE: Powering Precision Medicine through an International Consortium (GENIE Cohort v11.0-public [8]).
Figure 1.
Percentage of pancreatic cancer by type and the probability of related mutations. (a) The proportion of main subtypes in pancreatic cancer. (b) Frequencies of mutations in individual genes contained in PDAC. All charts were constructed based on data from AACR Project GENIE: Powering Precision Medicine through an International Consortium (GENIE Cohort v11.0-public [8]).

Figure 2.
Structures of RAS proteins and inhibitors. Protein is indicated by surface representation, and compounds and nucleotides are shown in stick models. The carbon and hydrogen atoms of the inhibitor are marked in yellow to highlight them. (a) KRASG12C and AMG510 (Protein Data Bank (PDB): 6OIM). (b) KRASG12C and MRTX849 (PDB: 6UT0). (c) KRASG12D and MRTX1133 (PDB: 7RPZ). (d) HRASG60A and NSC290956 [14].
Figure 2.
Structures of RAS proteins and inhibitors. Protein is indicated by surface representation, and compounds and nucleotides are shown in stick models. The carbon and hydrogen atoms of the inhibitor are marked in yellow to highlight them. (a) KRASG12C and AMG510 (Protein Data Bank (PDB): 6OIM). (b) KRASG12C and MRTX849 (PDB: 6UT0). (c) KRASG12D and MRTX1133 (PDB: 7RPZ). (d) HRASG60A and NSC290956 [14].

Figure 3.
Schematic representation of the RAS signaling pathway and relevant targets for combination therapy regimens. T symbol in red: therapeutic targets combined with KRAS G12 inhibitors in clinical trials (Table 1). RTKs: receptor tyrosine kinases; AUKRA: aurora kinase A; FAK: focal adhesion kinase.
Figure 3.
Schematic representation of the RAS signaling pathway and relevant targets for combination therapy regimens. T symbol in red: therapeutic targets combined with KRAS G12 inhibitors in clinical trials (Table 1). RTKs: receptor tyrosine kinases; AUKRA: aurora kinase A; FAK: focal adhesion kinase.


{kind=link}
{kind=link}
{kind=link}
Table 1.
Registered trials of KRASG12C inhibitor combination therapy on clinicaltrials.gov.
| ClinicalTrials.Gov Identifier | Title | Phase | Drugs | Targets |
|---|---|---|---|---|
| NCT05374538 | VIC-1911 Monotherapy in Combination With Sotorasib for the Treatment of KRAS G12C-Mutant Non-Small Cell Lung Cancer | 1 | Sotorasib VIC-1911 | KRASG12C Aurora Kinase A |
| NCT05067283 | A Study of MK-1084 as Monotherapy and in Combination With Pembrolizumab (MK-3475) in Participants With KRASG12C Mutant Advanced Solid Tumors (MK-1084-001) | 1 | MK-1084 Pembrolizumab | KRASG12C PD-1 |
| NCT05379946 | Study to Evaluate D-1553 in Combination With IN10018 in Subjects With Solid Tumors | 1/2 | D-1553 IN10018 | KRASG12C FAK |
| NCT05074810 | Phase 1/2 Study of VS-6766 + Sotorasib in G12C NSCLC Patients (RAMP203) | 1/2 | Sotorasib VS-6766 | KRASG12C RAF/MEK |
| NCT05054725 | Combination Study of RMC-4630 and Sotorasib for NSCLC Subjects With KRASG12C Mutation After Failure of Prior Standard Therapies | 2 | Sotorasib RMC-4630 | KRASG12C SHP2 |
| NCT05313009 | Tarlox and Sotorasib in Patients With KRAS G12C Mutations | 1/2 | Sotorasib Tarloxotinib | KRASG12C EGFR/HER2/HER3 |
| NCT05198934 | Sotorasib and Panitumumab Versus Investigator’s Choice for Participants With Kirsten Rat Sarcoma (KRAS) p.G12C Mutation (CodeBreak 300) | 3 | Sotorasib Panitumumab | KRASG12C EGFR |
| NCT04613596 | Phase 2 Trial of MRTX849 Monotherapy and in Combination With Pembrolizumab for NSCLC With KRAS G12C Mutation KRYSTAL-7 | 2 | MRTX849 Pembrolizumab | KRASG12C PD-1 |
| NCT04330664 | Adagrasib in Combination With TNO155 in Patients With Cancer (KRYSTAL 2) | 1/2 | MRTX849 TNO155 | KRASG12C SHP2 |
| NCT05375994 | Study of VS-6766 + Adagrasib in KRAS G12C NSCLC Patients (RAMP204) | 1/2 | MRTX849 VS-6766 | KRASG12C RAF/MEK |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
He, Q.; Liu, Z.; Wang, J. Targeting KRAS in PDAC: A New Way to Cure It? Cancers 2022, 14, 4982. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14204982
AMA Style
He Q, Liu Z, Wang J. Targeting KRAS in PDAC: A New Way to Cure It? Cancers. 2022; 14(20):4982. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14204982
Chicago/Turabian StyleHe, Qianyu, Zuojia Liu, and Jin Wang. 2022. "Targeting KRAS in PDAC: A New Way to Cure It?" Cancers 14, no. 20: 4982. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14204982
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.