
Tumor-Derived Extracellular Vesicles in Cancer Immunoediting and Their Potential as Oncoimmunotherapeutics

Abstract
:Simple Summary
Abstract
1. Introduction
2. Tumor-Derived Extracellular Vesicles and Tumor-Supportive Cells
2.1. TEVs Modulate Macrophage Activity in TME

{kind=link}
{kind=link}
| Cancer Type | Cellular Source | Vesicular Cargo | The Main Result | Refs. |
|---|---|---|---|---|
| Breast cancer | MCF10A MCF10AT MCF10CA1a MDA-MB-231 | Anx II | Activated NF-B, p38MAPK, and STAT3 pathways in macrophages, leading to increased IL-6 and TNF-α secretion | [31] |
| C57BL/6 EO771 | gp130 | Caused macrophages to shift from a normal to a polarized phenotype such as TAM via activation of the IL-6 response pathway and STAT3. | [28] | |
| 4T1 | miR-125b-1-3p, miR-100-5p, and miR-183-5p | Inhibited the expression of PPP2CA, which could promote the release of pro-inflammatory cytokines such as IL-1b, IL-6, and TNF-a from macrophages stimulating tumor invasion. | [20] | |
| MDA-MB-231 | Vesicular CD63 protein | Polarized and activated macrophages, in which CD206 (a marker for M2) was expressed more than NOS2 (a marker for M1). | [36] | |
| Prostate cancer | PC3 | miRNA Let-7b | Prostate-derived extracellular vesicles had more miRNA Let-7b than cellular miRNA Let-7b can lead to macrophage polarization. | [37] |
| Lung cancer | A549 | Vesicular cargoes | Altered transcriptomic and bioenergetic profiles of macrophages, forced them to polarize to an M2 phenotype. | [38] |
| NCI-H1437 NCI-H1792 NCI-H2087 | miR-103a | Polarized monocytes toward immunosuppressive M2-type macrophages. | [39] | |
| A549 H1299 | Vesicular cargoes | Enhanced the levels of MMP2, MMP9 CD163, TNF-, IL-8, IL-6, and IL-10 and decreased expression of iNOS which led macrophages to exhibit a dual M1/M2 phenotype | [40] | |
| A549 H1299 | Vesicular PRPS2 | Induced M2 polarization and led to drug resistance of cancer cells. | [41] | |
| Hepatocellular carcinoma (HCC) | PLC/PRF/5 | Long non-coding RNAs (lncRNA) TUC339 | Caused macrophage polarization to be more immunosuppressive. | [42] |
| Hepa1-6 H22 | miR-146a-5p | Enhanced M2 polarization by triggering NF-B signaling and producing pro-inflammatory proteins | [34] | |
| Colorectal cancer(CRC) | DLD-1 | miR-145 | Induced M2 polarization via upregulation of IL-10 and downregulation of HDAC11. | [43] |
| Blood samples from CRC patients HCT116 HT29 | miR-106b | Contributed to M2 polarization of macrophages via significant increase in the miR-106b level in macrophages. It directly suppressed programmed cell death 4 (PDCD4) at a post-transcription level that led to an activated PI3Kγ, AKT, and mTOR signaling cascade. | [35] | |
| Blood samples from CRC patients HCT-8 LoVo HT-29 Caco-2 | miR-934 | Induced M2 macrophage polarization by activating the PI3K/AKT signaling pathway and downregulating PTEN. | [30] | |
| CT-26 SW620 | Cytoskeleton-centric proteins | In macrophages, caused cytoskeleton reorganization via promoting elongation and F-actin polarization. | [44] | |
| Blood samples from CRC patients HCT116 DLD-1 HT29 | miR-1246 | Reprogrammed macrophages into the cancer-promoting state after macrophage uptake. | [45] | |
| Blood samples from CRC patients DLD1 HCT116 Lovo SW480 SW620 HT29 CaR-1 RKO Colo205 Colo320DM | miR-203 | Promoted M2 polarization, which modulated liver metastasis of colon cancer cells. | [46] | |
| Epithelial ovarian cancer | SKOV3 | miR-21-3p, miR-181d-5p, and miR-125b-5p | Promoted M2 macrophage polarization results in epithelial ovarian cancer cell proliferation and migration under hypoxic circumstances. | [47] |
| Glioblastoma | GSC20 GSC276 U87 | Vesicular cargoes | The presence of phospho-STAT3 in TEVs switched monocytes toward the tumor-supportive M2 phenotype | [33] |
| U87MG SBN19 U251 | FasL, TRAIL, CTLA-4, CD39, and CD73 | Promoted M2 polarization by activating the NF-κB pathway in macrophages | [48] | |
| U251 | Vesicular cargo | Induced M2 polarization leading to tumor growth via promoting TAM Arginase-1+ exosome secretion | [49] | |
| Oral squamous cell carcinoma | SCC-9 CAL-27 | miR-29a-3p | Targeted macrophages directly, and activated p-STAT1 to promote M2 expression | [32] |
| Cal-27 | CMTM6 | Delivered CMTM6 to macrophages and induced M2-like macrophage polarization by activating ERK1/2 signaling | [29] | |
| Ovarian cancer | Blood samples from overian cancer patients Skov3 A2780 | miR-222 | Induced M2 polarization of macrophages by activating STAT3 pathway | [50] |
2.2. TEVs Modulate Fibroblast Activity in TME
2.3. TEVs Effect on MDSC Formation in TME
| Cancer Type | Cellular Source | Vesicular Cargo | The Main Result | Refs. |
|---|---|---|---|---|
| Breast cancer | 4T1 tumor model in BALB/c mice | PGE2 and TGF-β | Induced the differentiation of IMCs to MDSC expressing IL-6, Cox2, VEGF, and arginase-1. | [75] |
| MCF-7 4T1 MDA-MB-231 | PD-L1+ | Boosted tumor growth and accumulation of MDSCs and M2 in the TME. | [76] | |
| 4T1 | Vesicular cargoes | Differentiated bone marrow cells into MDSCs | [79] | |
| 4T1 tumor-bearing mice plasma 4T1 | miR-181a and miR-9 | Stimulated MDSC differentiation by inhibiting SOCS3 and PIAS3 (regulators of the JAK/STAT signaling pathway). | [80] | |
| Gastric cancer | MKN-28 MKN-45 SGC-7901 | Vesicular cargo | Increased frequency of MDSC, and decreased CD8+ T and NK cells. | [22] |
| Renal cancer | RenCa | HSP 70 | Antigen-specific immunosuppression effect on CTL | [78] |
| Glioblastoma | Blood samples from glioma patients | miR-1246 | Induced MDSCs via specificity phosphatase 3 (DUSP3) and ERK-dependent manner. | [81] |
| P3 G422 GL261 U87 | miR-29a | Increased MDSCs via interaction with high-mobility group box transcription factor 1 (Hbp1) and protein kinase cAMP-dependent type I regulatory subunit alpha (Prkar1a). | [82] | |
| Blood samples from glioma patient Human astrocytes supernatant | Vesicular cargo | Acting on MDSC, reduced T-cell immune response in an indirect manner. | [83] | |
| Blood samples from glioma patient | PD-L1 | Induced immunosuppressive monocytes, including MDSCs and nonclassical monocytes. | [77] | |
| Lung cancer (LC) | 95D H292 H358 | miR-21a | Induced MDSC expression by downregulation of the PDCD4 protein. | [84] |
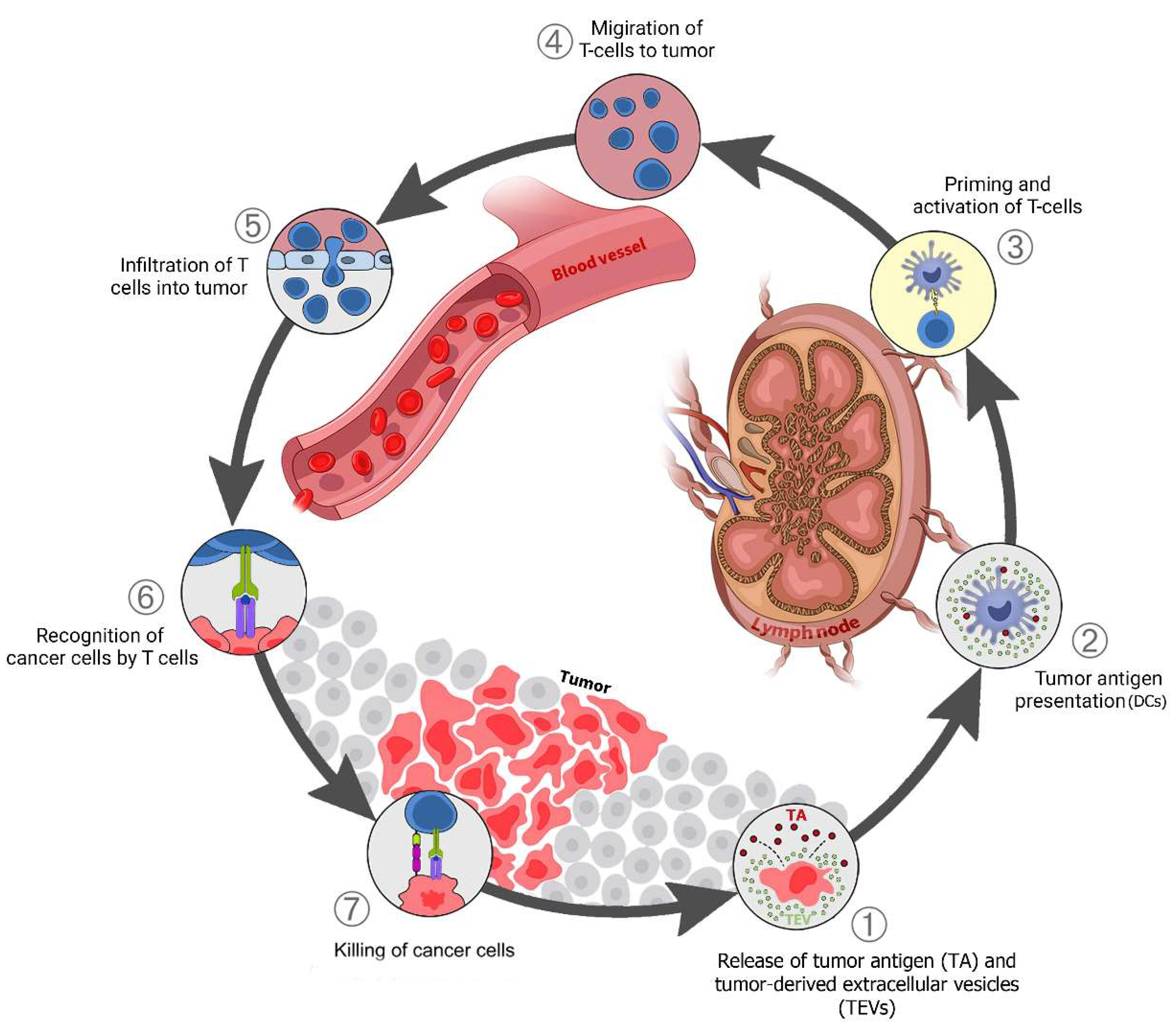
3. TEVs-Mediated Communication between Tumor and Immune Cells
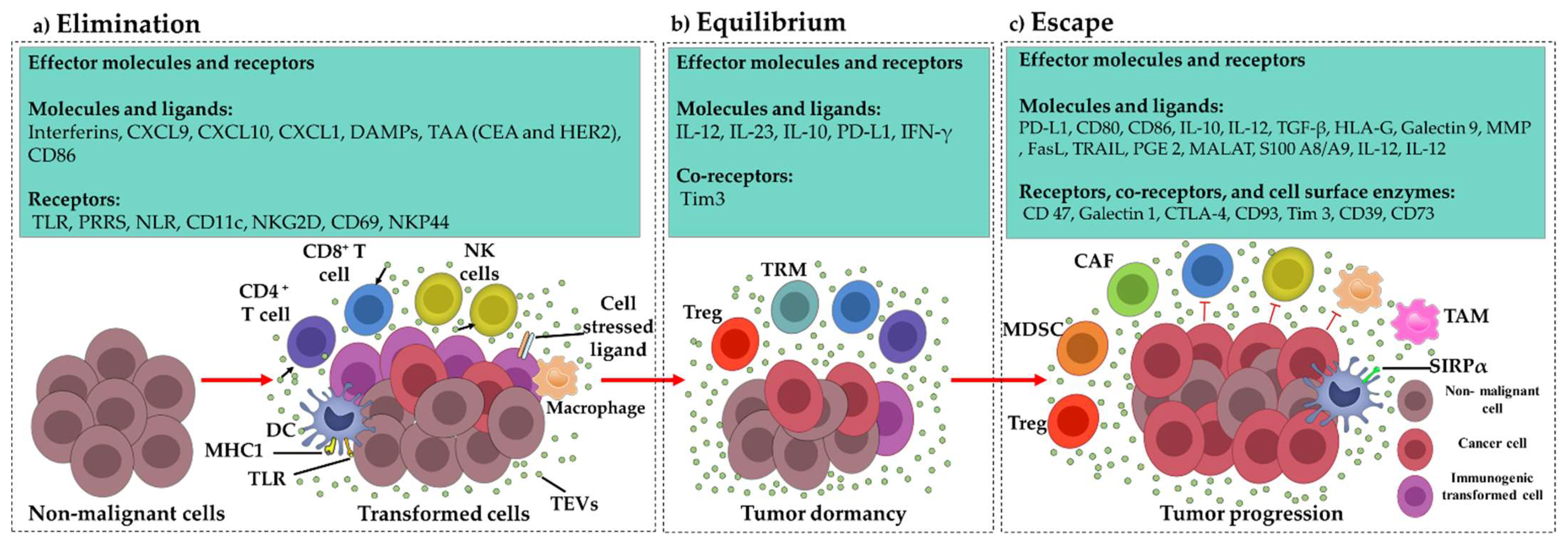
3.1. Elimination Phase TEV Involvement
Tumor-Derived Immunostimulatory Vesicular DAMPs
3.2. Equilibrium Phase TEV Involvement
3.3. Escape Phase TEV Involvement
3.3.1. Effect of TEVs on Dendritic Cells
3.3.2. Effect of TEVs on T Cells
3.3.3. Effect of TEVs on NK Cells
4. Potentials of EVs in Cancer Therapy
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Dai, S.; Wei, D.; Wu, Z.; Zhou, X.; Wei, X.; Huang, H.; Li, G. Phase I clinical trial of autologous ascites-derived exosomes combined with GM-CSF for colorectal cancer. Mol. Ther. 2008, 16, 782–790. [Google Scholar] [CrossRef] [PubMed]
- Hussain, S.P.; Amstad, P.; Raja, K.; Ambs, S.; Nagashima, M.; Bennett, W.P.; Shields, P.G.; Ham, A.-J.; Swenberg, J.A.; Marrogi, A.J. Increased p53 mutation load in noncancerous colon tissue from ulcerative colitis: A cancer-prone chronic inflammatory disease. Cancer Res. 2000, 60, 3333–3337. [Google Scholar] [PubMed]
- Quail, D.F.; Joyce, J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013, 19, 1423–1437. [Google Scholar] [CrossRef]
- Hanahan, D.; Coussens, L.M. Accessories to the crime: Functions of cells recruited to the tumor microenvironment. Cancer Cell 2012, 21, 309–322. [Google Scholar] [CrossRef] [Green Version]
- Meurette, O.; Mehlen, P. Notch Signaling in the Tumor Microenvironment. Cancer Cell 2018, 34, 536–548. [Google Scholar] [CrossRef] [Green Version]
- Danenberg, E.; Bardwell, H.; Zanotelli, V.R.; Provenzano, E.; Chin, S.-F.; Rueda, O.M.; Green, A.; Rakha, E.; Aparicio, S.; Ellis, I.O. Breast tumor microenvironment structures are associated with genomic features and clinical outcome. Nat. Genet. 2022, 54, 660–669. [Google Scholar] [CrossRef]
- Robbins, P.D.; Morelli, A.E. Regulation of immune responses by extracellular vesicles. Nat. Rev. Immunol. 2014, 14, 195–208. [Google Scholar] [CrossRef] [Green Version]
- Théry, C.; Witwer, K.W.; Aikawa, E.; Alcaraz, M.J.; Anderson, J.D.; Andriantsitohaina, R.; Antoniou, A.; Arab, T.; Archer, F.; Atkin-Smith, G.K. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): A position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles 2018, 7, 1535750. [Google Scholar] [CrossRef] [Green Version]
- Kalluri, R. The biology and function of exosomes in cancer. J. Clin. Investig. 2016, 126, 1208–1215. [Google Scholar] [CrossRef]
- Melo, S.A.; Luecke, L.B.; Kahlert, C.; Fernandez, A.F.; Gammon, S.T.; Kaye, J.; LeBleu, V.S.; Mittendorf, E.A.; Weitz, J.; Rahbari, N. Glypican-1 identifies cancer exosomes and detects early pancreatic cancer. Nature 2015, 523, 177–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Donnell, J.S.; Teng, M.W.; Smyth, M.J. Cancer immunoediting and resistance to T cell-based immunotherapy. Nat. Rev. Clin. Oncol. 2019, 16, 151–167. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Bruce, A.T.; Ikeda, H.; Old, L.J.; Schreiber, R.D. Cancer immunoediting: From immunosurveillance to tumor escape. Nat. Immunol. 2002, 3, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Schreiber, R.D.; Old, L.J.; Smyth, M.J. Cancer immunoediting: Integrating immunity’s roles in cancer suppression and promotion. Science 2011, 331, 1565–1570. [Google Scholar] [CrossRef] [Green Version]
- von Locquenghien, M.; Rozalén, C.; Celià-Terrassa, T. Interferons in cancer immunoediting: Sculpting metastasis and immunotherapy response. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.-G.; Zhou, C.-F.; Zhang, Y.-M.; Yan, R.-M.; Wei, W.-F.; Chen, X.-J.; Yi, H.-Y.; Liang, L.-J.; Fan, L.-s.; Liang, L. Cancer-derived exosomal miR-221-3p promotes angiogenesis by targeting THBS2 in cervical squamous cell carcinoma. Angiogenesis 2019, 22, 397–410. [Google Scholar] [CrossRef] [PubMed]
- Dorayappan, K.D.P.; Wanner, R.; Wallbillich, J.J.; Saini, U.; Zingarelli, R.; Suarez, A.A.; Cohn, D.E.; Selvendiran, K. Hypoxia-induced exosomes contribute to a more aggressive and chemoresistant ovarian cancer phenotype: A novel mechanism linking STAT3/Rab proteins. Oncogene 2018, 37, 3806–3821. [Google Scholar] [CrossRef]
- Lopatina, T.; Koni, M.; Grange, C.; Cedrino, M.; Femminò, S.; Lombardo, G.; Favaro, E.; Brizzi, M.F. IL-3 signalling in the tumour microenvironment shapes the immune response via tumour endothelial cell-derived extracellular vesicles. Pharm. Res. 2022, 179, 106206. [Google Scholar] [CrossRef]
- Yeon, J.H.; Jeong, H.E.; Seo, H.; Cho, S.; Kim, K.; Na, D.; Chung, S.; Park, J.; Choi, N.; Kang, J.Y. Cancer-derived exosomes trigger endothelial to mesenchymal transition followed by the induction of cancer-associated fibroblasts. Acta Biomater. 2018, 76, 146–153. [Google Scholar] [CrossRef]
- Guo, J.; Duan, Z.; Zhang, C.; Wang, W.; He, H.; Liu, Y.; Wu, P.; Wang, S.; Song, M.; Chen, H. Mouse 4T1 breast cancer cell–derived exosomes induce proinflammatory cytokine production in macrophages via miR-183. J. Immunol. 2020, 205, 2916–2925. [Google Scholar] [CrossRef]
- Zhou, M.; Wang, S.; Liu, D.; Zhou, J. LINC01915 Facilitates the Conversion of Normal Fibroblasts into Cancer-Associated Fibroblasts Induced by Colorectal Cancer-Derived Extracellular Vesicles through the miR-92a-3p/KLF4/CH25H Axis. ACS Biomater. Sci. Eng. 2021, 7, 5255–5268. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Wu, S.; Zheng, X.; Zheng, P.; Fu, Y.; Wu, C.; Lu, B.; Ju, J.; Jiang, J. Immune suppressed tumor microenvironment by exosomes derived from gastric cancer cells via modulating immune functions. Sci. Rep. 2020, 10, 14749. [Google Scholar] [CrossRef] [PubMed]
- Guan, H.; Peng, R.; Fang, F.; Mao, L.; Chen, Z.; Yang, S.; Dai, C.; Wu, H.; Wang, C.; Feng, N. Tumor-associated macrophages promote prostate cancer progression via exosome-mediated miR-95 transfer. J. Cell Physiol. 2020, 235, 9729–9742. [Google Scholar] [CrossRef] [PubMed]
- Gordon, S. Alternative activation of macrophages. Nat. Rev. Immunol. 2003, 3, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Jiang, M.; Li, X.; Zhang, J.; Lu, Y.; Shi, Y.; Zhu, C.; Liu, Y.; Qin, B.; Luo, Z.; Du, Y. Dual Inhibition of Endoplasmic Reticulum Stress and Oxidation Stress Manipulates the Polarization of Macrophages under Hypoxia to Sensitize Immunotherapy. ACS Nano 2021, 15, 14522–14534. [Google Scholar] [CrossRef]
- Mantovani, A.; Marchesi, F.; Malesci, A.; Laghi, L.; Allavena, P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017, 14, 399–416. [Google Scholar] [CrossRef]
- Scott, E.M.; Jacobus, E.J.; Lyons, B.; Frost, S.; Freedman, J.D.; Dyer, A.; Khalique, H.; Taverner, W.K.; Carr, A.; Champion, B.R. Bi-and tri-valent T cell engagers deplete tumour-associated macrophages in cancer patient samples. J. Immunother. Cancer 2019, 7, 1–18. [Google Scholar] [CrossRef]
- Ham, S.; Lima, L.G.; Chai, E.P.Z.; Muller, A.; Lobb, R.J.; Krumeich, S.; Wen, S.W.; Wiegmans, A.P.; Möller, A. Breast cancer-derived exosomes alter macrophage polarization via gp130/STAT3 signaling. Front. Immunol. 2018, 9, 871. [Google Scholar] [CrossRef] [Green Version]
- Pang, X.; Wang, S.-s.; Zhang, M.; Jiang, J.; Fan, H.-y.; Wu, J.-s.; Wang, H.-f.; Liang, X.-h.; Tang, Y.-l. OSCC cell-secreted exosomal CMTM6 induced M2-like macrophages polarization via ERK1/2 signaling pathway. Cancer Immunol. Immunother. 2021, 70, 1015–1029. [Google Scholar] [CrossRef]
- Zhao, S.; Mi, Y.; Guan, B.; Zheng, B.; Wei, P.; Gu, Y.; Zhang, Z.; Cai, S.; Xu, Y.; Li, X. Tumor-derived exosomal miR-934 induces macrophage M2 polarization to promote liver metastasis of colorectal cancer. J. Hematol. Oncol. 2020, 13, 156. [Google Scholar] [CrossRef]
- Maji, S.; Chaudhary, P.; Akopova, I.; Nguyen, P.M.; Hare, R.J.; Gryczynski, I.; Vishwanatha, J.K. Exosomal annexin II promotes angiogenesis and breast cancer metastasis. Mol. Cancer Res. 2017, 15, 93–105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, J.; Qiao, B.; Gao, N.; Lin, N.; He, W. Oral squamous cell carcinoma-derived exosomes promote M2 subtype macrophage polarization mediated by exosome-enclosed miR-29a-3p. Am. J. Physiol.-Cell Physiol. 2019, 316, C731–C740. [Google Scholar] [CrossRef] [PubMed]
- Gabrusiewicz, K.; Li, X.; Wei, J.; Hashimoto, Y.; Marisetty, A.L.; Ott, M.; Wang, F.; Hawke, D.; Yu, J.; Healy, L.M. Glioblastoma stem cell-derived exosomes induce M2 macrophages and PD-L1 expression on human monocytes. Oncoimmunology 2018, 7, e1412909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, C.; Han, Q.; Xu, D.; Zheng, B.; Zhao, X.; Zhang, J. SALL4-mediated upregulation of exosomal miR-146a-5p drives T-cell exhaustion by M2 tumor-associated macrophages in HCC. Oncoimmunology 2019, 8, e1601479. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Dou, R.; Wei, C.; Liu, K.; Shi, D.; Zhang, C.; Liu, Q.; Wang, S.; Xiong, B. Tumor-derived exosomal microRNA-106b-5p activates EMT-cancer cell and M2-subtype TAM interaction to facilitate CRC metastasis. Mol. Ther. 2021, 29, 2088–2107. [Google Scholar] [CrossRef]
- Piao, Y.J.; Kim, H.S.; Hwang, E.H.; Woo, J.; Zhang, M.; Moon, W.K. Breast cancer cell-derived exosomes and macrophage polarization are associated with lymph node metastasis. Oncotarget 2018, 9, 7398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costanzi, E.; Romani, R.; Scarpelli, P.; Bellezza, I. Extracellular Vesicles-Mediated Transfer of miRNA Let-7b from PC3 Cells to Macrophages. Genes 2020, 11, 1495. [Google Scholar] [CrossRef]
- Pritchard, A.; Tousif, S.; Wang, Y.; Hough, K.; Khan, S.; Strenkowski, J.; Chacko, B.K.; Darley-Usmar, V.M.; Deshane, J.S. Lung tumor cell-derived exosomes promote M2 macrophage polarization. Cells 2020, 9, 1303. [Google Scholar] [CrossRef]
- Hsu, Y.-L.; Hung, J.-Y.; Chang, W.-A.; Jian, S.-F.; Lin, Y.-S.; Pan, Y.-C.; Wu, C.-Y.; Kuo, P.-L. Hypoxic lung-cancer-derived extracellular vesicle microRNA-103a increases the oncogenic effects of macrophages by targeting PTEN. Mol. Ther. 2018, 26, 568–581. [Google Scholar] [CrossRef] [Green Version]
- Chen, L.; Yang, Y.; Huang, C.; Cao, P.; Wu, Q.; Chen, S.; Chen, F. Human THP-1 macrophages activated by exosomes derived from lung adenocarcinoma cells promote lung cancer cell invasion. Chin. J. Cell Mol. Immunol. 2019, 35, 967–972. [Google Scholar]
- Liu, G.; Luo, Y.; Hou, P. PRPS2 Enhances Resistance to Cisplatin via Facilitating Exosomes-mediated Macrophage M2 Polarization in Non-small Cell Lung Cancer. Immunol. Investig. 2021, 51, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Lei, Y.; Wu, M.; Li, N. Regulation of macrophage activation and polarization by HCC-derived exosomal lncRNA TUC339. Int. J. Mol. Sci. 2018, 19, 2958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shinohara, H.; Kuranaga, Y.; Kumazaki, M.; Sugito, N.; Yoshikawa, Y.; Takai, T.; Taniguchi, K.; Ito, Y.; Akao, Y. Regulated polarization of tumor-associated macrophages by mir-145 via colorectal cancer–derived extracellular vesicles. J. Immunol. 2017, 199, 1505–1515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Z.; Yang, L.; Cui, Y.; Zhou, Y.; Yin, X.; Guo, J.; Zhang, G.; Wang, T.; He, Q.-Y. Cytoskeleton-centric protein transportation by exosomes transforms tumor-favorable macrophages. Oncotarget 2016, 7, 67387. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cooks, T.; Pateras, I.S.; Jenkins, L.M.; Patel, K.M.; Robles, A.I.; Morris, J.; Forshew, T.; Appella, E.; Gorgoulis, V.G.; Harris, C.C. Mutant p53 cancers reprogram macrophages to tumor supporting macrophages via exosomal miR-1246. Nat. Commun. 2018, 9, 771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takano, Y.; Masuda, T.; Iinuma, H.; Yamaguchi, R.; Sato, K.; Tobo, T.; Hirata, H.; Kuroda, Y.; Nambara, S.; Hayashi, N. Circulating exosomal microRNA-203 is associated with metastasis possibly via inducing tumor-associated macrophages in colorectal cancer. Oncotarget 2017, 8, 78598. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Zhou, J.; Li, X.; Wang, X.; Lin, Y.; Wang, X. Exosomes derived from hypoxic epithelial ovarian cancer cells deliver microRNAs to macrophages and elicit a tumor-promoted phenotype. Cancer Lett. 2018, 435, 80–91. [Google Scholar] [CrossRef]
- Azambuja, J.H.; Ludwig, N.; Yerneni, S.; Rao, A.; Braganhol, E.; Whiteside, T.L. Molecular profiles and immunomodulatory activities of glioblastoma-derived exosomes. Neuro-Oncol. Adv. 2020, 2, vdaa056. [Google Scholar] [CrossRef]
- Azambuja, J.H.; Ludwig, N.; Yerneni, S.S.; Braganhol, E.; Whiteside, T.L. Arginase-1+ exosomes from reprogrammed macrophages promote glioblastoma progression. Int. J. Mol. Sci. 2020, 21, 3990. [Google Scholar] [CrossRef]
- Ying, X.; Wu, Q.; Wu, X.; Zhu, Q.; Wang, X.; Jiang, L.; Chen, X.; Wang, X. Epithelial ovarian cancer-secreted exosomal miR-222-3p induces polarization of tumor-associated macrophages. Oncotarget 2016, 7, 43076. [Google Scholar] [CrossRef] [Green Version]
- Kalluri, R. The biology and function of fibroblasts in cancer. Nat. Rev. Cancer 2016, 16, 582–598. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.-f.; Yu, Z.-l.; Lv, M.-y.; Cai, Z.-r.; Zou, Y.-f.; Lan, P.; Wu, X.-j.; Gao, F. Cancer-associated fibroblasts impact the clinical outcome and treatment response in colorectal cancer via immune system modulation: A comprehensive genome-wide analysis. Mol. Med. 2021, 27, 139. [Google Scholar] [CrossRef] [PubMed]
- Pelon, F.; Bourachot, B.; Kieffer, Y.; Magagna, I.; Mermet-Meillon, F.; Bonnet, I.; Costa, A.; Givel, A.-M.; Attieh, Y.; Barbazan, J. Cancer-associated fibroblast heterogeneity in axillary lymph nodes drives metastases in breast cancer through complementary mechanisms. Nat. Commun. 2020, 11, 404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, J.; Shi, X.; Yang, M.; Luo, J.; Gao, Q.; Wang, X.; Wu, Y.; Tian, Y.; Wu, F.; Zhou, H. Glycolysis reprogramming in cancer-associated fibroblasts promotes the growth of oral cancer through the lncRNA H19/miR-675-5p/PFKFB3 signaling pathway. Int. J. Oral Sci. 2021, 13, 12. [Google Scholar] [CrossRef] [PubMed]
- Mitra, A.K.; Zillhardt, M.; Hua, Y.; Tiwari, P.; Murmann, A.E.; Peter, M.E.; Lengyel, E. MicroRNAs reprogram normal fibroblasts into cancer-associated fibroblasts in ovarian cancer. Cancer Discov. 2012, 2, 1100–1108. [Google Scholar] [CrossRef] [Green Version]
- Shen, H.; Yu, X.; Yang, F.; Zhang, Z.; Shen, J.; Sun, J.; Choksi, S.; Jitkaew, S.; Shu, Y. Reprogramming of normal fibroblasts into cancer-associated fibroblasts by miRNAs-mediated CCL2/VEGFA signaling. PLoS Genet. 2016, 12, e1006244. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Motohara, T.; Masuda, K.; Morotti, M.; Zheng, Y.; El-Sahhar, S.; Chong, K.Y.; Wietek, N.; Alsaadi, A.; Karaminejadranjbar, M.; Hu, Z. An evolving story of the metastatic voyage of ovarian cancer cells: Cellular and molecular orchestration of the adipose-rich metastatic microenvironment. Oncogene 2019, 38, 2885–2898. [Google Scholar] [CrossRef] [Green Version]
- Baroni, S.; Romero-Cordoba, S.; Plantamura, I.; Dugo, M.; D’ippolito, E.; Cataldo, A.; Cosentino, G.; Angeloni, V.; Rossini, A.; Daidone, M. Exosome-mediated delivery of miR-9 induces cancer-associated fibroblast-like properties in human breast fibroblasts. Cell Death Dis. 2016, 7, e2312. [Google Scholar] [CrossRef] [Green Version]
- Wang, C.; Wang, Y.; Chang, X.; Ba, X.; Hu, N.; Liu, Q.; Fang, L.; Wang, Z. Melanoma-derived exosomes endow fibroblasts with an invasive potential via miR-21 target signaling pathway. Cancer Manag. Res. 2020, 12, 12965. [Google Scholar] [CrossRef]
- Cui, Y.; Wang, D.; Xie, M. Tumor-derived extracellular vesicles promote activation of carcinoma-associated fibroblasts and facilitate invasion and metastasis of ovarian cancer by carrying miR-630. Front. Cell Dev. Biol. 2021, 9, 1576. [Google Scholar] [CrossRef]
- Fan, J.; Xu, G.; Chang, Z.; Zhu, L.; Yao, J. miR-210 transferred by lung cancer cell-derived exosomes may act as proangiogenic factor in cancer-associated fibroblasts by modulating JAK2/STAT3 pathway. Clin. Sci. 2020, 134, 807–825. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Liu, T.; Chen, J.; Ni, H.; Li, W. Survivin in breast cancer–derived exosomes activates fibroblasts by up-regulating SOD1, whose feedback promotes cancer proliferation and metastasis. J. Biol. Chem. 2020, 295, 13737–13752. [Google Scholar] [CrossRef] [PubMed]
- Goulet, C.R.; Bernard, G.; Tremblay, S.; Chabaud, S.; Bolduc, S.; Pouliot, F. Exosomes induce fibroblast differentiation into cancer-associated fibroblasts through TGFβ signaling. Mol. Cancer Res. 2018, 16, 1196–1204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sung, J.S.; Kang, C.W.; Kang, S.; Jang, Y.; Chae, Y.C.; Kim, B.G.; Cho, N.H. ITGB4-mediated metabolic reprogramming of cancer-associated fibroblasts. Oncogene 2020, 39, 664–676. [Google Scholar] [CrossRef] [PubMed]
- Rai, A.; Greening, D.W.; Chen, M.; Xu, R.; Ji, H.; Simpson, R.J. Exosomes derived from human primary and metastatic colorectal cancer cells contribute to functional heterogeneity of activated fibroblasts by reprogramming their proteome. Proteomics 2019, 19, 1800148. [Google Scholar] [CrossRef] [PubMed]
- Hu, T.; Hu, J. Melanoma-derived exosomes induce reprogramming fibroblasts into cancer-associated fibroblasts via Gm26809 delivery. Cell Cycle 2019, 18, 3085–3094. [Google Scholar] [CrossRef] [PubMed]
- Mito, I.; Takahashi, H.; Kawabata-Iwakawa, R.; Horikawa, M.; Ida, S.; Tada, H.; Matsuyama, T.; Misawa, K.; Takeda, S.; Chikamatsu, K. Tumor-derived exosomes elicit cancer-associated fibroblasts shaping inflammatory tumor microenvironment in head and neck squamous cell carcinoma. Oral Oncol. 2023, 136, 106270. [Google Scholar] [CrossRef]
- Gabrilovich, D.I.; Nagaraj, S. Myeloid-derived suppressor cells as regulators of the immune system. Nat. Rev. Immunol. 2009, 9, 162–174. [Google Scholar] [CrossRef] [Green Version]
- Groth, C.; Hu, X.; Weber, R.; Fleming, V.; Altevogt, P.; Utikal, J.; Umansky, V. Immunosuppression mediated by myeloid-derived suppressor cells (MDSCs) during tumour progression. Br. J. Cancer 2019, 120, 16–25. [Google Scholar] [CrossRef] [Green Version]
- Alicea-Torres, K.; Sanseviero, E.; Gui, J.; Chen, J.; Veglia, F.; Yu, Q.; Donthireddy, L.; Kossenkov, A.; Lin, C.; Fu, S. Immune suppressive activity of myeloid-derived suppressor cells in cancer requires inactivation of the type I interferon pathway. Nat. Commun. 2021, 12, 1–13. [Google Scholar] [CrossRef]
- Ferrer, G.; Jung, B.; Chiu, P.Y.; Aslam, R.; Palacios, F.; Mazzarello, A.N.; Vergani, S.; Bagnara, D.; Chen, S.-S.; Yancopoulos, S. Myeloid-derived suppressor cell subtypes differentially influence T-cell function, T-helper subset differentiation, and clinical course in CLL. Leukemia 2021, 35, 3163–3175. [Google Scholar] [CrossRef] [PubMed]
- Lechner, M.G.; Liebertz, D.J.; Epstein, A.L. Characterization of cytokine-induced myeloid-derived suppressor cells from normal human peripheral blood mononuclear cells. J. Immunol. 2010, 185, 2273–2284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, M.; Chen, J.; Zhang, W.; Zhang, R.; Ye, Y.; Liu, P.; Yu, W.; Wei, F.; Ren, X.; Yu, J. Interleukin-6 trans-signaling pathway promotes immunosuppressive myeloid-derived suppressor cells via suppression of suppressor of cytokine signaling 3 in breast cancer. Front. Immunol. 2017, 8, 1840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, Y.-J.; Song, B.; Kim, Y.-S.; Kim, E.-K.; Lee, J.-M.; Lee, G.-E.; Kim, J.-O.; Kim, Y.-J.; Chang, W.-S.; Kang, C.-Y. Tumor microenvironmental conversion of natural killer cells into myeloid-derived suppressor cells. Cancer Res. 2013, 73, 5669–5681. [Google Scholar] [CrossRef] [Green Version]
- Xiang, X.; Poliakov, A.; Liu, C.; Liu, Y.; Deng, Z.b.; Wang, J.; Cheng, Z.; Shah, S.V.; Wang, G.J.; Zhang, L. Induction of myeloid-derived suppressor cells by tumor exosomes. Int. J. Cancer 2009, 124, 2621–2633. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Goliwas, K.F.; Severino, P.E.; Hough, K.P.; Van Vessem, D.; Wang, H.; Tousif, S.; Koomullil, R.P.; Frost, A.R.; Ponnazhagan, S. Mechanical strain induces phenotypic changes in breast cancer cells and promotes immunosuppression in the tumor microenvironment. Lab. Investig. 2020, 100, 1503–1516. [Google Scholar] [CrossRef]
- Himes, B.T.; Peterson, T.E.; de Mooij, T.; Garcia, L.M.C.; Jung, M.-Y.; Uhm, S.; Yan, D.; Tyson, J.; Jin-Lee, H.J.; Parney, D. The role of extracellular vesicles and PD-L1 in glioblastoma-mediated immunosuppressive monocyte induction. Neuro-Oncology 2020, 22, 967–978. [Google Scholar] [CrossRef]
- Gao, Y.; Xu, H.; Li, N.; Wang, H.; Ma, L.; Chen, S.; Liu, J.; Zheng, Y.; Zhang, Y. Renal cancer-derived exosomes induce tumor immune tolerance by MDSCs-mediated antigen-specific immunosuppression. Cell Commun. Signal. 2020, 18, 1–14. [Google Scholar] [CrossRef]
- Liu, Q.W.; Chen, Y.; Li, J.Y.; Xiao, L.; Zhang, W.J.; Zhao, J.L.; Gu, H.C.; Wu, H.Y.; Zuo, G.S.L.; Deng, K.Y. Bone marrow cells are differentiated into MDSCs by BCC-Ex through down-regulating the expression of CXCR4 and activating STAT3 signalling pathway. J. Cell. Mol. Med. 2021, 25, 5497–5510. [Google Scholar] [CrossRef]
- Jiang, M.; Zhang, W.; Zhang, R.; Liu, P.; Ye, Y.; Yu, W.; Guo, X.; Yu, J. Cancer exosome-derived miR-9 and miR-181a promote the development of early-stage MDSCs via interfering with SOCS3 and PIAS3 respectively in breast cancer. Oncogene 2020, 39, 4681–4694. [Google Scholar] [CrossRef]
- Qiu, W.; Guo, X.; Li, B.; Wang, J.; Qi, Y.; Chen, Z.; Zhao, R.; Deng, L.; Qian, M.; Wang, S. Exosomal miR-1246 from glioma patient body fluids drives the differentiation and activation of myeloid-derived suppressor cells. Mol. Ther. 2021, 29, 3449–3464. [Google Scholar] [CrossRef] [PubMed]
- Guo, X.; Qiu, W.; Wang, J.; Liu, Q.; Qian, M.; Wang, S.; Zhang, Z.; Gao, X.; Chen, Z.; Guo, Q. Glioma exosomes mediate the expansion and function of myeloid-derived suppressor cells through microRNA-29a/Hbp1 and microRNA-92a/Prkar1a pathways. Int. J. Cancer 2019, 144, 3111–3126. [Google Scholar] [CrossRef] [PubMed]
- Domenis, R.; Cesselli, D.; Toffoletto, B.; Bourkoula, E.; Caponnetto, F.; Manini, I.; Beltrami, A.P.; Ius, T.; Skrap, M.; Di Loreto, C. Systemic T cells immunosuppression of glioma stem cell-derived exosomes is mediated by monocytic myeloid-derived suppressor cells. PLoS ONE 2017, 12, e0169932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, X.; Li, F.; Tang, Y.; Ren, Q.; Xiao, B.; Wan, Y.; Jiang, S. miR-21a in exosomes from Lewis lung carcinoma cells accelerates tumor growth through targeting PDCD4 to enhance expansion of myeloid-derived suppressor cells. Oncogene 2020, 39, 6354–6369. [Google Scholar] [CrossRef] [PubMed]
- Krysko, D.V.; Garg, A.D.; Kaczmarek, A.; Krysko, O.; Agostinis, P.; Vandenabeele, P. Immunogenic cell death and DAMPs in cancer therapy. Nat. Rev. Cancer 2012, 12, 860–875. [Google Scholar] [CrossRef]
- Guo, Y.; Wang, S.-Z.; Zhang, X.; Jia, H.-R.; Zhu, Y.-X.; Zhang, X.; Gao, G.; Jiang, Y.-W.; Li, C.; Chen, X. In situ generation of micrometer-sized tumor cell-derived vesicles as autologous cancer vaccines for boosting systemic immune responses. Nat. Commun. 2022, 13, 1–20. [Google Scholar] [CrossRef]
- Hiam-Galvez, K.J.; Allen, B.M.; Spitzer, M.H. Systemic immunity in cancer. Nat. Rev. Cancer 2021, 21, 345–359. [Google Scholar] [CrossRef]
- Łuksza, M.; Sethna, Z.M.; Rojas, L.A.; Lihm, J.; Bravi, B.; Elhanati, Y.; Soares, K.; Amisaki, M.; Dobrin, A.; Hoyos, D. Neoantigen quality predicts immunoediting in survivors of pancreatic cancer. Nature 2022, 606, 389–395. [Google Scholar] [CrossRef]
- DuPage, M.; Mazumdar, C.; Schmidt, L.M.; Cheung, A.F.; Jacks, T. Expression of tumour-specific antigens underlies cancer immunoediting. Nature 2012, 482, 405–409. [Google Scholar] [CrossRef] [Green Version]
- Lakatos, E.; Williams, M.J.; Schenck, R.O.; Cross, W.C.; Househam, J.; Zapata, L.; Werner, B.; Gatenbee, C.; Robertson-Tessi, M.; Barnes, C.P. Evolutionary dynamics of neoantigens in growing tumors. Nat. Genet. 2020, 52, 1057–1066. [Google Scholar] [CrossRef]
- Rao, S.; Gharib, K.; Han, A. Cancer immunosurveillance by T cells. Int. Rev. Cell Mol. Biol. 2019, 342, 149–173. [Google Scholar] [PubMed]
- Hu, Q.; Hong, Y.; Qi, P.; Lu, G.; Mai, X.; Xu, S.; He, X.; Guo, Y.; Gao, L.; Jing, Z. Atlas of breast cancer infiltrated B-lymphocytes revealed by paired single-cell RNA-sequencing and antigen receptor profiling. Nat. Commun. 2021, 12, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Cole, K.E.; Strick, C.A.; Paradis, T.J.; Ogborne, K.T.; Loetscher, M.; Gladue, R.P.; Lin, W.; Boyd, J.G.; Moser, B.; Wood, D.E. Interferon–inducible T cell alpha chemoattractant (I-TAC): A novel Non-ELR CXC Chemokine with potent activity on activated T cells through selective high affinity binding to CXCR3. J. Exp. Med. 1998, 187, 2009–2021. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, Y.; Zheng, L.; Du, Q.; Yazdani, H.; Dong, K.; Guo, Y.; Geller, D.A. Interferon regulatory factor 1 (IRF-1) activates anti-tumor immunity via CXCL10/CXCR3 axis in hepatocellular carcinoma (HCC). Cancer Lett. 2021, 506, 95–106. [Google Scholar] [CrossRef] [PubMed]
- Dunn, G.P.; Bruce, A.T.; Sheehan, K.C.; Shankaran, V.; Uppaluri, R.; Bui, J.D.; Diamond, M.S.; Koebel, C.M.; Arthur, C.; White, J.M. A critical function for type I interferons in cancer immunoediting. Nat. Immunol. 2005, 6, 722–729. [Google Scholar] [CrossRef]
- Jan, A.T.; Rahman, S.; Khan, S.; Tasduq, S.A.; Choi, I. Biology, pathophysiological role, and clinical implications of exosomes: A critical appraisal. Cells 2019, 8, 99. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.-q.; Du, Q.; Varley, P.R.; Goswami, J.; Liang, Z.; Wang, R.; Li, H.; Stolz, D.B.; Geller, D.A. Interferon regulatory factor 1 priming of tumour-derived exosomes enhances the antitumour immune response. Br. J. Cancer 2018, 118, 62–71. [Google Scholar] [CrossRef] [Green Version]
- Hartman, Z.C.; Wei, J.; Glass, O.K.; Guo, H.; Lei, G.; Yang, X.-Y.; Osada, T.; Hobeika, A.; Delcayre, A.; Le Pecq, J.-B. Increasing vaccine potency through exosome antigen targeting. Vaccine 2011, 29, 9361–9367. [Google Scholar] [CrossRef] [Green Version]
- Logozzi, M.; Capasso, C.; Di Raimo, R.; Del Prete, S.; Mizzoni, D.; Falchi, M.; Supuran, C.T.; Fais, S. Prostate cancer cells and exosomes in acidic condition show increased carbonic anhydrase IX expression and activity. J. Enzym. Inhib. Med. Chem. 2019, 34, 272–278. [Google Scholar] [CrossRef] [Green Version]
- Shi, S.; Rao, Q.; Zhang, C.; Zhang, X.; Qin, Y.; Niu, Z. Dendritic cells pulsed with exosomes in combination with PD-1 antibody increase the efficacy of sorafenib in hepatocellular carcinoma model. Transl. Oncol. 2018, 11, 250–258. [Google Scholar] [CrossRef]
- Wolfers, J.; Lozier, A.; Raposo, G.; Regnault, A.; Thery, C.; Masurier, C.; Flament, C.; Pouzieux, S.; Faure, F.; Tursz, T. Tumor-derived exosomes are a source of shared tumor rejection antigens for CTL cross-priming. Nat. Med. 2001, 7, 297–303. [Google Scholar] [CrossRef] [PubMed]
- Hao, Q.; Wu, Y.; Wu, Y.; Wang, P.; Vadgama, J.V. Tumor-Derived Exosomes in Tumor-Induced Immune Suppression. Int. J. Mol. Sci. 2022, 23, 1461. [Google Scholar] [CrossRef] [PubMed]
- Segura, E.; Guérin, C.; Hogg, N.; Amigorena, S.; Théry, C. CD8+ dendritic cells use LFA-1 to capture MHC-peptide complexes from exosomes in vivo. J. Immunol. 2007, 179, 1489–1496. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Chen, L.; Peng, Y.; Yu, S.; Liu, J.; Wu, L.; Zhang, L.; Wu, Q.; Chang, X.; Yu, X. Dendritic cells loaded with tumor derived exosomes for cancer immunotherapy. Oncotarget 2018, 9, 2887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, G.; Wang, Y.; Yuan, S.; Wang, B. Dendritic cells loaded with HeLa-derived exosomes simulate an antitumor immune response. Oncol. Lett. 2018, 15, 6636–6640. [Google Scholar] [CrossRef] [PubMed]
- Dai, S.; Wan, T.; Wang, B.; Zhou, X.; Xiu, F.; Chen, T.; Wu, Y.; Cao, X. More efficient induction of HLA-A* 0201-restricted and carcinoembryonic antigen (CEA)–specific CTL response by immunization with exosomes prepared from heat-stressed CEA-positive tumor cells. Clin. Cancer Res. 2005, 11, 7554–7563. [Google Scholar] [CrossRef] [Green Version]
- Diamond, J.M.; Vanpouille-Box, C.; Spada, S.; Rudqvist, N.-P.; Chapman, J.R.; Ueberheide, B.M.; Pilones, K.A.; Sarfraz, Y.; Formenti, S.C.; Demaria, S. Exosomes shuttle TREX1-sensitive IFN-stimulatory dsDNA from irradiated cancer cells to DCs. Cancer Immunol. Res. 2018, 6, 910–920. [Google Scholar] [CrossRef] [Green Version]
- Hao, S.; Bai, O.; Yuan, J.; Qureshi, M.; Xiang, J. Dendritic cell-derived exosomes stimulate stronger CD8+ CTL responses and antitumor immunity than tumor cell-derived exosomes. Cell Mol. Immunol. 2006, 3, 205–211. [Google Scholar]
- Graner, M.W.; Alzate, O.; Dechkovskaia, A.M.; Keene, J.D.; Sampson, J.H.; Mitchell, D.A.; Bigner, D.D. Proteomic and immunologic analyses of brain tumor exosomes. FASEB J. 2009, 23, 1541–1557. [Google Scholar] [CrossRef] [Green Version]
- Garg, A.; Martin, S.; Golab, J.; Agostinis, P. Danger signalling during cancer cell death: Origins, plasticity and regulation. Cell Death Differ. 2014, 21, 26–38. [Google Scholar] [CrossRef] [Green Version]
- Zhang, G.; Liu, Z.; Ding, H.; Zhou, Y.; Doan, H.A.; Sin, K.W.T.; Zhu, Z.J.; Flores, R.; Wen, Y.; Gong, X. Tumor induces muscle wasting in mice through releasing extracellular Hsp70 and Hsp90. Nat. Commun. 2017, 8, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Yang, C.; Li, L.; Jin, X.; Zhang, Z.; Zheng, H.; Pan, J.; Shi, L.; Jiang, Z.; Su, K. Tumor-derived HMGB1 induces CD62L dim neutrophil polarization and promotes lung metastasis in triple-negative breast cancer. Oncogenesis 2020, 9, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Sachet, M.; Liang, Y.Y.; Oehler, R. The immune response to secondary necrotic cells. Apoptosis 2017, 22, 1189–1204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gamrekelashvili, J.; Ormandy, L.A.; Heimesaat, M.M.; Kirschning, C.J.; Manns, M.P.; Korangy, F.; Greten, T.F. Primary sterile necrotic cells fail to cross-prime CD8+ T cells. Oncoimmunology 2012, 1, 1017–1026. [Google Scholar] [CrossRef] [Green Version]
- Panaretakis, T.; Kepp, O.; Brockmeier, U.; Tesniere, A.; Bjorklund, A.C.; Chapman, D.C.; Durchschlag, M.; Joza, N.; Pierron, G.; Van Endert, P. Mechanisms of pre-apoptotic calreticulin exposure in immunogenic cell death. EMBO J. 2009, 28, 578–590. [Google Scholar] [CrossRef] [Green Version]
- Turubanova, V.D.; Balalaeva, I.V.; Mishchenko, T.A.; Catanzaro, E.; Alzeibak, R.; Peskova, N.N.; Efimova, I.; Bachert, C.; Mitroshina, E.V.; Krysko, O. Immunogenic cell death induced by a new photodynamic therapy based on photosens and photodithazine. J. Immunother. Cancer 2019, 7, 1–13. [Google Scholar] [CrossRef]
- Garg, A.D.; Krysko, D.V.; Vandenabeele, P.; Agostinis, P. The emergence of phox-ER stress induced immunogenic apoptosis. Oncoimmunology 2012, 1, 786–788. [Google Scholar] [CrossRef] [Green Version]
- Choi, M.E.; Price, D.R.; Ryter, S.W.; Choi, A.M. Necroptosis: A crucial pathogenic mediator of human disease. JCI Insight 2019, 4, e128834. [Google Scholar] [CrossRef] [Green Version]
- Hou, L.; Yang, Z.; Wang, Z.; Zhang, X.; Zhao, Y.; Yang, H.; Zheng, B.; Tian, W.; Wang, S.; He, Z. NLRP3/ASC-mediated alveolar macrophage pyroptosis enhances HMGB1 secretion in acute lung injury induced by cardiopulmonary bypass. Lab. Investig. 2018, 98, 1052–1064. [Google Scholar] [CrossRef]
- Yang, D.; He, Y.; Muñoz-Planillo, R.; Liu, Q.; Núñez, G. Caspase-11 requires the pannexin-1 channel and the purinergic P2X7 pore to mediate pyroptosis and endotoxic shock. Immunity 2015, 43, 923–932. [Google Scholar] [CrossRef] [Green Version]
- Li, W.; Deng, M.; Loughran, P.A.; Yang, M.; Lin, M.; Yang, C.; Gao, W.; Jin, S.; Li, S.; Cai, J. LPS induces active HMGB1 release from hepatocytes into exosomes through the coordinated activities of TLR4 and caspase-11/GSDMD signaling. Front. Immunol. 2020, 11, 229. [Google Scholar] [CrossRef] [PubMed]
- Nair, R.R.; Mazza, D.; Brambilla, F.; Gorzanelli, A.; Agresti, A.; Bianchi, M.E. LPS-challenged macrophages release microvesicles coated with histones. Front. Immunol. 2018, 9, 1463. [Google Scholar] [CrossRef] [PubMed]
- Vulpis, E.; Cecere, F.; Molfetta, R.; Soriani, A.; Fionda, C.; Peruzzi, G.; Caracciolo, G.; Palchetti, S.; Masuelli, L.; Simonelli, L. Genotoxic stress modulates the release of exosomes from multiple myeloma cells capable of activating NK cell cytokine production: Role of HSP70/TLR2/NF-kB axis. Oncoimmunology 2017, 6, e1279372. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jella, K.; Nasti, T.; Li, Z.; Lawson, D.; Ahmed, R.; Dynan, W.; Khan, M. Post-irradiated tumor-derived exosomes lead to melanoma tumor growth delay, potentially mediated by death associated molecular pattern (damps) proteins. Int. J. Radiat. Oncol. Biol. Phys. 2018, 102, S155. [Google Scholar] [CrossRef] [Green Version]
- Zhou, M.; Chen, J.; Zhou, L.; Chen, W.; Ding, G.; Cao, L. Pancreatic cancer derived exosomes regulate the expression of TLR4 in dendritic cells via miR-203. Cell Immunol. 2014, 292, 65–69. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Bai, O.; Zhang, H.; Yuan, J.; Zong, S.; Chibbar, R.; Slattery, K.; Qureshi, M.; Wei, Y.; Deng, Y. Membrane-bound HSP70-engineered myeloma cell-derived exosomes stimulate more efficient CD8+ CTL-and NK-mediated antitumour immunity than exosomes released from heat-shocked tumour cells expressing cytoplasmic HSP70. J. Cell Mol. Med. 2010, 14, 2655–2666. [Google Scholar] [CrossRef] [Green Version]
- Cho, J.-a.; Lee, Y.-S.; Kim, S.-H.; Ko, J.-K.; Kim, C.-W. MHC independent anti-tumor immune responses induced by Hsp70-enriched exosomes generate tumor regression in murine models. Cancer Lett. 2009, 275, 256–265. [Google Scholar] [CrossRef]
- Gastpar, R.; Gehrmann, M.; Bausero, M.A.; Asea, A.; Gross, C.; Schroeder, J.A.; Multhoff, G. Heat shock protein 70 surface-positive tumor exosomes stimulate migratory and cytolytic activity of natural killer cells. Cancer Res. 2005, 65, 5238–5247. [Google Scholar] [CrossRef] [Green Version]
- Lv, L.-H.; Wan, Y.-L.; Lin, Y.; Zhang, W.; Yang, M.; Li, G.-L.; Lin, H.-M.; Shang, C.-Z.; Chen, Y.-J.; Min, J. Anticancer drugs cause release of exosomes with heat shock proteins from human hepatocellular carcinoma cells that elicit effective natural killer cell antitumor responses in vitro. J. Biol. Chem. 2012, 287, 15874–15885. [Google Scholar] [CrossRef] [Green Version]
- Koebel, C.M.; Vermi, W.; Swann, J.B.; Zerafa, N.; Rodig, S.J.; Old, L.J.; Smyth, M.J.; Schreiber, R.D. Adaptive immunity maintains occult cancer in an equilibrium state. Nature 2007, 450, 903–907. [Google Scholar] [CrossRef]
- Neophytou, C.M.; Kyriakou, T.-C.; Papageorgis, P. Mechanisms of metastatic tumor dormancy and implications for cancer therapy. Int. J. Mol. Sci. 2019, 20, 6158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Zhao, X.; Wang, Y.; Ren, F.; Sun, D.; Yan, Y.; Kong, X.; Bu, J.; Liu, M.; Xu, S. circRNA-002178 act as a ceRNA to promote PDL1/PD1 expression in lung adenocarcinoma. Cell Death Dis. 2020, 11, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Grange, C.; Tapparo, M.; Tritta, S.; Deregibus, M.C.; Battaglia, A.; Gontero, P.; Frea, B.; Camussi, G. Role of HLA-G and extracellular vesicles in renal cancer stem cell-induced inhibition of dendritic cell differentiation. BMC Cancer 2015, 15, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ono, M.; Kosaka, N.; Tominaga, N.; Yoshioka, Y.; Takeshita, F.; Takahashi, R.-u.; Yoshida, M.; Tsuda, H.; Tamura, K.; Ochiya, T. Exosomes from bone marrow mesenchymal stem cells contain a microRNA that promotes dormancy in metastatic breast cancer cells. Sci. Signal. 2014, 7, ra63. [Google Scholar] [CrossRef]
- Sandiford, O.A.; Donnelly, R.J.; Markos, H.; Burgmeyer, L.M.; Sinha, G.; Pamarthi, S.H.; Sherman, L.S.; Ferrer, A.I.; DeVore, D.E.; Patel, S.A. Mesenchymal Stem Cell–Secreted Extracellular Vesicles Instruct Stepwise Dedifferentiation of Breast Cancer Cells into Dormancy at the Bone Marrow Perivascular Region. Cancer Res. 2021, 81, 1567–1582. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.; Huang, A.C.; Zhang, W.; Zhang, G.; Wu, M.; Xu, W.; Yu, Z.; Yang, J.; Wang, B.; Sun, H. Exosomal PD-L1 contributes to immunosuppression and is associated with anti-PD-1 response. Nature 2018, 560, 382–386. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liang, X.; Yin, X.; Lv, J.; Tang, K.; Ma, J.; Ji, T.; Zhang, H.; Dong, W.; Jin, X. Blockade of IDO-kynurenine-AhR metabolic circuitry abrogates IFN-γ-induced immunologic dormancy of tumor-repopulating cells. Nat. Commun. 2017, 8, 1–15. [Google Scholar] [CrossRef]
- Yang, Y.; Li, C.-W.; Chan, L.-C.; Wei, Y.; Hsu, J.-M.; Xia, W.; Cha, J.-H.; Hou, J.; Hsu, J.L.; Sun, L. Exosomal PD-L1 harbors active defense function to suppress T cell killing of breast cancer cells and promote tumor growth. Cell Res. 2018, 28, 862–864. [Google Scholar] [CrossRef] [Green Version]
- Gao, L.; Wang, L.; Dai, T.; Jin, K.; Zhang, Z.; Wang, S.; Xie, F.; Fang, P.; Yang, B.; Huang, H. Tumor-derived exosomes antagonize innate antiviral immunity. Nat. Immunol. 2018, 19, 233–245. [Google Scholar] [CrossRef]
- Bakhoum, S.F.; Ngo, B.; Laughney, A.M.; Cavallo, J.-A.; Murphy, C.J.; Ly, P.; Shah, P.; Sriram, R.K.; Watkins, T.B.; Taunk, N.K. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature 2018, 553, 467–472. [Google Scholar] [CrossRef] [Green Version]
- Dhatchinamoorthy, K.; Colbert, J.D.; Rock, K.L. Cancer Immune Evasion Through Loss of MHC Class I Antigen Presentation. Front. Immunol. 2021, 12, 469. [Google Scholar] [CrossRef] [PubMed]
- Wang, M.; Cai, Y.; Peng, Y.; Xu, B.; Hui, W.; Jiang, Y. Exosomal LGALS9 in the cerebrospinal fluid of glioblastoma patients suppressed dendritic cell antigen presentation and cytotoxic T-cell immunity. Cell Death Dis. 2020, 11, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Willingham, S.B.; Volkmer, J.-P.; Gentles, A.J.; Sahoo, D.; Dalerba, P.; Mitra, S.S.; Wang, J.; Contreras-Trujillo, H.; Martin, R.; Cohen, J.D. The CD47-signal regulatory protein alpha (SIRPa) interaction is a therapeutic target for human solid tumors. Proc. Natl. Acad. Sci. USA 2012, 109, 6662–6667. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mannino, M.H.; Zhu, Z.; Xiao, H.; Bai, Q.; Wakefield, M.R.; Fang, Y. The paradoxical role of IL-10 in immunity and cancer. Cancer Lett. 2015, 367, 103–107. [Google Scholar] [CrossRef]
- Flavell, R.A.; Sanjabi, S.; Wrzesinski, S.H.; Licona-Limón, P. The polarization of immune cells in the tumour environment by TGFβ. Nat. Rev. Immunol. 2010, 10, 554–567. [Google Scholar] [CrossRef] [Green Version]
- Pardoll, D.M. The blockade of immune checkpoints in cancer immunotherapy. Nat. Rev. Cancer 2012, 12, 252–264. [Google Scholar] [CrossRef] [Green Version]
- Dhani, S.; Zhao, Y.; Zhivotovsky, B. A long way to go: Caspase inhibitors in clinical use. Cell Death Dis. 2021, 12, 1–13. [Google Scholar] [CrossRef]
- Malchow, S.; Leventhal, D.S.; Nishi, S.; Fischer, B.I.; Shen, L.; Paner, G.P.; Amit, A.S.; Kang, C.; Geddes, J.E.; Allison, J.P. Aire-dependent thymic development of tumor-associated regulatory T cells. Science 2013, 339, 1219–1224. [Google Scholar] [CrossRef] [Green Version]
- Yu, S.; Liu, C.; Su, K.; Wang, J.; Liu, Y.; Zhang, L.; Li, C.; Cong, Y.; Kimberly, R.; Grizzle, W.E. Tumor exosomes inhibit differentiation of bone marrow dendritic cells. J. Immunol. 2007, 178, 6867–6875. [Google Scholar] [CrossRef] [Green Version]
- Maus, R.L.; Jakub, J.W.; Nevala, W.K.; Christensen, T.A.; Noble-Orcutt, K.; Sachs, Z.; Hieken, T.J.; Markovic, S.N. Human melanoma-derived extracellular vesicles regulate dendritic cell maturation. Front. Immunol. 2017, 8, 358. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Yin, Z.; Lu, P.; Ma, Y.; Luo, B.; Xiang, L.; Zhang, W.; He, Y.; Liang, X. Lung carcinoma cells secrete exosomal MALAT1 to inhibit dendritic cell phagocytosis, inflammatory response, costimulatory molecule expression and promote dendritic cell autophagy via AKT/mTOR pathway. OncoTargets Ther. 2020, 13, 10693. [Google Scholar] [CrossRef] [PubMed]
- Ning, Y.; Shen, K.; Wu, Q.; Sun, X.; Bai, Y.; Xie, Y.; Pan, J.; Qi, C. Tumor exosomes block dendritic cells maturation to decrease the T cell immune response. Immunol. Lett. 2018, 199, 36–43. [Google Scholar] [CrossRef] [PubMed]
- Yin, X.; Zeng, W.; Wu, B.; Wang, L.; Wang, Z.; Tian, H.; Wang, L.; Jiang, Y.; Clay, R.; Wei, X. PPARα inhibition overcomes tumor-derived exosomal lipid-induced dendritic cell dysfunction. Cell Rep. 2020, 33, 108278. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.; Xiao, C.; Evans, K.S.; Theivanthiran, T.; DeVito, N.; Holtzhausen, A.; Liu, J.; Liu, X.; Boczkowski, D.; Nair, S. Paracrine Wnt5a-β-catenin signaling triggers a metabolic program that drives dendritic cell tolerization. Immunity 2018, 48, 147–160.e147. [Google Scholar] [CrossRef] [Green Version]
- Wculek, S.K.; Khouili, S.C.; Priego, E.; Heras-Murillo, I.; Sancho, D. Metabolic control of dendritic cell functions: Digesting information. Front. Immunol. 2019, 10, 775. [Google Scholar] [CrossRef] [Green Version]
- Salimu, J.; Webber, J.; Gurney, M.; Al-Taei, S.; Clayton, A.; Tabi, Z. Dominant immunosuppression of dendritic cell function by prostate-cancer-derived exosomes. J. Extracell. Vesicles 2017, 6, 1368823. [Google Scholar] [CrossRef] [Green Version]
- Gao, J.; Qiu, X.; Li, X.; Fan, H.; Zhang, F.; Lv, T.; Song, Y. Expression profiles and clinical value of plasma exosomal Tim-3 and Galectin-9 in non-small cell lung cancer. Biochem. Biophys. Res. Commun. 2018, 498, 409–415. [Google Scholar] [CrossRef]
- Haderk, F.; Schulz, R.; Iskar, M.; Cid, L.L.; Worst, T.; Willmund, K.V.; Schulz, A.; Warnken, U.; Seiler, J.; Benner, A. Tumor-derived exosomes modulate PD-L1 expression in monocytes. Sci. Immunol. 2017, 2, eaah5509. [Google Scholar] [CrossRef] [Green Version]
- Maus, R.L.; Jakub, J.W.; Hieken, T.J.; Nevala, W.K.; Christensen, T.A.; Sutor, S.L.; Flotte, T.J.; Markovic, S.N. Identification of novel, immune-mediating extracellular vesicles in human lymphatic effluent draining primary cutaneous melanoma. Oncoimmunology 2019, 8, e1667742. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Kim, S.-H.; Bianco, N.R.; Robbins, P.D. Tumor-derived exosomes confer antigen-specific immunosuppression in a murine delayed-type hypersensitivity model. PLoS ONE 2011, 6, e22517. [Google Scholar] [CrossRef] [Green Version]
- Shen, Y.; Guo, D.; Weng, L.; Wang, S.; Ma, Z.; Yang, Y.; Wang, P.; Wang, J.; Cai, Z. Tumor-derived exosomes educate dendritic cells to promote tumor metastasis via HSP72/HSP105-TLR2/TLR4 pathway. Oncoimmunology 2017, 6, e1362527. [Google Scholar] [CrossRef] [PubMed]
- Prieto, D.; Sotelo, N.; Seija, N.; Sernbo, S.; Abreu, C.; Durán, R.; Gil, M.; Sicco, E.; Irigoin, V.; Oliver, C. S100-A9 protein in exosomes from chronic lymphocytic leukemia cells promotes NF-κB activity during disease progression. Blood J. Am. Soc. Hematol. 2017, 130, 777–788. [Google Scholar] [CrossRef] [PubMed]
- Cheng, P.; Corzo, C.A.; Luetteke, N.; Yu, B.; Nagaraj, S.; Bui, M.M.; Ortiz, M.; Nacken, W.; Sorg, C.; Vogl, T. Inhibition of dendritic cell differentiation and accumulation of myeloid-derived suppressor cells in cancer is regulated by S100A9 protein. J. Exp. Med. 2008, 205, 2235–2249. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.; Elkahloun, A.G.; Arakelyan, A.; Young, L.; Myers, T.G.; Otaizo-Carrasquero, F.; Wu, W.; Margolis, L.; Roberts, D.D. CD63, MHC class 1, and CD47 identify subsets of extracellular vesicles containing distinct populations of noncoding RNAs. Sci. Rep. 2018, 8, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, G.; Zhou, L.; Qian, Y.; Fu, M.; Chen, J.; Chen, J.; Xiang, J.; Wu, Z.; Jiang, G.; Cao, L. Pancreatic cancer-derived exosomes transfer miRNAs to dendritic cells and inhibit RFXAP expression via miR-212-3p. Oncotarget 2015, 6, 29877. [Google Scholar] [CrossRef] [Green Version]
- Thommen, D.S.; Schumacher, T.N. T cell dysfunction in cancer. Cancer Cell 2018, 33, 547–562. [Google Scholar] [CrossRef] [Green Version]
- Wieckowski, E.U.; Visus, C.; Szajnik, M.; Szczepanski, M.J.; Storkus, W.J.; Whiteside, T.L. Tumor-derived microvesicles promote regulatory T cell expansion and induce apoptosis in tumor-reactive activated CD8+ T lymphocytes. J. Immunol. 2009, 183, 3720–3730. [Google Scholar] [CrossRef] [Green Version]
- Huber, V.; Fais, S.; Iero, M.; Lugini, L.; Canese, P.; Squarcina, P.; Zaccheddu, A.; Colone, M.; Arancia, G.; Gentile, M. Human colorectal cancer cells induce T-cell death through release of proapoptotic microvesicles: Role in immune escape. Gastroenterology 2005, 128, 1796–1804. [Google Scholar] [CrossRef]
- Abusamra, A.J.; Zhong, Z.; Zheng, X.; Li, M.; Ichim, T.E.; Chin, J.L.; Min, W.-P. Tumor exosomes expressing Fas ligand mediate CD8+ T-cell apoptosis. Blood Cells Mol. Dis. 2005, 35, 169–173. [Google Scholar] [CrossRef]
- Li, L.; Cao, B.; Liang, X.; Lu, S.; Luo, H.; Wang, Z.; Wang, S.; Jiang, J.; Lang, J.; Zhu, G. Microenvironmental oxygen pressure orchestrates an anti-and pro-tumoral γδ T cell equilibrium via tumor-derived exosomes. Oncogene 2019, 38, 2830–2843. [Google Scholar] [CrossRef]
- Klibi, J.; Niki, T.; Riedel, A.; Pioche-Durieu, C.; Souquere, S.; Rubinstein, E.; Le Moulec, S.; Guigay, J.; Hirashima, M.; Guemira, F. Blood diffusion and Th1-suppressive effects of galectin-9–containing exosomes released by Epstein-Barr virus–infected nasopharyngeal carcinoma cells. Blood J. Am. Soc. Hematol. 2009, 113, 1957–1966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.W.; Wieckowski, E.; Taylor, D.D.; Reichert, T.E.; Watkins, S.; Whiteside, T.L. Fas ligand–positive membranous vesicles isolated from sera of patients with oral cancer induce apoptosis of activated T lymphocytes. Clin. Cancer Res. 2005, 11, 1010–1020. [Google Scholar] [CrossRef] [PubMed]
- Taylor, D.; Gercel-Taylor, C. Tumour-derived exosomes and their role in cancer-associated T-cell signalling defects. Br. J. Cancer 2005, 92, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Beccard, I.J.; Hofmann, L.; Schroeder, J.C.; Ludwig, S.; Laban, S.; Brunner, C.; Lotfi, R.; Hoffmann, T.K.; Jackson, E.K.; Schuler, P.J. Immune suppressive effects of plasma-derived exosome populations in head and neck cancer. Cancers 2020, 12, 1997. [Google Scholar] [CrossRef]
- Shao, Q.; Deng, L.; Liu, H.; Liu, Z.; Chen, J.; Jiang, F.; Yan, S.; Fu, R. Involvement of MM cell-derived exosomes in T lymphocytes immune responses. Oncol. Lett. 2020, 20, 1. [Google Scholar] [CrossRef]
- Kim, D.H.; Kim, H.; Choi, Y.J.; Kim, S.Y.; Lee, J.-E.; Sung, K.J.; Sung, Y.H.; Pack, C.-G.; Jung, M.-k.; Han, B. Exosomal PD-L1 promotes tumor growth through immune escape in non-small cell lung cancer. Exp. Mol. Med. 2019, 51, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Poggio, M.; Hu, T.; Pai, C.-C.; Chu, B.; Belair, C.D.; Chang, A.; Montabana, E.; Lang, U.E.; Fu, Q.; Fong, L. Suppression of exosomal PD-L1 induces systemic anti-tumor immunity and memory. Cell 2019, 177, 414–427.e413. [Google Scholar] [CrossRef] [Green Version]
- Zhou, K.; Guo, S.; Li, F.; Sun, Q.; Liang, G. Exosomal PD-L1: New insights into tumor immune escape mechanisms and therapeutic strategies. Front. Cell Dev. Biol. 2020, 8, 569219. [Google Scholar] [CrossRef]
- Taylor, D.D.; Gerçel-Taylor, Ç.; Lyons, K.S.; Stanson, J.; Whiteside, T.L. T-cell apoptosis and suppression of T-cell receptor/CD3-ζ by Fas ligand-containing membrane vesicles shed from ovarian tumors. Clin. Cancer Res. 2003, 9, 5113–5119. [Google Scholar]
- Söderberg, A.; Barral, A.M.; Söderström, M.; Sander, B.; Rosén, A. Redox-signaling transmitted in trans to neighboring cells by melanoma-derived TNF-containing exosomes. Free Radic. Biol. Med. 2007, 43, 90–99. [Google Scholar] [CrossRef]
- Shen, T.; Huang, Z.; Shi, C.; Pu, X.; Xu, X.; Wu, Z.; Ding, G.; Cao, L. Pancreatic cancer-derived exosomes induce apoptosis of T lymphocytes through the p38 MAPK-mediated endoplasmic reticulum stress. FASEB J. 2020, 34, 8442–8458. [Google Scholar] [CrossRef] [PubMed]
- Ye, S.-b.; Li, Z.-L.; Luo, D.-h.; Huang, B.-j.; Chen, Y.-S.; Zhang, X.-s.; Cui, J.; Zeng, Y.-x.; Li, J. Tumor-derived exosomes promote tumor progression and T-cell dysfunction through the regulation of enriched exosomal microRNAs in human nasopharyngeal carcinoma. Oncotarget 2014, 5, 5439. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, S.B.; Zhang, H.; Cai, T.T.; Liu, Y.N.; Ni, J.J.; He, J.; Peng, J.Y.; Chen, Q.Y.; Mo, H.Y.; Zhang, X.S. Exosomal miR-24-3p impedes T-cell function by targeting FGF11 and serves as a potential prognostic biomarker for nasopharyngeal carcinoma. J. Pathol. 2016, 240, 329–340. [Google Scholar] [CrossRef] [PubMed]
- Clayton, A.; Mitchell, J.P.; Mason, M.D.; Tabi, Z. Human tumor-derived exosomes selectively impair lymphocyte responses to interleukin-2. Cancer Res. 2007, 67, 7458–7466. [Google Scholar] [CrossRef] [Green Version]
- Maybruck, B.T.; Pfannenstiel, L.W.; Diaz-Montero, M.; Gastman, B.R. Tumor-derived exosomes induce CD8+ T cell suppressors. J. Immunother. Cancer 2017, 5, 1–15. [Google Scholar] [CrossRef]
- Jiang, Y.; Li, Y.; Zhu, B. T-cell exhaustion in the tumor microenvironment. Cell Death Dis. 2015, 6, e1792. [Google Scholar] [CrossRef] [Green Version]
- McLane, L.M.; Abdel-Hakeem, M.S.; Wherry, E.J. CD8 T cell exhaustion during chronic viral infection and cancer. Annu. Rev. Immunol. 2019, 37, 457–495. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Shen, H.; Zhangyuan, G.; Huang, R.; Zhang, W.; He, Q.; Jin, K.; Zhuo, H.; Zhang, Z.; Wang, J. 14-3-3ζ delivered by hepatocellular carcinoma-derived exosomes impaired anti-tumor function of tumor-infiltrating T lymphocytes. Cell Death Dis. 2018, 9, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Kalvala, A.; Wallet, P.; Yang, L.; Wang, C.; Li, H.; Nam, A.; Nathan, A.; Mambetsariev, I.; Poroyko, V.; Gao, H. Phenotypic switching of naive T cells to immune-suppressive Treg-like cells by mutant KRAS. J. Clin. Med. 2019, 8, 1726. [Google Scholar] [CrossRef] [Green Version]
- Szajnik, M.; Czystowska, M.; Szczepanski, M.J.; Mandapathil, M.; Whiteside, T.L. Tumor-derived microvesicles induce, expand and up-regulate biological activities of human regulatory T cells (Treg). PLoS ONE 2010, 5, e11469. [Google Scholar] [CrossRef] [Green Version]
- Czystowska-Kuzmicz, M.; Sosnowska, A.; Nowis, D.; Ramji, K.; Szajnik, M.; Chlebowska-Tuz, J.; Wolinska, E.; Gaj, P.; Grazul, M.; Pilch, Z. Small extracellular vesicles containing arginase-1 suppress T-cell responses and promote tumor growth in ovarian carcinoma. Nat. Commun. 2019, 10, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clayton, A.; Al-Taei, S.; Webber, J.; Mason, M.D.; Tabi, Z. Cancer exosomes express CD39 and CD73, which suppress T cells through adenosine production. J. Immunol. 2011, 187, 676–683. [Google Scholar] [CrossRef] [Green Version]
- Yamada, N.; Kuranaga, Y.; Kumazaki, M.; Shinohara, H.; Taniguchi, K.; Akao, Y. Colorectal cancer cell-derived extracellular vesicles induce phenotypic alteration of T cells into tumor-growth supporting cells with transforming growth factor-β1-mediated suppression. Oncotarget 2016, 7, 27033. [Google Scholar] [CrossRef]
- Hellwinkel, J.E.; Redzic, J.S.; Harland, T.A.; Gunaydin, D.; Anchordoquy, T.J.; Graner, M.W. Glioma-derived extracellular vesicles selectively suppress immune responses. Neuro-Oncology 2015, 18, 497–506. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mrizak, D.; Martin, N.; Barjon, C.; Jimenez-Pailhes, A.-S.; Mustapha, R.; Niki, T.; Guigay, J.; Pancré, V.; de Launoit, Y.; Busson, P. Effect of nasopharyngeal carcinoma-derived exosomes on human regulatory T cells. J. Natl. Cancer Inst. 2015, 107, dju363. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rong, L.; Li, R.; Li, S.; Luo, R. Immunosuppression of breast cancer cells mediated by transforming growth factor-β in exosomes from cancer cells. Oncol. Lett. 2016, 11, 500–504. [Google Scholar] [CrossRef] [Green Version]
- Dianat-Moghadam, H.; Mahari, A.; Heidarifard, M.; Parnianfard, N.; Pourmousavi-Kh, L.; Rahbarghazi, R.; Amoozgar, Z. NK cells-directed therapies target circulating tumor cells and metastasis. Cancer Lett. 2021, 497, 41–53. [Google Scholar] [CrossRef]
- Bottos, A.; Gotthardt, D.; Gill, J.W.; Gattelli, A.; Frei, A.; Tzankov, A.; Sexl, V.; Wodnar-Filipowicz, A.; Hynes, N.E. Decreased NK-cell tumour immunosurveillance consequent to JAK inhibition enhances metastasis in breast cancer models. Nat. Commun. 2016, 7, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Kruse, P.H.; Matta, J.; Ugolini, S.; Vivier, E. Natural cytotoxicity receptors and their ligands. Immunol. Cell Biol. 2014, 92, 221–229. [Google Scholar] [CrossRef]
- Hedlund, M.; Stenqvist, A.-C.; Nagaeva, O.; Kjellberg, L.; Wulff, M.; Baranov, V.; Mincheva-Nilsson, L. Human placenta expresses and secretes NKG2D ligands via exosomes that down-modulate the cognate receptor expression: Evidence for immunosuppressive function. J. Immunol. 2009, 183, 340–351. [Google Scholar] [CrossRef] [Green Version]
- Ashiru, O.; Boutet, P.; Fernández-Messina, L.; Agüera-González, S.; Skepper, J.N.; Valés-Gómez, M.; Reyburn, H.T. Natural killer cell cytotoxicity is suppressed by exposure to the human NKG2D ligand MICA* 008 that is shed by tumor cells in exosomes. Cancer Res. 2010, 70, 481–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labani-Motlagh, A.; Israelsson, P.; Ottander, U.; Lundin, E.; Nagaev, I.; Nagaeva, O.; Dehlin, E.; Baranov, V.; Mincheva-Nilsson, L. Differential expression of ligands for NKG2D and DNAM-1 receptors by epithelial ovarian cancer-derived exosomes and its influence on NK cell cytotoxicity. Tumor Biol. 2016, 37, 5455–5466. [Google Scholar] [CrossRef]
- Lundholm, M.; Schröder, M.; Nagaeva, O.; Baranov, V.; Widmark, A.; Mincheva-Nilsson, L.; Wikström, P. Prostate tumor-derived exosomes down-regulate NKG2D expression on natural killer cells and CD8+ T cells: Mechanism of immune evasion. PLoS ONE 2014, 9, e108925. [Google Scholar] [CrossRef] [PubMed]
- Clayton, A.; Mitchell, J.P.; Linnane, S.; Mason, M.D.; Tabi, Z. Human tumor-derived exosomes down-modulate NKG2D expression. J. Immunol. 2008, 180, 7249–7258. [Google Scholar] [CrossRef] [Green Version]
- López-Cobo, S.; Campos-Silva, C.; Moyano, A.; Oliveira-Rodríguez, M.; Paschen, A.; Yáñez-Mó, M.; Blanco-López, M.C.; Valés-Gómez, M. Immunoassays for scarce tumour-antigens in exosomes: Detection of the human NKG2D-Ligand, MICA, in tetraspanin-containing nanovesicles from melanoma. J. Nanobiotechnol. 2018, 16, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Admyre, C.; Johansson, S.M.; Qazi, K.R.; Filén, J.-J.; Lahesmaa, R.; Norman, M.; Neve, E.P.; Scheynius, A.; Gabrielsson, S. Exosomes with immune modulatory features are present in human breast milk. J. Immunol. 2007, 179, 1969–1978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Catalano, M.; O’Driscoll, L. Inhibiting extracellular vesicles formation and release: A review of EV inhibitors. J. Extracell. Vesicles 2020, 9, 1703244. [Google Scholar] [CrossRef] [Green Version]
- Kulshreshtha, A.; Singh, S.; Ahmad, M.; Khanna, K.; Ahmad, T.; Agrawal, A.; Ghosh, B. Simvastatin mediates inhibition of exosome synthesis, localization and secretion via multicomponent interventions. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Datta, A.; Kim, H.; Lal, M.; McGee, L.; Johnson, A.; Moustafa, A.A.; Jones, J.C.; Mondal, D.; Ferrer, M.; Abdel-Mageed, A.B. Manumycin A suppresses exosome biogenesis and secretion via targeted inhibition of Ras/Raf/ERK1/2 signaling and hnRNP H1 in castration-resistant prostate cancer cells. Cancer Lett. 2017, 408, 73–81. [Google Scholar] [CrossRef]
- Huda, M.N.; Nafiujjaman, M.; Deaguero, I.G.; Okonkwo, J.; Hill, M.L.; Kim, T.; Nurunnabi, M. Potential use of exosomes as diagnostic biomarkers and in targeted drug delivery: Progress in clinical and preclinical applications. ACS Biomater. Sci. Eng. 2021, 7, 2106–2149. [Google Scholar] [CrossRef]
- Chen, W.; Wang, J.; Shao, C.; Liu, S.; Yu, Y.; Wang, Q.; Cao, X. Efficient induction of antitumor T cell immunity by exosomes derived from heat-shocked lymphoma cells. Eur. J. Immunol. 2006, 36, 1598–1607. [Google Scholar] [CrossRef] [PubMed]
- Podolska, M.J.; Shan, X.; Janko, C.; Boukherroub, R.; Gaipl, U.S.; Szunerits, S.; Frey, B.; Muñoz, L.E. Graphene-induced hyperthermia (GIHT) combined with radiotherapy fosters immunogenic cell death. Front. Oncol. 2021, 11, 664615. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Chen, Y.; Wang, S.; Yu, L.; Shen, Y.; Zhong, H.; Yang, Y. Exosomes from heat-stressed tumour cells inhibit tumour growth by converting regulatory T cells to Th17 cells via IL-6. Immunology 2018, 154, 132–143. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Guo, J.; Yang, M.; Zhu, X.; Cao, X. Chemokine-containing exosomes are released from heat-stressed tumor cells via lipid raft-dependent pathway and act as efficient tumor vaccine. J. Immunol. 2011, 186, 2219–2228. [Google Scholar] [CrossRef] [Green Version]
- Toraya-Brown, S.; Fiering, S. Local tumour hyperthermia as immunotherapy for metastatic cancer. Int. J. Hyperth. 2014, 30, 531–539. [Google Scholar] [CrossRef]
- Guo, G.; Tan, Z.; Liu, Y.; Shi, F.; She, J. The therapeutic potential of stem cell-derived exosomes in the ulcerative colitis and colorectal cancer. Stem Cell Res. Ther. 2022, 13, 1–18. [Google Scholar] [CrossRef]
- Shi, X.; Sun, J.; Li, H.; Lin, H.; Xie, W.; Li, J.; Tan, W. Antitumor efficacy of interferon-γ-modified exosomal vaccine in prostate cancer. Prostate 2020, 80, 811–823. [Google Scholar] [CrossRef]
- Hu, W.; Huang, F.; Ning, L.; Hao, J.; Wan, J.; Hao, S. Enhanced immunogenicity of leukemia-derived exosomes via transfection with lentiviral vectors encoding costimulatory molecules. Cell Oncol. 2020, 43, 889–900. [Google Scholar] [CrossRef]
- Gu, X.; Erb, U.; Büchler, M.W.; Zöller, M. Improved vaccine efficacy of tumor exosome compared to tumor lysate loaded dendritic cells in mice. Int. J. Cancer 2015, 136, E74–E84. [Google Scholar] [CrossRef]
- Lee, Y.S.; Kim, S.H.; Cho, J.A.; Kim, C.W. Introduction of the CIITA gene into tumor cells produces exosomes with enhanced anti-tumor effects. Exp. Mol. Med. 2011, 43, 281–290. [Google Scholar] [CrossRef] [Green Version]
- Zuo, B.; Qi, H.; Lu, Z.; Chen, L.; Sun, B.; Yang, R.; Zhang, Y.; Liu, Z.; Gao, X.; You, A. Alarmin-painted exosomes elicit persistent antitumor immunity in large established tumors in mice. Nat. Commun. 2020, 11, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thakur, A.; Parra, D.C.; Motallebnejad, P.; Brocchi, M.; Chen, H.J. Exosomes: Small vesicles with big roles in cancer, vaccine development, and therapeutics. Bioact. Mater. 2022, 10, 281–294. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Martin, P.; Fogarty, B.; Brown, A.; Schurman, K.; Phipps, R.; Yin, V.P.; Lockman, P.; Bai, S. Exosome delivered anticancer drugs across the blood-brain barrier for brain cancer therapy in Danio rerio. Pharm. Res. 2015, 32, 2003–2014. [Google Scholar] [CrossRef] [PubMed]
- Yong, T.; Zhang, X.; Bie, N.; Zhang, H.; Zhang, X.; Li, F.; Hakeem, A.; Hu, J.; Gan, L.; Santos, H.A. Tumor exosome-based nanoparticles are efficient drug carriers for chemotherapy. Nat. Commun. 2019, 10, 1–16. [Google Scholar]
- Qiao, L.; Hu, S.; Huang, K.; Su, T.; Li, Z.; Vandergriff, A.; Cores, J.; Dinh, P.-U.; Allen, T.; Shen, D. Tumor cell-derived exosomes home to their cells of origin and can be used as Trojan horses to deliver cancer drugs. Theranostics 2020, 10, 3474. [Google Scholar] [CrossRef]


| Cancer Type | Cellular Source | Vesicular Cargo | The Main Result | Refs. |
|---|---|---|---|---|
| Triple-negative breast cancer | MDA-MB-231 BT-20 MDA-MB-453 MCF7 BT-474 SK-BR-3 | ITGB4 proteins | Enhanced mitophagy and lactate generation in CAFs in a BNIP3L-dependent manner. | [64] |
| TNBC MDA-MB-231 MDA-MB-468 | miR-9 | Influenced the properties of NFs and promoted the switch to CAF, thereby leading to tumor growth. | [58] | |
| Ovarian cancer | A2780 SKVO3 | miR-630 | Raised amounts of α-SMA and FAP in NFs resulted in the differentiation of NFs into CAFs via inhibiting KLF6 and activating the NFκB pathway. | [60] |
| Bladder cancer | RT4 T24 SW1710 | TGF-β | Triggered the differentiation of fibroblasts to CAFs by SMAD pathway activation | [63] |
| Colorectal cancer | SW620 SW480 | Vesicular cargo | Activated normal quiescent fibroblasts (α-SMA−, CAV+) into CAF-like fibroblasts (α-SMA+, CAV−) with pro-proliferative and pro-angiogenic features | [65] |
| Lung cancer (LC) | A549 H460 | miR-210 | Promoted the NFs transferring into CAFs via activating JAK2/STAT3 pathway, and ten-eleven translocation 2 (TET2) | [61] |
| Melanoma | B16BL6 | eTGF-β | Triggered TGF-β signaling in HUVECs and differentiated them into CAFs | [19] |
| B16F0 | Gm26809 | Stimulated conversion of fibroblast NIH3T3 cells into CAFs | [66] | |
| B16-F10 | miR-21 | Stimulated invasiveness of fibroblasts by increasing matrix metalloprotein (MMP) and down-regulation of tissue inhibitor of metalloproteinase 3 (TIMP3) expressions. | [59] | |
| Breast cancer | MCF-7 MDA-MB-231 | Vesicular survivin | Converted NFs into myofibroblasts by upregulating SOD1 and increased proliferation, EMT, and stemness. | [62] |
| Head and neck squamous cell carcinoma (HNSCC) | SAS HSC-3 | Vesicular cargo | Convert normal fibroblasts into CAF-like cells and raised fibroblast proliferation, migration and activation of 11 signaling pathways (IL-6- and IL-17-related signaling) | [67] |
| Cancer Type. | Cellular Source | Vesicular Cargo | The Main Result | Refs. |
|---|---|---|---|---|
| Prostate cancer | DU145 | PGE2 | Triggered the expression of CD73 and then CD39 on DCs, resulting in inhibition of TNFα- and IL-12-production via an ATP-dependent manner | [156] |
| NSCLC | Blood samples from NSCLC patients | Galectin-9 and Tim-3 | Interacted with TIM-3 on DCs | [157] |
| Renal cancer | CD105+ CSCs CD105− TCs | HLA-G | Disrupted maturation of DCs and T-cell immune responses | [133] |
| Glioblastoma | CSF samples from glioma patients GL261 U87MG U118 MG | Galectin-9 | Inhibited antigen recognition, processing, and presentation by interacting with TIM-3 on DCs | [142] |
| Ascites of glioma patients | PD-L1 | Impaired DCs maturation via formation of immunosuppressive monocytes | [77] | |
| Blood samples from glioma patients GSC20 GSC267 GSC17 MEC-1 | Vesicular cargo | Skewed monocytes toward an immune suppressive phenotype and induced programmed PD-L1 expression on monocytes through STAT3 phosphorylation and TLR7-dependent manner | [33,158] | |
| Melanoma | SKMEL28 A375 C32TG | S100, A8/A9 | Inhibited DCs maturation and reduced expression of CD83, CD86, Th1 polarizing chemokines (such as Flt3L, IL-15), and migration chemokines (MIP-1α and MIP-1β) | [150] |
| lymphatic fluid sample of melanoma patients ATCC | S100A9 | Inhibited DCs maturation and prepared metastatic niche in lymph nodes | [159] | |
| B16-F0 | TGF-β1 | Increased mRNA levels of IL-4 and TGF-β1 which inhibited DCs’ maturation | [160] | |
| Blood samples from melanoma patients B16-F0 | HSP72 and HSP105 | Induced secretion of IL-6 from DCs via TLR4- and TLR2-dependent manner activating STAT3-dependent MMP 9 activity | [161] | |
| lymphocytic leukemia | Blood samples from CLL patients | S100A8/A9 | CD83, CD86, IL-12, and IL-15 expressions were all downregulated via activating the NFκB pathway | [162,163] |
| lung carcinoma | LLC | PD-L1 | Myeloid precursor cells were unable to differentiate into CD11c+ DCs in the presence of vesicular PD-L1 and resulted in DCs death | [152] |
| LLC A549 | MALAT1 | Inhibited DC function and T-cell proliferation and increased DC autophagy via AKT/mTOR Pathway | [151] | |
| Breast cancer | MDA-MB-231 TS/A | Vesicular cargo | Inhibited the development of myeloid precursor cells into DCs by increasing IL-6 production and reducing CD83 and CD86 expression | [149] |
| 4T1 | PD-L1 | Myeloid precursor cells were unable to differentiate into CD11c+ DCs in the presence of vesicular PD-L1 and resulted in DC death | [152] | |
| Blood samples from melanoma patients 4T1 | HSP72 and HSP105 | Promoted DCs to IL-6 secretion in a TLR2- and TLR4-dependent manner which activated STAT3-dependent MMP 9 activity | [161] |
| Cancer Type | Cellular Source | Vesicular Cargo | Mechanism of Action | Refs. |
|---|---|---|---|---|
| Ovarian cancer | Ascites of ovarian patients OVCAR3 SKOV3 AD10 | TGF-β1, IL-10 | Increased IL-10, FasL, TGF-β1, CTLA-4, which promoted Treg proliferation, suppressor activity, and Treg cell survival. | [190] |
| Blood samples from ovarian patients Ascites of ovarian patients OVCAR-3 AD10 A2780 Skov3 CaOv-3 MDAH2774 OvCa-14 OVP-10 | Arginase-1 | Inhibited antigen-specific T-cell proliferation | [191] | |
| Prostate cancer | Pleural fluid samples of malignant pleural mesothelioma patients DU145 PC3 | PGE2 | T-cell inhibition was mediated through the adenosine A2A receptor | [192] |
| DU145 PC3 | TGF-β1 | Skewed IL-2 responses in T cells and suppressed cytotoxicity | [184] | |
| Melanoma | Blood samples from melanoma patients Blood samples from melanoma tumor-bearing mice WM1552C WM35 WM793 WM902B UACC-903 1205Lu WM9 WM164 | PD-L1 | Suppressed the function of CD8 T cells | [136] |
| Colorectal cancer | Blood samples from colorectal patients SW403 CRC28462 1869col | FasL, TRAIL | Induced T-cell apoptosis | [168] |
| DLD-1 WiDr | TGF-β1 | Induced differentiation of T cells to Treg-like cells via the TGF-β pathway while inactivating the SAPK signaling pathway | [193] | |
| Caco-2 | Galectin- 1 | Induced suppressor phenotype in human CD8+ T cells | [185] | |
| Head and neck cancer | Tu167 SCC0209 HN60 | Galectin- 1 | Induced suppressor phenotype in human CD8+ T cells | [185] |
| Blood samples from HNSCC patients | Vesicular cargo | Induced apoptosis in CD8+ T cells by converting CD4+ T cells to Treg | [174] | |
| Glioblastoma | Blood samples from glioma patients UPN933 E3-2 E6-5 | Vesicular cargo | Deactivated T cells by FasL-dependent mechanisms and inhibit secretion of IL-2 | [194] |
| Nasopharyngeal cancer (NPC) | Blood samples from NPC patients Blood samples from NPC tumor-bearing mice C15 C17 | Galectin- 9 | Induced huge apoptosis in T cells via membrane receptor Tim-3 | [171] |
| Blood samples from NPC patients C15 C17 | CCL20 | Facilitated Treg recruitment and expansion that increased secretion of immunosuppressive cytokines (IL10, TGFB1) | [195] | |
| Blood samples from NPC patients TW03 C666 CNE2 | miR- 24–3p | Blocked T-cell proliferation and Th1 and Th17 differentiation and promoted Treg induction via dephosphorylating ERK, STAT1, and STAT3 by reducing IL-2, IFNγ, and IL-17 secretion and phosphorylating STAT5 with increasing IL-6, IL-1β, and IL-10 secretion | [182,183] | |
| Oral squamous cell carcinoma(OSCC) | SCC-9 SCC-4 CAL-27 | HSP70 | Altered development and cytotoxicity of T cells in an HSP70-dependent way via miR-21/PTEN/PD-L1 regulatory pathway | [170] |
| Blood samples from OSCC patients PCI-13 | FasL | Induced apoptotic pathways in T cells through triggering caspase-3 cleavage, the release of cytochrome c that led to disrupting mitochondrial membrane, and decreased TCR-ζ chain production | [172] | |
| Breast cancer | MCF7 | CD73, CD39 | Inhibited T cells via the adenosine A2A receptor | [192] |
| BT-474 MDA-MB-231 | TGF-β1 | Suppressed T-cell proliferation | [196] | |
| Lung cancer | Blood samples from lung cancer patients A549 PC9 95D | circRNA- 002178 | Enhanced PDL1 expression led to induced T-cell exhaustion | [132] |
| Hepatocellular Carcinoma (HCC) | Blood samples from HCC patients MHCC97H | 14- 3- 3ζ | Inhibited the functions of T cells against cancer in the HCC microenvironment | [188] |
| Hepa1-6 H22 | SALL4/miR-146a- 5p | T cells were exhausted by reducing IFN-γ and TNF-α expression while increasing the expression of inhibitory receptors such as PD-1 and CTLA-4 | [34] | |
| Pancreatic cancer | BxPC-3 tdTomato-BxPC-3 | Vesicular cargo | Induced ER stress-mediated apoptosis via activating the p38 MAP kinase signaling | [181] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Najaflou, M.; Shahgolzari, M.; Khosroushahi, A.Y.; Fiering, S. Tumor-Derived Extracellular Vesicles in Cancer Immunoediting and Their Potential as Oncoimmunotherapeutics. Cancers 2023, 15, 82. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers15010082
Najaflou M, Shahgolzari M, Khosroushahi AY, Fiering S. Tumor-Derived Extracellular Vesicles in Cancer Immunoediting and Their Potential as Oncoimmunotherapeutics. Cancers. 2023; 15(1):82. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers15010082
Chicago/Turabian StyleNajaflou, Meysam, Mehdi Shahgolzari, Ahmad Yari Khosroushahi, and Steven Fiering. 2023. "Tumor-Derived Extracellular Vesicles in Cancer Immunoediting and Their Potential as Oncoimmunotherapeutics" Cancers 15, no. 1: 82. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers15010082