

Ewing Sarcoma as Secondary Malignant Neoplasm—Epidemiological and Clinical Analysis of an International Trial Registry
 ,
,  , , and
, , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Cohorts and Eligibility Criteria
2.2. Statistical Analysis
3. Results
3.1. Epidemiology of Primary Malignancies
3.2. Patient Characteristics and Clinical Features of Ewing Sarcoma as Secondary Malignant Neoplasms
3.3. Treatment Management of Primary Malignancies
3.4. Treatment Management of Ewing Sarcoma as Secondary Malignant Neoplasms
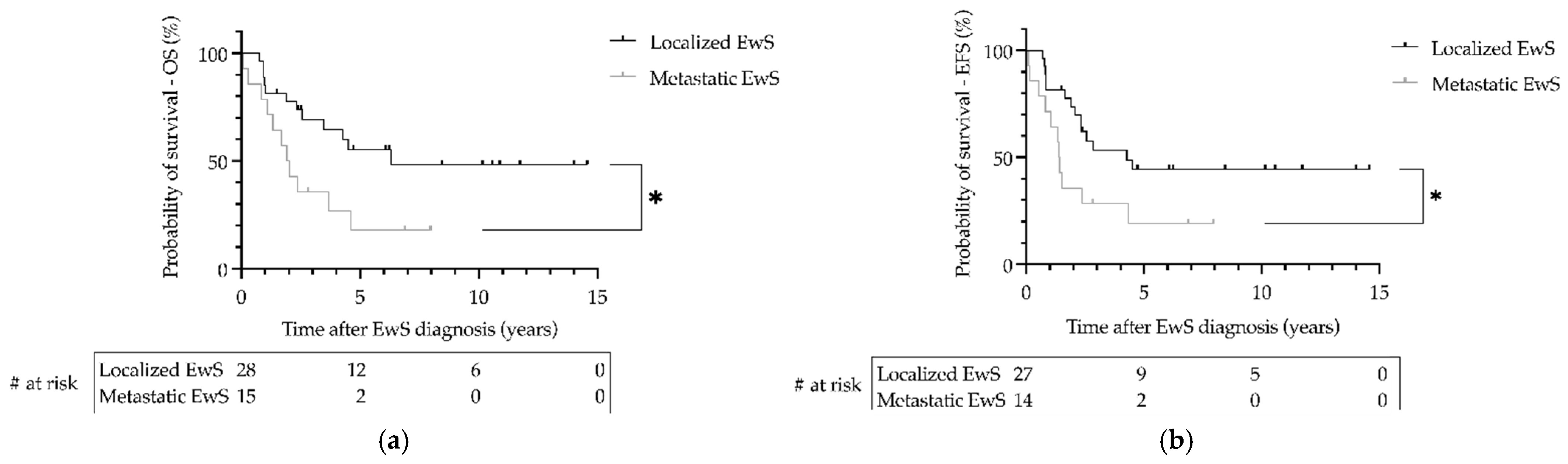
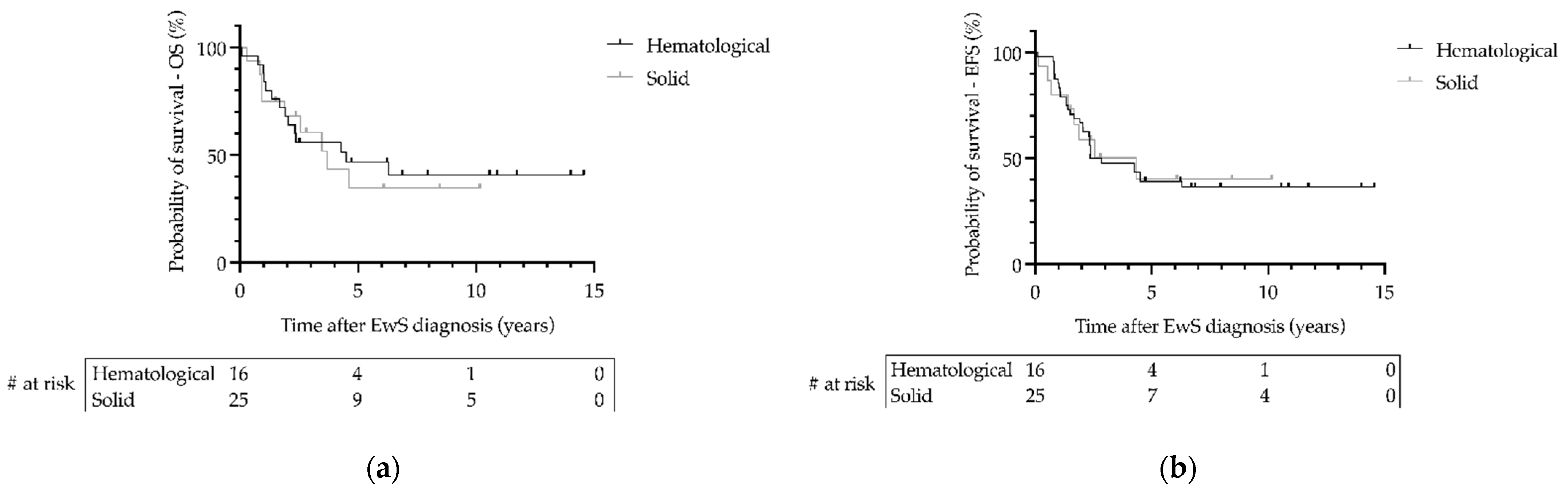
3.5. Outcome of Patients with Ewing Sarcoma as Secondary Malignant Neoplasms
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gatta, G.; Botta, L.; Rossi, S.; Aareleid, T.; Bielska-Lasota, M.; Clavel, J.; Dimitrova, N.; Jakab, Z.; Kaatsch, P.; Lacour, B.; et al. Childhood cancer survival in Europe 1999-2007: Results of EUROCARE-5--a population-based study. Lancet Oncol. 2014, 15, 35–47. [Google Scholar] [CrossRef] [PubMed]
- CCI Europe. Available online: https://ccieurope.eu/ (accessed on 19 July 2022).
- Bhakta, N.; Liu, Q.; Ness, K.K.; Baassiri, M.; Eissa, H.; Yeo, F.; Chemaitilly, W.; Ehrhardt, M.J.; Bass, J.; Bishop, M.W.; et al. The cumulative burden of surviving childhood cancer: An initial report from the St Jude Lifetime Cohort Study (SJLIFE). Lancet 2017, 390, 2569–2582. [Google Scholar] [CrossRef]
- Bhatia, S.; Sklar, C. Second cancers in survivors of childhood cancer. Nat. Rev. Cancer 2002, 2, 124–132. [Google Scholar] [CrossRef]
- Curtis, R.E.; Freedman, D.M.; Ron, E.; Ries, L.A.G.; Hacker, D.G.; Edwards, B.K.; Tucker, M.A.; Fraumeni, J.F., Jr. (Eds.) New Malignancies Among Cancer Survivors: SEER Cancer Registries, 1973-2000; NIH Publ. No. 05-5302; National Cancer Institute NIH: Bethesda, MD, USA, 2006. [Google Scholar]
- Ng, A.K.; Kenney, L.B.; Gilbert, E.S.; Travis, L.B. Secondary malignancies across the age spectrum. Semin. Radiat. Oncol. 2010, 20, 67–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grunewald, T.G.P.; Cidre-Aranaz, F.; Surdez, D.; Tomazou, E.M.; de Alava, E.; Kovar, H.; Sorensen, P.H.; Delattre, O.; Dirksen, U. Ewing sarcoma. Nat. Rev. Dis. Prim. 2018, 4, 5. [Google Scholar] [CrossRef] [PubMed]
- Sand, L.G.; Szuhai, K.; Hogendoorn, P.C. Sequencing Overview of Ewing Sarcoma: A Journey across Genomic, Epigenomic and Transcriptomic Landscapes. Int J. Mol. Sci. 2015, 16, 16176–16215. [Google Scholar] [CrossRef] [Green Version]
- Delattre, O.; Zucman, J.; Plougastel, B.; Desmaze, C.; Melot, T.; Peter, M.; Kovar, H.; Joubert, I.; de Jong, P.; Rouleau, G.; et al. Gene fusion with an ETS DNA-binding domain caused by chromosome translocation in human tumours. Nature 1992, 359, 162–165. [Google Scholar] [CrossRef]
- Gaspar, N.; Hawkins, D.S.; Dirksen, U.; Lewis, I.J.; Ferrari, S.; Le Deley, M.C.; Kovar, H.; Grimer, R.; Whelan, J.; Claude, L.; et al. Ewing Sarcoma: Current Management and Future Approaches Through Collaboration. J. Clin. Oncol. 2015, 33, 3036–3046. [Google Scholar] [CrossRef] [Green Version]
- Henderson, T.O.; Rajaraman, P.; Stovall, M.; Constine, L.S.; Olive, A.; Smith, S.A.; Mertens, A.; Meadows, A.; Neglia, J.P.; Hammond, S.; et al. Risk factors associated with secondary sarcomas in childhood cancer survivors: A report from the childhood cancer survivor study. Int. J. Radiat. Oncol. Biol. Phys. 2012, 84, 224–230. [Google Scholar] [CrossRef] [Green Version]
- Schiffman, J.D.; Wright, J. Ewing’s Sarcoma and Second Malignancies. Sarcoma 2011, 2011, 736841. [Google Scholar] [CrossRef]
- Wolpert, F.; Grotzer, M.A.; Niggli, F.; Zimmermann, D.; Rushing, E.; Bode-Lesniewska, B. Ewing’s Sarcoma as a Second Malignancy in Long-Term Survivors of Childhood Hematologic Malignancies. Sarcoma 2016, 2016, 5043640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Applebaum, M.A.; Worch, J.; Matthay, K.K.; Goldsby, R.; Neuhaus, J.; West, D.C.; Dubois, S.G. Clinical features and outcomes in patients with extraskeletal Ewing sarcoma. Cancer 2011, 117, 3027–3032. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hiramoto, N.; Kobayashi, Y.; Nomoto, J.; Maruyama, D.; Watanabe, T.; Tochigi, N.; Furuta, K.; Takeda, K.; Chuman, H.; Yagyu, S.; et al. Ewing sarcoma arising after treatment of diffuse large B-cell lymphoma. Jpn. J. Clin. Oncol. 2013, 43, 417–421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vanhapiha, N.; Knuutila, S.; Vettenranta, K.; Lohi, O. Burkitt lymphoma and Ewing sarcoma in a child with Williams syndrome. Pediatr. Blood Cancer 2014, 61, 1877–1879. [Google Scholar] [CrossRef] [Green Version]
- Applebaum, M.A.; Goldsby, R.; Neuhaus, J.; DuBois, S.G. Clinical features and outcomes in patients with secondary Ewing sarcoma. Pediatr. Blood Cancer 2013, 60, 611–615. [Google Scholar] [CrossRef] [Green Version]
- Choi, D.K.; Helenowski, I.; Hijiya, N. Secondary malignancies in pediatric cancer survivors: Perspectives and review of the literature. Int. J. Cancer 2014, 135, 1764–1773. [Google Scholar] [CrossRef]
- Srivastava, S.; Wang, S.; Tong, Y.A.; Pirollo, K.; Chang, E.H. Several mutant p53 proteins detected in cancer-prone families with Li-Fraumeni syndrome exhibit transdominant effects on the biochemical properties of the wild-type p53. Oncogene 1993, 8, 2449–2456. [Google Scholar]
- Armstrong, G.T.; Liu, W.; Leisenring, W.; Yasui, Y.; Hammond, S.; Bhatia, S.; Neglia, J.P.; Stovall, M.; Srivastava, D.; Robison, L.L. Occurrence of multiple subsequent neoplasms in long-term survivors of childhood cancer: A report from the childhood cancer survivor study. J. Clin. Oncol. 2011, 29, 3056–3064. [Google Scholar] [CrossRef]
- Robison, L.L.; Mertens, A. Second tumors after treatment of childhood malignancies. Hematol. Oncol. Clin. N. Am. 1993, 7, 401–415. [Google Scholar] [CrossRef]
- Erdmann, F.; Kaatsch, P.; Grabow, D.; Spix, C. German Childhood Cancer Registry—Annual Report 2019 (1980–2018); Institute of Medical Biostatistics, Epidemiology and Informatics (IMBEI) at the University Medical Center of the Johannes Gutenberg University Mainz: Mainz, Germany, 2020. [Google Scholar]
- Zöllner, S.K.; Amatruda, J.F.; Bauer, S.; Collaud, S.; de Alava, E.; DuBois, S.G.; Hardes, J.; Hartmann, W.; Kovar, H.; Metzler, M.; et al. Ewing Sarcoma-Diagnosis, Treatment, Clinical Challenges and Future Perspectives. J. Clin. Med. 2021, 10, 1685. [Google Scholar] [CrossRef]
- Spunt, S.L.; Rodriguez-Galindo, C.; Fuller, C.E.; Harper, J.; Krasin, M.J.; Billups, C.A.; Khoury, J.D. Ewing sarcoma-family tumors that arise after treatment of primary childhood cancer. Cancer 2006, 107, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Mirabello, L.; Troisi, R.J.; Savage, S.A. Osteosarcoma incidence and survival rates from 1973 to 2004: Data from the Surveillance, Epidemiology, and End Results Program. Cancer 2009, 115, 1531–1543. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henderson, T.O.; Whitton, J.; Stovall, M.; Mertens, A.C.; Mitby, P.; Friedman, D.; Strong, L.C.; Hammond, S.; Neglia, J.P.; Meadows, A.T.; et al. Secondary sarcomas in childhood cancer survivors: A report from the Childhood Cancer Survivor Study. J. Natl. Cancer Inst. 2007, 99, 300–308. [Google Scholar] [CrossRef] [PubMed]
- Zichova, A.; Eckschlager, T.; Ganevova, M.; Malinova, B.; Luks, A.; Kruseova, J. Subsequent neoplasms in childhood cancer survivors. Cancer Epidemiol. 2020, 68, 101779. [Google Scholar] [CrossRef] [PubMed]
- Cotterill, S.J.; Ahrens, S.; Paulussen, M.; Jurgens, H.F.; Voute, P.A.; Gadner, H.; Craft, A.W. Prognostic factors in Ewing’s tumor of bone: Analysis of 975 patients from the European Intergroup Cooperative Ewing’s Sarcoma Study Group. J. Clin. Oncol. 2000, 18, 3108–3114. [Google Scholar] [CrossRef]
- Ladenstein, R.; Potschger, U.; Le Deley, M.C.; Whelan, J.; Paulussen, M.; Oberlin, O.; van den Berg, H.; Dirksen, U.; Hjorth, L.; Michon, J.; et al. Primary disseminated multifocal Ewing sarcoma: Results of the Euro-EWING 99 trial. J. Clin. Oncol. 2010, 28, 3284–3291. [Google Scholar] [CrossRef]
- Kremeike, K.; Juergens, C.; Alz, H.; Reinhardt, D. Patients’ Adherence in the Maintenance Therapy of Children and Adolescents with Acute Lymphoblastic Leukemia. Klin. Padiatr. 2015, 227, 329–334. [Google Scholar] [CrossRef]
- Mody, R.; Li, S.; Dover, D.C.; Sallan, S.; Leisenring, W.; Oeffinger, K.C.; Yasui, Y.; Robison, L.L.; Neglia, J.P. Twenty-five-year follow-up among survivors of childhood acute lymphoblastic leukemia: A report from the Childhood Cancer Survivor Study. Blood 2008, 111, 5515–5523. [Google Scholar] [CrossRef] [Green Version]
- Mulrooney, D.A.; Dover, D.C.; Li, S.; Yasui, Y.; Ness, K.K.; Mertens, A.C.; Neglia, J.P.; Sklar, C.A.; Robison, L.L.; Davies, S.M.; et al. Twenty years of follow-up among survivors of childhood and young adult acute myeloid leukemia: A report from the Childhood Cancer Survivor Study. Cancer 2008, 112, 2071–2079. [Google Scholar] [CrossRef]
- Cooper, S.L.; Brown, P.A. Treatment of pediatric acute lymphoblastic leukemia. Pediatr. Clin. N. Am. 2015, 62, 61–73. [Google Scholar] [CrossRef] [Green Version]
- Moore, M.J. Clinical pharmacokinetics of cyclophosphamide. Clin. Pharm. 1991, 20, 194–208. [Google Scholar] [CrossRef] [PubMed]
- Flinn, I.W.; Neuberg, D.S.; Grever, M.R.; Dewald, G.W.; Bennett, J.M.; Paietta, E.M.; Hussein, M.A.; Appelbaum, F.R.; Larson, R.A.; Moore, D.F., Jr.; et al. Phase III trial of fludarabine plus cyclophosphamide compared with fludarabine for patients with previously untreated chronic lymphocytic leukemia: US Intergroup Trial E2997. J. Clin. Oncol. 2007, 25, 793–798. [Google Scholar] [CrossRef] [PubMed]
- Neglia, J.P.; Meadows, A.T.; Robison, L.L.; Kim, T.H.; Newton, W.A.; Ruymann, F.B.; Sather, H.N.; Hammond, G.D. Second neoplasms after acute lymphoblastic leukemia in childhood. N. Engl. J. Med. 1991, 325, 1330–1336. [Google Scholar] [CrossRef] [PubMed]
- Eulo, V.; Lesmana, H.; Doyle, L.A.; Nichols, K.E.; Hirbe, A.C. Secondary Sarcomas: Biology, Presentation, and Clinical Care. Am. Soc. Clin. Oncol. Educ. Book 2020, 40, 1–12. [Google Scholar] [CrossRef]
- Grunewald, T.G.; Bernard, V.; Gilardi-Hebenstreit, P.; Raynal, V.; Surdez, D.; Aynaud, M.M.; Mirabeau, O.; Cidre-Aranaz, F.; Tirode, F.; Zaidi, S.; et al. Chimeric EWSR1-FLI1 regulates the Ewing sarcoma susceptibility gene EGR2 via a GGAA microsatellite. Nat. Genet. 2015, 47, 1073–1078. [Google Scholar] [CrossRef] [Green Version]
- Tirode, F.; Surdez, D.; Ma, X.; Parker, M.; Le Deley, M.C.; Bahrami, A.; Zhang, Z.; Lapouble, E.; Grossetete-Lalami, S.; Rusch, M.; et al. Genomic landscape of Ewing sarcoma defines an aggressive subtype with co-association of STAG2 and TP53 mutations. Cancer Discov. 2014, 4, 1342–1353. [Google Scholar] [CrossRef] [Green Version]
- Randall, R.L.; Lessnick, S.L.; Jones, K.B.; Gouw, L.G.; Cummings, J.E.; Cannon-Albright, L.; Schiffman, J.D. Is There a Predisposition Gene for Ewing’s Sarcoma? J. Oncol. 2010, 2010, 397632. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| EwS Trial | EICESS 92 | EURO E.W.I.N.G. 99 | EWING 2008 | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Number of cycles | 14 | 14 | 14 | 8 | 8 | 14 | 14 | 8 | 14 | 15 | ||||||||
| Risk strata | SR | HR | R1 = SR | R2 = HR | R3 = VHR | R1 = SR | R2 = HR | R3 = VHR | ||||||||||
| Clinical criteria for risk strata | localized vol. < 100 mL | metastasized vol ≥ 100 mL | localized vol. ≥ 200 mL <10% viable cells * | locolazied/ pulmonary mets vol. ≥ 200 mL ≥10% viable cells * | metastasized | localized <10% viable cells * | localized/ pulmonary mets ≥10% viable cells * | metastasized | ||||||||||
| Chemotherapeutic regimen | VAIA + VACA | VAIA | VAIA | EVAIA | VIDE + VAC ♀ | VIDE + VAI ♂ | VIDE + VAI | VIDE + VAI + BU/MEL | VIDE + VAI + ME/ME | VIDE + VAI + TREO/MEL | VIDE + VAI + BU/MEL | VIDE + VAI | VIDE + VAC ♀ | VIDE + VAI ♂ | VIDE + VAI | VIDE + VAI + BU/MEL | VIDE + VAC | VIDE + VAC + TREO/MEL |
| Clinical Features at Diagnosis (Avaiblabe Data for EwS as SMN) | EICESS 92, Euro-E.W.I.N.G. 99, EWING 2008 | p Value | ||
|---|---|---|---|---|
| EwS as Primary Malignancy (%) | EwS as SMN (%) | |||
| Gender (n = 42) | Male | 2238 (58.9) | 20 (47.6) | 0.16 |
| Female | 1564 (41.1) | 22 (52.4) | ||
| Age (n = 42) | <14 years | 1609 (42.3) | 14 (33.3) | 0.27 |
| ≥14 years | 2193 (57.7) | 28 (66.7) | ||
| Tumor stage (n = 42) | Localized | 2496 (67.1) | 28 (66.7) | 1.00 |
| Metastasized | 1223 (32.9) | 14 (33.3) | ||
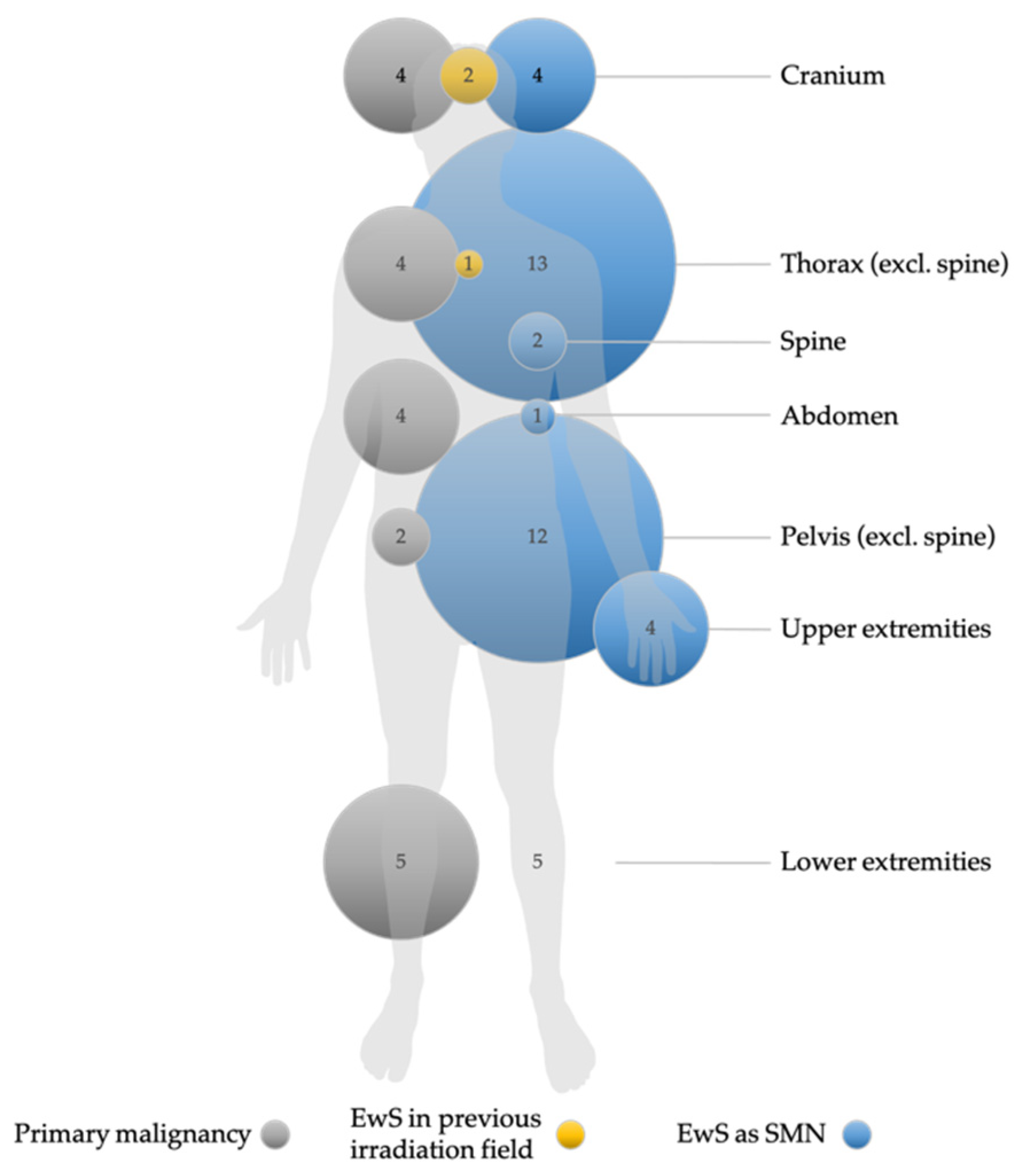
| Tumor localization (n = 41) # | Cranium | 152 (4.1) | 4 (9.75) | 0.03 |
| Thorax (excl. spine) | 628 (16.9) | 11 (26.8) | ||
| Spine | 252 (6.8) | 1 (2.4) | ||
| Extraosseous # (incl. abdomen) | 552 (14.9) | 9 (22.0) | ||
| Pelvis (excl. spine) | 841 (22.6) | 9 (22.0) | ||
| Upper extremities | 262 (7.0) | 3 (7.3) | ||
| Lower extremities | 1030 (27.7) | 4 (9.75) | ||
| Tumor volume (n = 38) | <200 mL | 2000 (57.7) | 23 (60.5) | 0.75 |
| ≥200 mL | 1468 (42.3) | 15 (39.5) | ||
| Response assessment | ||||
| Histological regression (Salzer-Kuntschik *) (n = 18) | <10% | 1375 (71.9) | 10 (55.6) | 0.19 |
| ≥10% | 538 (28.1) | 8 (44.4) | ||
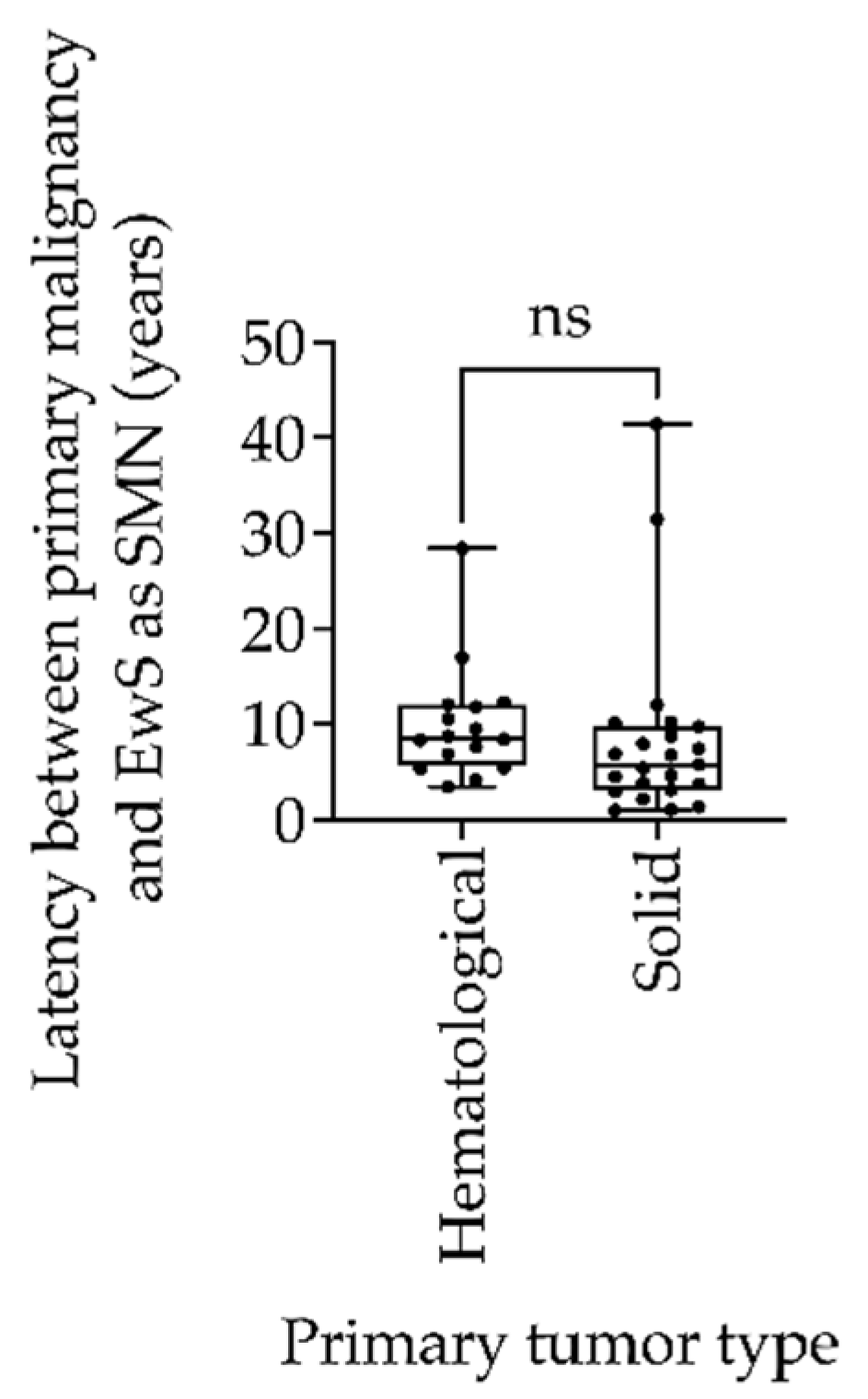
| Trial | Patient (n) | Primary Malignancy | Latency (Years) | Risk Strata | Follow-Up (Years) | Outcome |
|---|---|---|---|---|---|---|
| EICESS 92 | 1 | Retinoblastoma | 12 | R3 | 2.4 | 1 |
| 2 | Lymphoma | 7.3 | R2p | 4.1 | 1 | |
| 3 | Melanoma | 3.1 | SR | 10.9 | 0 | |
| 4 | Testicular teratoma | 6.8 | R3 | 1.7 | 1 | |
| 5 | Cervix carcinoma | 21 | SR | 4.3 | 1 | |
| 6 # | Rhabdoid tumor | 7.4 | R2p | 2.0 | 1 | |
| 7 | Malignant hemangiopericytoma | 1.3 | SR | 4.5 | 1 | |
| 8 | Osteosarcoma | 2.9 | SR | 14 | 0 | |
| Euro-E.W.I.N.G. 99 | 9 | ALL | 11.8 | R2pulm | 3.68 | 1 |
| 10 | ALL | 12.3 | R2loc | 0.93 | 1 | |
| 11 | ALL | 6.8 | R1 | 8.45 | 0 | |
| 12 | Retinoblastoma | 6.2 | R3 | 1.92 | 1 | |
| 13 | ALL | 8.3 | R1 | 10.15 | 0 | |
| 14 | Non-Hodgkin lymphoma | 8.75 | R1 | 14.6 | 0 | |
| 15 | Ovarian germ cell tumor | 3.8 | R1 | 10.6 | 0 | |
| 16 | ALL | 5.4 | R1 | 6.1 | 0 | |
| 17 | Nephroblastoma | 4.5 | R1 | 11.7 | 0 | |
| 18 | Hepatoblastoma | 6.9 | R3 | 1.1 | 1 | |
| 19 | ALL | 8.7 | R3 | 0.9 | 1 | |
| 20 | Hodgkin lymphoma | 17.0 | R2pulm | 0.3 | 1 | |
| 21 | Hodgkin lymphoma | 8.4 | R1 | 3.5 | 1 | |
| 22 | Renal cell carcinoma | 1.0 | R3 | 1.3 | 1 | |
| 23 | Hodgkin lymphoma | 28.4 | R1 | 2.6 | 1 | |
| 24 | Langerhans cell histiocytosis | 12.0 | R1 | 1.9 | 1 | |
| EWING 2008 | 25 | Neuroblastoma | 4.6 | R2loc | 2.33 | 1 |
| 26 | Osteosarcoma | 7.9 | R2loc | 1.0 | 1 | |
| 27 | Breast Cancer | 5.4 | R3 | 8.0 | 0 | |
| 28 | Sweat gland carcinoma | 10.1 | R2pulm | 6.9 | 0 | |
| 29 | Synovial sarcoma | 31.5 | R2loc | 1.0 | 1 | |
| 30 | Breast cancer | 3.8 | R1 | 6.2 | 0 | |
| 31 | Rhabdomyosarcoma | 10.2 | R1 | 6.3 | 1 | |
| 32 | ALL | 10.6 | R1 | 6.1 | 0 | |
| 33 | Seminoma | 41.4 | R2pulm | 0.1 | 1 | |
| 34 | Chronic myeloid leukemia | 3.5 | R1 | 0.9 | 1 | |
| 35 | Rhabdomyosarcoma | 5.8 | R2loc | 4.7 | 0 | |
| 36 | Non-Hodgkin lymphoma | 4.1 | R2pulm | 2.8 | 0 | |
| 37 | Non-Hodgkin lymphoma | 5.3 | R1 | 2.4 | 0 | |
| 38 | Hepatocellular carcinoma | 9.7 | NA | 2.5 | 0 | |
| 39 | Breast cancer | 2.1 | NA | 2.4 | 0 | |
| 40 | Hodgkin lymphoma | 9.5 | R1 | 1.5 | 0 | |
| 41 | Liposarcoma | 1.1 | NA | 0.8 | 1 | |
| 42 | Breast cancer | NA | NA | NA | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zöllner, S.K.; Kauertz, K.L.; Kaiser, I.; Kerkhoff, M.; Schaefer, C.; Tassius, M.; Jabar, S.; Jürgens, H.; Ladenstein, R.; Kühne, T.; et al. Ewing Sarcoma as Secondary Malignant Neoplasm—Epidemiological and Clinical Analysis of an International Trial Registry. Cancers 2022, 14, 5935. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14235935
Zöllner SK, Kauertz KL, Kaiser I, Kerkhoff M, Schaefer C, Tassius M, Jabar S, Jürgens H, Ladenstein R, Kühne T, et al. Ewing Sarcoma as Secondary Malignant Neoplasm—Epidemiological and Clinical Analysis of an International Trial Registry. Cancers. 2022; 14(23):5935. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14235935
Chicago/Turabian StyleZöllner, Stefan K., Katja L. Kauertz, Isabelle Kaiser, Maximilian Kerkhoff, Christiane Schaefer, Madita Tassius, Susanne Jabar, Heribert Jürgens, Ruth Ladenstein, Thomas Kühne, and et al. 2022. "Ewing Sarcoma as Secondary Malignant Neoplasm—Epidemiological and Clinical Analysis of an International Trial Registry" Cancers 14, no. 23: 5935. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers14235935