

Systematic Evaluation of Antigenic Stimulation in Chronic Lymphocytic Leukemia: Humoral Immunity as Biomarkers for Disease Evolution
 , , , , ,
, , , , ,  , , , and
, , , and
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Patient Cohort
2.2. Serum Sample
2.3. Evaluation of Immunoglobulin Quantitative Levels
2.3.1. Immunoglobulin Profiling
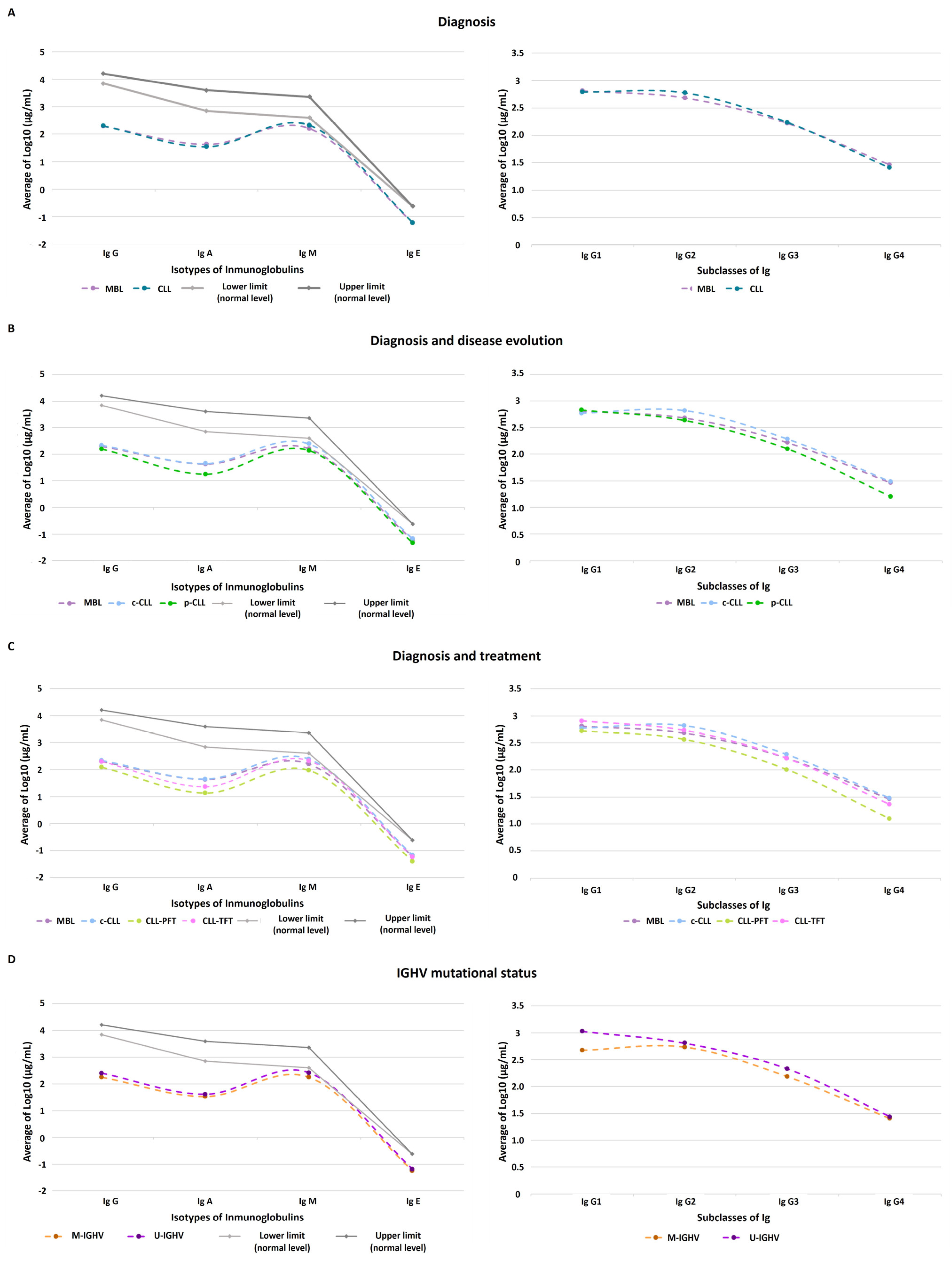
2.3.2. Data Analysis of Immunoglobulin Profiles
2.4. Evaluation of Autoantibody Serum Profiles
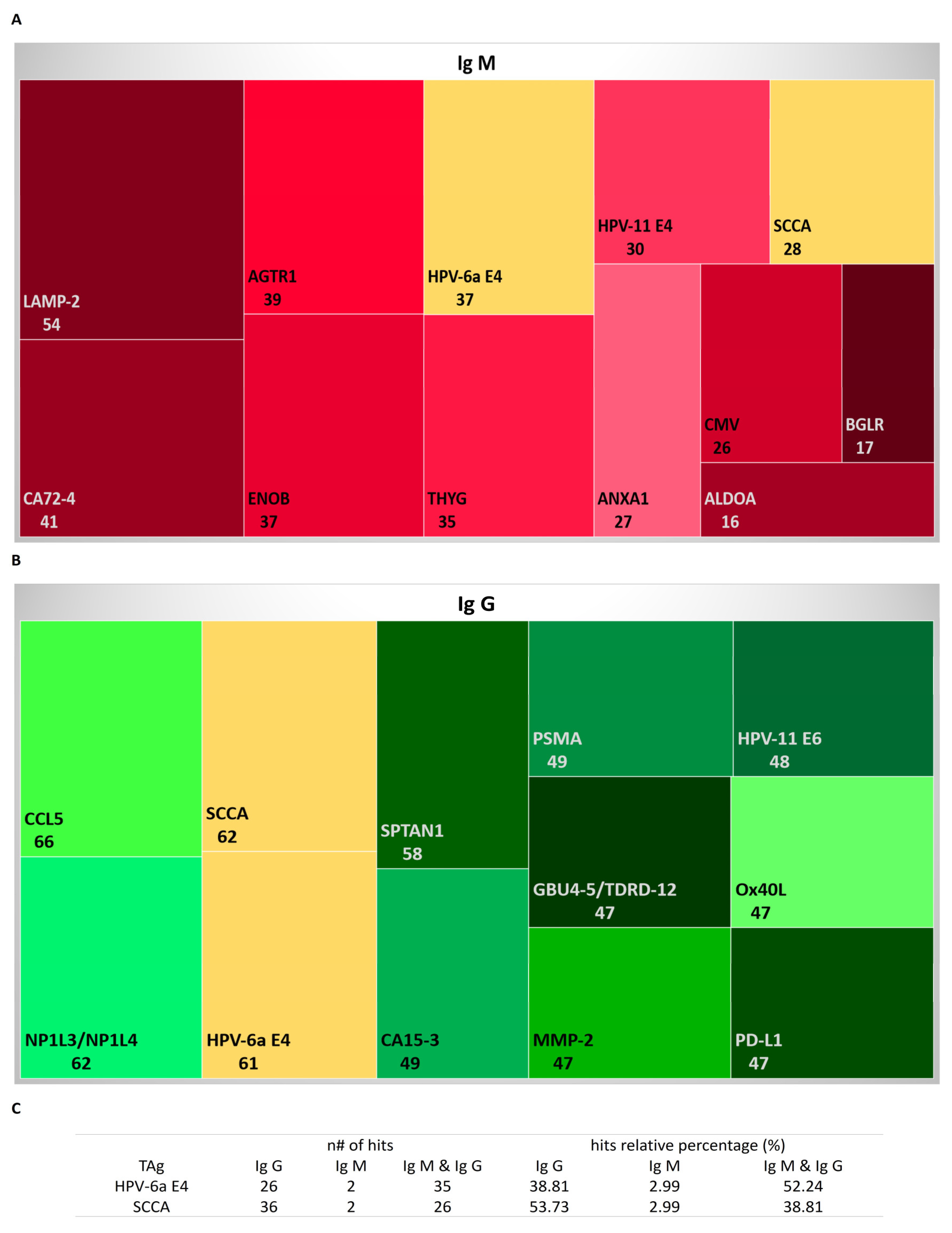
2.4.1. Autoantibodies to Known Tumoral and Neoplasm Antigen Proteins
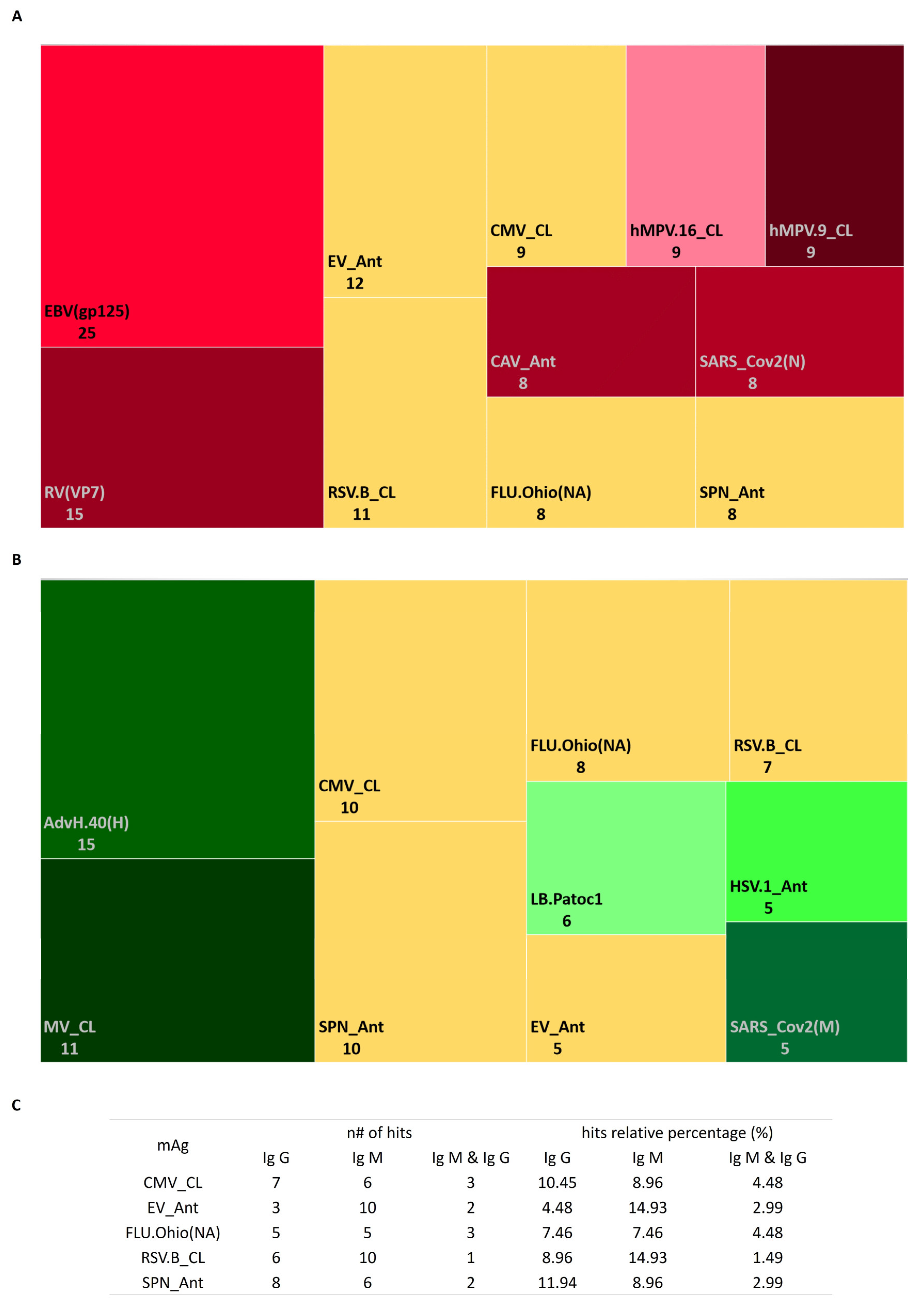
2.4.2. Profiling Antimicrobial Antibodies via Stimulation Persistent Antigenic Array
2.4.3. Image Acquisition
2.5. Data Analyses
2.5.1. Quantitative Determination of Immunoglobulin Level Profiles
2.5.2. Data Preprocessing and Dataset Analyses
- (i)
- Signal normalization ():where is the median signal intensity of the feature pixels, is the median signal intensity of the background pixels, and is the standard deviation for the background pixels at a selected wavelength ( [31].
- (ii)
- Negative control () signal intensity:where is the normalized signal for a selected spot (incubated with the negative control) and is the number of subarrays (belonging to the same incubation).
- (iii)
- Spot signal intensity ():where is the normalized signal for the spot and is the normalized signal for the corresponding negative control spot.
- (iv)
- Ag average signal intensity ():where is the signal intensity at a selected spot and is the number of replicas.
- (v)
- Spot selection with respect to the Ref.:
- (a)
- Only Ags with a signal intensity > 0 were considered.
- (b)
- Selection of Ag-positive signals above compared to Ref.:where and are the average signal intensity of the Ag for the sample and the Ref., respectively; and is the standard deviation signal intensity of the Ag for the Ref.
- (vi)
- Ag Z-score:
- (vii)
- Number (n#) of Ag hits for each Ab isotype or clinical groups (hits).
3. Results
3.1. Quantitative Immunoglobulin Levels
3.2. Autoantibodies to Known Tumoral and Neoplasm Antigen Proteins
3.3. Profile of Antimicrobial Antibodies according to Stimulation Persistent Antigenic Array (SPA Array)
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Haseeb, M.; Anwar, M.A.; Choi, S. Molecular Interactions Between Innate and Adaptive Immune Cells in Chronic Lymphocytic Leukemia and Their Therapeutic Implications. Front. Immunol. 2018, 9, 2720. [Google Scholar] [CrossRef] [PubMed]
- Allegra, A.; Musolino, C.; Tonacci, A.; Pioggia, G.; Casciaro, M.; Gangemi, S. Clinico-Biological Implications of Modified Levels of Cytokines in Chronic Lymphocytic Leukemia: A Possible Therapeutic Role. Cancers 2020, 12, 524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dubois, N.; Crompot, E.; Meuleman, N.; Bron, D.; Lagneaux, L.; Stamatopoulos, B. Importance of Crosstalk Between Chronic Lymphocytic Leukemia Cells and the Stromal Microenvironment: Direct Contact, Soluble Factors, and Extracellular Vesicles. Front. Oncol. 2020, 10, 1422. [Google Scholar] [CrossRef] [PubMed]
- Manouchehri-Doulabi, E.; Abbaspour, S.; Rostami, S.; Faranoush, M.; Ghahramanfard, F.; Pak, F.; Barati, M.; Kokhaei, P.; Momtazi-Borojeni, A.A. Evaluating the Mechanism Underlying Antitumor Effect of Interleukin 27 on B Cells of Chronic Lymphocytic Leukemia Patients. J. Cell. Physiol. 2020, 235, 9424–9431. [Google Scholar] [CrossRef]
- Forconi, F.; Lanham, S.A.; Chiodin, G. Biological and Clinical Insight from Analysis of the Tumor B-Cell Receptor Structure and Function in Chronic Lymphocytic Leukemia. Cancers 2022, 14, 663. [Google Scholar] [CrossRef]
- Purroy, N.; Wu, C.J. Coevolution of Leukemia and Host Immune Cells in Chronic Lymphocytic Leukemia. Cold Spring Harb. Perspect. Med. 2017, 7, a026740. [Google Scholar] [CrossRef] [Green Version]
- Griggio, V.; Perutelli, F.; Salvetti, C.; Boccellato, E.; Boccadoro, M.; Vitale, C.; Coscia, M. Immune Dysfunctions and Immune-Based Therapeutic Interventions in Chronic Lymphocytic Leukemia. Front. Immunol. 2020, 11, 594556. [Google Scholar] [CrossRef]
- Lin, K.; Rockliffe, N.; Johnson, G.G.; Sherrington, P.D.; Pettitt, A.R. Hsp90 Inhibition Has Opposing Effects on Wild-Type and Mutant P53 and Induces P21 Expression and Cytotoxicity Irrespective of P53/ATM Status in Chronic Lymphocytic Leukaemia Cells. Oncogene 2008, 27, 2445–2455. [Google Scholar] [CrossRef] [Green Version]
- Nückel, H.; Switala, M.; Sellmann, L.; Horn, P.A.; Dürig, J.; Dührsen, U.; Küppers, R.; Grosse-Wilde, H.; Rebmann, V. The Prognostic Significance of Soluble NKG2D Ligands in B-Cell Chronic Lymphocytic Leukemia. Leukemia 2010, 24, 1152–1159. [Google Scholar] [CrossRef]
- Nisticò, N.; Maisano, D.; Iaccino, E.; Vecchio, E.; Fiume, G.; Rotundo, S.; Quinto, I.; Mimmi, S. Role of Chronic Lymphocytic Leukemia (CLL)-Derived Exosomes in Tumor Progression and Survival. Pharmaceuticals 2020, 13, 244. [Google Scholar] [CrossRef]
- Vitale, C.; Boccellato, E.; Comba, L.; Jones, R.; Perutelli, F.; Griggio, V.; Coscia, M. Impact of Immune Parameters and Immune Dysfunctions on the Prognosis of Patients with Chronic Lymphocytic Leukemia. Cancers 2021, 13, 3856. [Google Scholar] [CrossRef] [PubMed]
- Landeira-Viñuela, A.; Arias-Hidalgo, C.; Juanes-Velasco, P.; Alcoceba, M.; Navarro-Bailón, A.; Pedreira, C.E.; Lecrevisse, Q.; Díaz-Muñoz, L.; Sánchez-Santos, J.M.; Hernández, Á.-P.; et al. Unravelling Soluble Immune Checkpoints in Chronic Lymphocytic Leukemia: Physiological Immunomodulators or Immune Dysfunction. Front. Immunol. 2022, 13, 965905. [Google Scholar] [CrossRef] [PubMed]
- Bojarska-Junak, A.; Waldowska, M.; Woś, J.; Chocholska, S.; Hus, I.; Tomczak, W.; Dzik, M.; Hus, M.; Roliński, J. Intracellular IL-4 and IFN-γ Expression in INKT Cells from Patients with Chronic Lymphocytic Leukemia. Oncol. Lett. 2018, 15, 1580–1590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sordo-Bahamonde, C.; Lorenzo-Herrero, S.; González-Rodríguez, A.P.; Payer, Á.R.; González-García, E.; López-Soto, A.; Gonzalez, S. LAG-3 Blockade with Relatlimab (BMS-986016) Restores Anti-Leukemic Responses in Chronic Lymphocytic Leukemia. Cancers 2021, 13, 2112. [Google Scholar] [CrossRef] [PubMed]
- Pascutti, M.F.; Jak, M.; Tromp, J.M.; Derks, I.A.M.; Remmerswaal, E.B.M.; Thijssen, R.; van Attekum, M.H.A.; van Bochove, G.G.; Luijks, D.M.; Pals, S.T.; et al. IL-21 and CD40L Signals from Autologous T Cells Can Induce Antigen-Independent Proliferation of CLL Cells. Blood 2013, 122, 3010–3019. [Google Scholar] [CrossRef] [Green Version]
- Davi, F.; Langerak, A.W.; de Septenville, A.L.; Kolijn, P.M.; Hengeveld, P.J.; Chatzidimitriou, A.; Bonfiglio, S.; Sutton, L.-A.; Rosenquist, R.; Ghia, P.; et al. Immunoglobulin Gene Analysis in Chronic Lymphocytic Leukemia in the Era of next Generation Sequencing. Leukemia 2020, 34, 2545–2551. [Google Scholar] [CrossRef]
- Higgins, B.W.; McHeyzer-Williams, L.J.; McHeyzer-Williams, M.G. Programming Isotype-Specific Plasma Cell Function. Trends Immunol. 2019, 40, 345–357. [Google Scholar] [CrossRef]
- Darwiche, W.; Gubler, B.; Marolleau, J.-P.; Ghamlouch, H. Chronic Lymphocytic Leukemia B-Cell Normal Cellular Counterpart: Clues from a Functional Perspective. Front. Immunol. 2018, 9, 683. [Google Scholar] [CrossRef]
- Stahl, D.; Lacroix-Desmazes, S.; Sibrowski, W.; Kazatchkine, M.D.; Kaveri, S.V. Broad Alterations of Self-Reactive Antibody-Repertoires of Plasma IgM and IgG in B-Cell Chronic Lymphocytic Leukemia (B-CLL) and B-CLL Related Target-Restricted Autoimmunity. Leuk. Lymphoma 2001, 42, 163–176. [Google Scholar] [CrossRef]
- Freeman, J.A.; Crassini, K.R.; Best, O.G.; Forsyth, C.J.; Mackinlay, N.J.; Han, P.; Stevenson, W.; Mulligan, S.P. Immunoglobulin G Subclass Deficiency and Infection Risk in 150 Patients with Chronic Lymphocytic Leukemia. Leuk. Lymphoma 2013, 54, 99–104. [Google Scholar] [CrossRef]
- Visentin, A.; Compagno, N.; Cinetto, F.; Imbergamo, S.; Zambello, R.; Piazza, F.; Semenzato, G.; Trentin, L.; Agostini, C. Clinical Profile Associated with Infections in Patients with Chronic Lymphocytic Leukemia. Protective Role of Immunoglobulin Replacement Therapy. Haematologica 2015, 100, e515–e518. [Google Scholar] [CrossRef] [Green Version]
- Kipps, T.J.; Stevenson, F.K.; Wu, C.J.; Croce, C.M.; Packham, G.; Wierda, W.G.; O’Brien, S.; Gribben, J.; Rai, K. Chronic Lymphocytic Leukaemia. Nat. Rev. Dis. Prim. 2017, 3, 16096. [Google Scholar] [CrossRef] [Green Version]
- Awan, F.T.; Byrd, J.C. 99-Chronic Lymphocytic Leukemia. In Abeloff’s Clinical Oncology, 6th ed.; Niederhuber, J.E., Armitage, J.O., Kastan, M.B., Doroshow, J.H., Tepper, J.E., Eds.; Elsevier: Amsterdam, The Netherlands, 2020; pp. 1850–1871.e5. ISBN 978-0-323-47674-4. [Google Scholar]
- Grywalska, E.; Zaborek, M.; Łyczba, J.; Hrynkiewicz, R.; Bębnowska, D.; Becht, R.; Sosnowska-Pasiarska, B.; Smok-Kalwat, J.; Pasiarski, M.; Góźdź, S.; et al. Chronic Lymphocytic Leukemia-Induced Humoral Immunosuppression: A Systematic Review. Cells 2020, 9, 2398. [Google Scholar] [CrossRef]
- Hallek, M.; Cheson, B.D.; Catovsky, D.; Caligaris-Cappio, F.; Dighiero, G.; Döhner, H.; Hillmen, P.; Keating, M.; Montserrat, E.; Chiorazzi, N.; et al. IwCLL Guidelines for Diagnosis, Indications for Treatment, Response Assessment, and Supportive Management of CLL. Blood 2018, 131, 2745–2760. [Google Scholar] [CrossRef] [Green Version]
- Rai, K.; Sawitsky, A.; Cronkite, E.; Chanana, A.; Levy, R.; Pasternack, B. Clinical Staging of Chronic Lymphocytic Leukemia. Blood 1975, 46, 219–234. [Google Scholar] [CrossRef] [Green Version]
- Binet, J.L.; Auquier, A.; Dighiero, G.; Chastang, C.; Piguet, H.; Goasguen, J.; Vaugier, G.; Potron, G.; Colona, P.; Oberling, F.; et al. A New Prognostic Classification of Chronic Lymphocytic Leukemia Derived from a Multivariate Survival Analysis. Cancer 1981, 48, 198–206. [Google Scholar] [CrossRef]
- González-González, M.; Bartolome, R.; Jara-Acevedo, R.; Casado-Vela, J.; Dasilva, N.; Matarraz, S.; García, J.; Alcazar, J.A.; Sayagues, J.M.; Orfao, A.; et al. Evaluation of Homo- and Hetero-Functionally Activated Glass Surfaces for Optimized Antibody Arrays. Anal. Biochem. 2014, 450, 37–45. [Google Scholar] [CrossRef]
- Dong, M.P.; Enomoto, M.; Thuy, L.T.T.; Hai, H.; Hieu, V.N.; Hoang, D.V.; Iida-Ueno, A.; Odagiri, N.; Amano-Teranishi, Y.; Hagihara, A.; et al. Clinical Significance of Circulating Soluble Immune Checkpoint Proteins in Sorafenib-Treated Patients with Advanced Hepatocellular Carcinoma. Sci. Rep. 2020, 10, 3392. [Google Scholar] [CrossRef] [Green Version]
- Landeira-Viñuela, A.; Díez, P.; Juanes-Velasco, P.; Lécrevisse, Q.; Orfao, A.; De Las Rivas, J.; Fuentes, M. Deepening into Intracellular Signaling Landscape through Integrative Spatial Proteomics and Transcriptomics in a Lymphoma Model. Biomolecules 2021, 11, 1776. [Google Scholar] [CrossRef]
- Axon Instruments GenePix® Pro 6.0 Microarray Acquisition and Analysis Software for GenePix Microarray Scanners. User’s Guide & Tutorial. 2005. Available online: https://support.moleculardevices.com/s/article/GenePix-Pro-Software-Manual-Download-Page (accessed on 10 July 2022).
- Zahn-Zabal, M.; Michel, P.-A.; Gateau, A.; Nikitin, F.; Schaeffer, M.; Audot, E.; Gaudet, P.; Duek, P.D.; Teixeira, D.; Rech de Laval, V.; et al. The NeXtProt Knowledgebase in 2020: Data, Tools and Usability Improvements. Nucleic Acids Res. 2020, 48, D328–D334. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Gable, A.L.; Lyon, D.; Junge, A.; Wyder, S.; Huerta-Cepas, J.; Simonovic, M.; Doncheva, N.T.; Morris, J.H.; Bork, P.; et al. STRING V11: Protein-Protein Association Networks with Increased Coverage, Supporting Functional Discovery in Genome-Wide Experimental Datasets. Nucleic Acids Res. 2019, 47, D607–D613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillespie, M.; Jassal, B.; Stephan, R.; Milacic, M.; Rothfels, K.; Senff-Ribeiro, A.; Griss, J.; Sevilla, C.; Matthews, L.; Gong, C.; et al. The Reactome Pathway Knowledgebase 2022. Nucleic Acids Res. 2022, 50, D687–D692. [Google Scholar] [CrossRef] [PubMed]
- Uhlén, M.; Fagerberg, L.; Hallström, B.M.; Lindskog, C.; Oksvold, P.; Mardinoglu, A.; Sivertsson, Å.; Kampf, C.; Sjöstedt, E.; Asplund, A.; et al. Tissue-Based Map of the Human Proteome. Science 2015, 347, 1260419. [Google Scholar] [CrossRef] [PubMed]
- Thul, P.J.; Åkesson, L.; Wiking, M.; Mahdessian, D.; Geladaki, A.; Ait Blal, H.; Alm, T.; Asplund, A.; Björk, L.; Breckels, L.M.; et al. A Subcellular Map of the Human Proteome. Science 2017, 356, aal3321. [Google Scholar] [CrossRef] [PubMed]
- Uhlen, M.; Zhang, C.; Lee, S.; Sjöstedt, E.; Fagerberg, L.; Bidkhori, G.; Benfeitas, R.; Arif, M.; Liu, Z.; Edfors, F.; et al. A Pathology Atlas of the Human Cancer Transcriptome. Science 2017, 357, eaan2507. [Google Scholar] [CrossRef] [Green Version]
- Ishdorj, G.; Streu, E.; Lambert, P.; Dhaliwal, H.S.; Mahmud, S.M.; Gibson, S.B.; Banerji, V.; Marshall, A.J.; Johnston, J.B. IgA Levels at Diagnosis Predict for Infections, Time to Treatment, and Survival in Chronic Lymphocytic Leukemia. Blood Adv. 2019, 3, 2188–2198. [Google Scholar] [CrossRef] [Green Version]
- Hamblin, T.J. 7 Chronic Lymphocytic Leukaemia. Baillière’s Clin. Haematol. 1987, 1, 449–491. [Google Scholar] [CrossRef]
- Schroeder, H.W.; Cavacini, L. Structure and Function of Immunoglobulins. J. Allergy Clin. Immunol. 2010, 125, S41–S52. [Google Scholar] [CrossRef] [Green Version]
- Vidarsson, G.; Dekkers, G.; Rispens, T. IgG Subclasses and Allotypes: From Structure to Effector Functions. Front. Immunol. 2014, 5, 520. [Google Scholar] [CrossRef] [Green Version]
- Shvidel, L.; Tadmor, T.; Braester, A.; Bairey, O.; Rahimi-Levene, N.; Herishanu, Y.; Klepfish, A.; Ruchlemer, R.; Berrebi, A.; Polliack, A.; et al. Serum Immunoglobulin Levels at Diagnosis Have No Prognostic Significance in Stage a Chronic Lymphocytic Leukemia: A Study of 1113 Cases from the Israeli CLL Study Group. Eur. J. Haematol. 2014, 93, 29–33. [Google Scholar] [CrossRef]
- Kretschmer, A.; Schwanbeck, R.; Valerius, T.; Rösner, T. Antibody Isotypes for Tumor Immunotherapy. Transfus. Med. Hemother. 2017, 44, 320–326. [Google Scholar] [CrossRef] [Green Version]
- Jones, K.; Savulescu, A.F.; Brombacher, F.; Hadebe, S. Immunoglobulin M in Health and Diseases: How Far Have We Come and What Next? Front. Immunol. 2020, 11, 595535. [Google Scholar] [CrossRef]
- Peppas, I.; George, G.; Sollie, S.; Josephs, D.H.; Hammar, N.; Walldius, G.; Karagiannis, S.N.; Van Hemelrijck, M. Association of Serum Immunoglobulin Levels with Solid Cancer: A Systematic Review and Meta-Analysis. Cancer Epidemiol. Biomarkers Prev. 2020, 29, 527–538. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Wang, G.; Li, N.; Wang, Y.; Zhu, Q.; Chu, H.; Wu, W.; Tan, Y.; Yu, F.; Su, X.-D.; et al. Structural Insights into Immunoglobulin M. Science 2020, 367, 1014–1017. [Google Scholar] [CrossRef]
- Khan, S.R.; van der Burgh, A.C.; Peeters, R.P.; van Hagen, P.M.; Dalm, V.A.S.H.; Chaker, L. Determinants of Serum Immunoglobulin Levels: A Systematic Review and Meta-Analysis. Front. Immunol. 2021, 12, 664526. [Google Scholar] [CrossRef]
- Bournazos, S.; Wang, T.T.; Dahan, R.; Maamary, J.; Ravetch, J.V. Signaling by Antibodies: Recent Progress. Annu. Rev. Immunol. 2017, 35, 285–311. [Google Scholar] [CrossRef] [Green Version]
- Copson, E.R.; Ellis, B.A.; Westwood, N.B.; Majumdar, G. IgG Subclass Levels in Patients with B Cell Chronic Lymphocytic Leukaemia. Leuk. Lymphoma 1994, 14, 471–473. [Google Scholar] [CrossRef]
- Lacombe, C.; Gombert, J.; Dreyfus, B.; Brizard, A.; Preud’Homme, J.L. Heterogeneity of Serum IgG Subclass Deficiencies in B Chronic Lymphocytic Leukemia. Clin. Immunol. 1999, 90, 128–132. [Google Scholar] [CrossRef]
- Shome, M.; Chung, Y.; Chavan, R.; Park, J.G.; Qiu, J.; LaBaer, J. Serum Autoantibodyome Reveals That Healthy Individuals Share Common Autoantibodies. Cell Rep. 2022, 39, 110873. [Google Scholar] [CrossRef]
- Buus, S.; Rockberg, J.; Forsström, B.; Nilsson, P.; Uhlen, M.; Schafer-Nielsen, C. High-Resolution Mapping of Linear Antibody Epitopes Using Ultrahigh-Density Peptide Microarrays. Mol. Cell. Proteom. 2012, 11, 1790–1800. [Google Scholar] [CrossRef]
- Fujinami, R.S.; Oldstone, M.B.; Wroblewska, Z.; Frankel, M.E.; Koprowski, H. Molecular Mimicry in Virus Infection: Crossreaction of Measles Virus Phosphoprotein or of Herpes Simplex Virus Protein with Human Intermediate Filaments. Proc. Natl. Acad Sci. USA 1983, 80, 2346–2350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suurmond, J.; Diamond, B. Autoantibodies in Systemic Autoimmune Diseases: Specificity and Pathogenicity. J. Clin. Investig. 2015, 125, 2194–2202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.; Fu, Z.; Yan, F.; Ju, H. Biomedical and Clinical Applications of Immunoassays and Immunosensors for Tumor Markers. TrAC Trends Anal. Chem. 2007, 26, 679–688. [Google Scholar] [CrossRef]
- Sharma, S. Tumor Markers in Clinical Practice: General Principles and Guidelines. Indian J. Med. Paediatr. Oncol. 2009, 30, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dasgupta, A.; Wahed, A. Chapter 13-Tumor Markers. In Clinical Chemistry, Immunology and Laboratory Quality Control; Dasgupta, A., Wahed, A., Eds.; Elsevier: San Diego, CA, USA, 2014; pp. 229–247. ISBN 978-0-12-407821-5. [Google Scholar]
- Duffy, M.J.; Evoy, D.; McDermott, E.W. CA 15-3: Uses and Limitation as a Biomarker for Breast Cancer. Clin. Chim. Acta 2010, 411, 1869–1874. [Google Scholar] [CrossRef]
- Xu, Y.; Zhang, P.; Zhang, K.; Huang, C. The Application of CA72-4 in the Diagnosis, Prognosis, and Treatment of Gastric Cancer. Biochim. Biophys. Acta Rev. Cancer 2021, 1876, 188634. [Google Scholar] [CrossRef]
- Svanberg, R.; Janum, S.; Patten, P.E.M.; Ramsay, A.G.; Niemann, C.U. Targeting the Tumor Microenvironment in Chronic Lymphocytic Leukemia. Haematologica 2021, 106, 2312–2324. [Google Scholar] [CrossRef]
- Lauko, A.; Volovetz, J.; Turaga, S.M.; Bayik, D.; Silver, D.J.; Mitchell, K.; Mulkearns-Hubert, E.E.; Watson, D.C.; Desai, K.; Midha, M.; et al. SerpinB3 Drives Cancer Stem Cell Survival in Glioblastoma. Cell Rep. 2022, 40, 111348. [Google Scholar] [CrossRef]
- Pittayapruek, P.; Meephansan, J.; Prapapan, O.; Komine, M.; Ohtsuki, M. Role of Matrix Metalloproteinases in Photoaging and Photocarcinogenesis. Int. J. Mol. Sci. 2016, 17, 868. [Google Scholar] [CrossRef] [Green Version]
- Chang, Y.-C.; Yang, Y.-C.; Tien, C.-P.; Yang, C.-J.; Hsiao, M. Roles of Aldolase Family Genes in Human Cancers and Diseases. Trends Endocrinol. Metab. 2018, 29, 549–559. [Google Scholar] [CrossRef]
- Perini, M.V.; Dmello, R.S.; Nero, T.L.; Chand, A.L. Evaluating the Benefits of Renin-Angiotensin System Inhibitors as Cancer Treatments. Pharmacol. Ther. 2020, 211, 107527. [Google Scholar] [CrossRef]
- Kallunki, T.; Olsen, O.D.; Jäättelä, M. Cancer-Associated Lysosomal Changes: Friends or Foes? Oncogene 2013, 32, 1995–2004. [Google Scholar] [CrossRef] [Green Version]
- Rizwani, W. Enolase. In Handbook of Oxidative Stress in Cancer: Mechanistic Aspects; Chakraborti, S., Ray, B.K., Roychoudhury, S., Eds.; Springer Nature: Singapore, 2022; pp. 2473–2491. ISBN 9789811594113. [Google Scholar]
- Griggio, V.; Mandili, G.; Vitale, C.; Capello, M.; Macor, P.; Serra, S.; Castella, B.; Peola, S.; Foglietta, M.; Drandi, D.; et al. Humoral Immune Responses toward Tumor-Derived Antigens in Previously Untreated Patients with Chronic Lymphocytic Leukemia. Oncotarget 2017, 8, 3274–3288. [Google Scholar] [CrossRef] [Green Version]
- Morrison, V.A. Infectious Complications in Patients with Chronic Lymphocytic Leukemia: Pathogenesis, Spectrum of Infection, and Approaches to Prophylaxis. Clin. Lymphoma Myeloma 2009, 9, 365–370. [Google Scholar] [CrossRef]
- Nosari, A. Infectious Complications in Chronic Lymphocytic Leukemia. Mediterr. J. Hematol. Infect Dis. 2012, 4, e2012070. [Google Scholar] [CrossRef] [Green Version]
- Rivera, D.; Ferrajoli, A. Managing the Risk of Infection in Chronic Lymphocytic Leukemia in the Era of New Therapies. Curr. Oncol. Rep. 2022, 24, 1003–1014. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Clinical Information | Frequency (n) | Percentage (%) | |
|---|---|---|---|
| Gender | Female | 29 | 43.3 |
| Male | 38 | 56.7 | |
| Age | ≤65 | 24 | 35.8 |
| >65 | 43 | 64.2 | |
| Diagnosis | MBL | 10 | 14.9 |
| CLL | 57 | 85.1 | |
| CLL status | Stable/constant | 42 | 73.7 |
| Progression | 15 | 26.3 | |
| Binet stage | A | 54 | 80.6 |
| B | 6 | 9 | |
| C | 7 | 10.4 | |
| Rai stage | 0 | 45 | 67.2 |
| I | 5 | 7.5 | |
| II | 10 | 14.9 | |
| III | 1 | 1.5 | |
| IV | 6 | 9 | |
| Treatment status | Previous to first-line | 9 | 60 |
| Time from first-line | 5 | 33.3 | |
| IGHV gene status | Mutated | 45 | 67.2 |
| Unmutated | 22 | 32.8 | |
| Cytogenetic | Normality | 24 | 35.8 |
| Abnormality | 43 | 64.2 | |
| Total | 67 | 100 | |
| mAgs | |||||||
|---|---|---|---|---|---|---|---|
| Concentration (µg/µL) | S < Ref. | Ref. > S | |||||
| n# of mAgs | IgM | IgG | IgM and IgG | IgM | IgG | IgM and IgG | |
| 0.08 and 0.06 | 11 | 11 | 7 | 8 | 3 | 0 | 3 |
| 0.01 | 3 | 26 | 1 | 7 | 7 | 0 | 3 |
| 0.001 | 8 | 16 | 1 | 12 | 4 | 0 | 5 |
| 0.0001 | 12 | 10 | 3 | 12 | 1 | 0 | 5 |
| 0.00001 | 5 | 12 | 1 | 19 | 5 | 0 | 5 |
| mAgs (n#) | Top 5 mAgs (n#) | ||||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Landeira-Viñuela, A.; Alcoceba-Sanchez, M.; Navarro-Bailón, A.; Arias-Hidalgo, C.; Juanes-Velasco, P.; Sánchez-Santos, J.M.; Lecrevisse, Q.; Pedreira, C.E.; García-Vaquero, M.L.; Hernández, Á.-P.; et al. Systematic Evaluation of Antigenic Stimulation in Chronic Lymphocytic Leukemia: Humoral Immunity as Biomarkers for Disease Evolution. Cancers 2023, 15, 891. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers15030891
Landeira-Viñuela A, Alcoceba-Sanchez M, Navarro-Bailón A, Arias-Hidalgo C, Juanes-Velasco P, Sánchez-Santos JM, Lecrevisse Q, Pedreira CE, García-Vaquero ML, Hernández Á-P, et al. Systematic Evaluation of Antigenic Stimulation in Chronic Lymphocytic Leukemia: Humoral Immunity as Biomarkers for Disease Evolution. Cancers. 2023; 15(3):891. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers15030891
Chicago/Turabian StyleLandeira-Viñuela, Alicia, Miguel Alcoceba-Sanchez, Almudena Navarro-Bailón, Carlota Arias-Hidalgo, Pablo Juanes-Velasco, José Manuel Sánchez-Santos, Quentin Lecrevisse, Carlos Eduardo Pedreira, Marina L. García-Vaquero, Ángela-Patricia Hernández, and et al. 2023. "Systematic Evaluation of Antigenic Stimulation in Chronic Lymphocytic Leukemia: Humoral Immunity as Biomarkers for Disease Evolution" Cancers 15, no. 3: 891. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers15030891