Ceria-Based Catalysts Studied by Near Ambient Pressure X-ray Photoelectron Spectroscopy: A Review


 , ,
, , 
Abstract
:1. Ceria in Catalysis
2. Near Ambient Pressure XPS
3. Examples for the Application of NAP-XPS to Ceria Catalysts
3.1. Fundamental Studies
3.2. Gas-Solid Catalysis
3.2.1. CO Oxidation and Preferential CO Oxidation (PROX)
3.2.2. Water-Gas Shift Reaction (WGS)
3.2.3. Soot Oxidation
3.2.4. CO2 Hydrogenation
3.2.5. Hydrocarbons
Methane Partial and Complete Oxidation
Dry Reforming of Hydrocarbons
3.2.6. Alcohols
Steam Reforming of Alcohols
Methanol Oxidation
3.2.7. Hydrogenation Reactions
3.3. Gas-Solid Electrocatalysis
3.3.1. H2O Electrolysis/H2 Electro-Oxidation
3.3.2. CH4 Electro-Oxidation
3.3.3. CO2 Electrolysis/CO Electro-Oxidation
4. Summary and Outlook
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| AFM | atomic force microscopy | POX | partial oxidation |
| AP-IRRAS | ambient pressure IRRAS | PROX | preferential CO oxidation |
| CE | counter electrode | RWGS | reverse water-gas shift |
| CO-TPR | CO temperature-programmed reduction | ScSZ | scandia-stabilized zirconia |
| DFT | density-functional theory | SDC | samaria-doped ceria |
| DRB | dry reforming of butane | SEM | scanning electron microscopy |
| DRE | dry reforming of ethane | SMSI | strong metal-support interaction |
| DRIFTS | diffuse reflectance infrared Fourier transform spectroscopy | SOC | solid oxide electrochemical cell |
| DRM | dry reforming of methane | SOEC | solid oxide electrolysis cell |
| EXAFS | extended X-ray absorption fine structure | SOFC | solid oxide fuel cell |
| FT | Fischer-Tropsch | SRE | steam reforming of ethanol |
| GDC | gadolinium-doped ceria | SRM | steam reforming of methanol |
| INS | inelastic neutron scattering | STM | scanning tunnelling microscopy |
| IRRAS | infrared reflection absorption spectroscopy | STO | strontium titanate (SrTiO3) |
| LB | Langmuir-Blodgett | TR-XRD | time-resolved XRD |
| LSCM | lanthanum strontium chromium manganite | TWC | three-way catalyst |
| MIEC | mixed ionic-electronic conductor | UHV | ultra-high vacuum |
| ML | monolayer | VOC | volatile organic compounds |
| MPO | methane partial oxidation | WE | working electrode |
| MPs | microparticles | WGS | water-gas shift |
| NAP-XPS | near ambient pressure XPS | XANES | X-ray absorption near edge structure |
| NEXAFS | near-edge X-ray absorption fine structure | XAS | X-ray absorption spectroscopy |
| NPs | nanoparticles | XPS | X-ray photoelectron spectroscopy |
| OSC | oxygen storage capacity | XRD | X-ray diffraction |
| OSR | oxidative steam reforming | YSZ | yttria-stabilized zirconia |
| PEMFC | proton-exchange membrane fuel cell | Θ | coverage |
References
- Vivier, L.; Duprez, D. Ceria-Based Solid Catalysts for Organic Chemistry. ChemSusChem 2010, 3, 654–678. [Google Scholar] [CrossRef]
- Gorte, R.J. Ceria in Catalysis: From Automotive Applications to the Water-Gas Shift Reaction. AICHE J. 2010, 56, 1126–1135. [Google Scholar] [CrossRef]
- Montini, T.; Melchionna, M.; Monai, M.; Fornasiero, P. Fundamentals and Catalytic Applications of CeO2-Based Materials. Chem. Rev. 2016, 116, 5987–6041. [Google Scholar] [CrossRef] [PubMed]
- Trovarelli, A.; Llorca, J. Ceria Catalysts at Nanoscale: How Do Crystal Shapes Shape Catalysis? ACS Catal. 2017, 7, 4716–4735. [Google Scholar] [CrossRef]
- Wang, Q.; Yeung, K.L.; Bañares, M.A. Ceria and its related materials for VOC catalytic combustion: A review. Catal. Today 2019. [Google Scholar] [CrossRef]
- Liu, S.; Wu, X.; Weng, D.; Ran, R. Ceria-based catalysts for soot oxidation: A review. J. Rare Earths 2015, 33, 567–590. [Google Scholar] [CrossRef]
- Aneggi, E.; Wiater, D.; De Leitenburg, C.; Llorca, J.; Trovarelli, A. Shape-Dependent Activity of Ceria in Soot Combustion. ACS Catal. 2014, 4, 172–181. [Google Scholar] [CrossRef]
- Mai, H.; Sun, L.; Zhang, Y.; Si, R.; Feng, W.; Zhang, H.; Liu, H.-C.; Yan, C.-H. Shape-Selective Synthesis and Oxygen Storage Behavior of Ceria Nanopolyhedra, Nanorods, and Nanocubes. J. Phys. Chem. B 2005, 109, 24380–24385. [Google Scholar] [CrossRef]
- Conesa, J.C. Computer modeling of surfaces and defects on cerium dioxide. Surf. Sci. 1995, 339, 337–352. [Google Scholar] [CrossRef]
- Huang, W.; Gao, Y. Morphology-dependent surface chemistry and catalysis of CeO2 nanocrystals. Catal. Sci. Technol. 2014, 4, 3772–3784. [Google Scholar] [CrossRef]
- Wu, K.; Sun, L.; Yan, C. Recent Progress in Well-Controlled Synthesis of Ceria-Based Nanocatalysts towards Enhanced Catalytic Performance. Adv. Energy Mater. 2016, 6, 1600501. [Google Scholar] [CrossRef]
- Wang, Y.; Wöll, C. IR spectroscopic investigations of chemical and photochemical reactions on metal oxides: Bridging the materials gap. Chem. Soc. Rev. 2017, 46, 1875–1932. [Google Scholar] [CrossRef] [PubMed]
- Nolan, M.; Grigoleit, S.; Sayle, D.C.; Parker, S.C.; Watson, G.W. Density functional theory studies of the structure and electronic structure of pure and defective low index surfaces of ceria. Surf. Sci. 2005, 576, 217–229. [Google Scholar] [CrossRef]
- Branda, M.M.; Ferullo, R.M.; Caus, M.; Illas, F. Relative Stabilities of Low Index and Stepped CeO2 Surfaces from Hybrid and GGA+U Implementations of Density Functional Theory. J. Phys. Chem. C 2011, 115, 3716–3721. [Google Scholar] [CrossRef]
- Yang, C.; Yu, X.; Heißler, S.; Nefedov, A.; Colussi, S.; Llorca, J.; Trovarelli, A.; Wang, Y.; Wöll, C. Surface Faceting and Reconstruction of Ceria Nanoparticles. Angew. Chem. Int. Ed. 2017, 56, 375–379. [Google Scholar] [CrossRef]
- Aneggi, E.; Rico-Perez, V.; De Leitenburg, C.; Maschio, S.; Soler, L.; Llorca, J.; Trovarelli, A. Ceria-Zirconia Particles Wrapped in a 2D Carbon Envelope: Improved Low-Temperature Oxygen Transfer and Oxidation Activity. Angew. Chem. Int. Ed. 2015, 54, 14040–14043. [Google Scholar] [CrossRef] [Green Version]
- Paier, J.; Penschke, C.; Sauer, J. Oxygen Defects and Surface Chemistry of Ceria: Quantum Chemical Studies Compared to Experiment. Chem. Rev. 2013, 113, 3949–3985. [Google Scholar] [CrossRef]
- Aneggi, E.; Llorca, J.; Boaro, M.; Trovarelli, A. Surface-structure sensitivity of CO oxidation over polycrystalline ceria powders. J. Catal. 2005, 234, 88–95. [Google Scholar] [CrossRef]
- Zhou, X.; Huebner, W. Size-induced lattice relaxation in CeO2 nanoparticles. Appl. Phys. Lett. 2001, 79, 3512–3514. [Google Scholar] [CrossRef]
- Migani, A.; Vayssilov, G.N.; Bromley, S.T.; Illas, F.; Neyman, K.M. Dramatic reduction of the oxygen vacancy formation energy in ceria particles: A possible key to their remarkable reactivity at the nanoscale. J. Mater. Chem. 2010, 20, 10535–10546. [Google Scholar] [CrossRef]
- Bruix, A.; Neyman, K.M. Modeling Ceria-Based Nanomaterials for Catalysis and Related Applications. Catal. Lett. 2016, 146, 2053–2080. [Google Scholar] [CrossRef]
- Gatica, J.M.; Gómez, D.M.; Hernández-Garrido, J.C.; Calvino, J.J.; Cifredo, G.A.; Vidal, H. Experimental evidences of the relationship between reducibility and micro- and nanostructure in commercial high surface area ceria. Appl. Catal. A Gen. 2014, 479, 35–44. [Google Scholar] [CrossRef]
- Aneggi, E.; Llorca, J.; Trovarelli, A.; Aouine, M.; Vernoux, P. In situ environmental HRTEM discloses low temperature carbon soot oxidation by ceria-zirconia at the nanoscale. Chem. Commun. 2019, 55, 3876–3878. [Google Scholar] [CrossRef] [PubMed]
- Kullgren, J.; Hermansson, K.; Broqvist, P. Supercharged low-temperature oxygen storage capacity of ceria at the nanoscale. J. Phys. Chem. Lett. 2013, 4, 604–608. [Google Scholar] [CrossRef] [PubMed]
- Guzman, J.; Carrettin, S.; Corma, A. Spectroscopic evidence for the supply of reactive oxygen during CO oxidation catalyzed by gold supported on nanocrystalline CeO2. J. Am. Chem. Soc. 2005, 127, 3286–3287. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Harmer, J.; Li, G.; Chapman, T.; Collier, P.; Longworth, S.; Tsang, S.C. Size dependent oxygen buffering capacity of ceria nanocrystals. Chem. Commun. 2010, 46, 1887–1889. [Google Scholar] [CrossRef]
- Liu, X.; Zhou, K.; Wang, L.; Wang, B.; Li, Y. Oxygen Vacancy Clusters Promoting Reducibility and Activity of Ceria Nanorods. J. Am. Chem. Soc. 2009, 131, 3140–3141. [Google Scholar] [CrossRef]
- Wang, L.; Wang, Y.; Zhang, Y.; Yu, Y.; He, H.; Qin, X.; Wang, B. Shape dependence of nanoceria on complete catalytic oxidation of o-xylene. Catal. Sci. Technol. 2016, 6, 4840–4848. [Google Scholar] [CrossRef]
- Carrettin, S.; Concepción, P.; Corma, A.; López Nieto, J.M.; Puntes, V.F. Nanocrystalline CeO2 increases the activity of Au for CO oxidation by two orders of magnitude. Angew. Chem. Int. Ed. 2004, 43, 2538–2540. [Google Scholar] [CrossRef]
- Fu, Q.; Saltsburg, H.; Flytzani-stephanopoulos, M. Active Nonmetallic Au and Pt Species on Ceria-Based Water-Gas Shift Catalysts. Science 2003, 301, 935–939. [Google Scholar] [CrossRef] [PubMed]
- Danielis, M.; Colussi, S.; De Leitenburg, C.; Soler, L.; Llorca, J.; Trovarelli, A. Outstanding Methane Oxidation Performance of Palladium-Embedded Ceria Catalysts Prepared by a One-Step Dry Ball-Milling Method. Angew. Chem. Int. Ed. 2018, 57, 10212–10216. [Google Scholar] [CrossRef]
- Farmer, J.A.; Campbell, C.T. Ceria Maintains Smaller Metal Catalyst Particles by Strong Metal-Support Bonding. Science 2010, 329, 933–936. [Google Scholar] [CrossRef]
- Jones, J.; Xiong, H.; DeLaRiva, A.T.; Peterson, E.J.; Pham, H.; Challa, S.R.; Qi, G.; Oh, S.; Wiebenga, M.H.; Hernández, X.I.P.; et al. Thermally stable single-atom platinum-on-ceria catalysts via atom trapping. Science 2016, 353, 150–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cargnello, M.; Doan-nguyen, V.V.T.; Gordon, T.R.; Diaz, R.E.; Stach, E.A.; Gorte, R.J.; Fornasiero, P.; Murray, C.B. Control of Metal Nanocrystal Size Reveals Metal-Support Interface Role for Ceria Catalysts. Science 2013, 341, 771–774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, J.A.; Grinter, D.C.; Liu, Z.; Palomino, R.M.; Senananayake, S.D. Ceria-based model catalysts: Fundamental studies on the importance of the metal-ceria interface in CO oxidation, the water-gas shift, CO2 hydrogenation, and methane and alcohol reforming. Chem. Soc. Rev. 2017, 46, 1824–1841. [Google Scholar] [CrossRef] [PubMed]
- Trovarelli, A. Catalysis by Ceria and Related Materials; Imperial College Press: London, UK, 2002. [Google Scholar]
- Rodriguez, J.A.; Senanayake, S.D.; Stacchiola, D.; Liu, P.; Hrbek, J. The activation of gold and the water-gas shift reaction: Insights from studies with model catalysts. Acc. Chem. Res. 2014, 47, 773–782. [Google Scholar] [CrossRef]
- Vayssilov, G.N.; Lykhach, Y.; Migani, A.; Staudt, T.; Petrova, G.P.; Tsud, N.; Skála, T.; Bruix, A.; Illas, F.; Prince, K.C.; et al. Support nanostructure boosts oxygen transfer to catalytically active platinum nanoparticles. Nat. Mater. 2011, 10, 310–315. [Google Scholar] [CrossRef]
- Mudiyanselage, K.; Senanayake, S.D.; Feria, L.; Kundu, S.; Baber, A.E.; Graciani, J.; Vidal, A.B.; Agnoli, S.; Evans, J.; Chang, R.; et al. Importance of the metal-oxide interface in catalysis: In situ studies of the water-gas shift reaction by ambient-pressure X-ray photoelectron spectroscopy. Angew. Chem. Int. Ed. 2013, 52, 5101–5105. [Google Scholar] [CrossRef]
- Soler, L.; Casanovas, A.; Urrich, A.; Angurell, I.; Llorca, J. CO oxidation and COPrOx over preformed Au nanoparticles supported over nanoshaped CeO2. Appl. Catal. B Environ. 2016, 197, 47–55. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.Y.; Lee, H.M.; Henkelman, G. CO Oxidation Mechanism on CeO2-Supported Au Nanoparticles. J. Am. Chem. Soc. 2012, 134, 1560–1570. [Google Scholar] [CrossRef] [Green Version]
- Patarroyo, J.; Delgado, J.A.; Merkoçi, F.; Genç, A.; Sauthier, G.; Llorca, J.; Arbiol, J.; Bastus, N.G.; Godard, C.; Claver, C.; et al. Hollow PdAg-CeO2 heterodimer nanocrystals as highly structured heterogeneous catalysts. Sci. Rep. 2019, 9, 18776. [Google Scholar] [CrossRef] [PubMed]
- Nordling, C.; Sokolowski, E.; Siegbahn, K. Precision Method for Obtaining Absolute Values of Atomic Binding Energies. Phys. Rev. 1957, 105, 1676–1677. [Google Scholar] [CrossRef]
- Siegbahn, K. ESCA: Atomic, Molecular and Solid State Structure Studied by Means of Electron Spectroscopy; Kunglia Vetenskap-Societeten Uppsala: Nova acta Regiae Societate’s Uppsaliensis; Almqvist & Wiksells: Uppsala, Sweden, 1967. [Google Scholar]
- Rupprechter, G.; Weilach, C. Spectroscopic studies of surface-gas interactions and catalyst restructuring at ambient pressure: Mind the gap! J. Phys. Condens. Matter 2008, 20, 184019. [Google Scholar] [CrossRef]
- Somorjai, G.A.; Frei, H.; Park, J.Y. Advancing the frontiers in nanocatalysis, biointerfaces, and renewable energy conversion by innovations of surface techniques. J. Am. Chem. Soc. 2009, 131, 16589–16605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Escudero, C.; Salmeron, M. From solid-vacuum to solid-gas and solid-liquid interfaces: In situ studies of structure and dynamics under relevant conditions. Surf. Sci. 2013, 607, 2–9. [Google Scholar] [CrossRef]
- Siegbahn, K.; Nordling, C.; Johansson, G.; Hedman, J.; Hedén, P.F.; Hamrin, K.; Gelius, U.; Bergmark, T.; Werme, L.O.; Manne, R.; et al. ESCA Applied to Free Molecules; North-Holland Publishing Company: Amsterdam, The Netherlands, 1969. [Google Scholar]
- Siegbahn, H.; Siegbahn, K. ESCA applied to liquids. J. Electron Spectros. Relat. Phenom. 1973, 2, 319–325. [Google Scholar] [CrossRef]
- Salmeron, M.; Schlögl, R. Ambient pressure photoelectron spectroscopy: A new tool for surface science and nanotechnology. Surf. Sci. Rep. 2008, 63, 169–199. [Google Scholar] [CrossRef] [Green Version]
- Ogletree, D.F.; Bluhm, H.; Lebedev, G.; Fadley, C.S.; Hussain, Z.; Salmeron, M. A differentially pumped electrostatic lens system for photoemission studies in the millibar range. Rev. Sci. Instrum. 2002, 73, 3872. [Google Scholar] [CrossRef]
- Shavorskiy, A.; Karslioglu, O.; Zegkinoglou, I.; Bluhm, H. Synchrotron-based Ambient Pressure X-ray Photoelectron Spectroscopy. Synchrotron Radiat. News 2014, 27, 14–23. [Google Scholar] [CrossRef]
- Arble, C.; Jia, M.; Newberg, J.T. Lab-based ambient pressure X-ray photoelectron spectroscopy from past to present. Surf. Sci. Rep. 2018, 73, 37–57. [Google Scholar] [CrossRef]
- Trotochaud, L.; Head, A.R.; Karslioǧlu, O.; Kyhl, L.; Bluhm, H. Ambient pressure photoelectron spectroscopy: Practical considerations and experimental frontiers. J. Phys. Condens. Matter 2017, 29, 053002. [Google Scholar] [CrossRef]
- Lundgren, E.; Zhang, C.; Merte, L.R.; Shipilin, M.; Blomberg, S.; Hejral, U.; Zhou, J.; Zetterberg, J.; Gustafson, J. Novel in Situ Techniques for Studies of Model Catalysts. Acc. Chem. Res. 2017, 50, 2326–2333. [Google Scholar] [CrossRef] [PubMed]
- Dou, J.; Sun, Z.; Opalade, A.A.; Wang, N.; Fu, W.; Tao, F. Operando chemistry of catalyst surfaces during catalysis. Chem. Soc. Rev. 2017, 46, 2001–2027. [Google Scholar] [CrossRef] [PubMed]
- Alayoglu, S.; Somorjai, G.A. Ambient Pressure X-ray Photoelectron Spectroscopy for Probing Monometallic, Bimetallic and Oxide-Metal Catalysts under Reactive Atmospheres and Catalytic Reaction Conditions. Top. Catal. 2016, 59, 420–438. [Google Scholar] [CrossRef]
- Wu, C.H.; Eren, B.; Salmeron, M.B. Structure and Dynamics of Reactant Coadsorption on Single Crystal Model Catalysts by HP-STM and AP-XPS: A Mini Review. Top. Catal. 2016, 59, 405–419. [Google Scholar] [CrossRef]
- Toyoshima, R.; Kondoh, H. In-situ observations of catalytic surface reactions with soft x-rays under working conditions. J. Phys. Condens. Matter 2015, 27, 83003. [Google Scholar] [CrossRef]
- Yoshida, M.; Kondoh, H. In Situ Observation of Model Catalysts under Reaction Conditions Using X-ray Core-Level Spectroscopy. Chem. Rec. 2014, 14, 806–818. [Google Scholar] [CrossRef] [PubMed]
- Starr, D.E.; Bluhm, H.; Liu, Z.; Knop-Gericke, A.; Hävecker, M. Application of Ambient-Pressure X-ray Photoelectron Spectroscopy for the In-situ Investigation of Heterogeneous Catalytic Reactions. In In-situ Characterization of Heterogeneous Catalysts; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2013; pp. 315–343. [Google Scholar]
- Starr, D.E.; Liu, Z.; Hävecker, M.; Knop-Gericke, A.; Bluhm, H. Investigation of solid/vapor interfaces using ambient pressure X-ray photoelectron spectroscopy. Chem. Soc. Rev. 2013, 42, 5833–5857. [Google Scholar] [CrossRef] [Green Version]
- Palomino, R.M.; Hamlyn, R.; Liu, Z.; Grinter, D.C.; Waluyo, I.; Rodriguez, J.A.; Senanayake, S.D. Interfaces in heterogeneous catalytic reactions: Ambient pressure XPS as a tool to unravel surface chemistry. J. Electron. Spectros. Relat. Phenom. 2017, 221, 28–43. [Google Scholar] [CrossRef]
- Nguyen, L.; Tao, F.F.; Tang, Y.; Dou, J.; Bao, X. Understanding Catalyst Surfaces during Catalysis through Near Ambient Pressure X-ray Photoelectron Spectroscopy. Chem. Rev. 2019, 119, 6822–6905. [Google Scholar] [CrossRef]
- Zhong, L.; Chen, D.; Zafeiratos, S. A mini review of in situ near-ambient pressure XPS studies on non-noble, late transition metal catalysts. Catal. Sci. Technol. 2019, 9, 3851–3867. [Google Scholar] [CrossRef]
- Van Spronsen, M.A.; Frenken, J.W.; Groot, I.M. Surface science under reaction conditions: CO oxidation on Pt and Pd model catalysts. Chem. Soc. Rev. 2017, 46, 4347–4374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, F.; Grass, M.E.; Zhang, Y.; Butcher, D.R.; Renzas, J.R.; Liu, Z.; Chung, J.Y.; Mun, B.S.; Salmeron, M.; Somorjai, G.A. Reaction-driven restructuring of Rh-Pd and Pt-Pd core-shell nanoparticles. Science 2008, 322, 932–934. [Google Scholar] [CrossRef] [Green Version]
- Divins, N.J.; Angurell, I.; Escudero, C.; Pérez-Dieste, V.; Llorca, J. Influence of the support on surface rearrangements of bimetallic nanoparticles in real catalysts. Science 2014, 346, 620–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soler, L.; Casanovas, A.; Ryan, J.; Angurell, I.; Escudero, C.; Pérez-Dieste, V.; Llorca, J. Dynamic Reorganization of Bimetallic Nanoparticles under Reaction Depending on the Support Nanoshape: The Case of RhPd over Ceria Nanocubes and Nanorods under Ethanol Steam Reforming. ACS Catal. 2019, 9, 3641–3647. [Google Scholar] [CrossRef]
- Crumlin, E.J.; Mutoro, E.; Hong, W.T.; Biegalski, M.D.; Christen, H.M.; Liu, Z.; Bluhm, H.; Shao-Horn, Y. In situ Ambient Pressure X-ray Photoelectron Spectroscopy of Cobalt Perovskite Surfaces under Cathodic Polarization at High Temperatures. J. Phys. Chem. C 2013, 117, 16087–16094. [Google Scholar] [CrossRef]
- Knop-gericke, A.A.; Pfeifer, V.; Jones, T.; Arrigo, R.; Michael, H. In situ X-ray Photoelectron Spectroscopy of Electrochemically Active Solid-Gas and Solid-Liquid Interfaces. J. Electron Spectros. Relat. Phenom. 2017, 221, 10–17. [Google Scholar] [CrossRef] [Green Version]
- Saveleva, V.A.; Savinova, E.R. Insights into electrocatalysis from ambient pressure photoelectron spectroscopy. Curr. Opin. Electrochem. 2019, 17, 79–89. [Google Scholar] [CrossRef]
- Friebel, D.; Ogasawara, H.; Nilsson, A. X-ray Spectroscopy at Electro-catalytic Interfaces. In Surface and Interface Science; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2020; pp. 733–772. [Google Scholar]
- Starr, D.E.; Favaro, M.; Abdi, F.F.; Bluhm, H.; Crumlin, E.J.; Van de Krol, R. Combined soft and hard X-ray ambient pressure photoelectron spectroscopy studies of semiconductor/electrolyte interfaces. J. Electron. Spectros. Relat. Phenom. 2017, 221, 106–115. [Google Scholar] [CrossRef] [Green Version]
- DeCaluwe, S.C.; Jackson, G.S.; Farrow, R.; McDaniel, A.; El Gabaly, F.; McCarty, K.; Nie, S.; Linne, M.; Bluhm, H.; Newberg, J.; et al. In Situ XPS for Evaluating Ceria Oxidation States in SOFC Anodes. ECS Trans. 2009, 16, 253–263. [Google Scholar]
- Trovarelli, A. Catalytic properties of ceria and CeO2-containing materials. Catal. Rev. 1996, 38, 439–520. [Google Scholar] [CrossRef]
- Tauster, S.J. Strong Metal-Support Interaction. Acc. Chem. Res. 1987, 20, 389–394. [Google Scholar] [CrossRef]
- Datye, A.K.; Kalakkad, D.S.; Yao, M.H.; Smith, D.J. Comparison of metal-support interactions in Pt/TiO2 and Pt/CeO2. J. Catal. 1995, 155, 148–153. [Google Scholar] [CrossRef]
- De Leitenburg, C.; Trovarelli, A. Metal-support interactions in Rh/CeO2, Rh/TiO2, and Rh/Nb2O5 catalysts as inferred from CO2 methanation activity. J. Catal. 1995, 156, 171–174. [Google Scholar] [CrossRef]
- Mogensen, M.; Sammes, N.M.; Tompsett, G.A. Physical, chemical and electrochemical properties of pure and doped ceria. Solid State Ion. 2000, 129, 63–94. [Google Scholar] [CrossRef]
- Caballero, A.; Holgado, J.P.; Gonzalez-Delacruz, V.M.; Habas, S.E.; Herranz, T.; Salmeron, M. In situ spectroscopic detection of SMSI effect in a Ni/CeO2 system: Hydrogen-induced burial and dig out of metallic nickel. Chem. Commun. 2010, 46, 1097–1099. [Google Scholar] [CrossRef] [Green Version]
- Bernardi, F.; Grass, M.E.; Hong, Y.P.; Chang, R.; Jabeen, N.; Zhang, C.; Eichhorn, B.W.; Seo, B.; Alayoglu, S.; Hussain, Z.; et al. Control of the surface atomic population of Rh0.5Pd0.5 bimetallic nanoparticles supported on CeO2. Catal. Today 2016, 260, 95–99. [Google Scholar] [CrossRef] [Green Version]
- Figueiredo, W.T.; Della Mea, G.B.; Segala, M.; Baptista, D.L.; Escudero, C.; Pérez-Dieste, V.; Bernardi, F. Understanding the Strong Metal–Support Interaction (SMSI) Effect in Cu x Ni 1–x /CeO2 (0 < x <1) Nanoparticles for Enhanced Catalysis. ACS Appl. Nano Mater. 2019, 2, 2559–2573. [Google Scholar]
- Carrasco, J.; Lõpez-Durán, D.; Liu, Z.; Duchoň, T.; Evans, J.; Senanayake, S.D.; Crumlin, E.J.; Matolín, V.; Rodríguez, J.A.; Ganduglia-Pirovano, M.V. In situ and theoretical studies for the dissociation of water on an active Ni/CeO2 Catalyst: Importance of strong metal-support interactions for the cleavage of O-H bonds. Angew. Chem. Int. Ed. 2015, 54, 3917–3921. [Google Scholar] [CrossRef]
- Sharma, S.; Hu, Z.; Zhang, P.; Mcfarland, E.W.; Metiu, H. CO2 methanation on Ru-doped ceria. J. Catal. 2011, 278, 297–309. [Google Scholar] [CrossRef]
- Bunluesin, T.; Gorte, R.J.; Graham, G.W. Studies of the water-gas-shift reaction on ceria-supported Pt, Pd, and Rh: Implications for oxygen-storage properties. Appl. Catal. B Environ. 1998, 15, 107–114. [Google Scholar] [CrossRef]
- Bernal, S.; Calvino, J.J.; Cauqui, M.A.; Gatica, J.M.; Larese, C.; Pérez Omil, J.A.; Pintado, J.M. Some recent results on metal/support interaction effects in NM/CeO2 (NM: Noble metal) catalysts. Catal. Today 1999, 50, 175–206. [Google Scholar] [CrossRef]
- Hilaire, S.; Wang, X.; Luo, T.; Gorte, R.J.; Wagner, J. A comparative study of water-gas-shift reaction over ceria supported metallic catalysts. Appl. Catal. A Gen. 2001, 215, 271–278. [Google Scholar] [CrossRef]
- Sudarsanam, P.; Mallesham, B.; Reddy, P.S.; Großmann, D.; Grünert, W.; Reddy, B.M. Nano-Au / CeO2 catalysts for CO oxidation: Influence of dopants (Fe, La and Zr) on the physicochemical properties and catalytic activity. Appl. Catal. B Environ. 2014, 144, 900–908. [Google Scholar] [CrossRef]
- Aguilar-guerrero, V.; Gates, B.C. Kinetics of CO oxidation catalyzed by highly dispersed CeO2 -supported gold. J. Catal. 2008, 260, 351–357. [Google Scholar] [CrossRef]
- Fonseca, J.; Royer, S.; Bion, N.; Pirault-Roy, L.; Do Carmo Range, M.; Duprez, D.; Epron, F. Preferential CO oxidation over nanosized gold catalysts supported on ceria and amorphous ceria-alumina. Appl. Catal. B Environ. 2012, 128, 10–20. [Google Scholar] [CrossRef]
- Liotta, L.F.; Pantaleo, G.; Puleo, F.; Venezia, A.M. Au/CeO2 -SBA-15 catalysts for CO oxidation: Effect of ceria loading on physic-chemical properties and catalytic performances. Catal. Today 2012, 187, 10–19. [Google Scholar] [CrossRef]
- Russo, N.; Fino, D.; Saracco, G.; Specchia, V. Supported gold catalysts for CO oxidation. Catal. Today 2006, 117, 214–219. [Google Scholar] [CrossRef]
- Meyer, R.; Lemire, C.; Shaikhutdinov, S.K.; Freund, H. Surface Chemistry of Catalysis by Gold. Gold Bull. 2004, 37, 72–124. [Google Scholar] [CrossRef] [Green Version]
- Kang, M.; Song, M.W.; Lee, C.H. Catalytic carbon monoxide oxidation over CoOx/CeO2 composite catalysts. Appl. Catal. A Gen. 2003, 251, 143–156. [Google Scholar] [CrossRef]
- Qi, X.; Flytzani-Stephanopoulos, M. Activity and stability of Cu–CeO2 catalysts in high-temperature water-gas shift for fuel-cell applications. Ind. Eng. Chem. Res. 2004, 43, 3055–3062. [Google Scholar] [CrossRef]
- Lin, L.; Yao, S.; Liu, Z.; Zhang, F.; Li, N.; Vovchok, D.; Martínez-Arias, A.; Castaneda, R.; Lin, J.; Senanayake, S.D.; et al. In Situ Characterization of Cu/CeO2 Nanocatalysts for CO2 Hydrogenation: Morphological Effects of Nanostructured Ceria on the Catalytic Activity. J. Phys. Chem. C 2018, 122, 12934–12943. [Google Scholar] [CrossRef]
- Li, Y.; Fu, Q.; Flytzani-stephanopoulos, M. Low-temperature water-gas shift reaction over Cu- and Ni-loaded cerium oxide catalysts. Appl. Catal. B Environ. 2000, 27, 179–191. [Google Scholar] [CrossRef] [Green Version]
- Alayoglu, S.; An, K.; Melaet, G.; Chen, S.; Bernardi, F.; Wang, L.W.; Lindeman, A.E.; Musselwhite, N.; Guo, J.; Liu, Z.; et al. Pt-mediated reversible reduction and expansion of CeO2 in Pt nanoparticle/mesoporous CeO2 catalyst: In situ X-ray spectroscopy and diffraction studies under redox (H2 and O2) atmospheres. J. Phys. Chem. C 2013, 117, 26608–26616. [Google Scholar] [CrossRef]
- Kato, S.; Ammann, M.; Huthwelker, T.; Paun, C.; Lampimäki, M.; Lee, M.T.; Rothensteiner, M.; Van Bokhoven, J.A. Quantitative depth profiling of Ce3+ in Pt/CeO2 by in situ high-energy XPS in a hydrogen atmosphere. Phys. Chem. Chem. Phys. 2015, 17, 5078–5083. [Google Scholar] [CrossRef]
- Sohn, H.; Celik, G.; Gunduz, S.; Dogu, D.; Zhang, S.; Shan, J.; Tao, F.F.; Ozkan, U.S. Oxygen Mobility in Pre-Reduced Nano- and Macro-Ceria with Co Loading: An AP-XPS, In-Situ DRIFTS and TPR Study. Catal. Lett. 2017, 147, 2863–2876. [Google Scholar] [CrossRef]
- Li, P.; Chen, X.; Li, Y.; Schwank, J.W. A review on oxygen storage capacity of CeO2 -based materials: Influence factors, measurement techniques, and applications in reactions related to catalytic automotive emissions control. Catal. Today 2019, 327, 90–115. [Google Scholar] [CrossRef]
- Singh, P.; Hegde, M.S.; Gopalakrishnan, J. Ce2/3Cr1/3O2+y: A new oxygen storage material based on the fluorite structure. Chem. Mater. 2008, 20, 7268–7273. [Google Scholar] [CrossRef]
- Deng, C.; Li, M.; Qian, J.; Hu, Q.; Huang, M.; Lin, Q.; Ruan, Y.; Dong, L.; Li, B.; Fan, M. A Study of Different Doped Metal Cations on the Physicochemical Properties and Catalytic Activities of Ce20M1Ox (M = Zr, Cr, Mn, Fe, Co, Sn) Composite Oxides for Nitric Oxide Reduction by Carbon Monoxide. Chem. An. Asian J. 2016, 11, 2144–2156. [Google Scholar] [CrossRef]
- Jampaiah, D.; Venkataswamy, P.; Coyle, V.E.; Reddy, B.M.; Bhargava, S.K. Low-temperature CO oxidation over manganese, cobalt, and nickel doped CeO2 nanorods. RSC Adv. 2016, 6, 80541–80548. [Google Scholar] [CrossRef]
- Lee, K.J.; Kim, Y.; Lee, J.H.; Cho, S.J.; Kwak, J.H.; Moon, H.R. Facile Synthesis and Characterization of Nanostructured Transition Metal/Ceria Solid Solutions (TMxCe1-xO2-δ, TM = Mn, Ni, Co, or Fe) for CO Oxidation. Chem. Mater. 2017, 29, 2874–2882. [Google Scholar] [CrossRef]
- Beckers, J.; Rothenberg, G. Redox properties of doped and supported copper-ceria catalysts. Dalt. Trans. 2008, 6573–6578. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Y.; Li, L.; Zhang, L.; Cao, Y.; Yu, S.; Tang, C.; Dong, L. Migration of copper species in CexCu1-xO2 catalyst driven by thermal treatment and the effect on CO oxidation. Phys. Chem. Chem. Phys. 2017, 19, 21840–21847. [Google Scholar] [CrossRef] [PubMed]
- Colón, G.; Pijolat, M.; Valdivieso, F.; Vidal, H.; Kašpar, J.; Finocchio, E.; Daturi, M.; Bernal, M.; Pijolat, F.; Valdivieso, H.; et al. Surface and structural characterization of CexZr1-xO2 CEZIRENCAT mixed oxides as potential three-way catalyst promoters. J. Chem. Soc. Faraday Trans. 1998, 94, 3717–3726. [Google Scholar] [CrossRef]
- Finocchio, E.; Daturi, M.; Binet, C.; Lavalley, J.C.; Flly, F.; Perrichon, V.; Vidal, H.; Kaspar, J.; Graziani, M.; Blanchard, G. Oxygen storage capacity improvement using CeO2-ZrO2 mixed oxides in three way catalysts. In Science and Technology in Catalysis 1998; Hattori, H., Otsuka, K., Eds.; Studies in Surface Science and Catalysis; Elsevier: Amsterdam, The Netherlands, 1999; Volume 121, pp. 257–262. [Google Scholar]
- Daturi, M.; Finocchio, E.; Binet, C.; Lavalley, J.C.; Fally, F.; Perrichon, V. Study of Bulk and Surface Reduction by Hydrogen of CexZr1-xO2 Mixed Oxides Followed by FTIR Spectroscopy and Magnetic Balance. J. Phys. Chem. B 1999, 103, 4884–4891. [Google Scholar] [CrossRef]
- Wang, Y.; Liang, S.; Cao, A.; Thompson, R.L.; Veser, G. Au-mixed lanthanum/cerium oxide catalysts for water gas shift. Appl. Catal. B Environ. 2010, 99, 89–95. [Google Scholar] [CrossRef]
- Liang, S.; Broitman, E.; Wang, Y.; Cao, A.; Veser, G. Highly stable, mesoporous mixed lanthanum-cerium oxides with tailored structure and reducibility. J. Mater. Sci. 2011, 46, 2928–2937. [Google Scholar] [CrossRef] [Green Version]
- Orge, C.A.; Órfão, J.J.M.; Pereira, M.F.R.; Duarte de Farias, A.M.; Fraga, M.A. Ceria and cerium-based mixed oxides as ozonation catalysts. Chem. Eng. J. 2012, 200–202, 499–505. [Google Scholar] [CrossRef]
- Singh, P.; Hegde, M.S. Ce1-xRuxO2-δ (x = 0.05, 0.10): A new high oxygen storage material and Pt, Pd-free three-way catalyst. Chem. Mater. 2009, 21, 3337–3345. [Google Scholar] [CrossRef]
- Gayen, A.; Priolkar, K.R.; Sarode, P.R.; Jayaram, V.; Hegde, M.S.; Subbanna, G.N.; Emura, S. Ce1-xRhxO2-δ solid solution formation in combustion-synthesized Rh/CeO2 catalyst studied by XRD, TEM, XPS, and EXAFS. Chem. Mater. 2004, 16, 2317–2328. [Google Scholar] [CrossRef]
- Kurnatowska, M.; Schuster, M.E.; Mista, W.; Kepinski, L. Self-regenerative property of nanocrystalline Ce0.89M0.11O2-y (M = Pd, Rh) mixed oxides. ChemCatChem 2014, 6, 3125–3131. [Google Scholar] [CrossRef]
- Hiley, C.I.; Fisher, J.M.; Thompsett, D.; Kashtiban, R.J.; Sloan, J.; Walton, R.I. Incorporation of square-planar Pd2+ in fluorite CeO2: Hydrothermal preparation, local structure, redox properties and stability. J. Mater. Chem. A 2015, 3, 13072–13079. [Google Scholar] [CrossRef] [Green Version]
- Hegde, M.S.; Bera, P. Noble metal ion substituted CeO2 catalysts: Electronic interaction between noble metal ions and CeO2 lattice. Catal. Today 2015, 253, 40–50. [Google Scholar] [CrossRef]
- Ikemoto, S.; Huang, X.; Muratsugu, S.; Nagase, S.; Koitaya, T.; Matsui, H.; Yokota, G.; Sudoh, T.; Hashimoto, A.; Tan, Y.; et al. Reversible low-temperature redox activity and selective oxidation catalysis derived from the concerted activation of multiple metal species on Cr and Rh-incorporated ceria catalysts. Phys. Chem. Chem. Phys. 2019, 21, 20868–20877. [Google Scholar] [CrossRef] [PubMed]
- Della Mea, G.B.; Matte, L.P.; Thill, A.S.; Lobato, F.O.; Benvenutti, E.V.; Arenas, L.T.; Jürgensen, A.; Hergenröder, R.; Poletto, F.; Bernardi, F. Tuning the oxygen vacancy population of cerium oxide (CeO2−x, 0 < x <0.5) nanoparticles. Appl. Surf. Sci. 2017, 422, 1102–1112. [Google Scholar]
- Pereira-Hernández, X.I.; DeLaRiva, A.; Muravev, V.; Kunwar, D.; Xiong, H.; Sudduth, B.; Engelhard, M.; Kovarik, L.; Hensen, E.J.M.; Wang, Y.; et al. Tuning Pt-CeO2 interactions by high-temperature vapor-phase synthesis for improved reducibility of lattice oxygen. Nat. Commun. 2019, 10, 1–10. [Google Scholar] [CrossRef]
- Sayle, T.X.T.; Caddeo, F.; Zhang, X.; Sakthivel, T.; Das, S.; Seal, S.; Ptasinska, S.; Sayle, D.C. Structure-Activity Map of Ceria Nanoparticles, Nanocubes, and Mesoporous Architectures. Chem. Mater. 2016, 28, 7287–7295. [Google Scholar] [CrossRef]
- Gopal, C.B.; García-Melchor, M.; Lee, S.C.; Shi, Y.; Shavorskiy, A.; Monti, M.; Guan, Z.; Sinclair, R.; Bluhm, H.; Vojvodic, A.; et al. Equilibrium oxygen storage capacity of ultrathin CeO2-δ depends non-monotonically on large biaxial strain. Nat. Commun. 2017, 8, 1–12. [Google Scholar]
- Bluhm, H.; Hävecker, M.; Knop-gericke, A.; Kiskinova, M.; Schlögl, R.; Salmeron, M. In Situ X-Ray Photoelectron Studies of Gas – Solid Interfaces at Near- Ambient Conditions. MRS Bull. 2007, 32, 1022–1030. [Google Scholar] [CrossRef] [Green Version]
- Pozdnyakova, O.; Teschner, D.; Wootsch, A.; Kröhnert, J.; Steinhauer, B.; Sauer, H.; Toth, L.; Jentoft, F.C.; Knop-Gericke, A.; Paál, Z.; et al. Preferential CO oxidation in hydrogen (PROX) on ceria-supported catalysts, part I: Oxidation state and surface species on Pt/CeO2 under reaction conditions. J. Catal. 2006, 237, 1–16. [Google Scholar] [CrossRef]
- Pozdnyakova, O.; Teschner, D.; Wootsch, A.; Kröhnert, J.; Steinhauer, B.; Sauer, H.; Toth, L.; Jentoft, F.C.; Knop-Gericke, A.; Paál, Z.; et al. Preferential CO oxidation in hydrogen (PROX) on ceria-supported catalysts, part II: Oxidation states and surface species on Pd/CeO2 under reaction conditions, suggested reaction mechanism. J. Catal. 2006, 237, 17–28. [Google Scholar] [CrossRef] [Green Version]
- Teschner, D.; Wootsch, A.; Pozdnyakova-Tellinger, O.; Kröhnert, J.; Vass, E.M.; Hävecker, M.; Zafeiratos, S.; Schnörch, P.; Jentoft, P.C.; Knop-Gericke, A.; et al. Partial pressure dependent in situ spectroscopic study on the preferential CO oxidation in hydrogen (PROX) over Pt/ceria catalysts. J. Catal. 2007, 249, 318–327. [Google Scholar] [CrossRef]
- Holgado, J.P.; Ternero, F.; Gonzalez-Delacruz, V.M.; Caballero, A. Promotional effect of the base metal on bimetallic Au-Ni/CeO2 catalysts prepared from core-shell nanoparticles. ACS Catal. 2013, 3, 2169–2180. [Google Scholar] [CrossRef]
- Monte, M.; Munuera, G.; Costa, D.; Conesa, J.C.; Martínez-Arias, A. Near-ambient XPS characterization of interfacial copper species in ceria-supported copper catalysts. Phys. Chem. Chem. Phys. 2015, 17, 29995–30004. [Google Scholar] [CrossRef]
- Lukashuk, L.; Föttinger, K.; Kolar, E.; Rameshan, C.; Teschner, D.; Hävecker, M.; Knop-Gericke, A.; Yigit, N.; Li, H.; McDermott, E.; et al. Operando XAS and NAP-XPS studies of preferential CO oxidation on Co3O4 and CeO2-Co3O4 catalysts. J. Catal. 2016, 344, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Artiglia, L.; Orlando, F.; Roy, K.; Kopelent, R.; Safonova, O.; Nachtegaal, M.; Huthwelker, T.; Van Bokhoven, J.A. Introducing Time Resolution to Detect Ce3+ Catalytically Active Sites at the Pt/CeO2 Interface through Ambient Pressure X-ray Photoelectron Spectroscopy. J. Phys. Chem. Lett. 2017, 8, 102–108. [Google Scholar] [CrossRef]
- Wen, C.; Zhu, Y.; Ye, Y.; Zhang, S.; Cheng, F.; Liu, Y.; Wang, P.; Tao, F. Water-gas shift reaction on metal nanoclusters encapsulated in mesoporous ceria studied with ambient-pressure X-ray photoelectron spectroscopy. ACS Nano 2012, 6, 9305–9313. [Google Scholar] [CrossRef]
- Cámara, A.L.; Monte, M.; Martínez-Arias, A.; Conesa, J.C. XPS and DRIFTS operando studies of an inverse CeO2/CuO WGS catalyst: Deactivating role of interfacial carbonates in redox activity. Catal. Sci. Technol. 2012, 2, 2436–2439. [Google Scholar] [CrossRef]
- Soler, L.; Casanovas, A.; Escudero, C.; Pérez-Dieste, V.; Aneggi, E.; Trovarelli, A.; Llorca, J. Ambient Pressure Photoemission Spectroscopy Reveals the Mechanism of Carbon Soot Oxidation in Ceria-Based Catalysts. ChemCatChem 2016, 8, 2748–2751. [Google Scholar] [CrossRef] [Green Version]
- Graciani, J.; Mudiyanselage, K.; Xu, F.; Baber, A.E.; Evans, J.; Senanayake, S.D.; Stacchiola, D.J.; Liu, P.; Hrbek, J.; Fernández-Sanz, J.; et al. Highly active copper-ceria and copper-ceria-titania catalysts for methanol synthesis from CO2. Science 2014, 345, 2–6. [Google Scholar] [CrossRef]
- Senanayake, S.D.; Ramírez, P.J.; Waluyo, I.; Kundu, S.; Mudiyanselage, K.; Liu, Z.; Liu, Z.; Axnanda, S.; Stacchiola, D.J.; Evans, J.; et al. Hydrogenation of CO2 to Methanol on CeOx/Cu(111) and ZnO/Cu(111) Catalysts: Role of the Metal-Oxide Interface and Importance of Ce3+ Sites. J. Phys. Chem. C 2016, 120, 1778–1784. [Google Scholar] [CrossRef] [Green Version]
- Winter, L.R.; Chen, R.; Chen, X.; Chang, K.; Liu, Z.; Senanayake, S.D.; Ebrahim, A.M.; Chen, J.G. Elucidating the roles of metallic Ni and oxygen vacancies in CO2 hydrogenation over Ni/CeO2 using isotope exchange and in situ measurements. Appl. Catal. B Environ. 2019, 245, 360–366. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhang, S.; Shan, J.J.; Nguyen, L.; Zhan, S.; Gu, X.; Tao, F. In situ surface chemistries and catalytic performances of ceria doped with palladium, platinum, and rhodium in methane partial oxidation for the production of syngas. ACS Catal. 2013, 3, 2627–2639. [Google Scholar] [CrossRef]
- Dou, J.; Tang, Y.; Nie, L.; Andolina, C.M.; Zhang, X.; House, S.; Li, Y.; Yang, J.; Tao, F. (Feng) Complete Oxidation of Methane on Co3O4/CeO2 Nanocomposite: A Synergic Effect. Catal. Today 2018, 311, 48–55. [Google Scholar] [CrossRef]
- Zhang, X.; House, S.D.; Tang, Y.; Nguyen, L.; Li, Y.; Opalade, A.A.; Yang, J.C.; Sun, Z.; Tao, F.F. Complete Oxidation of Methane on NiO Nanoclusters Supported on CeO2 Nanorods through Synergistic Effect. ACS Sustain. Chem. Eng. 2018, 6, 6467–6477. [Google Scholar] [CrossRef]
- Gonzalez-DelaCruz, V.M.; Holgado, J.P.; Pereñíguez, R.; Caballero, A. Morphology changes induced by strong metal-support interaction on a Ni-ceria catalytic system. J. Catal. 2008, 257, 307–314. [Google Scholar] [CrossRef]
- Liu, Z.; Grinter, D.C.; Lustemberg, P.G.; Nguyen-Phan, T.D.; Zhou, Y.; Luo, S.; Waluyo, I.; Crumlin, E.J.; Stacchiola, D.J.; Zhou, J.; et al. Dry Reforming of Methane on a Highly-Active Ni-CeO2 Catalyst: Effects of Metal-Support Interactions on C-H Bond Breaking. Angew. Chem. Int. Ed. 2016, 55, 7455–7459. [Google Scholar] [CrossRef] [Green Version]
- Lustemberg, P.G.; Ramírez, P.J.; Liu, Z.; Gutiérrez, R.A.; Grinter, D.G.; Carrasco, J.; Senanayake, S.D.; Rodriguez, J.A.; Ganduglia-Pirovano, M.V. Room-Temperature Activation of Methane and Dry Re-forming with CO2 on Ni-CeO2(111) Surfaces: Effect of Ce3+ Sites and Metal-Support Interactions on C-H Bond Cleavage. ACS Catal. 2016, 6, 8184–8191. [Google Scholar] [CrossRef]
- Yan, B.; Yang, X.; Yao, S.; Wan, J.; Myint, M.N.Z.; Gomez, E.; Xie, Z.; Kattel, S.; Xu, W.; Chen, J.G. Dry Reforming of Ethane and Butane with CO2 over PtNi/CeO2 Bimetallic Catalysts. ACS Catal. 2016, 6, 7283–7292. [Google Scholar] [CrossRef]
- Liu, Z.; Lustemberg, P.; Gutiérrez, R.A.; Carey, J.J.; Palomino, R.M.; Vorokhta, M.; Grinter, D.C.; Ramírez, P.J.; Matolín, V.; Nolan, M.; et al. In Situ Investigation of Methane Dry Reforming on Metal/Ceria(111) Surfaces: Metal-Support Interactions and C−H Bond Activation at Low Temperature. Angew. Chem. Int. Ed. 2017, 56, 13041–13046. [Google Scholar] [CrossRef]
- Zhang, F.; Liu, Z.; Zhang, S.; Akter, N.; Palomino, R.M.; Vovchok, D.; Orozco, I.; Salazar, D.; Rodriguez, J.A.; Llorca, J.; et al. In Situ Elucidation of the Active State of Co-CeOx Catalysts in the Dry Reforming of Methane: The Important Role of the Reducible Oxide Support and Interactions with Cobalt. ACS Catal. 2018, 8, 3550–3560. [Google Scholar] [CrossRef]
- Xie, Z.; Yan, B.; Kattel, S.; Lee, J.H.; Yao, S.; Wu, Q.; Rui, N.; Gomez, E.; Liu, Z.; Xu, W.; et al. Dry reforming of methane over CeO2-supported Pt-Co catalysts with enhanced activity. Appl. Catal. B Environ. 2018, 236, 280–293. [Google Scholar] [CrossRef]
- Liu, Z.; Zhang, F.; Rui, N.; Li, X.; Lin, L.; Betancourt, L.E.; Su, D.; Xu, W.; Cen, J.; Attenkofer, K.; et al. Highly Active Ceria-Supported Ru Catalyst for the Dry Reforming of Methane: In Situ Identification of Ruδ+-Ce3+ Interactions for Enhanced Conversion. ACS Catal. 2019, 9, 3349–3359. [Google Scholar] [CrossRef]
- Óvári, L.; Krick Calderon, S.; Lykhach, Y.; Libuda, J.; Erdohelyi, A.; Papp, C.; Kiss, J.; Steinrück, H.P. Near ambient pressure XPS investigation of the interaction of ethanol with Co/CeO2(1 1 1). J. Catal. 2013, 307, 132–139. [Google Scholar] [CrossRef]
- Turczyniak, S.; Teschner, D.; Machocki, A.; Zafeiratos, S. Effect of the surface state on the catalytic performance of a Co/CeO2 ethanol steam-reforming catalyst. J. Catal. 2016, 340, 321–330. [Google Scholar] [CrossRef]
- Turczyniak, S.; Luo, W.; Papaefthimiou, V.; Ramgir, N.S.; Haevecker, M.; MacHocki, A.; Zafeiratos, S. A Comparative Ambient Pressure X-ray Photoelectron and Absorption Spectroscopy Study of Various Cobalt-Based Catalysts in Reactive Atmospheres. Top. Catal. 2016, 59, 532–542. [Google Scholar] [CrossRef]
- Sohn, H.; Soykal, I.I.; Zhang, S.; Shan, J.; Tao, F.; Miller, J.T.; Ozkan, U.S. Effect of cobalt on reduction characteristics of ceria under ethanol steam reforming conditions: AP-XPS and XANES studies. J. Phys. Chem. C 2016, 120, 14631–14642. [Google Scholar] [CrossRef]
- Liu, Z.; Duchoň, T.; Wang, H.; Grinter, D.C.; Waluyo, I.; Zhou, J.; Liu, Q.; Jeong, B.; Crumlin, E.J.; Matolín, V.; et al. Ambient pressure XPS and IRRAS investigation of ethanol steam reforming on Ni-CeO2(111) catalysts: An in situ study of C-C and O-H bond scission. Phys. Chem. Chem. Phys. 2016, 18, 16621–16628. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Yao, S.; Johnston-Peck, A.; Xu, W.; Rodriguez, J.A.; Senanayake, S.D. Methanol steam reforming over Ni-CeO2 model and powder catalysts: Pathways to high stability and selectivity for H2/CO2 production. Catal. Today 2018, 311, 74–80. [Google Scholar] [CrossRef]
- Mullins, D.R. Variations in the surface chemistry of methanol with CeO2(100) at low and elevated pressures. Surf. Interface Anal. 2018, 50, 913–920. [Google Scholar] [CrossRef]
- Wu, Z.; Cheng, Y.; Tao, F.; Daemen, L.; Foo, G.S.; Nguyen, L.; Zhang, X.; Beste, A.; Ramirez-Cuesta, A.J. Direct Neutron Spectroscopy Observation of Cerium Hydride Species on a Cerium Oxide Catalyst. J. Am. Chem. Soc. 2017, 139, 9721–9727. [Google Scholar] [CrossRef] [PubMed]
- Mueanngern, Y.; Yang, X.; Tang, Y.; Tao, F.; Baker, L.R. Catalysis at Multiple Length Scales: Crotonaldehyde Hydrogenation at Nanoscale and Mesoscale Interfaces in Platinum-Cerium Oxide Catalysts. J. Phys. Chem. C 2017, 121, 13765–13776. [Google Scholar] [CrossRef]
- Armor, J.N. The multiple roles for catalysis in the production of H2. Appl. Catal. A Gen. 1999, 176, 159–176. [Google Scholar] [CrossRef]
- Dudfield, C.D.; Chen, R.; Adcock, P.L. A carbon monoxide PROX reactor for PEM fuel cell automotive application. Int. J. Hydrog. Energy 2001, 26, 763–775. [Google Scholar] [CrossRef]
- Kahlich, M.J.; Gasteiger, H.A.; Behm, R.J. Kinetics of the selective low-temperature oxidation of CO in H2-rich gas over Au/α-Fe2O3. J. Catal. 1999, 182, 430–440. [Google Scholar] [CrossRef]
- Schubert, M.M.; Kahlich, M.J.; Gasteiger, H.A.; Behm, R.J. Correlation between CO surface coverage and selectivity/kinetics for the preferential CO oxidation over Pt/γ-Al2O3 and Au/α-Fe2O3: An in-situ DRIFTS study. J. Power Sources 1999, 84, 175–182. [Google Scholar] [CrossRef]
- Bethke, G.K.; Kung, H.H. Selective CO oxidation in a hydrogen-rich stream over Au/γ-Al2O3 catalysts. Appl. Catal. A Gen. 2000, 194–195, 43–53. [Google Scholar] [CrossRef]
- Avgouropoulos, G.; Ioannides, T.; Papadopoulou, C.; Batista, J.; Hocevar, S.; Matralis, H.K. A comparative study of Pt/γ-Al2O3, Au/α-Fe2O3 and CuO-CeO2 catalysts for the selective oxidation of carbon monoxide in excess hydrogen. Catal. Today 2002, 75, 157–167. [Google Scholar] [CrossRef]
- Oh, S.H.; Sinkevitch, R.M. Carbon Monoxide Removal from Hydrogen-Rich Fuel Cell Feedstreams by Selective Catalytic Oxidation. J. Catal. 1993, 142, 254–262. [Google Scholar] [CrossRef]
- Kahlich, M.J.; Gasteiger, H.A.; Behm, R.J. Kinetics of the Selective CO Oxidation in H2-Rich Gas on Pt/Al2O3. J. Catal. 1997, 171, 93–105. [Google Scholar] [CrossRef]
- Özkara, Ş.; Aksoylu, A.E. Selective low temperature carbon monoxide oxidation in H2-rich gas streams over activated carbon supported catalysts. Appl. Catal. A Gen. 2003, 251, 75–83. [Google Scholar] [CrossRef]
- Han, Y.-F.; Kahlich, M.J.; Kinne, M.; Behm, R.J. Kinetic study of selective CO oxidation in H2-rich gas on a Ru/γ-Al2O3 catalyst. Phys. Chem. Chem. Phys. 2002, 4, 389–397. [Google Scholar] [CrossRef]
- Jacobs, G.; Williams, L.; Graham, U.; Sparks, D.; Davis, B.H. Low-Temperature Water-Gas Shift: In-Situ DRIFTS−Reaction Study of a Pt/CeO2 Catalyst for Fuel Cell Reformer Applications. J. Phys. Chem. B 2003, 107, 10398–10404. [Google Scholar] [CrossRef]
- Johansson, S.; Österlund, L.; Kasemo, B. CO Oxidation Bistability Diagrams for Pt/CeOx and Pt/SiO2 Model Catalysts Prepared by Electron-Beam Lithography. J. Catal. 2001, 201, 275–285. [Google Scholar] [CrossRef]
- Bunluesin, T.; Cordatos, H.; Gorte, R.J. Study of CO Oxidation Kinetics on Rh/Ceria. J. Catal. 1995, 157, 222–226. [Google Scholar] [CrossRef]
- Bekyarova, E.; Fornasiero, P.; Kašpar, J.; Graziani, M. CO oxidation on Pd/CeO2–ZrO2 catalysts. Catal. Today 1998, 45, 179–183. [Google Scholar] [CrossRef]
- Kaspar, J.; Fornasiero, P.; Graziani, M. Use of CeO2 -based oxides in the three-way catalysis. Catal. Today 1999, 50, 285–298. [Google Scholar] [CrossRef]
- Kopelent, R.; Van Bokhoven, J.A.; Szlachetko, J.; Edebeli, J.; Paun, C.; Nachtegaal, M.; Safonova, O. V Catalytically Active and Spectator Ce3+ in Ceria-Supported Metal Catalysts. Angew. Chem. Int. Ed. 2015, 54, 8728–8731. [Google Scholar] [CrossRef]
- Penkala, B.; Aubert, D.; Kaper, H.; Tardivat, C.; Conder, K.; Paulus, W. The role of lattice oxygen in CO oxidation over Ce18O2-based catalysts revealed under operando conditions. Catal. Sci. Technol. 2015, 5, 4839–4848. [Google Scholar] [CrossRef]
- Wang, X.; Rodriguez, J.A.; Hanson, J.C.; Gamarra, D.; Martínez-Arias, A.; Fernández-García, M. In Situ Studies of the Active Sites for the Water Gas Shift Reaction over Cu-CeO2 Catalysts: Complex Interaction between Metallic Copper and Oxygen Vacancies of Ceria. J. Phys. Chem. B 2006, 110, 428–434. [Google Scholar] [CrossRef] [Green Version]
- Gamarra, D.; Belver, C.; Fernández-García, M.; Martínez-Arias, A. Selective CO Oxidation in Excess H2 over Copper−Ceria Catalysts: Identification of Active Entities/Species. J. Am. Chem. Soc. 2007, 129, 12064–12065. [Google Scholar] [CrossRef] [PubMed]
- Jia, A.-P.; Jiang, S.-Y.; Lu, J.-Q.; Luo, M.-F. Study of Catalytic Activity at the CuO−CeO2 Interface for CO Oxidation. J. Phys. Chem. C 2010, 114, 21605–21610. [Google Scholar] [CrossRef]
- Gamarra, D.; Cámara, A.L.; Monte, M.; Rasmussen, S.B.; Chinchilla, L.E.; Hungría, A.B.; Munuera, G.; Gyorffy, N.; Schay, Z.; Corberán, V.C.; et al. Preferential oxidation of CO in excess H2 over CuO/CeO2 catalysts: Characterization and performance as a function of the exposed face present in the CeO2 support. Appl. Catal. B Environ. 2013, 130–131, 224–238. [Google Scholar] [CrossRef]
- Arango-Díaz, A.; Moretti, E.; Talon, A.; Storaro, L.; Lenarda, M.; Núñez, P.; Marrero-Jerez, J.; Jiménez-Jiménez, J.; Jiménez-López, A.; Rodríguez-Castellón, E. Preferential CO oxidation (CO-PROX) catalyzed by CuO supported on nanocrystalline CeO2 prepared by a freeze-drying method. Appl. Catal. A Gen. 2014, 477, 54–63. [Google Scholar] [CrossRef] [Green Version]
- Teng, Y.; Sakurai, H.; Ueda, A.; Kobayashi, T. Oxidative removal of CO contained in hydrogen by using metal oxide catalysts. Int. J. Hydrog. Energy 1999, 24, 355–358. [Google Scholar] [CrossRef]
- Zhao, Z.; Yung, M.M.; Ozkan, U.S. Effect of support on the preferential oxidation of CO over cobalt catalysts. Catal. Commun. 2008, 9, 1465–1471. [Google Scholar] [CrossRef]
- Woods, M.P.; Gawade, P.; Tan, B.; Ozkan, U.S. Preferential oxidation of carbon monoxide on Co/CeO2 nanoparticles. Appl. Catal. B Environ. 2010, 97, 28–35. [Google Scholar] [CrossRef]
- Gawade, P.; Bayram, B.; Alexander, A.-M.C.; Ozkan, U.S. Preferential oxidation of CO (PROX) over CoOx/CeO2 in hydrogen-rich streams: Effect of cobalt loading. Appl. Catal. B Environ. 2012, 128, 21–30. [Google Scholar] [CrossRef]
- Gorte, R.J.; Zhao, S. Studies of the water-gas-shift reaction with ceria-supported precious metals. Catal. Today 2005, 104, 18–24. [Google Scholar] [CrossRef]
- Fu, Q.; Weber, A.; Flytzani-Stephanopoulos, M. Nanostructured Au-CeO2 catalysts for low-temperature water-gas shift. Catal. Lett. 2001, 77, 87–95. [Google Scholar] [CrossRef]
- Rodriguez, J.A.; Graciani, J.; Evans, J.; Park, J.B.; Yang, F.; Stacchiola, D.; Senanayake, S.D.; Ma, S.; Perez, M.; Liu, P.; et al. Water-gas shift reaction on a highly active inverse CeOx /Cu(111) catalyst: Unique role of ceria nanoparticles. Angew. Chem. Int. Ed. 2009, 48, 8047–8050. [Google Scholar] [CrossRef] [PubMed]
- Barrio, L.; Estrella, M.; Zhou, G.; Wen, W.; Hanson, J.C.; Hungría, A.B.; Hornés, A.; Fernández-García, M.; Martínez-Arias, A.; Rodriguez, J.A. Unraveling the Active Site in Copper-Ceria Systems for the Water-Gas Shift Reaction: In Situ Characterization of an Inverse Powder CeO2-x/CuO-Cu Catalyst. J. Phys. Chem. C 2010, 114, 3580–3587. [Google Scholar] [CrossRef]
- Senanayake, S.D.; Stacchiola, D.; Evans, J.; Estrella, M.; Barrio, L.; Pérez, M.; Hrbek, J.; Rodriguez, J.A. Probing the reaction intermediates for the water-gas shift over inverse CeOx /Au(111) catalysts. J. Catal. 2010, 271, 392–400. [Google Scholar] [CrossRef]
- Stanmore, B.R.; Brilhac, J.F.; Gilot, P. The oxidation of soot: A review of experiments, mechanisms and models. Carbon 2001, 39, 2247–2268. [Google Scholar] [CrossRef]
- Van Setten, B.A.; Makkee, M.; Moulijn, J.A. Science and technology of catalytic diesel particulate filters. Catal. Rev. 2001, 43, 489–564. [Google Scholar] [CrossRef]
- Bueno-López, A. Diesel soot combustion ceria catalysts. Appl. Catal. B Environ. 2014, 146, 1–11. [Google Scholar] [CrossRef] [Green Version]
- Fino, D.; Specchia, V. Open issues in oxidative catalysis for diesel particulate abatement. Powder Technol. 2008, 180, 64–73. [Google Scholar] [CrossRef]
- Darcy, P.; Da Costa, P.; Mellottée, H.; Trichard, J.-M.; Djéga-Mariadassou, G. Kinetics of catalyzed and non-catalyzed oxidation of soot from a diesel engine. Catal. Today 2007, 119, 252–256. [Google Scholar] [CrossRef]
- Kim, G. Ceria-Promoted Three-Way Catalysts for Auto Exhaust Emission Control. Ind. Eng. Chem. Prod. Res. Dev. 1982, 21, 267–274. [Google Scholar] [CrossRef]
- Yao, H.C.; Yao, Y.Y. Ceria in Automotive Exhaust Catalysts: I. Oxygen storage. J. Catal. 1984, 86, 254–265. [Google Scholar] [CrossRef]
- Trovarelli, A.; De Leitenburg, C.; Boaro, M.; Dolcetti, G. The utilization of ceria in industrial catalysis. Catal. Today 1999, 50, 353–367. [Google Scholar] [CrossRef]
- Aneggi, E.; De Leitenburg, C.; Trovarelli, A. Ceria-Based Formulations for Catalysis for Diesel Soot Combustion. In Catalysis by Ceria and Related Materials; Trovarelli, A., Fornasiero, P., Eds.; World Scientific: Singapore, 2013; pp. 565–621. [Google Scholar]
- Harada, K.; Oishi, T.; Hamamoto, S.; Ishihara, T. Lattice Oxygen Activity in Pr- and La-Doped CeO2 for Low-Temperature Soot Oxidation. J. Phys. Chem. C 2014, 118, 559–568. [Google Scholar] [CrossRef]
- Pushkarev, V.V.; Kovalchuk, V.I.; Itri, J.L. Probing Defect Sites on the CeO2 Surface with Dioxygen. J. Phys. Chem. B 2004, 108, 5341–5348. [Google Scholar] [CrossRef]
- Saab, E.; Abi-Aad, E.; Bokova, M.N.; Zhilinskaya, E.A.; Aboukaı, A. EPR characterisation of carbon black in loose and tight contact with Al2O3 and CeO2 catalysts. Carbon 2007, 45, 561–567. [Google Scholar] [CrossRef]
- Preda, G.; Migani, A.; Neyman, K.M.; Bromley, S.T.; Illas, F.; Pacchioni, G.; Milano, I. Formation of Superoxide Anions on Ceria Nanoparticles by Interaction of Molecular Oxygen with Ce3+ Sites. J. Phys. Chem. C 2011, 115, 5817–5822. [Google Scholar] [CrossRef]
- Machida, M.; Murata, Y.; Kishikawa, K.; Zhang, D.; Ikeue, K. On the Reasons for High Activity of CeO2 Catalyst for Soot Oxidation. Chem. Mater. 2008, 20, 4489–4494. [Google Scholar] [CrossRef]
- Gross, M.S.; Ulla, M.A.; Querini, C.A. Diesel particulate matter combustion with CeO2 as catalyst. Part I: System characterization and reaction mechanism. J. Mol. Catal. A Chem. 2012, 352, 86–94. [Google Scholar] [CrossRef]
- Holgado, J.P.; Munuera, G.; Espinos, J.P.; González-Elipe, A.R. XPS study of oxidation processes of CeOx defective layers. Appl. Surf. Sci. 2000, 158, 164–171. [Google Scholar] [CrossRef]
- Behrens, M.; Studt, F.; Kasatkin, I.; Kühl, S.; Hävecker, M.; Abild-Pedersen, F.; Zander, S.; Girgsdies, F.; Kurr, P.; Kniep, B.-L.; et al. The Active Site of Methanol Synthesis over Cu/ZnO/Al2O3 Industrial Catalysts. Science 2012, 336, 893–897. [Google Scholar] [CrossRef]
- Yang, Y.; Evans, J.; Rodriguez, J.A.; White, M.G.; Liu, P. Fundamental studies of methanol synthesis from CO2 hydrogenation on Cu(111), Cu clusters, and Cu/ZnO(000). Phys. Chem. Chem. Phys. 2010, 12, 9909–9917. [Google Scholar] [CrossRef]
- Rasmussen, P.B.; Kazuta, M.; Chorkendorff, I. Synthesis of methanol from a mixture of H2 and CO2 on Cu(100). Surf. Sci. 1994, 318, 267–280. [Google Scholar] [CrossRef]
- Yoshihara, J.; Campbell, C.T. Methanol Synthesis and Reverse Water-Gas Shift Kinetics over Cu(110) Model Catalysts: Structural Sensitivity. J. Catal. 1996, 161, 776–782. [Google Scholar] [CrossRef]
- Yang, Y.; White, M.G.; Liu, P. Theoretical Study of Methanol Synthesis from CO2 Hydrogenation on Metal-Doped Cu(111) Surfaces. J. Phys. Chem. C 2012, 116, 248–256. [Google Scholar] [CrossRef]
- Kondratenko, E.V.; Mul, G.; Baltrusaitis, J.; Larrazábal, G.O.; Pérez-Ramírez, J. Status and perspectives of CO2 conversion into fuels and chemicals by catalytic, photocatalytic and electrocatalytic processes. Energy Environ. Sci. 2013, 6, 3112–3135. [Google Scholar] [CrossRef] [Green Version]
- Quadrelli, E.A.; Centi, G.; Duplan, J.-L.; Perathoner, S. Carbon Dioxide Recycling: Emerging Large-Scale Technologies with Industrial Potential. ChemSusChem 2011, 4, 1194–1215. [Google Scholar] [CrossRef] [PubMed]
- Preti, D.; Resta, C.; Squarcialupi, S.; Fachinetti, G. Carbon Dioxide Hydrogenation to Formic Acid by Using a Heterogeneous Gold Catalyst. Angew. Chem. Int. Ed. 2011, 50, 12551–12554. [Google Scholar] [CrossRef]
- Ansari, M.B.; Min, B.-H.; Mo, Y.-H.; Park, S.-E. CO2 activation and promotional effect in the oxidation of cyclic olefins over mesoporous carbon nitrides. Green Chem. 2011, 13, 1416–1421. [Google Scholar] [CrossRef]
- Vidal, A.B.; Feria, L.; Evans, J.; Takahashi, Y.; Liu, P.; Nakamura, K.; Illas, F.; Rodriguez, J.A. CO2 Activation and Methanol Synthesis on Novel Au/TiC and Cu/TiC Catalysts. J. Phys. Chem. Lett. 2012, 3, 2275–2280. [Google Scholar] [CrossRef]
- Miguel, C.V.; Soria, M.A.; Mendes, A.; Madeira, L.M. Direct CO2 hydrogenation to methane or methanol from post-combustion exhaust streams—A thermodynamic study. J. Nat. Gas. Sci. Eng. 2015, 22, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Torrente-Murciano, L.; Mattia, D.; Jones, M.D.; Plucinski, P.K. Formation of hydrocarbons via CO2 hydrogenation—A thermodynamic study. J. CO2 Util. 2014, 6, 34–39. [Google Scholar] [CrossRef]
- Medford, A.J.; Lausche, A.C.; Abild-Pedersen, F.; Temel, B.; Schjødt, N.C.; Nørskov, J.K.; Studt, F. Activity and Selectivity Trends in Synthesis Gas Conversion to Higher Alcohols. Top. Catal. 2014, 57, 135–142. [Google Scholar] [CrossRef]
- Studt, F.; Behrens, M.; Kunkes, E.L.; Thomas, N.; Zander, S.; Tarasov, A.; Schumann, J.; Frei, E.; Varley, J.B.; Abild-Pedersen, F.; et al. The Mechanism of CO and CO2 Hydrogenation to Methanol over Cu-Based Catalysts. ChemCatChem 2015, 7, 1105–1111. [Google Scholar] [CrossRef] [Green Version]
- Lunkenbein, T.; Schumann, J.; Behrens, M.; Schlögl, R.; Willinger, M.G. Formation of a ZnO Overlayer in Industrial Cu/ZnO/Al2O3 Catalysts Induced by Strong Metal–Support Interactions. Angew. Chem. Int. Ed. 2015, 54, 4544–4548. [Google Scholar] [CrossRef] [PubMed]
- Slaa, J.C.; Van Ommen, J.G.; Ross, J.R.H. The synthesis of higher alcohols using modified Cu/ZnO/Al2O3 catalysts. Catal. Today 1992, 15, 129–148. [Google Scholar] [CrossRef] [Green Version]
- Nakamura, J.; Nakamura, I.; Uchijima, T.; Kanai, Y.; Watanabe, T.; Saito, M.; Fujitani, T. A Surface Science Investigation of Methanol Synthesis over a Zn-Deposited Polycrystalline Cu Surface. J. Catal. 1996, 160, 65–75. [Google Scholar] [CrossRef]
- Schumann, J.; Eichelbaum, M.; Lunkenbein, T.; Thomas, N.; Álvarez Galván, M.C.; Schlögl, R.; Behrens, M. Promoting Strong Metal Support Interaction: Doping ZnO for Enhanced Activity of Cu/ZnO:M (M = Al, Ga, Mg) Catalysts. ACS Catal. 2015, 5, 3260–3270. [Google Scholar] [CrossRef]
- Kandemir, T.; Kasatkin, I.; Girgsdies, F.; Zander, S.; Kühl, S.; Tovar, M.; Schlögl, R.; Behrens, M. Microstructural and Defect Analysis of Metal Nanoparticles in Functional Catalysts by Diffraction and Electron Microscopy: The Cu/ZnO Catalyst for Methanol Synthesis. Top. Catal. 2014, 57, 188–206. [Google Scholar] [CrossRef] [Green Version]
- Rostrup-Nielsen, J.R. New aspects of syngas production and use. Catal. Today 2000, 63, 159–164. [Google Scholar] [CrossRef]
- Reyes, S.C.; Sinfelt, J.H.; Feeley, J.S. Evolution of Processes for Synthesis Gas Production: Recent Developments in an Old Technology. Ind. Eng. Chem. Res. 2003, 42, 1588–1597. [Google Scholar] [CrossRef]
- Liander, H. The utilisation of natural gases for the ammonia process. Trans. Faraday Soc. 1929, 25, 462. [Google Scholar] [CrossRef]
- Goetsch, D.A.; Schmidt, L.D. Microsecond Catalytic Partial Oxidation of Alkanes. Science 1996, 271, 1560–1562. [Google Scholar] [CrossRef]
- Tsang, S.C.; Claridge, J.B.; Green, M.L. Recent advances in the conversion of methane to synthesis gas. Catal. Today 1995, 23, 3–15. [Google Scholar] [CrossRef]
- York, A.P.E.; Xiao, T.; Green, M.L.H. Brief overview of the partial oxidation of methane to synthesis gas. Top. Catal. 2003, 22, 345–358. [Google Scholar] [CrossRef]
- Wilson, J.N.; Pedigo, R.A.; Zaera, F. Kinetics and Mechanism of Catalytic Partial Oxidation Reactions of Alkanes on Rhodium Surfaces. J. Am. Chem. Soc. 2008, 130, 15796–15797. [Google Scholar] [CrossRef]
- Christian Enger, B.; Lødeng, R.; Holmen, A. A review of catalytic partial oxidation of methane to synthesis gas with emphasis on reaction mechanisms over transition metal catalysts. Appl. Catal. A Gen. 2008, 346, 1–27. [Google Scholar] [CrossRef]
- Basile, F.; Benito, P.; Fornasari, G.; Monti, M.; Scavetta, E.; Tonelli, D.; Vaccari, A. Novel Rh-based structured catalysts for the catalytic partial oxidation of methane. Catal. Today 2010, 157, 183–190. [Google Scholar] [CrossRef]
- Ashcroft, A.T.; Cheetham, A.K.; Foord, J.S.; Green, M.L.H.; Grey, C.P.; Murrell, A.J.; Vernon, P.D.F. Selective oxidation of methane to synthesis gas using transition metal catalysts. Nature 1990, 344, 319–321. [Google Scholar] [CrossRef]
- Hargreaves, J.S.J.; Hutchings, G.J.; Joyner, R.W. Control of product selectivity in the partial oxidation of methane. Nature 1990, 348, 428–429. [Google Scholar] [CrossRef]
- Hickman, D.A.; Schmidt, L.D. Production of Syngas by Direct Catalytic Oxidation of Methane. Science 1993, 259, 343–346. [Google Scholar] [CrossRef]
- Pino, L.; Recupero, V.; Beninati, S.; Shukla, A.K.; Hegde, M.S.; Bera, P. Catalytic partial-oxidation of methane on a ceria-supported platinum catalyst for application in fuel cell electric vehicles. Appl. Catal. A Gen. 2002, 225, 63–75. [Google Scholar] [CrossRef]
- Tang, W.; Hu, Z.; Wang, M.; Stucky, G.D.; Metiu, H.; McFarland, E.W. Methane complete and partial oxidation catalyzed by Pt-doped CeO2. J. Catal. 2010, 273, 125–137. [Google Scholar] [CrossRef]
- Gélin, P.; Primet, M. Complete oxidation of methane at low temperature over noble metal based catalysts: A review. Appl. Catal. B Environ. 2002, 39, 1–37. [Google Scholar] [CrossRef]
- Honkanen, M.; Kärkkäinen, M.; Viitanen, V.; Jiang, H.; Kallinen, K.; Huuhtanen, M.; Vippola, M.; Lahtinen, J.; Keiski, R.; Lepistö, T. Structural Characteristics of Natural-Gas-Vehicle-Aged Oxidation Catalyst. Top. Catal. 2013, 56, 576–585. [Google Scholar] [CrossRef]
- Choudhary, T.V.; Banerjee, S.; Choudhary, V.R. Catalysts for combustion of methane and lower alkanes. Appl. Catal. A Gen. 2002, 234, 1–23. [Google Scholar] [CrossRef]
- Chen, J.; Arandiyan, H.; Gao, X.; Li, J. Recent Advances in Catalysts for Methane Combustion. Catal. Surv. Asia 2015, 19, 140–171. [Google Scholar] [CrossRef]
- Farrauto, R.J. Low-Temperature Oxidation of Methane. Science 2012, 337, 659–660. [Google Scholar] [CrossRef]
- Sekizawa, K.; Widjaja, H.; Maeda, S.; Ozawa, Y.; Eguchi, K. Low temperature oxidation of methane over Pd catalyst supported on metal oxides. Catal. Today 2000, 59, 69–74. [Google Scholar] [CrossRef]
- Datye, A.K.; Bravo, J.; Nelson, T.R.; Atanasova, P.; Lyubovsky, M.; Pfefferle, L. Catalyst microstructure and methane oxidation reactivity during the Pd↔PdO transformation on alumina supports. Appl. Catal. A Gen. 2000, 198, 179–196. [Google Scholar] [CrossRef]
- Chin, Y.-H.; Buda, C.; Neurock, M.; Iglesia, E. Consequences of Metal-Oxide Interconversion for C-H Bond Activation during CH4 Reactions on Pd Catalysts. J. Am. Chem. Soc. 2013, 135, 15425–15442. [Google Scholar] [CrossRef]
- Ramírez-López, R.; Elizalde-Martinez, I.; Balderas-Tapia, L. Complete catalytic oxidation of methane over Pd/CeO2–Al2O3: The influence of different ceria loading. Catal. Today 2010, 150, 358–362. [Google Scholar] [CrossRef]
- Tippayawong, N.; Thanompongchart, P. Biogas quality upgrade by simultaneous removal of CO2 and H2S in a packed column reactor. Energy 2010, 35, 4531–4535. [Google Scholar] [CrossRef]
- Lavoie, J.M. Review on dry reforming of methane, a potentially more environmentally-friendly approach to the increasing natural gas exploitation. Front. Chem. 2014, 2, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Ghorbanzadeh, A.M.; Norouzi, S.; Mohammadi, T. High energy efficiency in syngas and hydrocarbon production from dissociation of CH4-CO2 mixture in a non-equilibrium pulsed plasma. J. Phys. D. Appl. Phys. 2005, 38, 3804–3811. [Google Scholar] [CrossRef]
- Dry, M.E.; Steynberg, A.P. Chapter 5—Commercial FT Process Applications. In Fischer-Tropsch Technology; Steynberg, A., Dry, M., Eds.; Studies in Surface Science and Catalysis; Elsevier: Amsterdam, The Netherlands, 2004; pp. 406–481. [Google Scholar]
- Wang, S.; Lu, G.Q.; Millar, G.J. Carbon dioxide reforming of methane to produce synthesis gas over metal-supported catalysts: State of the art. Energy Fuels 1996, 10, 896–904. [Google Scholar] [CrossRef]
- Bradford, M.C.J.; Vannice, M.A. CO2 reforming of CH4. Catal. Rev. Sci. Eng. 1999, 41, 1–42. [Google Scholar] [CrossRef]
- Rodhe, H. A Comparison of the Contribution of Various Gases to the Greenhouse Effect. Science 1990, 248, 1217–1219. [Google Scholar] [CrossRef]
- Huang, A.; Xia, G.; Wang, J.; Suib, S.L.; Hayashi, Y.; Matsumoto, H. CO2 Reforming of CH4 by Atmospheric Pressure ac Discharge Plasmas. J. Catal. 2000, 189, 349–359. [Google Scholar] [CrossRef]
- Wei, J.; Iglesia, E. Mechanism and Site Requirements for Activation and Chemical Conversion of Methane on Supported Pt Clusters and Turnover Rate Comparisons among Noble Metals. J. Phys. Chem. B 2004, 108, 4094–4103. [Google Scholar] [CrossRef]
- Rostrupnielsen, J.R.; Hansen, J.H.B. CO2-Reforming of Methane over Transition Metals. J. Catal. 1993, 144, 38–49. [Google Scholar] [CrossRef]
- Hou, Z.; Chen, P.; Fang, H.; Zheng, X.; Yashima, T. Production of synthesis gas via methane reforming with CO2 on noble metals and small amount of noble-(Rh-) promoted Ni catalysts. Int. J. Hydrog. Energy 2006, 31, 555–561. [Google Scholar] [CrossRef]
- Pakhare, D.; Spivey, J. A review of dry (CO2) reforming of methane over noble metal catalysts. Chem. Soc. Rev. 2014, 43, 7813–7837. [Google Scholar] [CrossRef] [PubMed]
- Bradford, M.C.J.; Vannice, M.A. Catalytic reforming of methane with carbon dioxide over nickel catalysts I. Catalyst characterization and activity. Appl. Catal. A Gen. 1996, 142, 73–96. [Google Scholar] [CrossRef]
- Budiman, A.W.; Song, S.-H.; Chang, T.-S.; Shin, C.-H.; Choi, M.-J. Dry Reforming of Methane Over Cobalt Catalysts: A Literature Review of Catalyst Development. Catal. Surv. Asia 2012, 16, 183–197. [Google Scholar] [CrossRef]
- Kim, D.K.; Stöwe, K.; Müller, F.; Maier, W.F. Mechanistic study of the unusual catalytic properties of a new NiCe mixed oxide for the CO2 reforming of methane. J. Catal. 2007, 247, 101–111. [Google Scholar] [CrossRef]
- Wei, J.; Iglesia, E. Reaction Pathways and Site Requirements for the Activation and Chemical Conversion of Methane on Ru−Based Catalysts. J. Phys. Chem. B 2004, 108, 7253–7262. [Google Scholar] [CrossRef] [Green Version]
- Mimura, N.; Takahara, I.; Inaba, M.; Okamoto, M.; Murata, K. High-performance Cr/H-ZSM-5 catalysts for oxidative dehydrogenation of ethane to ethylene with CO2 as an oxidant. Catal. Commun. 2002, 3, 257–262. [Google Scholar] [CrossRef]
- Porosoff, M.D.; Myint, M.N.Z.; Kattel, S.; Xie, Z.; Gomez, E.; Liu, P.; Chen, J.G. Identifying Different Types of Catalysts for CO2 Reduction by Ethane through Dry Reforming and Oxidative Dehydrogenation. Angew. Chem. Int. Ed. 2015, 54, 15501–15505. [Google Scholar] [CrossRef]
- Huber, G.W.; Iborra, S.; Corma, A. Synthesis of Transportation Fuels from Biomass: Chemistry, Catalysts, and Engineering. Chem. Rev. 2006, 106, 4044–4098. [Google Scholar] [CrossRef] [Green Version]
- Mattos, L.V.; Jacobs, G.; Davis, B.H.; Noronha, F.B. Production of Hydrogen from Ethanol: Review of Reaction Mechanism and Catalyst Deactivation. Chem. Rev. 2012, 112, 4094–4123. [Google Scholar] [CrossRef]
- Banach, B.; Machocki, A.; Rybak, P.; Denis, A.; Grzegorczyk, W.; Gac, W. Selective production of hydrogen by steam reforming of bio-ethanol. Catal. Today 2011, 176, 28–35. [Google Scholar] [CrossRef]
- Breen, J.P.; Burch, R.; Coleman, H.M. Metal-catalysed steam reforming of ethanol in the production of hydrogen for fuel cell applications. Appl. Catal. B Environ. 2002, 39, 65–74. [Google Scholar] [CrossRef]
- Liguras, D.K.; Kondarides, D.I.; Verykios, X.E. Production of hydrogen for fuel cells by steam reforming of ethanol over supported noble metal catalysts. Appl. Catal. B Environ. 2003, 43, 345–354. [Google Scholar] [CrossRef]
- Frusteri, F.; Freni, S.; Spadaro, L.; Chiodo, V.; Bonura, G.; Donato, S.; Cavallaro, S. H2 production for MC fuel cell by steam reforming of ethanol over MgO supported Pd, Rh, Ni and Co catalysts. Catal. Commun. 2004, 5, 611–615. [Google Scholar] [CrossRef]
- Ni, M.; Leung, D.Y.C.; Leung, M.K.H. A review on reforming bio-ethanol for hydrogen production. Int. J. Hydrog. Energy 2007, 32, 3238–3247. [Google Scholar] [CrossRef]
- Scott, M.; Goeffroy, M.; Chiu, W.; Blackford, M.A.; Idriss, H. Hydrogen Production from Ethanol over Rh-Pd/CeO2 Catalysts. Top. Catal. 2008, 51, 13–21. [Google Scholar] [CrossRef]
- Contreras, J.L.; Salmones, J.; Colín-Luna, J.A.; Nuño, L.; Quintana, B.; Córdova, I.; Zeifert, B.; Tapia, C.; Fuentes, G.A. Catalysts for H2 production using the ethanol steam reforming (a review). Int. J. Hydrog. Energy 2014, 39, 18835–18853. [Google Scholar] [CrossRef]
- Mariño, F.J.; Cerrella, E.G.; Duhalde, S.; Jobbagy, M.; Laborde, M.A. Hydrogen from steam reforming of ethanol. characterization and performance of copper-nickel supported catalysts. Int. J. Hydrog. Energy 1998, 23, 1095–1101. [Google Scholar] [CrossRef]
- Llorca, J.; Homs, N.; Sales, J.; De la Piscina, P.R. Efficient Production of Hydrogen over Supported Cobalt Catalysts from Ethanol Steam Reforming. J. Catal. 2002, 209, 306–317. [Google Scholar] [CrossRef]
- Zhang, B.; Tang, X.; Li, Y.; Cai, W.; Xu, Y.; Shen, W. Steam reforming of bio-ethanol for the production of hydrogen over ceria-supported Co, Ir and Ni catalysts. Catal. Commun. 2006, 7, 367–372. [Google Scholar] [CrossRef]
- Denis, A.; Grzegorczyk, W.; Gac, W.; Machocki, A. Steam reforming of ethanol over Ni/support catalysts for generation of hydrogen for fuel cell applications. Catal. Today 2008, 137, 453–459. [Google Scholar] [CrossRef]
- Bayram, B.; Soykal, I.I.; Von Deak, D.; Miller, J.T.; Ozkan, U.S. Ethanol steam reforming over Co-based catalysts: Investigation of cobalt coordination environment under reaction conditions. J. Catal. 2011, 284, 77–89. [Google Scholar] [CrossRef]
- Dan, M.; Mihet, M.; Tasnadi-Asztalos, Z.; Imre-Lucaci, A.; Katona, G.; Lazar, M.D. Hydrogen production by ethanol steam reforming on nickel catalysts: Effect of support modification by CeO2 and La2O3. Fuel 2015, 147, 260–268. [Google Scholar] [CrossRef]
- Laosiripojana, N.; Assabumrungrat, S. Catalytic steam reforming of ethanol over high surface area CeO2: The role of CeO2 as an internal pre-reforming catalyst. Appl. Catal. B Environ. 2006, 66, 29–39. [Google Scholar] [CrossRef]
- Lin, S.S.-Y.; Kim, D.H.; Ha, S.Y. Metallic phases of cobalt-based catalysts in ethanol steam reforming: The effect of cerium oxide. Appl. Catal. A Gen. 2009, 355, 69–77. [Google Scholar] [CrossRef]
- Virginie, M.; Araque, M.; Roger, A.-C.; Vargas, J.C.; Kiennemann, A. Comparative study of H2 production by ethanol steam reforming on Ce2Zr1.5Co0.5O8−δ and Ce2Zr1.5Co0.47Rh0.07O8−δ: Evidence of the Rh role on the deactivation process. Catal. Today 2008, 138, 21–27. [Google Scholar] [CrossRef]
- Llorca, J.; Ramírez de la Piscina, P.; Dalmon, J.-A.; Homs, N. Transformation of Co3O4 during Ethanol Steam-Re-forming. Activation Process for Hydrogen Production. Chem. Mater. 2004, 16, 3573–3578. [Google Scholar] [CrossRef]
- Raskó, J.; Dömök, M.; Baán, K.; Erdőhelyi, A. FTIR and mass spectrometric study of the interaction of ethanol and ethanol–water with oxide-supported platinum catalysts. Appl. Catal. A Gen. 2006, 299, 202–211. [Google Scholar] [CrossRef]
- Palo, D.R.; Dagle, R.A.; Holladay, J.D. Methanol Steam Reforming for Hydrogen Production. Chem. Rev. 2007, 107, 3992–4021. [Google Scholar] [CrossRef]
- Peppley, B.A.; Amphlett, J.C.; Kearns, L.M.; Mann, R.F. Methanol–steam reforming on Cu/ZnO/Al2O3. Part 1: The reaction network. Appl. Catal. A Gen. 1999, 179, 21–29. [Google Scholar] [CrossRef]
- Lee, J.K.; Ko, J.B.; Kim, D.H. Methanol steam reforming over Cu/ZnO/Al2O3 catalyst: Kinetics and effectiveness factor. Appl. Catal. A Gen. 2004, 278, 25–35. [Google Scholar] [CrossRef]
- Shishido, T.; Yamamoto, Y.; Morioka, H.; Takaki, K.; Takehira, K. Active Cu/ZnO and Cu/ZnO/Al2O3 catalysts prepared by homogeneous precipitation method in steam reforming of methanol. Appl. Catal. A Gen. 2004, 263, 249–253. [Google Scholar] [CrossRef]
- Laosiripojana, N.; Assabumrungrat, S. Catalytic steam reforming of methane, methanol, and ethanol over Ni/YSZ: The possible use of these fuels in internal reforming SOFC. J. Power Sources 2007, 163, 943–951. [Google Scholar] [CrossRef]
- Lu, J.; Li, X.; He, S.; Han, C.; Wan, G.; Lei, Y.; Chen, R.; Liu, P.; Chen, K.; Zhang, L.; et al. Hydrogen production via methanol steam reforming over Ni-based catalysts: Influences of Lanthanum (La) addition and supports. Int. J. Hydrog. Energy 2017, 42, 3647–3657. [Google Scholar] [CrossRef]
- Tatibouët, J.M. Methanol oxidation as a catalytic surface probe. Appl. Catal. A Gen. 1997, 148, 213–252. [Google Scholar] [CrossRef]
- Vilé, G.; Bridier, B.; Wichert, J.; Pérez-Ramírez, J. Ceria in Hydrogenation Catalysis: High Selectivity in the Conversion of Alkynes to Olefins. Angew. Chem. Int. Ed. 2012, 51, 8620–8623. [Google Scholar] [CrossRef] [PubMed]
- Vilé, G.; Colussi, S.; Krumeich, F.; Trovarelli, A.; Pérez-Ramírez, J. Opposite Face Sensitivity of CeO2 in Hydrogenation and Oxidation Catalysis. Angew. Chem. Int. Ed. 2014, 53, 12069–12072. [Google Scholar] [CrossRef]
- Sohlberg, K.; Pantelides, S.T.; Pennycook, S.J. Interactions of Hydrogen with CeO2. J. Am. Chem. Soc. 2001, 123, 6609–6611. [Google Scholar] [CrossRef]
- Watkins, M.B.; Foster, A.S.; Shluger, A.L. Hydrogen Cycle on CeO2 (111) Surfaces: Density Functional Theory Calculations. J. Phys. Chem. C 2007, 111, 15337–15341. [Google Scholar] [CrossRef]
- Brisse, A.; Schefold, J.; Zahid, M. High temperature water electrolysis in solid oxide cells. Int. J. Hydrog. Energy 2008, 33, 5375–5382. [Google Scholar] [CrossRef]
- Laguna-Bercero, M.A. Recent advances in high temperature electrolysis using solid oxide fuel cells: A review. J. Power Sources 2012, 203, 4–16. [Google Scholar] [CrossRef] [Green Version]
- El Gabaly, F.; Grass, M.; McDaniel, A.H.; Farrow, R.L.; Linne, M.A.; Hussain, Z.; Bluhm, H.; Liu, Z.; McCarty, K.F. Measuring individual overpotentials in an operating solid-oxide electrochemical cell. Phys. Chem. Chem. Phys. 2010, 12, 12138–12145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeCaluwe, S.C.; Grass, M.E.; Zhang, C.; Gabaly, F.E.; Bluhm, H.; Liu, Z.; Jackson, G.S.; McDaniel, A.H.; McCarty, K.F.; Farrow, R.L.; et al. In situ characterization of ceria oxidation states in high-temperature electrochemical cells with ambient pressure XPS. J. Phys. Chem. C 2010, 114, 19853–19861. [Google Scholar] [CrossRef]
- Crumlin, E.J.; Bluhm, H.; Liu, Z. In situ investigation of electrochemical devices using ambient pressure photoelectron spectroscopy. J. Electron. Spectros. Relat. Phenom. 2013, 190, 84–92. [Google Scholar] [CrossRef]
- Stoerzinger, K.A.; Hong, W.T.; Crumlin, E.J.; Bluhm, H.; Shao-Horn, Y. Insights into Electrochemical Reactions from Ambient Pressure Photoelectron Spectroscopy. Acc. Chem. Res. 2015, 48, 2976–2983. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, Y.; Mao, B.; Geller, A.; Chang, R.; Gaskell, K.; Eichhorn, B.W. CO2 activation and carbonate intermediates: An operando AP-XPS study of CO2 electrolysis reactions on solid oxide electrochemical cells. Phys. Chem. Chem. Phys. 2014, 16, 11633–11639. [Google Scholar] [CrossRef]
- Jacobson, A.J. Materials for solid oxide fuel cells. Chem. Mater. 2010, 22, 660–674. [Google Scholar] [CrossRef]
- Orera, A.; Slater, P.R. New chemical systems for solid oxide fuel cells. Chem. Mater. 2010, 22, 675–690. [Google Scholar] [CrossRef]
- Rafique, M.; Nawaz, H.; Rafique, M.S.; Tahir, M.B.; Nabi, G.; Khalid, N.R. Material and method selection for efficient solid oxide fuel cell anode: Recent advancements and reviews. Int. J. Energy Res. 2019, 43, 2423–2446. [Google Scholar] [CrossRef]
- Haile, S.M. Fuel cell materials and components. Acta Mater. 2003, 51, 5981–6000. [Google Scholar] [CrossRef]
- Kharton, V.V.; Figueiredo, F.M.; Navarro, L.; Naumovich, E.N.; Kovalevsky, A.V.; Yaremchenko, A.A.; Viskup, A.P.; Carneiro, A.; Marques, F.M.B.; Frade, J.R. Ceria-based materials for solid oxide fuel cells. J. Mater. Sci. 2001, 36, 1105–1117. [Google Scholar] [CrossRef]
- Zhang, C.; Grass, M.E.; McDaniel, A.H.; Decaluwe, S.C.; El Gabaly, F.; Liu, Z.; McCarty, K.F.; Farrow, R.L.; Linne, M.A.; Hussain, Z.; et al. Measuring fundamental properties in operating solid oxide electrochemical cells by using in situ X-ray photoelectron spectroscopy. Nat. Mater. 2010, 9, 944–949. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Grass, M.E.; Yu, Y.; Gaskell, K.J.; Decaluwe, S.C.; Chang, R.; Jackson, G.S.; Hussain, Z.; Bluhm, H.; Eichhorn, B.W.; et al. Multielement activity mapping and potential mapping in solid oxide electrochemical cells through the use of operando XPS. ACS Catal. 2012, 2, 2297–2304. [Google Scholar] [CrossRef]
- Chueh, W.C.; Mcdaniel, A.H.; Grass, M.E.; Hao, Y.; Jabeen, N.; Liu, Z.; Haile, S.M.; Mccarty, K.F.; Bluhm, H.; El Gabaly, F. Highly Enhanced Concentration and Stability of Reactive Ce3+ on Doped CeO2 Surface Revealed In Operando. Chem. Mater. 2012, 24, 1876–1882. [Google Scholar] [CrossRef] [Green Version]
- Zhang, C.; Yu, Y.; Grass, M.E.; Dejoie, C.; Ding, W.; Gaskell, K.; Jabeen, N.; Hong, Y.P.; Shavorskiy, A.; Bluhm, H.; et al. Mechanistic studies of water electrolysis and hydrogen electro-oxidation on high temperature ceria-based solid oxide electrochemical cells. J. Am. Chem. Soc. 2013, 135, 11572–11579. [Google Scholar] [CrossRef]
- Gopal, C.B.; El Gabaly, F.; McDaniel, A.H.; Chueh, W.C. Origin and Tunability of Unusually Large Surface Capacitance in Doped Cerium Oxide Studied by Ambient-Pressure X-Ray Photoelectron Spectroscopy. Adv. Mater. 2016, 28, 4692–4697. [Google Scholar] [CrossRef]
- Papaefthimiou, V.; Niakolas, D.K.; Paloukis, F.; Dintzer, T.; Zafeiratos, S. Is Steam an Oxidant or a Reductant for Nickel/Doped-Ceria Cermets? ChemPhysChem 2017, 18, 164–170. [Google Scholar] [CrossRef]
- Papaefthimiou, V.; Niakolas, D.K.; Paloukis, F.; Teschner, D.; Knop-Gericke, A.; Haevecker, M.; Zafeiratos, S. Operando observation of nickel/ceria electrode surfaces during intermediate temperature steam electrolysis. J. Catal. 2017, 352, 305–313. [Google Scholar] [CrossRef]
- Nurk, G.; Kooser, K.; Urpelainen, S.; Käämbre, T.; Joost, U.; Kodu, M.; Kivi, I.; Kanarbik, R.; Kukk, E.; Lust, E. Near ambient pressure X-ray photoelectron-and impedance spectroscopy study of NiO-Ce0.9Gd0.1O2-Δ anode reduction using a novel dual-chamber spectroelectrochemical cell. J. Power Sources 2018, 378, 589–596. [Google Scholar] [CrossRef]
- Wang, L.; Jackson, G.S. Evaluating the behavior of CO/CO2 in Ni/GDC solid oxide fuel cell anodes. ECS Trans. 2015, 68, 1193–1205. [Google Scholar] [CrossRef]
- Papaefthimiou, V.; Shishkin, M.; Niakolas, D.K.; Athanasiou, M.; Law, Y.T.; Arrigo, R.; Teschner, D.; Hävecker, M.; Knop-Gericke, A.; Schlögl, R.; et al. On the active surface state of nickel-ceria solid oxide fuel cell anodes during methane electrooxidation. Adv. Energy Mater. 2013, 3, 762–769. [Google Scholar] [CrossRef]
- Graves, C.; Ebbesen, S.D.; Mogensen, M.; Lackner, K.S. Sustainable hydrocarbon fuels by recycling CO2 and H2O with renewable or nuclear energy. Renew. Sustain. Energy Rev. 2011, 15, 1–23. [Google Scholar] [CrossRef]
- Sridhar, K.R.; Vaniman, B.T. Oxygen production on Mars using solid oxide electrolysis. Solid State Ion. 1997, 93, 321–328. [Google Scholar] [CrossRef]










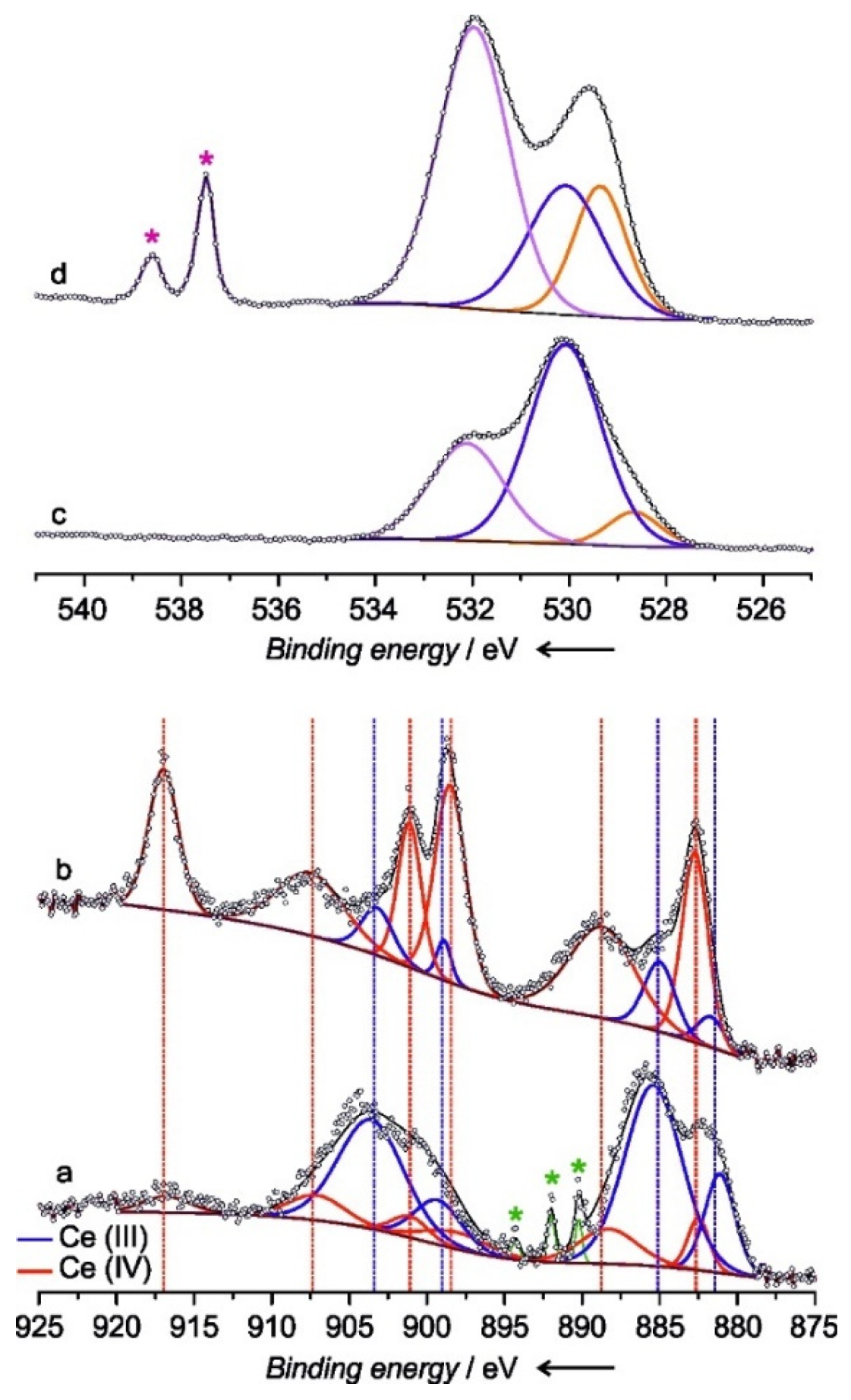{kind=link}
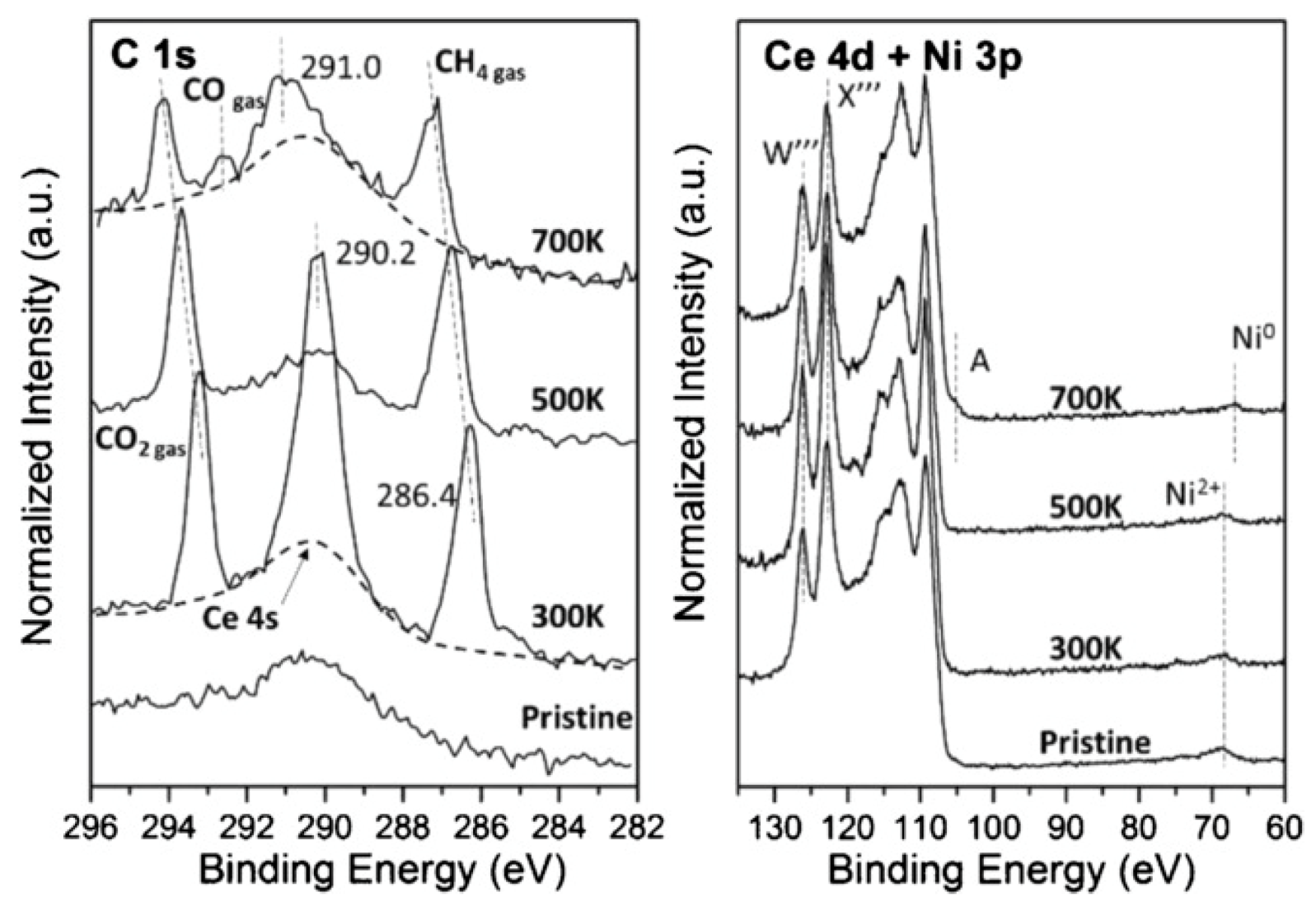{kind=link}
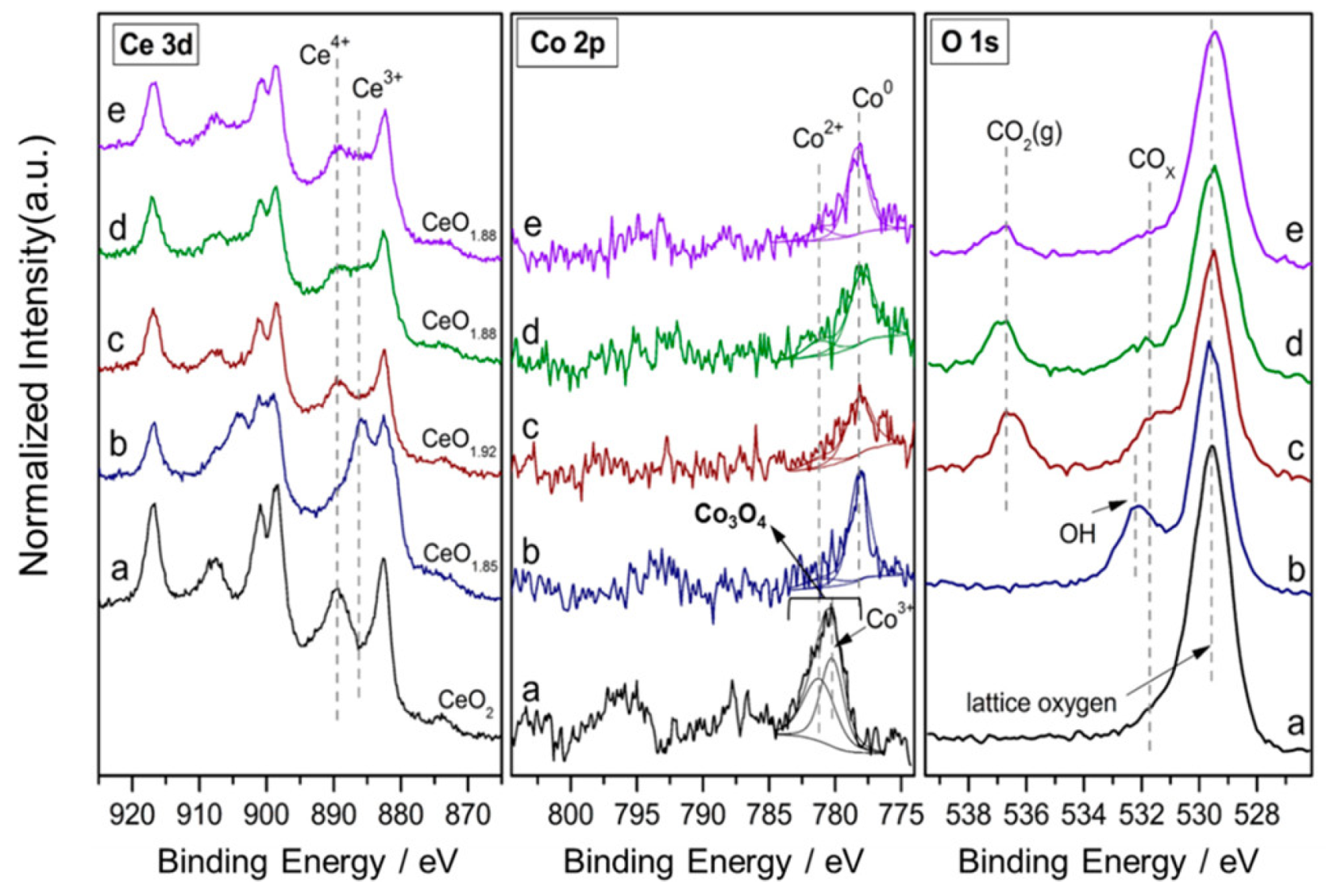{kind=link}
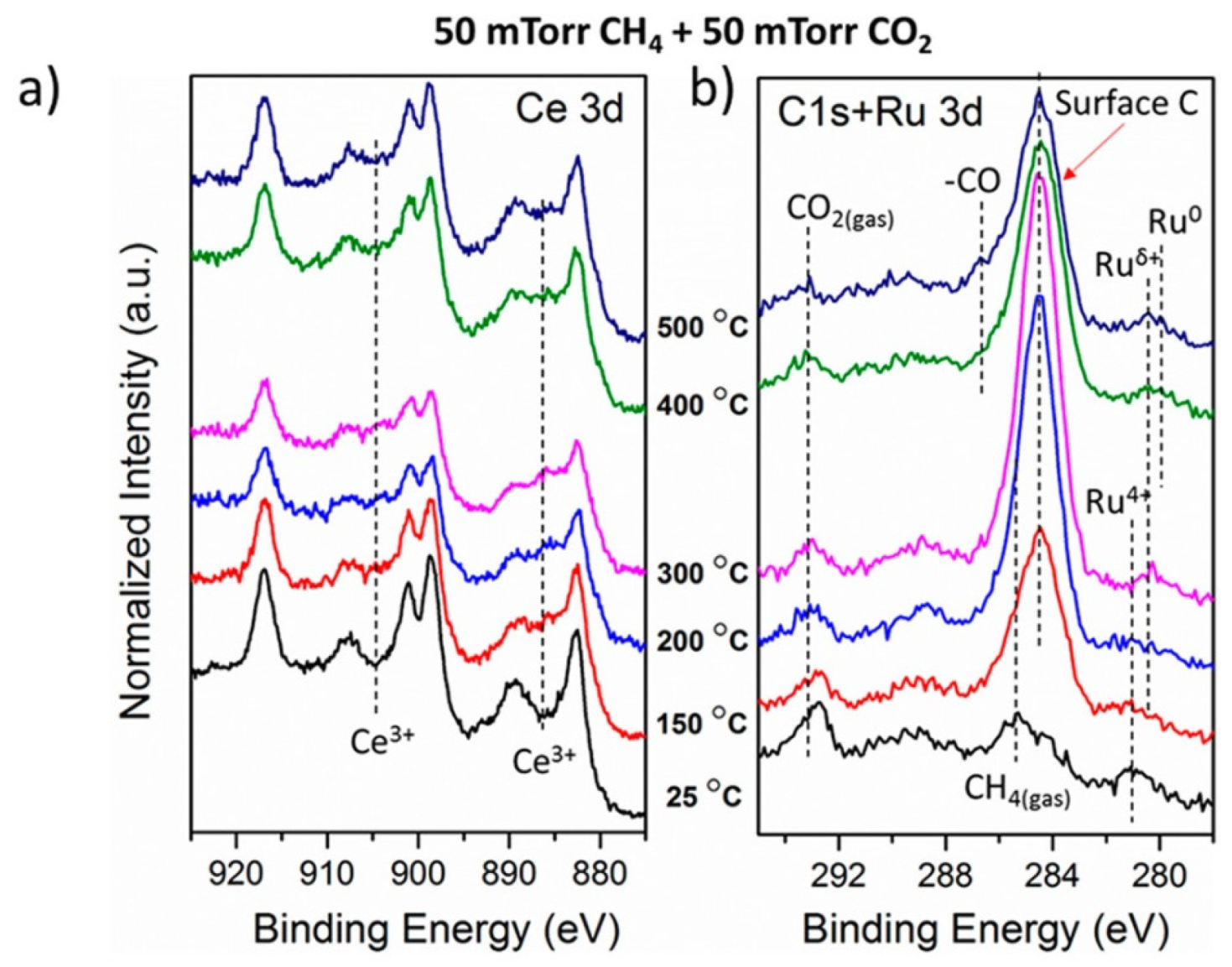{kind=link}
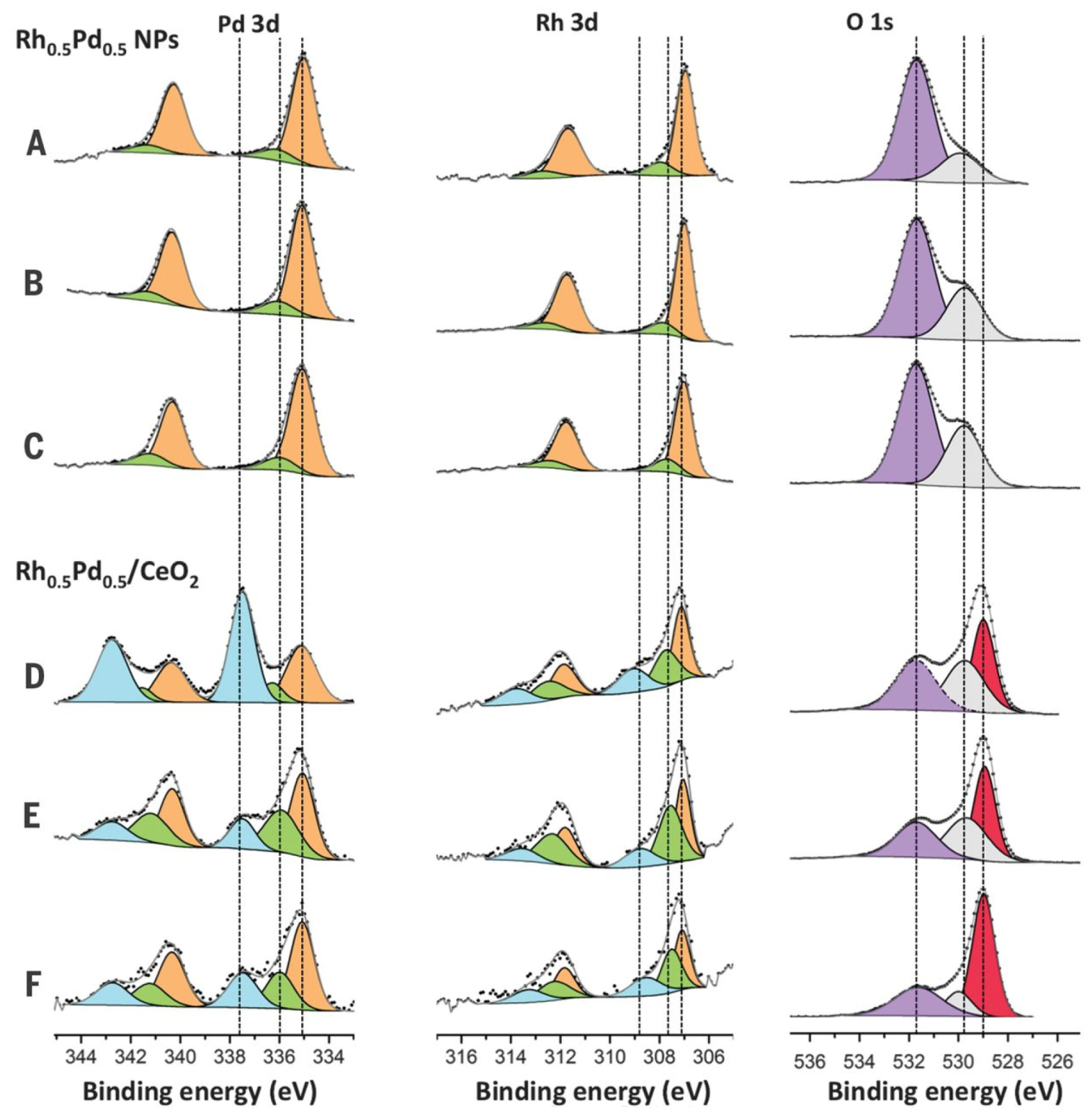{kind=link}
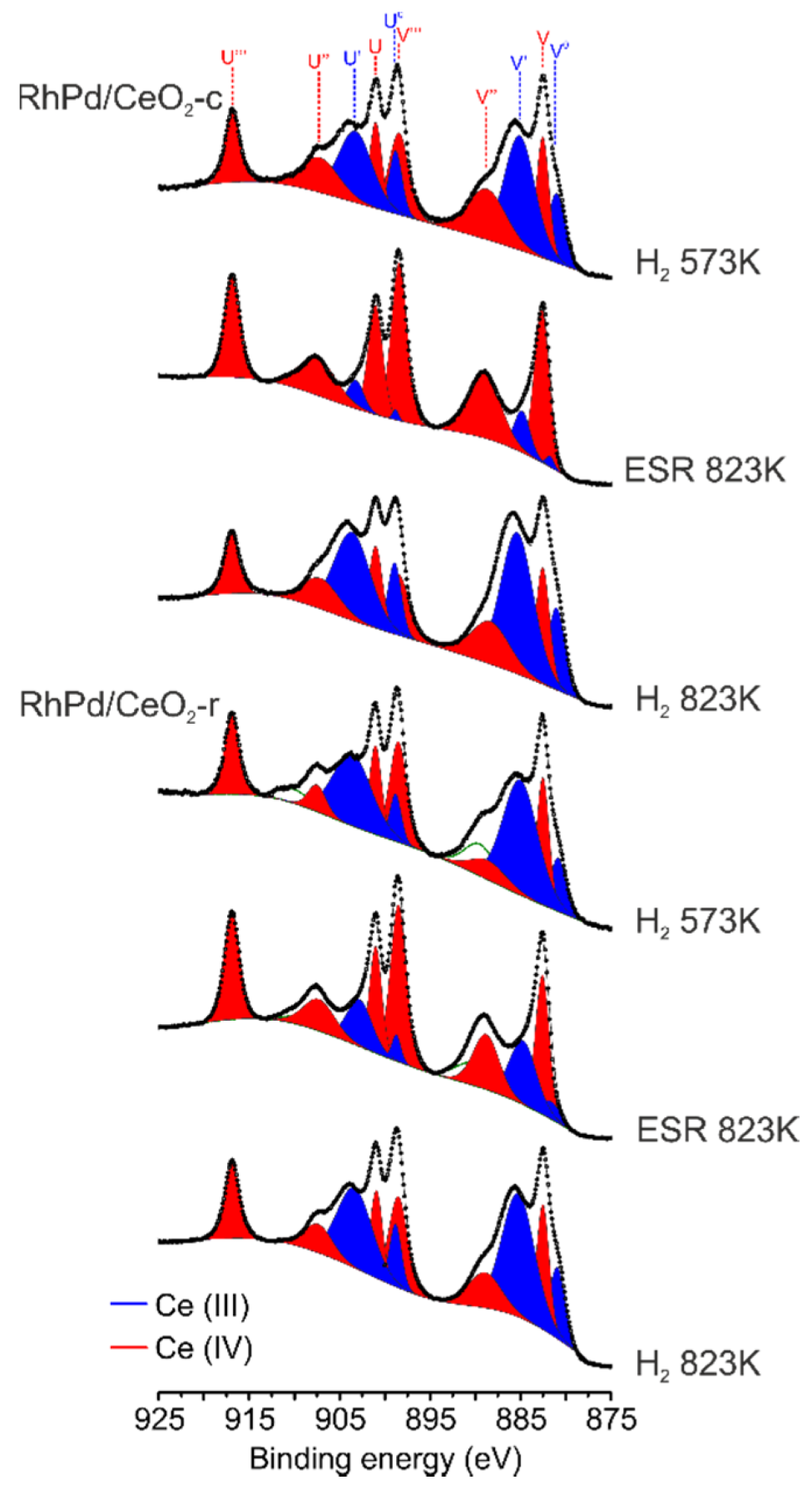{kind=link}
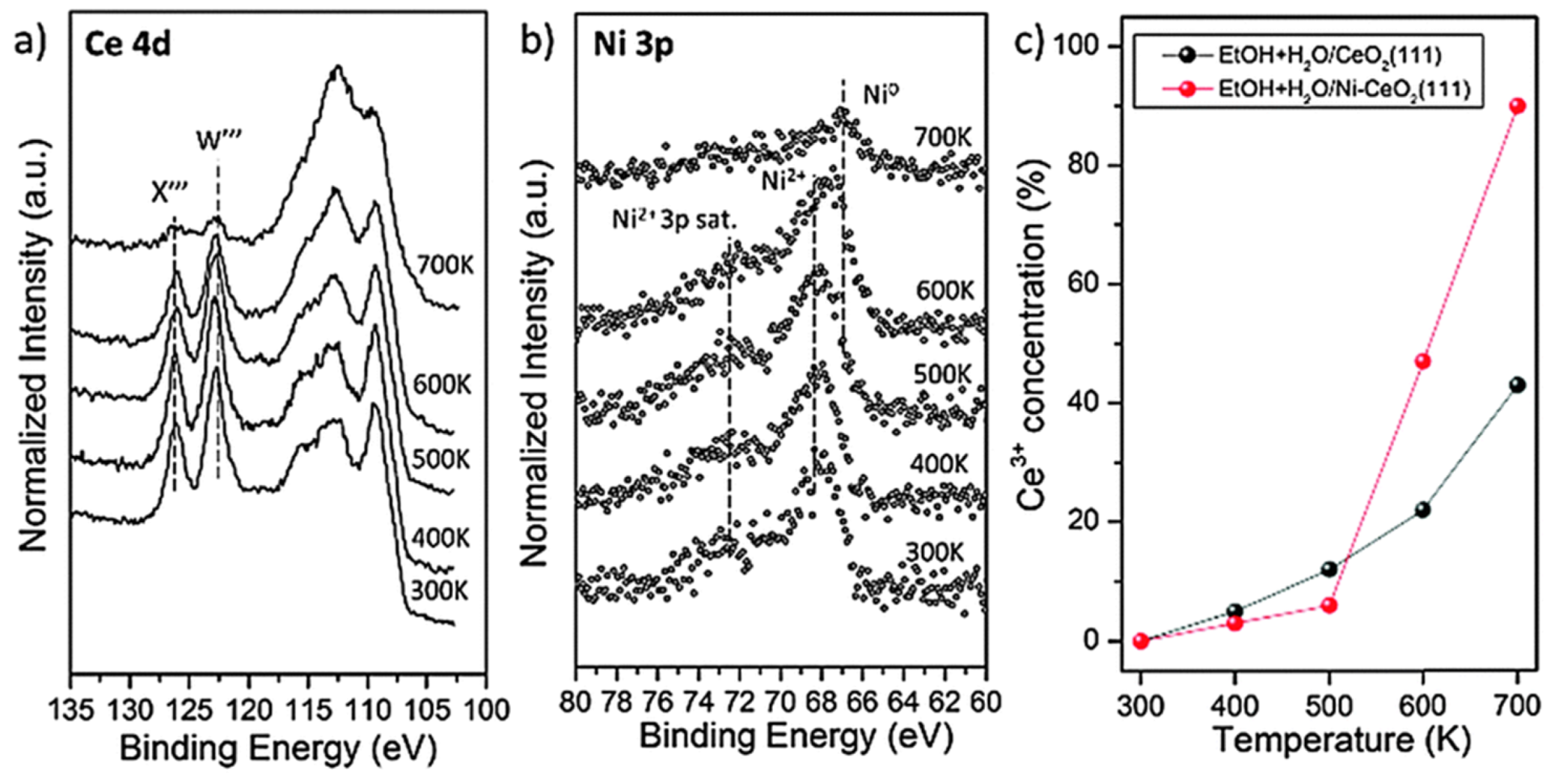{kind=link}
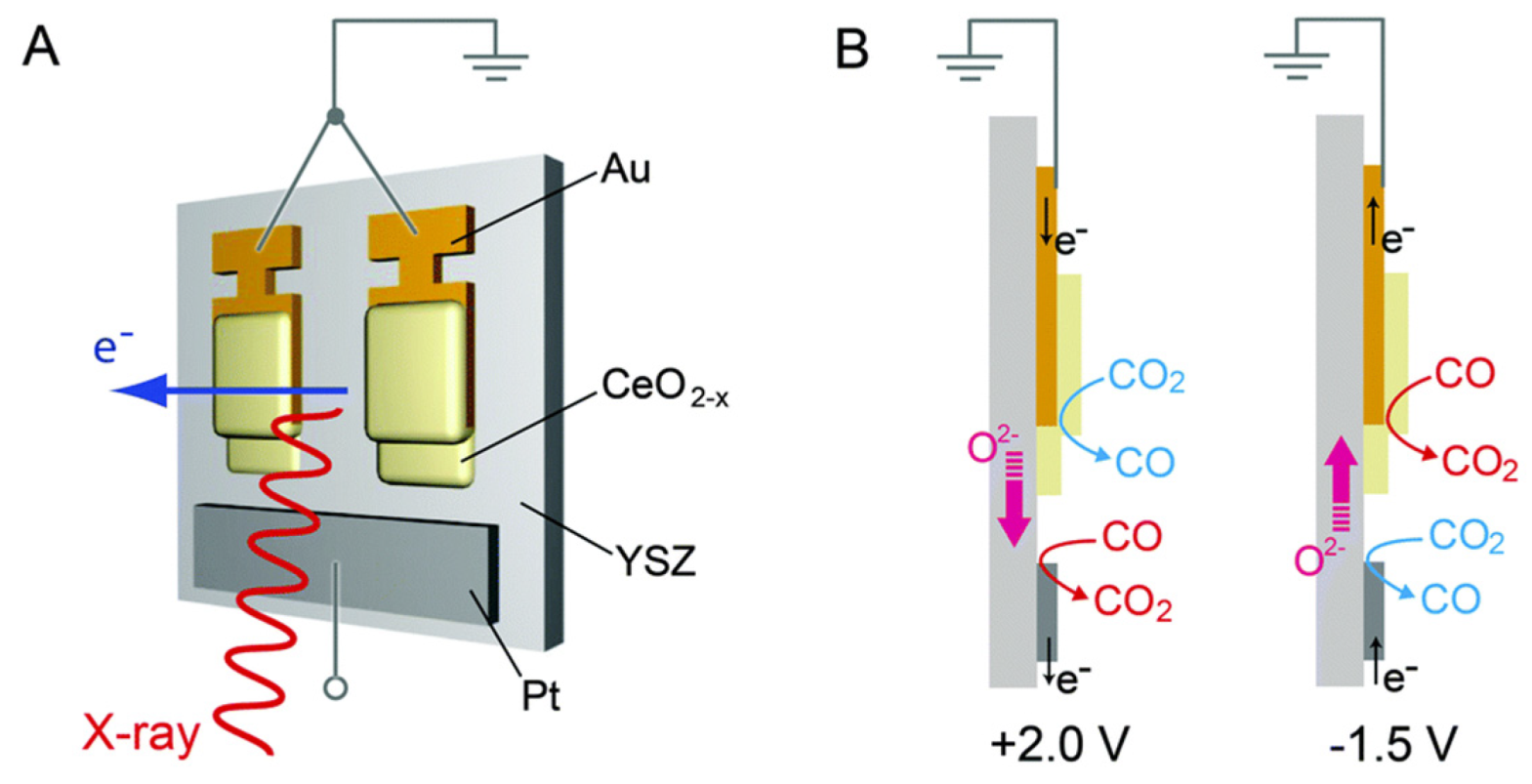{kind=link}
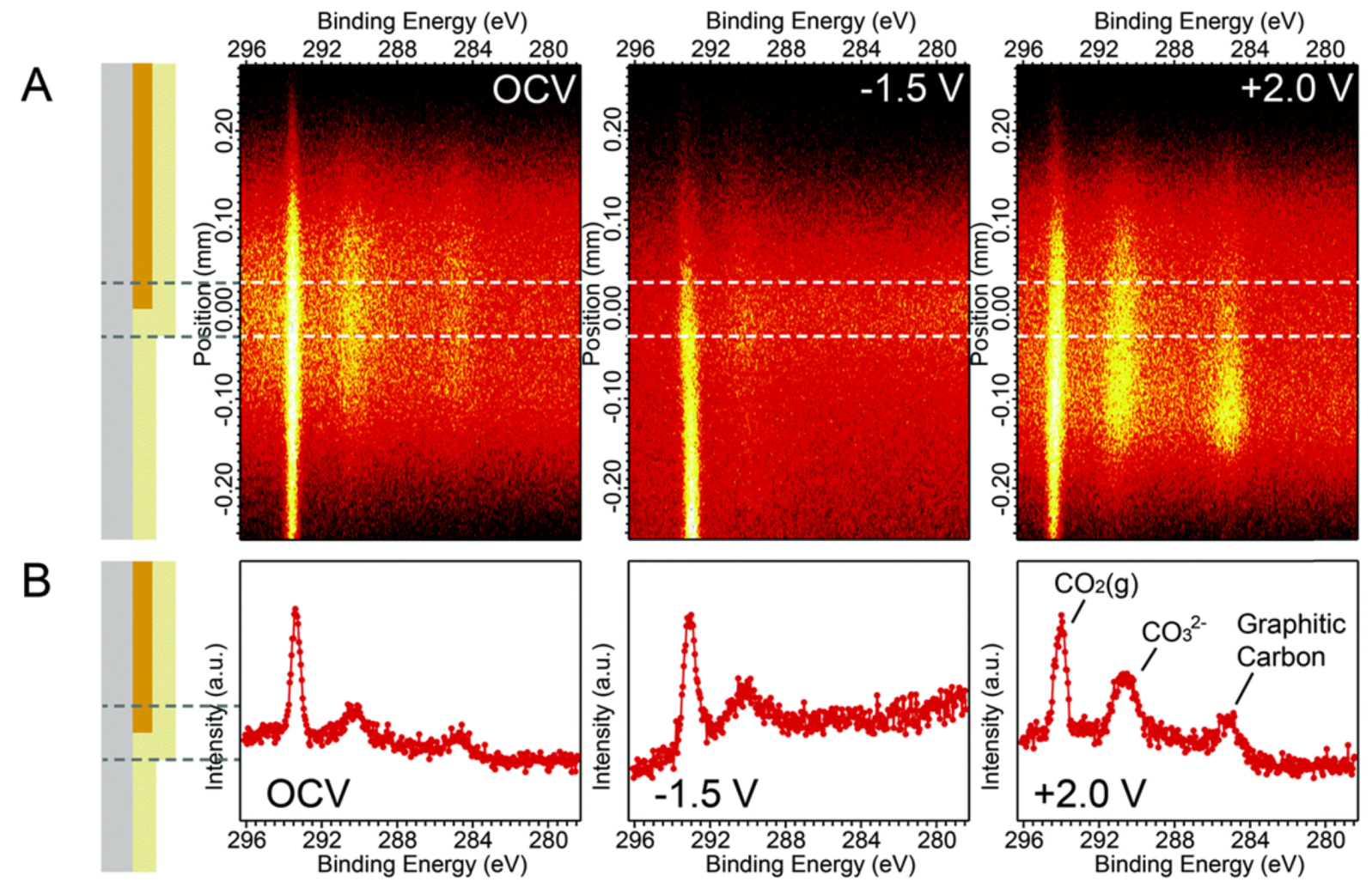{kind=link}
| Reaction | Catalyst | Pmax (mbar) | T (K) | X-ray Source | Year | Ref. | |
|---|---|---|---|---|---|---|---|
| CO oxidation and preferential CO oxidation (PROX) | 5% Pt/CeO2 | ~ 0.5 | 358−523 | Synchrotron | 2006 | [126] | |
| 5% Pd/CeO2 | ~ 0.5 | 358−523 | Synchrotron | 2006 | [127] | ||
| 5% Pt/CeO2 | ~ 1 | 393 | Synchrotron | 2007 | [128] | ||
| Au-Ni/CeO2 | ~ 1 | 523 | Synchrotron | 2013 | [129] | ||
| 1% Cu/CeO2 nanospheres 1% Cu/CeO2 nanocubes | ~ 1 | 300−553 | Synchrotron | 2015 | [130] | ||
| 10% CeO2/Co3O4 | 0.5 | 300−573 | Synchrotron | 2016 | [131] | ||
| 3% Pt/CeO2 | 1 | 373 | Synchrotron | 2017 | [132] | ||
| CeO2 nanoparticles | ~ 1 | 373−773 | Lab-based | 2017 | [121] | ||
| 1% Pt/CeO2 | ~ 2 | 323−373 | Lab-based | 2019 | [122] | ||
| Water-gas shift reaction (WGS) | 2.5% M@mesoporous-CeO22.5% M/rod-CeO2 (M = Au, Pt, Pd, Cu) | 1.95 | 403−543 | Lab-based | 2012 | [133] | |
| CeO2/CuO | 0.5 | 473−523 | Synchrotron | 2012 | [134] | ||
| CeOx/Cu(111) CeOx/Au(111) | ~ 0.5 | 300−573 | Synchrotron | 2013 | [39] | ||
| Soot oxidation | CeO2 and Ce0.8Zr0.2O2 nanoparticles | 1 | 300−823 | Synchrotron | 2016 | [135] | |
| CO2 hydrogenation | CeOx/Cu(111) | 0.39 | 473 | Synchrotron | 2014 | [136] | |
| CeOx/Cu(111) | ~ 0.4 | 300−500 | Synchrotron | 2016 | [137] | ||
| 5% Cu/CeO2 nanospheres 5% Cu/CeO2 nanorods | 0.06 | 300−723 | Lab-based | 2018 | [97] | ||
| 0.5, 1.5 and 5% Ni/CeO2 | ~ 0.06 | 300−623 | Lab-based | 2019 | [138] | ||
| Hydrocarbons | Oxidation and partial oxidation | 5% M/CeO2 (M = Pd, Pt, Rh) | 3.9 | 300−873 | Lab-based | 2013 | [139] |
| Co3O4/CeO2 nanorods | 1.56 | 423−773 | Lab-based | 2018 | [140] | ||
| 15% NiO/CeO2 nanorods | 1.56 | 423−773 | Lab-based | 2018 | [141] | ||
| Dry reforming | 26% Ni/CeO2 | 1.3 | 773−923 | Synchrotron | 2008 | [142] | |
| Ni/CeO2(111) | 0.26 | 300−700 | Synchrotron | 2016 | [143] | ||
| Ni/CeO2(111) | 0.26 | 700 | Synchrotron | 2016 | [144] | ||
| 1.5% Ni, 1.7% Pt and PtNi/CeO2/TiO2(110) | 0.39 | 303−723 | Synchrotron | 2016 | [145] | ||
| M/CeO2(111)/Ru(0001) (M = Co, Ni, Cu) | 0.13 | 300−700 | Lab-based | 2017 | [146] | ||
| 10% Co/CeO2 | ~ 0.2 | 300−773 | Lab-based | 2018 | [147] | ||
| PtCo/CeO2(Pt = 1.67%, Co = 1.51%) | 0.052 | 823 | Lab-based | 2018 | [148] | ||
| 0.5% Ru (NC)/CeO2 | 0.13 | 300−773 | Lab-based | 2019 | [149] | ||
| Alcohols | Steam reforming (SR) | CeO2(111)/Cu(111) Co/CeO2(111)/Cu(111) | 1 | 320−600 | Lab-based | 2013 | [150] |
| 3% Rh0.5Pd0.5/CeO2 | 0.05 | 823 | Synchrotron | 2014 | [68] | ||
| 8% Co/CeO2 | 0.2 | 523−693 | Synchrotron | 2016 | [151] | ||
| 23% Co/CeO2 | 0.3 | 693 | Synchrotron | 2016 | [152] | ||
| 10% Co/CeO2-NP a 10% Co/CeO2-MP b | 1.43 | 623−723 | Lab-based | 2016 | [153] | ||
| Ni/CeO2(111)/Ru(0001) | ~ 0.3 | 300−700 | Synchrotron | 2016 | [154] | ||
| Co/CeO2-NP Co/CeO2-MP | 1.43 | 623−723 | Lab-based | 2017 | [101] | ||
| Ni/CeO2 Ni/CeO2−x/Ru(0001) | 2 L * | 300−700 | Synchrotron | 2018 | [155] | ||
| 3% Rh0.5Pd0.5/CeO2 nanocubes 3% Rh0.5Pd0.5/CeO2 nanorods | 0.05 | 823 | Synchrotron | 2019 | [69] | ||
| Oxidation | CeO2(100)/STO(100) | 0.39 | 523−723 | Synchrotron | 2018 | [156] | |
| Hydrogenation reactions | CeO2 nanorods | 0.65 | 533−673 | Lab-based | 2017 | [157] | |
| CeO2/Pt | 0.39 | 300−473 | Lab-based | 2017 | [158] | ||
| Reaction | Catalyst | Pmax (mbar) | T (K) | X-ray Source | Year | Ref. |
|---|---|---|---|---|---|---|
| H2O electrolysis/ H2 electro-oxidation | WE = CeO2−x CE = Pt | 0.66 | 973 | Synchrotron | 2009 | [75] |
| WE = CeO2−x CE = Pt | 1.06 | 923–1013 | Synchrotron | 2010 | [300] | |
| WE = CeO2−x CE = Pt | 1 | 1023 | Synchrotron | 2010 | [309] | |
| WE = CeO2 CE = Pt | 0.65 | 973–1023 | Synchrotron | 2012 | [310] | |
| SDC a | 1.06 | 739–923 | Synchrotron | 2012 | [311] | |
| WE=CeO2−x CE=Pt | 0.65 | 973 | Synchrotron | 2013 | [312] | |
| CeO2−x and SDC | 0.37 | 763–923 | Synchrotron | 2016 | [313] | |
| Ni/GDC b | 0.2 | 773–973 | Synchrotron | 2017 | [314] | |
| WE = NiO/GDC CE = Pt | 0.2 | 973 | Synchrotron | 2017 | [315] | |
| WE = Ni/GDC CE = LSC c | 0.1 | 923 | Synchrotron | 2018 | [316] | |
| CO2 electrolysis/ CO electro-oxidation | WE = CeO2−x CE = Pt | 0.65 | 873 | Synchrotron | 2014 | [303] |
| WE = Ni/GDC CE = Pt | 0.53 | 893 | Synchrotron | 2015 | [317] | |
| CH4 electro-oxidation | WE = Ni/GDC CE = Pt | 0.1 | 973 | Synchrotron | 2013 | [318] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garcia, X.; Soler, L.; Divins, N.J.; Vendrell, X.; Serrano, I.; Lucentini, I.; Prat, J.; Solano, E.; Tallarida, M.; Escudero, C.; et al. Ceria-Based Catalysts Studied by Near Ambient Pressure X-ray Photoelectron Spectroscopy: A Review. Catalysts 2020, 10, 286. https://0-doi-org.brum.beds.ac.uk/10.3390/catal10030286
Garcia X, Soler L, Divins NJ, Vendrell X, Serrano I, Lucentini I, Prat J, Solano E, Tallarida M, Escudero C, et al. Ceria-Based Catalysts Studied by Near Ambient Pressure X-ray Photoelectron Spectroscopy: A Review. Catalysts. 2020; 10(3):286. https://0-doi-org.brum.beds.ac.uk/10.3390/catal10030286
Chicago/Turabian StyleGarcia, Xènia, Lluís Soler, Núria J. Divins, Xavier Vendrell, Isabel Serrano, Ilaria Lucentini, Jordi Prat, Eduardo Solano, Massimo Tallarida, Carlos Escudero, and et al. 2020. "Ceria-Based Catalysts Studied by Near Ambient Pressure X-ray Photoelectron Spectroscopy: A Review" Catalysts 10, no. 3: 286. https://0-doi-org.brum.beds.ac.uk/10.3390/catal10030286