
A Novel Family of Lysosomotropic Tetracyclic Compounds for Treating Leukemia
 , , , , ,
, , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Primary Samples
2.2. Chemoinformatic Screening
2.3. Cytotoxicity Assays
2.4. In Vivo Studies
2.5. Lysosomal Studies
2.6. Autophagy Analysis
2.7. TFEB Activation
2.8. Mitochondrial Analysis
2.9. ADMET and PK Profiles
2.10. Synergy Calculation
3. Results
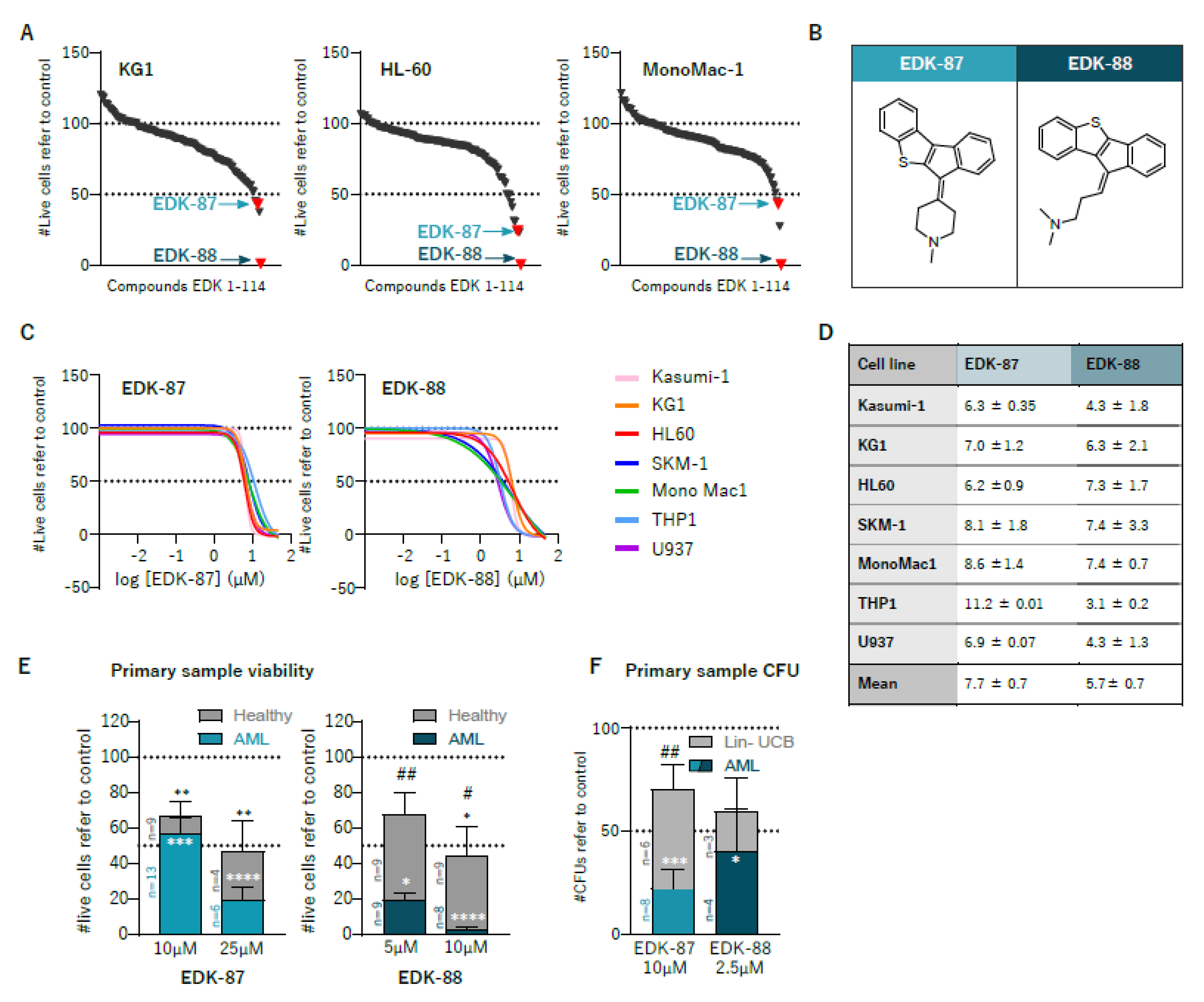
3.1. Identification of Novel Antileukemic Molecules
3.2. In Vivo Effectivity of EDK-87/EDK-88
3.3. Dual Targeting of Lysosomes and Mitochondria
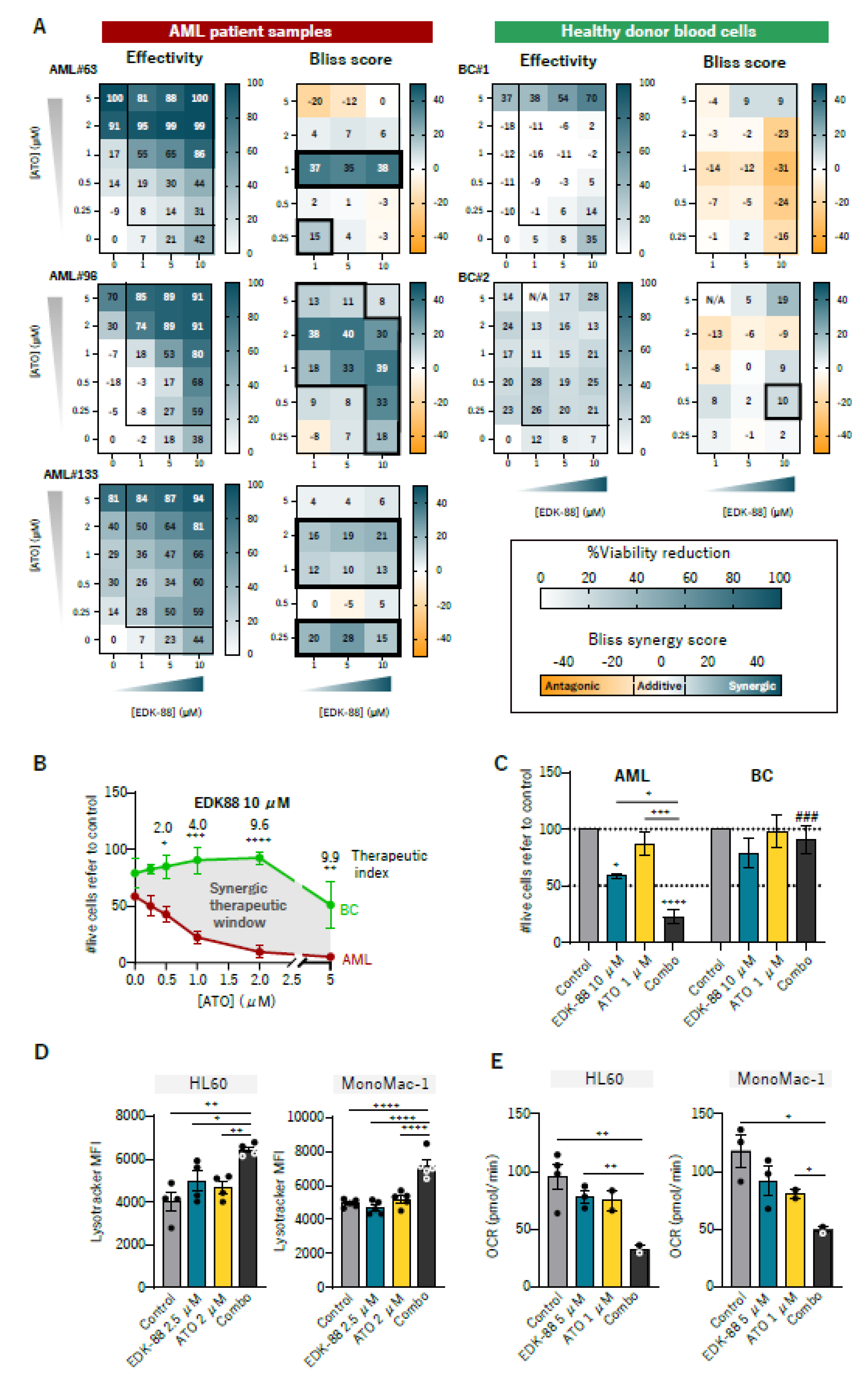
3.4. Synergism with Arsenic Trioxide
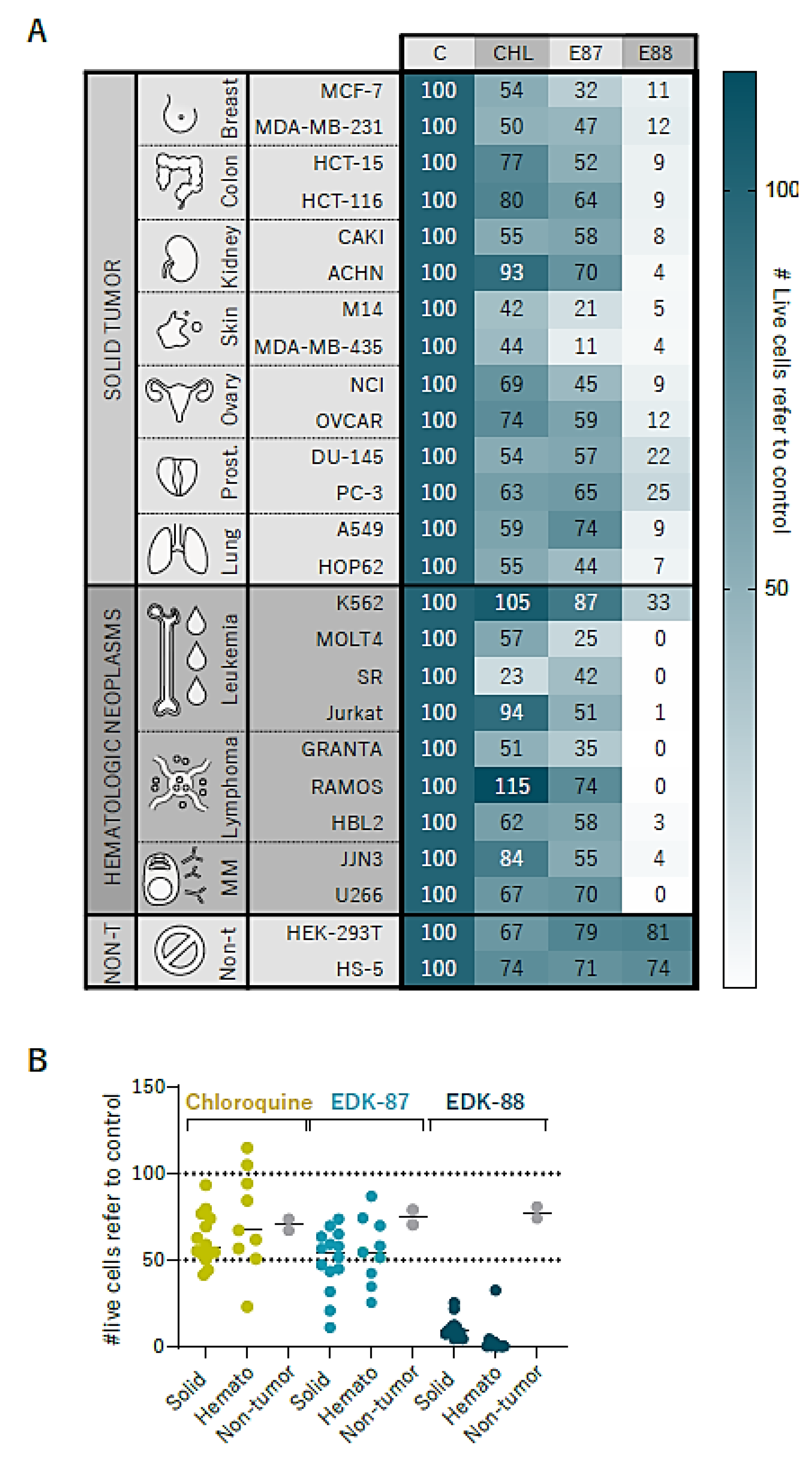
3.5. Pan-Cancer Effectivity of EDK-87/EDK-88
3.6. Pharmacokinetic and ADMET Profiles
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknoledgements
Conflicts of Interest
References
- Estey, E.; Karp, J.E.; Emadi, A.; Othus, M.; Gale, R.P. Recent Drug Approvals for Newly Diagnosed Acute Myeloid Leukemia: Gifts or a Trojan Horse? Leukemia 2020, 34, 671–681. [Google Scholar] [CrossRef]
- Kopmar, N.E.; Estey, E.H. New Drug Approvals in Acute Myeloid Leukemia: An Unprecedented Paradigm Shift. Clin. Adv. Hematol. Oncol. 2019, 17, 569–575. [Google Scholar] [PubMed]
- Thol, F. What to Use to Treat AML: The Role of Emerging Therapies. Hematology 2021, 2021, 16–23. [Google Scholar] [CrossRef]
- Fiorentini, A.; Capelli, D.; Saraceni, F.; Menotti, D.; Poloni, A.; Olivieri, A. The Time Has Come for Targeted Therapies for AML: Lights and Shadows. Oncol. Ther. 2020, 8, 13–32. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paunovic, V.; Kosic, M.; Misirkic-Marjanovic, M.; Trajkovic, V.; Harhaji-Trajkovic, L. Dual Targeting of Tumor Cell Energy Metabolism and Lysosomes as an Anticancer Strategy. Biochim. Biophys. Acta Mol. Cell. Res. 2021, 1868, 118944. [Google Scholar] [CrossRef]
- Sukhai, M.A.; Prabha, S.; Hurren, R.; Rutledge, A.C.; Lee, A.Y.; Sriskanthadevan, S.; Sun, H.; Wang, X.; Skrtic, M.; Seneviratne, A.; et al. Lysosomal Disruption Preferentially Targets Acute Myeloid Leukemia Cells and Progenitors. J. Clin. Investig. 2013, 123, 315–328. [Google Scholar] [CrossRef]
- Kitatani, K.; Taniguchi, M.; Okazaki, T. Role of Sphingolipids and Metabolizing Enzymes in Hematological Malignancies. Mol. Cells 2015, 38, 482–495. [Google Scholar] [CrossRef] [Green Version]
- Aasebø, E.; Bartaula-Brevik, S.; Hernandez-Valladares, M.; Bruserud, Ø. Vacuolar ATPase as a Possible Therapeutic Target in Human Acute Myeloid Leukemia. Expert. Rev. Hematol. 2018, 11, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Farge, T.; Saland, E.; de Toni, F.; Aroua, N.; Hosseini, M.; Perry, R.; Bosc, C.; Sugita, M.; Stuani, L.; Fraisse, M.; et al. Chemotherapy-Resistant Human Acute Myeloid Leukemia Cells Are Not Enriched for Leukemic Stem Cells but Require Oxidative Metabolism. Cancer Discov. 2017, 7, 716–735. [Google Scholar] [CrossRef] [Green Version]
- Goto, M.; Miwa, H.; Shikami, M.; Tsunekawa-Imai, N.; Suganuma, K.; Mizuno, S.; Takahashi, M.; Mizutani, M.; Hanamura, I.; Nitta, M. Importance of Glutamine Metabolism in Leukemia Cells by Energy Production through TCA Cycle and by Redox Homeostasis. Cancer Invest. 2014, 32, 241–247. [Google Scholar] [CrossRef]
- Lee, E.A.; Angka, L.; Rota, S.G.; Hanlon, T.; Mitchell, A.; Hurren, R.; Wang, X.M.; Gronda, M.; Boyaci, E.; Bojko, B.; et al. Targeting Mitochondria with Avocatin B Induces Selective Leukemia Cell Death. Cancer Res. 2015, 75, 2478–2488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Škrtić, M.; Sriskanthadevan, S.; Jhas, B.; Gebbia, M.; Wang, X.; Wang, Z.; Hurren, R.; Jitkova, Y.; Gronda, M.; Maclean, N.; et al. Inhibition of Mitochondrial Translation as a Therapeutic Strategy for Human Acute Myeloid Leukemia. Cancer Cell. 2011, 20, 674–688. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mondet, J.; Lo Presti, C.; Chevalier, S.; Bertrand, A.; Tondeur, S.; Blanchet, S.; Mc Leer, A.; Pernet-Gallay, K.; Mossuz, P. Mitochondria in Human Acute Myeloid Leukemia Cell Lines Have Ultrastructural Alterations Linked to Deregulation of Their Respiratory Profiles. Exp. Hematol. 2021, 98, 53–62.e3. [Google Scholar] [CrossRef] [PubMed]
- Cornet-Masana, J.M.; Banús-Mulet, A.; Carbó, J.M.; Torrente, M.Á.; Guijarro, F.; Cuesta-Casanovas, L.; Esteve, J.; Risueño, R.M. Dual Lysosomal-Mitochondrial Targeting by Antihistamines to Eradicate Leukaemic Cells. EBioMedicine 2019, 47, 221–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirali, S.; Botham, A.; Voisin, V.; Xu, C.; St-Germain, J.; Sharon, D.; Hoff, F.W.; Qiu, Y.; Hurren, R.; Gronda, M.; et al. The Mitochondrial Peptidase, Neurolysin, Regulates Respiratory Chain Supercomplex Formation and Is Necessary for AML Viability. Sci. Transl. Med. 2020, 12, 1–17. [Google Scholar] [CrossRef]
- Molina, J.R.; Sun, Y.; Protopopova, M.; Gera, S.; Bandi, M.; Bristow, C.; McAfoos, T.; Morlacchi, P.; Ackroyd, J.; Agip, A.-N.N.A.; et al. An Inhibitor of Oxidative Phosphorylation Exploits Cancer Vulnerability. Nat. Med. 2018, 24, 1036–1046. [Google Scholar] [CrossRef] [Green Version]
- Nelson, M.A.; McLaughlin, K.L.; Hagen, J.T.; Coalson, H.S.; Schmidt, C.; Kassai, M.; Kew, K.A.; McClung, J.M.; Neufer, P.D.; Brophy, P.; et al. Intrinsic OXPHOS Limitations Underlie Cellular Bioenergetics in Leukemia. eLife 2021, 10, 1–31. [Google Scholar] [CrossRef]
- Ishii, S.; Matsuura, A.; Itakura, E. Identification of a Factor Controlling Lysosomal Homeostasis Using a Novel Lysosomal Trafficking Probe. Sci. Rep. 2019, 9, 11635. [Google Scholar] [CrossRef] [Green Version]
- Long, J.; Jia, M.-Y.; Fang, W.-Y.; Chen, X.-J.; Mu, L.-L.; Wang, Z.-Y.; Shen, Y.; Xiang, R.-F.; Wang, L.-N.; Wang, L.; et al. FLT3 Inhibition Upregulates HDAC8 via FOXO to Inactivate P53 and Promote Maintenance of FLT3-ITD+ Acute Myeloid Leukemia. Blood 2020, 135, 1472–1483. [Google Scholar] [CrossRef]
- Cuesta-Casanovas, L.; Delgado-Martínez, J.; Cornet-Masana, J.M.; Carbó, J.M.; Clément-Demange, L.; Risueño, R.M. Lysosome-Mediated Chemoresistance in Acute Myeloid Leukemia. Cancer Drug. Resist. 2022, 5, 233–244. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Abdel-Aziz, A.K.; Abdelfatah, S.; Abdellatif, M.; Abdoli, A.; Abel, S.; Abeliovich, H.; Abildgaard, M.H.; Abudu, Y.P.; Acevedo-Arozena, A.; et al. Full Article: Guidelines for the Use and Interpretation of Assays for Monitoring Autophagy (4th Edition)1. Autophagy 2021, 17, 1–382. [Google Scholar] [CrossRef]
- Aits, S. Methods to Detect Loss of Lysosomal Membrane Integrity. In Methods in Molecular Biology; Humana Press: New York, NY, USA, 2019; Volume 1880, pp. 315–329. [Google Scholar]
- Anand, A.; Liu, B.; Giacobini, J.D.; Maeda, K.; Rohde, M.; Jaattela, M. Cell Death Induced by Cationic Amphiphilic Drugs Depends on Lysosomal Ca2+ Release and Cyclic AMP. Mol. Cancer Ther. 2019, 18, 1602–1614. [Google Scholar] [CrossRef] [Green Version]
- Zhitomirsky, B.; Yunaev, A.; Kreiserman, R.; Kaplan, A.; Stark, M.; Assaraf, Y.G. Lysosomotropic Drugs Activate TFEB via Lysosomal Membrane Fluidization and Consequent Inhibition of MTORC1 Activity. Cell. Death Dis. 2018, 9, 1191. [Google Scholar] [CrossRef] [Green Version]
- Medina, D.L.; Di Paola, S.; Peluso, I.; Armani, A.; De Stefani, D.; Venditti, R.; Montefusco, S.; Scotto-Rosato, A.; Prezioso, C.; Forrester, A.; et al. Lysosomal Calcium Signalling Regulates Autophagy through Calcineurin and TFEB. Nat. Cell. Biol. 2015, 17, 288–299. [Google Scholar] [CrossRef] [Green Version]
- Yun, S.; Vincelette, N.D.; Yu, X.; Watson, G.W.; Fernandez, M.R.; Yang, C.; Hitosugi, T.; Cheng, C.-H.H.; Freischel, A.R.; Zhang, L.; et al. TFEB Links MYC Signaling to Epigenetic Control of Myeloid Differentiation and Acute Myeloid Leukemia. Cancer Discov. 2021, 2, 162–185. [Google Scholar] [CrossRef]
- Sachs, L. Control of Normal Cell Differentiation and the Phenotypic Reversion of Malignancy in Myeloid Leukaemia. Nature 1978, 274, 535–539. [Google Scholar] [CrossRef] [PubMed]
- Madan, V.; Koeffler, H.P. Differentiation Therapy of Myeloid Leukemia: Four Decades of Development. Haematologica 2020, 106, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Sriskanthadevan, S.; Jeyaraju, D.V.; Chung, T.E.; Prabha, S.; Xu, W.; Skrtic, M.; Jhas, B.; Hurren, R.; Gronda, M.; Wang, X.; et al. AML Cells Have Low Spare Reserve Capacity in Their Respiratory Chain That Renders Them Susceptible to Oxidative Metabolic Stress. Blood 2015, 125, 2120–2130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Petrova-Benedict, R.; Buncic, J.R.; Wallace, D.C.; Robinson, B.H. Selective Killing of Cells with Oxidative Defects in Galactose Medium: A Screening Test for Affected Patient Fibroblasts. J. Inherit. Metab. Dis. 1992, 15, 943–944. [Google Scholar] [CrossRef]
- Kashif, M.; Andersson, C.; Åberg, M.; Nygren, P.; Sjöblom, T.; Hammerling, U.; Larsson, R.; Gustafsson, M.G. A Pragmatic Definition of Therapeutic Synergy Suitable for Clinically Relevant in Vitro Multicompound Analyses. Mol. Cancer Ther. 2014, 13, 1964–1976. [Google Scholar] [CrossRef] [Green Version]
- Kannan, S.; Sutphin, R.M.; Hall, M.G.; Golfman, L.S.; Fang, W.; Nolo, R.M.; Akers, L.J.; Hammitt, R.A.; McMurray, J.S.; Kornblau, S.M.; et al. Notch Activation Inhibits AML Growth and Survival: A Potential Therapeutic Approach. J. Exp. Med. 2013, 210, 321–337. [Google Scholar] [CrossRef]
- Yang, Y.P.; Liang, Z.Q.; Gao, B.; Jia, Y.L.; Qin, Z.H. Dynamic Effects of Autophagy on Arsenic Trioxide-Induced Death of Human Leukemia Cell Line HL60 Cells. Acta Pharm. Sin. 2008, 29, 123–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, X.B.; Zhang, Y.; Yang, D.Y.; Yang, Y.Z.; Wu, F.C.; Ning, Y.P.; Wu, K. Analysis of First-Episode and Chronic Schizophrenia Using Multi-Modal Magnetic Resonance Imaging. Eur. Rev. Med. Pharm. Sci. 2018, 22, 6422–6435. [Google Scholar] [CrossRef]
- Ashton, T.M.; Gillies McKenna, W.; Kunz-Schughart, L.A.; Higgins, G.S. Oxidative Phosphorylation as an Emerging Target in Cancer Therapy. Clin. Cancer Res. 2018, 24, 2482–2490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohsen, S.; Sobash, P.T.; Algwaiz, G.F.; Nasef, N.; Al-Zeidaneen, S.A.; Karim, N.A. Autophagy Agents in Clinical Trials for Cancer Therapy: A Brief Review. Curr. Oncol. 2022, 29, 1695–1708. [Google Scholar] [CrossRef]
- Joshi, S.K.; Nechiporuk, T.; Bottomly, D.; Piehowski, P.D.; Reisz, J.A.; Pittsenbarger, J.; Kaempf, A.; Gosline, S.J.C.; Wang, Y.-T.T.; Hansen, J.R.; et al. The AML Microenvironment Catalyzes a Stepwise Evolution to Gilteritinib Resistance. Cancer Cell. 2021, 39, 999–1014.e8. [Google Scholar] [CrossRef] [PubMed]
- Cremer, A.; Ellegast, J.M.; Alexe, G.; Frank, E.S.; Ross, L.; Chu, S.H.; Pikman, Y.; Robichaud, A.; Goodale, A.; Häupl, B.; et al. Resistance Mechanisms to SYK Inhibition in Acute Myeloid Leukemia. Cancer Discov. 2020, 10, 214–231. [Google Scholar] [CrossRef]
- Pei, S.; Pollyea, D.A.; Gustafson, A.; Stevens, B.M.; Minhajuddin, M.; Fu, R.; Riemondy, K.A.; Gillen, A.E.; Sheridan, R.M.; Kim, J.; et al. Monocytic Subclones Confer Resistance to Venetoclax-Based Therapy in Patients with Acute Myeloid Leukemia. Cancer Discov. 2020, 10, 536–551. [Google Scholar] [CrossRef] [PubMed]
- Short, N.J.; Konopleva, M.; Kadia, T.M.; Borthakur, G.; Ravandi, F.; DiNardo, C.D.; Daver, N. Advances in the Treatment of Acute Myeloid Leukemia: New Drugs and New Challenges. Cancer Discov. 2020, 10, 506–525. [Google Scholar] [CrossRef] [Green Version]
- Eppert, K.; Takenaka, K.; Lechman, E.R.; Waldron, L.; Nilsson, B.; van Galen, P.; Metzeler, K.H.; Poeppl, A.; Ling, V.; Beyene, J.; et al. Stem Cell Gene Expression Programs Influence Clinical Outcome in Human Leukemia. Nat. Med. 2011, 17, 1086–1093. [Google Scholar] [CrossRef]
- Short, N.J.; Benton, C.B.; Chen, H.H.C.; Qiu, P.; Gu, L.; Pierce, S.; Brandt, M.; Maiti, A.; Min, T.L.; Naqvi, K.; et al. Sphingolipid Metabolism Determines the Therapeutic Efficacy of Nanoliposomal Ceramide in Acute Myeloid Leukemia. Autophagy 2016, 3, 154–157. [Google Scholar] [CrossRef]
- Koenig, K.; Mims, A. Relapsed or Primary Refractory AML: Moving Past MEC and FLAG-Ida. Curr. Opin. Hematol. 2020, 27, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Appelqvist, H.; Wäster, P.; Kågedal, K.; Öllinger, K. The Lysosome: From Waste Bag to Potential Therapeutic Target. J. Mol. Cell. Biol. 2013, 5, 214–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffman, B.; Amanullah, A.; Shafarenko, M.; Liebermann, D.A. The Proto-Oncogene c-Myc in Hematopoietic Development and Leukemogenesis. Oncogene 2002, 21, 3414–3421. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amaya, M.L.; Inguva, A.; Pei, S.; Jones, C.; Krug, A.; Ye, H.; Minhajuddin, M.; Winters, A.; Furtek, S.L.; Gamboni, F.; et al. The STAT3-MYC Axis Promotes Survival of Leukemia Stem Cells by Regulating SLC1A5 and Oxidative Phosphorylation. Blood 2022, 139, 584–596. [Google Scholar] [CrossRef]
- Taglialatela, M.; Pannaccione, A.; Castaldo, P.; Giorgio, G.; Zhou, Z.; January, C.T.; Genovese, A.; Marone, G.; Annunziato, L. Molecular Basis for the Lack of HERG K+ Channel Block-Related Cardiotoxicity by the H1 Receptor Blocker Cetirizine Compared with Other Second-Generation Antihistamines. Mol. Pharm. 1998, 54, 113–121. [Google Scholar] [CrossRef] [Green Version]
- Gupta, D.; Bhatia, D.; Dave, V.; Sutariya, V.; Gupta, S.V. Salts of Therapeutic Agents: Chemical, Physicochemical, and Biological Considerations. Molecules 2018, 23, 1719. [Google Scholar] [CrossRef] [Green Version]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Carbó, J.M.; Cornet-Masana, J.M.; Cuesta-Casanovas, L.; Delgado-Martínez, J.; Banús-Mulet, A.; Clément-Demange, L.; Serra, C.; Catena, J.; Llebaria, A.; Esteve, J.; et al. A Novel Family of Lysosomotropic Tetracyclic Compounds for Treating Leukemia. Cancers 2023, 15, 1912. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers15061912
Carbó JM, Cornet-Masana JM, Cuesta-Casanovas L, Delgado-Martínez J, Banús-Mulet A, Clément-Demange L, Serra C, Catena J, Llebaria A, Esteve J, et al. A Novel Family of Lysosomotropic Tetracyclic Compounds for Treating Leukemia. Cancers. 2023; 15(6):1912. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers15061912
Chicago/Turabian StyleCarbó, José M., Josep M. Cornet-Masana, Laia Cuesta-Casanovas, Jennifer Delgado-Martínez, Antònia Banús-Mulet, Lise Clément-Demange, Carme Serra, Juanlo Catena, Amadeu Llebaria, Jordi Esteve, and et al. 2023. "A Novel Family of Lysosomotropic Tetracyclic Compounds for Treating Leukemia" Cancers 15, no. 6: 1912. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers15061912