Simple Environmentally-Friendly Reduction of 4-Nitrophenol


1
Empa, Swiss Federal Laboratories for Materials Science and Technology, Laboratory for Mechanics of Materials and Nanostructures, Feuerwerkerstrasse 39, CH-3602 Thun, Switzerland
2
Grup d’Electrodeposició de Capes Primes i Nanoestructures (GE-CPN), Departament de Ciència de Materials i Química Física, Universitat de Barcelona, Martí i Franquès 1, E-08028 Barcelona, Catalonia, Spain
3
Institute of Nanoscience and Nanotechnology (IN2UB), Universitat de Barcelona, E-08028 Barcelona, Catalonia, Spain
4
Grup de Materials Orgànics, Departament de Química Inorgànica i Orgànica (Secció de Química Orgànica), Universitat de Barcelona, Martí i Franquès 1, E-08028 Barcelona, Catalonia, Spain
*
Author to whom correspondence should be addressed.
Catalysts 2020, 10(4), 458; https://0-doi-org.brum.beds.ac.uk/10.3390/catal10040458
Submission received: 21 March 2020
/
Revised: 17 April 2020
/
Accepted: 21 April 2020
/
Published: 23 April 2020
(This article belongs to the Special Issue Electrocatalysis in Energy and Green Chemistry)
Abstract
:The low molecular-mass organic compound 4-nitrophenol is involved in many chemical processes and is commonly present in soils and in surface and ground waters, thereby causing severe environmental impact and health risk. Several methods have been proposed for its transformation (bio and chemical degradation). However, these strategies not only produce equally or more toxic aromatic species but also require harsh operating conditions and/or time-consuming treatments. In this context, we report a comprehensive and systematic study of the electrochemical reduction of 4-nitrophenol as a viable alternative. We have explored the electrochemical reduction of this pollutant over different metallic and carbonaceous substrata. Specifically, we have focused on the use of gold and silver working electrodes since they combine a high electrocatalytic activity for 4-nitrophenol reduction and a low electrocatalytic capacity for hydrogen evolution. The influence of the pH, temperature, and applied potential have also been considered as crucial parameters in the overall optimization of the process. While acidic media and high temperatures favor the clean reduction of 4-nitrophenol to 4-aminophenol, the simultaneous hydrogen evolution is pernicious for this purpose. Herein, a simple and effective electrochemical method for the transformation of 4-nitrophenol into 4-aminophenol is proposed with virtually no undesired by-products.
1. Introduction
Industries constantly release tons of hazardous chemicals into the atmosphere, rivers and seas, causing immense environmental devastation. Particularly, 4-nitrophenol (4-NP in Scheme 1) is a simple organic aromatic compound, widely used in manufacturing many products required in the modern society such as dyes, pesticides, drugs, etc. [1,2,3]. Its widespread industrial use makes 4-NP a common pollutant in soils and surface and ground waters. It has a severe environmental impact and health risks due to its toxicity and mutagenic potential in humans and other living organisms [4]. Moreover, the degradation rate of 4-NP in urban wastewater is slow. This causes it to accumulate in the environment [5]. Indeed, this is the reason for the United States Environmental Protection Agency (US EPA) including 4-NP and its derivatives to the list of top priority pollutants [6,7,8]. Consequently, removing this compound from the sources of manufacturing is mandatory to allow the efficient and safe elimination of this hazardous organic compound. This is an essential step for conserving the ecosystem and human health.
Several methods have been proposed for the partial or total oxidation of 4-NP, including microbial catabolism [9,10], photocatalytic degradation [10,11,12,13], biodegradation [14,15,16], Fenton [17,18] and electrochemical methods [19,20,21]. Alternatively, chemical or electrochemical reduction of 4-NP to 4-aminophenol (4-AP in Scheme 1) is also suggested. Nonetheless, these strategies can produce distinct aromatic species that are equally or more toxic than 4-NP. For example, p-benzoquinone, hydroquinone, 4-nitrocathechol or phenol [17,22,23,24]. Currently, chemical reduction is the most common methodology to degrade 4-NP. However, this protocol implies the combined use of strong reducing agents and toxic transition metals to catalyze the reduction process [25,26]. Additionally, most of the chemical reduction methods require harsh operating conditions, and/or time-consuming treatments, thereby considerably increasing the total economic demand [25,26,27].
In this context, the remediation of 4-NP through electrochemical reduction emerges as a viable alternative for efficient and green elimination and revalorization of this hazardous pollutant which creates value-added product: 4-AP. The compound 4-AP is also toxic (i.e., harmful if swallowed or inhaled, very toxic for aquatic biota and suspected to cause genetic effects). However, its toxicity is significantly lower than 4-NP. This is corroborated by its no-observed-adverse-effect level (NOAEL) on aquatic biota and on the oral, dermal or inhaled median lethal dose (LD50) administrated to rats or rabbits: oral LD50 of 230 mg kg−1 (4-NP) and 375 mg kg−1 (4-AP) on rats; dermal LD50 of 5000 mg kg−1 (4-NP) and 8,000–10,000 mg kg−1 (4-AP) on rabbits or inhalation LD50 (for 1 h) of 1,175 mg m−3 (4-NP) and 5.91 mg m−3 (4-AP) on rats. Importantly, 4-AP is not listed in the priority pollutant list of US EPA [28,29,30,31]. In fact, 4-AP can be utilized as a raw material in various industrial processes and applications [32,33] including the production of dyes and medicines (e.g., as a chemical required for the synthesis of paracetamol). Consequently, electrochemical reduction can be the most suitable path for the electroreduction of this persistent organic pollutant as well as for the electrosynthesis of 4-AP, since it requires a relatively simple operation.
We report on a systematic study of the electrochemical reduction of 4-NP to 4-AP under different experimental conditions. The nature of the electrode plays a key role in the effectiveness of the redox process. The electrochemical reduction of 4-NP has been explored over different metallic and carbonaceous substrata, e.g., glassy carbon (GC), Ag, Au, Pt and Ni. The pH and temperature of the electrochemical bath and the applied potential have also been considered as parameters in the overall optimization of the process to potentially implement in further. Overall, we propose a simple and effective electrochemical method for transforming 4-NP into 4-AP. The kinetics of the reduction process have been directly monitored through high performance liquid chromatography (HPLC) and indirectly monitored following 4-NPO by UV-vis spectroscopy after deprotonation of the 4-NP. The structural identity of the formed products has been confirmed by 1H nuclear magnetic resonance (NMR). Indeed, the electrochemical methodology might pave the way to the further design of continuous processes, combining a pre-concentration step of a 4-NP solution and the subsequent electrochemical reduction of the pollutant. This will yield significant amounts of 4-AP with virtually no undesired by-products.
2. Results and Discussion
2.1. Preliminary Voltammetric Study
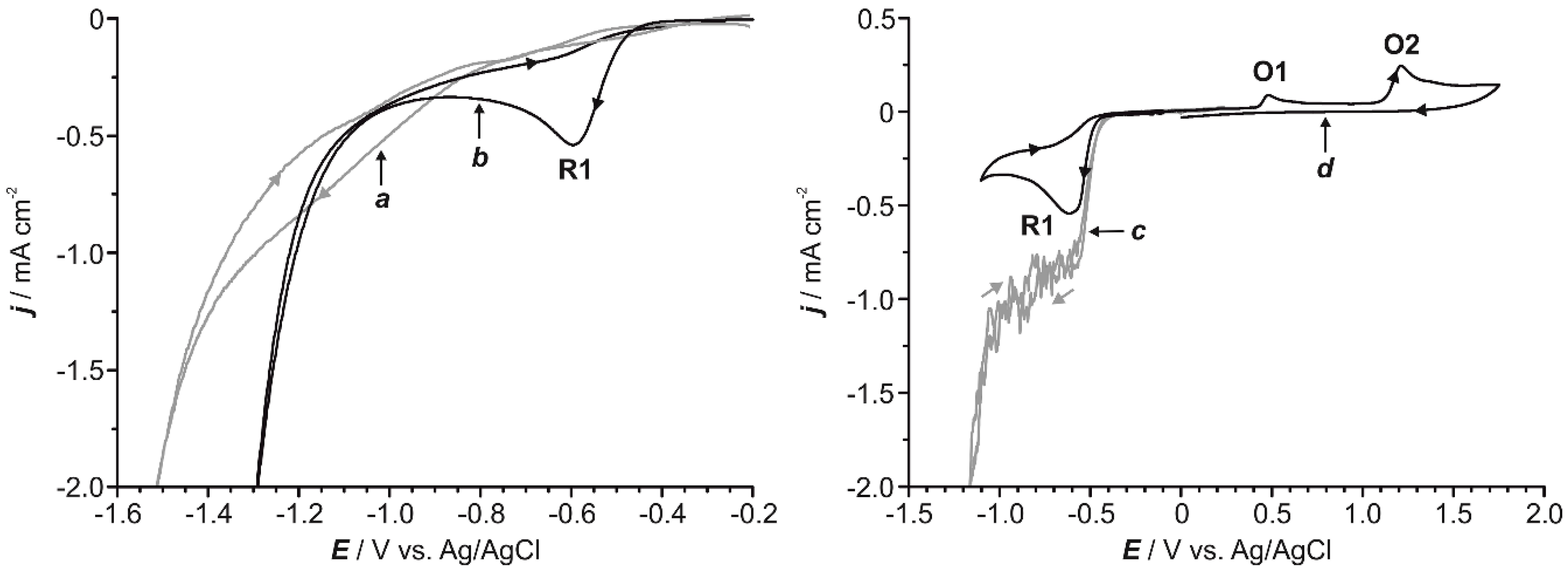
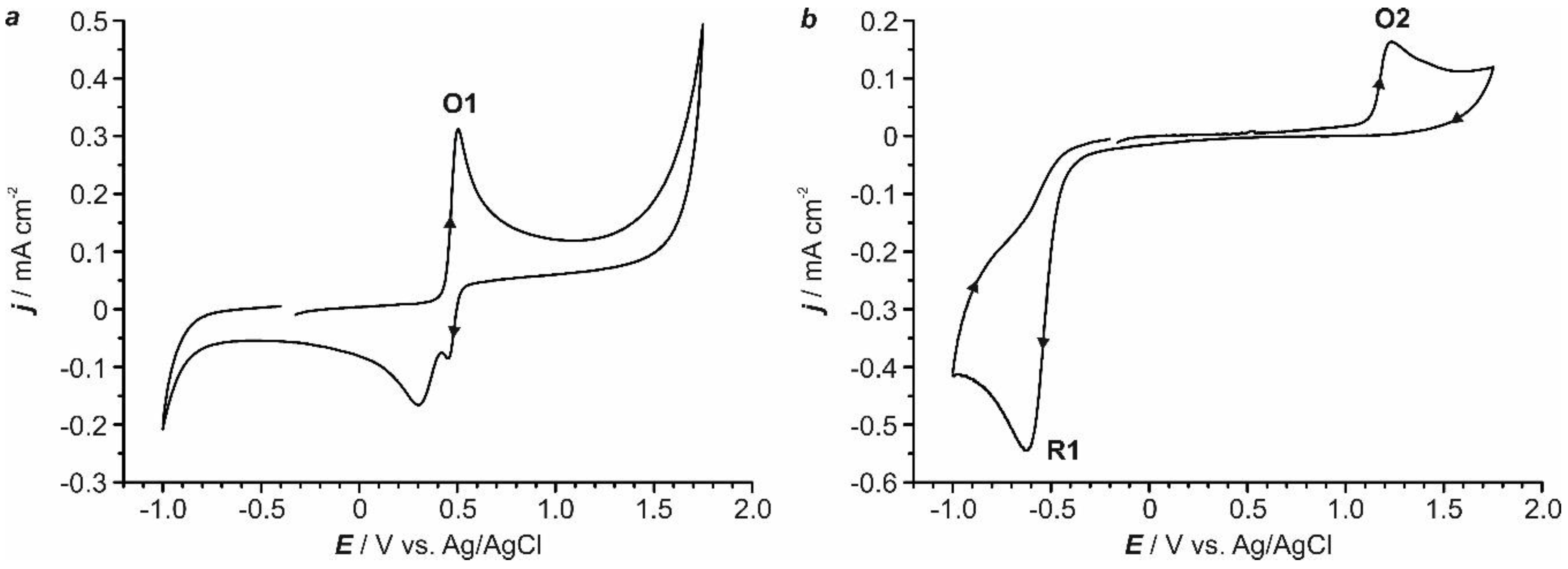
In order to establish the general behavior of the 4-NP reduction process, voltammetric experiments were conducted on GC with a 0.6 mM aqueous solution of 4-NP at two different pH values, namely, 2.0 and 5.3. The voltammogram recorded at pH = 5.3 (a in Figure 1) did not show any clear reduction wave but revealed a reduction band, which initiated at ca. −0.75 V and widened up to around −1.40 V, prior to the reduction of the solvent. At pH = 2.0 (b in Figure 1), current started at a less negative potential (−0.40 V) and a clear reduction peak (R1 in Figure 1), centered at −0.59 V, was observed. These results evidence that an acidic medium favors the reduction process and, therefore, the pH of the electrochemical bath was set at 2.0. Additional experiments performed under continuous stirring of the solution (Figure 1c), using a forced argon flow, showed an important current increase, highlighting the significance of mass transport in the process. Importantly, if the voltammetric scan at pH = 2 was started towards negative potentials and then reversed (Figure 1d), two different oxidation peaks (O1 and O2 in Figure 1) were detected (vide infra).
The voltammetric study was extended to different metallic electrodes in order to select the optimal conditions for the reduction of 4-NP in each case (Figure S1 and Figure 2). Electrodes active for the hydrogen evolution reaction (i.e. nickel and more especially platinum) catalyzed and advanced the protons reduction in such a way that both 4-NP and solvent diminished almost simultaneously (Figure S1). Such a feature precluded using those electrodes for practical purposes. Gold and silver (Figure 2a,b) shifted the reduction peak of 4-NP toward more positive potentials with respect to GC (Figure 2c) and prevented its overlap with the primary current related to hydrogen evolution. The general electrochemical behavior observed on gold and silver working electrodes was similar to the one recorded on GC (Figure S2). It should be also stressed that, for those two electrodes, the potential range at which the reduction of 4-NP occurred without any contribution from secondary reactions was wide enough for practical purposes. According to the literature, the clear irreversible reduction peak R1 in Figure 1 and Figure 2 can be ascribed to the direct reduction of 4-NP to 4-HAP (Scheme 1), which involves a four-electron and four-proton transfer process. Once 4-HAP is formed in acidic media, it can rearrange itself into 4-AP. Therefore, acidic conditions favors the electroreduction of 4-NP to 4-HAP as well as the transformation of 4-HAP to 4-AP [34,35,36,37].
2.2. Electrochemical Synthesis
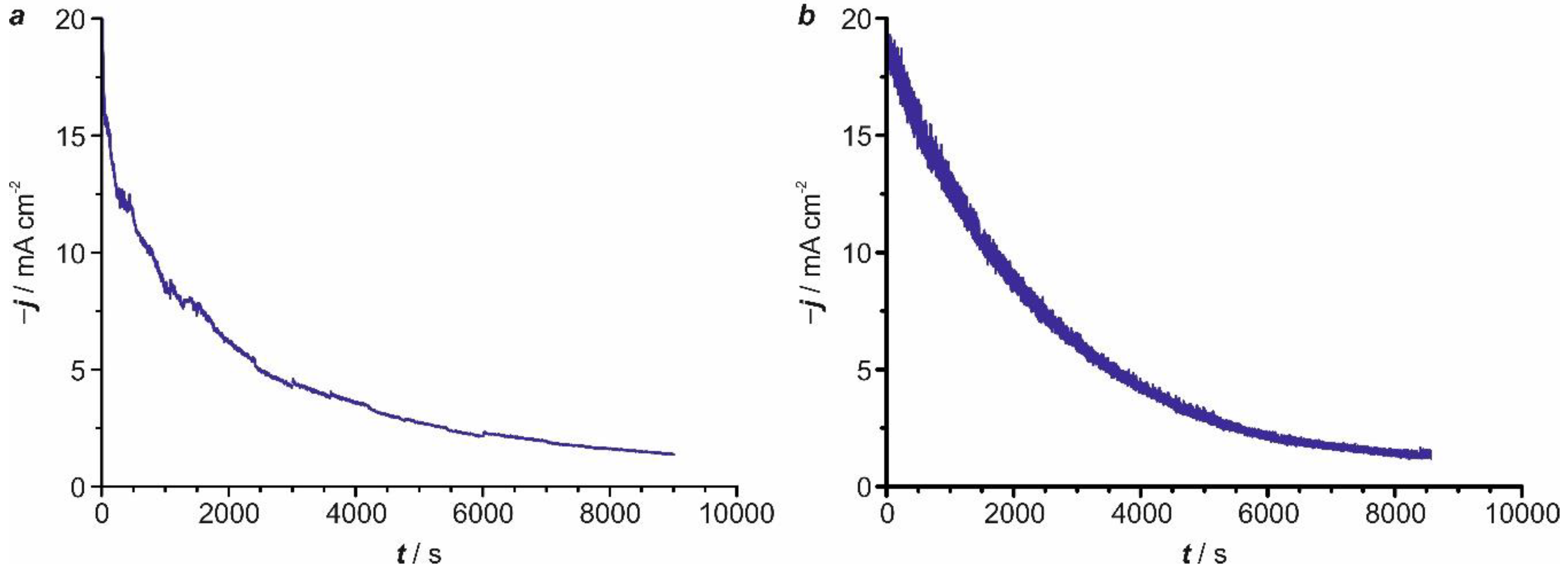
Electrochemical reduction of 4-NP under the application of a constant potential was evaluated only with those metals that showed the highest electroactivity, i.e., silver and gold. The potentials were selected between the peak potential of the irreversible peak (Figure 2, R1) ascribed to the reduction of 4-NP to 4-HAP and the sharp current by the hydrogen evolution reaction. First, we focused our attention in the electrolysis of 4-NP ([4-NP] = 5.25 mM) on silver at pH = 2.0, which was carried out at E = −0.550 V, in agreement with the voltammetric study. For an effective electrolysis, the electrochemical bath was stirred during the reduction process, thereby observing a significant decrease in current over time (Figure 3a). In order to asses if this behavior was due to passivation of the substrate produced by some of the intermediate reduction products, the electrode was removed and electrochemically activated. The current recorded before and after this treatment remained virtually unchanged, confirming that the decrease observed in the j-t curve comes from the consumption of the organic pollutant due to its electrocatalytic reduction. A similar current decrease over time was detected with gold working electrodes at pH = 2.0, using a reduction potential of E = −0.465 V (Figure 3b).
2.3. Structural and Electrochemical Characterization of the Formed Products
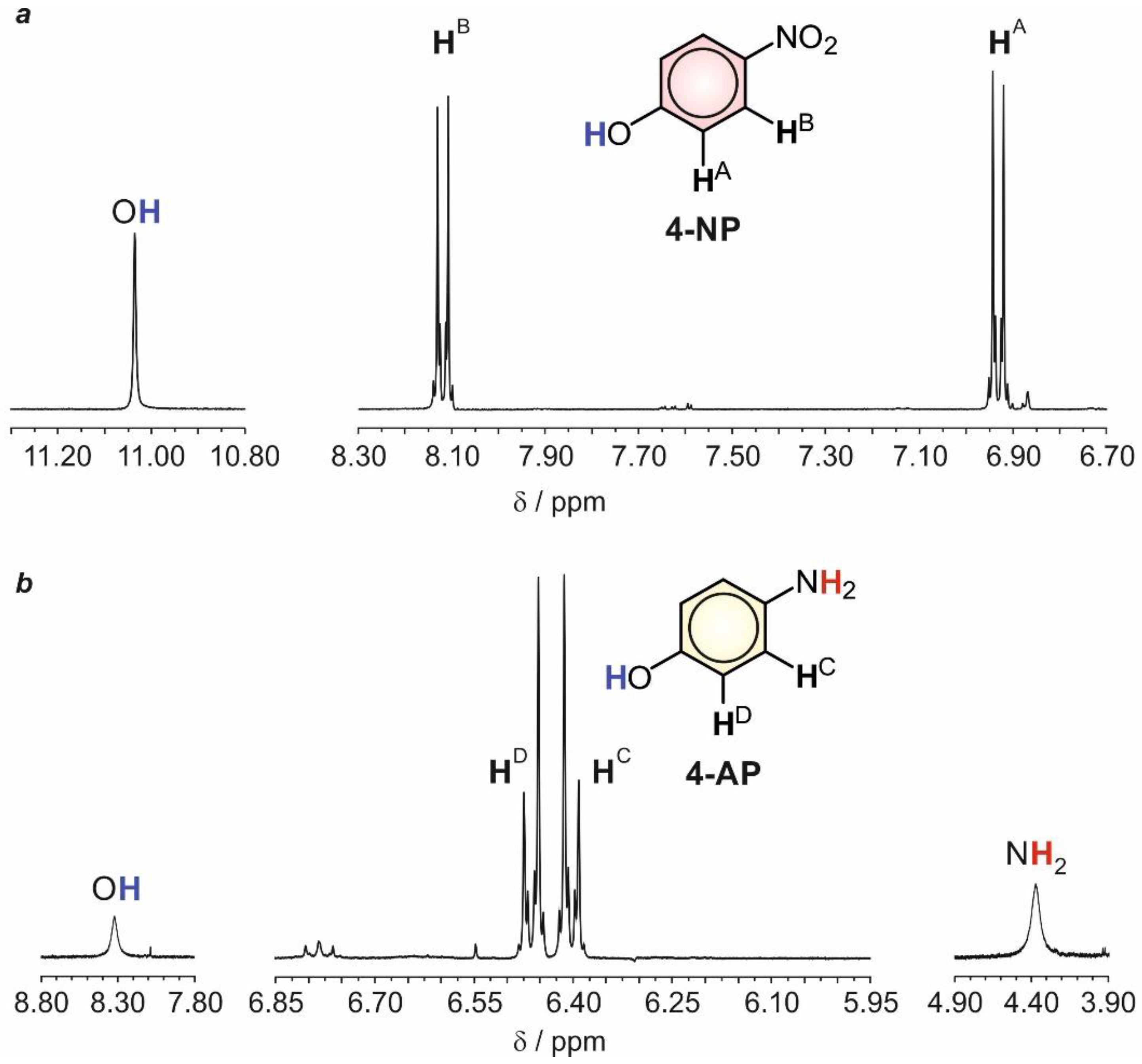
The structural identities of the organic products present in the electrochemical bath after the electrolysis under applied potentials in the range of R1, using either silver or gold working electrodes, were analyzed by 1H NMR spectroscopy. For this purpose, the electrolyzed solution, with a pH of 2.0, was transferred to a separatory funnel and extracted with diethyl ether. Under these conditions, 4-NP was selectively transferred to the organic layer and isolated after distilling off the solvent under reduced pressure. The 1H NMR spectrum of the residue, dissolved in d6-DMSO, showed two sets of signals: (1) two doublet resonances centered at 6.93 and 8.11 ppm (3J = 8.8 Hz), which can be ascribed to HA and HB, respectively (Figure 4a), and (2) a broad singlet peaking at 11.04 ppm belonging to the hydrogen atom of the phenol function. On this basis, 1H NMR spectroscopy revealed the presence of 4-NP in the electrochemical bath after the electrolysis and, thus, evidenced that its reduction to 4-AP was not complete after the selected duration for the electrolytic process.
Next, the remaining aqueous layer was neutralized (pH ~7.0) and extracted, once again, with diethyl ether. In this instance, 4-AP was separated and isolated. The corresponding 1H NMR spectrum (Figure 4b), recorded in the same deuterated solvent, shows two doublets centered at 6.40 and 6.46 ppm (3J = 8.8 Hz), corresponding to the two pairs of aromatic protons of the molecule, HC and HD, respectively. In addition, two broad singlets peaking at 4.36 and 8.32 ppm are also detected, ascribable to the amino and phenol groups, respectively. Thus, 1H NMR spectroscopy confirmed the successful reduction of 4-NP to 4-AP.
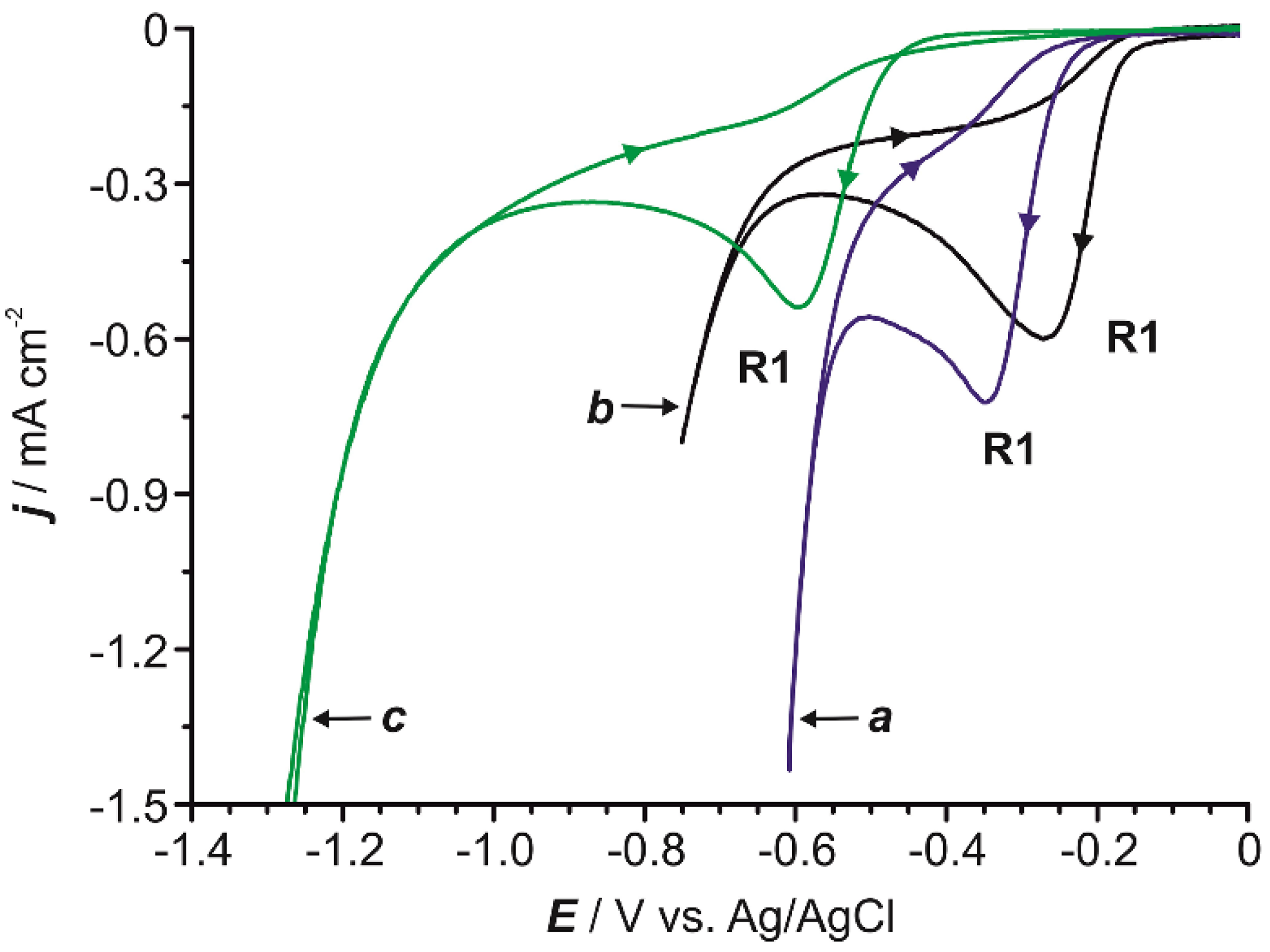
Furthermore, the monitoring of the voltammetric response enables a fast and in situ detection of the electro-generated product. The voltammetric curve obtained scanning towards positive potentials for a 0.6 mM solution of commercially available 4-AP at pH = 2 revealed a clear oxidation peak (O1 in Figure 5a) that initiated at around +0.35 V. No other significant oxidation processes were observed before the massive solvent reaction (Figure 5a). On the other hand, the voltammetric scan for a 4-NP solution, started towards positive potentials, displayed only one oxidation peak (O2 in Figure 5b), centered at +1.23 V, previous to the oxygen evolution. This experimental result demonstrated that the two oxidation peaks detected in curve d in Figure 1 (see above) can be assigned as follows: O1 to the oxidation of the 4-AP produced during the reduction of 4-NP (R1) and O2 to the oxidation of pristine 4-NP.
As a whole, the combination of 1H NMR spectroscopy and voltammetric experiments have allowed us to confirm the electrochemical transformation of the pollutant into 4-AP when either gold or silver working electrodes are used.
2.4. Kinetic Analysis
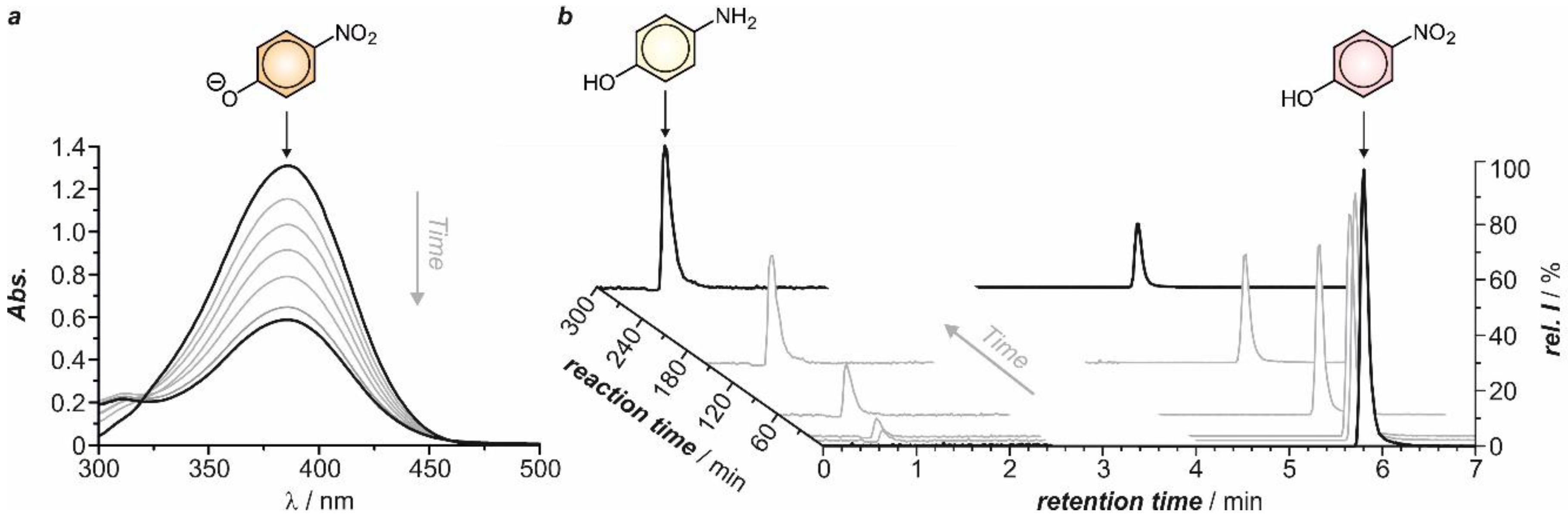
Once the feasibility of the reduction of 4-NP has been established, we addressed our efforts to the monitoring of the reduction process as a function of the electrode nature, the temperature and the applied potential in order to evaluate the viability of the proposed electrochemical conversion of this pollutant into a valuable compound (4-AP). The disappearance of 4-NP over time was quantified by means of conventional time-resolved UV-vis spectroscopy and HPLC. The spectroscopic monitoring of the process was evaluated by measuring the temporal evolution of the absorbance of 4-NPO at λmax = 400 nm, taking advantage of the significant absorption of this species in the visible region of the electromagnetic spectrum in alkaline medium (Figure 6a).
For the chromatographic monitoring, solutions prepared with commercially available 4-NP and 4-AP were first injected in order to establish their retention times (tR) under the chromatographic conditions. The retention time for 4-NP and 4-AP was 5.8 min and 0.8 min, respectively (Figure S3). Thus, the kinetics of the process was evaluated from the diminishment of the integrated area of the peak at 5.8 min, corresponding to 4-NP, over time (Figure 6b). Importantly, the experimental conditions investigated in this study offered both excellent conversion yields (higher than 80% after 150 min of reaction for both silver and gold working electrodes) and chemoselectivity.
According to the Langmuir–Hinshelwood (L-H) model, when the concentration of the reactant is low, a pseudo-first-order reaction can be used to determine the kinetics of the process. Therefore, the apparent rate constant normalized by geometrical surface area (k in Table 1) was calculated from the slope of the corresponding −ln(Xt/X0) versus t plot, where X0 and Xt is the monitored magnitude—absorbance at λmax for UV-vis spectroscopy or integrated area for HPLC—before any potential was applied and at a specific reaction time, t, respectively. In all instances, the experimental error associated with rate constants was of less than 10%.
2.5. Influence of the Electrode Nature
As abovementioned, the electrochemical reduction under the application of a constant potential has been only studied with those metals that showed the highest electroactivity in the preliminary voltammetric study avoiding other parallel processes, i.e., silver and gold. In both instances, the applied potential was more negative than the one of the corresponding cathodic peak R1 (EAg = −0.550 V and EAu = −0.465 V, Figure 2a,b). For both metals, the electrochemical transformation of 4-NP was monitored by UV-vis spectroscopy and HPLC. In all the cases, linear X-t relationships were obtained, thereby confirming the pseudo-first-order kinetics. From the corresponding −ln(Xt/X0) versus t plots, apparent rate constants of 1.2 and 1.2 × 10−3 min−1 cm−2 were obtained using silver working electrodes at 25 °C by spectrophotometry and liquid chromatography, respectively (Table 1). In the case of gold, similar k values of 1.2 and 1.4 × 10−3 min−1 cm−2 were registered. The appearance of 4-AP over time, monitored through the area of the HPLC peak with a retention time of 0.8 min, followed also a pseudo-first-order kinetics. Fitting of the experimental data to a monoexponential growing function yielded the very same rate constant than the one determined chromatographically for the disappearance of 4-NP (see above). Under these experimental conditions, by-products are not formed during the reduction process, i.e., 4-NP is cleanly transformed into 4-AP. This observation is in accordance with the good retention of the isosbestic points observed in the time-resolved absorption spectra shown in Figure 6a.
2.6. Influence of the Temperature
In order to explore if an increase in temperature is beneficial for the degradation of 4-NP, chronoamperometric experiments were performed at different temperatures (25, 40 and 50 °C) on gold working electrodes (E = −0.465 V) under stirring. Experimental data show that an increase in temperature enhances the progress of the reduction reaction as expected (Table 1). Indeed, the recorded absorbance at a determined reaction time is lower at higher temperatures. Importantly, the electrokinetic performance for 4-NP reduction is sufficiently high at room temperature, which is fundamental for relevant technological applications in order to minimize the waste energetic issues and economic demand.
2.7. Influence of the Applied Potential
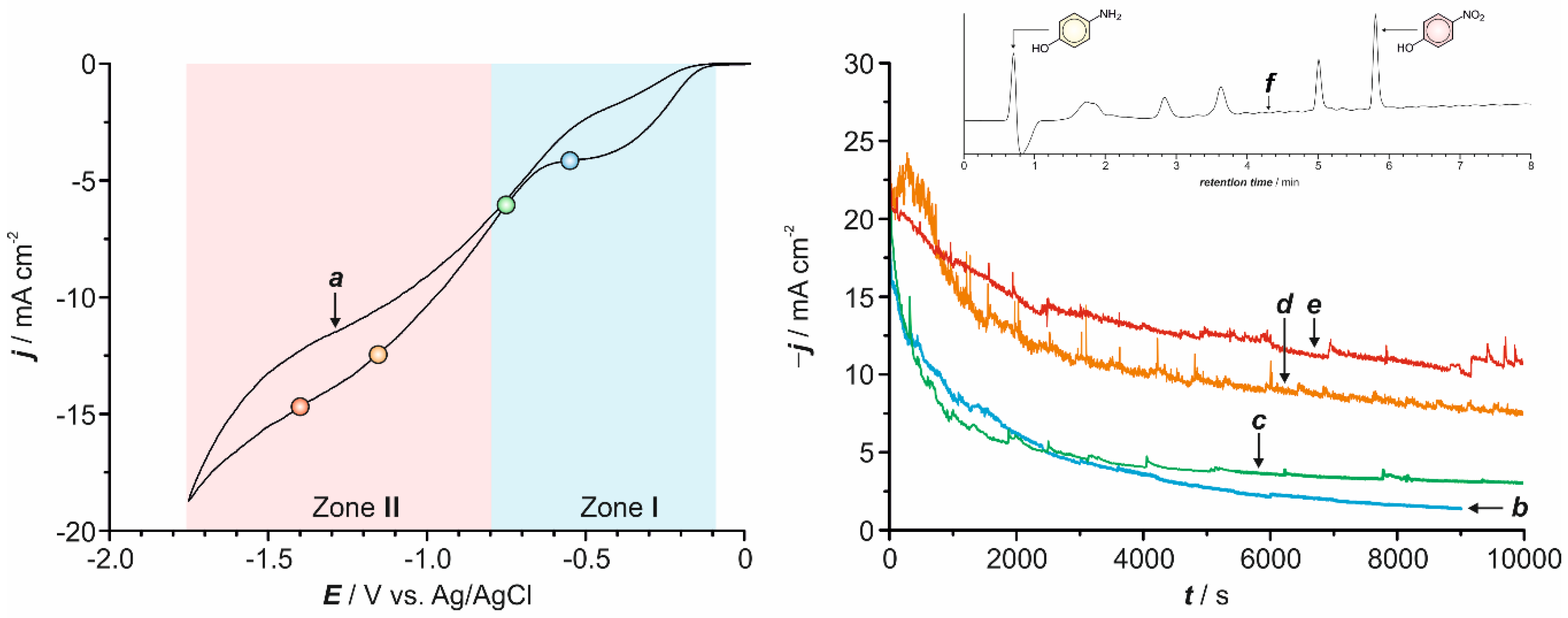
Literature findings reveal that 4-NP hydrogenation is a viable alternative to produce 4-AP [20,34,35,36,37,38,39]. With this mind, we have also analyzed the effect of simultaneous hydrogen evolution during the reduction of 4-NP. Therefore, two different potential ranges have been studied: one at which only the reduction of 4-NP occurs (Zone I in Figure 7a) and another where the reduction of the solvent is an overlapped process (Zone II in Figure 7a).
Using silver substrata when working with potentials falling within Zone I, i.e., where only 4-NP reduction takes place, the current diminishes progressively (Figure 7b,c), being the initial current recorded greater as more negative potentials were applied. Simultaneous monitoring of the process by means of UV-vis spectroscopy and HPLC confirmed that the only product obtained was 4-AP. Indeed, within this potential range, the electrolyzed solution changed from colorless to pale pink and progressively gains color intensity due to its continuous reduction in the electrochemical bath (Figure S4a). Fitting of the experimental data confirmed an apparent first-order kinetics within this potential range, being the process faster as the reduction process was carried out at more negative potentials (Table 1).
When potentials corresponding to Zone II were applied instead, a significant gas bubbling was observed due to the simultaneous hydrogen evolution. Consequently, the current involved in the process was considerably greater than the one observed in Zone I. In addition, and as a main difference with that observed within Zone I, the j-t curves registered showed a non-negligible current value at long reaction times due to the additional contribution of the simultaneous reduction of the solvent (Figure 7d,e). The recorded current values oscillated randomly thereby evidencing the punctual blocking of the electrode surface by the gas bubbles, which presence perturbs the available surface.
Under these experimental conditions, the spectrophotometric monitoring of the process evidences that, during the early stages of the reaction, the decrease of 4-NP concentration was faster than the one in Zone I. As the reaction progresses, the absorbance decrease stopped and the absorption maximum shifted to longer wavelengths, evidencing the formation of undesired by-products. A similar behavior was observed using gold substrates as working electrodes. Indeed, the solution color changed dramatically over time, from yellow to dark pink to orange (Figure S4b). Accordingly, the chromatographic monitoring of the reduction reaction revealed the appearance of new peaks with retention times of ca. 1.8, 2.8, 3.6 and 5.0 min (Figure 7f). The electrochemical reduction of 4-NP is strongly dependent on pH, solvent and catalytic nature and conditions. The high hydrogen evolution can dramatically modify the pH on the electrode interface, promoting the formation mainly of azoxybenzene or aniline. Note that acid conditions are required to assure the formation of 4-AP as a result of the nucleophilic attack by water at the protonated 4-HAP [1,20,34,35,36,37,38,39]. However, the structural elucidation of the formed by-products is out of the scope of the present study as they are formed in non-adequate working conditions for 4-NP reduction. In this case, only the data corresponding to short reaction times were considered for the kinetics quantification by both monitoring methods (Table 1). On the basis of the above considerations, the combined effect of higher overpotentials and hydrogen evolution leads to the operation of secondary reactions, which transform 4-NP to more reduced species precluding, in turn, a clean transformation of 4-NP into 4-AP within these range of potentials. Therefore, a possible viable method for achieving an improved reduction of 4-NP would involve the moderate formation of hydrogen during the first reaction times applying potentials located within Zone II and, after a short period of time, either shift to less negative potentials comprised within Zone I or apply a pulse signal.
3. Materials and Methods
3.1. Materials
The solid compounds needed to prepare the different solutions were purchased from Panreac (NaOH), Merck (Na2SO4), Fluka (4-nitrophenol) and Sigma Aldrich (4-aminophenol). Sulfuric and nitric acids were purchased from Panreac. All solutions were prepared using Millipore-MilliQ water. The pH of the solutions was adjusted by adding either sulfuric acid or sodium hydroxide as required.
3.2. General Methods and Instrumentation
Electrochemical experiments were performed in a typical three-electrode cell, using an Autolab potentiostat/galvanostat PGSTAT30 (Metrohm Autolab BV, Utrecht, Netherlands) equipped with the GPES software (version 4.9). The working electrodes used were GC, platinum, gold, silver and nickel. A spiral of platinum was used as the counter electrode. The surface of the GC electrode was polished using aluminum oxide (Al2O3) particles with two different diameters, 3.75 and 1.87 µm; afterwards, the electrode was rinsed with Millipore-MilliQ water under sonication. Gold and platinum electrodes were treated with a nitric acid aqueous solution (1:1 v/v) and cleaned by flame-annealing. Silver and nickel electrodes were deepened in diluted nitric acid (1:1 v/v) and rinsed with water. The reference electrode was a Metrohm Ag/AgCl/KCl (3M)/SO42− electrode mounted in a Luggin capillary containing a 0.1 M solution of sodium sulfate, which was the supporting electrolyte.
Electrochemical characterization was carried out using 0.6 mM aqueous solutions of 4-NP or 4-AP as required. Electrolysis was performed using 5.25 mM solutions of 4-NP.
Absorption spectra were recorded with a Shimadzu UV-1800 spectrophotometer (Shimadzu, Kyoto, Japan) equipped with the software UVprobe (version 5.1). For the spectrophotometric monitoring of the reduction process, 40 µL aliquots of the electrolytic bath were collected over time. The different aliquots were alkalinized by adding 3 mL of a 0.1 M NaOH aqueous solution (final pH = 13) and homogenized to ensure the quantitative transformation of 4-NP into 4-NPO. This anionic species absorbs significantly within the visible region of the electromagnetic spectrum in comparison with its neutral counterpart 4-NP, thereby enabling the monitoring of the reduction process by UV-vis spectroscopy.
1H NMR (400 MHz) spectra were registered with a Varian Mercury spectrometer (Varian Inc., California, USA). 1H NMR spectra have been processed with the MestRec Nova software (version 10.0).
HPLC chromatograms were recorded with a Waters 2795 Alliance HT Separations Module (Waters Corporation, Massachusetts, USA) equipped with a Waters 2996 photodiode array detector. The HPLC instrument was coupled to a ZQ micromass 2000 mass spectrometer. The column used was an X-bridge Shield RP18 (particle size: 3.5 μm, internal diameter: 2.1 mm, length: 100 mm). The mobile phase flowed at 0.5 mL min−1. The mobile phase used was a gradient from CH3CN and H2O 95:5 v/v (both containing a 0.1% v/v of formic acid) to pure CH3CN. This gradient was applied over a period of 9 min. HPLC solutions were prepared using Millipore-MilliQ water and acetonitrile of HPLC-MS grade (VWR-BDH Prolabo).
4. Conclusions
A simple and clean electrochemical method, as a possible alternative to chemical and biological methods, is presented for the reduction of 4-NP to 4-AP. This protocol promotes, in a simple way, the removal of 4-NP from wastewater or, depending on the potential interest, its massive transformation into 4-AP, an added-value product. For this purpose, we have conducted a comprehensive and systematic study of the electrochemical transformation of 4-NP into 4-AP. Specifically, the electrochemical process has been tested on different metallic substrata, being those noble metals with lower electrocatalytic activity for hydrogen formation, i.e. silver and gold, the ones more attractive for this purpose. 1H NMR spectroscopy and HPLC corroborate the successful conversion of 4-NP into 4-AP with virtually no undesired by-products. Moreover, cyclic voltammetry enables a straightforward detection of 4-AP and stands up a useful technique to monitor the process in situ.
Supplementary Materials
The following are available online at https://0-www-mdpi-com.brum.beds.ac.uk/2073-4344/10/4/458/s1, Figure S1: Linear sweep voltammetry curves for a 0.6 mM aqueous solution of 4-NP at pH = 2.0 on nickel and platinum working electrodes at a scan rate of 50 mV s−1. Figure S2: Cyclic voltammetry for a 0.6 mM aqueous solution of 4-NP at pH = 2.0 on silver and gold working electrodes at a scan rate of 50 mV s−1. Figure S3: HPLC chromatograms and mass spectra of commercially-available samples of 4-NP and 4-AP, Figure S4: Color changes observed during the electrochemical reduction of a 5.25 mM aqueous solution of 4-NP.
Author Contributions
Conceptualization, A.S., J.G.-A. and E.G.; methodology, A.S., R.A., M.P., J.G.-A. and E.G.; validation, A.S., J.G.-A. and E.G.; formal analysis, A.S., R.A., M.P., J.G.-A. and E.G.; investigation, M.P.; resources, E.G.; writing—original draft preparation, R.A., J.G.-A. and E.G.; writing—review and editing, A.S., J.G.-A. and E.G.; supervision, J.G.-A. and E.G.; project administration, E.G.; funding acquisition, E.G. All authors have read and agreed to the published version of the manuscript.
Funding
This work has been received funding from the TEC2017-85059-C3-2-R project (co-financed by the Fondo Europeo de Desarrollo Regional, FEDER) from the Spanish Ministerio de Economía y Competitividad (MINECO). Albert Serrà would like to acknowledge funding from the EMPAPOSTDOCS-II program. The EMPAPOSTDOCS-II programme has received funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Skłodowska-Curie grant agreement number 754364.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Payra, S.; Challagulla, S.; Chakraborty, C.; Roy, S. A hydrogen evolution reaction induced unprecedentedly rapid electrocatalytic reduction of 4-nitrophenol over ZIF-67 compare to ZIF-8. J. Electroanal. Chem. 2019, 853, 113545. [Google Scholar] [CrossRef]
- Li, J.; Kuang, D.; Feng, Y.; Zhang, F.; Xu, Z.; Liu, M. A graphene oxide-based electrochemical sensor for sensitive determination of 4-nitrophenol. J. Hazard. Mater. 2012, 201, 250–259. [Google Scholar] [CrossRef]
- Serrà, A.; Gómez, E.; Philippe, L. Bioinspired ZnO-based solar photocatalysts for the efficient decontamination of persistent organic pollutants and hexavalent chromium in wastewater. Catalysts 2019, 9, 974. [Google Scholar] [CrossRef] [Green Version]
- WWAP (UNESCO World Water Assessment Programme). The United Nations World Water Development Report 2019: Leaving No One Behind; UNESCO: Paris, France, 2019. [Google Scholar]
- Zheng, Y.; Liu, D.; Liu, S.; Xu, S.; Yuan, Y.; Xiong, L. Kinetics and mechanisms of p-nitrophenol biodegradation by Pseudomonas aeruginosa HS-D38. J. Environ. Sci. 2009, 21, 1194–1199. [Google Scholar] [CrossRef]
- United States Environmental Protection Agency. Health and Environmental Effect Profiles; United States Environmental Protection Agency: Washington, DC, USA, 1980.
- United States Environmental Protection Agency. Health Effects Assessment for Nitrophenols; United States Environmental Protection Agency: Washington, DC, USA, 1987.
- Keith, L.H.; Telliard, W.A. Priority pollutants. I. A perspective view. Environ. Sci. Technol. 1979, 13, 416–423. [Google Scholar] [CrossRef]
- Min, J.; Xu, L.; Fang, S.; Chen, W.; Hu, X. Molecular and biochemical characterization of 2-chloro-4-nitrophenol degradation via the 1,2,4-benzenetriol pathway in a Gram-negative bacterium. Appl. Microbiol. Biotechnol. 2019, 103, 7741–7750. [Google Scholar] [CrossRef]
- Sj, S. The gene cluster for para-nitrophenol catabolism is responsible for 2-chloro-4-nitrophenol degradation in Burkholderia sp. Strain SJ98. Appl. Environ. Microbiol. 2014, 80, 6212–6222. [Google Scholar]
- Wang, J.C.; Li, Y.; Li, H.; Cui, Z.H.; Hou, Y.; Shi, W.; Jiang, K.; Qu, L.; Zhang, Y.P. A novel synthesis of oleophylic Fe2O3/polystyrene fibers by Γ-Ray irradiation for the enhanced photocatalysis of 4-chlorophenol and 4-nitrophenol degradation. J. Hazard. Mater. 2019, 379, 120806. [Google Scholar] [CrossRef]
- Serrà, A.; Artal, R.; García-Amorós, J.; Sepúlveda, B.; Gómez, E.; Nogués, J.; Philippe, L. Hybrid Ni@ZnO@ZnS-Microalgae for Circular Economy: A Smart Route to the Efficient Integration of Solar Photocatalytic Water Decontamination and Bioethanol Production. Adv. Sci. 2020, 7, 1–9. [Google Scholar] [CrossRef]
- Osin, O.A.; Yu, T.; Cai, X.; Jiang, Y.; Peng, G.; Cheng, X.; Li, R.; Qin, Y.; Lin, S. Photocatalytic degradation of 4-nitrophenol by C, N-TiO2: Degradation efficiency vs. embryonic toxicity of the resulting compounds. Front. Chem. 2018, 6, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Salehi, Z.; Rasouli, A.; Doosthosseini, H. p-nitrophenol Degradation Kinetics and Mass Transfer Study by Ralstonia eutropha as a Whole Cell Biocatalyst. Polycycl. Aromat. Compd. 2019, 5, 1–14. [Google Scholar] [CrossRef]
- Kulkarni, M.; Chaudhari, A. Biodegradation of p-nitrophenol by P. putida. Bioresour. Technol. 2006, 97, 982–988. [Google Scholar] [CrossRef]
- Tomei, M.C.; Annesini, M.C. 4-Nitrophenol biodegradation in a sequencing batch reactor operating with aerobic-anoxic cycles. Environ. Sci. Technol. 2005, 39, 5059–5065. [Google Scholar] [CrossRef]
- Oturan, M.A.; Peiroten, J.; Chartrin, P.; Acher, A.J. Complete destruction of p-Nitrophenol in aqueous medium by electro-fenton method. Environ. Sci. Technol. 2000, 34, 3474–3479. [Google Scholar] [CrossRef]
- Kiwi, J.; Pulgarin, C.; Peringer, P. Effect of Fenton and photo-Fenton reactions on the degradation and biodegradability of 2 and 4-nitrophenols in water treatment. Appl. Catal. B Environ. 1994, 3, 335–350. [Google Scholar] [CrossRef]
- Xie, F.; Xu, Y.; Xia, K.; Jia, C.; Zhang, P. Alternate pulses of ultrasound and electricity enhanced electrochemical process for p-nitrophenol degradation. Ultrason. Sonochem. 2016, 28, 199–206. [Google Scholar] [CrossRef]
- Serrà, A.; Alcobé, X.; Sort, J.; Nogués, J.; Vallés, E. Highly efficient electrochemical and chemical hydrogenation of 4-nitrophenol using recyclable narrow mesoporous magnetic CoPt nanowires. J. Mater. Chem. A 2016, 4, 15676–15687. [Google Scholar] [CrossRef] [Green Version]
- Chen, G. Electrochemical technologies in wastewater treatment. Sep. Purif. Technol. 2004, 38, 11–41. [Google Scholar] [CrossRef]
- Arora, P.K.; Srivastava, A.; Singh, V.P. Bacterial degradation of nitrophenols and their derivatives. J. Hazard. Mater. 2014, 266, 42–59. [Google Scholar] [CrossRef]
- Yuan, S.; Tian, M.; Cui, Y.; Lin, L.; Lu, X. Treatment of nitrophenols by cathode reduction and electro-Fenton methods. J. Hazard. Mater. 2006, 137, 573–580. [Google Scholar] [CrossRef]
- Wang, T.C.; Lu, N.; Li, J.; Wu, Y. Plasma-TiO2 catalytic method for high-efficiency remediation of p-nitrophenol contaminated soil in pulsed discharge. Environ. Sci. Technol. 2011, 45, 9301–9307. [Google Scholar] [CrossRef]
- Ruan, M.; Song, P.; Liu, J.; Li, E.; Xu, W. Highly Efficient Regeneration of Deactivated Au/C Catalyst for 4-Nitrophenol Reduction. J. Phys. Chem. C 2017, 121, 25882–25887. [Google Scholar] [CrossRef]
- Wang, C.; Salmon, L.; Li, Q.; Igartua, M.E.; Moya, S.; Ciganda, R.; Ruiz, J.; Astruc, D. From Mono to Tris-1,2,3-triazole-Stabilized Gold Nanoparticles and Their Compared Catalytic Efficiency in 4-Nitrophenol Reduction. Inorg. Chem. 2016, 55, 6776–6780. [Google Scholar] [CrossRef]
- Dutta, S.; Sarkar, S.; Ray, C.; Roy, A.; Sahoo, R.; Pal, T. Mesoporous gold and palladium nanoleaves from liquid-liquid interface: Enhanced catalytic activity of the palladium analogue toward hydrazine-assisted room-temperature 4-nitrophenol reduction. ACS Appl. Mater. Interfaces 2014, 6, 9134–9143. [Google Scholar] [CrossRef]
- Song, H.; Chen, T.S. P-Aminophenol-induced liver toxicity: Tentative evidence of a role for acetaminophen. J. Biochem. Mol. Toxicol. 2001, 15, 34–40. [Google Scholar] [CrossRef]
- SCCS (Scientific Committee on Consumer Safety). Opinion on P-Aminophenol; European Commission: Brussels, Belgium, 2011. [Google Scholar]
- United States Environmental Protection Agency. Provisional Peer Reviewed Toxicity Values for p-Aminophenol (CASRN 123-30-8); United States Environmental Protection Agency: Washington, DC, USA, 2005.
- Megharaj, M.; Pearson, H.W.; Venkateswarlu, K. Toxicity of p-aminophenol and p-nitrophenol to Chlorella vulgaris and two species of Nostoc isolated from soil. Pestic. Biochem. Physiol. 1991, 40, 266–273. [Google Scholar] [CrossRef]
- Saran, S.; Manjari, G.; Devipriya, S.P. Synergistic eminently active catalytic and recyclable Ag, Cu and Ag-Cu alloy nanoparticles supported on TiO2 for sustainable and cleaner environmental applications: A phytogenic mediated synthesis. J. Clean. Prod. 2018, 177, 134–143. [Google Scholar] [CrossRef]
- Corbett, J.F. An historical review of the use of dye precursors in the formulation of commercial oxidation hair dyes. Dye. Pigment. 1999, 41, 127–136. [Google Scholar] [CrossRef]
- He, Q.; Tian, Y.; Wu, Y.; Liu, J.; Li, G.; Deng, P.; Chen, D. Facile and ultrasensitive determination of 4-nitrophenol based on acetylene black paste and graphene hybrid electrode. Nanomaterials 2019, 9, 429. [Google Scholar] [CrossRef] [Green Version]
- Marquez, J.; Pletcher, D. A study of the electrochemical reduction of nitrobenzene to p-aminophenol. J. Appl. Electrochem. 1980, 10, 567–573. [Google Scholar] [CrossRef]
- Cyr, A.; Huot, P.; Marcoux, J.F.; Belot, G.; Laviron, E.; Lessard, J. The electrochemical reduction of nitrobenzene and azoxybenzene in neutral and basic aqueous methanolic solutions at polycrystalline copper and nickel electrodes. Electrochim. Acta 1989, 34, 439–445. [Google Scholar] [CrossRef]
- Wu, T.; Wang, G.; Zhang, Y.; Kang, S.; Zhang, H. Electrochemical deposition of Pt on carbon fiber cloth utilizing Pt mesh counter electrode during hydrogen evolution reaction for electrocatalytic hydrogenation reduction of p-nitrophenol. New J. Chem. 2017, 41, 7012–7019. [Google Scholar] [CrossRef]
- Silvester, D.S.; Wain, A.J.; Aldous, L.; Hardacre, C.; Compton, R.G. Electrochemical reduction of nitrobenzene and 4-nitrophenol in the room temperature ionic liquid [C4dmim][N(Tf)2]. J. Electroanal. Chem. 2006, 596, 131–140. [Google Scholar] [CrossRef]
- Karthik, R.; Hou, Y.S.; Chen, S.M.; Elangovan, A.; Ganesan, M.; Muthukrishnan, P. Eco-friendly synthesis of Ag-NP s using Cerasus serrulata plant extract—Its catalytic, electrochemical reduction of 4-NPh and antibacterial activity. J. Ind. Eng. Chem. 2016, 37, 330–339. [Google Scholar] [CrossRef]
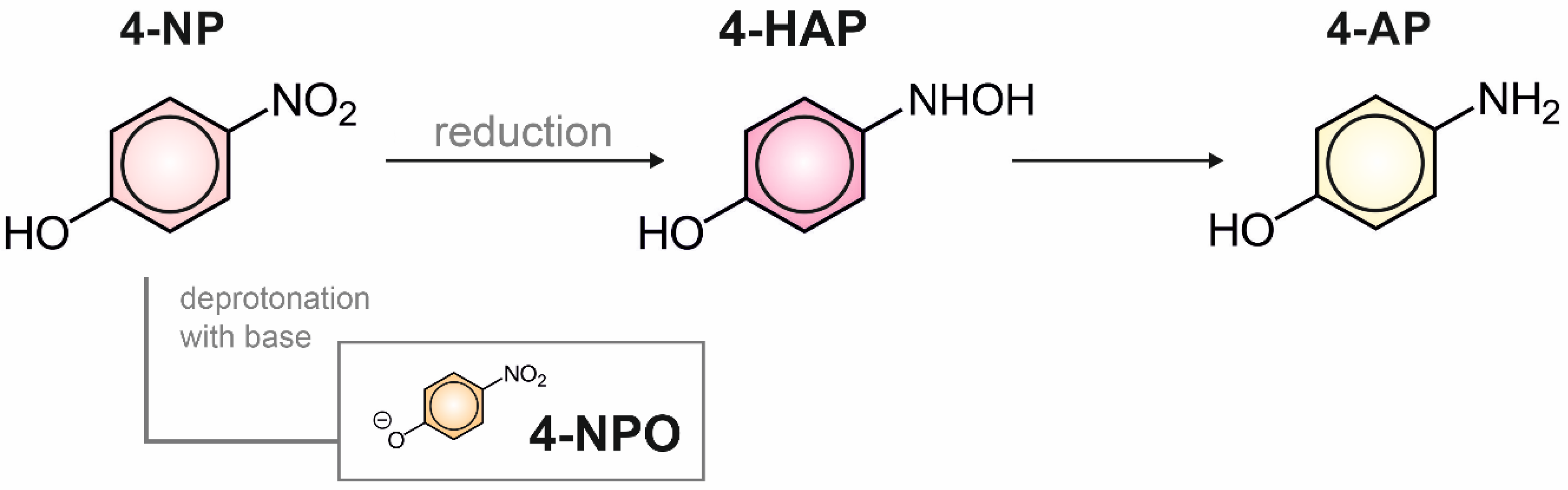
Scheme 1.
Electrosynthesis of 4-aminophenol (4-AP) through the reduction of 4-nitrophenol (4-NP) to 4-hydroxyaminophenol (4-HAP). Deprotonation of 4-NP to 4-nitrophenoxide (4-NPO) after the addition of base.
Scheme 1.
Electrosynthesis of 4-aminophenol (4-AP) through the reduction of 4-nitrophenol (4-NP) to 4-hydroxyaminophenol (4-HAP). Deprotonation of 4-NP to 4-nitrophenoxide (4-NPO) after the addition of base.

Figure 1.
Cyclic voltammograms for a 0.6 mM aqueous solution of 4-NP (0.1 M Na2SO4) at pH = 5.3 (a) and 2.0 (b) under stagnant conditions. Cyclic voltammetry for a 0.6 mM aqueous solution of 4-NP (0.1 M Na2SO4) at pH = 2.0 under argon-flow agitation (c) and global scan, started towards negative potentials, under quiescent conditions (d). In all instances, the voltammograms were recorded on a glassy carbon (GC) electrode and the scan rate was 50 mV s−1.
Figure 1.
Cyclic voltammograms for a 0.6 mM aqueous solution of 4-NP (0.1 M Na2SO4) at pH = 5.3 (a) and 2.0 (b) under stagnant conditions. Cyclic voltammetry for a 0.6 mM aqueous solution of 4-NP (0.1 M Na2SO4) at pH = 2.0 under argon-flow agitation (c) and global scan, started towards negative potentials, under quiescent conditions (d). In all instances, the voltammograms were recorded on a glassy carbon (GC) electrode and the scan rate was 50 mV s−1.

Figure 2.
Cyclic voltammograms for a 0.6 mM aqueous solution of 4-NP (0.1 M Na2SO4) at pH = 2.0 using gold (a), silver (b) and GC (c) working electrodes under quiescent conditions. In all instances, the scan rate was 50 mV s−1.
Figure 2.
Cyclic voltammograms for a 0.6 mM aqueous solution of 4-NP (0.1 M Na2SO4) at pH = 2.0 using gold (a), silver (b) and GC (c) working electrodes under quiescent conditions. In all instances, the scan rate was 50 mV s−1.

Figure 3.
Chronoamperometric (j-t) curves registered for the electrochemical reduction of a 5.25 mM aqueous solution of 4-NP (0.1 M Na2SO4) at pH = 2 on silver (a) (E = −0.550 V) and gold (b) (E = −0.465 V) working electrodes.
Figure 3.
Chronoamperometric (j-t) curves registered for the electrochemical reduction of a 5.25 mM aqueous solution of 4-NP (0.1 M Na2SO4) at pH = 2 on silver (a) (E = −0.550 V) and gold (b) (E = −0.465 V) working electrodes.

Figure 4.
1H NMR spectra, recorded in d6-DMSO, of the remaining 4-NP (a) and the electro-generated 4-AP (b) in the electrolytic bath.
Figure 4.
1H NMR spectra, recorded in d6-DMSO, of the remaining 4-NP (a) and the electro-generated 4-AP (b) in the electrolytic bath.

Figure 5.
Cyclic voltammograms, started towards positive potentials, for a 0.6 mM aqueous solution of 4-AP (a) and 4-NP (b) (0.1 M Na2SO4) at pH = 2.0 under quiescent conditions. In both instances, the voltammogram was recorded on a GC electrode and the scan rate was 50 mV s−1.
Figure 5.
Cyclic voltammograms, started towards positive potentials, for a 0.6 mM aqueous solution of 4-AP (a) and 4-NP (b) (0.1 M Na2SO4) at pH = 2.0 under quiescent conditions. In both instances, the voltammogram was recorded on a GC electrode and the scan rate was 50 mV s−1.

Figure 6.
Evolution of the absorption spectrum (a) of 4-NPO during the electrochemical reduction of a 5.25 mM aqueous solution of 4-NP (0.1 M Na2SO4) at pH = 2 on a silver working electrode at 25 °C (E = −0.550 V). The aliquots were alkalinized (pH = 13) with 0.1 M NaOH prior registering the absorption spectra. Evolution of the HPLC chromatogram (b) during the electrolysis of the same solution on a gold working electrode at 25 °C (E = −0.465 V).
Figure 6.
Evolution of the absorption spectrum (a) of 4-NPO during the electrochemical reduction of a 5.25 mM aqueous solution of 4-NP (0.1 M Na2SO4) at pH = 2 on a silver working electrode at 25 °C (E = −0.550 V). The aliquots were alkalinized (pH = 13) with 0.1 M NaOH prior registering the absorption spectra. Evolution of the HPLC chromatogram (b) during the electrolysis of the same solution on a gold working electrode at 25 °C (E = −0.465 V).

Figure 7.
Cyclic voltammogram (a) of a 5.25 mM solution of 4-NP (0.1 M Na2SO4) at pH = 2.0 under quiescent conditions (scan rate = 50 mV s−1). Chronoamperometric curves for the reduction of the same solution at −0.550 V (b), −0.750 V (c), −1.150 (d) and −1.400 V (e). HPLC chromatogram of the electrolytic bath after 130 min of electrolysis at E = −1.400 V (f). All experiments have been performed using a silver working electrode.
Figure 7.
Cyclic voltammogram (a) of a 5.25 mM solution of 4-NP (0.1 M Na2SO4) at pH = 2.0 under quiescent conditions (scan rate = 50 mV s−1). Chronoamperometric curves for the reduction of the same solution at −0.550 V (b), −0.750 V (c), −1.150 (d) and −1.400 V (e). HPLC chromatogram of the electrolytic bath after 130 min of electrolysis at E = −1.400 V (f). All experiments have been performed using a silver working electrode.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Apparent kinetic constant for 4-nitrophenol electro-reduction.
| k/min−1 cm−2 | ||||
|---|---|---|---|---|
| Electrode | T/°C | E/V | UV-vis Spectroscopy | HPLC |
| Ag | 25 | −0.550 | 1.2 × 10−3 | 1.2 × 10−3 |
| 25 | −0.750 | 1.9 × 10−3 | 1.8 × 10−3 | |
| 25 | −1.150 | 3.2 × 10−3 * | 4.0 × 10−3 * | |
| 25 | −1.400 | 3.9 × 10−3 * | 2.9 × 10−3 * | |
| Au | 25 | −0.465 | 1.2 × 10−3 | 1.4 × 10−3 |
| 40 | −0.465 | 2.5 × 10−3 | 2.5 × 10−3 | |
| 50 | −0.465 | 4.2 × 10−3 | 4.3 × 10−3 | |
* Note that at these potentials the hydrogen evolution reaction and other secondary reactions become important and compete with the reduction of 4-NP, leading to a complex competitive process on the electrode interface, in which various chemical and electrochemical reactions can take place, changing the local conditions on the electrode interface (e.g., local pH on the electrode interface). Under these conditions, the assumption of the L-H model gets weak.
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Serrà, A.; Artal, R.; Pozo, M.; Garcia-Amorós, J.; Gómez, E. Simple Environmentally-Friendly Reduction of 4-Nitrophenol. Catalysts 2020, 10, 458. https://0-doi-org.brum.beds.ac.uk/10.3390/catal10040458
AMA Style
Serrà A, Artal R, Pozo M, Garcia-Amorós J, Gómez E. Simple Environmentally-Friendly Reduction of 4-Nitrophenol. Catalysts. 2020; 10(4):458. https://0-doi-org.brum.beds.ac.uk/10.3390/catal10040458
Chicago/Turabian StyleSerrà, Albert, Raül Artal, Maria Pozo, Jaume Garcia-Amorós, and Elvira Gómez. 2020. "Simple Environmentally-Friendly Reduction of 4-Nitrophenol" Catalysts 10, no. 4: 458. https://0-doi-org.brum.beds.ac.uk/10.3390/catal10040458
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.