
T Cells Directed against the Metastatic Driver Chondromodulin-1 in Ewing Sarcoma: Comparative Engineering with CRISPR/Cas9 vs. Retroviral Gene Transfer for Adoptive Transfer

 , , , ,
, , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines
2.2. Expansion of TCR-Transgenic T Cells
2.3. Functional Characterization of CHM1319/HLA-A*02:01-Specific TCR Transgenic T Cells
2.4. Western Blot
2.5. TCR DNA Template Design
2.6. CRISPR/Cas9 Mediated TCR KI and Retrovirus Transduction
2.7. Analysis of Published Chip-Sequence Data and Microarray
2.8. Statistical Analysis
3. Results
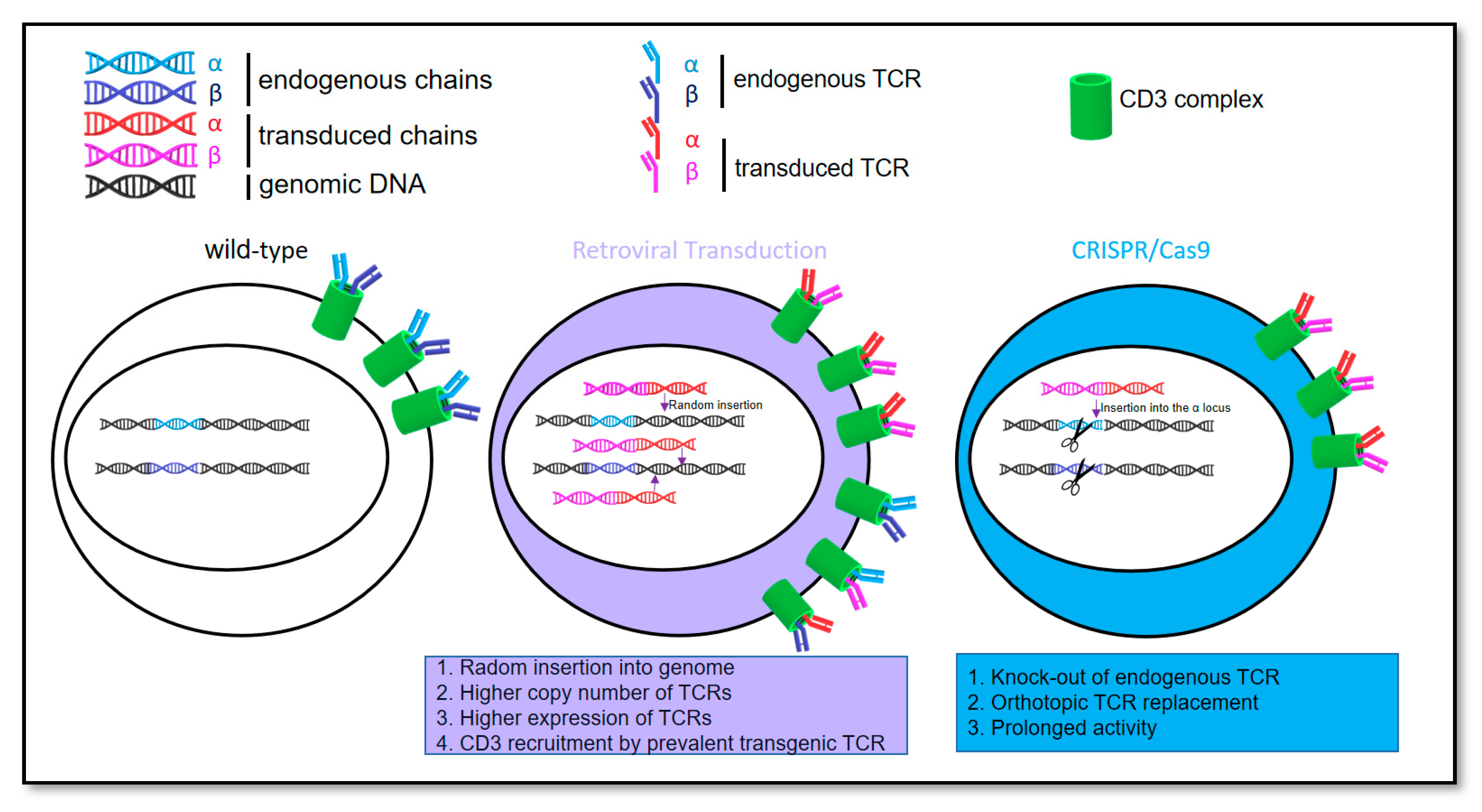
3.1. Feasibility of Orthotopic Replacement of the Endogenous T Cell Receptor with a T Cell Receptor Containing Chondromodulin-1 Targeting Sequence
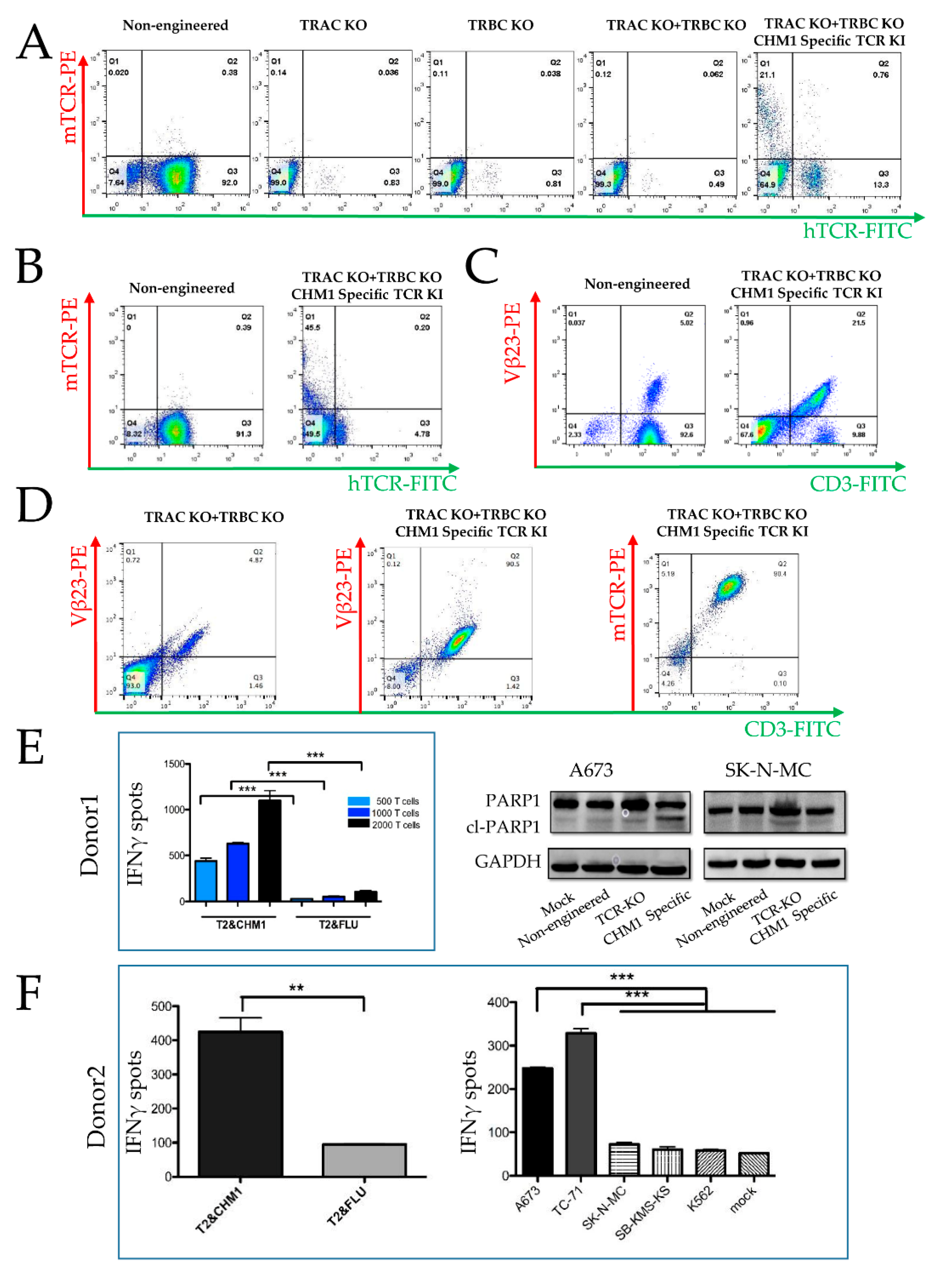
3.1.1. CRISPR/Cas9-Engineered Orthotopic TCR Replacement
3.1.2. Tumor Recognition and Cytotoxicity by CRISPR/Cas9-Engineered T Cells
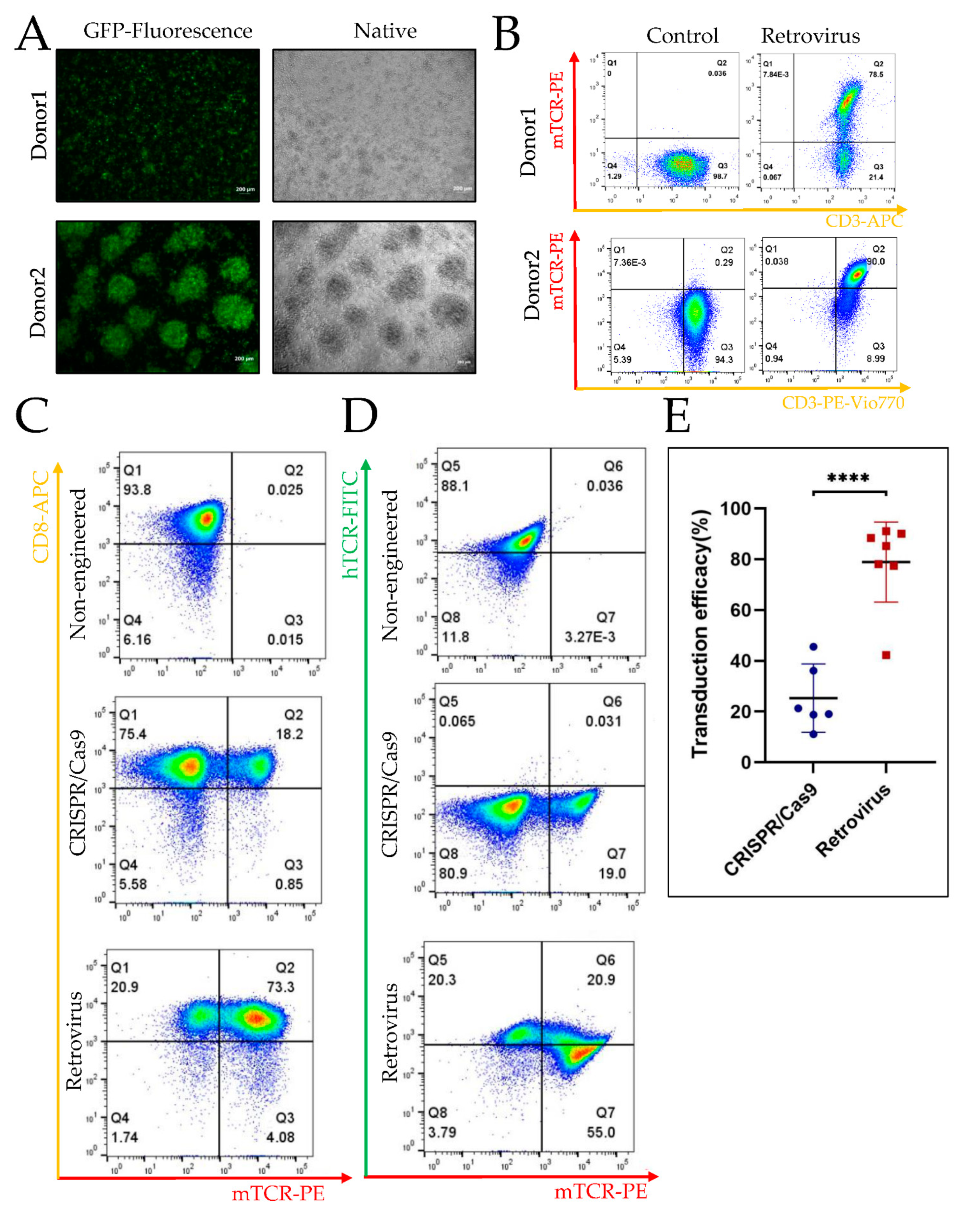
3.2. Higher Efficiency of Retroviral Transduction Compared to Gene Editing by CRISPR/Cas9
3.2.1. TCR Transgenic T Cells Engineered by CRISPR/cas9 vs. Retroviral Gene Transduction
3.2.2. Higher Efficiency of Retroviral Transduction Compared to Gene Editing by CRISPR/Cas9
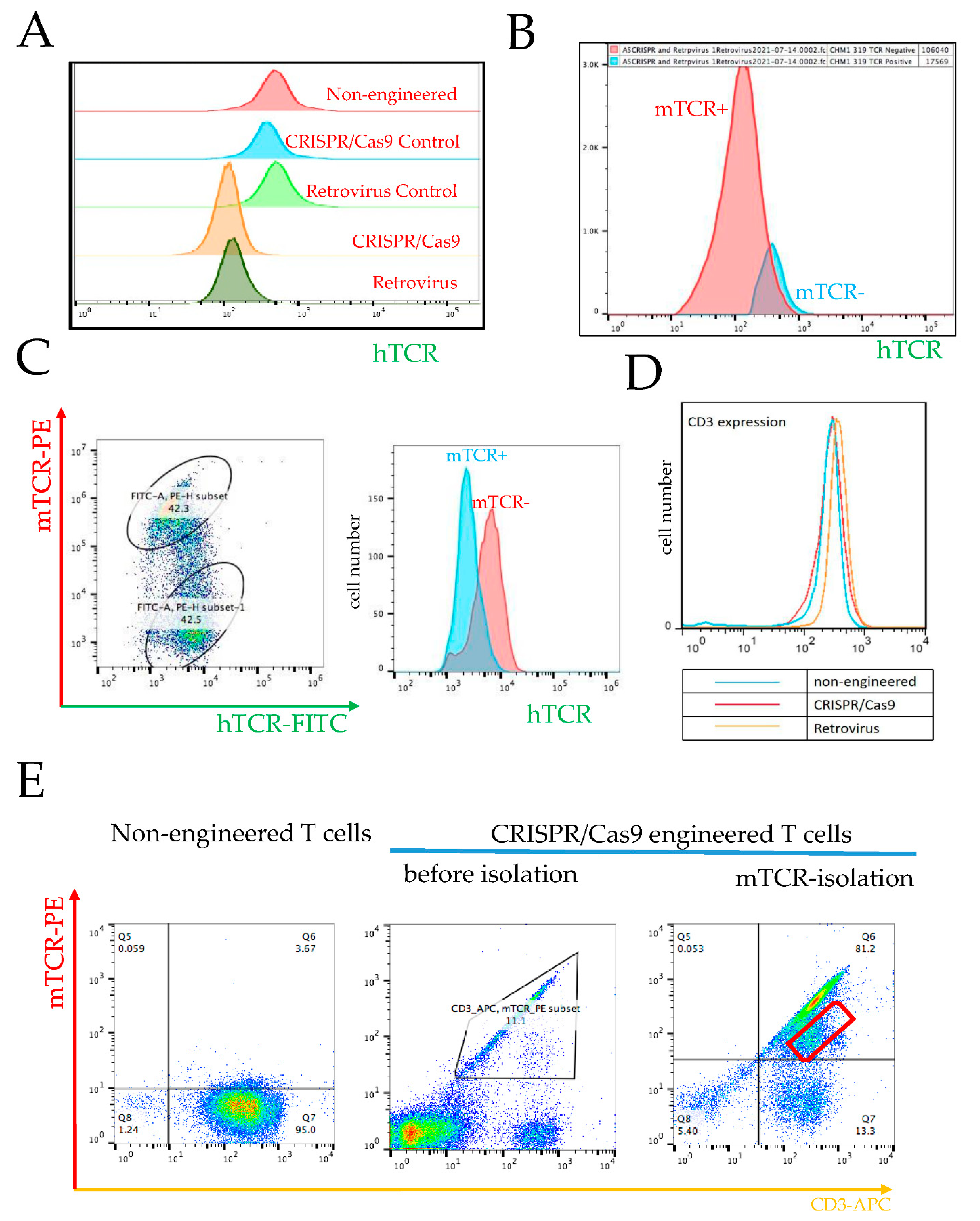
3.3. Prevention of Endogenous TCR Expression in CRISPR/Cas9 vs. Retrovirally Engineered T Cells
3.3.1. Requirement of High Retroviral Gene Transduction Efficacy and High CRISPR/Cas9 KO Efficacy for Prevention of Endogenous TCR Expression and TCR Chain Mispairing
3.3.2. Failure in KO of Endogenous β Chain Generates a Subpopulation with TCR Misparing
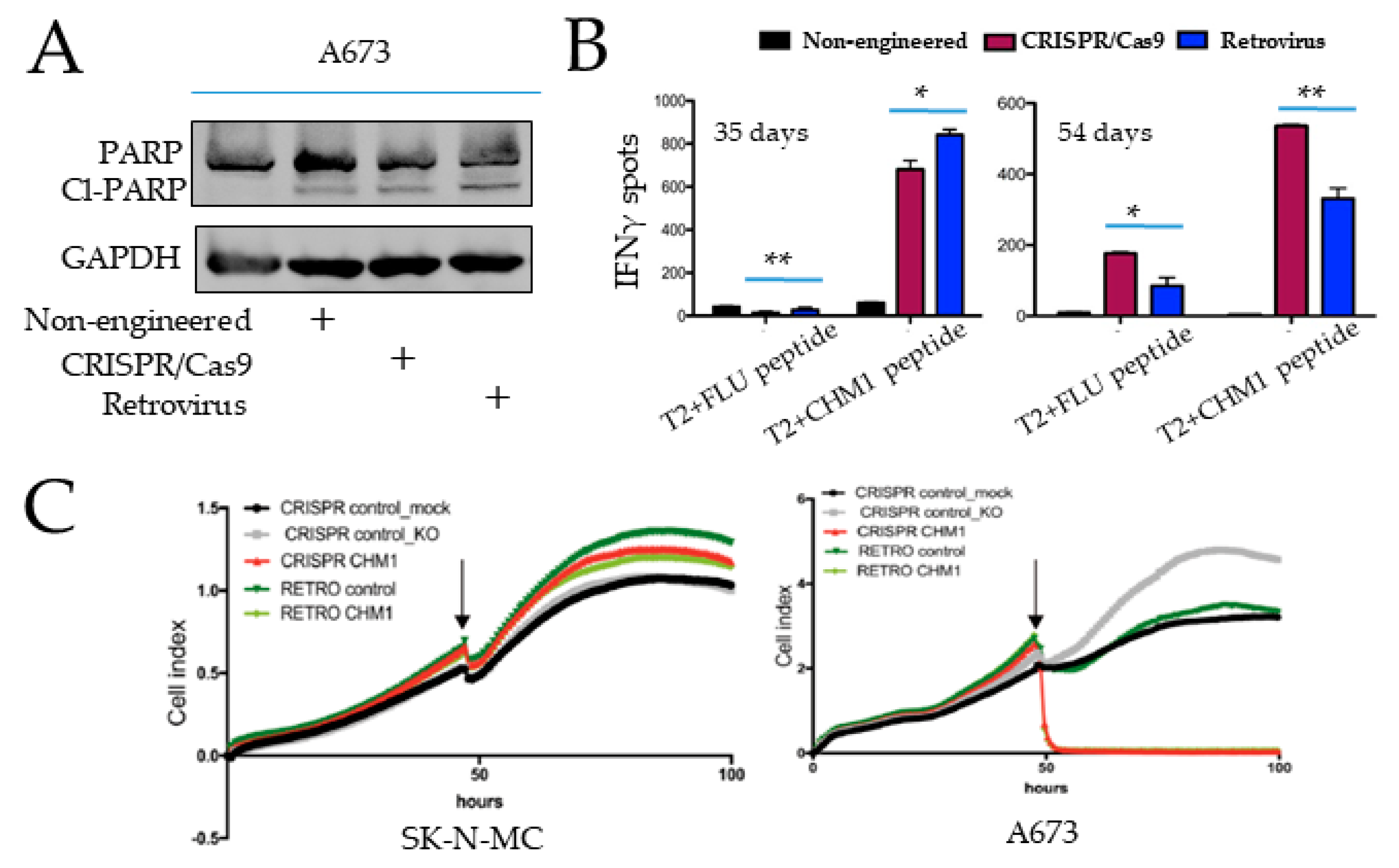
3.4. Specific Tumor Cell Recognition and Cytotoxicity In Vitro by Both T Cell Products with Better Prolonged Activity of CRISPR/Cas9-Engineered T Cells
3.5. CHM1 as a Unique Immunotherapy Target in EwS
CHM1 Is a Direct Target of EWS-FLI1 Selectively Expressed in EwS
4. Discussion
4.1. TCR-Based Immunotherapy of Ewing Sarcoma
4.2. Orthotopic Replacement of TCR with Cytotoxic Functionality
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Grünewald, T.G.; Cidre-Aranaz, F.; Surdez, D.; Tomazou, E.M.; de Álava, E.; Kovar, H.; Sorensen, P.H.; Delattre, O.; Dirksen, U. Ewing sarcoma. Nat. Rev. Dis. Prim. 2018, 4, 5. [Google Scholar] [PubMed]
- Riggi, N.; Suvà, M.L.; Stamenkovic, I. Ewing’s sarcoma. N. Engl. J. Med. 2021, 384, 154–164. [Google Scholar] [PubMed]
- Thacker, M.M.; Temple, H.; Scully, S.P. Current treatment for Ewing’s sarcoma. Expert Rev. Anticancer Ther. 2005, 5, 319–331. [Google Scholar] [CrossRef] [PubMed]
- Burdach, S.; Jürgens, H. High-dose chemoradiotherapy (HDC) in the Ewing family of tumors (EFT). Crit. Rev. Oncol. 2002, 41, 169–189. [Google Scholar] [CrossRef]
- Burdach, S.T.; Jürgens, H.; Peters, C.; Nürnberger, W.; Mauz-Körholz, C.; Körholz, D.; Paulussen, M.; Pape, H.; Dilloo, D.; Koscielniak, E. Myeloablative radiochemotherapy and hematopoietic stem-cell rescue in poor-prognosis Ewing’s sarcoma. J. Clin. Oncol. 1993, 11, 1482–1488. [Google Scholar] [CrossRef]
- Burdach, S.; Thiel, U.; Schöniger, M.; Haase, R.; Wawer, A.; Nathrath, M.; Kabisch, H.; Urban, C.; Laws, H.J.; Dirksen, U.; et al. Total body MRI-governed involved compartment irradiation combined with high-dose chemotherapy and stem cell rescue improves long-term survival in Ewing tumor patients with multiple primary bone metastases. Bone Marrow Transpl. 2009, 45, 483–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhakta, N.; Liu, Q.; Ness, K.K.; Baassiri, M.; Eissa, H.; Yeo, F.; Chemaitilly, W.; Ehrhardt, M.J.; Bass, J.; Bishop, M.W.; et al. The cumulative burden of surviving childhood cancer: An initial report from the St Jude Lifetime Cohort Study (SJLIFE). Lancet 2017, 390, 2569–2582. [Google Scholar]
- Gaspar, N.; Hawkins, D.S.; Dirksen, U.; Lewis, I.J.; Ferrari, S.; Le Deley, M.-C.; Kovar, H.; Grimer, R.; Whelan, J.; Claude, L.; et al. Ewing Sarcoma: Current Management and Future Approaches Through Collaboration. J. Clin. Oncol. 2015, 33, 3036–3046. [Google Scholar]
- Burdach, S.; Kolb, H.J. The vigor of defense against non-self: Potential superiority of allorestricted T cells in immunotherapy of cancer? Front. Oncol. 2013, 3, 100. [Google Scholar] [CrossRef] [Green Version]
- Rudolph, M.G.; Stanfield, R.L.; Wilson, I.A. How TCRs bind MHCs, peptides, and coreceptors. Annu. Rev. Immunol. 2006, 24, 419–466. [Google Scholar] [CrossRef]
- Davis, M.M.; Bjorkman, P.J. T-cell antigen receptor genes and T-cell recognition. Nature 1988, 334, 395–402. [Google Scholar]
- Manfredi, F.; Cianciotti, B.C.; Potenza, A.; Tassi, E.; Noviello, M.; Biondi, A.; Ciceri, F.; Bonini, C.; Ruggiero, E. TCR Redirected T Cells for Cancer Treatment: Achievements, Hurdles, and Goals. Front. Immunol. 2020, 11, 1689. [Google Scholar] [CrossRef] [PubMed]
- Howe, S.J.; Mansour, M.; Schwarzwaelder, K.; Bartholomae, C.; Hubank, M.; Kempski, H.; Brugman, M.; Pike-Overzet, K.; Chatters, S.J.; De Ridder, D.; et al. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J. Clin. Investig. 2008, 118, 3143–3150. [Google Scholar] [CrossRef] [PubMed]
- Fraietta, J.A.; Nobles, C.L.; Sammons, M.A.; Lundh, S.; Carty, S.A.; Reich, T.J.; Cogdill, A.P.; Morrissette, J.J.D.; DeNizio, J.E.; Reddy, S.; et al. Disruption of TET2 promotes the therapeutic efficacy of CD19-targeted T cells. Nature 2018, 558, 307–312. [Google Scholar] [PubMed]
- Schober, K.; Müller, T.R.; Gökmen, F.; Grassmann, S.; Effenberger, M.; Poltorak, M.; Stemberger, C.; Schumann, K.; Roth, T.L.; Marson, A.; et al. Orthotopic replacement of T-cell receptor alpha- and beta-chains with preservation of near-physiological T-cell function. Nat. Biomed. Eng. 2019, 3, 974–984. [Google Scholar] [CrossRef] [PubMed]
- Sarukhan, A.; Garcia, C.; Lanoue, A.; von Boehmer, H. Allelic Inclusion of T Cell Receptor α Genes Poses an Autoimmune Hazard Due to Low-Level Expression of Autospecific Receptors. Immunity 1998, 8, 563–570. [Google Scholar] [CrossRef] [Green Version]
- Coulie, P.G.; Karanikas, V.; Colau, D.; Lurquin, C.; Landry, C.; Marchand, M.; Dorval, T.; Brichard, V.; Boon, T. A monoclonal cytolytic T-lymphocyte response observed in a melanoma patient vaccinated with a tumor-specific antigenic peptide encoded by gene MAGE-3. Proc. Natl. Acad. Sci. USA 2001, 98, 10290–10295. [Google Scholar] [CrossRef] [Green Version]
- Thiel, U.; Schober, S.J.; Einspieler, I.; Kirschner, A.; Thiede, M.; Schirmer, D.; Gall, K.; Blaeschke, F.; Schmidt, O.; Jabar, S.; et al. Ewing sarcoma partial regression without GvHD by chondromodulin-I/HLA-A*02:01-specific allorestricted T cell receptor transgenic T cells. Oncoimmunology 2017, 6, e1312239. [Google Scholar]
- Graef, P.; Buchholz, V.R.; Stemberger, C.; Flossdorf, M.; Henkel, L.; Schiemann, M.; Drexler, I.; Höfer, T.; Riddell, S.R.; Busch, D.H. Serial Transfer of Single-Cell-Derived Immunocompetence Reveals Stemness of CD8+ Central Memory T Cells. Immunity 2014, 41, 116–126. [Google Scholar] [CrossRef] [Green Version]
- Kaneko, S.; Mastaglio, S.; Bondanza, A.; Ponzoni, M.; Sanvito, F.; Aldrighetti, L.; Radrizzani, M.; La Seta-Catamancio, S.; Provasi, E.; Mondino, A.; et al. IL-7 and IL-15 allow the generation of suicide gene–modified alloreactive self-renewing central memory human T lymphocytes. Blood 2009, 113, 1006–1015. [Google Scholar] [CrossRef] [Green Version]
- Buchholz, V.R.; Flossdorf, M.; Hensel, I.; Kretschmer, L.; Weissbrich, B.; Gräf, P.; Verschoor, A.; Schiemann, M.; Höfer, T.; Busch, D.H. Disparate Individual Fates Compose Robust CD8 + T Cell Immunity. Science 2013, 340, 630–635. [Google Scholar] [CrossRef] [PubMed]
- Blaeschke, F.; Thiel, U.; Kirschner, A.; Thiede, M.; Rubio, R.A.; Schirmer, D.; Kirchner, T.; Richter, G.H.; Mall, S.; Klar, R.; et al. Human HLA-A*02:01/CHM1+ allo-restricted T cell receptor transgenic CD8+ T cells specifically inhibit Ewing sarcoma growth in vitro and in vivo. Oncotarget 2016, 7, 43267–43280. [Google Scholar] [CrossRef] [PubMed]
- Schirmer, D.; Grünewald, T.G.P.; Klar, R.; Schmidt, O.; Wohlleber, D.; Rubio, R.A.; Uckert, W.; Thiel, U.; Bohne, F.; Busch, D.H.; et al. Transgenic antigen-specific, HLA-A*02:01-allo-restricted cytotoxic T cells recognize tumor-associated target antigen STEAP1 with high specificity. OncoImmunology 2016, 5, e1175795. [Google Scholar]
- Schober, S.J.; Thiede, M.; Gassmann, H.; Prexler, C.; Xue, B.; Schirmer, D.; Wohlleber, D.; Stein, S.; Grünewald, T.G.P.; Busch, D.H.; et al. MHC Class I-Restricted TCR-Transgenic CD4+ T Cells Against STEAP1 Mediate Local Tumor Control of Ewing Sarcoma In Vivo. Cells 2020, 9, 1581. [Google Scholar]
- Robinson, J.T.; Thorvaldsdóttir, H.; Wenger, A.M.; Zehir, A.; Mesirov, J.P. Variant Review with the Integrative Genomics Viewer. Cancer Res. 2017, 77, e31–e34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehár, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, M.; Schlegl, J.; Hahne, H.; Gholami, A.M.; Lieberenz, M.; Savitski, M.M.; Ziegler, E.; Butzmann, L.; Gessulat, S.; Marx, H.; et al. Mass-spectrometry-based draft of the human proteome. Nature 2014, 509, 582–587. [Google Scholar] [CrossRef] [PubMed]
- Acuto, O.; Reinherz, E.L. The human T-cell receptor. Structure and function. N. Engl. J. Med. 1985, 312, 1100–1111. [Google Scholar]
- von Heyking, K.; Calzada-Wack, J.; Göllner, S.; Neff, F.; Schmidt, O.; Hensel, T.; Schirmer, D.; Fasan, A.; Esposito, I.; Müller-Tidow, C.; et al. The endochondral bone protein CHM1 sustains an undifferentiated, invasive phenotype, promoting lung metastasis in Ewing sarcoma. Mol. Oncol. 2017, 11, 1288–1301. [Google Scholar]
- Biele, E.; Schober, S.J.; Prexler, C.; Thiede, M.; Heyking, K.V.; Gassmann, H.; Eck, J.; Xue, B.; Burdach, S.; Thiel, U. Monocyte Maturation Mediators Upregulate CD83, ICAM-1 and MHC Class 1 Expression on Ewing’s Sarcoma, Enhancing T Cell Cytotoxicity. Cells 2021, 10, 3070. [Google Scholar] [CrossRef]
- Ghandi, M.; Huang, F.W.; Jané-Valbuena, J.; Kryukov, G.V.; Lo, C.C.; McDonald, E.R., III; Barretina, J.; Gelfand, E.T.; Bielski, C.M.; Li, H.; et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 2019, 569, 503–508. [Google Scholar] [CrossRef] [PubMed]
- Staege, M.S.; Hutter, C.; Neumann, I.; Foja, S.; Hattenhorst, U.E.; Hansen, G.; Afar, D.; Burdach, S.E.G. DNA Microarrays Reveal Relationship of Ewing Family Tumors to Both Endothelial and Fetal Neural Crest-Derived Cells and Define Novel Targets. Cancer Res. 2004, 64, 8213–8221. [Google Scholar] [CrossRef] [Green Version]
- Thiel, U.; Pirson, S.; Müller-Spahn, C.; Conrad, H.; Busch, D.H.; Bernhard, H.; Burdach, S.; Richter, G.H.S. Specific recognition and inhibition of Ewing tumour growth by antigen-specific allo-restricted cytotoxic T cells. Br. J. Cancer 2011, 104, 948–956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, T.; Samaras, P.; Frejno, M.; Gessulat, S.; Barnert, M.; Kienegger, H.; Krcmar, H.; Schlegl, J.; Ehrlich, H.C.; Aiche, S.; et al. ProteomicsDB. Nucleic Acids Res. 2018, 46, D1271–D1281. [Google Scholar] [PubMed] [Green Version]
- Samaras, P.; Schmidt, T.; Frejno, M.; Gessulat, S.; Reinecke, M.; Jarzab, A.; Zecha, J.; Mergner, J.; Giansanti, P.; Ehrlich, H.-C.; et al. ProteomicsDB: A multi-omics and multi-organism resource for life science research. Nucleic Acids Res. 2019, 48, D1153–D1163. [Google Scholar] [CrossRef]
- Koch, R.; Gelderblom, H.; Haveman, L.; Brichard, B.; Jürgens, H.; Cyprova, S.; Berg, H.V.D.; Hassenpflug, W.; Raciborska, A.; Ek, T.; et al. High-Dose Treosulfan and Melphalan as Consolidation Therapy Versus Standard Therapy for High-Risk (Metastatic) Ewing Sarcoma. J. Clin. Oncol. 2022, 40, 2307–2320. [Google Scholar]
- Copelan, E.A. Hematopoietic stem-cell transplantation. N. Engl. J. Med. 2006, 354, 1813–1826. [Google Scholar]
- Thiel, U.; Schober, S.J.; Ranft, A.; Gassmann, H.; Jabar, S.; Gall, K.; von Lüttichau, I.; Wawer, A.; Koscielniak, E.; Diaz, M.A.; et al. No difference in survival after HLA mismatched versus HLA matched allogeneic stem cell transplantation in Ewing sarcoma patients with advanced disease. Bone Marrow Transpl. 2021, 56, 1550–1557. [Google Scholar] [CrossRef]
- Thiel, U.; Wawer, A.; Wolf, P.; Badoglio, M.; Santucci, A.; Klingebiel, T.; Basu, O.; Borkhardt, A.; Laws, H.-J.; Kodera, Y.; et al. No improvement of survival with reduced- versus high-intensity conditioning for allogeneic stem cell transplants in Ewing tumor patients. Ann. Oncol. 2011, 22, 1614–1621. [Google Scholar] [CrossRef]
- Nicolini, A.; Rossi, G.; Ferrari, P.; Carpi, A. Minimal residual disease in advanced or metastatic solid cancers: The G0-G1 state and immunotherapy are key to unwinding cancer complexity. Semin. Cancer Biol. 2020, 79, 68–82. [Google Scholar]
- Altvater, B.; Kailayangiri, S.; Lanuza, L.P.; Urban, K.; Greune, L.; Flügge, M.; Meltzer, J.; Farwick, N.; König, S.; Görlich, D.; et al. HLA-G and HLA-E Immune Checkpoints Are Widely Expressed in Ewing Sarcoma but Have Limited Functional Impact on the Effector Functions of Antigen-Specific CAR T Cells. Cancers 2021, 13, 2857. [Google Scholar] [CrossRef]
- Kirschner, A.; Thiede, M.; Grünewald, T.G.; Alba Rubio, R.; Richter, G.H.; Kirchner, T.; Busch, D.H.; Burdach, S.; Thiel, U. Pappalysin-1 T cell receptor transgenic allo-restricted T cells kill Ewing sarcoma in vitro and in vivo. Oncoimmunology 2017, 6, e1273301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brohl, A.S.; Sindiri, S.; Wei, J.S.; Milewski, D.; Chou, H.-C.; Song, Y.K.; Wen, X.; Kumar, J.; Reardon, H.V.; Mudunuri, U.S.; et al. Immuno-transcriptomic profiling of extracranial pediatric solid malignancies. Cell Rep. 2021, 37, 110047. [Google Scholar] [PubMed]
- Robbins, P.F.; Kassim, S.H.; Tran, T.L.N.; Crystal, J.S.; Morgan, R.A.; Feldman, S.A.; Yang, J.C.; Dudley, M.E.; Wunderlich, J.R.; Sherry, R.M.; et al. A Pilot Trial Using Lymphocytes Genetically Engineered with an NY-ESO-1–Reactive T-cell Receptor: Long-term Follow-up and Correlates with Response. Clin. Cancer Res. 2015, 21, 1019–1027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Loenen, M.M.; de Boer, R.; Amir, A.L.; Hagedoorn, R.S.; Volbeda, G.L.; Willemze, R.; van Rood, J.J.; Falkenburg, J.F.; Heemskerk, M.H. Mixed T cell receptor dimers harbor potentially harmful neoreactivity. Proc. Natl. Acad. Sci. USA 2010, 107, 10972–10977. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, C.J.; Zhao, Y.; Zheng, Z.; Rosenberg, S.A.; Morgan, R.A. Enhanced Antitumor Activity of Murine-Human Hybrid T-Cell Receptor (TCR) in Human Lymphocytes Is Associated with Improved Pairing and TCR/CD3 Stability. Cancer Res. 2006, 66, 8878–8886. [Google Scholar] [CrossRef] [Green Version]
- Sommermeyer, D.; Uckert, W. Minimal Amino Acid Exchange in Human TCR Constant Regions Fosters Improved Function of TCR Gene-Modified T Cells. J. Immunol. 2010, 184, 6223–6231. [Google Scholar] [CrossRef] [Green Version]
- Scholten, K.B.; Kramer, D.; Kueter, E.W.; Graf, M.; Schoedl, T.; Meijer, C.J.; Schreurs, M.W.; Hooijberg, E. Codon modification of T cell receptors allows enhanced functional expression in transgenic human T cells. Clin. Immunol. 2006, 119, 135–145. [Google Scholar] [CrossRef]
- Cohen, C.J.; Li, Y.F.; El-Gamil, M.; Robbins, P.F.; Rosenberg, S.A.; Morgan, R.A. Enhanced Antitumor Activity of T Cells Engineered to Express T-Cell Receptors with a Second Disulfide Bond. Cancer Res. 2007, 67, 3898–3903. [Google Scholar] [CrossRef] [Green Version]
- Provasi, E.; Genovese, P.; Lombardo, A.L.; Magnani, Z.I.; Liu, P.-Q.; Reik, A.; Chu, V.; Paschon, D.E.; Zhang, L.; Kuball, J.; et al. Editing T cell specificity towards leukemia by zinc finger nucleases and lentiviral gene transfer. Nat. Med. 2012, 18, 807–815. [Google Scholar] [CrossRef]
- Eyquem, J.; Mansilla-Soto, J.; Giavridis, T.; van der Stegen, S.J.C.; Hamieh, M.; Cunanan, K.M.; Odak, A.; Gönen, M.; Sadelain, M. Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 2017, 543, 113–117. [Google Scholar] [PubMed]
- Roth, T.L.; Puig-Saus, C.; Yu, R.; Shifrut, E.; Carnevale, J.; Li, P.J.; Hiatt, J.; Saco, J.; Krystofinski, P.; Li, H.; et al. Reprogramming human T cell function and specificity with non-viral genome targeting. Nature 2018, 559, 405–409. [Google Scholar] [PubMed]
- Müller, T.R.; Jarosch, S.; Hammel, M.; Leube, J.; Grassmann, S.; Bernard, B.; Effenberger, M.; Andrä, I.; Chaudhry, M.Z.; Käuferle, T.; et al. Targeted T cell receptor gene editing provides predictable T cell product function for immunotherapy. Cell Rep. Med. 2021, 2, 100374. [Google Scholar] [CrossRef]
- Moosmann, C.; Müller, T.R.; Busch, D.H.; Schober, K. Orthotopic T-cell receptor replacement in primary human T cells using CRISPR-Cas9-mediated homology-directed repair. STAR Protoc. 2022, 3, 101031. [Google Scholar]
- Santeramo, I.; Bagnati, M.; Harvey, E.J.; Hassan, E.; Surmacz-Cordle, B.; Marshall, D.; Di Cerbo, V. Vector Copy Distribution at a Single-Cell Level Enhances Analytical Characterization of Gene-Modified Cell Therapies. Mol. Ther.-Methods Clin. Dev. 2020, 17, 944–956. [Google Scholar] [CrossRef] [PubMed]
- Aiuti, A.; Cossu, G.; De Felipe, P.; Galli, M.C.; Narayanan, G.; Renner, M.; Stahlbom, A.; Schneider, C.K.; Voltz-Girolt, C. The Committee for Advanced Therapies’ of the European Medicines Agency Reflection Paper on Management of Clinical Risks Deriving from Insertional Mutagenesis. Hum. Gene Ther. Clin. Dev. 2013, 24, 47–54. [Google Scholar] [PubMed] [Green Version]
- Monjezi, R.; Miskey, C.; Gogishvili, T.; Schleef, M.; Schmeer, M.; Einsele, H.; Ivics, Z.; Hudecek, M. Enhanced CAR T-cell engineering using non-viral Sleeping Beauty transposition from minicircle vectors. Leukemia 2016, 31, 186–194. [Google Scholar] [CrossRef]
- Leibowitz, M.L.; Papathanasiou, S.; Doerfler, P.A.; Blaine, L.J.; Sun, L.; Yao, Y.; Zhang, C.-Z.; Weiss, M.J.; Pellman, D. Chromothripsis as an on-target consequence of CRISPR–Cas9 genome editing. Nat. Genet. 2021, 53, 895–905. [Google Scholar] [CrossRef]
- Cullot, G.; Boutin, J.; Toutain, J.; Prat, F.; Pennamen, P.; Rooryck, C.; Teichmann, M.; Rousseau, E.; Lamrissi-Garcia, I.; Guyonnet-Duperat, V.; et al. CRISPR-Cas9 genome editing induces megabase-scale chromosomal truncations. Nat. Commun. 2019, 10, 1136. [Google Scholar] [CrossRef] [Green Version]
- Slaymaker, I.M.; Gao, L.; Zetsche, B.; Scott, D.A.; Yan, W.X.; Zhang, F. Rationally engineered Cas9 nucleases with improved specificity. Science 2016, 351, 84–88. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xue, B.; von Heyking, K.; Gassmann, H.; Poorebrahim, M.; Thiede, M.; Schober, K.; Mautner, J.; Hauer, J.; Ruland, J.; Busch, D.H.; et al. T Cells Directed against the Metastatic Driver Chondromodulin-1 in Ewing Sarcoma: Comparative Engineering with CRISPR/Cas9 vs. Retroviral Gene Transfer for Adoptive Transfer. Cancers 2022, 14, 5485. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14225485
Xue B, von Heyking K, Gassmann H, Poorebrahim M, Thiede M, Schober K, Mautner J, Hauer J, Ruland J, Busch DH, et al. T Cells Directed against the Metastatic Driver Chondromodulin-1 in Ewing Sarcoma: Comparative Engineering with CRISPR/Cas9 vs. Retroviral Gene Transfer for Adoptive Transfer. Cancers. 2022; 14(22):5485. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14225485
Chicago/Turabian StyleXue, Busheng, Kristina von Heyking, Hendrik Gassmann, Mansour Poorebrahim, Melanie Thiede, Kilian Schober, Josef Mautner, Julia Hauer, Jürgen Ruland, Dirk H. Busch, and et al. 2022. "T Cells Directed against the Metastatic Driver Chondromodulin-1 in Ewing Sarcoma: Comparative Engineering with CRISPR/Cas9 vs. Retroviral Gene Transfer for Adoptive Transfer" Cancers 14, no. 22: 5485. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14225485