CO2 Hydrogenation to Methanol over La2O3-Promoted CuO/ZnO/Al2O3 Catalysts: A Kinetic and Mechanistic Study


Department of Chemical Engineering, University of Patras, 26504 Patras, Greece
*
Author to whom correspondence should be addressed.
Catalysts 2020, 10(2), 183; https://0-doi-org.brum.beds.ac.uk/10.3390/catal10020183
Submission received: 21 December 2019
/
Revised: 22 January 2020
/
Accepted: 31 January 2020
/
Published: 3 February 2020
(This article belongs to the Special Issue Nanomaterials in Catalysis Applications)
Abstract
:The hydrogenation of CO2 to methanol has been investigated over CuO/ZnO/Al2O3 (CZA) catalysts, where a part of the Al2O3 (0, 25, 50, 75, or 100%) was substituted by La2O3. Results of catalytic performance tests obtained at atmospheric pressure showed that the addition of La2O3 generally resulted in a decrease of CO2 conversion and in an increase of methanol selectivity. Optimal results were obtained for the CZA-La50 catalyst, which exhibited a 30% higher yield of methanol, compared to the un-promoted sample. This was attributed to the relatively high specific surface area and porosity of this material, the creation of basic sites of moderate strength, which enhance adsorption of CO2 and intermediates that favor hydrogenation steps, and the ability of the catalyst to maintain a large part of the copper in its metallic form under reaction conditions. The reaction mechanism was studied with the use of in situ infrared spectroscopy (DRIFTS). It was found that the reaction proceeded with the intermediate formation of surface formate and methoxy species and that both methanol and CO were mainly produced via a common formate intermediate species. The kinetic behavior of the best performing CZA-La50 catalyst was investigated in the temperature range 190–230 °C as a function of the partial pressures of H2 (0.3–0.9 atm) and CO2 (0.05–0.20 atm), and a kinetic model was developed, which described the measured reaction rates satisfactorily.
1. Introduction
Carbon dioxide, which is one of the main greenhouse gases and a major contributor to global warming, has been identified as a promising feedstock for producing valuable chemicals [1], and its efficient utilization is expected to contribute toward the development of a carbon-neutral energy cycle [2]. One of the most investigated processes aiming at this direction is hydrogenation of CO2 to methanol (Equation (1)) using H2 coming from renewable energy sources.
Methanol is one of the most important chemicals as it can be used directly as a fuel, as an energy carrier in fuel cells, or as a raw material for the synthesis of products such as formaldehyde, acetic acid, and olefins [3]. Methanol is traditionally produced from syngas. However, for obvious economic and environmental reasons, there is great interest in developing a process where methanol is produced from abundant and inexpensive CO2. The main problem that is currently hindering the wider application of the latter process is the low methanol selectivity due to the occurrence of the competitive reverse water-gas shift (RWGS) reaction (Equation (2)) [4]. Thus, it is important to develop catalytic materials characterized by high activity and selectivity toward methanol.
The state-of-the-art catalyst for the title reaction is CuO/ZnO/Al2O3 (CZA), where CuO and ZnO catalyze both the methanol synthesis and the WGS reaction [5]. A plethora of studies have been reported dealing with the reaction kinetics and mechanism over CZA-based materials, and several models have been proposed to explain the experimental results [6,7,8,9]. However, there is still debate regarding the reaction mechanism and catalyst requirements [4,10,11]. As far as catalysts are concerned, efforts have been made to improve the performance of CZA by either replacing/doping Cu with noble metals (e.g., Pt, Pd) [12,13,14,15] or by adding oxides in order to enhance the adsorption properties of the support and to increase the dispersion of the Cu phases [16,17,18,19,20,21]. This is the reason why the majority of studies pinpoint some sort of coupling in the mechanism between the copper phase and the support oxide, with the latter being the anchor point for one of the oxygen atoms of CO2 [22,23]. In this respect, several promoters have been studied, mainly oxides of metals such as Mn, La, Zr, Cr, and B [21,24,25,26,27,28]. In several cases, the catalytic performance of CZA catalysts was enhanced following substitution of part or all of Al2O3 by oxides, such as ZrO2, Cr2O3, Ga2O3, or SiO2 [29,30,31]. Results were explained considering that, for example, ZrO2 improves the dispersion of copper particles, SiO2 enhances the long-term stability of the catalyst, whereas dopants such as Cr2O3 or Ga2O3 increase the specific activity per unit Cu surface area [29,30,31].
In the present work, a series of La-promoted CZA catalysts (CZA-Lax) of variable La2O3-to-Al2O3 ratios was synthesized, characterized, and tested for the title reaction. Results showed that the addition of La2O3 increased the selectivity toward methanol and that the yield of methanol was maximized for the CZA-La50 catalyst, for which 50% of Al2O3 was replaced by La2O3. This was explained considering that the addition of La2O3 affected the distribution and strength of basic sites on the catalyst surface and promoted the formation of metallic copper under the reaction conditions. Results of in situ DRIFTS experiments indicated that the reaction proceeds via the intermediate formation of surface formate and methoxy species. The kinetic behavior of the best performing catalyst was investigated as a function or the reaction temperature and partial pressures of H2 and CO2, and a kinetic model was developed, which described the measured reaction rates satisfactorily.
2. Results
2.1. Characteristics of the Synthesized Catalysts
CuO/ZnO/Al2O3 and La-promoted CZA catalysts were synthesized by a co-precipitation method, as described in Section 4.1. The molar composition of the unpromoted CZA catalyst was 61.7% CuO, 30.1% ZnO, and 8.2% Al2O3. The La-promoted samples were prepared by substituting different portions of Al2O3 by La2O3. The catalysts thus prepared are denoted in the following as CZA-Lax, where x (0, 25, 50, 75, or 100) indicates the molar fraction (%) of Al2O3 replaced by La2O3 (Table 1).
The textural properties of the as-synthesized CZA-Lax catalysts are summarized in Table 1. It was observed that, as a general trend, the increase of the La2O3 content resulted in a decrease of the specific surface area (SSA), which dropped from 111 m2 g−1 for the un-promoted sample to 47 m2 g−1 for the CZA-La100 catalyst, for which Al2O3 was fully substituted by La2O3. This confirmed the higher ability of Al2O3 to act as a structural stabilizing agent of the CuO/ZnO catalyst, compared to La2O3. It is of interest to note, however, that the CZA-La50 sample retained a relatively large SSA and exhibited the highest pore volume and pore size among the catalysts investigated (Table 1).
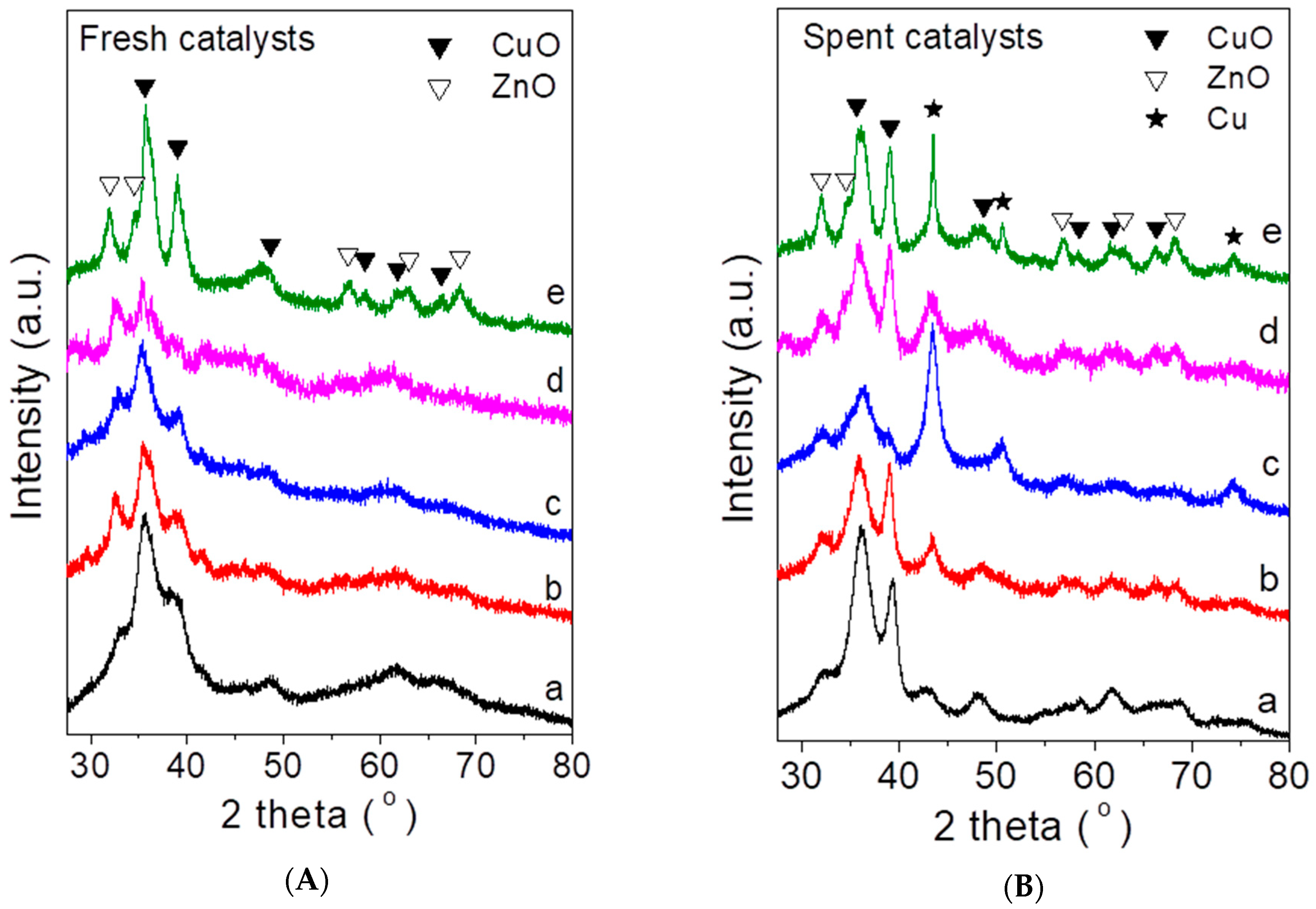
In Figure 1A are shown the X-ray diffraction (XRD) patterns of the freshly prepared catalysts. It was observed that all materials were characterized by peaks attributable to CuO and ZnO phases, with the most intense ones located at 2θ = 35.5°, 38.6°, 31.8°, and 34.3°, respectively. The absence of clearly resolved peaks corresponding to Al2O3 and La2O3 phases may be explained considering the low content of these oxides and indicated that they exist in an amorphous or microcrystalline state [32]. Qualitatively similar results have been reported for La-modified CuO-ZnO-Al2O3/HZSM-5 catalysts synthesized by the co-precipitation method [21].
The XRD patterns obtained following the exposure of the catalyst samples to the reaction conditions are shown in Figure 1B. It was observed that, in all cases, the diffractograms were characterized by the presence of additional peaks located at 2θ = 43.5° and 50.5°, attributable to metallic copper (Cu0), which is known to be an active component for methanol synthesis [33,34]. It is of interest to note that the relative intensity of the Cu0 peaks, compared to those of CuO, was generally higher for the La-promoted catalysts and was maximized for the CZA-La50 sample (c), which, as discussed below, exhibited the highest yield toward methanol.
The surface basicity of the pre-reduced catalysts was studied by temperature-programmed desorption of CO2 (CO2-TPD), and the results obtained are shown in Figure 2. It was observed that the desorption profile of the unpromoted CZA-La0 catalyst (a) was characterized by an intense low-temperature (LT) peak centered at ca. 85 °C and a broad high-temperature (HT) shoulder extending up to 300 °C. The LT peak could be assigned to weak basic sites related to surface hydroxyl groups, whereas the HT shoulder could be attributed to basic sites of moderate strength corresponding to metal-oxygen pairs (Al–O, Zn–O, and/or Cu–O) [35,36].
Substitution of 50% or 100% of Al2O3 by La2O3 (b and c, respectively) resulted in a substantial decrease of the intensity of the LT peak, indicating that weak basic sites were mainly related to the OH groups of alumina. This was accompanied by a progressive growth and shift of the HT shoulder toward higher temperatures. The total amount of desorbed CO2, calculated by integration of the corresponding TPD curves, decreased from 25.9 μmol/g for CZA-La0 to 23.1 μmol/g for CZA-La50 and to 18.1 μmol/g for the CZA-La100 catalyst. This indicated that the total number of basic sites decreased following the addition of La2O3. On the other hand, it is evident from Figure 2 that both the relative concentration and strength of the HT basic sites increased with increasing La content. This showed that the La-promoted catalysts were characterized by a higher amount of basic sites of moderate strength, the number and strength of which increased with increasing La2O3 content. Qualitatively similar results have been reported for Cu/ZnO catalysts promoted with Fe or Ga [36].
2.2. Catalytic Performance Tests
The catalytic performance of the CZA-Lax samples was investigated at atmospheric pressure in the temperature range of 160–260 °C. Under these reaction conditions, the only carbon-containing products detected were methanol and CO, and no catalyst deactivation was observed with time-on-stream. The yield of methanol was typically lower than 1% because, according to the Le Chatelier principle, Reaction (1) is favored at high pressures. Typical results obtained over the best performing CZA-La50 catalyst are shown in Figure 3 and are compared with those of the CZA-La0 and CZA-La100 samples. It was observed that, in all cases, conversion of CO2 (XCO2) increased with the increase of the reaction temperature (Figure 3A). Substitution of Al2O3 by La2O3 resulted in a decrease of XCO2, which took its lowest values for the CZA-La100 catalyst. Selectivity toward methanol (SCH3OH) followed the opposite trend, i.e., it was higher than 90% at 160 °C for all samples and decreased with the increase of temperature, dropping close to zero at ca. 260 °C (Figure 3B) where CO2 was selectively converted to CO. Interestingly, SCH3OH was found to be higher for all La-containing catalysts, compared to un-promoted CZA, and was maximized for the CZA-La100 sample, where Al2O3 was fully substituted by La2O3. The beneficial effect of La2O3 on methanol selectivity could be attributed to the increase of the concentration of surface basic sites of moderate strength (Figure 2), which are believed to be responsible for catalyzing the C–O bond activation of adsorbed CO2 species [26,27,28]. La2O3 also stabilized copper in its metallic form under reaction conditions (Figure 1), which is known to promote the title reaction. A similar beneficial effect on methanol selectivity was reported for La-promoted Cu/ZnO/ZrO2 catalysts [37], as well as for Cu/ZnO catalysts promoted with other basic oxides, such as CeO2 [38].
As a result of the counteracting effects of temperature on CO2 conversion and methanol selectivity, the yield of methanol went through a maximum at temperatures around 210 °C (Figure 3C). As mentioned above, optimal results were obtained for the CZA-La50 catalyst, which combined high activity and selectivity, leading to a yield of methanol of 0.9% at 210 °C, which was ca. 30% higher than that of the un-promoted CZA-La0 sample (0.7%).
2.3. In Situ DRIFTS Studies
The in situ diffuse reflectance infrared Fourier-transform (DRIFT) spectra obtained following the interaction of carbon monoxide with CZA-La0 and CZA-La50 catalysts are presented in Figure 4. Adsorption and desorption of CO was investigated not only because CO is a reaction product, but also because it is a suitable probe molecule to study the nature and strength of active sites. As shown in Figure 4A, exposure of the pre-reduced CZA-La0 catalyst to CO at 25 °C for 30 min (a) resulted in the appearance of bands located at ca. 2170 and 2108 cm−1, which were attributed to gas-phase CO [39,40]. The latter peak also contained a contribution from carbonyl species linearly adsorbed on metallic Cu, which are known to vibrate in this region [41]. Switching the feed to He resulted in the removal of the bands due to gas-phase CO and in the appearance of the Cu-CO peak at 2105 cm−1, which slightly decreased in intensity with time-on-stream (b–d). The increase of temperature under He flow at 100 °C resulted in a substantial decrease of the peak intensity (e). A further increase of temperature resulted in the disappearance of the peak (f,g), indicating that carbonyl species were weakly adsorbed on the catalyst surface.
Similar DRIFT spectra obtained over the CZA-La50 catalyst are presented in Figure 4B. It was observed that the carbonyl peak (b) appeared at a higher wavenumber (2110 cm−1) and was of lower intensity, compared to CZA-La0 (Figure 4A), indicating that it was less strongly adsorbed on Cu. This was verified by the disappearance of the peak upon increasing the temperature to 100 °C (e). Comparison of the spectra obtained over the un-promoted (Figure 4A) and the La-containing sample (Figure 4B) indicated that the presence of La2O3 resulted in a decrease of the adsorption strength of Cu toward CO.
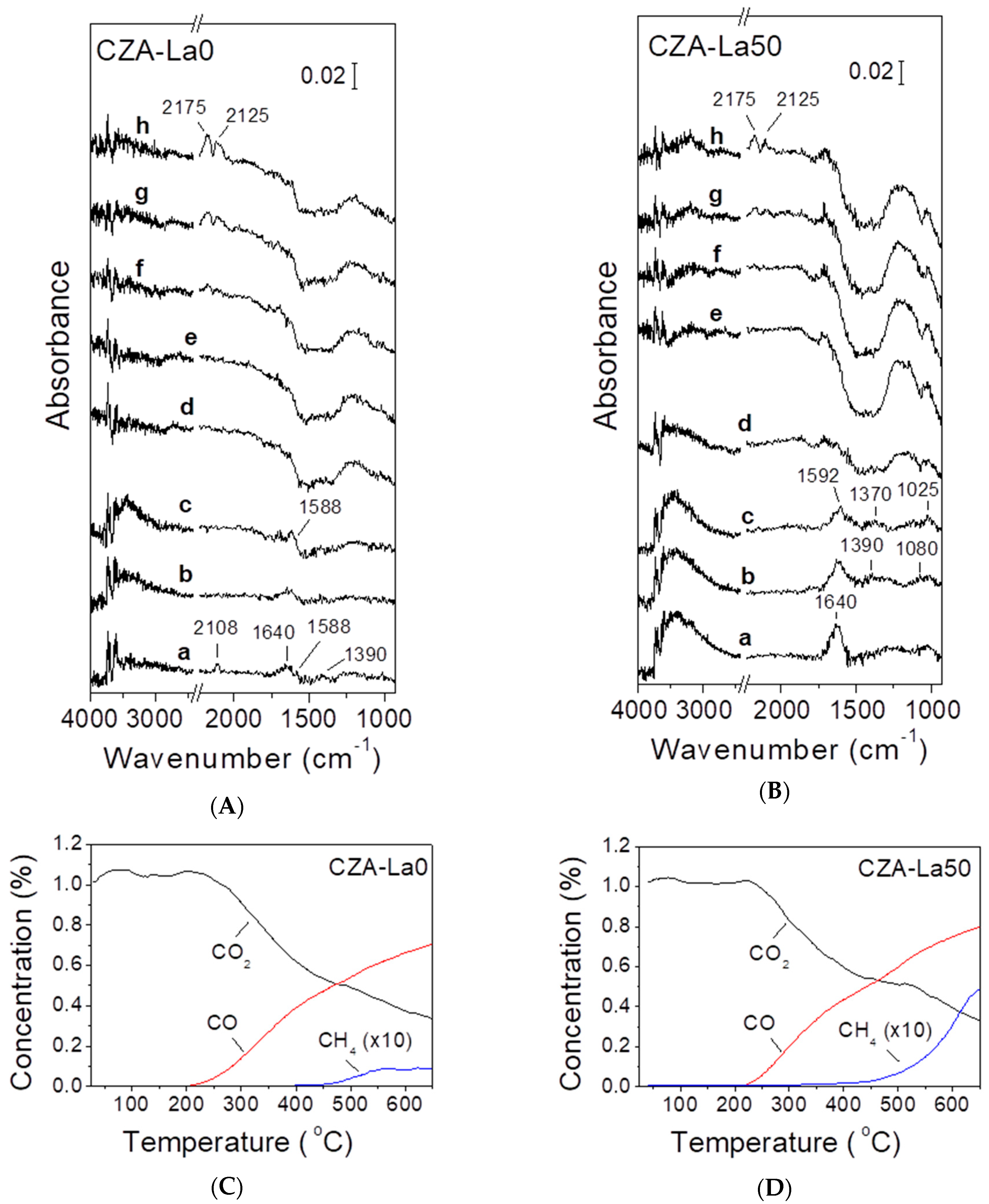
The formation and fate of surface species formed under reaction conditions was investigated over the CZA-La0 and the CZA-La50 catalysts by in situ DRIFT spectroscopy in the temperature range 25–400 °C, and the results obtained are presented in Figure 5A,B. In the same figure is also shown the evolution of products in the gas-phase, which was monitored in separate experiments performed under the same reaction conditions with the use of the transient-MS technique (Figure 5C,D).
Regarding the CZA-La0 catalyst (Figure 5A), it was observed that exposure of the pre-reduced sample to the CO2/H2 mixture at 25 °C (a) resulted in the appearance of a peak located at 2108 cm−1, attributed to CO adsorbed on metallic Cu (see above), a band centered at ca. 1640 cm−1 attributed to adsorbed water [39,42], and a broad feature centered at ca. 3450 cm−1, which was due to adsorbed OH species [41]. The appearance of these bands indicated that the gas-phase CO2 adsorbed dissociatively on the catalyst surface at room temperature to yield adsorbed CO and adsorbed oxygen species. The latter combined with dissociatively adsorbed hydrogen to form hydroxyl species and then H2O [39]. Two additional bands located at 1588 and 1390 cm−1 could be assigned to adsorbed formate (HCOO) species [42].
The stepwise increase of temperature at 200 °C (b–d) resulted in the disappearance of all spectral features mentioned above. The fact that gas-phase products started evolving at this temperature (Figure 5C) indicated that these surface species were active intermediates for the title reaction. It should be noted that, in addition to CO, small amounts of methanol were also produced in the gas-phase at temperatures around 200 °C, but due to the low concentration of reactants and the transient nature of the experiments presented in Figure 5C, the methanol concentration was close to the detection limit of the apparatus and could not be quantified. A further increase of temperature at 400 °C (e–h) resulted in the development of two bands at 2125 and 2175 cm−1 due to the gas-phase CO, the production of which was favored at high temperatures because of the endothermic nature of the RWGS reaction. At temperatures higher than ca. 450 °C, measurable amounts of CH4 also appeared in the gas-phase (Figure 5C). It should be noted that the DRIFT spectra obtained at temperatures higher than 200 °C (Figure 5A,d–h) were characterized by a change of the background signal in the range 1250–1600 cm−1, which resulted in the appearance of a “negative” curve in this region. This was most possibly due to the (partial) oxidation of Cu by product H2O, which is a strong oxidant at elevated temperatures. A qualitatively similar behavior was observed for the CZA-La50 sample (Figure 5B).
The in situ DRIFT spectra obtained over the CZA-La50 catalyst under reaction conditions are shown in Figure 5B. Similar to the un-promoted sample, exposure to the CO2/H2 mixture at 25 °C (a) resulted in the development of bands attributed to adsorbed water (1640 cm−1) and OH species (broad band at 3450 cm−1). However, the carbonyl band at ca. 2110 cm−1 did not appear over this catalyst, probably because of the weak adsorption of this species on the CZA-L50 catalyst, compared to CZA-La0 (see Figure 4). The increase of the reaction temperature to 100 °C (b) and 150 °C (c) resulted in the development of bands at 1592, 1390, and 1370 cm−1 due to adsorbed formate (HCOO) species, as well as in the appearance of bands at ca. 1025 and 1080 cm−1, attributable to surface methoxy (CH3O) species [41,42]. The intensity of these bands decreased upon further increasing temperature to 200 °C (d), i.e., in the temperature range where methanol was produced (Figure 3) and diminished at higher temperatures (e–h). This indicated that CO2 hydrogenation to methanol occurred with the intermediate formation of formate and methoxy species, in agreement with the results of previous studies [42]. For the reasons mentioned above, the amount of methanol produced in the gas-phase at temperatures around 200 °C was very low and, therefore, w not shown in the transient-MS results presented in Figure 5D. On the other hand, CO evolved in the gas-phase at temperatures higher than 200 °C and was accompanied by the formation of relatively small amounts of CH4 at temperatures above ca. 350 °C (Figure 5D).
2.4. Reaction Kinetics
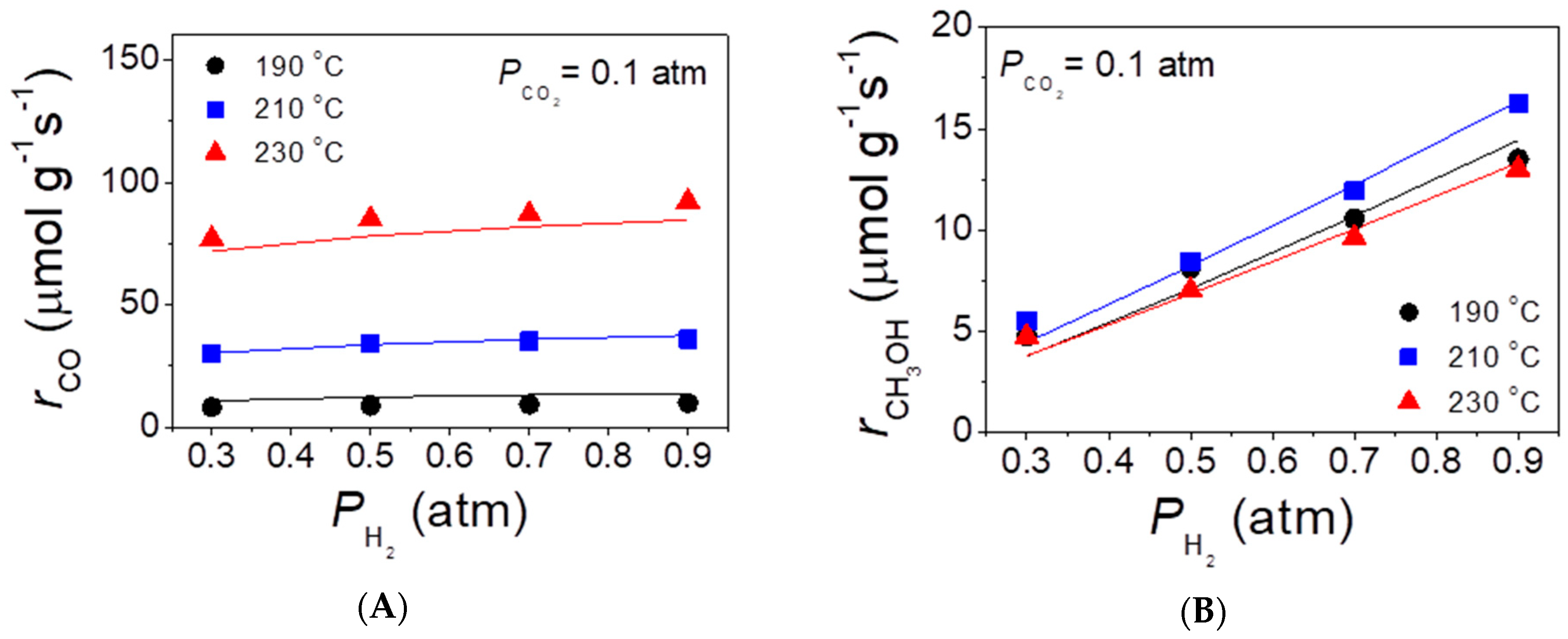
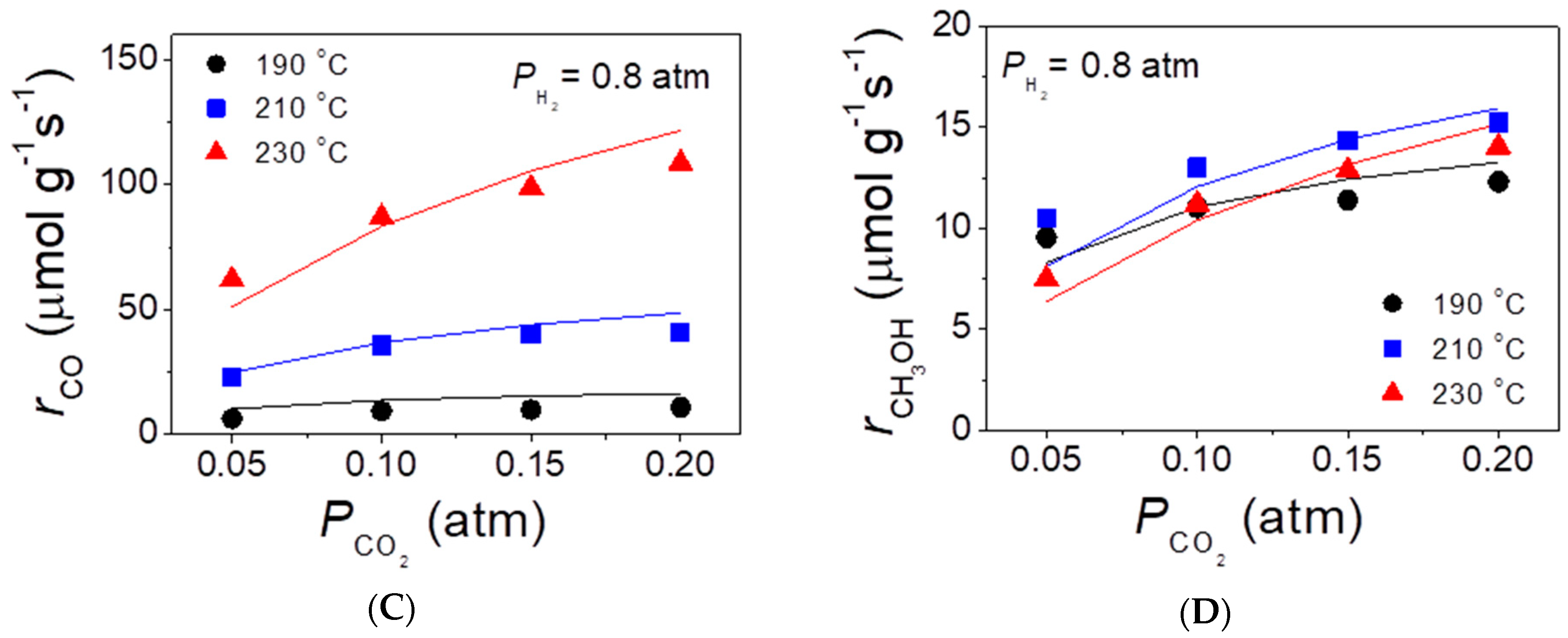
The initial rates of methanol and CO evolution were measured at atmospheric pressure over the best performing CZA-La50 catalyst in the temperature range of 190–230 °C. The results obtained with the use of different feed compositions are summarized in Figure 6, where symbols correspond to the measured values and solid curves represent their fitting according to the kinetic model discussed below (see Section 3.2). It was observed that the increase of the partial pressure of H2 (PH2) in the range 0.3–0.9 atm at a constant partial pressure of CO2 (PCO2 = 0.1 atm) did not affect appreciably the rate of CO evolution at a given temperature (Figure 6A), but resulted in an almost linear increase of the methanol formation rate (Figure 6B). On the other hand, the increase of PCO2 in the range 0.05–0.20 atm at constant PH2 = 0.8 atm resulted in a progressive increase of both the rate of CO (Figure 6C) and methanol (Figure 6D) production.
3. Discussion
3.1. Effects of La2O3 Addition on the Characteristics and Performance of Catalysts
The results of the catalyst characterization experiments listed in Table 1 showed that, although replacement of Al2O3 by La2O3 generally resulted in a decrease of the specific surface area, the CZA-La50 sample was characterized by a relatively large SSA and exhibited comparatively high pore volume and pore size. These parameters are known to affect positively the catalytic activity because the increase of SSA results in higher dispersion of the active copper phase, whereas enhanced porosity improves the diffusion of reactants/products to/from the active sites. As a result, conversion of CO2 over the CZA-La50 catalyst was comparable to that of the un-promoted CZA-La0 sample (Figure 3A).
Regarding selectivity toward methanol, it was substantially improved over the La2O3-promoted catalysts in the whole temperature range investigated (Figure 3B). This could be related to the ability of La2O3 to enhance the surface basicity of the catalyst [26] and was in agreement with previous results obtained with the use of other basic oxides as dopants [43,44], which showed that basic sites enhanced the adsorption of CO2 and intermediates and favored hydrogenation of surface formate to methoxy species [26]. As a general trend, a linear relationship was found to exist between the yield of methanol and the CO2 adsorption capacity of the catalyst [45]. The results of the present CO2-TPD experiments (Figure 2) and catalytic performance tests (Figure 3B) indicated that selectivity to methanol increased monotonically with increasing the surface concentration of basic sites of moderate strength. Overall, although Al2O3 elevated CO2 conversion, it did not benefit the methanol selectivity, probably because it interacted strongly with H2O molecules produced during the methanol synthesis reaction, thereby promoting the RWGS reaction to produce CO [38]. On the other hand, the addition of La2O3 improved the selectivity toward methanol, and as a result, the yield of methanol was enhanced over the LCZA-La50 catalyst (Figure 3C).
It is of interest to note that exposure of the CZA-La50 sample to the reaction conditions resulted in the transformation of a larger part of CuO to metallic Cu, compared to the un-promoted CZA-La0 sample (Figure 1B). A similar promotion of the in situ reduction of oxidized copper species (CuOx) to Cu0 was reported for Zr-doped CZA catalysts [46]. This could be explained considering that under reaction conditions, Cu0 is oxidized to CuOx by water, and Cu0 sites are regenerated by CO and H2 [47]. When the regeneration rate of Cu0 sites is not high enough, most of the surface copper sites will be oxidized, thereby resulting in decreased activity [46]. The relatively high content of Cu0 observed over the “spent” La-promoted samples (Figure 1B) indicated that La2O3 favored the latter reactions and maintained a larger portion of copper in its metallic form. It may then be suggested that the improved methanol selectivities obtained over the La-promoted catalysts could be related, at least in part, to their ability to keep the content of Cu0 at high levels under reaction conditions. In this respect, it was proposed that both Cu+ and Cu0 were involved in the methanol formation reaction and that catalytic activity depended on the Cu+/Cu0 ratio [8,47,48,49,50]. The results of the theoretical calculations indicated that CO2 hydrogenation to methanol was favored over metallic Cu0 sites, whereas Cu+ sites were mainly responsible for the hydrogenation of CO [49]. Further investigation of this issue is beyond the scope of the present work and may be the subject of our future investigation.
3.2. Mechanistic Implications and Development of the Kinetic Model
The results presented in Figure 5 showed that exposure of the CZA-La50 catalyst to the CO2/H2 reaction mixture resulted in the formation of surface oxygenated species, in particular formate and methoxy species. Methoxy species, which can be formed by stepwise hydrogenation of formates, are considered as intermediates for the formation of methanol [39,42]. On the other hand, carbon monoxide can be also produced from formate species [39] through decomposition or via the reverse water-gas shift reaction [41]. In the present work, it was suggested that both methanol and CO were produced predominantly via a common formate intermediate, in agreement with the results of previous experimental [51] and theoretical [52] studies.
The results of kinetic measurements obtained over the best performing CZA-La50 catalyst (Figure 6) were modeled using a dual-site Langmuir–Hinshelwood mechanism similar to that proposed by Graaf et al. [53,54], taking into account two reactions representing the CO2 hydrogenation to methanol (Equation (1)) and the water-gas shift reaction (Equation (2)). According to this mechanistic model, CO and CO2 adsorb competitively on one site (s1), whereas H2 and H2O adsorb competitively on another site (s2). The participation of two different active sites in the CO2 hydrogenation reaction over Cu/ZnO-based catalysts has been well demonstrated in the literature. It is generally believed that the role of copper is to adsorb hydrogen and deliver atomic hydrogen by spillover, whereas the “support” serves not only as an agent that controls the catalyst texture, but also to adsorb and activate CO2 [21,26,38,48,50,55,56]. The latter assumption was supported by the present experimental results, which indicated that the addition of La2O3 improved methanol selectivity by increasing the fraction of basic sites of moderate strength on the catalyst surface. This was in agreement with literature results showing that methanol yield was closely related to the number and strength of moderate/strong basic sites, which could be tuned by modification of the support with suitable metal oxides [26,36,57].
According to the above model [53,54], hydrogen adsorbs dissociatively, whereas adsorption of methanol is considered to be negligible. The elementary reaction steps corresponding to this model are described below.
(i) Equilibrium adsorption of reactants and products:
(ii) Water-gas shift reaction (Equation (2)):
(iii) CO2 hydrogenation to methanol (Equation (1)):
Assuming that the total number of sites s1 and s2 was constant, kinetic expressions were derived by choosing different combinations of surface reactions as rate-determining steps (RDS). The best results were obtained assuming that the RDS were Reaction (7) for the WGS and Reaction (11) for the CO2 conversion to methanol and that all other reactions were at equilibrium. Accordingly, the reaction rate of the WGS reaction is given by the following expression:
where k5 is the kinetic constant of the WGS reaction, Pi is the partial pressure of compound I (CO2, H2, CO2, H2O), and KA is the equilibrium constant of the WGS reaction. Regarding the reaction rate of CO2 conversion to methanol, it is given by:
where k9 is the kinetic constant of the reaction, Pi is the partial pressure of compound i (CO2, H2, CO2, H2O, CH3OH), and KB is the equilibrium constant of the CO2 conversion to methanol reaction. Equilibrium constants KA and KB can be determined from data available in the literature (e.g., [58]).
Since kinetic measurements were obtained in the absence of products in the feed, the initial partial pressures of CH3OH, CO, and H2O were zero, and, assuming that the terms in the denominator of Equations (15) and (16) were much higher than unity, the above two equations can be simplified as follows to give the initial reaction rates of CO and methanol production:
The kinetic constants, ki, and the equilibrium constants, Ki, depend on temperature as follows:
where Ai and Ei correspond to the pre-exponential factor and the activation energy, respectively, of reaction step i and Bi and ΔHi are the pre-exponential factor and the adsorption enthalpy, respectively, of the adsorption of species i.
The introduction of Equations (19) and (20) to the rate Equations (17) and (18) allows the expression of the reaction rates of CO and methanol production as functions of temperature. The activation energies, pre-exponential factors, and adsorption enthalpies of the reactants were estimated by fitting the experimentally measured reaction rates (Figure 6) using MATLAB, and the values obtained are presented in Table 2. The values of the model parameters satisfied the thermodynamic constraints [59] and were in good agreement with the literature results. For example, the activation energies calculated for CO formation (126.6 kJ/mol) and methanol formation (66.3 kJ/mol) were close to those reported for Cu/ZnO-based catalysts [60].
The fitting of the experimental results made according to the rate equations developed for CO and methanol production (Equations (17) and (18)) is shown in Figure 6 (solid lines). It was observed that the proposed kinetic model described the measured reaction rates well. It is the intention of the authors to further test the catalytic performance of the CZA-La50 catalyst at high pressures, relevant to practical applications, and to test the model under these conditions. This will be the subject of our future investigation.
4. Materials and Methods
4.1. Catalyst Preparation
The CZA-Lax catalysts were synthesized by the co-precipitation method, using Cu(NO3)2.3H2O, Zn(NO3)2.6H2O, Al(NO3)3.9H2O, and La(NO3)3.6H2O (Alfa Aesar, Thermo Fisher (Kandel) GmbH, Kandel, Germany) as metal precursor salts. Weighed amounts of the precursors, calculated to yield the desired metal oxide composition, were dissolved in distilled water, and the solution was heated at 65 °C. This was followed by dropwise addition of the precipitating agent (Na2CO3 1M) at pH 6.2 ± 0.1 under continuous stirring. After aging for 1 h, the precipitate was filtered, washed with distilled water, dried overnight at 80 °C, and finally calcined in air at 300 °C for 3 h.
4.2. Catalyst Characterization
The textural properties of the catalysts (specific surface area and porosity) were studied employing nitrogen adsorption at the temperature of liquid N2 using a Micromeritics TriStar 3000 instrument. Specific surface areas were calculated using the BET equation for nitrogen relative pressures in the range 0.06 < P/Po < 0.20. The pore size distribution was estimated with the Barret–Joyner–Halenda (BJH) method.
X-ray diffraction (XRD) patterns were obtained on a Bruker D8 Advance diffractometer (Billerica, MA, USA) equipped with a Ni-filtered Cu radiation and a LynxEye detector. The diffractograms were recorded in the range 20° < 2θ < 80° with a scan speed of 0.3 s/step.
4.3. In Situ DRIFTS Experiments
The in situ diffuse reflectance infrared Fourier-transform (DRIFT) experiments were performed on a Nicolet 6700 FTIR spectrometer (Waltham, MA, USA) equipped with a DRIFT cell (Spectra Tech), an MCT detector, and a KBr beam splitter. Typically, the catalyst sample was heated under He flow (30 cm3 min−1) at 450 °C for 10 min and then reduced with 20% H2 in He (30 cm3 min−1) at 300 °C for 1 h. The adsorbed species were desorbed by heating at 450 °C for 10 min, and the sample was cooled to room temperature. During the cooling stage, background spectra were recorded under He flow at the desired temperatures. The flow was finally switched to the reaction mixture (1% CO2 + 9% H2 in He, 30 cm3 min−1), and the first spectrum was recorded at 25 °C after 15 min on-stream. The temperature was then step-wise increased up to 400 °C, and spectra were obtained at the desired temperatures after equilibration for 15 min on-stream.
4.4. Transient-MS and CO2-TPD Experiments
Temperature-programmed surface reaction experiments coupled with mass spectrometry (transient-MS) were carried out employing the equipment and following the procedures described elsewhere [40]. Briefly, 100 mg of the catalyst sample were placed in a quartz reactor, heated under He flow at 450 °C for 15 min, and then reduced with H2 at 300 °C for 1 h. The sample was purged with He at the same temperature for 15 min and finally cooled to room temperature under He flow. Transient experiments were then carried out by switching the flow to the feed composition (1% CO2 + 9% H2 in He). In all experiments, the total flow rate through the reactor was 30 cm3 min−1. The effluent gas composition was monitored on-line with an Omnistar/Pfeiffer Vacuum mass spectrometer (Asslar, Germany).
The temperature-programmed desorption of CO2 (CO2-TPD) was studied over catalyst samples previously reduced with hydrogen at 300 °C following the procedure described above. Adsorption of CO2 was carried out at room temperature following exposure of the sample to a 1% CO2/He mixture for 1 h. The gas stream was then switched to He (30 cm3 min−1), and the temperature was increased to 400 °C with a heating rate of 10 °C/min.
4.5. Catalytic Performance Tests and Kinetic Measurements
The catalytic performance of the synthesized catalysts for the title reaction was evaluated at atmospheric pressure. The fixed-bed reactor employed consisted of a quartz tube (6 mm O.D.) with an expanded 1 cm long section in the middle (8 mm I.D.), containing the catalyst sample. The reaction temperature was measured in the middle of the catalyst bed by means of a K-type thermocouple running through the reactor. The feed composition was controlled with the use of mass-flow controllers. A gas chromatograph (Shimadzu, Kyoto, Japan) equipped with two packed columns (Carboxen, Carbovax) and two detectors (TCD, FID), operating with He as the carrier gas, was used for the analysis of the reactor effluent.
In a typical experiment, 200 mg of a fresh catalyst sample (0.18 mm < d < 0.25 mm) were placed in the reactor and reduced in situ at 300 °C for 1 h under a hydrogen flow of 50 cm3 min−1. The temperature was then lowered at 260 °C, and the flow was switched to the reaction mixture (10% CO2 + 90% H2, total flow rate: 50 cm3 min−1). The catalyst was conditioned at 260 °C for 30 min, and then, the concentrations of reactants and products were determined by gas chromatography. Similar measurements were carried out following a step-wise lowering of temperature down to 160 °C. Carbon dioxide conversion (XCO2) and selectivities (Si) toward product i (CH4, CO, CH3OH) were calculated using the following expressions:
where and are the concentrations of CO2 at the inlet and the outlet of the reactor, respectively.
Intrinsic reaction rates were measured in separate experiments where the conversion of carbon dioxide was kept below 15% so that differential reaction conditions could be assumed, with negligible heat and mass transfer effects. Kinetic experiments were conducted by varying the partial pressure of one reactant, while keeping the partial pressure of the other reactant constant using He as the balance gas. The procedure followed prior to kinetic measurements was similar to that described above for catalytic performance test.
5. Conclusions
A set of five La-doped CuO/ZnO/Al2O3 catalysts with variable La2O3 content was synthesized by the co-precipitation method and evaluated for CO2 hydrogenation toward methanol under atmospheric pressure. The results of catalyst characterization showed that substitution of Al2O3 by La2O3 resulted in a decrease of the specific surface area, which was more evident for samples with high La2O3 content, and in changes of the pore volume and pore size, which were maximized for the CZA-La50 sample where 50% of Al2O3 was replaced by La2O3. Furthermore, La-promotion resulted in an increase of the relative population and strength of basic sites of moderate strength.
The addition of La2O3 generally resulted in a decrease of CO2 conversion, which was less pronounced for the CZA-La50 sample, and in an increase of selectivity to methanol. The CZA-La50 catalyst exhibited the highest yields of methanol, which was 30% higher than that of the un-promoted catalyst at 210 °C. This could be attributed to the La-induced modification of the distribution of basic sites on the catalyst surface and the concomitant enhancement of the adsorption of CO2 and intermediate species that were involved in the methanol formation reaction. In addition, the CZA-La50 catalyst was found to maintain a relatively large part of copper in its metallic form under reaction conditions, indicating that the presence of this phase in high relative concentrations may be related to the enhanced methanol selectivities of the La-promoted catalysts.
The results of in situ DRIFTS experiments performed in the temperature range of 25–400 °C showed that the reaction proceeded with the intermediate formation of surface formate and methoxy species. These species, which were more clearly resolved over the CZA-La50 catalyst compared to the un-promoted sample, existed on the catalyst surface at temperatures up to 200 °C, where methanol and CO formation started taking place. Based on these observations and literature results, it was proposed that the adsorbed formate was the common surface intermediate species for both methanol and CO production.
The kinetics of the reaction were studied over the best performing CZA-La50 catalyst in the temperature range of 190–230 °C under different partial pressures of CO2 and H2. It was found that the increase of the partial pressure of H2 at a constant partial pressure of CO2 did not affect the rate of CO evolution appreciably, but resulted in an almost linear increase of the methanol formation rate. On the other hand, the increase of PCO2 at constant PH2 resulted in a progressive increase of the formation rates of both products. The kinetic measurements were modeled using a dual-site Langmuir–Hinshelwood mechanism according to which CO and CO2 adsorbed competitively on one site (s1), whereas H2 and H2O adsorbed competitively on a different site (s2). Based on the results of DRIFTS experiments, it was suggested that both methanol and CO were produced with the intermediate formation of formate species, which may either hydrogenate to yield methoxy species and, eventually, methanol, or convert to CO via the RWGS reaction. The rate expressions developed assuming that the rate-determining step (RDS) for CO production was the formation of the formate species and the RDS for methanol production was the formation of methoxy species provided the best fitting to the experimental kinetic results.
Author Contributions
M.K., K.K., and D.I.K. conceived of and designed the experiments; M.K. and K.K. performed the experiments; M.K., K.K., and D.I.K. analyzed the results and wrote the paper. All authors have read and agreed to the published version of the manuscript.
Funding
This research was performed under the research project entitled “Development of Catalysts for CO2 Hydrogenation to Methanol”, which is implemented through/co-financed by the Operational Program “Human Resources Development, Education and Lifelong Learning” and is co-financed by the European Union (European Social Fund) and Greek national funds.
Acknowledgments
The assistance of Theodora Ramantani, Ph.D. student at the Department of Chemical Engineering of the University of Patras, Greece, is gratefully acknowledged.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; nor in the decision to publish the results.
References
- Álvarez, A.; Bansode, A.; Urakawa, A.; Bavykina, A.V.; Wezendonk, T.A.; Makkee, M.; Gascon, J.; Kapteijn, F. Challenges in the greener production of formates/formic acid, methanol, and DME by heterogeneously catalyzed CO2 hydrogenation processes. Chem. Rev. 2017, 117, 9804–9838. [Google Scholar] [CrossRef]
- Kobayashi, H.; Taylor, J.M.; Mitsuka, Y.; Ogiwara, N.; Yamamoto, T.; Toriyama, T.; Matsumura, S.; Kitagawa, H. Charge transfer dependence on CO2 hydrogenation activity to methanol in Cu nanoparticles covered with metal–organic framework systems. Chem. Sci. 2019, 10, 3289–3294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.; Wang, S.; Ma, X.; Gong, J. Recent advances in catalytic hydrogenation of carbon dioxide. Chem. Soc. Rev. 2011, 40, 3703–3727. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowker, M. Methanol synthesis from CO2 hydrogenation. ChemCatChem 2019, 11, 4238–4246. [Google Scholar] [CrossRef] [Green Version]
- Tabatabaei, J.; Sakakini, B.H.; Waugh, K.C. On the mechanism of methanol synthesis and the water-gas shift reaction on ZnO. Catal. Lett. 2006, 110, 77–84. [Google Scholar] [CrossRef]
- Behrens, M.; Studt, F.; Kasatkin, I.; Kuhl, S.; Havecker, M.; Abild-Pedersen, F.; Zander, S.; Girgsdies, F.; Kurr, P.; Kniep, B.-L.; et al. The active site of methanol synthesis over Cu/ZnO/Al2O3 industrial catalysts. Science 2012, 336, 893–897. [Google Scholar] [CrossRef]
- Kattel, S.; Ramírez, P.J.; Chen, J.G.; Rodriguez, J.A.; Liu, P. Active sites for CO2 hydrogenation to methanol on Cu/ZnO catalysts. Science 2017, 355, 1296–1299. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, J.; Uchijima, T.; Kanai, Y.; Fujitani, T. The role of ZnO in Cu/ZnO methanol synthesis catalysts. Catal. Today 1996, 28, 223–230. [Google Scholar] [CrossRef]
- Yoshihara, J.; Parker, S.C.; Schafer, A.; Campbell, C.T. Methanol synthesis and reverse water-gas shift kinetics over clean polycrystalline copper. Catal. Lett. 1995, 31, 313–324. [Google Scholar] [CrossRef]
- Grabow, L.C.; Mavrikakis, M. Mechanism of methanol synthesis on Cu through CO2 and CO hydrogenation. ACS Catal. 2011, 1, 365–384. [Google Scholar] [CrossRef]
- Huš, M.; Kopač, D.; Štefančič, N.S.; Jurković, D.L.; Dasireddy, V.D.B.C.; Likozar, B. Unravelling the mechanisms of CO2 hydrogenation to methanol on Cu-based catalysts using first-principles multiscale modelling and experiments. Catal. Sci. Technol. 2017, 7, 5900–5913. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Yao, H.; Jiang, Z.; Fang, T. Theoretical study of methanol synthesis from CO2 hydrogenation on PdCu3(111) surface. Appl. Surf. Sci. 2018, 451, 333–345. [Google Scholar] [CrossRef]
- Kakumoto, T.; Watanabe, T. A theoretical study for methanol synthesis by CO2 hydrogenation. Catal. Today 1997, 36, 39–44. [Google Scholar] [CrossRef]
- Liu, L.; Fan, F.; Bai, M.; Xue, F.; Ma, X.; Jiang, Z.; Fang, T. Mechanistic study of methanol synthesis from CO2 hydrogenation on Rh-doped Cu(111) surfaces. Mol. Catal. 2019, 466, 26–36. [Google Scholar] [CrossRef]
- Bahruji, H.; Bowker, M.; Hutchings, G.J.; Dimitratos, N.; Wells, P.; Gibson, E.; Jones, W.; Brookes, C.; Morgan, D.; Lalev, G.M. Pd/ZnO catalysts for direct CO2 hydrogenation to methanol. J. Catal. 2016, 343, 133–146. [Google Scholar] [CrossRef] [Green Version]
- Li, K.; Chen, J.G. CO2 hydrogenation to methanol over ZrO2-containing catalysts: Insights into ZrO2 induced synergy. ACS Catal. 2019, 9, 7840–7861. [Google Scholar] [CrossRef]
- Phongamwong, T.; Chantaprasertporn, U.; Witoon, T.; Numpilai, T.; Poo-arporn, Y.; Limphirat, W.; Donphai, W.; Dittanet, P.; Chareonpanich, M.; Limtrakul, J. CO2 hydrogenation to methanol over CuO–ZnO–ZrO2–SiO2 catalysts: Effects of SiO2 contents. Chem. Eng. J. 2017, 316, 692–703. [Google Scholar] [CrossRef]
- Słoczyński, J.; Grabowski, R.; Olszewski, P.; Kozłowska, A.; Stoch, J.; Lachowska, M.; Skrzypek, J. Effect of metal oxide additives on the activity and stability of Cu/ZnO/ZrO2 catalysts in the synthesis of methanol from CO2 and H2. Appl. Catal. A 2006, 310, 127–137. [Google Scholar] [CrossRef]
- Hu, X.; Zhao, C.; Guan, Q.; Hu, X.; Li, W.; Chen, J. Selective hydrogenation of CO2 over a Ce promoted Cu-based catalyst confined by SBA-15. Inorg. Chem. Front. 2019, 6, 1799–1812. [Google Scholar] [CrossRef]
- Allam, D.; Bennici, S.; Limousy, L.; Hocine, S. Improved Cu- and Zn-based catalysts for CO2 hydrogenation to methanol. C. R. Chim. 2019, 22, 227–237. [Google Scholar] [CrossRef]
- Gao, P.; Li, F.; Zhao, N.; Xiao, F.; Wei, W.; Zhong, L.; Sun, Y. Influence of modifier (Mn, La, Ce, Zr and Y) on the performance of Cu/Zn/Al catalysts via hydrotalcite-like precursors for CO2 hydrogenation to methanol. Appl. Catal. A 2013, 468, 442–452. [Google Scholar] [CrossRef]
- Lam, E.; Corral-Pérez, J.J.; Larmier, K.; Noh, G.; Wolf, P.; Comas-Vives, A.; Urakawa, A.; Copéret, C. CO2 hydrogenation on Cu/Al2O3: Role of the metal/support interface in driving activity and selectivity of a bifunctional catalyst. Angew. Chem. Int. Ed. 2019, 58, 13989–13996. [Google Scholar] [CrossRef]
- Natesakhawat, S.; Lekse, J.W.; Baltrus, J.P.; Ohodnicki, P.R.; Howard, B.H.; Deng, X.; Matranga, C. Active sites and structure–activity relationships of copper-based catalysts for carbon dioxide hydrogenation to methanol. ACS Catal. 2012, 2, 1667–1676. [Google Scholar] [CrossRef]
- Zhan, H.; Li, F.; Gao, P.; Zhao, N.; Xiao, F.; Wei, W.; Zhong, L.; Sun, Y. Methanol synthesis from CO2 hydrogenation over La–M–Cu–Zn–O (M = Y, Ce, Mg, Zr) catalysts derived from perovskite-type precursors. J. Power Sources 2014, 251, 113–121. [Google Scholar] [CrossRef]
- Hayward, J.S.; Smith, P.J.; Kondrat, S.A.; Bowker, M.; Hutchings, G.J. The effects of secondary oxides on copper-based catalysts for green methanol synthesis. ChemCatChem 2017, 9, 1655–1662. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Mao, D.; Lu, G.; Wang, S.; Wu, G. The influence of La doping on the catalytic behavior of Cu/ZrO2 for methanol synthesis from CO2 hydrogenation. J. Mol. Catal. A 2011, 345, 60–68. [Google Scholar] [CrossRef]
- Yang, C.; Ren, J.; Sun, Y. Role of La2O3 in Pd-supported catalysts for methanol decomposition. Catal. Lett. 2002, 84, 123–129. [Google Scholar] [CrossRef]
- Borodko, Y.; Somorjai, G.A. Catalytic hydrogenation of carbon oxides—A 10-year perspective. Appl. Catal. A 1999, 186, 355–362. [Google Scholar] [CrossRef]
- Toyir, J.; Miloua, R.; Elkadri, N.E.; Nawdali, M.; Toufik, H.; Miloua, F.; Saito, M. Sustainable process for the production of methanol from CO2 and H2 using Cu/ZnO-based multicomponent catalyst. Phys. Procedia 2009, 2, 1075–1079. [Google Scholar] [CrossRef] [Green Version]
- Ma, J.; Sun, N.; Zhang, X.; Zhao, N.; Xiao, F.; Wei, W.; Sun, Y. A short review of catalysis for CO2 conversion. Catal. Today 2009, 148, 221–231. [Google Scholar] [CrossRef]
- Bonura, G.; Cordaro, M.; Cannilla, C.; Arena, F.; Frusteri, F. The changing nature of the active site of Cu-Zn-Zr catalysts for the CO2 hydrogenation reaction to methanol. Appl. Catal. B 2014, 152–153, 152–161. [Google Scholar] [CrossRef]
- An, X.; Li, J.; Zuo, Y.; Zhang, Q.; Wang, D.; Wang, J. A Cu/Zn/Al/Zr fibrous catalyst that is an improved CO2 hydrogenation to methanol catalyst. Catal. Lett. 2007, 118, 264–269. [Google Scholar] [CrossRef]
- Yoshihara, J.; Campbell, C.T. Methanol synthesis and reverse water–gas shift kinetics over Cu(110) model catalysts: Structural sensitivity. J. Catal. 1996, 161, 776–782. [Google Scholar] [CrossRef]
- Yang, Y.; Mims, C.A.; Disselkamp, R.S.; Kwak, J.-H.; Peden, C.H.F.; Campbell, C.T. (Non)formation of methanol by direct hydrogenation of formate on copper catalysts. J. Phys. Chem. C 2010, 114, 17205–17211. [Google Scholar] [CrossRef]
- Ramli, M.Z.; Syed-Hassan, S.S.A.; Hadi, A. Performance of Cu-Zn-Al-Zr catalyst prepared by ultrasonic spray precipitation technique in the synthesis of methanol via CO2 hydrogenation. Fuel Process. Technol. 2018, 169, 191–198. [Google Scholar] [CrossRef]
- Cai, W.; Chen, Q.; Wang, F.; Li, Z.; Yu, H.; Zhang, S.; Cui, L.; Li, C. Comparison of the promoted CuZnMxOy (M: Ga, Fe) catalysts for CO2 hydrogenation to methanol. Catal. Lett. 2019, 149, 2508–2518. [Google Scholar] [CrossRef]
- Ban, H.; Li, C.; Asami, K.; Fujimoto, K. Influence of rare-earth elements (La, Ce, Nd and Pr) on the performance of Cu/Zn/Zr catalyst for CH3OH synthesis from CO2. Catal. Commun. 2014, 54, 50–54. [Google Scholar] [CrossRef]
- Arena, F.; Mezzatesta, G.; Zafarana, G.; Trunfio, G.; Frusteri, F.; Spadaro, L. How oxide carriers control the catalytic functionality of the Cu–ZnO system in the hydrogenation of CO2 to methanol. Catal. Today 2013, 210, 39–46. [Google Scholar] [CrossRef]
- Sun, Q.; Liu, C.-W.; Pan, W.; Zhu, Q.-M.; Deng, J.-F. In situ IR studies on the mechanism of methanol synthesis over an ultrafine Cu/ZnO/Al2O3 catalyst. Appl. Catal. A 1998, 171, 301–308. [Google Scholar] [CrossRef]
- Panagiotopoulou, P.; Kondarides, D.I.; Verykios, X.E. Mechanistic study of the selective methanation of CO over Ru/TiO2 catalyst. Identification of active surface species and reaction pathways. J. Phys. Chem. C 2011, 115, 1220–1230. [Google Scholar] [CrossRef]
- Le Peltier, F.; Chaumette, P.; Saussey, J.; Bettahar, M.M.; Lavalley, J.C. In situ FT-IR and kinetic study of methanol synthesis from CO/H2 over ZnAl2O4 and Cu–ZnAl2O4 catalysts. J. Mol. Catal. A 1998, 132, 91–100. [Google Scholar] [CrossRef]
- Sanchez-Escribano, V.; Larrubia Vargas, M.A.; Finocchio, E.; Busca, G. On the mechanisms and the selectivity determining steps in syngas conversion over supported metal catalysts: An IR study. Appl. Catal. A 2007, 316, 68–74. [Google Scholar] [CrossRef]
- Schumann, J.; Eichelbaum, M.; Lunkenbein, T.; Thomas, N.; Alvarez Galvan, M.C.; Schlogl, R.; Behrens, M. Promoting strong metal support interaction: Doping ZnO for enhanced activity of Cu/ZnO:M (M=Al, Ga, Mg) catalysts. ACS Catal. 2015, 5, 3260–3270. [Google Scholar] [CrossRef]
- Fang, X.; Men, Y.; Wu, F.; Zhao, Q.; Singh, R.; Xiao, P.; Du, T.; Webley, P.A. Moderate-pressure conversion of H2 and CO2 to methanol via adsorption enhanced hydrogenation. Int. J. Hydrogen Energy 2019, 44, 21913–21925. [Google Scholar] [CrossRef]
- Xiao, J.; Mao, D.; Guo, X.; Yu, J. Effect of TiO2, ZrO2, and TiO2–ZrO2 on the performance of CuO–ZnO catalyst for CO2 hydrogenation to methanol. Appl. Surf. Sci. 2015, 338, 146–153. [Google Scholar] [CrossRef]
- Li, C.; Yuan, X.; Fujimoto, K. Development of highly stable catalyst for methanol synthesis from carbon dioxide. Appl. Catal. A 2014, 469, 306–311. [Google Scholar] [CrossRef]
- Din, I.U.; Shaharun, M.S.; Alotaibi, M.A.; Alharthi, A.I.; Naeem, A. Recent developments on heterogeneous catalytic CO2 reduction to methanol. J. CO₂ Util. 2019, 34, 20–33. [Google Scholar] [CrossRef]
- Saito, M.; Fujitani, T.; Takeuchi, M.; Watanabe, T. Development of copper/zinc oxide-based multicomponent catalysts for methanol synthesis from carbon dioxide and hydrogen. Appl. Catal. A 1996, 138, 311–318. [Google Scholar] [CrossRef]
- Liu, Y.-M.; Liu, J.-T.; Liu, S.-Z.; Li, J.; Gao, Z.-H.; Zuo, Z.-J.; Huang, W. Reaction mechanisms of methanol synthesis from CO/CO2 hydrogenation on Cu2O(111): Comparison with Cu(111). J. CO₂ Util. 2017, 20, 59–65. [Google Scholar] [CrossRef]
- Dasireddy, V.B.C.D.; Likozar, B. The role of copper oxidation state in Cu/ZnO/Al2O3 catalysts in CO2 hydrogenation and methanol productivity. Renew. Energy 2019, 140, 452–460. [Google Scholar] [CrossRef]
- Fisher, I.A.; Woo, H.C.; Bell, A.T. Effects of zirconia promotion on the activity of Cu/SiO2 for methanol synthesis from CO/H2 and CO2/H2. Catal. Lett. 1997, 44, 11–17. [Google Scholar] [CrossRef]
- Hong, Q.J.; Liu, Z.P. Mechanism of CO2 hydrogenation over Cu/ZrO2(212) interface from first-principles kinetics Monte Carlo simulations. Surf. Sci. 2010, 604, 1869–1876. [Google Scholar]
- Graaf, G.H.; Stamhuis, E.J.; Beenackers, A.A.C.M. Kinetics of low-pressure methanol synthesis. Chem. Eng. Sci. 1988, 43, 3185–3195. [Google Scholar] [CrossRef]
- Graaf, G.H.; Scholtens, H.; Stamhuis, E.J.; Beenackers, A.A.C.M. Intra-particle diffusion limitations in low-pressure methanol synthesis. Chem. Eng. Sci. 1988, 45, 773–783. [Google Scholar] [CrossRef]
- Tisseraud, C.; Comminges, C.; Belin, T.; Ahouari, H.; Soualah, A.; Pouilloux, Y.; Le Valant, A. The Cu–ZnO synergy in methanol synthesis from CO2, Part 2: Origin of the methanol and CO selectivities explained by experimental studies and a sphere contact quantification model in randomly packed binary mixtures on Cu–ZnO coprecipitate catalysts. J. Catal. 2015, 330, 533–544. [Google Scholar] [CrossRef]
- Dasireddy, V.D.B.C.; Štefančič, N.S.; Huš, M.; Likozar, B. Effect of alkaline earth metal oxide (MO) Cu/MO/Al2O3 catalysts on methanol synthesis activity and selectivity via CO2 reduction. Fuel 2018, 233, 103–112. [Google Scholar] [CrossRef]
- Liu, C.; Guo, X.; Guo, Q.; Mao, D.; Yu, J.; Lu, G. Methanol synthesis from CO2 hydrogenation over copper catalysts supported on MgO-modified TiO2. J. Mol. Catal. A 2016, 425, 86–93. [Google Scholar] [CrossRef]
- Graaf, G.H.; Sijtsema, P.J.J.M.; Stamhuis, E.J.; Joosten, G.E.H. Chemical equilibria in methanol synthesis. Chem. Eng. Sci. 1986, 41, 2883–2890. [Google Scholar] [CrossRef]
- Boudart, M.; Djega-Mariadassou, G. Kinetics of Heterogeneous Catalytic Reactions; Princeton University Press: Princeton, NJ, USA, 1984. [Google Scholar]
- Karelovic, A.; Ruiz, P. The role of copper particle size in low pressure methanol synthesis via CO2 hydrogenation over Cu/ZnO catalysts. Catal. Sci. Technol. 2015, 5, 869–881. [Google Scholar] [CrossRef]
Figure 1.
XRD patterns obtained for (A) the freshly prepared and (B) the spent CZA-Lax catalysts: (a) x = 0; (b) x = 25; (c) x = 50; (d) x = 75; (e) x = 100.
Figure 1.
XRD patterns obtained for (A) the freshly prepared and (B) the spent CZA-Lax catalysts: (a) x = 0; (b) x = 25; (c) x = 50; (d) x = 75; (e) x = 100.

Figure 2.
CO2-temperature-programmed desorption (TPD) profiles obtained for the pre-reduced CZA-Lax catalysts: (a) x = 0; (b) x = 50; (c) x = 100.
Figure 2.
CO2-temperature-programmed desorption (TPD) profiles obtained for the pre-reduced CZA-Lax catalysts: (a) x = 0; (b) x = 50; (c) x = 100.

Figure 3.
(A) Conversion of CO2, (B) selectivity to methanol, and (C) yield of methanol as functions of the reaction temperature obtained over CZA-Lax catalysts with x = 0, 50, or 100. Experimental conditions: mass of catalyst: 200 mg; total flow rate: 50 cm3 min−1; feed composition: 10% CO2 and 90% H2.
Figure 3.
(A) Conversion of CO2, (B) selectivity to methanol, and (C) yield of methanol as functions of the reaction temperature obtained over CZA-Lax catalysts with x = 0, 50, or 100. Experimental conditions: mass of catalyst: 200 mg; total flow rate: 50 cm3 min−1; feed composition: 10% CO2 and 90% H2.

Figure 4.
DRIFT spectra obtained over (A) the CZA-La0 and (B) the CZA-La50 catalyst following interaction with 1% CO (in He) at room temperature (a), purging with He for 30 min (b–d), and subsequent stepwise heating under He flow at temperatures up to 200 °C (e–g).
Figure 4.
DRIFT spectra obtained over (A) the CZA-La0 and (B) the CZA-La50 catalyst following interaction with 1% CO (in He) at room temperature (a), purging with He for 30 min (b–d), and subsequent stepwise heating under He flow at temperatures up to 200 °C (e–g).

Figure 5.
In situ DRIFT spectra obtained over (A) the CZA-La0 and (B) the CZA-La50 catalyst following interaction with a reaction mixture consisting of 1% CO2 and 9% H2 (in He) at (a) 25 °C and stepwise heating at: (b) 100 °C; (c) 150 °C; (d) 200 °C; (e) 250 °C; (f) 300 °C; (g) 350 °C; (h) 400 °C. The transient-MS spectra obtained over the CZA-La0 and CZA-La50 catalyst under the same conditions are shown in (C) and (D), respectively.
Figure 5.
In situ DRIFT spectra obtained over (A) the CZA-La0 and (B) the CZA-La50 catalyst following interaction with a reaction mixture consisting of 1% CO2 and 9% H2 (in He) at (a) 25 °C and stepwise heating at: (b) 100 °C; (c) 150 °C; (d) 200 °C; (e) 250 °C; (f) 300 °C; (g) 350 °C; (h) 400 °C. The transient-MS spectra obtained over the CZA-La0 and CZA-La50 catalyst under the same conditions are shown in (C) and (D), respectively.

Figure 6.
Effects of the partial pressure of H2 in the feed at the constant partial pressure of CO2 (A,B) and of the partial pressure of CO2 at constant partial pressure of H2 (C,D) on the rates of CO (A,C) and methanol (B,D) evolution at temperatures of 190, 210, and 230 °C. Solid lines correspond to fitting of experimental results (symbols) with the use of the kinetic model described in Section 3.2.
Figure 6.
Effects of the partial pressure of H2 in the feed at the constant partial pressure of CO2 (A,B) and of the partial pressure of CO2 at constant partial pressure of H2 (C,D) on the rates of CO (A,C) and methanol (B,D) evolution at temperatures of 190, 210, and 230 °C. Solid lines correspond to fitting of experimental results (symbols) with the use of the kinetic model described in Section 3.2.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Textural properties of the synthesized CuO/ZnO/Al2O3 (CZA)-Lax catalysts.
| Catalyst | Specific Surface Area (m2 g−1) | Pore Volume (cm3 g−1) | Pore Size (nm) |
|---|---|---|---|
| CZA-La0 | 111 | 0.70 | 20 |
| CZA-La25 | 88 | 0.78 | 30 |
| CZA-La50 | 96 | 0.79 | 30 |
| CZA-La75 | 61 | 0.45 | 27 |
| CZA-La100 | 47 | 0.32 | 25 |
Table 2.
Values of the parameters estimated by fitting the reaction rates with Equations (17) and (18).
Table 2.
Values of the parameters estimated by fitting the reaction rates with Equations (17) and (18).
| k5 | k9 | K1 | K2 | K = K7.K8 |
|---|---|---|---|---|
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kourtelesis, M.; Kousi, K.; Kondarides, D.I. CO2 Hydrogenation to Methanol over La2O3-Promoted CuO/ZnO/Al2O3 Catalysts: A Kinetic and Mechanistic Study. Catalysts 2020, 10, 183. https://0-doi-org.brum.beds.ac.uk/10.3390/catal10020183
AMA Style
Kourtelesis M, Kousi K, Kondarides DI. CO2 Hydrogenation to Methanol over La2O3-Promoted CuO/ZnO/Al2O3 Catalysts: A Kinetic and Mechanistic Study. Catalysts. 2020; 10(2):183. https://0-doi-org.brum.beds.ac.uk/10.3390/catal10020183
Chicago/Turabian StyleKourtelesis, Marios, Kalliopi Kousi, and Dimitris I. Kondarides. 2020. "CO2 Hydrogenation to Methanol over La2O3-Promoted CuO/ZnO/Al2O3 Catalysts: A Kinetic and Mechanistic Study" Catalysts 10, no. 2: 183. https://0-doi-org.brum.beds.ac.uk/10.3390/catal10020183
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.