
Developmental Drift and the Role of Wnt Signaling in Aging


Abstract
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction: Wnt and Aging
2. Wnt, a Developmental Signaling Pathway
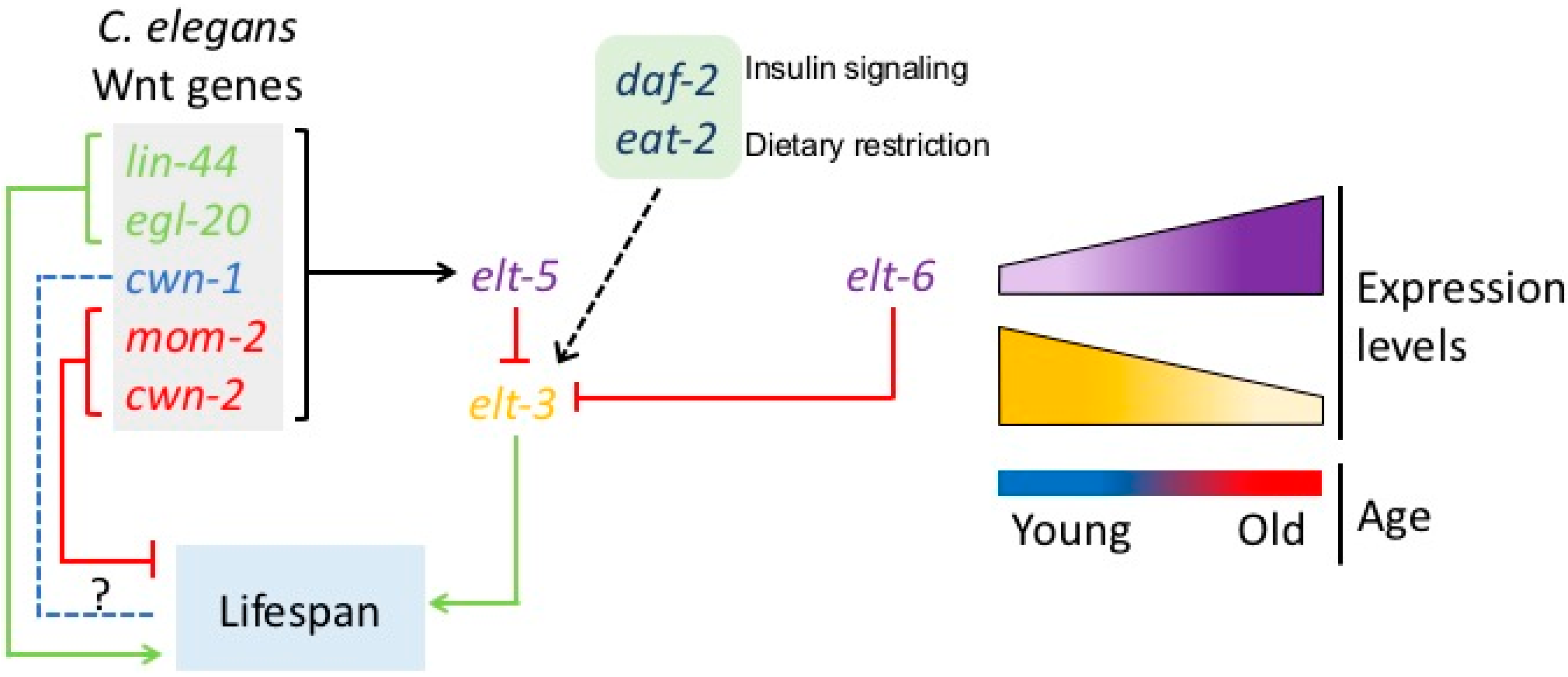
3. Developmental Drift Theory of Aging
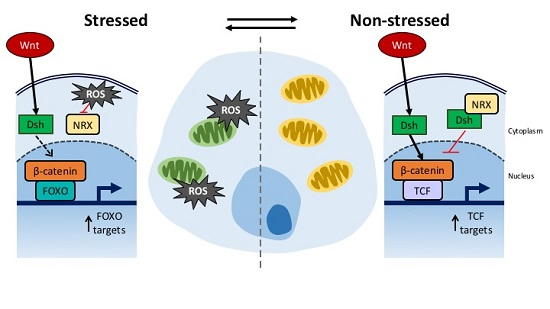
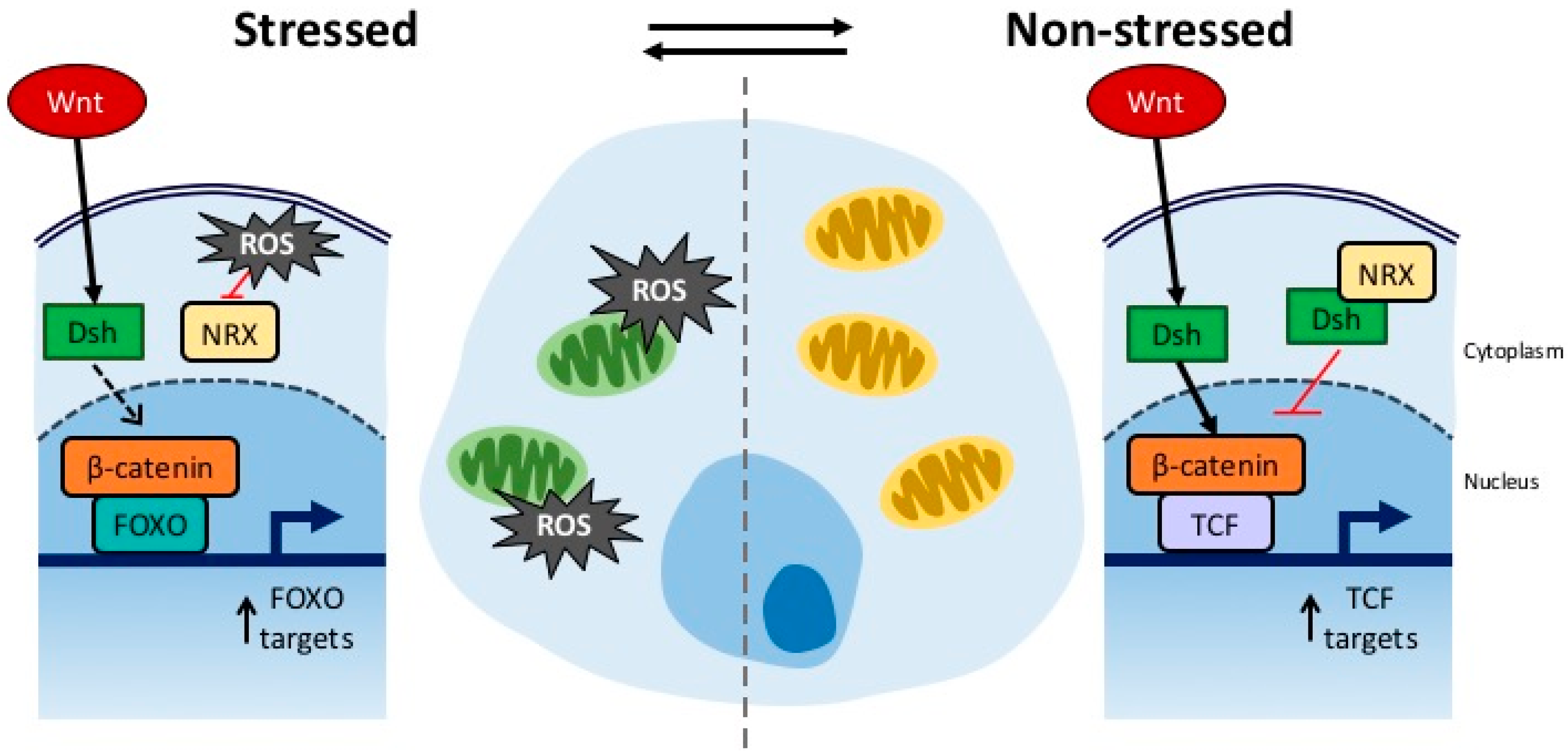
4. Connection between Developmental Drift and ROS
5. Conclusions: The Potential for Future Translation
Acknowledgements
Conflicts of Interest
References
- Clevers, H.; Loh, K.M.; Nusse, R. An integral program for tissue renewal and regeneration: Wnt signaling and stem cell control. Science 2014. [Google Scholar] [CrossRef] [PubMed]
- Clevers, H.; Nusse, R. Wnt/beta-catenin signaling and disease. Cell 2012, 149, 1192–1205. [Google Scholar] [CrossRef] [PubMed]
- Schlessinger, K.; Hall, A.; Tolwinski, N. Wnt signaling pathways meet rho gtpases. Genes Dev. 2009, 23, 265–277. [Google Scholar] [CrossRef] [PubMed]
- Cadigan, K.M.; Nusse, R. Wnt signaling: A common theme in animal development. Genes Dev. 1997, 11, 3286–3305. [Google Scholar] [CrossRef] [PubMed]
- Logan, C.Y.; Nusse, R. The wnt signaling pathway in development and disease. Annu. Rev. Cell Dev. Biol. 2004, 20, 781–810. [Google Scholar] [CrossRef] [PubMed]
- Reya, T.; Clevers, H. Wnt signalling in stem cells and cancer. Nature 2005, 434, 843–850. [Google Scholar] [CrossRef] [PubMed]
- Lezzerini, M.; Budovskaya, Y. A dual role of the wnt signaling pathway during aging in Caenorhabditis elegans. Aging Cell 2014, 13, 8–18. [Google Scholar] [CrossRef] [PubMed]
- World Fertility Patterns 2015-Data Booklet. Available online: http://www.un.org/en/development/desa/population/publications/pdf/fertility/world-fertility-patterns-2015.pdf (accessed on 20 March 2016).
- Gruber, J.; Chen, C.B.; Fong, S.; Ng, L.F.; Teo, E.; Halliwell, B. Caenorhabditis elegans: What we can and cannot learn from aging worms. Antioxid. Redox Signal. 2015, 23, 256–279. [Google Scholar] [CrossRef] [PubMed]
- Medawar, P.B. An Unsolved Problem of Biology, H.K. Lewis and Company: London, UK, 1952.
- Balaban, R.S.; Nemoto, S.; Finkel, T. Mitochondria, oxidants, and aging. Cell 2005, 120, 483–495. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T. Radical medicine: Treating ageing to cure disease. Nat. Rev. Mol. Cell Biol. 2005, 6, 971–976. [Google Scholar] [CrossRef] [PubMed]
- Finkel, T.; Serrano, M.; Blasco, M.A. The common biology of cancer and ageing. Nature 2007, 448, 767–774. [Google Scholar] [CrossRef] [PubMed]
- Rattan, S. Aging is not a disease: Implications for intervention. Aging Dis. 2014, 5, 196–202. [Google Scholar] [CrossRef] [PubMed]
- Houtkooper, R.H.; Argmann, C.; Houten, S.M.; Canto, C.; Jeninga, E.H.; Andreux, P.A.; Thomas, C.; Doenlen, R.; Schoonjans, K.; Auwerx, J. The metabolic footprint of aging in mice. Sci. Rep. 2011. [Google Scholar] [CrossRef] [PubMed]
- Schaffer, S.; Gruber, J.; Ng, L.F.; Fong, S.; Wong, Y.T.; Tang, S.Y.; Halliwell, B. The effect of dichloroacetate on health- and lifespan in C. elegans. Biogerontology 2010, 12, 195–209. [Google Scholar] [CrossRef] [PubMed]
- Martin, G.M.; Bergman, A.; Barzilai, N. Genetic determinants of human health span and life span: Progress and new opportunities. PLoS Genet. 2007, 3, e125. [Google Scholar] [CrossRef] [PubMed]
- Longo, V.D.; Antebi, A.; Bartke, A.; Barzilai, N.; Brown-Borg, H.M.; Caruso, C.; Curiel, T.J.; de Cabo, R.; Franceschi, C.; Gems, D.; et al. Interventions to slow aging in humans: Are we ready? Aging Cell 2015, 14, 497–510. [Google Scholar] [CrossRef] [PubMed]
- Hayflick, L. Biological aging is no longer an unsolved problem. Ann. N. Y. Acad. Sci. 2007, 1100, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Goldman, D.P.; Cutler, D.; Rowe, J.W.; Michaud, P.C.; Sullivan, J.; Peneva, D.; Olshansky, S.J. Substantial health and economic returns from delayed aging may warrant a new focus for medical research. Health Aff. 2013, 32, 1698–1705. [Google Scholar] [CrossRef] [PubMed]
- Partridge, L.; Gems, D. Mechanisms of ageing: Public or private? Nat. Rev. Genet. 2002, 3, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Kirkwood, T. Understanding ageing from an evolutionary perspective. J. Intern. Med. 2008, 263, 117–127. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.J.; Zhao, C.; Pleasure, S.J. Wnt signaling mutants have decreased dentate granule cell production and radial glial scaffolding abnormalities. J. Neurosci. 2004, 24, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Whetstone, H.C.; Lin, A.C.; Nadesan, P.; Wei, Q.; Poon, R.; Alman, B.A. Beta-catenin signaling plays a disparate role in different phases of fracture repair: Implications for therapy to improve bone healing. PLoS Med. 2007, 4, e249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, M.; Yang, Z.; Andl, T.; Cui, C.; Kim, N.; Millar, S.E.; Cotsarelis, G. Wnt-dependent de novo hair follicle regeneration in adult mouse skin after wounding. Nature 2007, 447, 316–320. [Google Scholar] [CrossRef] [PubMed]
- DeCarolis, N.A.; Wharton, K.A., Jr.; Eisch, A.J. Which way does the wnt blow? Exploring the duality of canonical wnt signaling on cellular aging. Bioessays 2008, 30, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Brack, A.S.; Conboy, M.J.; Roy, S.; Lee, M.; Kuo, C.J.; Keller, C.; Rando, T.A. Increased wnt signaling during aging alters muscle stem cell fate and increases fibrosis. Science 2007, 317, 807–810. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Fergusson, M.M.; Castilho, R.M.; Liu, J.; Cao, L.; Chen, J.; Malide, D.; Rovira, I.I.; Schimel, D.; Kuo, C.J.; et al. Augmented wnt signaling in a mammalian model of accelerated aging. Science 2007, 317, 803–806. [Google Scholar] [CrossRef] [PubMed]
- Dobzhansky, T. Nothing in biology makes sense except in the light of evolution. Am. Biol. Teach. 1973, 35, 125–129. [Google Scholar] [CrossRef]
- Kirkwood, T.B.; Holliday, R. The evolution of ageing and longevity. Proc. R. Soc. Lond. B Biol. Sci. 1979, 205, 531–546. [Google Scholar] [CrossRef] [PubMed]
- Williams, G.C. Pleiotropy, natural selection, and the evolution of senescence. Sci. Aging Knowl. Environ. 2001. [Google Scholar] [CrossRef]
- Holliday, R.; Rattan, S.I.S. Longevity mutants do not establish any “new science” of ageing. Biogerontology 2010, 11, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Hughes, K.A.; Reynolds, R.M. Evolutionary and mechanistic theories of aging. Annu. Rev. Entomol. 2005, 50, 421–445. [Google Scholar] [CrossRef] [PubMed]
- Friedman, D.B.; Johnson, T.E. Three mutants that extend both mean and maximum life span of the nematode, Caenorhabditis elegans, define the age-1 gene. J. Gerontol. 1988, 43, B102–B109. [Google Scholar] [CrossRef] [PubMed]
- Kenyon, C. The plasticity of aging: Insights from long-lived mutants. Cell 2005, 120, 449–460. [Google Scholar] [CrossRef] [PubMed]
- Kenyon, C. The first long-lived mutants: Discovery of the insulin/igf-1 pathway for ageing. Philos. Trans. R. Soc. B Biol. Sci. 2010, 366, 9–16. [Google Scholar] [CrossRef] [PubMed]
- Ayyadevara, S.; Alla, R.; Thaden, J.J.; Shmookler Reis, R.J. Remarkable longevity and stress resistance of nematode pi3k-null mutants. Aging Cell 2008, 7, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Van Heemst, D.; Beekman, M.; Mooijaart, S.P.; Heijmans, B.T.; Brandt, B.W.; Zwaan, B.J.; Slagboom, P.E.; Westendorp, R.G.J. Reduced insulin/igf-1 signalling and human longevity. Aging Cell 2005, 4, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Van der Spoel, E.; Rozing, M.P.; Houwing-Duistermaat, J.J.; Slagboom, P.E.; Beekman, M.; de Craen, A.J.M.; Westendorp, R.G.J.; van Heemst, D. Association analysis of insulin-like growth factor-1 axis parameters with survival and functional status in nonagenarians of the leiden longevity study. Aging 2015, 7, 956–963. [Google Scholar] [CrossRef] [PubMed]
- Guevara-Aguirre, J.; Rosenbloom, A.L. Obesity, diabetes and cancer: Insight into the relationship from a cohort with growth hormone receptor deficiency. Diabetologia 2014, 58, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, N.L.; McColl, G.; Lithgow, G.J. Fitness cost of extended lifespan in Caenorhabditis elegans. Proc. R. Soc. Biol. Sci. 2004, 271, 2523–2526. [Google Scholar] [CrossRef] [PubMed]
- Walker, D.W.; McColl, G.; Jenkins, N.L.; Harris, J.; Lithgow, G.J. Evolution of lifespan in C. elegans. Nature 2000, 405, 296–297. [Google Scholar] [PubMed]
- Heitman, J.; Movva, N.; Hall, M. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science 1991, 253, 905–909. [Google Scholar] [CrossRef] [PubMed]
- Jia, K. The tor pathway interacts with the insulin signaling pathway to regulate C. elegans larval development, metabolism and life span. Development 2004, 131, 3897–3906. [Google Scholar] [CrossRef] [PubMed]
- Kapahi, P.; Zid, B.M.; Harper, T.; Koslover, D.; Sapin, V.; Benzer, S. Regulation of lifespan in drosophila by modulation of genes in the tor signaling pathway. Curr. Biol. 2004, 14, 885–890. [Google Scholar] [CrossRef] [PubMed]
- Harrison, D.E.; Strong, R.; Sharp, Z.D.; Nelson, J.F.; Astle, C.M.; Flurkey, K.; Nadon, N.L.; Wilkinson, J.E.; Frenkel, K.; Carter, C.S.; et al. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 2009, 460, 392–395. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.A.; Harrison, D.E.; Astle, C.M.; Baur, J.A.; Boyd, A.R.; de Cabo, R.; Fernandez, E.; Flurkey, K.; Javors, M.A.; Nelson, J.F.; et al. Rapamycin, but not resveratrol or simvastatin, extends life span of genetically heterogeneous mice. J. Gerontol. A: Biol. Sci. Med. Sci. 2010, 66, 191–201. [Google Scholar] [CrossRef] [PubMed]
- Blagosklonny, M.V. Revisiting the antagonistic pleiotropy theory of aging: Tor-driven program and quasi-program. Cell Cycle 2010, 9, 3171–3176. [Google Scholar] [CrossRef] [PubMed]
- Rattan, S.I. Gerontogenes: Real or virtual? FASEB J. 1995, 9, 284–286. [Google Scholar] [PubMed]
- Budovskaya, Y.V.; Wu, K.; Southworth, L.K.; Jiang, M.; Tedesco, P.; Johnson, T.E.; Kim, S.K. An elt-3/elt-5/elt-6 gata transcription circuit guides aging in C. elegans. Cell 2008, 134, 291–303. [Google Scholar] [CrossRef] [PubMed]
- Gems, D.; de la Guardia, Y. Alternative perspectives on aging in Caenorhabditis elegans: Reactive oxygen species or hyperfunction? Antioxid. Redox Signal. 2013, 19, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Pincus, Z.; Slack, F.J. Transcriptional (dys)regulation and aging in Caenorhabditis elegans. Genome Biol. 2008. [Google Scholar] [CrossRef] [PubMed]
- Cassata, G.; Shemer, G.; Morandi, P.; Donhauser, R.; Podbilewicz, B.; Baumeister, R. Ceh-16/engrailed patterns the embryonic epidermis of Caenorhabditis elegans. Development 2005, 132, 739–749. [Google Scholar] [CrossRef] [PubMed]
- Gorrepati, L.; Thompson, K.W.; Eisenmann, D.M. C. elegans gata factors egl-18 and elt-6 function downstream of wnt signaling to maintain the progenitor fate during larval asymmetric divisions of the seam cells. Development 2013, 140, 2093–2102. [Google Scholar] [CrossRef] [PubMed]
- Koh, K.; Peyrot, S.M.; Wood, C.G.; Wagmaister, J.A.; Maduro, M.F.; Eisenmann, D.M.; Rothman, J.H. Cell fates and fusion in the C. elegans vulval primordium are regulated by the egl-18 and elt-6 gata factors—apparent direct targets of the lin-39 hox protein. Development 2002, 129, 5171–5180. [Google Scholar] [PubMed]
- Lezzerini, M.; Smith, R.L.; Budovskaya, Y. Developmental drift as a mechanism for aging: Lessons from nematodes. Biogerontology 2013, 14, 693–701. [Google Scholar] [CrossRef] [PubMed]
- Adamska, M.; Degnan, S.M.; Green, K.M.; Adamski, M.; Craigie, A.; Larroux, C.; Degnan, B.M. Wnt and tgf-beta expression in the sponge amphimedon queenslandica and the origin of metazoan embryonic patterning. PLoS ONE 2007, 2, e1031. [Google Scholar] [CrossRef] [PubMed]
- Van Amerongen, R.; Mikels, A.; Nusse, R. Alternative wnt signaling is initiated by distinct receptors. Sci. Signal. 2008. [Google Scholar] [CrossRef] [PubMed]
- Van Amerongen, R.; Nusse, R. Towards an integrated view of wnt signaling in development. Development 2009, 136, 3205–3214. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B.; Gutteridge, J.M.C. Free Radicals in Biology and Medicine; Oxford University Press: Oxford, UK, 2015. [Google Scholar]
- Harman, D. Aging: A theory based on free radical and radiation chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef] [PubMed]
- Harman, D. The biologic clock: The mitochondria? J. Am. Geriatr. Soc. 1972, 20, 145–147. [Google Scholar] [CrossRef] [PubMed]
- Gems, D.; Doonan, R. Antioxidant defense and aging in C. elegans: Is the oxidative damage theory of aging wrong? Cell Cycle 2009, 8, 1681–1687. [Google Scholar] [CrossRef] [PubMed]
- Gruber, J. The mitochondrial free radical theory of ageing—Where do we stand? Front. Biosci. 2008, 13, 6554–6579. [Google Scholar] [CrossRef] [PubMed]
- Lapointe, J.; Hekimi, S. When a theory of aging ages badly. Cell. Mol. Life Sci. 2009, 67, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ristow, M.; Zarse, K. How increased oxidative stress promotes longevity and metabolic health: The concept of mitochondrial hormesis (mitohormesis). Exp. Gerontol. 2010, 45, 410–418. [Google Scholar] [CrossRef] [PubMed]
- Wanagat, J.; Dai, D.F.; Rabinovitch, P. Mitochondrial oxidative stress and mammalian healthspan. Mech. Ageing Dev. 2010, 131, 527–535. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B.; Gutteridge, J.M.C. Free Radicals in Biology and Medicine, 5th ed.; Oxford University Press (OUP): Oxford, UK, 2015. [Google Scholar]
- Speakman, J.R.; Blount, J.D.; Bronikowski, A.M.; Buffenstein, R.; Isaksson, C.; Kirkwood, T.B.; Monaghan, P.; Ozanne, S.E.; Beaulieu, M.; Briga, M. Oxidative stress and life histories: Unresolved issues and current needs. Ecol. Evol. 2015, 5, 5745–5757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoogeboom, D.; Burgering, B.M. Should I stay or should I go: Beta-catenin decides under stress. Biochim. Biophys. Acta 2009, 1796, 63–74. [Google Scholar] [CrossRef] [PubMed]
- Funato, Y.; Miki, H. Redox regulation of wnt signalling via nucleoredoxin. Free Radical Res. 2010, 44, 379–388. [Google Scholar] [CrossRef] [PubMed]
- Love, N.R.; Chen, Y.; Ishibashi, S.; Kritsiligkou, P.; Lea, R.; Koh, Y.; Gallop, J.L.; Dorey, K.; Amaya, E. Amputation-induced reactive oxygen species are required for successful xenopus tadpole tail regeneration. Nat. Cell Biol. 2013, 15, 222–228. [Google Scholar] [CrossRef] [PubMed]
- Wen, J.W.; Hwang, J.T.; Kelly, G.M. Reactive oxygen species and wnt signalling crosstalk patterns mouse extraembryonic endoderm. Cell. Signal. 2012, 24, 2337–2348. [Google Scholar] [CrossRef] [PubMed]
- Eijkelenboom, A.; Burgering, B.M. Foxos: Signalling integrators for homeostasis maintenance. Nat. Rev. Mol. Cell Biol. 2013, 14, 83–97. [Google Scholar] [CrossRef] [PubMed]
- Labbadia, J.; Morimoto, R.I. Repression of the heat shock response is a programmed event at the onset of reproduction. Mol. Cell 2015, 59, 639–650. [Google Scholar] [CrossRef] [PubMed]
- David, D.C.; Ollikainen, N.; Trinidad, J.C.; Cary, M.P.; Burlingame, A.L.; Kenyon, C. Widespread protein aggregation as an inherent part of aging in C. elegans. PLoS Biol. 2010, 8, e1000450. [Google Scholar] [CrossRef] [PubMed]
- McCay, C.; Crowell, M.F.; Maynard, L. The effect of retarded growth upon the length of life span and upon the ultimate body size. J. Nutr. 1935, 10, 63–79. [Google Scholar]
- Osborne, T.B.; Mendel, L.B.; Ferry, E.L. The effect of retardation of growth upon the breeding period and duration of life of rats. Science 1917, 45, 294–295. [Google Scholar] [CrossRef] [PubMed]
- Rose, M.R.; Rauser, C.L.; Benford, G.; Matos, M.; Mueller, L.D. Hamilton’s forces of natural selection after forty years. Evolution 2007, 61, 1265–1276. [Google Scholar] [CrossRef] [PubMed]
- Check, H.E. Anti-ageing pill pushed as bona fide drug. Nature 2015, 522, 265–266. [Google Scholar] [CrossRef] [PubMed]
- Madan, B.; Virshup, D.M. Targeting wnts at the source—New mechanisms, new biomarkers, new drugs. Mol. Cancer Ther. 2015, 14, 1087–1094. [Google Scholar] [CrossRef] [PubMed]


© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gruber, J.; Yee, Z.; Tolwinski, N.S. Developmental Drift and the Role of Wnt Signaling in Aging. Cancers 2016, 8, 73. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers8080073
Gruber J, Yee Z, Tolwinski NS. Developmental Drift and the Role of Wnt Signaling in Aging. Cancers. 2016; 8(8):73. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers8080073
Chicago/Turabian StyleGruber, Jan, Zhuangli Yee, and Nicholas S. Tolwinski. 2016. "Developmental Drift and the Role of Wnt Signaling in Aging" Cancers 8, no. 8: 73. https://0-doi-org.brum.beds.ac.uk/10.3390/cancers8080073