
NAFLD-Related HCC: Focus on the Latest Relevant Preclinical Models

1
Centre de Recherche des Cordeliers, INSERM, Sorbonne Université, Université de Paris, 75006 Paris, France
2
Genomic Instability, Metabolism, Immunity and Liver Tumorigenesis Laboratory, Equipe Labellisée Ligue Contre le Cancer, 75005 Paris, France
*
Authors to whom correspondence should be addressed.
†
These authors contributed equally to this work.
Cancers 2023, 15(14), 3723; https://0-doi-org.brum.beds.ac.uk/10.3390/cancers15143723
Submission received: 16 June 2023
/
Revised: 17 July 2023
/
Accepted: 18 July 2023
/
Published: 22 July 2023
(This article belongs to the Collection Primary Liver Cancer)
Abstract
:Simple Summary
Non-alcoholic fatty liver disease (NAFLD) is a spectrum of disease ranging from simple steatosis (NAFL) to non-alcoholic steatohepatitis (NASH), which can progress to fibrosis and cirrhosis and ultimately lead to hepatocarcinoma (HCC). Due to its increasing prevalence, NAFLD has become a major public health problem. Given the partial understanding of the complex pathological mechanisms of NAFLD-induced human HCC and the lack of effective treatment, relevant pre-clinical models are still urgently needed to better recapitulate and investigate the process and mechanism of human NAFLD/HCC. This review discusses a selection of the most relevant mouse models in the study of NAFLD/HCC, with their specific advantages and disadvantages, and also the emergence of new ex vivo technologies, which will greatly accelerate the transition from basic science to clinical discoveries.
Abstract
Hepatocellular carcinoma (HCC) is the most common type of primary liver cancer and one of the deadliest cancers worldwide. Despite extensive research, the biological mechanisms underlying HCC’s development and progression remain only partially understood. Chronic overeating and/or sedentary-lifestyle-associated obesity, which promote Non-Alcoholic Fatty Liver Disease (NAFLD), have recently emerged as worrying risk factors for HCC. NAFLD is characterized by excessive hepatocellular lipid accumulation (steatosis) and affects one quarter of the world’s population. Steatosis progresses in the more severe inflammatory form, Non-Alcoholic Steatohepatitis (NASH), potentially leading to HCC. The incidence of NASH is expected to increase by up to 56% over the next 10 years. Better diagnoses and the establishment of effective treatments for NAFLD and HCC will require improvements in our understanding of the fundamental mechanisms of the disease’s development. This review describes the pathogenesis of NAFLD and the mechanisms underlying the transition from NAFL/NASH to HCC. We also discuss a selection of appropriate preclinical models of NAFLD for research, from cellular models such as liver-on-a-chip models to in vivo models, focusing particularly on mouse models of dietary NAFLD-HCC.
1. Introduction
Non-alcoholic fatty liver disease (NAFLD) is becoming the most common cause of chronic liver disease worldwide and is thus a public health problem of great concern [1,2,3,4,5,6]. NAFLD is associated with several metabolic disorders, such as obesity, type 2 diabetes mellitus, hypertension, and dyslipidemia. The prevalence of NAFLD is currently estimated at 25–35% in the general population, up to 50% in type 2 diabetes patients, and up to 90% in morbidly obese patients [6,7,8]. This disease encompasses a spectrum of liver conditions, ranging from simple hepatic steatosis or non-alcoholic fatty liver (NAFL) to the concomitant presence of hepatocellular damage (ballooning), Mallory-Denk body formation, and lobular necro-inflammation, defining non-alcoholic steatohepatitis (NASH), which can lead to various degrees of fibrosis [9,10,11,12]. NAFL is associated with a low risk of adverse outcomes, but the chronic liver injury occurring in NASH markedly increases the risk of end-stage liver diseases, such as cirrhosis and hepatocellular carcinoma [1,13,14]. The incidence of NAFLD-related HCC is highly variable [15]. The estimated annual incidence of HCC ranges from 0.5% to 2.6% among patients with NASH cirrhosis. HCC can develop in NAFLD patients without cirrhosis [1], and it has been estimated that almost half of the patients with NAFLD-related HCC have no marked liver fibrosis [16,17,18]. This clinical observation poses a challenge for HCC surveillance. The epidemiological features of HCC are changing, with the increase in vaccination coverage for HBV and the effective antiviral therapy for HCV infection decreasing the burden of virus-associated liver cancer worldwide [13]. Given the global increase in obesity and type 2 diabetes, it has been predicted that NAFLD will become the most common underlying etiological risk factor for HCC [13,19]. Unsurprisingly, NASH is already one of the major indications for liver transplantation worldwide [20,21,22]. There is currently no effective treatment for NAFLD, and most management approaches are based on preventive measures involving regular physical activity, low-calorie eating, and weight loss [23]. NAFLD is a complex disease, the development and progression of which require multi-hit combinations of various risk factors, including age, sex, ethnicity, dietary habits, hormonal status, genetic predisposition (e.g., polymorphisms of PNPLA3, TM6SF2, MBOAT1, or HSD17B13), epigenetic factors, and associated comorbid conditions (e.g., obesity, type 2 diabetes mellitus, obstructive sleep apnea, and gut dysbiosis) [4,5,24,25,26]. The individual impacts of these factors probably change over time, determining the phenotype and natural course of the disease.
Triglyceride accumulation (e.g., steatosis) is believed to be relatively benign, whereas hepatocyte lipotoxicity results chiefly from the accumulation of free fatty acids, free cholesterol, and other lipid metabolites [5,27,28]. These changes within the liver place extra metabolic stress on various organelles, such as the mitochondria and endoplasmic reticulum, triggering a cascade of stress-induced responses, including the generation of reactive oxygen species (ROS) [29,30,31]. This leads to further cell injury and programmed cell death via apoptosis, necrosis, or necroptosis, with the release of damage-associated molecular patterns (DAMPs) [32]. Cellular senescence has also been identified as a major actor in NAFLD progression through changes in mitochondrial β-oxidation and the release of inflammatory signals, the so-called “senescence-associated secretory phenotype”, in particular [33,34]. Local inflammation mediated by Kupffer cells and the influx of platelets, neutrophils, and inflammatory monocytes have been shown to play major roles in the inflammatory mechanism [35,36,37,38]. Over time, various adaptive immune cells (e.g., CD8+PD−1+ T cells) and innate immune cells (e.g., CXCR1+ cDCs) infiltrate the liver, supporting the development of autoaggressive CD8+ T cells [35,39].
The development of experimental models accurately reproducing the mechanisms underlying human NAFLD remains highly challenging. Nevertheless, diverse in vitro and in vivo models of NAFLD have been developed and have significantly advanced our understanding of NAFLD’s pathophysiology, making it possible to test new therapeutic targets [40,41,42,43]. In this review, we focus on the most widely used preclinical mouse models of NAFLD and in vitro human cell models of NAFLD crucial for drug development.
2. Mouse Models of Non-Alcoholic Fatty Liver Disease
NAFLD is considered to be a multiple-hit multisystem disease. This complexity makes it difficult to develop comprehensive models that fully reproduce the mechanisms of human NAFLD. Over the last two decades, investigators have addressed key questions about NAFLD with various rodent models, mostly in mice (Figure 1).
2.1. Murine Models of NAFL
2.1.1. High-Fat Diet (HFD)
One of the most widely used methods for obtaining overweight animals and inducing steatosis is the administration of a high-fat diet (HFD), generally with fats accounting for 45 to 60% of the total calorie content [44,45]. The fats included in this diet may include saturated, monosaturated, or polysaturated fatty acids, separately or in various combinations. HFD-induced metabolic syndrome in rodents can be modified by many factors, including the duration of the dietary exposure and the species, strain, age, and sex of the animals [46]. Mice fed a HFD for at least 10 to 14 weeks generally develop hyperlipidemia, insulin resistance, and glucose intolerance, the key features of metabolic syndrome and obesity in humans [44,47,48]. Their hepatocytes display fat accumulation, ballooning, and Mallory-Denk bodies. After nine months on a HFD, mice may display a significant increase in their circulating levels of liver enzymes, such as alanine aminotransferase (ALT) and aspartate aminotransferase (AST) [44,49]. However, they present only minor signs of inflammation and fibrosis [49,50]. Velazquez and coworkers investigated the effect of prolonged feeding on a HFD (80 weeks). They reported a significant increase in steatosis, inflammation, cell injury, fibrosis, and ER stress, mimicking the effects on the microbiota observed in NAFLD patients [51]. Mice fed a HFD are generally considered to be a model of non-alcoholic fatty liver (NAFL).
2.1.2. Lepob/Lepob (ob/ob) Mice
Leptin, a peptide hormone secreted principally by white adipose tissue, acts via the hypothalamus to decrease food intake and increase energy expenditure [52,53]. Lepob/Lepob (ob/ob) mice carry an autosomal recessive mutation of the leptin gene. Homozygosity for this mutation results in a redistribution of fat from the adipose tissue to the liver and other non-adipose tissues. Ob/ob mice are hyperphagic, inactive, sluggish, and gradually become severely obese, hyperlipidemic, hyperglycemic, hyperinsulinemic, and insulin-resistant with age (3–4 weeks old) [53]. In ob/ob mice, lipotoxicity and lipoapoptosis are induced in the hepatocytes within the liver parenchyma. However, these obese mice rarely develop severe liver damage or fibrosis, and do not, therefore, develop spontaneous steatohepatitis [54,55]. Nevertheless, NASH without progressive fibrosis can be induced by a second hit, such as a non-chow diet (e.g., MCD diet) or exposure to small doses of lipopolysaccharide endotoxin or ethanol [56,57,58].
2.1.3. Leptin-Receptor-Deficient (db/db) Mice
Db/db mice, or leptin-receptor-deficient mice, are homozygous for an autosomal recessive point mutation of a diabetes gene (db), resulting in an absence of the long isoform of the leptin receptor [49,59,60]. Leptin levels are normal or high in db/db mice, but their leptin signaling is defective [49,61]. The phenotype of db/db mice, which have an abnormally strong appetite, obesity, hyperglycemia, hyperinsulinemia, insulin resistance, and macrovesicular hepatic steatosis, is very similar to that of ob/ob mice. However, microvesicular steatosis, a feature of impaired mitochondrial function [62], is more frequently observed in db⁄db mice than in ob/ob mice [61]. The development of NASH features also requires additional stimulation in Db/db mice [61,63]. Interestingly, after three months on a high-calorie diet, db/db mice present steatohepatitis (NAS ≥ 5), whereas ob/ob mice do not develop NASH in the same conditions [61]. Iron overload in db/db mice has been shown to induce key features of NASH, such as an increase in hepatic oxidative stress and fibrogenesis [64].
2.2. Murine Models of NASH
2.2.1. Methionine- and Choline-Deficient Diet (MCD)
The MCD model is one of the most widely used dietary models in NASH research. The MCD contains a high proportion of sucrose (40%), moderate amounts of fat (10%), and is deficient in methionine and choline [65,66]. Choline is the precursor of phosphatidylcholine, which is essential for very-low-density lipoprotein (VLDL) production. Methionine plays a major role in the synthesis of glutathione, a crucial antioxidant protein. The MCD induces NAFLD through high levels of fatty acid intake and low levels of VLDL secretion in the context of strong oxidative stress. After four to six weeks on the MCD, the liver parenchyma presents steatosis, hepatocyte ballooning, cell death, inflammation, oxidative/ER stress, and fibrosis [43,66], without the development of insulin resistance or related comorbid conditions [67,68]. One of the drawbacks of using the MCD to induce NASH development is the severe weight loss and liver atrophy caused by this diet, which are not characteristics of human NASH [69,70]. As with the HFD, the sensitivity to the MCD differs between mouse strains, with A/J mice displaying significantly higher serum alanine aminotransferase (ALT) levels and a greater weight loss than other strains [71]. The MCD model mimics the histological phenotype of human NASH relatively quickly, but the use of this model is limited by the known metabolic profile disparities of this model relative to human NASH [43].
2.2.2. Choline-Deficient L-Amino Acid-Defined (CDAA) Diet
The CDAA diet contains 30% fat in terms of calorie content (mostly Primex fat as a source of trans fat) and 0.17% methionine to compensate for the choline deficiency [72,73]. Histological analyses of liver samples from C57BL/6J mice fed with the CDAA diet revealed steatosis (score 3) and focal lobular inflammation (score 1) after six weeks on the diet, followed by mild features of NASH after 12 weeks on the diet. Chronic feeding on this diet for at least 20 weeks is required for the development of fibrosis [74]. As in the MCD model, mice on the CDAA diet do not gain weight and do not develop insulin resistance [75,76]. After long-term feeding on this diet (65–84 weeks), male mice develop hepatocellular preneoplastic foci, adenomas (incidence: 65%), and carcinomas (incidence: 25%) associated with fibrosis and oxidative DNA damage [75]. It remains unclear whether the progression of NASH with fibrosis alone is sufficient to influence HCC development, or whether aging is also involved. These findings suggest that this model, like the MCD model, displays marked differences from human NAFLD.
2.2.3. High-Fructose Diet
There is now clear evidence that high-fructose diets are a major risk for the development of obesity and NAFLD [77,78]. A number of studies on rodent models have evaluated the influence of fructose on NAFLD development through the addition of fructose to their drinking water (55% fructose) or directly to their pelleted diet (60% fructose) [79,80,81]. C57BL/6 mice fed with a high-fat diet, without (HFD) or with high (HFHF) fructose in their drinking water, presented similar increases in body weight, body fat mass, and fasting glucose after 16 weeks, and both groups of mice displayed insulin resistance relative to mice fed with standard chow [80]. However, the HFHF mice displayed an increase in hepatic ROS production and a NASH-like phenotype, with significant fibrosis not observed in the HFD mice. The underlying mechanisms for this fibrosis seemed to involve the induction, by fructose, of an increase in ROS production, associated with CD11b+F4/80+Gr1+ hepatic macrophage aggregation, resulting in transforming growth factor beta-1 signaling-mediated collagen deposition [80]. Nigro and coworkers showed that fructose consumption also induces an amplifying loop involving lipogenesis, palmitate, Nrf2, and Nlrp3, leading to a higher risk of NAFLD progressing to NASH [81]. Fructose has been shown to induce alterations to the tight junction proteins that affect the gut permeability, with the translocation of bacteria and bacterial endotoxins into the bloodstream [82]. Karin and coworkers recently showed that a short-term high-fructose diet had no effect on intestinal permeability. However, long-term feeding on such a diet induced barrier deterioration and intestinal epithelial ER stress [83]. Notably, fructose-elicited endotoxemia activated Toll-like-receptor (TLR) signaling and TNF production via liver macrophages, inducing lipogenic enzymes [83].
2.2.4. The American-Lifestyle-Induced Obesity Syndrome (ALIOS) Diet
The American-lifestyle-induced obesity syndrome (ALIOS) mouse model is a dietary intervention based on the nutritional content of fast foods commonly consumed in the Western world [84]. Mice are fed with high-fat chow (45%), including trans fats, with high-fructose corn syrup added to their drinking water. The animals become obese, have a high HOMA index and impaired glucose tolerance, and develop hepatic steatosis, with a necroinflammatory response and fibrosis occurring within 16–26 weeks [84,85]. This diet has been demonstrated to reproduce the histopathological characteristics of human NASH [84,85]. Interestingly, the ALIOS diet induces NASH in both male and female rodents [86]. Hepatic transcriptomic analyses have revealed changes in the expression of multiple genes associated with inflammation and tissue repair in ALIOS-diet-fed mice [86]. In the past, this diet was widely used to model liver dysfunction, but its availability has decreased due to the current ban on trans fats. Alternatives to the ALIOS diet are currently being explored, including the Gubra Amylin NASH model and Non-Trans Fat Western Diet (WD-NTF), which has yielded promising results for the induction of NASH in mouse models (for a review see [43]).
2.3. Murine NASH-HCC Models
2.3.1. Choline-Deficient L-Amino Acid-Defined, High-Fat Diet (CDA-HFD)
The CDA-HFD (choline-deficient, L-amino acid-defined, high-fat diet, with 60% of the calorie content as fat and 0.1% methionine by weight) model resembles human NAFLD more closely than the CDAA diet model [76]. The CDA-HFD induces IR, increases hepatic steatosis, and alters the levels of the enzymes involved in carbohydrate and lipid metabolism. Histological changes similar to those seen in human NASH are observed, including hepatocyte ballooning and severe fibrosis [76,87]. After 24 weeks on this diet, an increase in the expression of carcinoembryonic markers (e.g., Afp and Gpc3) is observed, and HCC develops after 36 weeks [87,88]. Even after switching back to a standard diet at 37 weeks, many mice display a progression of fibrosis, with the development of HCC at 48 weeks [88]. Interestingly, NAFLD development clearly differs between the sexes in this diet-induced model. CDAHFD-fed male and female mice initially display similar hepatic damage after 6 weeks on the diet, but differences between the sexes are observed after 12–36 weeks. Male mice fed the CDAHFD present more severe hepatic damage, with greater TG accumulation, hepatocyte death, inflammation, fibrosis, and even tumorigenesis, in comparison to female mice fed the same diet [89]. This higher prevalence of NASH/HCC in male mice matches observations in humans [90]. Lee and collaborators recently demonstrated that formyl peptide receptor 2 (FRP2), an important mediator of inflammatory and immune responses [91], mediates the sex-specific responses to CDAHFD-diet-induced NAFLD/NASH [89].
2.3.2. Choline-Deficient, High-Fat Diet (CD-HFD)
Mathias Heikenwalder and coworkers used a choline-deficient high-fat diet (CD-HFD) to study the combined long-term effects of this diet on obesity/metabolic syndrome/NASH/HCC [35,39,43]. Male C57BL6/J mice fed with the CD-HFD presented progressive body weight gain, glucose intolerance, and insulin resistance. After 16 weeks on the CD-HFD, the mice displayed ballooning hepatocytes, ER/oxidative stress, immune cell infiltration, satellitosis, Mallory-Denk body (MDB) formation, and glycogenated nuclei, all of which are features of human NASH. This diet induced the activation of intrahepatic CD8(+) T cells, NKT cells, and inflammatory cytokines, as observed in NASH patients [35]. Only mild fibrosis developed in this model. The females gained less weight than the males and presented milder liver damage, steatosis, and inflammation [35]. Prolonged exposure to the CD-HFD leads to tumor development. Macroscopically visible tumors were detected in ~25% of mice after 12 months on the diet, and in ~50–70% of mice after 15 months [35]. Using this model, Malehmir et al. demonstrated that platelet recruitment to the liver contributes to the development of nonalcoholic steatohepatitis (NASH) and hepatocellular carcinoma (HCC) via platelet glycoprotein Ibα (GPIbα) [92]. Moreover, Pfister et al. showed that CD8+PD1+ T cells have a pro-tumorigenic effect in NASH, driving HCC formation [39]. This model is also particularly suitable for testing the efficacy of HCC treatments [39,92].
2.3.3. Western Diet (WD) + Carbon Tetrachloride (CCl4)
Carbon tetrachloride (CCl4) is a hepatotoxic chemical used to induce liver damage, fibrosis, and cirrhosis in experimental animals. Friedman and coworkers developed a mouse model of NASH with extensive fibrosis by combining a high-fat, high-fructose, high-cholesterol Western diet (WD) with weekly low-dose intraperitoneal CCl4 [93]. Histological analyses revealed that CCl4 exacerbated the hepatocyte ballooning, proliferation, inflammation, and fibrosis induced by the WD. In this model, NASH was induced within 12 weeks, with severe steatohepatitis, stage-three bridging fibrosis, and subsequent stage-four cirrhosis and HCC being observed by 24 weeks [93]. The dysregulation of the molecular pathways in WD/CCl4 mice at 12 weeks resembled that in early/mild human NASH [93]. Moreover, the HCCs collected from the WD/CCl4 mice at 24 weeks were similar to human HCC molecular subclasses, highlighting the ability of this model to parallel human disease progression [93]. This model has also been shown to be valuable as a system for investigating the NASH microbiome. Carter et al. revealed that microbiome remodeling was completed within 12 weeks in this model, consistent with the evidence of advanced fibrosis, hepatocellular injury, inflammation, and intestinal barrier dysfunction [94]. One limitation of this model is that the CCl4 treatment attenuated the increases in body weight, cholesterol, and insulin/glucose levels typically observed on the WD [93]. Nevertheless, these mice remain a good model, reproducing the progressive stages of human fatty liver disease, from simple steatosis to inflammation, fibrosis, and cancer.
2.3.4. MUP-uPA + HFD
Karin and coworkers developed a model for NASH-driven HCC based on MUP-uPA transgenic mice fed with a HFD [40,50]. The MUP-uPA mice experienced transient hepatic ER stress early in life due to the high levels of uPA expression sustained by the HFD. The full range of NASH-like pathological features are induced in this model, including hepatocyte ballooning, inflammatory infiltrates, and pericellular and bridging fibrosis within four months on the diet, with continuous hepatocyte death and compensatory proliferation. High levels of TNF production, ER stress, and the hepatic expression of p62, all of which have been implicated in human disease, are involved in the progression of both NASH and HCC [50,95]. A spontaneous progression to HCC was observed in 60–85% of the male mice within 40 weeks of this diet. As these mice can be studied from the onset of NASH until they develop tumors, this model can be used for biomarker discovery and the identification of the molecular drivers of HCC progression. The immunopathogenesis of the progression of NASH to HCC in MUP-uPA mice is very similar to that in human NASH-driven HCC, with the precancerous liver accumulating PD-L1- and IL-10-expressing IgA+ cells in parallel with the appearance of inflammation-induced HCC progenitor cells [96]. HFD-fed MUP-uPA mice may be a model of choice for testing treatments. Feng He et al. recently showed that ferroptosis inhibitors or ATF4 activators may be useful for preventing the development of NASH and its progression to HCC [97].
2.3.5. DIAMOND Mice
Sanyal and coworkers described a diet-induced animal model of NAFLD (DIAMOND) based on an isogenic strain derived from a cross between two common mouse strains, 129S1/SvImJ and C57BL/6J [98]. Mice (at 8–12 weeks of age) were fed with a high-fat, high-carbohydrate diet (WD, 42% of calories from fat and 0.1% cholesterol by weight) with ad libitum access to glucose/fructose in their drinking water. B6/129 mice developed obesity, insulin resistance, hypertriglyceridemia, and an increase in their LDL-cholesterol levels following the introduction of this diet. They subsequently also developed steatosis (4–8 weeks), steatohepatitis (16–24 weeks), progressive fibrosis (16 weeks onwards), and 90% went on to develop spontaneous HCC. The histopathological and transcriptional characteristics of DIAMOND mice resemble those of human NASH patients, with lipogenic, ER/oxidative stress, and the activation of the inflammatory and apoptotic signaling pathways [98]. Moreover, the transcriptomic HCC gene signature is similar to that of the S1 and S2 subclasses of human HCC [98,99].
3. Ex Vivo Models of Non-Alcoholic Fatty Liver Disease
In recent years, the critical need to develop effective therapies for the treatment of NAFLD/NASH has led to the emergence of new in vitro models for studies of the mechanisms involved in the development of the disease and for drug screening. Until recently, basic models consisting of two-dimensional (2D) monolayer cultures of single cell types were used to mimic part of the pathogenic process in NAFLD (e.g., steatosis) and to investigate the lipid-lowering effects of anti-steatotic compounds [60,100,101]. However, it proved challenging to model NAFLD fully with such conventional 2D models, due to the chronic nature of the disease. The complete modeling of NAFLD requires long-term stable cultures and an intricate interplay between parenchymal and nonparenchymal liver cells to mediate the disease progression and inflammatory responses [102]. Efforts have been made to overcome these shortcomings by developing new models more closely resembling the architectural and functional properties of in vivo tissues and by promoting alternatives to animal experiments in line with the “3Rs” concept: three-dimensional (3D) models, such as spheroids, organoids, liver-on-chip, and precision-cut liver slices [60,101,103,104] (Figure 2).
3.1. Spheroids
Spheroids were first introduced in the early 1970s by Sutherland and coworkers [105] and are now among the most widely used 3D culture models. Spheroids are formed when simple clusters of cells stick to each other. Three-dimensional floating spheres can be obtained with or without the support of a scaffold [106]. Scaffold-free methods are widely used as they are relatively simple, inexpensive, and rapidly generate spheroids, with the single-cell suspension typically being maintained in ultra-low attachment plates [107]. Scaffold-based methods are generally used in tissue engineering and regenerative medicine applications [106]. Spheroids can remain viable in culture for long periods of time and can reproduce some of the functional properties of an organ [108,109]. Furthermore, depending on the aim of the research (e.g., drug safety screening or the development of antitumor strategies), specific protocols have emerged to obtain spheroids of specific dimensions, composed of cells in different proliferative and metabolic states [106,110,111].
Liver spheroids can be derived from immortalized hepatic cell lines (generally HepG2/HepaRG), differentiated embryonic stem cells (ESC), or pluripotent stem cells (PSC), but are mostly derived from primary human hepatocytes (PHH), which are generally obtained from human liver resections or non-transplantable organs [112]. PHH cultured as 3D spheroids are the model that most closely mimics the phenotype of a hepatocyte in vivo, as these spheroids maintain cell–cell interactions, a tissue-like architecture, and hepatocyte-specific functions over periods of culture of at least five weeks [108]. They have been successfully used to model and study the liver’s metabolic pathophysiology [108,113,114,115,116].
The simple supplementation of spheroids with a mixture of palmitic and oleic acids, two common dietary long-chain FFAs that accumulate in excess in the liver during human steatosis [117], reproduces hallmarks of NAFLD-NASH (e.g., cytoplasmic accumulation of TG in hepatocytes, ER stress, inflammation, and cell death [60,101,104,118]). Kozyra and coworkers described the use of spheroids cultured in a lipotoxic environment as a model for steatosis and insulin resistance [115]. They also demonstrated a reversal of hepatic steatosis following treatment with various antisteatotic agents (metformin, or the antioxidant vitamin E), providing support for the view that 3D spheroids open up new perspectives for studies of potential pharmaceutical targets [115]. Furthermore, the use of multilineage 3D spheroids (coculture of hepatocytes with non-parenchymal cells) was found to provide a better characterization of the relevant mechanistic steps connected to liver steatosis and fibrosis during the progression of NAFLD to NASH. Following exposure to FFA or cyclosporine A, cocultured spheroids displayed enhanced steatogenesis and collagen production, an upregulation of the genes associated with fibrosis progression, such as TIMP metallopeptidase inhibitor 1 (TIMP1) or actin alpha 2 (ACTA2), HSC activation, and the induction of pro-inflammatory cytokines [108,119,120]. Furthermore, intraspheroid steatosis and fibrosis were attenuated by treatment with drugs (Cenicriviroc, Liraglutide, Selonsertib, or Firsocostat) currently under evaluation in clinical trials for the treatment of NASH [119,120,121]. PHH spheroids have also been used in studies on the human genetic variants associated with altered lipid biosynthesis, the establishment and progression of steatosis [119,120,122,123], inflammation [124], and fibrosis [119,123], and have been proved promising for the investigation of clinically relevant associations in NAFLD.
The 3D liver spheroid system elicits many of the phenomena observed in vivo, such as insulin resistance and, importantly, the reversibility of steatosis, making this system suitable both for studies of NAFLD’s pathophysiology and for global therapeutic drug screening. However, size variability and the formation of larger aggregates, hindering both nutrient supply and oxygen diffusion [125], are major shortcomings that will need to be improved if spheroid models are to be more widely used.
3.2. Organoids
Until recently, the terms “organoid” and “spheroid” were used interchangeably [126], but these two entities are produced differently and originate from different cells. Organoids are three-dimensional assemblies of one or more cell types that partially resemble the organ modeled and can perform one or more of its functions [106,127]. Organoids can develop from stem cells (pluripotent, fetal, or adult) [127] or organ-specific progenitors [128,129] through a self-organization process [130].
Liver organoids are generated by the incorporation of tissue stem cells, progenitor cells, or tissue-resident cells isolated from liver samples into an extracellular scaffold environment, such as Matrigel or collagen [106,131]. Unlike induced pluripotent stem cell (iPSC)-derived liver organoids, which are generated by a stepwise differentiation process, human liver-tissue-derived organoids may proliferate and can be screened for gene function [132]. Human liver organoids consist of a spherical monolayer of polarized hepatocytes with a bile canaliculus-like architecture; they can maintain directional bile acid excretion for several weeks [133,134]. With a view of reproducing some of the key features of steatosis and steatohepatitis, Ouchi and coworkers used pluripotent stem cell lines to develop a multicellular human liver organoid (HLO) composed of hepatocyte-, stellate-, and Kupffer-like cells, with a transcriptome similar to that of the tissues from which they were derived in vivo [135]. The exposure of these triple-lineage iPSC-derived organoids to various doses of FFA resulted in a dose-dependent accumulation of intracellular lipids, increases in the secretion of inflammatory cytokines and collagen, and hepatocyte ballooning. The authors proposed organoid stiffness as a potential readout for evaluating the fibrosis severity and for a direct assessment of the efficacy of potential anti-fibrogenic drug candidates [135]. Personalized studies of liver function in patients with NAFLD/NASH-specific differences are essential for predicting the efficacy of novel therapies. Gurevich and coworkers developed a specific differentiation protocol for the generation of cryopreservable hepatocytes from a panel of induced pluripotent stem cells from humans with NASH [136]. They found that the hepatocytes from donors with NASH successfully maintained and reproduced steatosis. They also reported that these hepatocytes were able to be integrated into 3D liver organoids, together with iPSC from NASH patients differentiated into analogs of Kupffer cells and hepatic stellate cell precursors, with the maintenance of hepatic function for at least 10 days [136]. Thus, this study highlighted a powerful new model based on cocultures of cells derived from NASH patients that mimics some of the hallmarks of the disease, which could help to guide general and personalized treatments. Using a similar approach, Raabe’s group demonstrated that irreversibly damaged livers from NASH patients consistently give rise to long-term expandable bipotent ductal organoids that readily undergo hepatic differentiation and functionally reproduce inflammation and fibrosis [137]. The combination of 3D structures of this type with methylomic, metabolomic, and transcriptomic analyses may make it possible to implement organoid-based phenotyping in the personalization of disease modeling and drug development. Furthermore, efforts have been made to make use of organoid technology to develop new strategies for improving patient stratification, notably by considering the interaction between genetic and environmental risk factors. Hendriks et al. developed a scalable, personalized hiPSC-organoid platform for investigating the etiology of steatosis (e.g., exogenous (overload nutritional diet) and genetic origins (PNPLA3 I148M high-risk variant and monogenic predisposition to lipid disorders)), for use with a drug-CRISPR toolkit for NAFLD target identification and testing [132,138,139]. Kimura et al. proposed the development of an organoid-level “forward cellomics” platform [140]. They created a genetically diverse population organoid panel (POP) by mixing cryopreserved foregut progenitors from multiple donors. The POP was treated with FFA (oleic acid) to induce a steatohepatitis phenotype and the genotype–phenotype associations were analyzed “en masse” to capture the pathological genetic variation associated with NAFLD [140]. These approaches are innovative and very promising, but there is still considerable room for improvement. For example, there is a need for cocultures with nonparenchymal cells to extend the application of POP models and CRISPR, and drug-screening approaches for the later stages of the NAFLD-NASH spectrum.
Liver organoids are among the most advanced human-cell-based 3D liver models and their potential for use in NAFLD studies is considerable, as they can be used not only for patient- or gene-mutation-specific lipid metabolism studies, but also for large-scale drug testing to predict the clinical outcomes in personalized medicine and drug efficacy.
3.3. Liver-on-a-Chip
Until recently, spheroids/organoids were considered to be the 3D in vitro models of choice for studying NAFLD [60]. However, the automated control of critical parameters, such as temperature, fluid pressures, cell shear stress, nutrient supply, and waste removal, is not possible with these static platforms. Efforts have been made to overcome these limitations by producing powerful “organ-on-a chip” (OOC) platforms for dealing with these parameters and for the dynamic modeling of diseases and drug testing [141,142,143]. OOC systems bear some similarity to spheroids/organoids. They consist of hollow channels lined with living cells and tissues grown under a more tightly regulated environment, so as to reproduce organ-level and even whole body-level functions [141,144,145,146,147]. Through the integration of cell biology with microengineering and microfluidics, OOCs model physiological and pathological tissue microenvironments, thereby overcoming the limitations of conventional in vitro and in vivo approaches [147].
For the liver, such chips are generated by using hepatocytes to seed a polymeric scaffold consisting of tiny tubes, replicating the microarchitecture of the liver. The device is organized into a long-donut-shaped structure closely resembling a hepatic lobule, through which a microfluid can be passed to recreate the dynamic physicochemical environment of the liver, the gradients involved in zonation, and hepatic functions, rendering this device relevant as a model for studying NAFLD [101,148,149,150,151,152]. Gori and coworkers developed the first liver-on-a-chip (LOC) model by culturing HepG2 cells in a central chamber surrounded by closely spaced parallel microchannels, simulating endothelial cells to mimic the endothelial–parenchymal interface of a liver sinusoid [153]. More gradual and milder intracellular triglyceride accumulation is possible, with a higher hepatic cell viability than that in static 3D models, making it possible to more accurately reproduce the conditions of chronic steatosis observed in vivo. The exposure of this microfluidic device to FFA supplementation for 48 h has been shown to lead to a significant accumulation of intracellular lipids, indicating that this device can readily reproduce the establishment of steatosis [153]. The power of liver-on-chip analyses has been greatly increased since this initial work, through the integration of multiple cell types to reflect the complexity of the liver microenvironment and its role in NAFLD progression more accurately [148]. For example, Lasli and coworkers created a 3D device composed of HepG2 cells and HUVECs transferred onto a chip platform to establish a “steatosis disease-on-a-chip model” [154]. They demonstrated the reversibility of steatosis by treating their LOC device with antisteatotic drugs (metformin or pioglitazone); this treatment triggered a return of intracellular lipid concentrations to basal levels [154]. Du et al. developed a liver lobule chip based on human hepatocytes (HepaRG cells), LSECs, and HSCs that mimicked liver zonation; they demonstrated a change in lipid zonation within a single liver lobule during the early stages of NAFLD progression [149]. Moreover, the treatment of the device with obeticholic acid and elafibranor, which are known to have beneficial effects on lipid metabolism [155,156,157], was shown to prevent or reverse steatosis [149]. Freag and coworkers developed a NASH-on-a-chip model to investigate the NAFL/NASH transition; this model was based on coculturing the four major types of human primary liver cells (hepatocytes, Kupffer cells, liver sinusoidal endothelial cells, and hepatic stellate cells) under microfluidic dynamics and exposing the device to lipotoxic stimuli (FFAs with and without LPS) [151]. Not only did Freag and coworkers demonstrate that it was possible to maintain their microstructured liver tissue under disease-inducing conditions for at least 10 days, but they also showed that this led to the gradual development of the key phenotypic characteristics of human NASH, including the accumulation of intracellular lipids, hepatocellular ballooning, and the expression of inflammatory and profibrotic markers. Furthermore, the exposure of the chip to elafibranor inhibited the development of NASH-specific hallmarks [151].
As mentioned above, several genetic polymorphisms are associated with NAFLD risk [158]. As a means of investigating the effect of the PNPLA3 I148M mutation, Kostrzewski et al. cultured human hepatocytes, KCs, and mutated HSCs in perfused LOC platforms [159]. They observed that the IL-6 secretion in FFA-treated LOCs was enhanced by the presence of this mutation, thus demonstrating the utility of the LOC as a tool for predicting the effects of the genetic polymorphisms associated with NASH progression [159].
The interactions between multiple organs, such as those of the gut-adipose tissue-liver axis, play important roles in NAFL pathogenesis [160,161]. LOC complexity has increased in the last few years and researchers are now focusing their efforts on multi-organ platforms to connect different organs-on-a-chip, in order to assess the role of other tissues in NAFLD development. Lee and coworkers developed a gut-liver-on-a-chip device to reproduce the absorption of fatty acids in the gut and the subsequent accumulation of lipids in hepatocytes [162]. They showed that the presence of gut cells in their model modified drug efficacies with respect to the monoculture model, suggesting that the gut–liver chip can partially reflect the dynamic interactions of drugs with the gut and the liver, thereby improving the predictive value for drug efficacy [163]. Kamei’s group recently described an enhanced gut-liver-on-a-chip platform with integrated microvalves and a pump providing access to individual cell-culture chambers without undesirable cross-contamination, and closed circulation of the medium to mimic the human gut–liver axis [103]. Slaughter et al. developed an adipose tissue-liver-on-a-chip system consisting of both hepatocytes and white adipocytes, which they used to model NAFLD phenotypes in both the liver and adipose modules, together with crosstalk between the organs [164]. This model made it possible to explore the roles of adipocyte lipolysis and insulin resistance in NAFLD, and the exchange of cytokines and adipokines between organs [164].
Liver-on-a-chip systems are highly promising for studies of liver pathophysiology in the context of NAFLD. A large number of liver-on-a-chip models are currently commercially available [148] and, undoubtedly, future developments will endow these liver-on-a-chip systems with an even greater potential to improve our understanding of liver pathophysiology and for use in the development of treatments for NAFLD-related disease.
3.4. Precision Cut Liver Slices
Precision-cut liver slices (PCLS) have been used since the 1980s [165] as in vitro liver models with a tremendous potential to reproduce the complex multicellular histoarchitecture of the hepatic environment, including liver-infiltrating immune cells, whilst maintaining complex biochemical and molecular processes [166]. These slices are obtained by cutting fresh liver tissue with a Krumdieck tissue slicer or automated vibratome [167] to thicknesses of as little as 100 µm [168], although the most widely used thickness is 250 µm, generally with a diameter of 5–8 mm. These slices are cultured in regular tissue culture plates in static, dynamic, or bioreactor-based systems [104,167,169,170]. Liver slices are reproducible, cheap, and maintain the viability of hepatocytes, Kupffer, endothelial, and hepatic stellate cells for five days in controlled culture conditions [167,168], and up to 15 days under certain conditions [171]. Interestingly, one of the main advantages of PCLS cultures is that multiple readouts can be collected from a single slice, as both the slice itself and the culture medium can be analyzed [167]. The tissue for PCLS preparation is usually obtained via partial hepatectomy, from discarded surgical waste, explanted tissue, or non-transplantable tissue. PCLS cultures were initially used for metabolic studies and toxicity testing, but over the last 20 years, the focus of PCLS experiments has shifted towards studies on chronic liver disease, such as fibrosis [167,172]. Indeed, a spontaneous fibrogenic process has been reported to occur during the prolonged incubation of liver slices, as the procedure induces tissue repair and regenerative responses [172,173].
Some of the early processes leading to NAFLD can be investigated in slices from healthy human livers. Indeed, PCLS challenged by the addition of toxic fats (a mixture of oleic and linoleic acids) to the culture medium reproduce the hepatic steatosis, lipid deposition, and lipotoxicity observed in patients during the early stages of NAFLD progression [174]. Using hPCLS, Janssen and coworkers obtained proof-of-concept that PPARα, through its immunosuppressive/anti-inflammatory effect in the human liver, may be relevant to the treatment of non-alcoholic fatty liver disease [175]. Furthermore, the spontaneous induction of fibrogenesis observed in healthy PCLS facilitates evaluations of the efficacy of antifibrotic compounds [176]. Interestingly, the use of this system has underlined clear differences in the fibrotic process and the efficacy of antifibrotic compounds between species, explaining why several drugs with proven antifibrotic effects in animal studies are not effective in humans [176,177]. Aoudjehane and coworkers went further, generating viable functional steatotic human PCLS [178] with a view of improving the quality of so-called “marginal” liver grafts, such as steatotic liver grafts. They demonstrated the efficacy of a “degreasing” cocktail for decreasing the number of intracellular fat droplets, TG content, ER, and oxidative stress in these steatotic hPCLS, thus paving the way for efforts toward increasing the number of usable liver grafts [178].
Finally, the wider use of hPCLS, reflecting inter-individual hepatic heterogeneity, would improve the prediction of treatment efficacy and might also provide insight into the factors (e.g., genetic and epigenetic) contributing to this heterogeneity [179].
4. Conclusions and Perspectives
NAFLD-HCC is undoubtedly a growing global health problem and its contribution to HCC morbidity and mortality is likely to escalate in the coming decades. However, due to the intricate and multifaceted pathophysiology of NAFLD, finding an ideal animal model that can comprehensively mimic the complete spectrum of NAFLD within a reasonable timeframe is challenging. In recent years, many excellent (in vitro/in vivo) models of NAFL/NASH-HCC have significantly advanced our understanding of the mechanistic basis of NAFL/NASH-HCC and have identified novel biomarkers, prognostic markers, and candidate treatment targets. Although in vitro models still require further technological advancements and cost reductions, their continuous improvement holds promise for achieving a comprehensive understanding of the pathogenesis of human NAFL/NASH-HCC and expediting the translation of basic scientific findings into clinical breakthroughs. As shown in this comprehensive review of various preclinical models, specific advantages and disadvantages are inherent to each mouse model (Table 1), and these models reproduce different aspects of human disease. Furthermore, the combination of in vitro and in vivo models could serve as a viable approach to accumulating sufficient knowledge, greatly assisting in understanding liver diseases, the development of new therapies, and the advancement of personalized medicine in hepatology. Accordingly, investigators should use their understanding of the disease they wish to study to ensure that they select the most appropriate model for their research objectives.
Author Contributions
J.F.: writing—original draft preparation, review and editing. S.C.-M.: conceptualization, writing—original draft preparation, review and editing. C.D.: conceptualization, writing—original draft preparation, review and editing. All authors have read and agreed to the published version of the manuscript.
Funding
The authors are supported by grants from the Institut National de la Santé et de la Recherche Médicale (INSERM), Equipe Labellisée LIGUE 2023, la Fondation pour la Recherche Médicale (Equipe FRM: EQU201903007824), l’Agence Nationale de la Recherche (ANR-22-CE14-0074), the SIRIC CARPEM, the Institut National du Cancer (PLBIO 2019-127), l’Association Française pour l’Etude du Foie (AFEF-SUBV 2022). J. Fang is a recipient of Chinese Scholarship Council.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Anstee, Q.M.; Reeves, H.L.; Kotsiliti, E.; Govaere, O.; Heikenwalder, M. From NASH to HCC: Current Concepts and Future Challenges. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 411–428. [Google Scholar] [CrossRef] [PubMed]
- Anstee, Q.M.; Targher, G.; Day, C.P. Progression of NAFLD to Diabetes Mellitus, Cardiovascular Disease or Cirrhosis. Nat. Rev. Gastroenterol. Hepatol. 2013, 10, 330–344. [Google Scholar] [CrossRef]
- Diehl, A.M.; Farpour-Lambert, N.J.; Zhao, L.; Tilg, H. Why We Need to Curb the Emerging Worldwide Epidemic of Nonalcoholic Fatty Liver Disease. Nat. Metab. 2019, 1, 1027–1029. [Google Scholar] [CrossRef] [PubMed]
- Eslam, M.; George, J. Genetic Contributions to NAFLD: Leveraging Shared Genetics to Uncover Systems Biology. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 40–52. [Google Scholar] [CrossRef]
- Friedman, S.L.; Neuschwander-Tetri, B.A.; Rinella, M.; Sanyal, A.J. Mechanisms of NAFLD Development and Therapeutic Strategies. Nat. Med. 2018, 24, 908–922. [Google Scholar] [CrossRef]
- Younossi, Z.; Anstee, Q.M.; Marietti, M.; Hardy, T.; Henry, L.; Eslam, M.; George, J.; Bugianesi, E. Global Burden of NAFLD and NASH: Trends, Predictions, Risk Factors and Prevention. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Estes, C.; Anstee, Q.M.; Arias-Loste, M.T.; Bantel, H.; Bellentani, S.; Caballeria, J.; Colombo, M.; Craxi, A.; Crespo, J.; Day, C.P.; et al. Modeling NAFLD Disease Burden in China, France, Germany, Italy, Japan, Spain, United Kingdom, and United States for the Period 2016–2030. J. Hepatol. 2018, 69, 896–904. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Golabi, P.; Paik, J.M.; Henry, A.; Van Dongen, C.; Henry, L. The Global Epidemiology of Nonalcoholic Fatty Liver Disease (NAFLD) and Nonalcoholic Steatohepatitis (NASH): A Systematic Review. Hepatology 2023, 77, 1335–1347. [Google Scholar] [CrossRef]
- Brunt, E.M.; Kleiner, D.E. Challenges in the Hepatic Histopathology in Non-Alcoholic Fatty Liver Disease. Gut 2017, 66, 1539–1540. [Google Scholar] [CrossRef]
- Brunt, E.M.; Kleiner, D.E.; Carpenter, D.H.; Rinella, M.; Harrison, S.A.; Loomba, R.; Younossi, Z.; Neuschwander-Tetri, B.A.; Sanyal, A.J. American Association for the Study of Liver Diseases NASH Task Force NAFLD: Reporting Histologic Findings in Clinical Practice. Hepatology 2021, 73, 2028–2038. [Google Scholar] [CrossRef]
- Kleiner, D.E.; Brunt, E.M.; Van Natta, M.; Behling, C.; Contos, M.J.; Cummings, O.W.; Ferrell, L.D.; Liu, Y.-C.; Torbenson, M.S.; Unalp-Arida, A.; et al. Design and Validation of a Histological Scoring System for Nonalcoholic Fatty Liver Disease. Hepatology 2005, 41, 1313–1321. [Google Scholar] [CrossRef]
- Singh, S.; Allen, A.M.; Wang, Z.; Prokop, L.J.; Murad, M.H.; Loomba, R. Fibrosis Progression in Nonalcoholic Fatty Liver vs Nonalcoholic Steatohepatitis: A Systematic Review and Meta-Analysis of Paired-Biopsy Studies. Clin. Gastroenterol. Hepatol. 2015, 13, 643–654.e1–9, quiz e39–40. [Google Scholar] [CrossRef] [Green Version]
- Huang, D.Q.; Singal, A.G.; Kono, Y.; Tan, D.J.H.; El-Serag, H.B.; Loomba, R. Changing Global Epidemiology of Liver Cancer from 2010 to 2019: NASH Is the Fastest Growing Cause of Liver Cancer. Cell Metab. 2022, 34, 969–977.e2. [Google Scholar] [CrossRef] [PubMed]
- Fingas, C.D.; Best, J.; Sowa, J.-P.; Canbay, A. Epidemiology of Nonalcoholic Steatohepatitis and Hepatocellular Carcinoma. Clin. Liver Dis. 2016, 8, 119–122. [Google Scholar] [CrossRef]
- Huang, D.Q.; El-Serag, H.B.; Loomba, R. Global Epidemiology of NAFLD-Related HCC: Trends, Predictions, Risk Factors and Prevention. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 223–238. [Google Scholar] [CrossRef]
- Piscaglia, F.; Svegliati-Baroni, G.; Barchetti, A.; Pecorelli, A.; Marinelli, S.; Tiribelli, C.; Bellentani, S. HCC-NAFLD Italian Study Group Clinical Patterns of Hepatocellular Carcinoma in Nonalcoholic Fatty Liver Disease: A Multicenter Prospective Study. Hepatology 2016, 63, 827–838. [Google Scholar] [CrossRef] [PubMed]
- Mittal, S.; El-Serag, H.B.; Sada, Y.H.; Kanwal, F.; Duan, Z.; Temple, S.; May, S.B.; Kramer, J.R.; Richardson, P.A.; Davila, J.A. Hepatocellular Carcinoma in the Absence of Cirrhosis in United States Veterans Is Associated With Nonalcoholic Fatty Liver Disease. Clin. Gastroenterol. Hepatol. 2016, 14, 124–131.e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paradis, V.; Zalinski, S.; Chelbi, E.; Guedj, N.; Degos, F.; Vilgrain, V.; Bedossa, P.; Belghiti, J. Hepatocellular Carcinomas in Patients with Metabolic Syndrome Often Develop without Significant Liver Fibrosis: A Pathological Analysis. Hepatology 2009, 49, 851–859. [Google Scholar] [CrossRef]
- Baffy, G.; Brunt, E.M.; Caldwell, S.H. Hepatocellular Carcinoma in Non-Alcoholic Fatty Liver Disease: An Emerging Menace. J. Hepatol. 2012, 56, 1384–1391. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Li, J.; Riaz, D.R.; Shi, G.; Liu, C.; Dai, Y. Outcomes of Liver Transplantation for Nonalcoholic Steatohepatitis: A Systematic Review and Meta-Analysis. Clin. Gastroenterol. Hepatol. 2014, 12, 394–402.e1. [Google Scholar] [CrossRef]
- Wong, R.J.; Aguilar, M.; Cheung, R.; Perumpail, R.B.; Harrison, S.A.; Younossi, Z.M.; Ahmed, A. Nonalcoholic Steatohepatitis Is the Second Leading Etiology of Liver Disease among Adults Awaiting Liver Transplantation in the United States. Gastroenterology 2015, 148, 547–555. [Google Scholar] [CrossRef] [PubMed]
- Haldar, D.; Kern, B.; Hodson, J.; Armstrong, M.J.; Adam, R.; Berlakovich, G.; Fritz, J.; Feurstein, B.; Popp, W.; Karam, V.; et al. Outcomes of Liver Transplantation for Non-Alcoholic Steatohepatitis: A European Liver Transplant Registry Study. J. Hepatol. 2019, 71, 313–322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vuppalanchi, R.; Noureddin, M.; Alkhouri, N.; Sanyal, A.J. Therapeutic Pipeline in Nonalcoholic Steatohepatitis. Nat. Rev. Gastroenterol. Hepatol. 2021, 18, 373–392. [Google Scholar] [CrossRef] [PubMed]
- Tilg, H.; Moschen, A. Update on Nonalcoholic Fatty Liver Disease: Genes Involved in Nonalcoholic Fatty Liver Disease and Associated Inflammation. Curr. Opin. Clin. Nutr. Metab. Care 2010, 13, 391–396. [Google Scholar] [CrossRef]
- Diehl, A.M.; Day, C. Cause, Pathogenesis, and Treatment of Nonalcoholic Steatohepatitis. N. Engl. J. Med. 2017, 377, 2063–2072. [Google Scholar] [CrossRef]
- Valenti, L.V.C.; Baselli, G.A. Genetics of Nonalcoholic Fatty Liver Disease: A 2018 Update. Curr. Pharm. Des. 2018, 24, 4566–4573. [Google Scholar] [CrossRef]
- Donnelly, K.L.; Smith, C.I.; Schwarzenberg, S.J.; Jessurun, J.; Boldt, M.D.; Parks, E.J. Sources of Fatty Acids Stored in Liver and Secreted via Lipoproteins in Patients with Nonalcoholic Fatty Liver Disease. J. Clin. Investig. 2005, 115, 1343–1351. [Google Scholar] [CrossRef]
- Mota, M.; Banini, B.A.; Cazanave, S.C.; Sanyal, A.J. Molecular Mechanisms of Lipotoxicity and Glucotoxicity in Nonalcoholic Fatty Liver Disease. Metabolism 2016, 65, 1049–1061. [Google Scholar] [CrossRef] [Green Version]
- Begriche, K.; Massart, J.; Robin, M.-A.; Bonnet, F.; Fromenty, B. Mitochondrial Adaptations and Dysfunctions in Nonalcoholic Fatty Liver Disease. Hepatology 2013, 58, 1497–1507. [Google Scholar] [CrossRef]
- Kim, J.Y.; Garcia-Carbonell, R.; Yamachika, S.; Zhao, P.; Dhar, D.; Loomba, R.; Kaufman, R.J.; Saltiel, A.R.; Karin, M. ER Stress Drives Lipogenesis and Steatohepatitis via Caspase-2 Activation of S1P. Cell 2018, 175, 133–145.e15. [Google Scholar] [CrossRef] [Green Version]
- Lebeaupin, C.; Vallée, D.; Hazari, Y.; Hetz, C.; Chevet, E.; Bailly-Maitre, B. Endoplasmic Reticulum Stress Signalling and the Pathogenesis of Non-Alcoholic Fatty Liver Disease. J. Hepatol. 2018, 69, 927–947. [Google Scholar] [CrossRef]
- Schwabe, R.F.; Luedde, T. Apoptosis and Necroptosis in the Liver: A Matter of Life and Death. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 738–752. [Google Scholar] [CrossRef]
- Ogrodnik, M.; Miwa, S.; Tchkonia, T.; Tiniakos, D.; Wilson, C.L.; Lahat, A.; Day, C.P.; Burt, A.; Palmer, A.; Anstee, Q.M.; et al. Cellular Senescence Drives Age-Dependent Hepatic Steatosis. Nat. Commun. 2017, 8, 15691. [Google Scholar] [CrossRef] [Green Version]
- Meijnikman, A.S.; Herrema, H.; Scheithauer, T.P.M.; Kroon, J.; Nieuwdorp, M.; Groen, A.K. Evaluating Causality of Cellular Senescence in Non-Alcoholic Fatty Liver Disease. JHEP Rep. 2021, 3, 100301. [Google Scholar] [CrossRef] [PubMed]
- Wolf, M.J.; Adili, A.; Piotrowitz, K.; Abdullah, Z.; Boege, Y.; Stemmer, K.; Ringelhan, M.; Simonavicius, N.; Egger, M.; Wohlleber, D.; et al. Metabolic Activation of Intrahepatic CD8+ T Cells and NKT Cells Causes Nonalcoholic Steatohepatitis and Liver Cancer via Cross-Talk with Hepatocytes. Cancer Cell 2014, 26, 549–564. [Google Scholar] [CrossRef] [Green Version]
- Pinter, M.; Pinato, D.J.; Ramadori, P.; Heikenwalder, M. NASH and Hepatocellular Carcinoma: Immunology and Immunotherapy. Clin. Cancer Res. 2023, 29, 513–520. [Google Scholar] [CrossRef] [PubMed]
- Peiseler, M.; Schwabe, R.; Hampe, J.; Kubes, P.; Heikenwälder, M.; Tacke, F. Immune Mechanisms Linking Metabolic Injury to Inflammation and Fibrosis in Fatty Liver Disease—Novel Insights into Cellular Communication Circuits. J. Hepatol. 2022, 77, 1136–1160. [Google Scholar] [CrossRef]
- Barreby, E.; Chen, P.; Aouadi, M. Macrophage Functional Diversity in NAFLD—More than Inflammation. Nat. Rev. Endocrinol. 2022, 18, 461–472. [Google Scholar] [CrossRef] [PubMed]
- Pfister, D.; Núñez, N.G.; Pinyol, R.; Govaere, O.; Pinter, M.; Szydlowska, M.; Gupta, R.; Qiu, M.; Deczkowska, A.; Weiner, A.; et al. NASH Limits Anti-Tumour Surveillance in Immunotherapy-Treated HCC. Nature 2021, 592, 450–456. [Google Scholar] [CrossRef]
- Febbraio, M.A.; Reibe, S.; Shalapour, S.; Ooi, G.J.; Watt, M.J.; Karin, M. Preclinical Models for Studying NASH-Driven HCC: How Useful Are They? Cell Metab. 2019, 29, 18–26. [Google Scholar] [CrossRef] [Green Version]
- Oligschlaeger, Y.; Shiri-Sverdlov, R. NAFLD Preclinical Models: More than a Handful, Less of a Concern? Biomedicines 2020, 8, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santhekadur, P.K.; Kumar, D.P.; Sanyal, A.J. Preclinical Models of Non-Alcoholic Fatty Liver Disease. J. Hepatol. 2018, 68, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Gallage, S.; Avila, J.E.B.; Ramadori, P.; Focaccia, E.; Rahbari, M.; Ali, A.; Malek, N.P.; Anstee, Q.M.; Heikenwalder, M. A Researcher’s Guide to Preclinical Mouse NASH Models. Nat. Metab. 2022, 4, 1632–1649. [Google Scholar] [CrossRef] [PubMed]
- Ito, M.; Suzuki, J.; Tsujioka, S.; Sasaki, M.; Gomori, A.; Shirakura, T.; Hirose, H.; Ito, M.; Ishihara, A.; Iwaasa, H.; et al. Longitudinal Analysis of Murine Steatohepatitis Model Induced by Chronic Exposure to High-Fat Diet. Hepatol. Res. 2007, 37, 50–57. [Google Scholar] [CrossRef]
- Hariri, N.; Thibault, L. High-Fat Diet-Induced Obesity in Animal Models. Nutr. Res. Rev. 2010, 23, 270–299. [Google Scholar] [CrossRef] [Green Version]
- Lai, M.; Chandrasekera, P.C.; Barnard, N.D. You Are What You Eat, or Are You? The Challenges of Translating High-Fat-Fed Rodents to Human Obesity and Diabetes. Nutr. Diabetes 2014, 4, e135. [Google Scholar] [CrossRef] [Green Version]
- Winzell, M.S.; Ahrén, B. The High-Fat Diet-Fed Mouse: A Model for Studying Mechanisms and Treatment of Impaired Glucose Tolerance and Type 2 Diabetes. Diabetes 2004, 53 (Suppl. S3), S215–S219. [Google Scholar] [CrossRef] [Green Version]
- Eccleston, H.B.; Andringa, K.K.; Betancourt, A.M.; King, A.L.; Mantena, S.K.; Swain, T.M.; Tinsley, H.N.; Nolte, R.N.; Nagy, T.R.; Abrams, G.A.; et al. Chronic Exposure to a High-Fat Diet Induces Hepatic Steatosis, Impairs Nitric Oxide Bioavailability, and Modifies the Mitochondrial Proteome in Mice. Antioxid. Redox Signal. 2011, 15, 447–459. [Google Scholar] [CrossRef] [Green Version]
- Flessa, C.-M.; Nasiri-Ansari, N.; Kyrou, I.; Leca, B.M.; Lianou, M.; Chatzigeorgiou, A.; Kaltsas, G.; Kassi, E.; Randeva, H.S. Genetic and Diet-Induced Animal Models for Non-Alcoholic Fatty Liver Disease (NAFLD) Research. Int. J. Mol. Sci. 2022, 23, 15791. [Google Scholar] [CrossRef]
- Nakagawa, H.; Umemura, A.; Taniguchi, K.; Font-Burgada, J.; Dhar, D.; Ogata, H.; Zhong, Z.; Valasek, M.A.; Seki, E.; Hidalgo, J.; et al. ER Stress Cooperates with Hypernutrition to Trigger TNF-Dependent Spontaneous HCC Development. Cancer Cell 2014, 26, 331–343. [Google Scholar] [CrossRef] [Green Version]
- Velázquez, K.T.; Enos, R.T.; Bader, J.E.; Sougiannis, A.T.; Carson, M.S.; Chatzistamou, I.; Carson, J.A.; Nagarkatti, P.S.; Nagarkatti, M.; Murphy, E.A. Prolonged High-Fat-Diet Feeding Promotes Non-Alcoholic Fatty Liver Disease and Alters Gut Microbiota in Mice. World J. Hepatol. 2019, 11, 619–637. [Google Scholar] [CrossRef]
- Moon, H.-S.; Dalamaga, M.; Kim, S.-Y.; Polyzos, S.A.; Hamnvik, O.-P.; Magkos, F.; Paruthi, J.; Mantzoros, C.S. Leptin’s Role in Lipodystrophic and Nonlipodystrophic Insulin-Resistant and Diabetic Individuals. Endocr. Rev. 2013, 34, 377–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Chandrasekera, P.C.; Pippin, J.J. Leptin- and Leptin Receptor-Deficient Rodent Models: Relevance for Human Type 2 Diabetes. Curr. Diabetes Rev. 2014, 10, 131–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, H.H.; Feigh, M.; Veidal, S.S.; Rigbolt, K.T.; Vrang, N.; Fosgerau, K. Mouse Models of Nonalcoholic Steatohepatitis in Preclinical Drug Development. Drug Discov. Today 2017, 22, 1707–1718. [Google Scholar] [CrossRef] [PubMed]
- Kristiansen, M.N.B.; Veidal, S.S.; Rigbolt, K.T.G.; Tølbøl, K.S.; Roth, J.D.; Jelsing, J.; Vrang, N.; Feigh, M. Obese Diet-Induced Mouse Models of Nonalcoholic Steatohepatitis-Tracking Disease by Liver Biopsy. World J. Hepatol. 2016, 8, 673–684. [Google Scholar] [CrossRef] [PubMed]
- Anstee, Q.M.; Goldin, R.D. Mouse Models in Non-Alcoholic Fatty Liver Disease and Steatohepatitis Research. Int. J. Exp. Pathol. 2006, 87, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Imajo, K.; Fujita, K.; Yoneda, M.; Nozaki, Y.; Ogawa, Y.; Shinohara, Y.; Kato, S.; Mawatari, H.; Shibata, W.; Kitani, H.; et al. Hyperresponsivity to Low-Dose Endotoxin during Progression to Nonalcoholic Steatohepatitis Is Regulated by Leptin-Mediated Signaling. Cell Metab. 2012, 16, 44–54. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.Q.; Lin, H.Z.; Lane, M.D.; Clemens, M.; Diehl, A.M. Obesity Increases Sensitivity to Endotoxin Liver Injury: Implications for the Pathogenesis of Steatohepatitis. Proc. Natl. Acad. Sci. USA 1997, 94, 2557–2562. [Google Scholar] [CrossRef]
- Hummel, K.P.; Dickie, M.M.; Coleman, D.L. Diabetes, a New Mutation in the Mouse. Science 1966, 153, 1127–1128. [Google Scholar] [CrossRef]
- Soret, P.-A.; Magusto, J.; Housset, C.; Gautheron, J. In Vitro and In Vivo Models of Non-Alcoholic Fatty Liver Disease: A Critical Appraisal. J. Clin. Med. 2020, 10, 36. [Google Scholar] [CrossRef]
- Trak-Smayra, V.; Paradis, V.; Massart, J.; Nasser, S.; Jebara, V.; Fromenty, B. Pathology of the Liver in Obese and Diabetic Ob/Ob and Db/Db Mice Fed a Standard or High-Calorie Diet. Int. J. Exp. Pathol. 2011, 92, 413–421. [Google Scholar] [CrossRef] [PubMed]
- Fromenty, B.; Pessayre, D. Inhibition of Mitochondrial Beta-Oxidation as a Mechanism of Hepatotoxicity. Pharmacol. Ther. 1995, 67, 101–154. [Google Scholar] [CrossRef]
- Sahai, A.; Malladi, P.; Pan, X.; Paul, R.; Melin-Aldana, H.; Green, R.M.; Whitington, P.F. Obese and Diabetic Db/Db Mice Develop Marked Liver Fibrosis in a Model of Nonalcoholic Steatohepatitis: Role of Short-Form Leptin Receptors and Osteopontin. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 287, G1035–G1043. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Handa, P.; Morgan-Stevenson, V.; Maliken, B.D.; Nelson, J.E.; Washington, S.; Westerman, M.; Yeh, M.M.; Kowdley, K.V. Iron Overload Results in Hepatic Oxidative Stress, Immune Cell Activation, and Hepatocellular Ballooning Injury, Leading to Nonalcoholic Steatohepatitis in Genetically Obese Mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2016, 310, G117–G127. [Google Scholar] [CrossRef]
- Caballero, F.; Fernández, A.; Matías, N.; Martínez, L.; Fucho, R.; Elena, M.; Caballeria, J.; Morales, A.; Fernández-Checa, J.C.; García-Ruiz, C. Specific Contribution of Methionine and Choline in Nutritional Nonalcoholic Steatohepatitis: Impact on Mitochondrial S-Adenosyl-L-Methionine and Glutathione. J. Biol. Chem. 2010, 285, 18528–18536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Itagaki, H.; Shimizu, K.; Morikawa, S.; Ogawa, K.; Ezaki, T. Morphological and Functional Characterization of Non-Alcoholic Fatty Liver Disease Induced by a Methionine-Choline-Deficient Diet in C57BL/6 Mice. Int. J. Clin. Exp. Pathol. 2013, 6, 2683–2696. [Google Scholar]
- Matsuzawa, N.; Takamura, T.; Kurita, S.; Misu, H.; Ota, T.; Ando, H.; Yokoyama, M.; Honda, M.; Zen, Y.; Nakanuma, Y.; et al. Lipid-Induced Oxidative Stress Causes Steatohepatitis in Mice Fed an Atherogenic Diet. Hepatology 2007, 46, 1392–1403. [Google Scholar] [CrossRef]
- Rinella, M.E.; Green, R.M. The Methionine-Choline Deficient Dietary Model of Steatohepatitis Does Not Exhibit Insulin Resistance. J. Hepatol. 2004, 40, 47–51. [Google Scholar] [CrossRef]
- Kashireddy, P.V.; Rao, M.S. Lack of Peroxisome Proliferator-Activated Receptor Alpha in Mice Enhances Methionine and Choline Deficient Diet-Induced Steatohepatitis. Hepatol. Res. 2004, 30, 104–110. [Google Scholar] [CrossRef]
- Rizki, G.; Arnaboldi, L.; Gabrielli, B.; Yan, J.; Lee, G.S.; Ng, R.K.; Turner, S.M.; Badger, T.M.; Pitas, R.E.; Maher, J.J. Mice Fed a Lipogenic Methionine-Choline-Deficient Diet Develop Hypermetabolism Coincident with Hepatic Suppression of SCD-1. J. Lipid Res. 2006, 47, 2280–2290. [Google Scholar] [CrossRef] [Green Version]
- Yamazaki, Y.; Kakizaki, S.; Takizawa, D.; Ichikawa, T.; Sato, K.; Takagi, H.; Mori, M. Interstrain Differences in Susceptibility to Non-Alcoholic Steatohepatitis. J. Gastroenterol. Hepatol. 2008, 23, 276–282. [Google Scholar] [CrossRef] [PubMed]
- Nakae, D.; Yoshiji, H.; Mizumoto, Y.; Horiguchi, K.; Shiraiwa, K.; Tamura, K.; Denda, A.; Konishi, Y. High Incidence of Hepatocellular Carcinomas Induced by a Choline Deficient L-Amino Acid Defined Diet in Rats. Cancer Res. 1992, 52, 5042–5045. [Google Scholar] [PubMed]
- Yoshiji, H.; Nakae, D.; Mizumoto, Y.; Horiguchi, K.; Tamura, K.; Denda, A.; Tsujii, T.; Konishi, Y. Inhibitory Effect of Dietary Iron Deficiency on Inductions of Putative Preneoplastic Lesions as Well as 8-Hydroxydeoxyguanosine in DNA and Lipid Peroxidation in the Livers of Rats Caused by Exposure to a Choline-Deficient L-Amino Acid Defined Diet. Carcinogenesis 1992, 13, 1227–1233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kodama, Y.; Kisseleva, T.; Iwaisako, K.; Miura, K.; Taura, K.; De Minicis, S.; Osterreicher, C.H.; Schnabl, B.; Seki, E.; Brenner, D.A. C-Jun N-Terminal Kinase-1 from Hematopoietic Cells Mediates Progression from Hepatic Steatosis to Steatohepatitis and Fibrosis in Mice. Gastroenterology 2009, 137, 1467–1477.e5. [Google Scholar] [CrossRef] [Green Version]
- Denda, A.; Kitayama, W.; Kishida, H.; Murata, N.; Tamura, K.; Kusuoka, O.; Tsutsumi, M.; Nishikawa, F.; Kita, E.; Nakae, D.; et al. Expression of Inducible Nitric Oxide (NO) Synthase but Not Prevention by Its Gene Ablation of Hepatocarcinogenesis with Fibrosis Caused by a Choline-Deficient, L-Amino Acid-Defined Diet in Rats and Mice. Nitric Oxide 2007, 16, 164–176. [Google Scholar] [CrossRef]
- Matsumoto, M.; Hada, N.; Sakamaki, Y.; Uno, A.; Shiga, T.; Tanaka, C.; Ito, T.; Katsume, A.; Sudoh, M. An Improved Mouse Model That Rapidly Develops Fibrosis in Non-Alcoholic Steatohepatitis. Int. J. Exp. Pathol. 2013, 94, 93–103. [Google Scholar] [CrossRef] [Green Version]
- Ouyang, X.; Cirillo, P.; Sautin, Y.; McCall, S.; Bruchette, J.L.; Diehl, A.M.; Johnson, R.J.; Abdelmalek, M.F. Fructose Consumption as a Risk Factor for Non-Alcoholic Fatty Liver Disease. J. Hepatol. 2008, 48, 993–999. [Google Scholar] [CrossRef] [Green Version]
- Abdelmalek, M.F.; Suzuki, A.; Guy, C.; Unalp-Arida, A.; Colvin, R.; Johnson, R.J.; Diehl, A.M. Nonalcoholic Steatohepatitis Clinical Research Network Increased Fructose Consumption Is Associated with Fibrosis Severity in Patients with Nonalcoholic Fatty Liver Disease. Hepatology 2010, 51, 1961–1971. [Google Scholar] [CrossRef] [Green Version]
- Radhakrishnan, S.; Ke, J.-Y.; Pellizzon, M.A. Targeted Nutrient Modifications in Purified Diets Differentially Affect Nonalcoholic Fatty Liver Disease and Metabolic Disease Development in Rodent Models. Curr. Dev. Nutr. 2020, 4, nzaa078. [Google Scholar] [CrossRef]
- Kohli, R.; Kirby, M.; Xanthakos, S.A.; Softic, S.; Feldstein, A.E.; Saxena, V.; Tang, P.H.; Miles, L.; Miles, M.V.; Balistreri, W.F.; et al. High-Fructose, Medium Chain Trans Fat Diet Induces Liver Fibrosis and Elevates Plasma Coenzyme Q9 in a Novel Murine Model of Obesity and Nonalcoholic Steatohepatitis. Hepatology 2010, 52, 934–944. [Google Scholar] [CrossRef] [Green Version]
- Nigro, D.; Menotti, F.; Cento, A.S.; Serpe, L.; Chiazza, F.; Dal Bello, F.; Romaniello, F.; Medana, C.; Collino, M.; Aragno, M.; et al. Chronic Administration of Saturated Fats and Fructose Differently Affect SREBP Activity Resulting in Different Modulation of Nrf2 and Nlrp3 Inflammasome Pathways in Mice Liver. J. Nutr. Biochem. 2017, 42, 160–171. [Google Scholar] [CrossRef]
- Lambertz, J.; Weiskirchen, S.; Landert, S.; Weiskirchen, R. Fructose: A Dietary Sugar in Crosstalk with Microbiota Contributing to the Development and Progression of Non-Alcoholic Liver Disease. Front. Immunol. 2017, 8, 1159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todoric, J.; Di Caro, G.; Reibe, S.; Henstridge, D.C.; Green, C.R.; Vrbanac, A.; Ceteci, F.; Conche, C.; McNulty, R.; Shalapour, S.; et al. Fructose Stimulated de Novo Lipogenesis Is Promoted by Inflammation. Nat. Metab. 2020, 2, 1034–1045. [Google Scholar] [CrossRef]
- Tetri, L.H.; Basaranoglu, M.; Brunt, E.M.; Yerian, L.M.; Neuschwander-Tetri, B.A. Severe NAFLD with Hepatic Necroinflammatory Changes in Mice Fed Trans Fats and a High-Fructose Corn Syrup Equivalent. Am. J. Physiol. Gastrointest. Liver Physiol. 2008, 295, G987–G995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trevaskis, J.L.; Griffin, P.S.; Wittmer, C.; Neuschwander-Tetri, B.A.; Brunt, E.M.; Dolman, C.S.; Erickson, M.R.; Napora, J.; Parkes, D.G.; Roth, J.D. Glucagon-like Peptide-1 Receptor Agonism Improves Metabolic, Biochemical, and Histopathological Indices of Nonalcoholic Steatohepatitis in Mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2012, 302, G762–G772. [Google Scholar] [CrossRef] [Green Version]
- Harris, S.E.; Poolman, T.M.; Arvaniti, A.; Cox, R.D.; Gathercole, L.L.; Tomlinson, J.W. The American Lifestyle-Induced Obesity Syndrome Diet in Male and Female Rodents Recapitulates the Clinical and Transcriptomic Features of Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 319, G345–G360. [Google Scholar] [CrossRef]
- De Minicis, S.; Agostinelli, L.; Rychlicki, C.; Sorice, G.P.; Saccomanno, S.; Candelaresi, C.; Giaccari, A.; Trozzi, L.; Pierantonelli, I.; Mingarelli, E.; et al. HCC Development Is Associated to Peripheral Insulin Resistance in a Mouse Model of NASH. PLoS ONE 2014, 9, e97136. [Google Scholar] [CrossRef] [PubMed]
- Ikawa-Yoshida, A.; Matsuo, S.; Kato, A.; Ohmori, Y.; Higashida, A.; Kaneko, E.; Matsumoto, M. Hepatocellular Carcinoma in a Mouse Model Fed a Choline-Deficient, L-Amino Acid-Defined, High-Fat Diet. Int. J. Exp. Pathol. 2017, 98, 221–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.; Kim, J.; Han, J.; Oh, D.; Kim, M.; Jeong, H.; Kim, T.-J.; Kim, S.-W.; Kim, J.N.; Seo, Y.-S.; et al. Formyl Peptide Receptor 2 Determines Sex-Specific Differences in the Progression of Nonalcoholic Fatty Liver Disease and Steatohepatitis. Nat. Commun. 2022, 13, 578. [Google Scholar] [CrossRef]
- Lonardo, A.; Nascimbeni, F.; Ballestri, S.; Fairweather, D.; Win, S.; Than, T.A.; Abdelmalek, M.F.; Suzuki, A. Sex Differences in Nonalcoholic Fatty Liver Disease: State of the Art and Identification of Research Gaps. Hepatology 2019, 70, 1457–1469. [Google Scholar] [CrossRef]
- Chen, T.; Xiong, M.; Zong, X.; Ge, Y.; Zhang, H.; Wang, M.; Won Han, G.; Yi, C.; Ma, L.; Ye, R.D.; et al. Structural Basis of Ligand Binding Modes at the Human Formyl Peptide Receptor 2. Nat. Commun. 2020, 11, 1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Malehmir, M.; Pfister, D.; Gallage, S.; Szydlowska, M.; Inverso, D.; Kotsiliti, E.; Leone, V.; Peiseler, M.; Surewaard, B.G.J.; Rath, D.; et al. Platelet GPIbα Is a Mediator and Potential Interventional Target for NASH and Subsequent Liver Cancer. Nat. Med. 2019, 25, 641–655. [Google Scholar] [CrossRef] [Green Version]
- Tsuchida, T.; Lee, Y.A.; Fujiwara, N.; Ybanez, M.; Allen, B.; Martins, S.; Fiel, M.I.; Goossens, N.; Chou, H.-I.; Hoshida, Y.; et al. A Simple Diet- and Chemical-Induced Murine NASH Model with Rapid Progression of Steatohepatitis, Fibrosis and Liver Cancer. J. Hepatol. 2018, 69, 385–395. [Google Scholar] [CrossRef] [PubMed]
- Carter, J.K.; Bhattacharya, D.; Borgerding, J.N.; Fiel, M.I.; Faith, J.J.; Friedman, S.L. Modeling Dysbiosis of Human NASH in Mice: Loss of Gut Microbiome Diversity and Overgrowth of Erysipelotrichales. PLoS ONE 2021, 16, e0244763. [Google Scholar] [CrossRef] [PubMed]
- Umemura, A.; He, F.; Taniguchi, K.; Nakagawa, H.; Yamachika, S.; Font-Burgada, J.; Zhong, Z.; Subramaniam, S.; Raghunandan, S.; Duran, A.; et al. P62, Upregulated during Preneoplasia, Induces Hepatocellular Carcinogenesis by Maintaining Survival of Stressed HCC-Initiating Cells. Cancer Cell 2016, 29, 935–948. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shalapour, S.; Lin, X.-J.; Bastian, I.N.; Brain, J.; Burt, A.D.; Aksenov, A.A.; Vrbanac, A.F.; Li, W.; Perkins, A.; Matsutani, T.; et al. Inflammation-Induced IgA+ Cells Dismantle Anti-Liver Cancer Immunity. Nature 2017, 551, 340–345. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, F.; Zhang, P.; Liu, J.; Wang, R.; Kaufman, R.J.; Yaden, B.C.; Karin, M. ATF4 Suppresses Hepatocarcinogenesis by Inducing SLC7A11 (XCT) to Block Stress-Related Ferroptosis. J. Hepatol. 2023, 79, 362–377. [Google Scholar] [CrossRef]
- Asgharpour, A.; Cazanave, S.C.; Pacana, T.; Seneshaw, M.; Vincent, R.; Banini, B.A.; Kumar, D.P.; Daita, K.; Min, H.-K.; Mirshahi, F.; et al. A Diet-Induced Animal Model of Non-Alcoholic Fatty Liver Disease and Hepatocellular Cancer. J. Hepatol. 2016, 65, 579–588. [Google Scholar] [CrossRef] [Green Version]
- Hoshida, Y.; Nijman, S.M.B.; Kobayashi, M.; Chan, J.A.; Brunet, J.-P.; Chiang, D.Y.; Villanueva, A.; Newell, P.; Ikeda, K.; Hashimoto, M.; et al. Integrative Transcriptome Analysis Reveals Common Molecular Subclasses of Human Hepatocellular Carcinoma. Cancer Res. 2009, 69, 7385–7392. [Google Scholar] [CrossRef] [Green Version]
- Tanner, N.; Kubik, L.; Luckert, C.; Thomas, M.; Hofmann, U.; Zanger, U.M.; Böhmert, L.; Lampen, A.; Braeuning, A. Regulation of Drug Metabolism by the Interplay of Inflammatory Signaling, Steatosis, and Xeno-Sensing Receptors in HepaRG Cells. Drug Metab. Dispos. 2018, 46, 326–335. [Google Scholar] [CrossRef] [Green Version]
- Ramos, M.J.; Bandiera, L.; Menolascina, F.; Fallowfield, J.A. In Vitro Models for Non-Alcoholic Fatty Liver Disease: Emerging Platforms and Their Applications. iScience 2022, 25, 103549. [Google Scholar] [CrossRef]
- Loomba, R.; Friedman, S.L.; Shulman, G.I. Mechanisms and Disease Consequences of Nonalcoholic Fatty Liver Disease. Cell 2021, 184, 2537–2564. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Hirai, Y.; Iida, K.; Ito, S.; Trumm, M.; Terada, S.; Sakai, R.; Tsuchiya, T.; Tabata, O.; Kamei, K. Integrated-Gut-Liver-on-a-Chip Platform as an in Vitro Human Model of Non-Alcoholic Fatty Liver Disease. Commun. Biol. 2023, 6, 310. [Google Scholar] [CrossRef]
- Kaur, S.; Kidambi, S.; Ortega-Ribera, M.; Thuy, L.T.T.; Nieto, N.; Cogger, V.C.; Xie, W.-F.; Tacke, F.; Gracia-Sancho, J. In Vitro Models for the Study of Liver Biology and Diseases: Advances and Limitations. Cell. Mol. Gastroenterol. Hepatol. 2023, 15, 559–571. [Google Scholar] [CrossRef] [PubMed]
- Sutherland, R.M.; McCredie, J.A.; Inch, W.R. Growth of Multicell Spheroids in Tissue Culture as a Model of Nodular Carcinomas. J. Natl. Cancer Inst. 1971, 46, 113–120. [Google Scholar] [PubMed]
- Gunti, S.; Hoke, A.T.K.; Vu, K.P.; London, N.R. Organoid and Spheroid Tumor Models: Techniques and Applications. Cancers 2021, 13, 874. [Google Scholar] [CrossRef]
- Ingelman-Sundberg, M.; Lauschke, V.M. 3D Human Liver Spheroids for Translational Pharmacology and Toxicology. Basic Clin. Pharmacol. Toxicol. 2022, 130 (Suppl. S1), 5–15. [Google Scholar] [CrossRef] [PubMed]
- Bell, C.C.; Hendriks, D.F.G.; Moro, S.M.L.; Ellis, E.; Walsh, J.; Renblom, A.; Fredriksson Puigvert, L.; Dankers, A.C.A.; Jacobs, F.; Snoeys, J.; et al. Characterization of Primary Human Hepatocyte Spheroids as a Model System for Drug-Induced Liver Injury, Liver Function and Disease. Sci. Rep. 2016, 6, 25187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tostões, R.M.; Leite, S.B.; Serra, M.; Jensen, J.; Björquist, P.; Carrondo, M.J.T.; Brito, C.; Alves, P.M. Human Liver Cell Spheroids in Extended Perfusion Bioreactor Culture for Repeated-Dose Drug Testing. Hepatology 2012, 55, 1227–1236. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Hartanto, Y.; Zhang, H. Advances in Multicellular Spheroids Formation. J. R. Soc. Interface 2017, 14, 20160877. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Jiang, T.; Chen, D.; Wang, Q.; Zhang, L.W. Three-Dimensional Liver Models: State of the Art and Their Application for Hepatotoxicity Evaluation. Crit. Rev. Toxicol. 2020, 50, 279–309. [Google Scholar] [CrossRef]
- Basu, A.; Dydowiczová, A.; Trosko, J.E.; Bláha, L.; Babica, P. Ready to Go 3D? A Semi-Automated Protocol for Microwell Spheroid Arrays to Increase Scalability and Throughput of 3D Cell Culture Testing. Toxicol. Mech. Methods 2020, 30, 590–604. [Google Scholar] [CrossRef]
- Damelin, L.H.; Coward, S.; Kirwan, M.; Collins, P.; Selden, C.; Hodgson, H.J.F. Fat-Loaded HepG2 Spheroids Exhibit Enhanced Protection from Pro-Oxidant and Cytokine Induced Damage. J. Cell. Biochem. 2007, 101, 723–734. [Google Scholar] [CrossRef]
- Chong, L.; Tsai, C.; Yang, K.; Liao, C.; Hsu, Y. Targeting Protein Palmitoylation Decreases Palmitate-induced Sphere Formation of Human Liver Cancer Cells. Mol. Med. Rep. 2020, 22, 939–947. [Google Scholar] [CrossRef]
- Kozyra, M.; Johansson, I.; Nordling, Å.; Ullah, S.; Lauschke, V.M.; Ingelman-Sundberg, M. Human Hepatic 3D Spheroids as a Model for Steatosis and Insulin Resistance. Sci. Rep. 2018, 8, 14297. [Google Scholar] [CrossRef] [Green Version]
- Eilenberger, C.; Selinger, F.; Rothbauer, M.; Lin, Y.; Limbeck, A.; Schädl, B.; Grillari, J.; Kavok, N.S.; Klochkov, V.K.; Malyukin, Y.V.; et al. Cytotoxicity, Retention, and Anti-Inflammatory Effects of a CeO2 Nanoparticle-Based Supramolecular Complex in a 3D Liver Cell Culture Model. ACS Pharmacol. Transl. Sci. 2021, 4, 101–106. [Google Scholar] [CrossRef]
- Mukherjee, S.; Zhelnin, L.; Sanfiz, A.; Pan, J.; Li, Z.; Yarde, M.; McCarty, J.; Jarai, G. Development and Validation of an in Vitro 3D Model of NASH with Severe Fibrotic Phenotype. Am. J. Transl. Res. 2019, 11, 1531–1540. [Google Scholar] [PubMed]
- Machado, M.V.; Diehl, A.M. Pathogenesis of Nonalcoholic Steatohepatitis. Gastroenterology 2016, 150, 1769–1777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hurrell, T.; Kastrinou-Lampou, V.; Fardellas, A.; Hendriks, D.F.G.; Nordling, Å.; Johansson, I.; Baze, A.; Parmentier, C.; Richert, L.; Ingelman-Sundberg, M. Human Liver Spheroids as a Model to Study Aetiology and Treatment of Hepatic Fibrosis. Cells 2020, 9, 964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pingitore, P.; Sasidharan, K.; Ekstrand, M.; Prill, S.; Lindén, D.; Romeo, S. Human Multilineage 3D Spheroids as a Model of Liver Steatosis and Fibrosis. Int. J. Mol. Sci. 2019, 20, 1629. [Google Scholar] [CrossRef] [Green Version]
- Ströbel, S.; Kostadinova, R.; Fiaschetti-Egli, K.; Rupp, J.; Bieri, M.; Pawlowska, A.; Busler, D.; Hofstetter, T.; Sanchez, K.; Grepper, S.; et al. A 3D Primary Human Cell-Based in Vitro Model of Non-Alcoholic Steatohepatitis for Efficacy Testing of Clinical Drug Candidates. Sci. Rep. 2021, 11, 22765. [Google Scholar] [CrossRef] [PubMed]
- Prill, S.; Caddeo, A.; Baselli, G.; Jamialahmadi, O.; Dongiovanni, P.; Rametta, R.; Kanebratt, K.P.; Pujia, A.; Pingitore, P.; Mancina, R.M.; et al. The TM6SF2 E167K Genetic Variant Induces Lipid Biosynthesis and Reduces Apolipoprotein B Secretion in Human Hepatic 3D Spheroids. Sci. Rep. 2019, 9, 11585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwartz, B.E.; Rajagopal, V.; Smith, C.; Cohick, E.; Whissell, G.; Gamboa, M.; Pai, R.; Sigova, A.; Grossman, I.; Bumcrot, D.; et al. Discovery and Targeting of the Signaling Controls of PNPLA3 to Effectively Reduce Transcription, Expression, and Function in Pre-Clinical NAFLD/NASH Settings. Cells 2020, 9, 2247. [Google Scholar] [CrossRef]
- Tanaka, Y.; Shimanaka, Y.; Caddeo, A.; Kubo, T.; Mao, Y.; Kubota, T.; Kubota, N.; Yamauchi, T.; Mancina, R.M.; Baselli, G.; et al. LPIAT1/MBOAT7 Depletion Increases Triglyceride Synthesis Fueled by High Phosphatidylinositol Turnover. Gut 2021, 70, 180–193. [Google Scholar] [CrossRef] [PubMed]
- Underhill, G.H.; Khetani, S.R. Advances in Engineered Human Liver Platforms for Drug Metabolism Studies. Drug Metab. Dispos. 2018, 46, 1626–1637. [Google Scholar] [CrossRef]
- Simian, M.; Bissell, M.J. Organoids: A Historical Perspective of Thinking in Three Dimensions. J. Cell Biol. 2017, 216, 31–40. [Google Scholar] [CrossRef]
- Lancaster, M.A.; Knoblich, J.A. Organogenesis in a Dish: Modeling Development and Disease Using Organoid Technologies. Science 2014, 345, 1247125. [Google Scholar] [CrossRef]
- Huch, M.; Koo, B.-K. Modeling Mouse and Human Development Using Organoid Cultures. Development 2015, 142, 3113–3125. [Google Scholar] [CrossRef] [Green Version]
- Cacciamali, A.; Villa, R.; Dotti, S. 3D Cell Cultures: Evolution of an Ancient Tool for New Applications. Front. Physiol. 2022, 13, 836480. [Google Scholar] [CrossRef]
- Clevers, H. Modeling Development and Disease with Organoids. Cell 2016, 165, 1586–1597. [Google Scholar] [CrossRef] [Green Version]
- Harrison, S.P.; Baumgarten, S.F.; Verma, R.; Lunov, O.; Dejneka, A.; Sullivan, G.J. Liver Organoids: Recent Developments, Limitations and Potential. Front. Med. 2021, 8, 574047. [Google Scholar] [CrossRef]
- Wei, J.; Zhang, W.; Zhao, B. Human Liver Organoid: Modeling Liver Steatosis and Beyond. Cell Regen. 2023, 12, 17. [Google Scholar] [CrossRef]
- Ramli, M.N.B.; Lim, Y.S.; Koe, C.T.; Demircioglu, D.; Tng, W.; Gonzales, K.A.U.; Tan, C.P.; Szczerbinska, I.; Liang, H.; Soe, E.L.; et al. Human Pluripotent Stem Cell-Derived Organoids as Models of Liver Disease. Gastroenterology 2020, 159, 1471–1486.e12. [Google Scholar] [CrossRef]
- Shinozawa, T.; Kimura, M.; Cai, Y.; Saiki, N.; Yoneyama, Y.; Ouchi, R.; Koike, H.; Maezawa, M.; Zhang, R.-R.; Dunn, A.; et al. High-Fidelity Drug-Induced Liver Injury Screen Using Human Pluripotent Stem Cell-Derived Organoids. Gastroenterology 2021, 160, 831–846.e10. [Google Scholar] [CrossRef]
- Ouchi, R.; Togo, S.; Kimura, M.; Shinozawa, T.; Koido, M.; Koike, H.; Thompson, W.; Karns, R.A.; Mayhew, C.N.; McGrath, P.S.; et al. Modeling Steatohepatitis in Humans with Pluripotent Stem Cell-Derived Organoids. Cell Metab. 2019, 30, 374–384.e6. [Google Scholar] [CrossRef] [PubMed]
- Gurevich, I.; Burton, S.A.; Munn, C.; Ohshima, M.; Goedland, M.E.; Czysz, K.; Rajesh, D. IPSC-Derived Hepatocytes Generated from NASH Donors Provide a Valuable Platform for Disease Modeling and Drug Discovery. Biol. Open 2020, 9, bio055087. [Google Scholar] [CrossRef] [PubMed]
- McCarron, S.; Bathon, B.; Conlon, D.M.; Abbey, D.; Rader, D.J.; Gawronski, K.; Brown, C.D.; Olthoff, K.M.; Shaked, A.; Raabe, T.D. Functional Characterization of Organoids Derived From Irreversibly Damaged Liver of Patients With NASH. Hepatology 2021, 74, 1825–1844. [Google Scholar] [CrossRef] [PubMed]
- Hendriks, D.; Brouwers, J.F.; Hamer, K.; Geurts, M.H.; Luciana, L.; Massalini, S.; López-Iglesias, C.; Peters, P.J.; Rodríguez-Colman, M.J.; Chuva de Sousa Lopes, S.; et al. Engineered Human Hepatocyte Organoids Enable CRISPR-Based Target Discovery and Drug Screening for Steatosis. Nat. Biotechnol. 2023, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Villanueva, M.T. Organoids Illuminate NAFLD Pathogenesis. Nat. Rev. Drug Discov. 2023, 22, 269. [Google Scholar] [CrossRef]
- Kimura, M.; Iguchi, T.; Iwasawa, K.; Dunn, A.; Thompson, W.L.; Yoneyama, Y.; Chaturvedi, P.; Zorn, A.M.; Wintzinger, M.; Quattrocelli, M.; et al. En Masse Organoid Phenotyping Informs Metabolic-Associated Genetic Susceptibility to NASH. Cell 2022, 185, 4216–4232.e16. [Google Scholar] [CrossRef]
- Ingber, D.E. Human Organs-on-Chips for Disease Modelling, Drug Development and Personalized Medicine. Nat. Rev. Genet. 2022, 23, 467–491. [Google Scholar] [CrossRef] [PubMed]
- Vargas, R.; Egurbide-Sifre, A.; Medina, L. Organ-on-a-Chip Systems for New Drugs Development. ADMET DMPK 2021, 9, 111–141. [Google Scholar] [CrossRef]
- Sun, W.; Luo, Z.; Lee, J.; Kim, H.-J.; Lee, K.; Tebon, P.; Feng, Y.; Dokmeci, M.R.; Sengupta, S.; Khademhosseini, A. Organ-on-a-Chip for Cancer and Immune Organs Modeling. Adv. Healthc. Mater. 2019, 8, e1900754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santos, A.K.; Scalzo, S.; de Souza, R.T.V.; Santana, P.H.G.; Marques, B.L.; Oliveira, L.F.; Filho, D.M.; Kihara, A.H.; da Costa Santiago, H.; Parreira, R.C.; et al. Strategic Use of Organoids and Organs-on-Chip as Biomimetic Tools. Semin. Cell Dev. Biol. 2023, 144, 3–10. [Google Scholar] [CrossRef] [PubMed]
- Palasantzas, V.E.J.M.; Tamargo-Rubio, I.; Le, K.; Slager, J.; Wijmenga, C.; Jonkers, I.H.; Kumar, V.; Fu, J.; Withoff, S. IPSC-Derived Organ-on-a-Chip Models for Personalized Human Genetics and Pharmacogenomics Studies. Trends Genet. 2023, 39, 268–284. [Google Scholar] [CrossRef]
- Polidoro, M.A.; Ferrari, E.; Marzorati, S.; Lleo, A.; Rasponi, M. Experimental Liver Models: From Cell Culture Techniques to Microfluidic Organs-on-Chip. Liver Int. 2021, 41, 1744–1761. [Google Scholar] [CrossRef]
- Wu, Q.; Liu, J.; Wang, X.; Feng, L.; Wu, J.; Zhu, X.; Wen, W.; Gong, X. Organ-on-a-Chip: Recent Breakthroughs and Future Prospects. BioMed Eng. OnLine 2020, 19, 9. [Google Scholar] [CrossRef] [Green Version]
- Hassan, S.; Sebastian, S.; Maharjan, S.; Lesha, A.; Carpenter, A.-M.; Liu, X.; Xie, X.; Livermore, C.; Zhang, Y.S.; Zarrinpar, A. Liver-on-a-Chip Models of Fatty Liver Disease. Hepatology 2020, 71, 733–740. [Google Scholar] [CrossRef]
- Du, K.; Li, S.; Li, C.; Li, P.; Miao, C.; Luo, T.; Qiu, B.; Ding, W. Modeling Nonalcoholic Fatty Liver Disease on a Liver Lobule Chip with Dual Blood Supply. Acta Biomater. 2021, 134, 228–239. [Google Scholar] [CrossRef]
- Deguchi, S.; Takayama, K. State-of-the-Art Liver Disease Research Using Liver-on-a-Chip. Inflamm. Regen. 2022, 42, 62. [Google Scholar] [CrossRef]
- Freag, M.S.; Namgung, B.; Reyna Fernandez, M.E.; Gherardi, E.; Sengupta, S.; Jang, H.L. Human Nonalcoholic Steatohepatitis on a Chip. Hepatol. Commun. 2021, 5, 217–233. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Wei, W.; Chen, Z.; Lin, B.; Zhao, W.; Luo, Y.; Zhang, X. Engineered Liver-on-a-Chip Platform to Mimic Liver Functions and Its Biomedical Applications: A Review. Micromachines 2019, 10, 676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gori, M.; Simonelli, M.C.; Giannitelli, S.M.; Businaro, L.; Trombetta, M.; Rainer, A. Investigating Nonalcoholic Fatty Liver Disease in a Liver-on-a-Chip Microfluidic Device. PLoS ONE 2016, 11, e0159729. [Google Scholar] [CrossRef] [Green Version]
- Lasli, S.; Kim, H.-J.; Lee, K.; Suurmond, C.-A.E.; Goudie, M.; Bandaru, P.; Sun, W.; Zhang, S.; Zhang, N.; Ahadian, S.; et al. A Human Liver-on-a-Chip Platform for Modeling Nonalcoholic Fatty Liver Disease. Adv. Biosyst. 2019, 3, e1900104. [Google Scholar] [CrossRef] [PubMed]
- Tacke, F.; Puengel, T.; Loomba, R.; Friedman, S.L. An Integrated View of Anti-Inflammatory and Antifibrotic Targets for the Treatment of NASH. J. Hepatol. 2023, 79, 552–566. [Google Scholar] [CrossRef]
- Tacke, F.; Weiskirchen, R. Non-Alcoholic Fatty Liver Disease (NAFLD)/Non-Alcoholic Steatohepatitis (NASH)-Related Liver Fibrosis: Mechanisms, Treatment and Prevention. Ann. Transl. Med. 2021, 9, 729. [Google Scholar] [CrossRef]
- Perazzo, H.; Dufour, J.-F. The Therapeutic Landscape of Non-Alcoholic Steatohepatitis. Liver Int. 2017, 37, 634–647. [Google Scholar] [CrossRef]
- Sulaiman, S.A.; Dorairaj, V.; Adrus, M.N.H. Genetic Polymorphisms and Diversity in Nonalcoholic Fatty Liver Disease (NAFLD): A Mini Review. Biomedicines 2022, 11, 106. [Google Scholar] [CrossRef]
- Kostrzewski, T.; Maraver, P.; Ouro-Gnao, L.; Levi, A.; Snow, S.; Miedzik, A.; Rombouts, K.; Hughes, D. A Microphysiological System for Studying Nonalcoholic Steatohepatitis. Hepatol. Commun. 2020, 4, 77–91. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Wang, H. Multiple Organs Involved in the Pathogenesis of Non-Alcoholic Fatty Liver Disease. Cell Biosci. 2020, 10, 140. [Google Scholar] [CrossRef]
- Bauer, K.C.; Littlejohn, P.T.; Ayala, V.; Creus-Cuadros, A.; Finlay, B.B. Nonalcoholic Fatty Liver Disease and the Gut-Liver Axis: Exploring an Undernutrition Perspective. Gastroenterology 2022, 162, 1858–1875.e2. [Google Scholar] [CrossRef]
- Lee, S.Y.; Sung, J.H. Gut-Liver on a Chip toward an in Vitro Model of Hepatic Steatosis. Biotechnol. Bioeng. 2018, 115, 2817–2827. [Google Scholar] [CrossRef]
- Jeon, J.-W.; Lee, S.H.; Kim, D.; Sung, J.H. In Vitro Hepatic Steatosis Model Based on Gut-Liver-on-a-Chip. Biotechnol. Prog. 2021, 37, e3121. [Google Scholar] [CrossRef]
- Slaughter, V.L.; Rumsey, J.W.; Boone, R.; Malik, D.; Cai, Y.; Sriram, N.N.; Long, C.J.; McAleer, C.W.; Lambert, S.; Shuler, M.L.; et al. Validation of an Adipose-Liver Human-on-a-Chip Model of NAFLD for Preclinical Therapeutic Efficacy Evaluation. Sci. Rep. 2021, 11, 13159. [Google Scholar] [CrossRef]
- Smith, P.F.; Gandolfi, A.J.; Krumdieck, C.L.; Putnam, C.W.; Zukoski, C.F.; Davis, W.M.; Brendel, K. Dynamic Organ Culture of Precision Liver Slices for in Vitro Toxicology. Life Sci. 1985, 36, 1367–1375. [Google Scholar] [CrossRef]
- Olinga, P.; Schuppan, D. Precision-Cut Liver Slices: A Tool to Model the Liver Ex Vivo. J. Hepatol. 2013, 58, 1252–1253. [Google Scholar] [CrossRef] [Green Version]
- Dewyse, L.; Reynaert, H.; van Grunsven, L.A. Best Practices and Progress in Precision-Cut Liver Slice Cultures. Int. J. Mol. Sci. 2021, 22, 7137. [Google Scholar] [CrossRef] [PubMed]
- de Graaf, I.A.M.; Olinga, P.; de Jager, M.H.; Merema, M.T.; de Kanter, R.; van de Kerkhof, E.G.; Groothuis, G.M.M. Preparation and Incubation of Precision-Cut Liver and Intestinal Slices for Application in Drug Metabolism and Toxicity Studies. Nat. Protoc. 2010, 5, 1540–1551. [Google Scholar] [CrossRef] [PubMed]
- Rezvani, M.; Vallier, L.; Guillot, A. Modeling Nonalcoholic Fatty Liver Disease in the Dish Using Human-Specific Platforms: Strategies and Limitations. Cell. Mol. Gastroenterol. Hepatol. 2023, 15, 1135–1145. [Google Scholar] [CrossRef] [PubMed]
- Paish, H.L.; Reed, L.H.; Brown, H.; Bryan, M.C.; Govaere, O.; Leslie, J.; Barksby, B.S.; Garcia Macia, M.; Watson, A.; Xu, X.; et al. A Bioreactor Technology for Modeling Fibrosis in Human and Rodent Precision-Cut Liver Slices. Hepatology 2019, 70, 1377–1391. [Google Scholar] [CrossRef]
- Wu, X.; Roberto, J.B.; Knupp, A.; Kenerson, H.L.; Truong, C.D.; Yuen, S.Y.; Brempelis, K.J.; Tuefferd, M.; Chen, A.; Horton, H.; et al. Precision-Cut Human Liver Slice Cultures as an Immunological Platform. J. Immunol. Methods 2018, 455, 71–79. [Google Scholar] [CrossRef] [PubMed]
- van de Bovenkamp, M.; Groothuis, G.M.M.; Meijer, D.K.F.; Olinga, P. Liver Slices as a Model to Study Fibrogenesis and Test the Effects of Anti-Fibrotic Drugs on Fibrogenic Cells in Human Liver. Toxicol. Vitr. 2008, 22, 771–778. [Google Scholar] [CrossRef] [PubMed]
- van de Bovenkamp, M.; Groothuis, G.M.M.; Draaisma, A.L.; Merema, M.T.; Bezuijen, J.I.; van Gils, M.J.; Meijer, D.K.F.; Friedman, S.L.; Olinga, P. Precision-Cut Liver Slices as a New Model to Study Toxicity-Induced Hepatic Stellate Cell Activation in a Physiologic Milieu. Toxicol. Sci. 2005, 85, 632–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palma, E.; Doornebal, E.J.; Chokshi, S. Precision-Cut Liver Slices: A Versatile Tool to Advance Liver Research. Hepatol. Int. 2019, 13, 51–57. [Google Scholar] [CrossRef] [Green Version]
- Janssen, A.W.F.; Betzel, B.; Stoopen, G.; Berends, F.J.; Janssen, I.M.; Peijnenburg, A.A.; Kersten, S. The Impact of PPARα Activation on Whole Genome Gene Expression in Human Precision Cut Liver Slices. BMC Genom. 2015, 16, 760. [Google Scholar] [CrossRef] [Green Version]
- Westra, I.M.; Mutsaers, H.A.M.; Luangmonkong, T.; Hadi, M.; Oosterhuis, D.; de Jong, K.P.; Groothuis, G.M.M.; Olinga, P. Human Precision-Cut Liver Slices as a Model to Test Antifibrotic Drugs in the Early Onset of Liver Fibrosis. Toxicol. Vitr. 2016, 35, 77–85. [Google Scholar] [CrossRef]
- Westra, I.M.; Oosterhuis, D.; Groothuis, G.M.M.; Olinga, P. Precision-Cut Liver Slices as a Model for the Early Onset of Liver Fibrosis to Test Antifibrotic Drugs. Toxicol. Appl. Pharmacol. 2014, 274, 328–338. [Google Scholar] [CrossRef]
- Aoudjehane, L.; Gautheron, J.; Le Goff, W.; Goumard, C.; Gilaizeau, J.; Nget, C.S.; Savier, E.; Atif, M.; Lesnik, P.; Morichon, R.; et al. Novel Defatting Strategies Reduce Lipid Accumulation in Primary Human Culture Models of Liver Steatosis. Dis. Model. Mech. 2020, 13, dmm042663. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharya, D.; Becker, C.; Readhead, B.; Goossens, N.; Novik, J.; Fiel, M.I.; Cousens, L.P.; Magnusson, B.; Backmark, A.; Hicks, R.; et al. Repositioning of a Novel GABA-B Receptor Agonist, AZD3355 (Lesogaberan), for the Treatment of Non-Alcoholic Steatohepatitis. Sci. Rep. 2021, 11, 20827. [Google Scholar] [CrossRef]
Figure 1.
Pre-clinical models mimicking the physiopathological characteristics of Non-Alcoholic Fatty Liver Disease/HCC. ALIOS: American-lifestyle-induced obesity syndrome; CCl4: Carbon tetrachloride; CDAA: Choline-deficient, L-amino defined diet; CDA-HFD: Choline-deficient, L-amino acid-defined, high-fat diet; CD-HFD: Choline-deficient, high-fat diet; Db/db: leptin-receptor-deficient; DIAMOND: Diet-induced animal model of non-alcoholic fatty liver disease; HCC: Hepatocellular carcinoma; HFD: High-fat diet; HFHF: High-fat high-fructose diet; MCD: Methionine and choline deficient diet; MUP-uPA: major urinary protein-urokinase-type plasminogen activator; NAFL: Non-alcoholic fatty liver; NASH: Non-alcoholic steatohepatitis; Ob/ob: leptin deficient; and WD: Western diet.
Figure 1.
Pre-clinical models mimicking the physiopathological characteristics of Non-Alcoholic Fatty Liver Disease/HCC. ALIOS: American-lifestyle-induced obesity syndrome; CCl4: Carbon tetrachloride; CDAA: Choline-deficient, L-amino defined diet; CDA-HFD: Choline-deficient, L-amino acid-defined, high-fat diet; CD-HFD: Choline-deficient, high-fat diet; Db/db: leptin-receptor-deficient; DIAMOND: Diet-induced animal model of non-alcoholic fatty liver disease; HCC: Hepatocellular carcinoma; HFD: High-fat diet; HFHF: High-fat high-fructose diet; MCD: Methionine and choline deficient diet; MUP-uPA: major urinary protein-urokinase-type plasminogen activator; NAFL: Non-alcoholic fatty liver; NASH: Non-alcoholic steatohepatitis; Ob/ob: leptin deficient; and WD: Western diet.

Figure 2.
Human in vitro models of Non-Alcoholic Fatty Liver Disease. −: inadequate; +: low; ++: medium; and +++: high.
Figure 2.
Human in vitro models of Non-Alcoholic Fatty Liver Disease. −: inadequate; +: low; ++: medium; and +++: high.


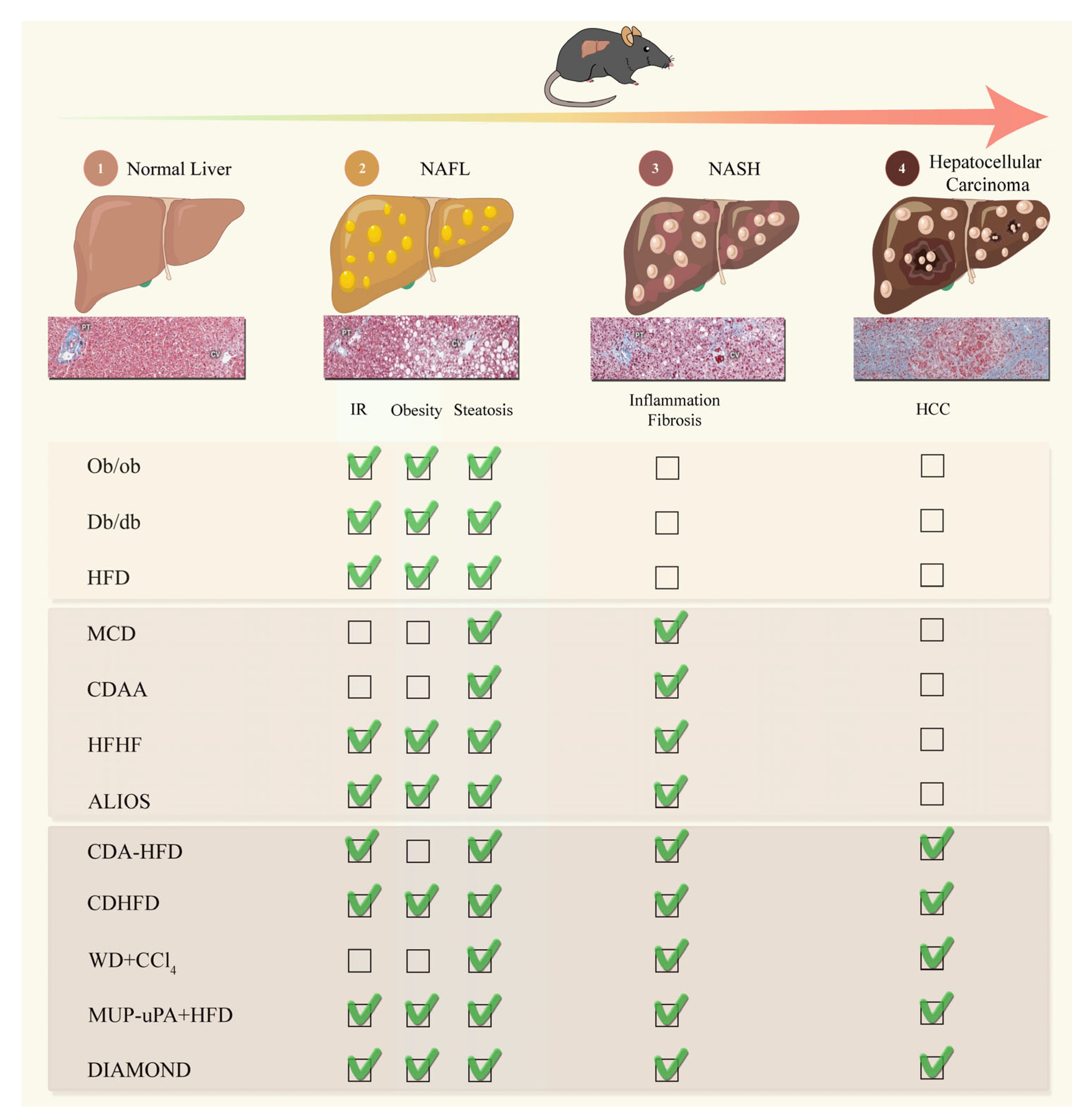{kind=link}
{kind=link}
Table 1.
Summary of the most relevant models of NAFL/NASH/HCC.
| Model Type or Name | Phenotype | Fibrosis | NASH | Human NASH Gene Signature | Time to Disease | Subsequent Development of HCC | Advantage | Disadvantage | References | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Insulin Resistance | Obesity | Steatosis | Inflammation/ER Stress | Ballooning | |||||||||
| NAFLD | |||||||||||||
| HFD | YES | YES | Strong | Weak | NO | Slight | NO | NO | Translocation of bacteria in 1 week, fibrosis after 9 weeks, hepatic inflammation after 19 weeks | No; requires additional insult | Low costs; easy to operate | Requires large sample size; difficult comparison between groups and protocols related to various dietary compositions | Ito et al., 2007; Nakagawa et al., 2014; Flessa et al., 2022 [44,49,50] |
| ob/ob (point mutation in leptin gene) | YES | YES | Strong | Weak | NO | Second insult | NO | ND | First signs of obesity recognizable at 4 weeks of age | No; requires additional insult | Features similar to human NAFLD | Leptin deficiency does not seem to play a role in NAFLD and NASH development in humans; Do not develop NASH or fibrosis without second insult | Kristiansen et al., 2016 [55] |
| db/db (point mutation in leptin receptor) | YES | YES | Strong | Weak | NO | Second insult | NO | ND | Develop NASH after the addition of MCD diet for 2 weeks | No; requires additional insult | Features similar to human NAFLD | Leptin deficiency does not seem to play a role in NAFLD and NASH development in humans; Do not develop NASH or fibrosis without second insult | Trak-Smayra et al., 2011 [61] |
| NASH | |||||||||||||
| MCD | NO | Weight loss | Strong | Strong | YES | YES | YES | ND | Steatohepatitis after 10 days and fibrosis after 8–10 weeks | No; requires additional insult | Short period; Easy to operate; High reproducibility, appropriate for the study of fibrosis mechanisms | No NAFLD-related metabolic syndrome; severe weight loss and liver atrophy | Caballero et al., 2010; Itagaki et al., 2013 [65,66] |
| CDAA | NO | Weight loss | Strong | Medium | YES | YES | YES | ND | Mild to moderate fibrosis ~22 weeks | ~25% prevalence of HCC 84 weeks | Robust; reproducible | Long-period; high costs; age-related systemic changes, No advanced fibrosis; Disease etiology does not mimic humans | Kodama et al., 2009; Denda et al., 2007; Matsumoto et al., 2013 [76,77,78] |
| High-Fructose Diet | YES | YES | Strong | Medium | YES | YES | YES | YES | 12~16 weeks can have fibrosis and NASH | Need other diet | Recapitulates histopathological characteristics of human from NAFLD to NASH | Do not develop HCC without second insult | Kohli et al., 2010; Nigro et al., 2017 [80,81] |
| ALIOS | YES | YES | Strong | Medium | YES | YES | YES | ND | NASH 12–16 weeks versus late NASH 24–30 weeks | Not relevant | Recapitulates histopathological characteristics of human NASH | No longer available as trans fats cannot be included in food | Tetri et al., 2008; Trevaskis et al., 2012; Gallage et al., 2022 [43,84,85] |
| NASH-HCC | |||||||||||||
| CDA-HFD | NO | NO | Strong | Weak | YES | YES | YES | ND | Lipid droplets with infiltration of inflammatory cells after 1 week;enlarged fatty liver with fibrosis in 6 week; NASH ~12 weeks | 24–36 weeks: 100% of mice | Accelerated model of NASH with severe fibrosis. High induction of hepatocellular adenomas and carcinomas | Does not induce obesity or metabolic syndrome | Matsumoto et al., 2013; De Minicis et al., 2014; Ikawa-Yoshida et al., 2017 [76,87,88] |
| CDHFD | YES | YES | Strong | Weak | YES | YES | YES | ND | NASH 16~18 weeks; HCA~20 weeks | 12 months: ~25–30% of mice; 15 months: ~50–70% of mice | ROS production, lipid peroxidation, and mitochondrial dysfunction. Fibrosis around central vein. Overexpression of cytokines TNF-α and IL-6 | Lack of choline is less physiological. Lower degree of fibrosis compared to WDs | Wolf et al., 2014; Pfister et al., 2021; Malehmir et al., 2019 [35,39,92] |
| WD+CCl4 | NO | NO | Strong | Strong | YES | YES | YES | YES | Microbiome remodeling within 12 weeks; F4 fibrosis and HCC ~24 weeks, | 24 weeks: 100% of mice | Recapitulates histopathological and transcriptional characteristics of human NASH | Does not induce obesity or metabolic syndrome. | Tsuchida et al., 2018 [93] |
| Mup-upA +HFD | YES | YES | Strong | Medium | YES | YES | YES | YES | Ballooning hepatocytes, pericellular and bridging fibrosis ~24 weeks; small HCC ~32 weeks | ~32–40 weeks: ~80% of mice | Can spontaneously mimic Human NASH to HCC; does not rely on the administration of liver toxins or carcinogens | Genetic background | Nakagawa et al., 2014; Febbraio et al., 2019 [40,50] |
| DIAMOND | YES | YES | Strong | Weak | YES | YES | YES | YES | Mice develop steatosis within 16 weeks; NASH ~16–24 weeks and nodule formation by 52 weeks (90%) | 52 weeks: 90% of mice | Histology and transcriptome mirror human NASH | Genetic background of DIAMOND mice is unique, making it difficult to cross them with other gene targeted mice | Asgharpour et al., 2016 [98] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Fang, J.; Celton-Morizur, S.; Desdouets, C. NAFLD-Related HCC: Focus on the Latest Relevant Preclinical Models. Cancers 2023, 15, 3723. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers15143723
AMA Style
Fang J, Celton-Morizur S, Desdouets C. NAFLD-Related HCC: Focus on the Latest Relevant Preclinical Models. Cancers. 2023; 15(14):3723. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers15143723
Chicago/Turabian StyleFang, Jing, Séverine Celton-Morizur, and Chantal Desdouets. 2023. "NAFLD-Related HCC: Focus on the Latest Relevant Preclinical Models" Cancers 15, no. 14: 3723. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers15143723
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.