
The Acquisition of Complement-Dependent Cytotoxicity by the Type II Anti-CD20 Therapeutic Antibody Obinutuzumab
 , , ,
, , , 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Cell Lines and Fresh Cultures of Chronic Lymphocytic Leukemia (CLL)
2.2. GoF Variant of Complement C2 Protein
2.3. CDC and Classical Complement Convertase Assays
2.4. XTT Assay
2.5. Whole Blood CDC Assay
3. Results
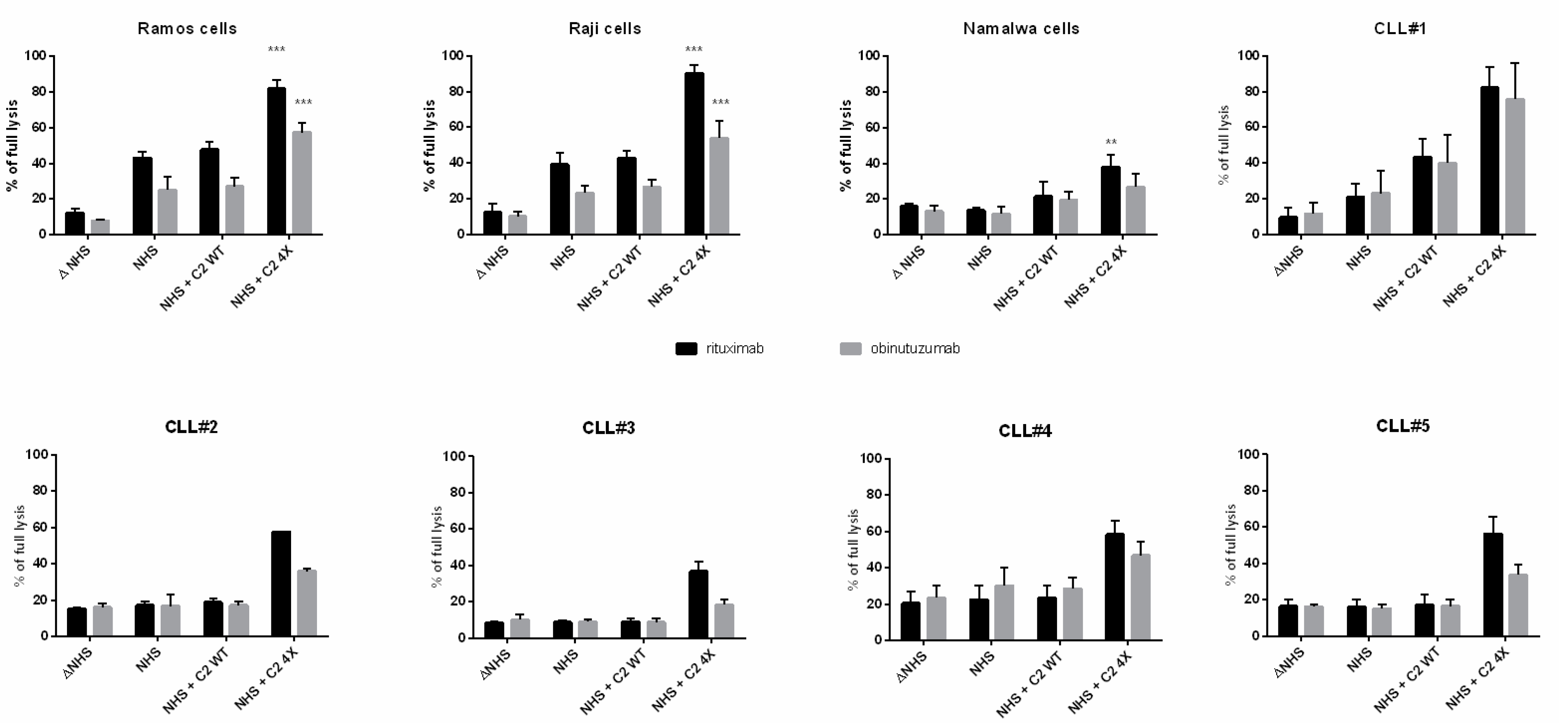
3.1. CDC Assays
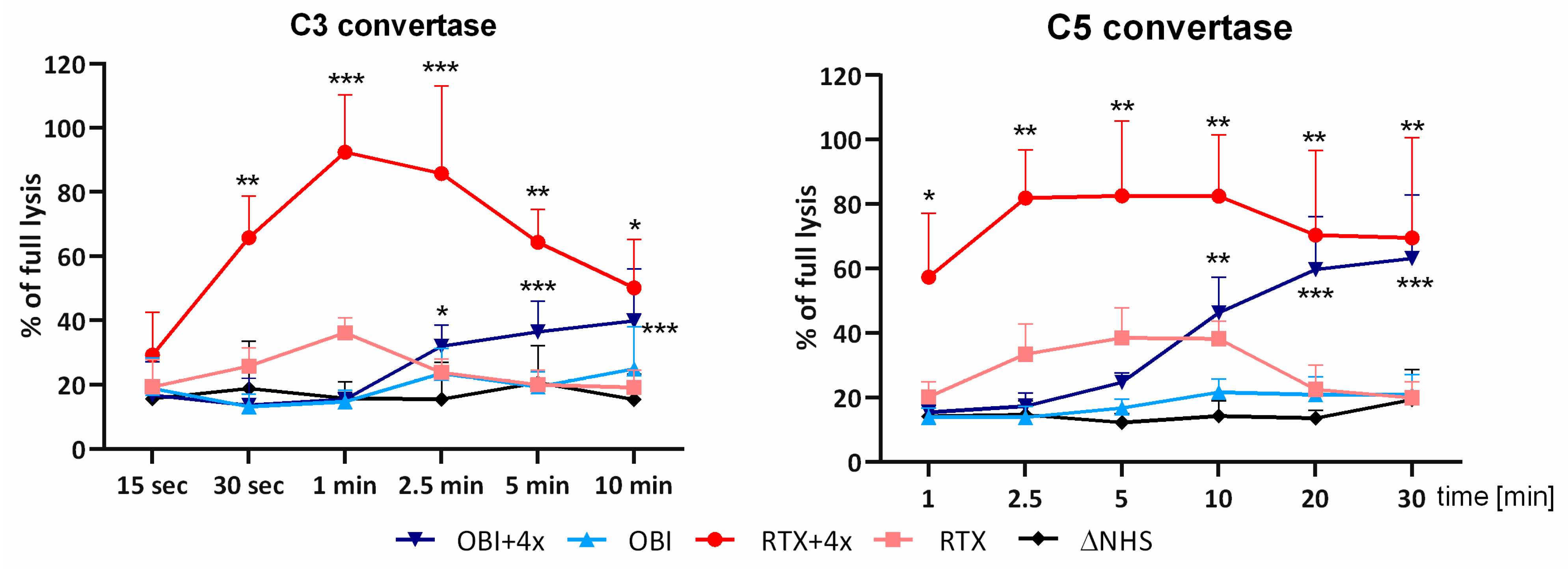
3.2. Convertase Assays
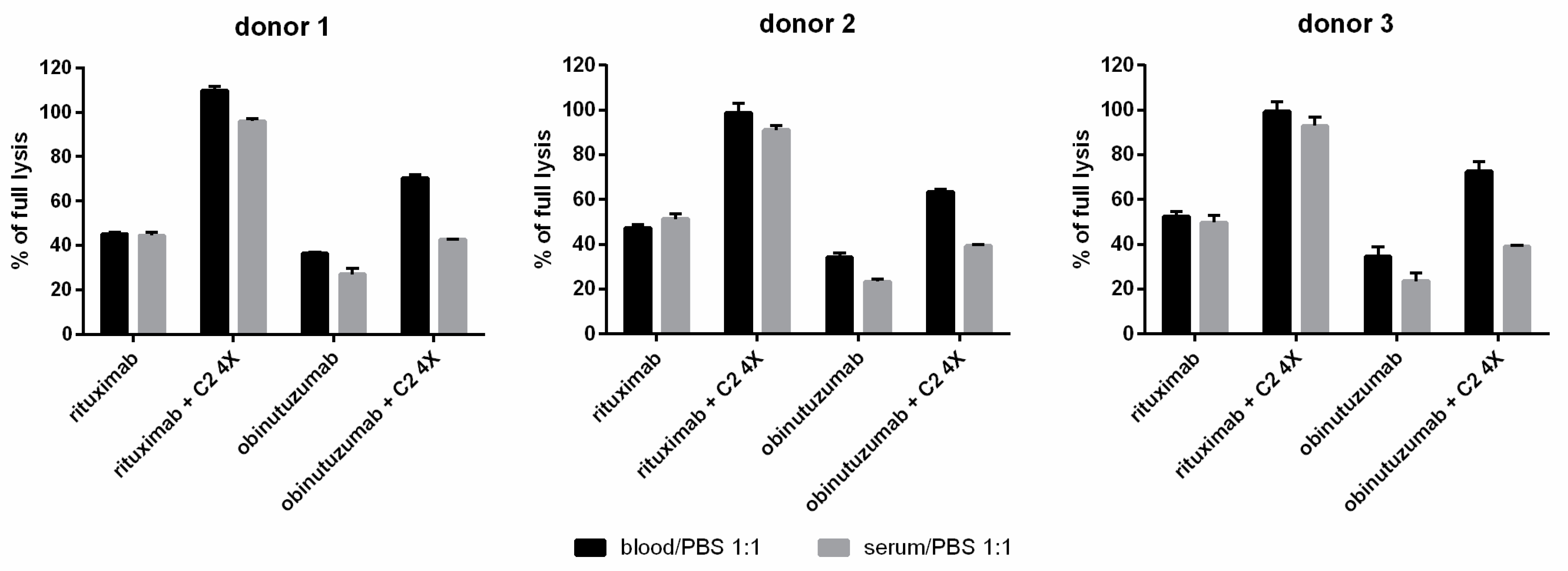
3.3. Whole Blood CDC Asssys
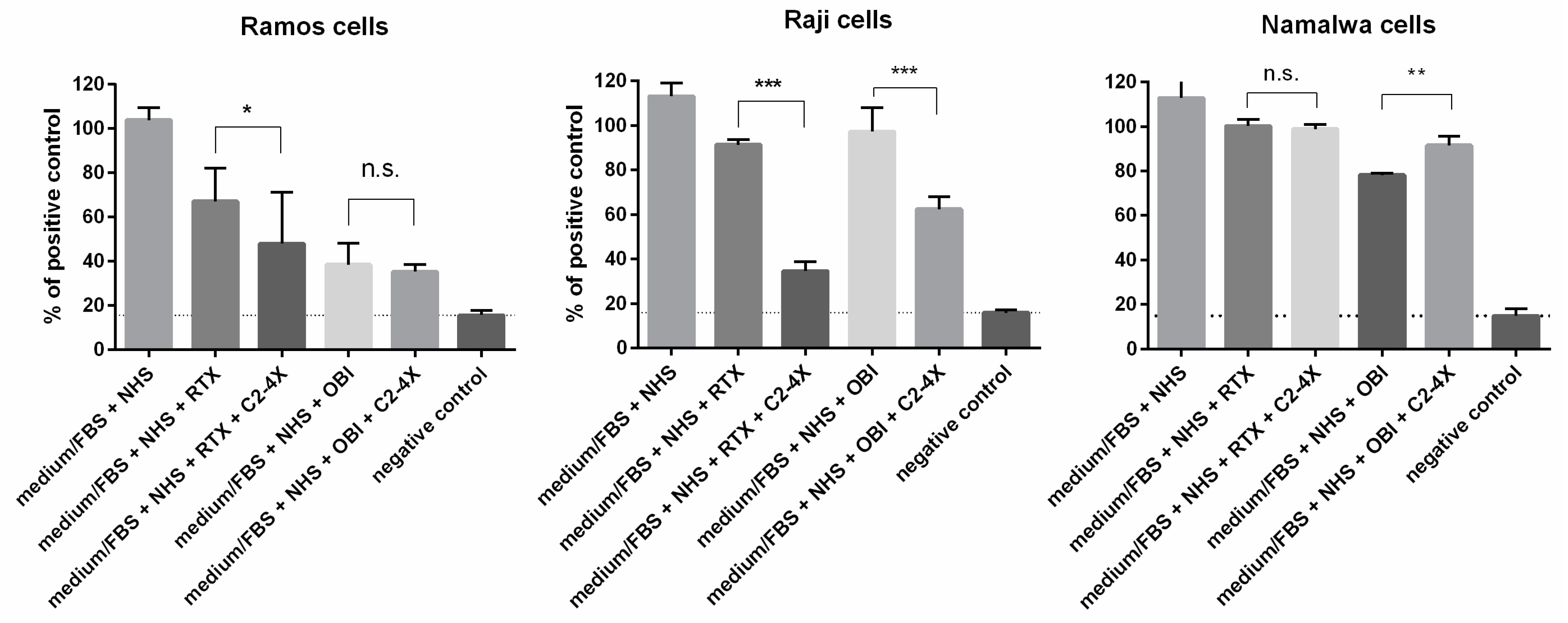
3.4. Long-Term Cytotoxicity
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Murawski, N.; Pfreundschuh, M. New drugs for aggressive B-cell and T-cell lymphomas. Lancet Oncol. 2010, 11, 1074–1085. [Google Scholar] [CrossRef]
- Maloney, D.G.; Grillo-López, A.J.; White, C.A.; Bodkin, D.; Schilder, R.J.; Neidhart, J.A.; Janakiraman, N.; Foon, K.A.; Liles, T.-M.; Dallaire, B.-K.; et al. IDEC-C2B8 (Rituximab) anti-CD20 monoclonal antibody therapy in patients with relapsed low-grade non-Hodgkin’s lymphoma. Blood 1997, 90, 2188–2195. [Google Scholar] [CrossRef] [PubMed]
- Delate, T.; Hansen, M.L.; Gutierrez, A.C.; Le, K.N. Indications for Rituximab Use in an Integrated Health Care Delivery System. J. Manag. Care Spec. Pharm. 2020, 26, 832–838. [Google Scholar] [CrossRef] [PubMed]
- Winiarska, M.; Glodkowska-Mrowka, E.; Bil, J.; Golab, J. Molecular mechanisms of the antitumor effects of anti-CD20 antibodies. Front. Biosci. 2011, 16, 277–306. [Google Scholar] [CrossRef] [PubMed]
- Okroj, M.; Österborg, A.; Blom, A.M. Effector mechanisms of anti-CD20 monoclonal antibodies in B cell malignancies. Cancer Treat. Rev. 2013, 39, 632–639. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Planchais, C.; Fronzes, R.; Mouquet, H.; Reyes, N. Binding mechanisms of therapeutic antibodies to human CD20. Science 2020, 369, 793–799. [Google Scholar] [CrossRef]
- Oostindie, S.C.; Van Der Horst, H.J.; Kil, L.P.; Strumane, K.; Overdijk, M.B.; van den Brink, E.N.; van den Brakel, J.H.N.; Rademaker, H.J.; Van Kessel, B.; van den Noort, J.; et al. DuoHexaBody-CD37®, a novel biparatopic CD37 antibody with enhanced Fc-mediated hexamerization as a potential therapy for B-cell malignancies. Blood Cancer J. 2020, 10, 30. [Google Scholar] [CrossRef]
- Urban, A.; Majeranowski, A.; Stasiłojć, G.; Koszałka, P.; Felberg, A.; Taszner, M.; Zaucha, J.M.; Okrój, M. In Silico Designed Gain-of-Function Variants of Complement C2 Support Cytocidal Activity of Anticancer Monoclonal Antibodies. Cancers 2022, 14, 1270. [Google Scholar] [CrossRef]
- Okroj, M.; Eriksson, I.; Österborg, A.; Blom, A.M. Killing of CLL and NHL cells by rituximab and ofatumumab under limited availability of complement. Med Oncol. 2013, 30, 759. [Google Scholar] [CrossRef]
- Stasiłojć, G.; Felberg, A.; Urban, A.; Kowalska, D.; Ma, S.; Blom, A.M.; Lundin, J.; Österborg, A.; Okrój, M. Calcein release assay as a method for monitoring serum complement activity during monoclonal antibody therapy in patients with B-cell malignancies. J. Immunol. Methods 2020, 476, 112675. [Google Scholar] [CrossRef]
- Mollnes, T.E.; Brekke, O.-L.; Fung, M.; Fure, H.; Christiansen, D.; Bergseth, G.; Videm, V.; Lappegård, K.T.; Köhl, J.; Lambris, J.D. Essential role of the C5a receptor in E coli-induced oxidative burst and phagocytosis revealed by a novel lepirudin-based human whole blood model of inflammation. Blood 2002, 100, 1869–1877. [Google Scholar] [PubMed]
- Blom, A.M.; Volokhina, E.B.; Fransson, V.; Strömberg, P.; Berghard, L.; Viktorelius, M.; Mollnes, T.E.; López-Trascasa, M.; van Den Heuvel, L.P.; Goodship, T.H.; et al. A novel method for direct measurement of complement convertases activity in human serum. Clin. Exp. Immunol. 2014, 178, 142–153. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.Y.; Veeramani, S.; Racila, E.; Cagley, J.; Fritzinger, D.C.; Vogel, C.W.; St John, W.; Weiner, G.J. Depletion of the C3 component of complement enhances the ability of rituximab-coated target cells to activate human NK cells and improves the efficacy of monoclonal antibody therapy in an in vivo model. Blood 2009, 114, 5322–5330. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.Y.; Racila, E.; Taylor, R.P.; Weiner, G.J. NK-cell activation and antibody-dependent cellular cytotoxicity induced by rituximab-coated target cells is inhibited by the C3b component of complement. Blood 2008, 111, 1456–1463. [Google Scholar] [CrossRef] [PubMed]
- van Meerten, T.; van Rijn, R.S.; Hol, S.; Hagenbeek, A.; Ebeling, S.B. Complement-Induced Cell Death by Rituximab Depends on CD20 Expression Level and Acts Complementary to Antibody-Dependent Cellular Cytotoxicity. Clin. Cancer Res. 2006, 12, 4027–4035. [Google Scholar] [CrossRef] [PubMed]
- Merle, N.S.; Church, S.E.; Fremeaux-Bacchi, V.; Roumenina, L.T. Complement System Part I—Molecular Mechanisms of Activation and Regulation. Front. Immunol. 2015, 6, 262. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez de Córdoba, S. Genetic variability shapes the alternative pathway complement activity and predisposition to complement-related diseases. Immunol. Rev. 2022, 313, 71–90. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, V.; Xu, Y.; Macon, K.; Volanakis, J.E.; Narayana, S.V.L. The structure of C2b, a fragment of complement component C2 produced during C3 convertase formation. Acta Crystallogr. Sect. D Biol. Crystallogr. 2009, 65 Pt 3, 266–274. [Google Scholar] [CrossRef]
- Mortensen, S.; Jensen, J.K.; Andersen, G.R. Solution Structures of Complement C2 and Its C4 Complexes Propose Pathway-specific Mechanisms for Control and Activation of the Complement Proconvertases. J. Biol. Chem. 2016, 291, 16494–16507. [Google Scholar] [CrossRef]
- Tobinai, K.; Klein, C.; Oya, N.; Fingerle-Rowson, G. A Review of Obinutuzumab (GA101), a Novel Type II Anti-CD20 Monoclonal Antibody, for the Treatment of Patients with B-Cell Malignancies. Adv. Ther. 2017, 34, 324–356. [Google Scholar] [CrossRef]
- Beum, P.V.; Lindorfer, M.A.; Beurskens, F.; Stukenberg, P.T.; Lokhorst, H.M.; Pawluczkowycz, A.W.; Parren, P.W.H.I.; van de Winkel, J.G.J.; Taylor, R.P. Complement Activation on B Lymphocytes Opsonized with Rituximab or Ofatumumab Produces Substantial Changes in Membrane Structure Preceding Cell Lysis. J. Immunol. 2008, 181, 822–832. [Google Scholar] [CrossRef] [PubMed]
- Stasiłojć, G.; Österborg, A.; Blom, A.M.; Okrój, M. New perspectives on complement mediated immunotherapy. Cancer Treat. Rev. 2016, 45, 68–75. [Google Scholar] [CrossRef] [PubMed]
- Hörl, S.; Bánki, Z.; Huber, G.; Ejaz, A.; Windisch, D.; Muellauer, B.; Willenbacher, E.; Steurer, M.; Stoiber, H. Reduction of complement factor H binding to CLL cells improves the induction of rituximab-mediated complement-dependent cytotoxicity. Leukemia 2013, 27, 2200–2208. [Google Scholar] [CrossRef] [PubMed]
- Gancz, D.; Fishelson, Z. Cancer resistance to complement-dependent cytotoxicity (CDC): Problem-oriented research and development. Mol. Immunol. 2009, 46, 2794–2800. [Google Scholar] [CrossRef] [PubMed]
- Li, B.; Zhao, L.; Guo, H.; Wang, C.; Zhang, X.; Wu, L.; Chen, L.; Tong, Q.; Qian, W.; Wang, H.; et al. Characterization of a rituximab variant with potent antitumor activity against rituximab-resistant B-cell lymphoma. Blood 2009, 114, 5007–5015. [Google Scholar] [CrossRef] [PubMed]
- Bornstein, G.G.; Quéva, C.; Tabrizi, M.; van Abbema, A.; Chavez, C.; Wang, P.; Foord, O.; Ahluwalia, K.; Laing, N.; Raja, S.; et al. Development of a new fully human anti-CD20 monoclonal antibody for the treatment of B-cell malignancies. Investig. New Drugs 2010, 28, 561–574. [Google Scholar] [CrossRef] [PubMed]
- Nishida, M.; Uematsu, N.; Kobayashi, H.; Matsunaga, Y.; Ishida, S.; Takata, M.; Niwa, O.; Padlan, E.A.; Newman, R. BM-ca is a newly defined type I/II anti-CD20 monoclonal antibody with unique biological properties. Int. J. Oncol. 2011, 38, 335–344. [Google Scholar]
- Stasiłojć, G.; Felberg, A.; Okrój, M. Parameters critical for the effector mechanism of anti-CD20 antibodies revisited. Br. J. Haematol. 2018, 180, 777–779. [Google Scholar] [CrossRef]
- Meyer, S.; Evers, M.; Jansen, J.H.M.; Buijs, J.; Broek, B.; Reitsma, S.E.; Moerer, P.; Amini, M.; Kretschmer, A.; ten Broeke, T.; et al. New insights in Type I and II CD20 antibody mechanisms-of-action with a panel of novel CD20 antibodies. Br. J. Haematol. 2018, 180, 808–820. [Google Scholar] [CrossRef]
- Liu, J.L.; Zabetakis, D.; Goldman, E.R.; Anderson, G.P. Selection and characterization of single domain antibodies against human CD20. Mol. Immunol. 2016, 78, 146–154. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuźniewska, A.; Majeranowski, A.; Henry, S.; Kowalska, D.; Stasiłojć, G.; Urban, A.; Zaucha, J.M.; Okrój, M. The Acquisition of Complement-Dependent Cytotoxicity by the Type II Anti-CD20 Therapeutic Antibody Obinutuzumab. Cancers 2024, 16, 49. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers16010049
Kuźniewska A, Majeranowski A, Henry S, Kowalska D, Stasiłojć G, Urban A, Zaucha JM, Okrój M. The Acquisition of Complement-Dependent Cytotoxicity by the Type II Anti-CD20 Therapeutic Antibody Obinutuzumab. Cancers. 2024; 16(1):49. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers16010049
Chicago/Turabian StyleKuźniewska, Alicja, Alan Majeranowski, Sara Henry, Daria Kowalska, Grzegorz Stasiłojć, Aleksandra Urban, Jan M. Zaucha, and Marcin Okrój. 2024. "The Acquisition of Complement-Dependent Cytotoxicity by the Type II Anti-CD20 Therapeutic Antibody Obinutuzumab" Cancers 16, no. 1: 49. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers16010049