
αV-Integrin-Dependent Inhibition of Glioblastoma Cell Migration, Invasion and Vasculogenic Mimicry by the uPAcyclin Decapeptide

 ,
,  , , and
, , and 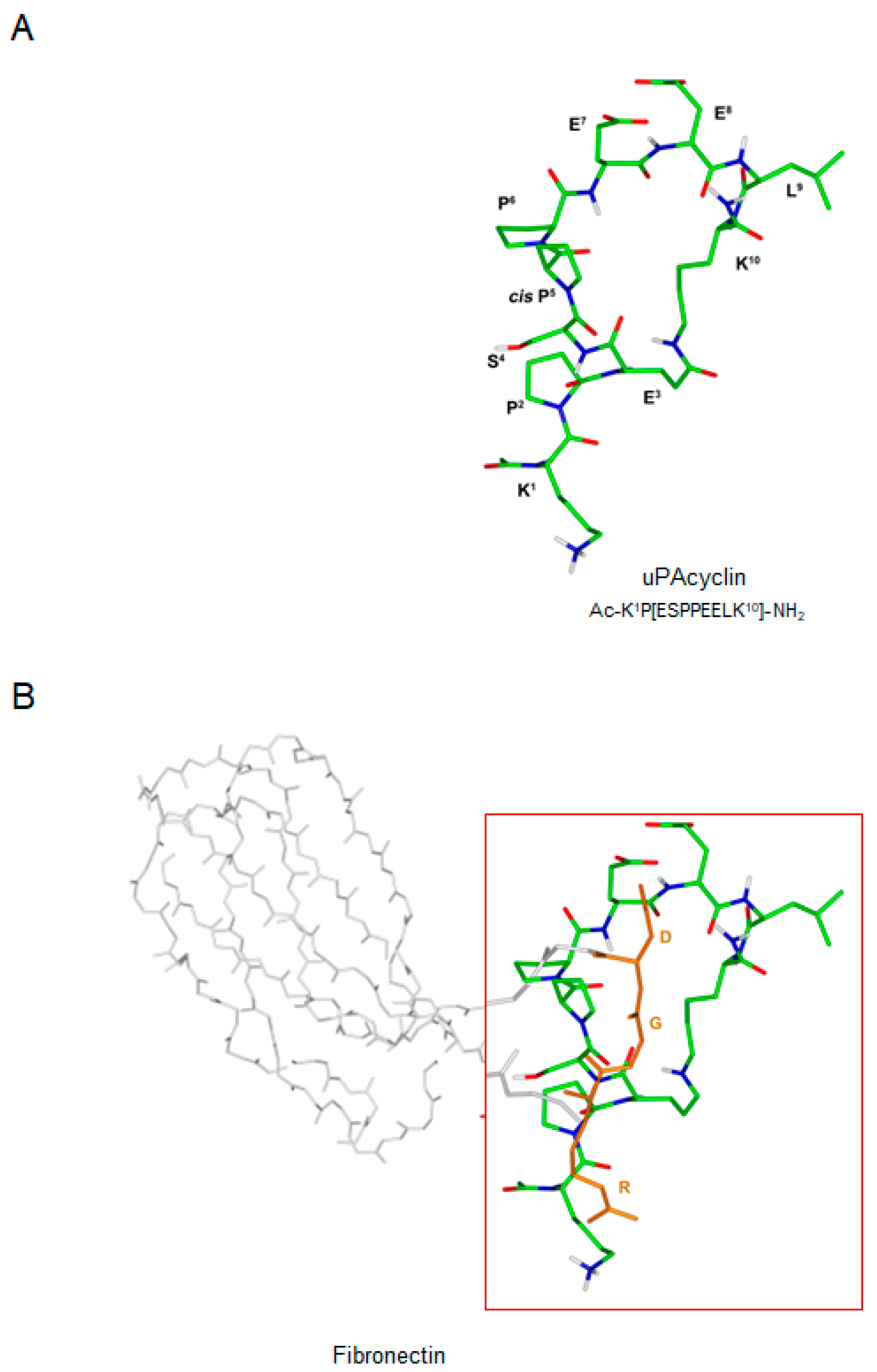{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Culture Conditions
2.2. Peptide Synthesis
2.3. NMR Conformational Analysis
2.4. Cell Viability Assay
2.5. Binding Assay
2.6. Protein Extraction and Western Blotting Analysis
2.7. Migration and Invasion Assays
2.8. Wound Healing Assay
2.9. Vasculogenic Mimicry Assay
2.10. Cells Recovery from 3D Matrix and Lysate Preparation
2.11. Statistical Analyses
3. Results
3.1. NMR Conformational Analysis of uPAcyclin
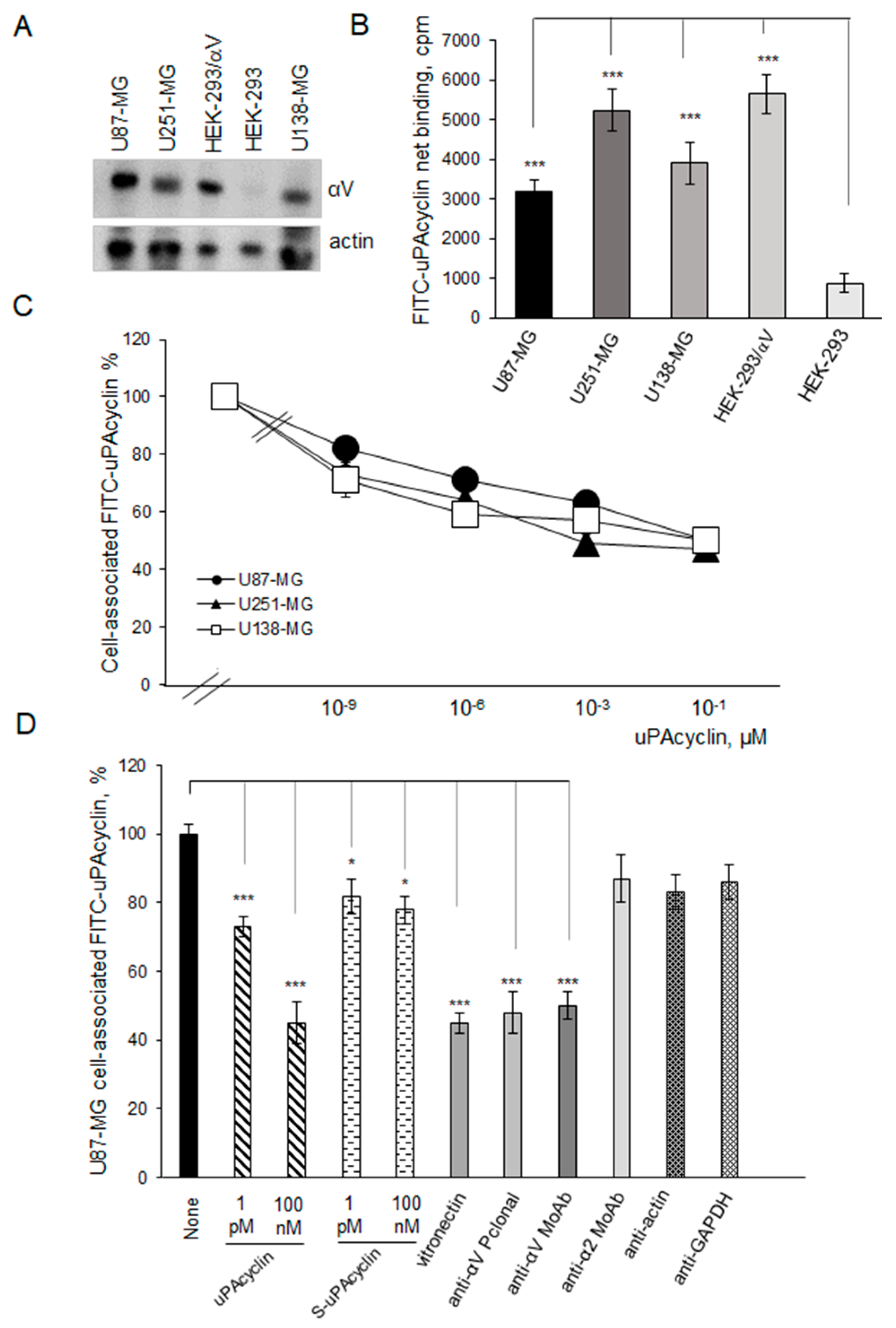
3.2. Specific Binding of FITC-uPAcyclin to U87-MG, U251-MG, and U138-MG GBM Cells
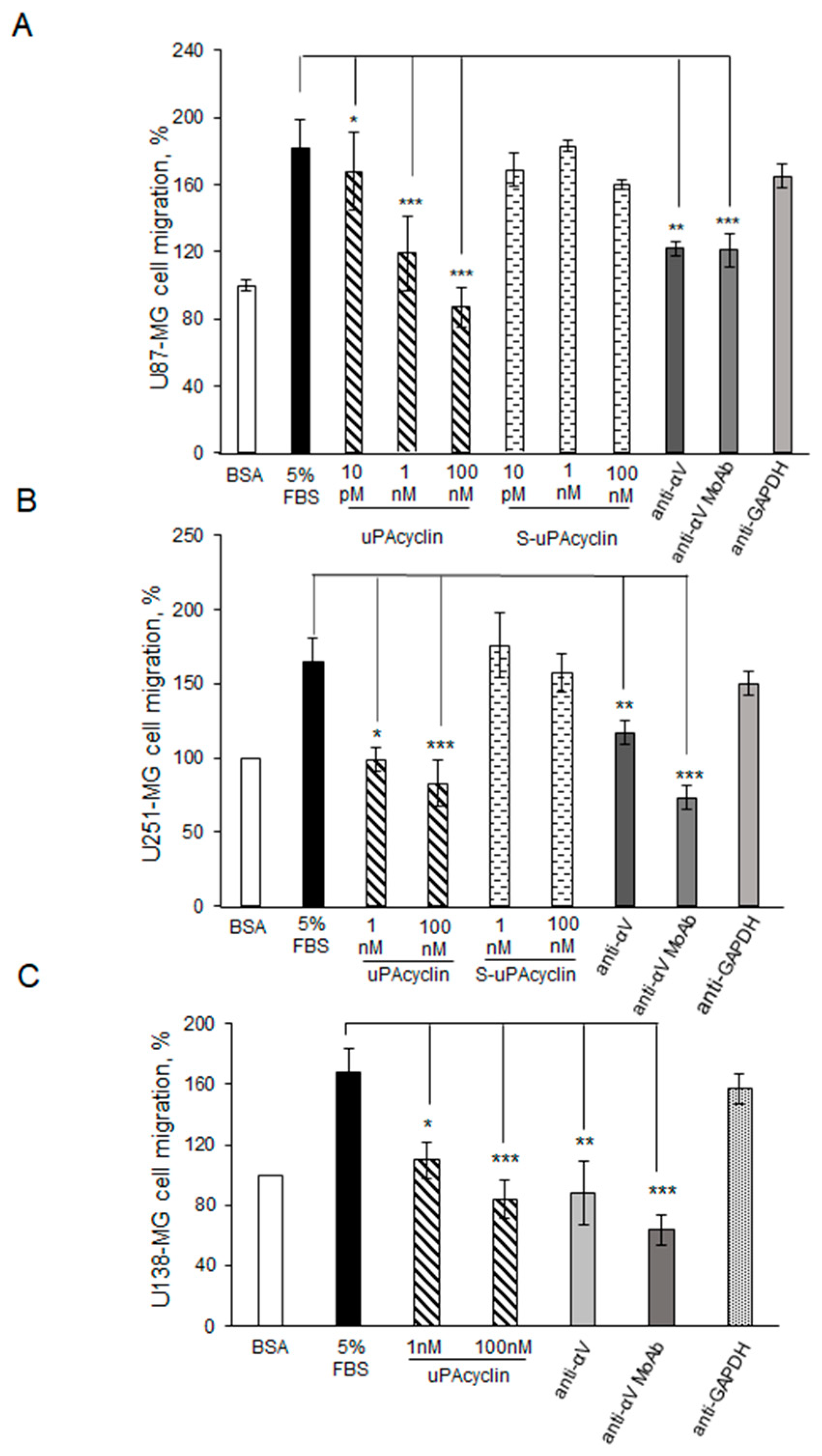
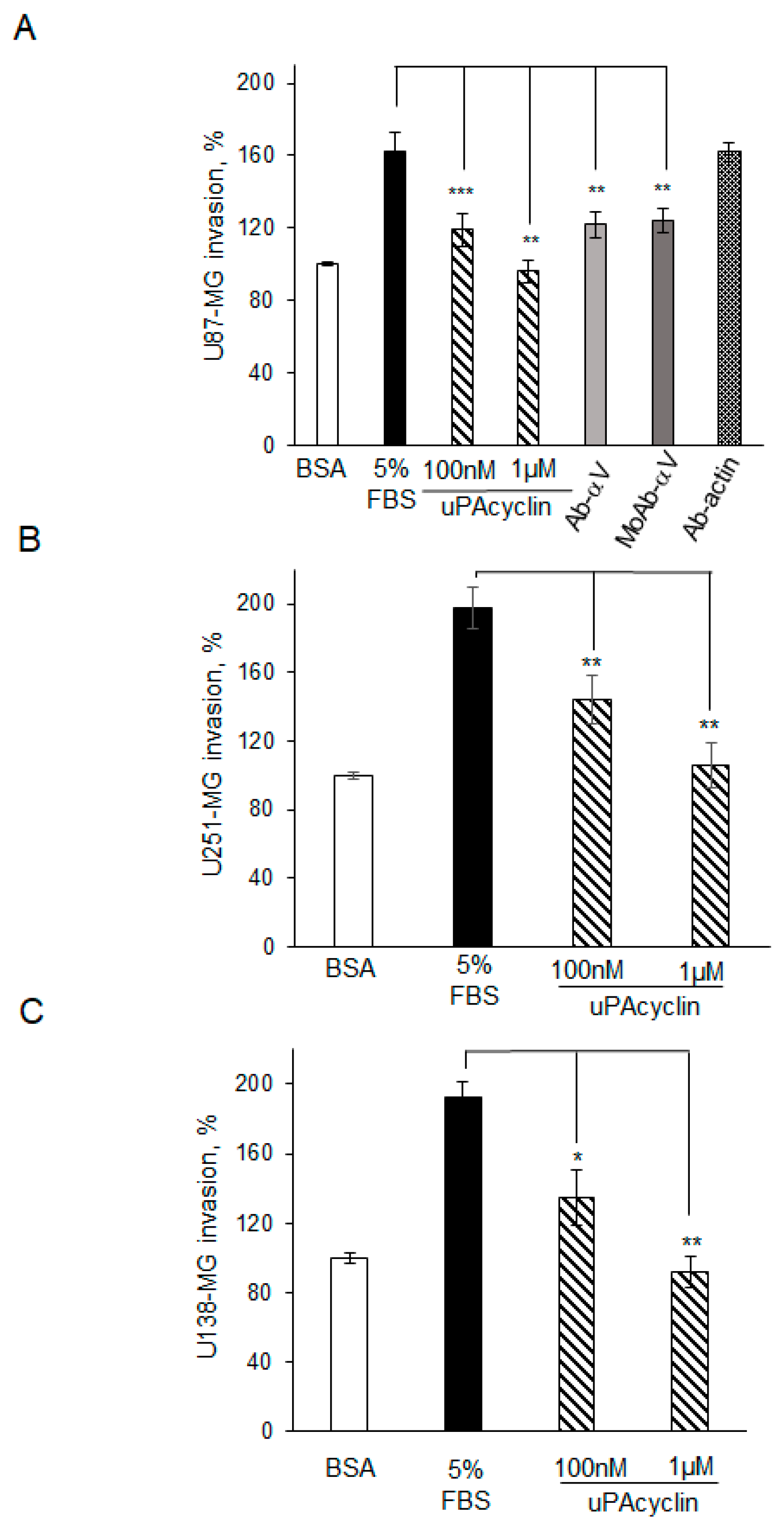
3.3. Inhibition of U87-MG, U251-MG, and U138 GBM Cell Migration and Invasion by uPAcyclin
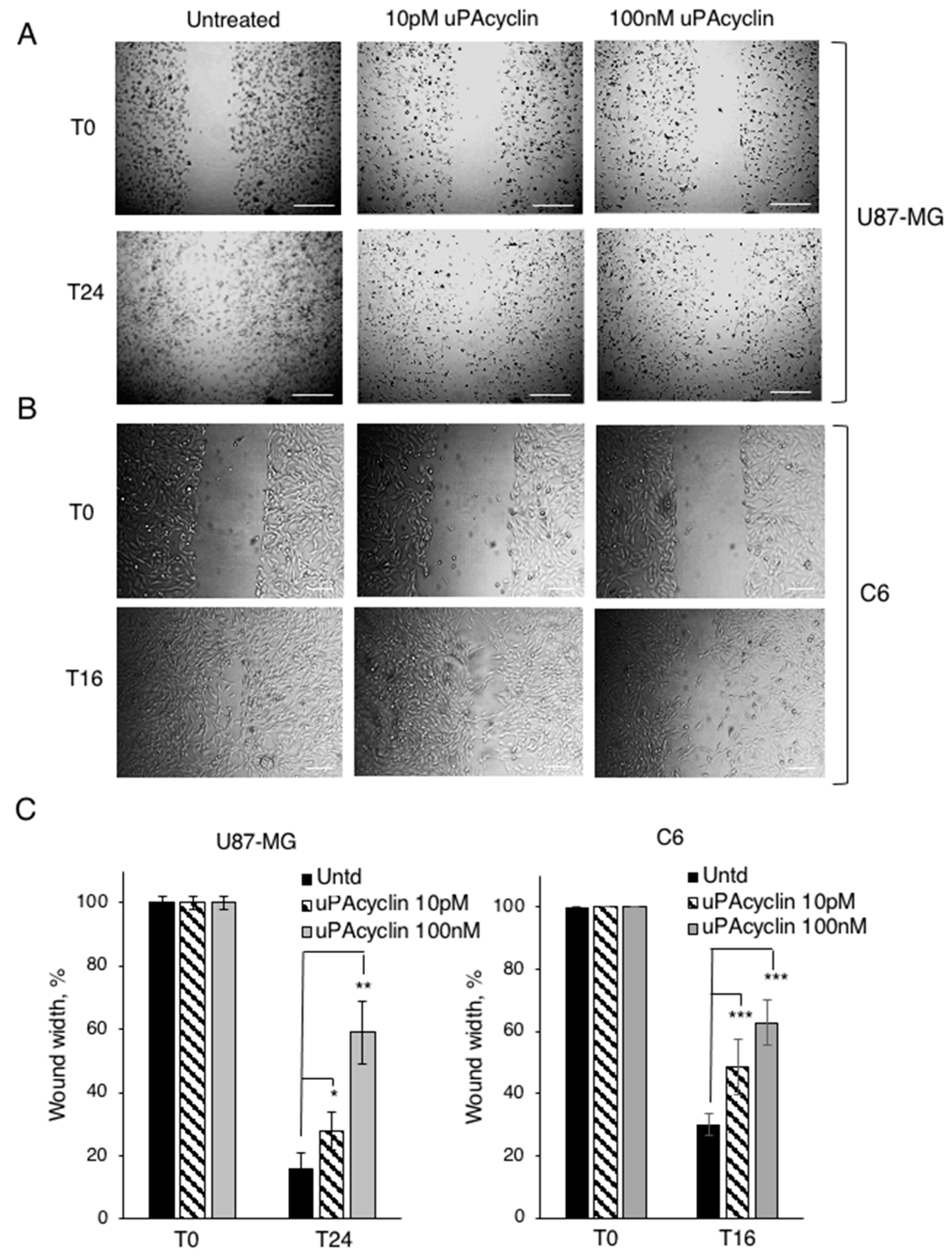
3.4. Effect of uPAcyclin in Wound Healing Assays of U87-MG and C6 Cells
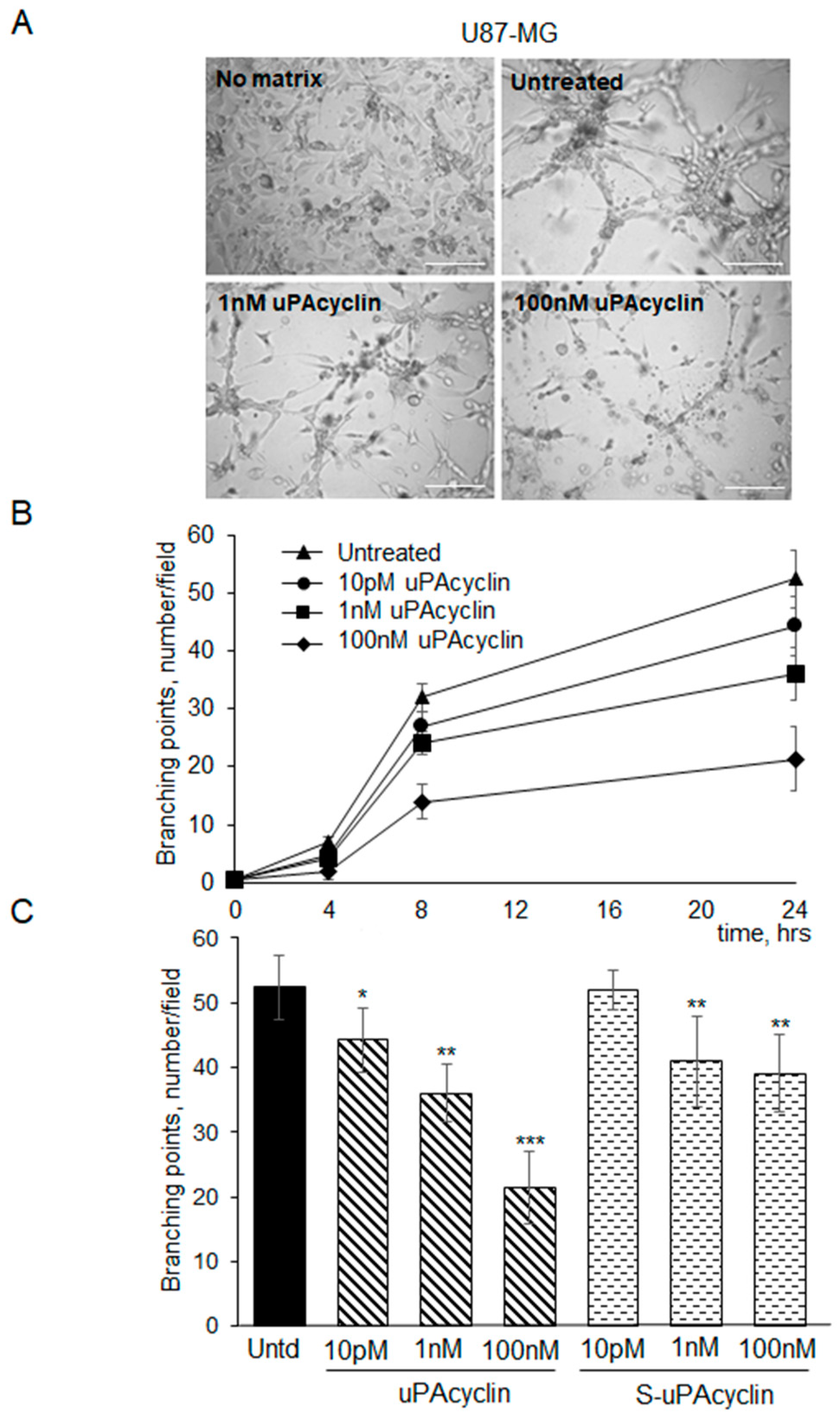
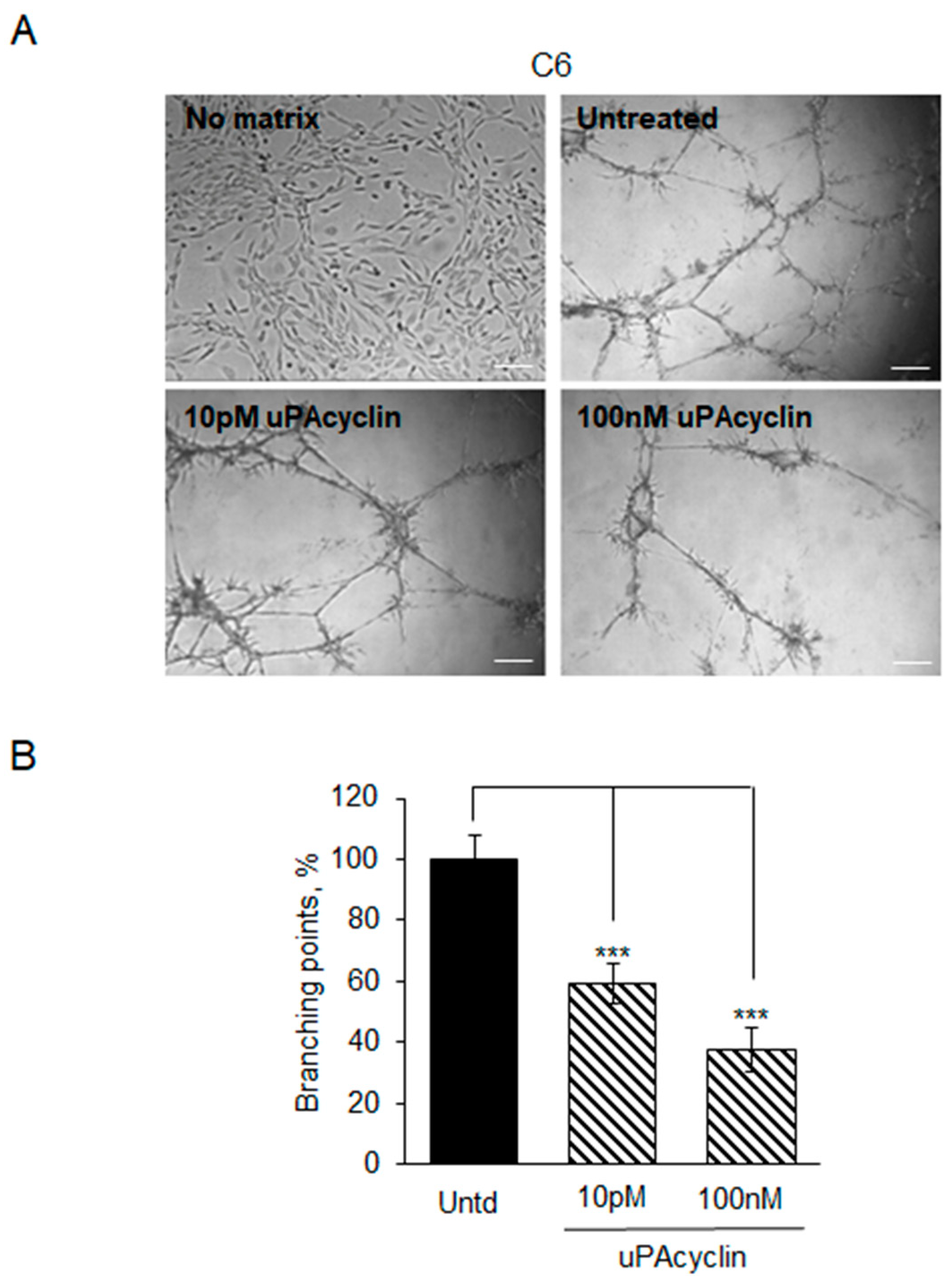
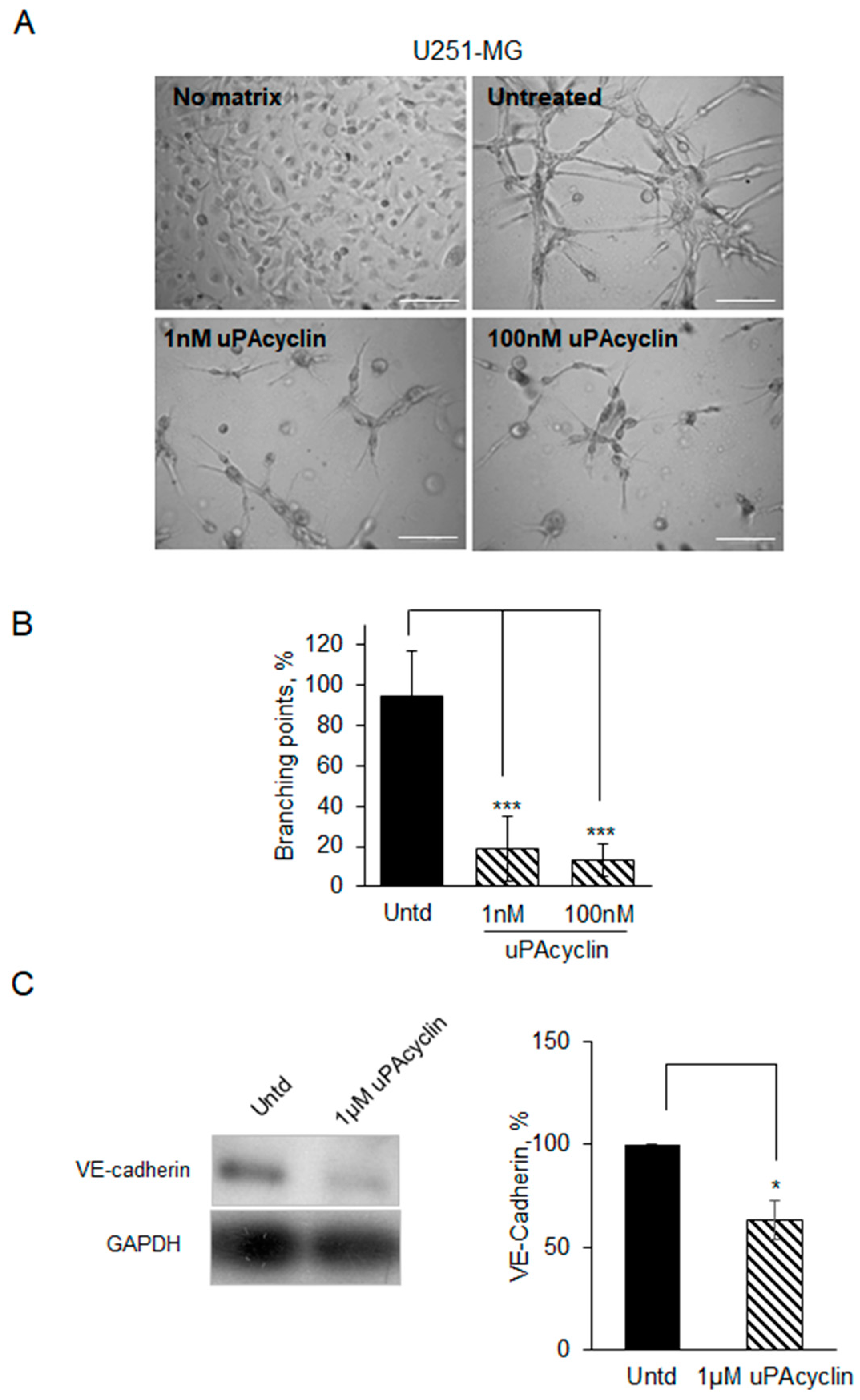
3.5. Vascular Mimicry Formation by U87-MG and U251-MG Glioblastoma Cell Lines
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Weller, M.; Wick, W.; Aldape, K.; Brada, M.; Berger, M.; Pfister, S.M.; Nishikawa, R.; Rosenthal, M.; Wen, P.Y.; Stupp, R.; et al. Glioma. Nat. Rev. Dis. Primers 2015, 1, 15040. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Perry, A.; Wesseling, P.; Brat, D.J.; Cree, I.A.; Figarella-Branger, D.; Hawkins, C.; Ng, H.K.; Pfister, S.M.; Reifenberger, G.; et al. The 2021 WHO Classification of Tumors of the Central Nervous System: A summary. Neuro Oncol. 2021, 23, 1231–1251. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.; Gueble, S.E.; Sundaram, R.K.; Huseman, E.D.; Bindra, R.S.; Herzon, S.B. Mechanism-based design of agents that selectively target drug-resistant glioma. Science 2022, 377, 502–511. [Google Scholar] [CrossRef]
- Silver, A.; Feier, D.; Ghosh, T.; Rahman, M.; Huang, J.; Sarkisian, M.R.; Deleyrolle, L.P. Heterogeneity of glioblastoma stem cells in the context of the immune microenvironment and geospatial organization. Front. Oncol. 2022, 12, 1022716. [Google Scholar] [CrossRef]
- Krishna Priya, S.; Nagare, R.P.; Sneha, V.S.; Sidhanth, C.; Bindhya, S.; Manasa, P.; Ganesan, T.S. Tumour angiogenesis-Origin of blood vessels. Int. J. Cancer 2016, 139, 729–735. [Google Scholar] [CrossRef]
- Döme, B.; Paku, S.; Somlai, B.; Tímár, J. Vascularization of cutaneous melanoma involves vessel co-option and has clinical significance. J. Pathol. 2002, 197, 355–362. [Google Scholar] [CrossRef]
- Patan, S.; Munn, L.L.; Jain, R.K. Intussusceptive microvascular growth in a human colon adenocarcinoma xenograft: A novel mechanism of tumor angiogenesis. Microvasc. Res. 1996, 51, 260–272. [Google Scholar] [CrossRef]
- El Hallani, S.; Boisselier, B.; Peglion, F.; Rousseau, A.; Colin, C.; Idbaih, A.; Marie, Y.; Mokhtari, K.; Thomas, J.L.; Eichmann, A.; et al. A new alternative mechanism in glioblastoma vascularization: Tubular vasculogenic mimicry. Brain 2010, 133 Pt 4, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Maddison, K.; Bowden, N.A.; Graves, M.C.; Tooney, P.A. Characteristics of vasculogenic mimicry and tumour to endothelial transdifferentiation in human glioblastoma: A systematic review. BMC Cancer 2023, 23, 185. [Google Scholar] [CrossRef]
- Maniotis, A.J.; Folberg, R.; Hess, A.; Seftor, E.A.; Gardner, L.M.; Pe’er, J.; Trent, J.M.; Meltzer, P.S.; Hendrix, M.J. Vascular channel formation by human melanoma cells in vivo and in vitro: Vasculogenic mimicry. Am. J. Pathol. 1999, 155, 739–752. [Google Scholar] [CrossRef] [PubMed]
- Scully, S.; Francescone, R.; Faibish, M.; Bentley, B.; Taylor, S.L.; Oh, D.; Schapiro, R.; Moral, L.; Yan, W.; Shao, R. Transdifferentiation of glioblastoma stem-like cells into mural cells drives vasculogenic mimicry in glioblastomas. J. Neurosci. 2012, 32, 12950–12960. [Google Scholar] [CrossRef] [PubMed]
- Ricci-Vitiani, L.; Pallini, R.; Biffoni, M.; Todaro, M.; Invernici, G.; Cenci, T.; Maira, G.; Parati, E.A.; Stassi, G.; Larocca, L.M.; et al. Tumour vascularization via endothelial differentiation of glioblastoma stem-like cells. Nature 2010, 468, 824–828. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Huang, Z.; Zhou, W.; Wu, Q.; Donnola, S.; Liu, J.K.; Fang, X.; Sloan, A.E.; Mao, Y.; Lathia, J.D.; et al. Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell 2013, 153, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Kreisl, T.N.; Kim, L.; Moore, K.; Duic, P.; Royce, C.; Stroud, I.; Garren, N.; Mackey, M.; Butman, J.A.; Camphausen, K.; et al. Phase II trial of single-agent bevacizumab followed by bevacizumab plus irinotecan at tumor progression in recurrent glioblastoma. J. Clin. Oncol. 2009, 27, 740–745. [Google Scholar] [CrossRef]
- Fernández-Cortés, M.; Delgado-Bellido, D.; Oliver, F.J. Vasculogenic Mimicry: Become an Endothelial Cell “But Not So Much”. Front. Oncol. 2019, 9, 803. [Google Scholar] [CrossRef]
- Broekman, M.L.; Maas, S.L.N.; Abels, E.R.; Mempel, T.R.; Krichevsky, A.M.; Breakefield, X.O. Multidimensional communication in the microenvirons of glioblastoma. Nat. Rev. Neurol. 2018, 14, 482–495. [Google Scholar] [CrossRef]
- Seguin, L.; Desgrosellier, J.S.; Weis, S.M.; Cheresh, D.A. Integrins and cancer: Regulators of cancer stemness, metastasis, and drug resistance. Trends Cell Biol. 2015, 25, 234–240. [Google Scholar] [CrossRef]
- Vogetseder, A.; Thies, S.; Ingold, B.; Roth, P.; Weller, M.; Schraml, P.; Goodman, S.L.; Moch, H. αv-Integrin isoform expression in primary human tumors and brain metastases. Int. J. Cancer 2013, 133, 2362–2371. [Google Scholar] [CrossRef]
- Desgrosellier, J.S.; Cheresh, D.A. Integrins in cancer: Biological implications and therapeutic opportunities. Nat. Rev. Cancer 2010, 10, 9–22. [Google Scholar] [CrossRef]
- Che, P.; Yu, L.; Friedman, G.K.; Wang, M.; Ke, X.; Wang, H.; Zhang, W.; Nabors, B.; Ding, Q.; Han, X. Integrin αvβ3 Engagement Regulates Glucose Metabolism and Migration through Focal Adhesion Kinase (FAK) and Protein Arginine Methyltransferase 5 (PRMT5) in Glioblastoma Cells. Cancers 2021, 13, 1111. [Google Scholar] [CrossRef]
- Franovic, A.; Elliott, K.C.; Seguin, L.; Camargo, M.F.; Weis, S.M.; Cheresh, D.A. Glioblastomas require integrin αvβ3/PAK4 signaling to escape senescence. Cancer Res. 2015, 75, 4466–4473. [Google Scholar] [CrossRef] [PubMed]
- Virga, J.; Szivos, L.; Hortobágyi, T.; Chalsaraei, M.K.; Zahuczky, G.; Steiner, L.; Tóth, J.; Reményi-Puskár, J.; Bognár, L.; Klekner, A. Extracellular matrix differences in glioblastoma patients with different prognoses. Oncol. Lett. 2019, 17, 797–806. [Google Scholar] [CrossRef]
- Minucci, S.; Pelicci, P.G. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nat. Rev. Cancer 2006, 6, 38–51. [Google Scholar] [CrossRef] [PubMed]
- Pastorino, O.; Gentile, M.T.; Mancini, A.; Del Gaudio, N.; Di Costanzo, A.; Bajetto, A.; Franco, P.; Altucci, L.; Florio, T.; Stoppelli, M.P.; et al. Histone Deacetylase Inhibitors Impair Vasculogenic Mimicry from Glioblastoma Cells. Cancers 2019, 11, 747. [Google Scholar] [CrossRef] [PubMed]
- Belli, S.; Franco, P.; Iommelli, F.; De Vincenzo, A.; Brancaccio, D.; Telesca, M.; Merlino, F.; Novellino, E.; Ranson, M.; Del Vecchio, S.; et al. Breast Tumor Cell Invasion and Pro-Invasive Activity of Cancer-Associated Fibroblasts Co-Targeted by Novel Urokinase-Derived Decapeptides. Cancers 2020, 12, 2404. [Google Scholar] [CrossRef]
- Franco, P.; Vocca, I.; Carriero, M.V.; Alfano, D.; Cito, L.; Longanesi-Cattani, I.; Grieco, P.; Ossowski, L.; Stoppelli, M.P. Activation of urokinase receptor by a novel interaction between the connecting peptide region of urokinase and alpha v beta 5 integrin. J. Cell Sci. 2006, 119 Pt 16, 3424–3434. [Google Scholar] [CrossRef]
- Franco, P.; Iaccarino, C.; Chiaradonna, F.; Brandazza, A.; Iavarone, C.; Mastronicola, M.R.; Nolli, M.L.; Stoppelli, M.P. Phosphorylation of human pro-urokinase on Ser138/303 impairs its receptor-dependent ability to promote myelomonocytic adherence and motility. J. Cell Biol. 1997, 137, 779–791. [Google Scholar] [CrossRef]
- Vocca, I.; Franco, P.; Alfano, D.; Votta, G.; Carriero, M.V.; Estrada, Y.; Caputi, M.; Netti, P.A.; Ossowski, L.; Stoppelli, M.P. Inhibition of migration and invasion of carcinoma cells by urokinase-derived antagonists of alphavbeta5 integrin activation. Int. J. Cancer 2009, 124, 316–325. [Google Scholar] [CrossRef]
- Franco, P.; Carotenuto, A.; Marcozzi, C.; Votta, G.; Sarno, C.; Iaccarino, I.; Brancaccio, D.; De Vincenzo, A.; Novellino, E.; Grieco, P.; et al. Opposite modulation of cell migration by distinct subregions of urokinase connecting peptide. Chembiochem 2013, 14, 882–889. [Google Scholar] [CrossRef]
- Stoppelli, M.P.; Franco, P.; Iommelli, F.; Carotenuto, A.; Grieco, P.; Novellino, E. Novel Peptides and Peptidomimetics as Potent Targeted Agents for Prevention and Treatment of Tumor Invasion and Metastasis. Italy Patent 102018000010511, 22 November 2018. [Google Scholar]
- Jullian, M.; Hernandez, A.; Maurras, A.; Puget, K.; Amblard, M.; Martinez, J.; Subra, G. N-terminus FITC labeling of peptides on solid support: The truth behind the spacer. Tetrahedron Lett. 2009, 50, 260–263. [Google Scholar] [CrossRef]
- Piantini, U.; Sorensen, O.W.; Ernst, R.R. Multiple quantum filters for elucidating NMR coupling network. J. Am. Chem. Soc. 1982, 104, 6800–6801. [Google Scholar] [CrossRef]
- Marion, D.; Wüthrich, K. Application of phase sensitive two-dimensional correlated spectroscopy (COSY) for measurements of 1H-1H spin-spin coupling constants in proteins. Biochem. Biophys. Res. Commun. 1983, 113, 967–974. [Google Scholar] [CrossRef] [PubMed]
- Braunschweiler, L.; Ernst, R.R. Coherence transfer by isotropic mixing: Application to proton correlation spectroscopy. J. Magn. Reson. 1983, 53, 521–528. [Google Scholar] [CrossRef]
- Jenner, J.; Meyer, B.H.; Bachman, P.; Ernst, R.R. Investigation of exchange processes by two-dimensional NMR spectroscopy. J. Chem. Phys. 1979, 71, 4546–4553. [Google Scholar] [CrossRef]
- States, D.J.; Haberkorn, R.A.; Ruben, D.J. A Two-dimensional nuclear Overhauser experiment with pure absorption phase in four quadrants. J. Magn. Reson. 1982, 48, 286–292. [Google Scholar] [CrossRef]
- Bartels, C.; Xia, T.H.; Billeter, M.; Güntert, P.; Wüthrich, K. The program XEASY for computer-supported NMR spectral analysis of biological macromolecules. J. Biomol. NMR 1995, 6, 1–10. [Google Scholar] [CrossRef]
- Wüthrich, K. NMR of Proteins and Nucleic Acids; John Wiley & Sons: New York, NY, USA, 1986; pp. 11–13. [Google Scholar]
- Güntert, P.; Buchner, L. Combined automated NOE assignment and structure calculation with CYANA. J. Biomol. NMR 2015, 62, 453–471. [Google Scholar] [CrossRef]
- Maple, J.R.; Dinur, U.; Hagler, A.T. Derivation of force fields for molecular mechanics and dynamics from ab initio energy surfaces. Proc. Natl. Acad. Sci. USA 1988, 85, 5350–5354. [Google Scholar] [CrossRef]
- Stoppelli, M.P.; Tacchetti, C.; Cubellis, M.V.; Corti, A.; Hearing, V.J.; Cassani, G.; Appella, E.; Blasi, F. Autocrine saturation of pro-urokinase receptors on human A431 cells. Cell 1986, 45, 675–684. [Google Scholar] [CrossRef]
- De Vincenzo, A.; Belli, S.; Franco, P.; Telesca, M.; Iaccarino, I.; Botti, G.; Carriero, M.V.; Ranson, M.; Stoppelli, M.P. Paracrine recruitment and activation of fibroblasts by c-Myc expressing breast epithelial cells through the IGFs/IGF-1R axis. Int. J. Cancer 2019, 145, 2827–2839. [Google Scholar] [CrossRef]
- Gentile, M.T.; Russo, R.; Pastorino, O.; Cioffi, S.; Barbieri, F.; Illingworth, E.A.; Grieco, M.; Chambery, A.; Colucci-D’Amato, L. Ruta graveolens water extract inhibits cell-cell network formation in human umbilical endothelial cells via MEK-ERK1/2 pathway. Exp. Cell Res. 2018, 364, 50–58. [Google Scholar] [CrossRef] [PubMed]
- Gentile, M.T.; Pastorino, O.; Bifulco, M.; Colucci-D’Amato, L. HUVEC Tube-formation Assay to Evaluate the Impact of Natural Products on Angiogenesis. J. Vis. Exp. 2019, 148, e58591. [Google Scholar] [CrossRef]
- Xie, M.; Liu, D.; Yang, Y. Anti-cancer peptides: Classification, mechanism of action, reconstruction and modification. Open Biol. 2020, 10, 200004. [Google Scholar] [CrossRef] [PubMed]
- Vadevoo, S.M.P.; Gurung, S.; Lee, H.S.; Gunassekaran, G.R.; Lee, S.M.; Yoon, J.W.; Lee, Y.K.; Lee, B. Peptides as multifunctional players in cancer therapy. Exp. Mol. Med. 2023, 55, 1099–1109. [Google Scholar] [CrossRef]
- Schnell, O.; Krebs, B.; Wagner, E.; Romagna, A.; Beer, A.J.; Grau, S.J.; Thon, N.; Goetz, C.; Kretzschmar, H.A.; Tonn, J.C.; et al. Expression of integrin alphavbeta3 in gliomas correlates with tumor grade and is not restricted to tumor vasculature. Brain Pathol. 2008, 18, 378–386. [Google Scholar] [CrossRef]
- Ghochani, Y.; Muthukrishnan, S.D.; Sohrabi, A.; Kawaguchi, R.; Condro, M.C.; Bastola, S.; Gao, F.; Qin, Y.; Mottahedeh, J.; Iruela-Arispe, M.L.; et al. A molecular interactome of the glioblastoma perivascular niche reveals integrin binding sialoprotein as a mediator of tumor cell migration. Cell Rep. 2022, 41, 111511. [Google Scholar] [CrossRef]
- Sung, V.; Stubbs, J.T., 3rd; Fisher, L.; Aaron, A.D.; Thompson, E.W. Bone sialoprotein supports breast cancer cell adhesion proliferation and migration through differential usage of the alpha(v)beta3 and alpha(v)beta5 integrins. J. Cell Physiol. 1998, 176, 482–494. [Google Scholar] [CrossRef]
- Cobb, D.A.; de Rossi, J.; Liu, L.; An, E.; Lee, D.W. Targeting of the alphav beta3 integrin complex by CAR-T cells leads to rapid regression of diffuse intrinsic pontine glioma and glioblastoma. J. Immunother. Cancer 2022, 10, e003816. [Google Scholar] [CrossRef]
- Weller, M.; Nabors, L.B.; Gorlia, T.; Leske, H.; Rushing, E.; Bady, P.; Hicking, C.; Perry, J.; Hong, Y.K.; Roth, P.; et al. Cilengitide in newly diagnosed glioblastoma: Biomarker expression and outcome. Oncotarget 2016, 7, 15018–15032. [Google Scholar] [CrossRef]
- Reardon, D.A.; Cheresh, D. Cilengitide: A prototypic integrin inhibitor for the treatment of glioblastoma and other malignancies. Genes Cancer 2011, 2, 1159–1165. [Google Scholar] [CrossRef]
- Stupp, R.; Hegi, M.E.; Gorlia, T.; Erridge, S.C.; Perry, J.; Hong, Y.K.; Aldape, K.D.; Lhermitte, B.; Pietsch, T.; Grujicic, D.; et al. Cilengitide combined with standard treatment for patients with newly diagnosed glioblastoma with methylated MGMT promoter (CENTRIC EORTC 26071-22072 study): A multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2014, 15, 1100–1108. [Google Scholar] [CrossRef] [PubMed]
- Tucci, M.; Stucci, S.; Silvestris, F. Does cilengitide deserve another chance? Lancet Oncol. 2014, 15, e584–e585. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Wang, X.; Li, B.; Hou, Y.; Yang, J.; Yi, L. Development of a novel morphological paclitaxel-loaded PLGA microspheres for effective cancer therapy: In vitro and in vivo evaluations. Drug Deliv. 2018, 25, 166–177. [Google Scholar] [CrossRef]
- Xin, H.; Sha, X.; Jiang, X.; Zhang, W.; Chen, L.; Fang, X. Anti-glioblastoma efficacy and safety of paclitaxel-loading Angiopep-conjugated dual targeting PEG-PCL nanoparticles. Biomaterials 2012, 33, 8167–8176. [Google Scholar] [CrossRef]
- Bhat, K.P.L.; Balasubramaniyan, V.; Vaillant, B.; Ezhilarasan, R.; Hummelink, K.; Hollingsworth, F.; Wani, K.; Heathcock, L.; James, J.D.; Goodman, L.D.; et al. Mesenchymal differentiation mediated by NF-κB promotes radiation resistance in glioblastoma. Cancer Cell. 2013, 24, 331–346. [Google Scholar] [CrossRef]
- Friedmann-Morvinski, D.; Narasimamurthy, R.; Xia, Y.; Myskiw, C.; Soda, Y.; Verma, I.M. Targeting NF-κB in glioblastoma: A therapeutic approach. Sci. Adv. 2016, 2, e1501292. [Google Scholar] [CrossRef]
- Severino, V.; Farina, A.; Colucci-D’Amato, L.; Reccia, M.G.; Volpicelli, F.; Parente, A.; Chambery, A. Secretome profiling of differentiated neural mes-c-myc A1 cell line endowed with stem cell properties. Biochim. Biophys. Acta 2013, 1834, 2385–2395. [Google Scholar] [CrossRef]
- Colucci-D’Amato, L.; Farina, A.; Vissers, J.P.; Chambery, A. Quantitative neuroproteomics: Classical and novel tools for studying neural differentiation and function. Stem Cell Rev. Rep. 2011, 7, 77–93. [Google Scholar] [CrossRef]
- Colucci-D’Amato, L.; Cicatiello, A.E.; Reccia, M.G.; Volpicelli, F.; Severino, V.; Russo, R.; Sandomenico, A.; Doti, N.; D’Esposito, V.; Formisano, P.; et al. A targeted secretome profiling by multiplexed immunoassay revealed that secreted chemokine ligand 2 (MCP-1/CCL2) affects neural differentiation in mesencephalic neural progenitor cells. Proteomics 2015, 15, 714–724. [Google Scholar] [CrossRef]
- Ruoslahti, E. RGD and other recognition sequences for integrins. Annu. Rev. Cell Dev. Biol. 1996, 12, 697–715. [Google Scholar] [CrossRef]
- Kapp, T.G.; Rechenmacher, F.; Neubauer, S.; Maltsev, O.V.; Cavalcanti-Adam, E.A.; Zarka, R.; Reuning, U.; Notni, J.; Wester, H.J.; Mas-Moruno, C.; et al. A Comprehensive Evaluation of the Activity and Selectivity Profile of Ligands for RGD-binding Integrins. Sci. Rep. 2017, 7, 39805. [Google Scholar] [CrossRef] [PubMed]
- Van Agthoven, J.F.; Xiong, J.P.; Alonso, J.L.; Rui, X.; Adair, B.D.; Goodman, S.L.; Arnaout, M.A. Structural basis for pure antagonism of integrin αVβ3 by a high-affinity form of fibronectin. Nat. Struct. Mol. Biol. 2014, 21, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Mazar, A.P.; Lebrun, J.; Rabbani, S.A. An antiangiogenic urokinase-derived pepide combined with tamoxifen decreases tumor growth and metastasis in a syngeneic model of breast cancer. Cancer Res. 2002, 62, 4678–4684. [Google Scholar]
- Ghamande, S.A.; Silverman, M.H.; Huh, W.; Behbakht, K.; Ball, G.; Cuasay, L.; Würtz, S.O.; Brunner, N.; Gold, M.A. A phase 2, randomized, double-blind, placebo-controlled trial of clinical activity and safety of subcutaneous Å6 in women with asymptomatic CA125 progression after first-line chemotherapy of epithelial ovarian cancer. Gynecol. Oncol. 2008, 111, 89–94. [Google Scholar] [CrossRef] [PubMed]
- Gold, M.A.; Brady, W.E.; Lankes, H.A.; Rose, P.G.; Kelley, J.L.; De Geest, K.; Crispens, M.A.; Resnick, K.E.; Howell, S.B. A phase II study of a urokinase-derived peptide (Å6) in the treatment of persistent or recurrent epithelial ovarian, fallopian tube, or primary peritoneal carcinomas: A Gynecologic Oncology Group study. Gynecol. Oncol. 2012, 125, 635–639. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.M.; Zhang, Q.P.; Mu, Y.G.; Zhang, X.H.; Sai, K.; Pang, J.C.; Ng, H.K.; Chen, Z.P. Clinical significance of vasculogenic mimicry in human gliomas. J. Neurooncol. 2011, 105, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Cai, H.; Liu, W.; Liu, X.; Li, Z.; Feng, T.; Xue, Y.; Liu, Y. Advances and Prospects of Vasculogenic Mimicry in Glioma: A Potential New Therapeutic Target? OncoTargets Ther. 2020, 13, 4473–4483. [Google Scholar] [CrossRef]
- Koh, I.; Cha, J.; Park, J.; Choi, J.; Kang, S.G.; Kim, P. The mode and dynamics of glioblastoma cell invasion into a decellularized tissue-derived extracellular matrix-based three-dimensional tumor model. Sci. Rep. 2018, 8, 4608. [Google Scholar] [CrossRef]
- Zhao, Z.; Ukidve, A.; Kim, J.; Mitragotri, S. Targeting Strategies for Tissue-Specific Drug Delivery. Cell 2020, 181, 151–167. [Google Scholar] [CrossRef] [PubMed]
- Graillon, T.; Sanson, M.; Campello, C.; Idbaih, A.; Peyre, M.; Peyrière, H.; Basset, N.; Autran, D.; Roche, C.; Kalamarides, M.; et al. Everolimus and Octreotide for Patients with Recurrent Meningioma: Results from the Phase II CEVOREM Trial. Clin. Cancer Res. 2020, 26, 552–557. [Google Scholar] [CrossRef]
- Van Solinge, T.S.; Nieland, L.; Chiocca, E.A.; Broekman, M.L.D. Advances in local therapy for glioblastoma—Taking the fight to the tumour. Nat. Rev. Neurol. 2022, 18, 221–236. [Google Scholar] [CrossRef] [PubMed]








Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Franco, P.; Camerino, I.; Merlino, F.; D’Angelo, M.; Cimmino, A.; Carotenuto, A.; Colucci-D’Amato, L.; Stoppelli, M.P. αV-Integrin-Dependent Inhibition of Glioblastoma Cell Migration, Invasion and Vasculogenic Mimicry by the uPAcyclin Decapeptide. Cancers 2023, 15, 4775. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers15194775
Franco P, Camerino I, Merlino F, D’Angelo M, Cimmino A, Carotenuto A, Colucci-D’Amato L, Stoppelli MP. αV-Integrin-Dependent Inhibition of Glioblastoma Cell Migration, Invasion and Vasculogenic Mimicry by the uPAcyclin Decapeptide. Cancers. 2023; 15(19):4775. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers15194775
Chicago/Turabian StyleFranco, Paola, Iolanda Camerino, Francesco Merlino, Margherita D’Angelo, Amelia Cimmino, Alfonso Carotenuto, Luca Colucci-D’Amato, and Maria Patrizia Stoppelli. 2023. "αV-Integrin-Dependent Inhibition of Glioblastoma Cell Migration, Invasion and Vasculogenic Mimicry by the uPAcyclin Decapeptide" Cancers 15, no. 19: 4775. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers15194775