A Kinetic Study on the Efficient Formation of High-Valent Mn(TPPS)-oxo Complexes by Various Oxidants


1
Jerzy Haber Institute of Catalysis and Surface Chemistry, Polish Academy of Sciences, Niezapominajek 8, 30-239 Kraków, Poland
2
Faculty of Chemistry, Jagiellonian University, Gronostajowa 2, 30-387 Kraków, Poland
3
Department of Chemistry and Pharmacy, University of Erlangen-Nuremberg, Egerlandstr 1, 91058 Erlangen, Germany
*
Author to whom correspondence should be addressed.
Catalysts 2020, 10(6), 610; https://0-doi-org.brum.beds.ac.uk/10.3390/catal10060610
Submission received: 10 May 2020
/
Revised: 27 May 2020
/
Accepted: 27 May 2020
/
Published: 1 June 2020
(This article belongs to the Special Issue Recent Developments on Catalysis by Metalloporphyrins and Analogues)
Abstract
:New, more efficient methods of wastewater treatment, which will limit the harmful effects of textile dyes on the natural environment, are still being sought. Significant research work suggests that catalysts based on transition metal complexes can be used in efficient and environmentally friendly processes. In this context, a number of compounds containing manganese have been investigated. A suitable catalyst should have the capacity to activate a selected oxidant or group of oxidants, in order to be used in industrial oxidation reactions. In the present study we investigated the ability of MnIII(TPPS), where TPPS = 5,10,15,20-tetrakis(4-sulphonatophenyl)-21H,23H-porphyrine, to activate five different oxidants, namely hydrogen peroxide, peracetic acid, sodium hypochlorite, potassium peroxomonosulfate and sodium perborate, via the formation of high valent Mn(TPPS)-oxo complexes. Kinetic and spectroscopic data showed that the oxidation process is highly pH dependent and is strongly accelerated by the presence of carbonate in the reaction mixture for three of the five oxidizing agents. The highest efficiency for the oxidation of MnIII(TPPS) to high-valent Mn(TPPS)-oxo complexes, was found for peracetic acid at pH ≈ 11 in 0.5 M carbonate solution, which is at least an order of magnitude higher than the rate constants found for the other tested oxidants under similar conditions.
1. Introduction
Estimations concerning the amount of textile dyes that are discharged into water are quite different. Some articles state that 2% of the produced pigments are liberated in an aqueous effluent and the next 10% comes from the coloration process [1]. Other sources claim that 280,000 tons are released yearly [2]. This does not change the fact that the scale of the problem is huge. Especially, since insignificant amounts of dye may disrupt the water transparency, gas solubility and entail formation of toxic side products [2,3,4,5].
Due to the non-biodegradable character of dyestuffs and the possible release of toxic and carcinogenic byproducts, new methods for the degradation of dyes are still being sought [6,7,8,9,10,11,12,13]. New environmentally benign processes should reduce energy consumption and minimize the amount of chemicals, which are used in the overall cleaning operations. The alternative seems to be the application of catalysts based on transition metal complexes combined with oxidation agents in efficient and environmentally friendly processes [14,15,16]. In this context a number of manganese complexes have been investigated [14,17,18].
The aforementioned approach requires the selection of adequate and harmless oxidants. One of the possible compounds is the well-known hydrogen peroxide. Besides its environmentally friendly nature, H2O2 is cheap and the only by-products released during the oxidation process are water and molecular oxygen [19]. Moreover, other peroxides are also involved in industrial oxidation reactions. Potassium peroxomonosulfate (Oxone®) is mainly used in the degradation of organic pollutants in wastewater treatment due to its ability to produce sulfate radicals [20,21]. On the other hand, peracetic acid (PAA) is also one of the most commonly used green oxidants. PAA seems to have become one of the alternatives for chlorine and non-chlorine bleaching reagents [22]. Furthermore, the only side products that are formed are water and acetic acid. In the literature, a wide range of applications of PAA can be found. For decades, it was used in oxidation reactions of organic compounds [23] or as a disinfectant in medicine, the food industry and wastewater treatment [24,25,26]. In the context of industrial processes, two more oxidants are commonly used: sodium perborate (PBS) and sodium hypochlorite. PBS has been used for a long time especially in laundry cleaning [27]. In turn sodium hypochlorite is an inexpensive, strong oxidant, which has been used for years mostly as a bleaching and disinfectant agent. Beyond the indicated applications, NaOCl is widely used in epoxidation reactions of olefins [28,29,30], which are commonly catalyzed by transition metal complexes [31,32,33,34,35,36].
The aim of the present work was to examine the ability of selected Mn(III) porphyrin complexes to activate several, commonly used oxidants. This issue is extremely important within the context of possible application in degradation processes. The conducted studies covered the reactions of different oxidants with a commercially available sulfonated derivative of a Mn(III) porphyrin, referred to as MnIII(TPPS), where TPPS = 5,10,15,20-tetrakis(4-sulphonatophenyl)-21H,23H- porphyrine. MnIII(TPPS) has previously been used as a catalyst for the oxidation of olefins [37,38,39] and as a model catalyst in studies on monooxygenase or peroxidase activities [40], with various oxygen atom donors. Very good solubility in water, as well as the presence of side substituents that do not affect the course of the studied reactions, were decisive in the selection of this compound for our study.
In our earlier work, we investigated the activation of MnIII(TPPS) to form high valent oxo complexes using hydrogen peroxide [41,42]. In the present study we extended our studies to another four oxidants, namely peracetic acid, sodium hypochlorite, potassium peroxomonosulfate and sodium perborate. The studies allowed us to compare the efficiency of the formation of high-valent Mn(TPPS)-oxo complexes for the selected MnIII(TPPS)/oxidant systems under different, carefully selected conditions, and to select the most promising bleaching reagents for further studies. By improving our understanding of the kinetic and mechanistic aspects of these reactions, it should lead to a more structured approach to design highly efficient homogeneous catalysts.
2. Results
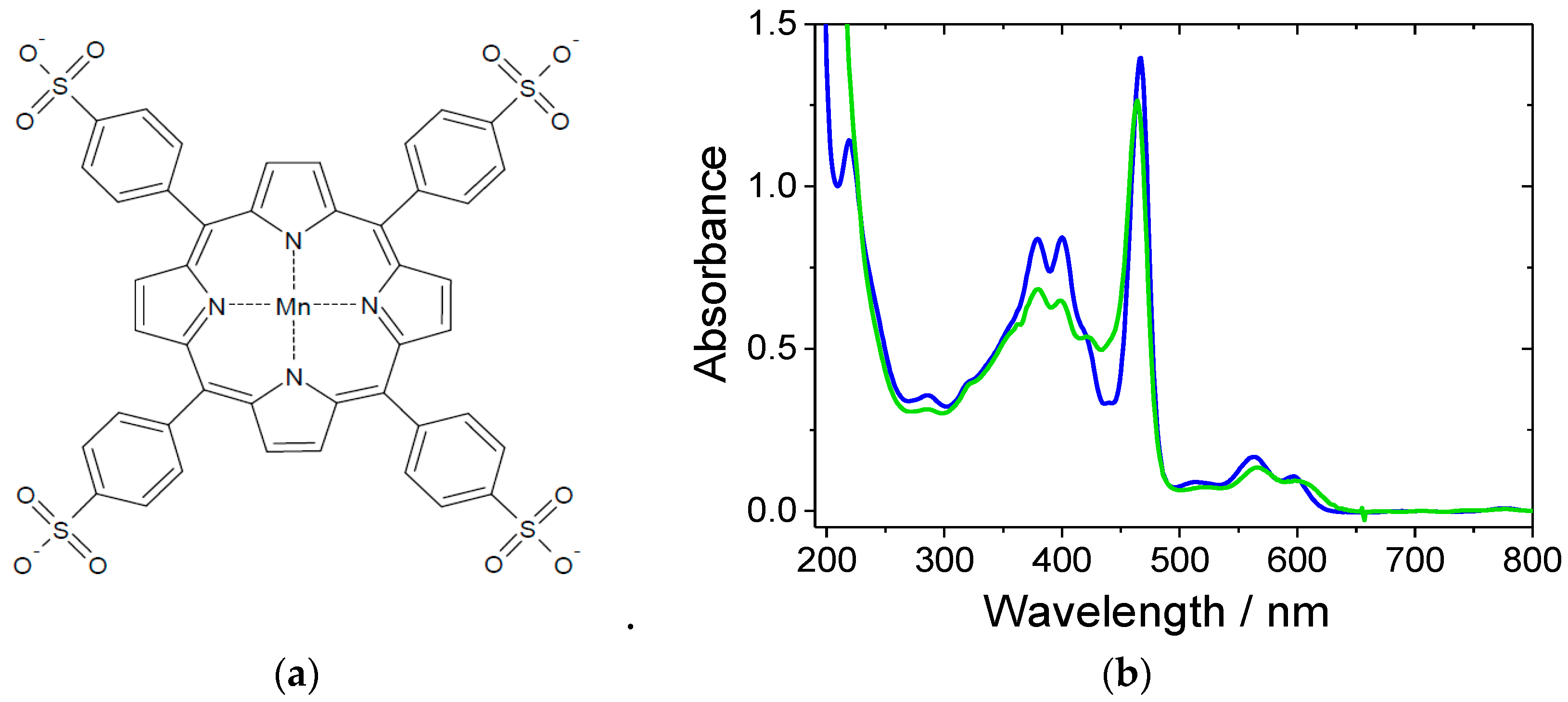
In this work, the oxidation reactions were carried out using a commercially available MnIII(TPPS) (see Figure 1a) porphyrin complex. The typical ultraviolet–visible (UV–Vis) spectrum of this complex can be described by the presence of a characteristic intense Soret band at 466 nm, two less intense bands at 379 and 401 nm, and weak Q bands at 564 and 596 nm (for a MnIII(TPPS) solution in water). Based on our previous studies, MnIII(TPPS) in aqueous solution has two pKa values (pK1 = 6.6, pK2 = 12.0) and hence can exist in three different forms, i.e., [MnIII(TPPS)(H2O)2]3−, [MnIII(TPPS)(H2O)(OH)]4− and [MnIII(TPPS)(OH)2]5−, depending on the pH of the solution [41]. Figure 1b shows how the pH affects the spectrum of MnIII(TPPS).
As shown in earlier studies, the oxidation of MnIII(TPPS) is connected with the hypsochromic shift of the most intense Soret band. The shift in the peak maximum from 466 nm to 422 nm is related to the formation of the high-valent (TPPS)MnV=O species. On the other hand, the maximum at 402 nm is associated with the presence of (TPPS)MnIV=O species [41]. Similar effects were also reported in the literature for other manganese porphyrin complexes [43,44,45].
It was shown before that the formation of higher oxidation states of manganese is not efficient under acidic and neutral pH conditions due to the fact that the MnIII(TPPS) complex is mainly present in a more inert form with both axial positions occupied by water molecules. However, the application of more basic solutions, where one of the coordinated water molecules is replaced by a hydroxo ligand, leads to the fast oxidation of the porphyrin to form Mn(IV)-oxo or Mn(V)-oxo complexes [41]. The most efficient oxidation was observed at pH around 11 [42]. This maximum was ascribed to the trans-labilization of the coordinated water molecule by hydroxide in [MnIII(TPPS)(H2O)(OH)]4− and the very inert bishydroxo complex [MnIII(TPPS)(OH)2]5−.
Based on this knowledge, we selected two pH values for which all spectroscopic and kinetic measurements were taken, namely pH 9.3 and 11. Also the careful selection of the type of buffer seemed to be essential. To prevent the unsuspected influence of buffer components on the kinetics of the scheduled reactions, we decided to follow the reactions in three types of solutions: in appropriately concentrated base, in 0.5 M solution of potassium nitrate (to keep the ionic strength at a constant level) with NaOH to adjust the pH, and in 0.5 M carbonate containing solution. Due to the fact that the oxidation of MnIII(TPPS) is too fast for using a standard UV–Vis spectrophotometer, all observed spectral changes and kinetic traces shown in this work were measured using the rapid-scan mode of a stopped-flow instrument.
Although the reactions of MnIII(TPPS) with H2O2 were studied before, we decided to perform new measurements to obtain observed rate constants under the same conditions for all oxidants. This allowed us to compare the ability to oxidize MnIII(TPPS) by different oxidants. The results obtained for the reaction between MnIII(TPPS) and H2O2 at pH = 9.3 and pH = 11 are presented in Figures S1 and S2 (Supporting Information).
At pH = 9.3 (Figure S1), the rate constants observed exhibit a linear dependence on the H2O2 concentration with a significant intercept for Figure S1a,b. The intercept can be ascribed to a parallel decomposition reaction at low hydrogen peroxide concentration. The second-order rate constants are 1.08 ± 0.04 (for Figure S1a), (3.2 ± 0.2) × 102 (for Figure S1b) and (7.2 ± 0.2) × 103 M−1 s−1 (for Figure S1c), respectively. The highest value was obtained for the reaction in carbonate buffer, which is in good agreement with our previous studies [41,42]. The reaction in the presence of carbonate is usually much faster due to the rapid formation of the peroxocarbonate ion (percarbonate), according to Equation (1), which is a more powerful oxidizing agent than H2O2 [46,47]. The equilibrium constant for this reaction is K = 0.32 ± 0.02 M−1 and the pKa value for HCO4−/CO42− is 10.6 [48,49].
In the case of measurements at pH = 11, also a linear dependence on hydrogen peroxide concentration was observed for reactions in NaOH and carbonate solutions (see Figure S2a,c). Both plots show a considerable intercept, which is associated with decomposition reactions. The slope of the plots in Figure S2a,c represent the second-order rate constants, which are (2.20 ± 0.06) × 103 and (9.3 ± 0.7) × 104 M−1 s−1 for Figure S2a,c, respectively.
The non-linear dependence of kobs on the H2O2 concentration observed in Figure S2b suggests that the reaction reaches a saturation level in terms of the observed rate constant. The second-order rate constant was taken from the initial slope and equals (4.1 ± 0.7) × 104 M−1 s−1. Similar to pH = 9.3, the highest value was obtained for the reaction containing carbonate. It is worth noting that experiments at pH = 11 show on average a one-order of magnitude higher rate constant than for the corresponding reactions at pH = 9.3. Due to the larger contribution of [MnIII(TPPS)(H2O)OH]4− in solutions with increasing basicity, the manganese complex becomes more labile and can be more efficiently oxidized by H2O2, OOH− or HCO4−.
Subsequently, we used peracetic acid (PAA) instead of hydrogen peroxide to oxidize MnIII(TPPS). PAA is produced in an equilibrium between hydrogen peroxide and acetic acid [50]. The reaction is catalyzed by sulfuric acid and proceeds according to Equation (2):
Due to this equilibration reaction, it is obvious that PAA solutions contain a certain concentration of H2O2. In the literature it is postulated that both peroxides are strong oxidants. On comparing PAA and H2O2, an advantage of the former is its lower tendency to undergo catalase reactions. Participation of the oxidants in catalase reactions causes faster decomposition of the oxidant and could be a problem in efficient degradation processes catalyzed by transition metal complexes [22,51]. Detailed spectroscopic and kinetic studies were performed to follow the oxidation of MnIII(TPPS) by PAA at pH 9.3 and 11. Typical spectral changes and a kinetic trace are shown in Figure 2.
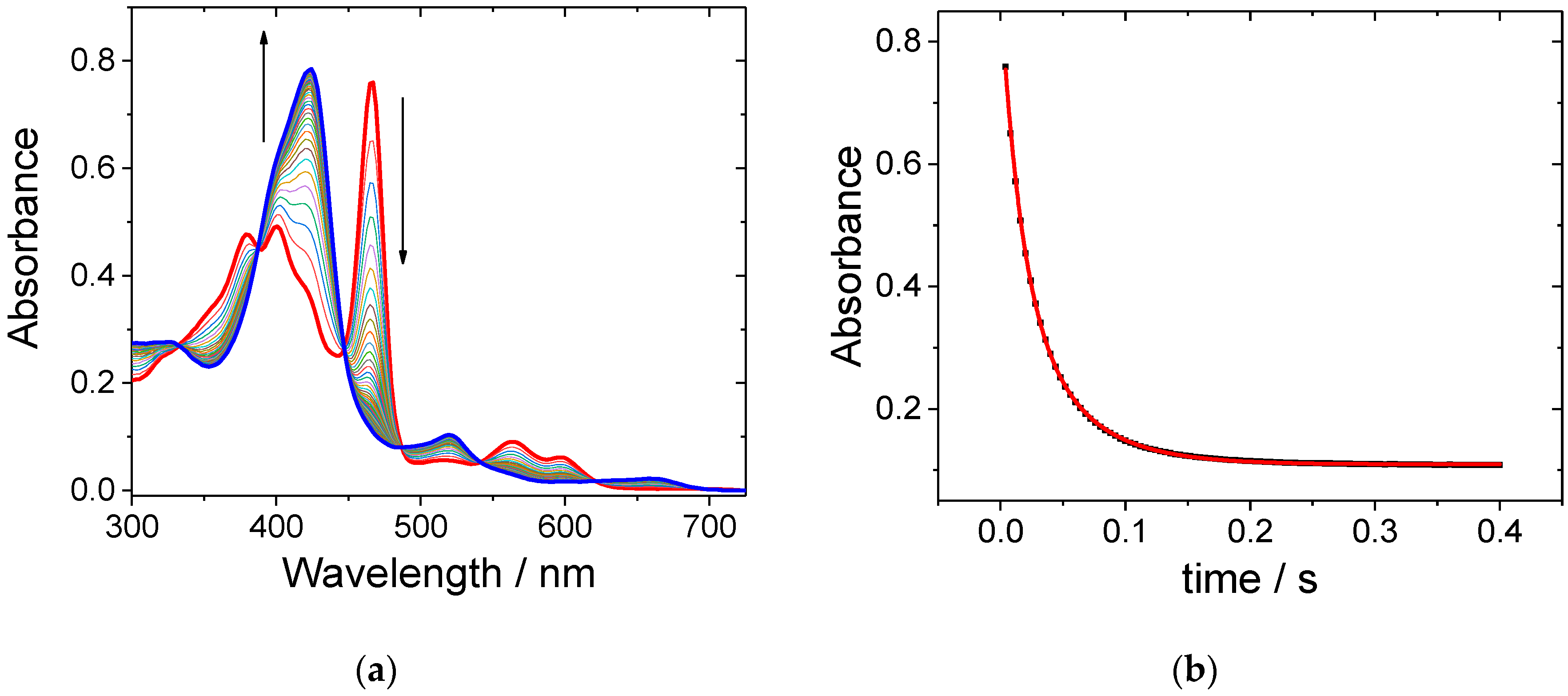
Upon addition of PAA to a solution of MnIII(TPPS) in basic aqueous media, an immediate decrease in the absorption of the Soret band at 466 nm was observed (see Figure 2a). Simultaneously, a new broad band at 422 nm was formed. The observed hypsochromic shift is ascribed to the oxidation of MnIII(TPPS) to (TPPS)MnV=O as described above. The formation of MnV=O species was completed in less than 0.4 s and revealed clean isosbestic points at 387, 447, 487 and 542 nm. The overall observed spectral changes are in good agreement with our earlier work dealing with the MnIII(TPPS)/H2O2 system. A typical kinetic trace presented in Figure 2b shows very good pseudo-first order behavior and can be fitted satisfactory with a single exponential function.
To gain more information on the reactivity of MnIII(TPPS) with PAA, we conducted experiments with different concentrations of PAA at pH = 9.3 and pH = 11. The obtained results are shown in Figure 3 and Figure S3.
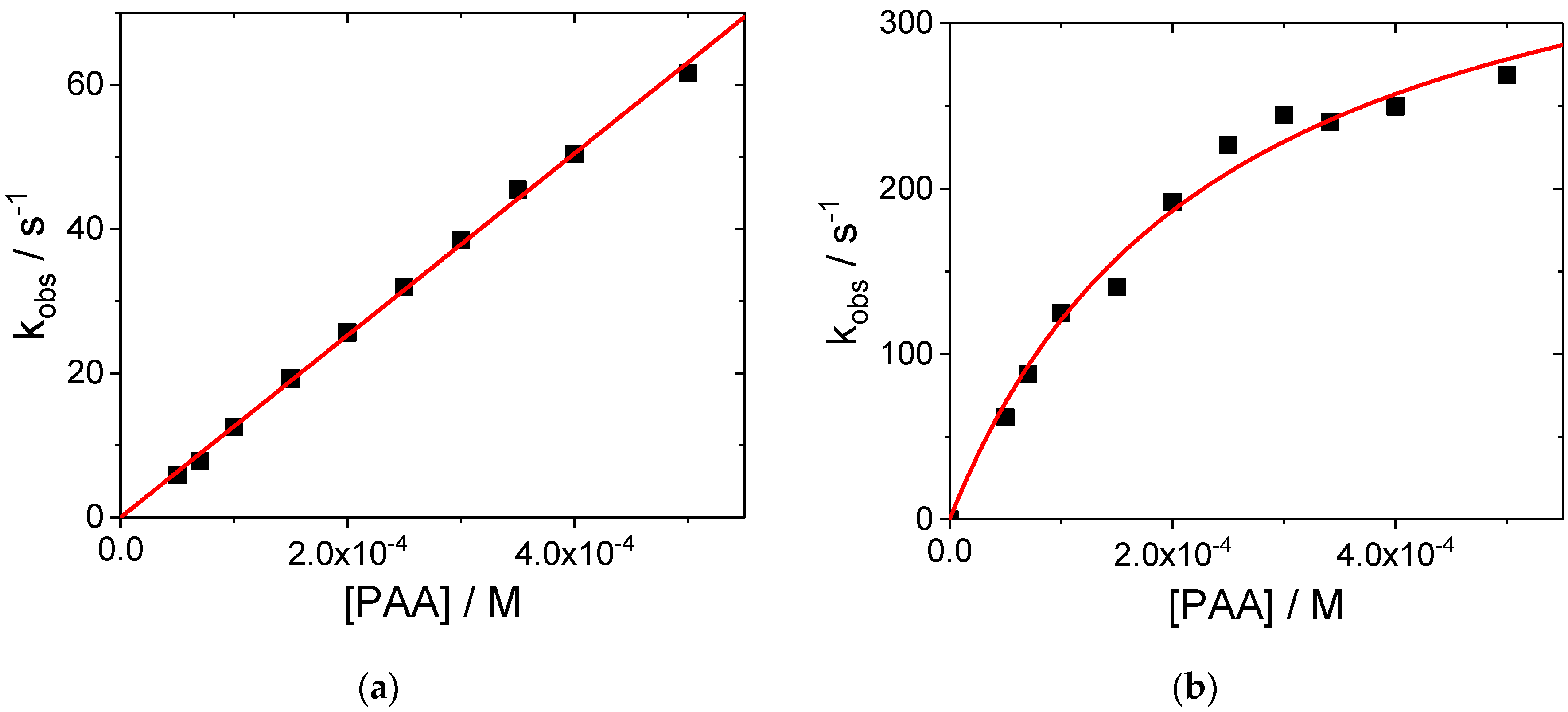
Plots of the observed rate constants vs. [PAA] in Figure 3a and Figure S3a exhibit a linear dependence with an almost unnoticeable intercept. The small intercept can be ascribed to a parallel decomposition of the porphyrin complex that occurs at low PAA concentrations. The obtained second-order rate constants for these reactions at pH 9.3 in carbonate solution (Figure 3a) and at pH 11 in basic solution (Figure S3a) are (1.26 ± 0.02) × 105 M−1 s−1 and (2.19 ± 0.06) × 104 M−1 s−1 at 25 °C, respectively. It differs for plots recorded at pH = 11 in solutions containing KNO3 and carbonate. For these plots, the data exhibit saturation kinetics (see Figure S3b and Figure 3b). The second-order rate constants were taken from the initial slope and equal (9.3 ± 2.0) × 104 M−1 s−1 and (1.7 ± 0.4) × 106 M−1 s−1, respectively. As in the case of the hydrogen peroxide system, the higher value was found for the reaction at pH = 11 in the presence of carbonate in solution. However, a comparison of all the values found for H2O2 as oxidant, with those obtained for the same reaction with PAA, leads to the conclusion that the efficiency of the formation of high-valent Mn(TPPS)-oxo complexes by PAA is significantly higher by an order of magnitude in terms of the second-order rate constant. This suggests that PAA is a more efficient oxidant in comparison to H2O2 under the selected conditions. Moreover, kinetic data showed that the oxidation by PAA is accelerated in carbonate solution. This can be accounted for in terms of the formation of peroxocarbonate in the presence of H2O2, which is released by the hydrolysis of PAA according to Equation (2).
It should be underlined that for pH = 9.3 only the reactions in the presence of carbonate buffer were successfully measured. Reactions in basic and KNO3 solution showed low reproducibility and were not taken into consideration. It is suggested in the literature that PAA itself can undergo three potential reactions, namely hydrolysis to H2O2 and acetic acid, spontaneous decomposition, and decomposition catalyzed by transition metal ions. These reactions may have an effect on the complexity of the observed processes and cause insufficient reproducibility of results especially at a pH close to the pKa value of PAA, which is 8.2 [22,51,52,53,54,55].
In a next step, we extended our work to elucidate how MnIII(TPPS) reacts with sodium hypochlorite. NaOCl exists in aqueous solution as an equilibrium between hypochlorous acid (HOCl) and hypochlorite ion (OCl−) at pH 5–10 [56]. The pKa value of HOCl is 7.53 [57]. Sodium hypochlorite undergoes an un-catalyzed two-step decomposition according to the reactions in Equations (3) and (4) [58]:
In the work by Lister it was shown that the reaction described in Equation (3) is the rate-determining slow step with the larger activation energy barrier. Moreover, the influence of sodium hydroxide, carbonate and chloride on the stability of NaOCl was studied. The data showed that the mentioned compounds do not exert a catalytic effect on the reactions outlined in Equations (3) and (4) [58].
A series of measurements between MnIII(TPPS) and NaOCl at pH 9.3 and 11 were conducted. Monitoring of the kinetic traces at 466 nm, which mostly showed very good pseudo-first order behavior, allowed the kinetic data shown in Figures S4 and S5 to be obtained. However, for the reaction at pH = 9.3 in base, it was necessary to fit the kinetic traces with a double exponential function. This effect was probably due to the unstable pH value during measurements, which was caused by the presence of only a diluted basic solution in the test system. Calculated rate constants exhibited a linear dependence on the NaOCl concentration. Only for pH = 9.3 with KNO3 (see Figure S4b) and pH = 11 with base (see Figure S5a), a significant intercept was observed. It can be associated with the back reaction in which (TPPS)MnV=O is reduced to the initial MnIII(TPPS) in a parallel reaction. The slope of the plots in Figures S4 and S5 represent the second-order rate constants for the oxidation of MnIII(TPPS), which are (4.1 ± 0.2) × 102, (1.42 ± 0.07) × 103 and (1.46 ± 0.04) × 103 M−1 s−1 for pH = 9.3 and (9.6 ± 0.4) × 102, (3.65 ± 0.06) × 103 and (1.19 ± 0.03) × 104 M−1 s−1 for pH = 11, respectively.
Analysis of the second-order rate constants for the oxidation of MnIII(TPPS) by NaOCl to form high-valent Mn(TPPS)-oxo complexes, indicates that the process is favored at pH ≈11. Furthermore, the reported rate constants are insignificantly higher than those for the MnIII(TPPS)/H2O2 system. However, they are lower than those obtained for the MnIII(TPPS)/PAA system. This suggests that NaOCl is a more efficient oxidant than H2O2, but a less efficient oxidant than PAA for the studied oxidation process. Moreover, the presence of carbonate in the solution has insignificant influence on the second-order rate constants. This demonstrates that the presence of carbonate does not affect the oxidation process as it was in the case for the MnIII(TPPS)/H2O2 and MnIII(TPPS)/PAA systems.
To gain more information about the reactions between MnIII(TPPS) and potassium peroxomonosulfate or sodium perborate, more detailed measurements using the rapid-scan mode of a stopped-flow instrument were conducted. Potassium peroxomonosulfate is commercially available as the stable triple salt KHSO5∙0.5 KHSO4∙0.5 K2SO4 (Oxone®). Oxone is non-toxic and has good water solubility. The pKa value of HSO5−/SO52− is 9.4. It is commonly used in synthesis and oxidation of organic compounds (such as alkenes, aldehydes, ketones, phenols) [59] and as an efficient disinfectant. The peroxomonosulfate anion (HSO5−) is a relatively strong oxidant with a standard electrode potential of 1.82 V (vs. NHE) according to Equation (5) [60]:
The peroxomonosulfate ion requires activation by transition metals, or irradiation, for the efficient degradation of pollutants. Activated HSO5− can easily generate sulfate radicals [60].
The perborate anion is produced when in aqueous solution boric acid and hydrogen peroxide are mixed together [61]. The pKa of boric acid is 8.98 [62], which indicates that above pH 9 the borate anion predominates according to Equation (6) with K(BOH)3 = 1.0 × 10−9 M.
In the presence of hydrogen peroxide, boric acid exists in equilibrium with two other forms, namely monoperoxoborate (also called perborate) and diperoxoborate ions, as shown below in Equations (7) and (8) with equilibrium constants KHOOB(OH)3− = 2.0 × 10−8 and K(HOO)2B(OH)2- = 2.0 M−1 [63]:
It is suggested in the literature that peroxoborate is the main species in the pH range 7–13, however, this depends on the concentrations of borate and hydrogen peroxide in solution. It is postulated that perborate is more reactive than percarbonate, but less reactive than persulfate and peroxycarboxylic acids [61,63,64]. The results of our kinetic studies dealing with the interactions between MnIII(TPPS) and persulfate or perborate are presented in Figures S6 and S7. All results were obtained using the stopped-flow technique.
It is worth noting that reproducible results were obtained only for measurements at pH = 11. The dependence of the determined kobs values on the peroxomonosulfate concentration in potassium nitrate solution (see Figure S6a) exhibits saturation kinetics with a second-order rate constant of (4.4 ± 1.1) × 104 M−1 s−1 calculated from the initial slope of the plot. It is different in carbonate-containing solution (see Figure S6b), where kobs exhibits a linear dependence on the peroxomonosulfate concentration with a second-order rate constant (1.17 ± 0.02) × 105 M−1 s−1. All kinetic traces showed very good pseudo-first order behavior and could be fitted with a single exponential function. Similarly, when perborate was used, plots of kobs versus perborate concentration are linear with a small intercept in Figure S7b, which is probably associated with a slow parallel decomposition of the catalyst that cannot be avoided. The second-order rate constants obtained were (8.8 ± 0.2) × 102 M−1 s−1 and (3.7 ± 0.2) × 104 M−1 s−1, respectively. It should be underlined that the presence of carbonate in solution significantly accelerates the reaction with perborate. This is probably due to the fact that perborate is always in equilibrium with an appropriate amount of hydrogen peroxide, which is able to rapidly generate the percarbonate ion in the reaction with carbonate as described earlier. In the case of persulfate, the difference between the rate of the reaction with and without carbonate was much smaller.
For clarity, all obtained values of second-order rate constants are collected in Table 1.
3. Discussion
In this study it was our aim to investigate the ability of MnIII(TPPS) to react with commercially available oxidants to form high-valent Mn(TPPS)-oxo complexes as catalytic active species in an efficient way. We examined five different oxidants, viz. hydrogen peroxide, peracetic acid, sodium hypochlorite, potassium peroxomonosulfate and sodium perborate, in their reactions with MnIII(TPPS). Although we studied the interaction of MnIII(TPPS) with H2O2 before, we repeated these measurements under the same conditions as for the other oxidants. It allowed us to minimize the influence of pH changes and small differences in the constitution of solutions between experiments with hydrogen peroxide and the remaining oxidants. The results showed that the second-order rate constants for the formation of high-valent Mn(TPPS)-oxo complexes are similar for the three oxidants: hydrogen peroxide, sodium hypochlorite and sodium perborate. Overall, significantly higher second-order rate constants were obtained for peracetic acid and potassium peroxomonosulfate. The most efficient formation of high-valent Mn(TPPS)-oxo complexes was found for PAA at pH ≈ 11 in carbonate solution, which is at least an order of magnitude higher than for the other tested oxidants on the basis of the reported second-order rate constants. It can be concluded that peracetic acid is the most efficient oxidant of MnIII(TPPS) among all tested oxidants under the selected conditions.
It is worth noting that the formation of higher oxidation state Mn(TPPS)-oxo species is faster and favored at pH = 11. This is due to the presence of the MnIII(TPPS) complex more in the [MnIII(TPPS)(H2O)OH]4− form than in the [MnIII(TPPS)(H2O)2]3- form, which is more labile as a result of the trans-labilization by the hydroxo ligand and therefore more reactive. Our studies also revealed that carbonate in solution significantly accelerated the reactions with H2O2, PAA and perborate. As shown before, in basic aqueous solutions containing carbonate and hydrogen peroxide, the rapid formation of peroxocarbonate (HCO4−) is observed. It is postulated that peroxocarbonate is a stronger oxidant than hydrogen peroxide and thus can easier oxidize MnIII(TPPS). Due to the fact that peracetic acid and perborate are always in an equilibrium with H2O2, also in basic solutions, HCO4− ions can be formed.
In some cases the plots of kobs versus oxidant concentration reached a limiting value (saturation kinetics, see Figure S2b, Figure 3b and Figure S3b) which can be ascribed to rapid precursor formation (K) between a specific MnIII(TPPS) complex and the oxidant as shown in Equations (9) and (10), followed by the rate-determining (k) formation of the high valent MnV=O oxo complex. Subsequently, the high-valent oxo complex oxidizes the substrate (S) in a fast reaction to reform the MnIII(TPPS) complex (to complete the catalytic cycle) and the oxidized substrate (SO) as shown in Equation (11).
Based on the mechanism outlined in Equations (9) to (11) for oxidant in excess, the observed first-order rate constant can be expressed as Equation (12),
which simplifies to kobs = kK [oxidant] at low [oxidant], and kobs = k at high [oxidant]. Thus, the initial slope of the curved concentration dependence represents kK, i.e., the second-order rate constant, and the maximum rate constant reached at saturation represents k, the rate-determining step of the suggested reaction mechanism.
Finally, it can be concluded that, in the longer term, the results presented in this work may have a positive impact on the design of new, efficient and environmentally friendly catalytic systems, which can be used in industrial processes for the purification of wastewater.
4. Materials and Methods
All chemicals were of analytical grade and used without further purification. Pure sodium hydroxide micro-pills and hydrochloric acid were provided by POCH (Avantor Performance Materials Poland S.A., Gliwice, Poland). MnIII(TPPS)Cl, where TPPS = 5,10,15,20-tetrakis(4-sulphonatophenyl)-21H,23H-porphyrin, sodium carbonate, sodium hydrogen carbonate, peracetic acid, sodium hypochlorite, sodium perborate, Oxone® and potassium nitrate, were supplied by Sigma–Aldrich (Merck KGaA, Darmstadt, Germany). Hydrogen peroxide (30% w/w) was purchased from Chempur (Chempur, Piekary Śląskie, Poland). The concentration of H2O2 was determined by standard titration with KMnO4 in an acidic medium. Water used in all the experiments was distilled and deionized (Mili-Q system).
The time-resolved visible spectra were recorded on a SX20 (Applied Photophysics, Leatherhead, Surrey, UK) stopped-flow spectrophotometer using a rapid scan photodiode array mode with 1 nm spectral resolution. The temperature was controlled by a Labo Plus, Polyscience (Labo Plus sp. Z o.o., Warsaw, Poland) thermostat bath. All measurements were done at 25 °C. One syringe of the stopped-flow instrument was filled with the MnIII(TPPS) complex and the second one with the appropriate oxidant solution. Both solutions were at the chosen pH. In the experiments dealing with the kinetics of the reactions, the traces were followed at 466 nm. Complete spectra and single wavelength kinetic traces were collected. Data were processed using the Pro-Data and OriginPro 2019 software. The pH measurements were carried out with a HI 221 (HANNA Instruments Polska, Olsztyn, Poland) pH-meter combined with a Hydromet ERH-13–6 electrode (Hydromat, Gliwice, Poland). The pH-meter was calibrated for two points with standard buffer solutions at pH 7, 9, 10 and 12.46, depending on the pH range required.
All solutions were freshly prepared before use by dissolving the starting material in pure, distilled water. Concentrations of MnIII(TPPS) were evaluated spectroscopically based on the absorption coefficients determined at different pH values. Different carbonate or NaOH solutions were used to adjust the pH of the experimental solutions. Carbonate buffer at pH 9.3 was prepared by dissolving and mixing NaHCO3 with Na2CO3 in water. For pH 11, dissolved Na2CO3 with the addition of NaOH was used. Sodium hydroxide solutions were obtained by dissolving suitable amounts of NaOH. In some cases, KNO3 was used to control the ionic strength. All reported concentrations are the final values obtained after mixing and dilution of the reactants.
5. Conclusions
The results presented in this paper indicate that MnIII(TPPS) can easily be activated by a wide range of oxidants to form a promising catalyst for many different oxidation reactions. We have shown that hydrogen peroxide, peracetic acid, sodium hypochlorite, potassium peroxomonosulfate and sodium perborate can oxidize MnIII(TPPS) to form high-valent Mn(V)-oxo species under appropriate conditions. All studied systems show high second-order rate constants for the formation of Mn(V)-oxo species. The highest value was found for the MnIII(TPPS)/PAA system at pH ≈ 11 in carbonate solution, which indicates that peracetic acid is the most efficient oxidant under the selected conditions. Moreover, it can be concluded that for all tested systems the oxidation of MnIII(TPPS) is much faster at pH = 11 compared to pH = 9.3, which is due to the presence of the most labile form of MnIII(TPPS), viz. [MnIII(TPPS)(H2O)(OH)]4−. It is worth noting that the use of carbonate solutions significantly accelerate the formation of higher oxidation states of MnIII(TPPS) in systems containing H2O2, PAA and sodium perborate.
Supplementary Materials
The following are available online at https://0-www-mdpi-com.brum.beds.ac.uk/2073-4344/10/6/610/s1: Figure S1: Plots of kobs versus H2O2 concentration for the reaction of MnIII(TPPS) with H2O2 at pH = 9.3; Figure S2: Plots of kobs versus H2O2 concentration for the reaction of MnIII(TPPS) with H2O2 at pH = 11; Figure S3: Dependence of kobs on [PAA] for the reaction of MnIII(TPPS) with PAA at pH = 11; Figure S4: Plots of kobs versus NaOCl concentration for the reaction of MnIII(TPPS) with H2O2 at pH = 9.3; Figure S5: Dependence of kobs on [NaOCl] for the reaction of MnIII(TPPS) with NaOCl at pH = 11; Figure S6: Plots of kobs versus persulfate concentration for the reaction of MnIII(TPPS) with peroxomonosulfate at pH = 11; Figure S7: Dependence of kobs on [perborate] for the reaction of MnIII(TPPS) with perborate at pH = 11.
Author Contributions
R.v.E. and M.P. conceived and designed the experiments; M.P. performed the experiments; M.P., R.v.E. and Ł.O. analyzed the data; M.P. and G.S. contributed reagents/materials/analytical tools; M.P., R.v.E., Ł.O. and G.S. wrote the manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the National Science Center in Poland under grant number DEC-2016/21/N/ST4/00172. The APC was funded by the Faculty of Chemistry of the Jagiellonian University in Kraków.
Acknowledgments
The Faculty of Chemistry of the Jagiellonian University is the beneficiary of structural funds from the European Union, grant no. POIG. 02.01.00-12-023/08 “Atomic Scale Science for Innovative Economy (ATOMIN)”.
Conflicts of Interest
The authors declare no conflict of interest. The founding sponsors had no role in the design of the study; in the collection, analyses, or interpretation of data; in the writing of the manuscript; and in the decision to publish the results.
References
- Pearce, C.I.; Lloyd, J.R.; Guthrie, J.T. The removal of colour from textile wastewater using whole bacterial cells: A review. Dyes Pigment. 2003, 58, 179–196. [Google Scholar] [CrossRef]
- Asghar, A.; Abdul Raman, A.A.; Wan Daud, W.M.A. Advanced oxidation processes for in-situ production of hydrogen peroxide/hydroxyl radical for textile wastewater treatment: A review. J. Clean. Prod. 2015, 87, 826–838. [Google Scholar] [CrossRef] [Green Version]
- Rajkumar, D.; Kim, J.G. Oxidation of various reactive dyes with in situ electro-generated active chlorine for textile dyeing industry wastewater treatment. J. Hazard. Mater. 2006, 136, 203–212. [Google Scholar] [CrossRef] [PubMed]
- Ghodbane, H.; Hamdaoui, O. Intensification of sonochemical decolorization of anthraquinonic dye Acid Blue 25 using carbon tetrachloride. Ultrason. Sonochem. 2009, 16, 455–461. [Google Scholar] [CrossRef]
- Lin, S.H.; Chen, M.L. Treatment of textile wastewater by chemical methods for reuse. Water Res. 1997, 31, 868–876. [Google Scholar] [CrossRef]
- Can, O.T.; Kobya, M.; Demirbas, E.; Bayramoglu, M. Treatment of the textile wastewater by combined electrocoagulation. Chemosphere 2006, 62, 181–187. [Google Scholar] [CrossRef]
- Chen, G. Electrochemical technologies in wastewater treatment. Sep. Purif. Technol. 2004, 38, 11–41. [Google Scholar] [CrossRef]
- Chang, W.; Hong, S.; Park, J. Effect of zeolite media for the treatment of textile wastewater in a biological aerated filter. Process Biochem. 2002, 37, 693–698. [Google Scholar] [CrossRef]
- Ellouze, E.; Tahri, N.; Amar, R. Enhancement of textile wastewater treatment process using Nanofiltration. Desalination 2012, 286, 16–23. [Google Scholar] [CrossRef]
- Jiang, J.; Lloyd, B. Progress in the development and use of ferrate(VI) salt as an oxidant and coagulant for water and wastewater treatment. Water Res. 2002, 36, 1397–1408. [Google Scholar] [CrossRef]
- Bulc, T.G.; Ojstršek, A. The use of constructed wetland for dye-rich textile wastewater treatment. J. Hazard. Mater. 2008, 155, 76–82. [Google Scholar] [CrossRef] [PubMed]
- Lin, S.H.; Peng, C.F. Treatment of textile wastewater by electrochemical method. Water Res. 1994, 28, 277–282. [Google Scholar] [CrossRef]
- Perkowski, J.; Kos, L.; Ledakowicz, S. Application of Ozone in Textile Wastewater Treatment. Ozone Sci. Eng. 1996, 18, 73–85. [Google Scholar] [CrossRef]
- Hage, R.; Lienke, A. Applications of Transition-Metal Catalysts to Textile and Wood-Pulp Bleaching. Angew. Chem. Int. Ed. 2006, 45, 206–222. [Google Scholar] [CrossRef] [PubMed]
- Bäckvall, J.E. Modern Oxidation Methods; WILEY-VCH: Weinheim, Germany, 2010; pp. 371–381. [Google Scholar]
- Chatterjee, D.; Ember, E.; Pal, U.; Ghosh, S.; van Eldik, R. Remarkably high catalytic activity of the RuIII(edta)/H2O2 system towards degradation of the azo-dye Orange II. Dalton Trans. 2011, 40, 10473–10480. [Google Scholar] [CrossRef]
- Rothbart, S.; van Eldik, R. Manganese Compounds as Versatile Catalysts for the Oxidative Degradation of Organic Dyes. Adv. Inorg. Chem. 2013, 65, 165–215. [Google Scholar] [CrossRef]
- Gilbert, B.C.; Lindsay Smith, J.R.; Newton, M.S.; Oakes, J.; Prats, R.P.i. Azo dye oxidation with hydrogen peroxide catalysed by manganese 1,4,7-triazacyclononane complexes in aqueous solutions. Org. Biomol. Chem. 2003, 1, 1568–1577. [Google Scholar] [CrossRef]
- Zaharia, C.; Suteu, D.; Muresan, A.; Muresan, R.; Popescu, A. Textile wastewater treatment by homogeneous oxidation with hydrogen peroxide. Environ. Eng. Manag. J. 2009, 8, 1359–1369. [Google Scholar] [CrossRef]
- Pagano, M.; Ciannarella, R.; Locaputo, V.; Mascolo, G.; Volpe, A. Oxidation of azo and anthraquinonic dyes by peroxymonosulphate activated by UV light. J. Environ. Sci. Health A 2018, 53, 393–404. [Google Scholar] [CrossRef]
- Lin, K.A.; Lin, T. Degradation of Acid Azo Dyes Using Oxone Activated by Cobalt Titanate Perovskite. Water Air Soil Pollut. 2017, 229, 10. [Google Scholar] [CrossRef]
- Yuan, Z.; Ni, Y.; Van Heiningen, A.R.P. Kinetics of peracetic acid decomposition: Part I: Spontaneous decomposition at typical pulp bleaching conditions. Can. J. Chem. Eng. 1997, 75, 37–41. [Google Scholar] [CrossRef]
- Emmons, W.D. The Oxidation of Amines with Peracetic Acid. J. Am. Chem. Soc. 1957, 79, 5528–5530. [Google Scholar] [CrossRef]
- Appels, L.; Assche, A.V.; Willems, K.; Degrève, J.; Van Impe, J.; Dewil, R. Peracetic acid oxidation as an alternative pre-treatment for the anaerobic digestion of waste activated sludge. Bioresour.Technol. 2011, 102, 4124–4130. [Google Scholar] [CrossRef] [PubMed]
- Kitis, M. Disinfection of wastewater with peracetic acid: A review. Environ. Int. 2004, 30, 47–55. [Google Scholar] [CrossRef]
- Wagner, M.; Brumelis, D.; Gehr, R. Disinfection of Wastewater by Hydrogen Peroxide or Peracetic Acid: Development of Procedures for Measurement of Residual Disinfectant and Application to a Physicochemically Treated Municipal Effluent. Water Environ. Res. 2002, 74, 33–50. [Google Scholar] [CrossRef] [PubMed]
- Suess, H.U. Pulp Bleaching Today; De Gruyter: Berlin, Germany, 2010. [Google Scholar] [CrossRef]
- Zhao, R.; Tang, Y.; Wei, S.J.; Xu, X.; Shi, X.; Zhang, G. Electrosynthesis of sodium hypochlorite in room temperature ionic liquids and in situ electrochemical epoxidation of olefins. React. Kinet. Mech. Cat. 2012, 106, 37–47. [Google Scholar] [CrossRef]
- Page, P.C.B.; Parker, P.; Buckley, B.R.; Rassias, G.A.; Bethell, D. Organocatalysis of asymmetric epoxidation by iminium salts using sodium hypochlorite as the stoichiometric oxidant. Tetrahedron 2009, 65, 2910–2915. [Google Scholar] [CrossRef]
- Kirihara, M.; Okada, T.; Sugiyama, Y.; Akiyoshi, M.; Matsunaga, T.; Kimura, Y. Sodium Hypochlorite Pentahydrate Crystals (NaOCl·5H2O): A Convenient and Environmentally Benign Oxidant for Organic Synthesis. Org. Process Res. Dev. 2017, 21, 1925–1937. [Google Scholar] [CrossRef]
- Zhang, W.; Jacobsen, E.N. Asymmetric olefin epoxidation with sodium hypochlorite catalyzed by easily prepared chiral manganese(III) salen complexes. J. Org. Chem. 1991, 56, 2296–2298. [Google Scholar] [CrossRef]
- Gonsalvi, L.; Arends, I.W.C.E.; Moilanen, P.; Sheldon, R.A. The Effect of pH Control on the Selective Ruthenium-Catalyzed Oxidation of Ethers and Alcohols with Sodium Hypochlorite. Adv. Synth. Catal. 2003, 345, 1321–1328. [Google Scholar] [CrossRef]
- Chellamani, A.; Harikengaram, S. Mechanism of oxidation of aryl methyl sulfoxides with sodium hypochlorite catalyzed by (salen)MnIII complexes. J. Mol. Catal. A Chem. 2006, 247, 260–267. [Google Scholar] [CrossRef]
- Chellamani, A.; Harikengaram, S. Mechanism of (Salen)manganese(III)-Catalyzed Oxidation of Aryl Phenyl Sulfides with Sodium Hypochlorite. Helv. Chim. Acta 2011, 94, 453–463. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhou, Q.; Ma, W.; Zhao, J. Enantioselective oxidation of racemic secondary alcohols catalyzed by chiral Mn(III)-salen complex with sodium hypochlorite as oxidant. Catal. Commun. 2014, 45, 114–117. [Google Scholar] [CrossRef]
- Patil, R.D.; Sasson, Y. Naphthalenes Oxidation by Aqueous Sodium Hypochlorite Catalyzed by Ruthenium Salts Under Phase-Transfer Catalytic Conditions. Catal. Lett. 2016, 146, 991–997. [Google Scholar] [CrossRef]
- Mirkhani, V.; Tangestaninejad, S.; Moghadam, M.; Yadollahi, B. Efficient and selective epoxidation of alkenes by supported manganese porphyrin under ultrasonic irradiation. J. Chem. Res. 2000, 2000, 515–517. [Google Scholar] [CrossRef]
- Tangestaninejad, S.; Moghadam, M.; Mirkhani, V.; Mohammadpoor-Baltork, I.; Hoseini, N. Efficient and Selective Hydrocarbon Oxidation with Sodium Periodate Catalyzed by Supported Manganese(III) Porphyrin. J. Iran. Chem. Soc. 2010, 7, 663–672. [Google Scholar] [CrossRef]
- Zakavi, S.; Ebadi, S.; Javanmard, M. Nanosized cationic and anionic manganese porphyrins as mesoporous catalysts for the oxidation of olefins: Nano versus bulk aggregates. Appl. Organomet. Chem. 2018, 32, e4175. [Google Scholar] [CrossRef]
- Song, R.; Sorokin, A.; Bernadou, J.; Meunier, B. Metalloporphyrin-Catalyzed Oxidation of 2-Methylnaphthalene to Vitamin K3 and 6-Methyl-1,4-naphthoquinone by Potassium Monopersulfate in Aqueous Solution. J. Org. Chem. 1997, 62, 673–678. [Google Scholar] [CrossRef]
- Procner, M.; Orzeł, Ł.; Stochel, G.; van Eldik, R. Spectroscopic and kinetic evidence for redox cycling, catalase and degradation activities of MnIII(TPPS) in a basic aqueous peroxide medium. Chem. Commun. 2016, 52, 5297–5300. [Google Scholar] [CrossRef] [Green Version]
- Procner, M.; Orzeł, Ł.; Stochel, G.; van Eldik, R. Catalytic Degradation of Orange II by MnIII(TPPS) in Basic Hydrogen Peroxide Medium: A Detailed Kinetic Analysis. Eur. J. Inorg. Chem. 2018, 2018, 3462–3471. [Google Scholar] [CrossRef]
- Arasasingham, R.D.; He, G.X.; Bruice, T.C. Mechanism of manganese porphyrin-catalyzed oxidation of alkenes. Role of manganese(IV)-oxo species. J. Am. Chem. Soc. 1993, 115, 7985–7991. [Google Scholar] [CrossRef]
- Nam, W.; Kim, I.; Lim, M.H.; Choi, H.J.; Lee, J.S.; Jang, H.G. Isolation of an oxomanganese(v) porphyrin intermediate in the reaction of a manganese(III) porphyrin complex and H2O2 in aqueous solution. Chem.-Eur. J. 2002, 8, 2067–2071. [Google Scholar] [CrossRef]
- Lahaye, T.; Groves, J.T. Modeling the haloperoxidases: Reversible oxygen atom transfer between bromide ion and an oxo-Mn(V) porphyrin. J. Inorg. Biochem. 2007, 101, 1786–1797. [Google Scholar] [CrossRef] [PubMed]
- Ember, E.; Rothbart, S.; Puchta, R.; van Eldik, R. Metal ion-catalyzed oxidative degradation of Orange II by H 2O2. High catalytic activity of simple manganese salts. New J. Chem. 2009, 33, 34–49. [Google Scholar] [CrossRef]
- Rothbart, S.; Ember, E.; van Eldik, R. Comparative study of the catalytic activity of [MnII(bpy)2Cl2] and [Mn2III/IV(μ-O)2(bpy)4](ClO4)3 in the H2O2 induced oxidation of organic dyes in carbonate buffered aqueous solution. Dalton Trans. 2010, 39, 3264–3272. [Google Scholar] [CrossRef]
- Richardson, D.E.; Yao, H.; Frank, K.M.; Bennett, D.A. Equilibria, Kinetics, and Mechanism in the Bicarbonate Activation of Hydrogen Peroxide: Oxidation of Sulfides by Peroxymonocarbonate. J. Am. Chem. Soc. 2000, 122, 1729–1739. [Google Scholar] [CrossRef]
- Bakhmutova-Albert, E.V.; Yao, H.; Denevan, D.E.; Richardson, D.E. Kinetics and mechanism of peroxymonocarbonate formation. Inorg. Chem. 2010, 49, 11287–11296. [Google Scholar] [CrossRef]
- Swern, D. Organic Peroxides; Wiley: New York, NY, USA, 1970; Volume I. [Google Scholar] [CrossRef]
- Rothbart, S.; Ember, E.; van Eldik, R. Mechanistic studies on the oxidative degradation of Orange II by peracetic acid catalyzed by simple manganese(ii) salts. Tuning the lifetime of the catalyst. New J. Chem. 2012, 36, 732–748. [Google Scholar] [CrossRef]
- Evans, D.F.; Upton, M.W. Studies on singlet oxygen in aqueous solution. Part 3. The decomposition of peroxy-acids. J. Chem. Soc. Dalton Trans. 1985, 1151–1153. [Google Scholar] [CrossRef]
- Koubek, E.; Haggett, M.L.; Battaglia, C.J.; Ibne-Rasa, K.M.; Pyun, H.Y.; Edwards, J.O. Kinetics and Mechanism of the Spontaneous Decompositions of Some Peroxoacids, Hydrogen Peroxide and t-Butyl Hydroperoxide. J. Am. Chem. Soc. 1963, 85, 2263–2268. [Google Scholar] [CrossRef]
- Ball, D.L.; Edwards, J.O. The Kinetics and Mechanism of the Decomposition of Caro’s Acid. I. J. Am. Chem. Soc. 1956, 78, 1125–1129. [Google Scholar] [CrossRef]
- Awad, M.I.; Harnoode, C.; Tokuda, K.; Ohsaka, T. Simultaneous Electroanalysis of Peroxyacetic Acid and Hydrogen Peroxide. Anal. Chem. 2001, 73, 1839–1843. [Google Scholar] [CrossRef]
- Urano, H.; Fukuzaki, S. The Mode of Action of Sodium Hypochlorite in the Decolorization of Azo Dye Orange II in Aqueous Solution. Biocontrol Sci. 2011, 16, 123–126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morris, J.C. The acid ionization constant of HOCl from 5 to 35. J. Phys. Chem. 1966, 70, 3798–3805. [Google Scholar] [CrossRef]
- Lister, M.W. Decomposition of sodium hypochlorite: The uncatalyzed reaction. Can. J. Chem. 1956, 34, 465–478. [Google Scholar] [CrossRef]
- Zheng, T.; Richardson, D.E. Homogeneous aqueous oxidation of organic molecules by Oxone® and catalysis by a water-soluble manganese porphyrin complex. Tetrahedron Lett. 1995, 36, 833–836. [Google Scholar] [CrossRef]
- Ghanbari, F.; Moradi, M. Application of peroxymonosulfate and its activation methods for degradation of environmental organic pollutants: Review. Chem. Eng. J. 2017, 310, 41–62. [Google Scholar] [CrossRef]
- Durrant, M.C.; Davies, D.M.; Deary, M.E. Dioxaborirane: A highly reactive peroxide that is the likely intermediate in borate catalysed electrophilic reactions of hydrogen peroxide in alkaline aqueous solution. Org. Biomol. Chem. 2011, 9, 7249–7254. [Google Scholar] [CrossRef]
- Pizer, R.; Tihal, C. Peroxoborates. Interaction of boric acid and hydrogen peroxide in aqueous solution. Inorg. Chem. 1987, 26, 3639–3642. [Google Scholar] [CrossRef]
- Deary, M.E.; Durrant, M.C.; Davies, D.M. A kinetic and theoretical study of the borate catalysed reactions of hydrogen peroxide: The role of dioxaborirane as the catalytic intermediate for a wide range of substrates. Org. Biomol. Chem. 2013, 11, 309–317. [Google Scholar] [CrossRef] [Green Version]
- Davies, D.M.; Deary, M.E.; Quill, K.; Smith, R.A. Borate-Catalyzed Reactions of Hydrogen Peroxide: Kinetics and Mechanism of the Oxidation of Organic Sulfides by Peroxoborates. Chem. Eur. J. 2005, 11, 3552–3558. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
(a) Schematic structure of MnIII(TPPS); (b) ultraviolet–visible (UV–Vis) spectra of MnIII(TPPS) in water, pH = 7 (blue line) and in 0.01 M NaOH, pH = 12 (green line). Experimental conditions: [MnIII(TPPS)] = 12 μM, T = 25 °C.
Figure 1.
(a) Schematic structure of MnIII(TPPS); (b) ultraviolet–visible (UV–Vis) spectra of MnIII(TPPS) in water, pH = 7 (blue line) and in 0.01 M NaOH, pH = 12 (green line). Experimental conditions: [MnIII(TPPS)] = 12 μM, T = 25 °C.

Figure 2.
(a) Spectral changes recorded during the reaction of MnIII(TPPS) and peracetic acid (PAA) at pH = 11 in the presence of carbonate in solution. Spectrum just after mixing the reactants (red line) and after 0.4 s (blue line); (b) typical kinetic trace recorded at 466 nm. Experimental conditions: [MnIII(TPPS)] = 6 μM, [PAA] = 50 μM, [Na2CO3 + NaHCO3] = 0.5 M, T = 25 °C, total reaction time = 0.4 s.
Figure 2.
(a) Spectral changes recorded during the reaction of MnIII(TPPS) and peracetic acid (PAA) at pH = 11 in the presence of carbonate in solution. Spectrum just after mixing the reactants (red line) and after 0.4 s (blue line); (b) typical kinetic trace recorded at 466 nm. Experimental conditions: [MnIII(TPPS)] = 6 μM, [PAA] = 50 μM, [Na2CO3 + NaHCO3] = 0.5 M, T = 25 °C, total reaction time = 0.4 s.

Figure 3.
Plot of kobs versus PAA concentration for the reaction of MnIII(TPPS) with PAA: (a) at pH = 9.3 in carbonate buffer; (b) at pH = 11 in 0.5 M Na2CO3 + NaHCO3 solution. Experimental conditions: [MnIII(TPPS)] = 6 μM, [PAA] = 50–500 μM, T = 25 °C.
Figure 3.
Plot of kobs versus PAA concentration for the reaction of MnIII(TPPS) with PAA: (a) at pH = 9.3 in carbonate buffer; (b) at pH = 11 in 0.5 M Na2CO3 + NaHCO3 solution. Experimental conditions: [MnIII(TPPS)] = 6 μM, [PAA] = 50–500 μM, T = 25 °C.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Rate constants for the oxidation of MnIII(TPPS) by studied oxidants at 25 °C.
| Oxidant | Experimental Conditions | Second-Order Rate Constant M−1 s−1 |
|---|---|---|
| H2O2 | pH = 9.3, NaOH | 1.08 ± 0.04 |
| pH = 9.3, 0.5 M KNO3 + NaOH | (3.2 ± 0.2) × 102 | |
| pH = 9.3, 0.5 M NaHCO3 + Na2CO3 | (7.2 ± 0.2) × 103 | |
| pH = 11, NaOH | (2.20 ± 0.06) × 103 | |
| pH = 11, 0.5 M KNO3 + NaOH | (4.1 ± 0.7) × 104 [a] | |
| pH = 11, 0.5 M NaHCO3 + Na2CO3 | (9.3 ± 0.7) × 104 | |
| PAA | pH = 9.3, 0.5 M NaHCO3 + Na2CO3 | (1.26 ± 0.02) × 105 |
| pH = 11, NaOH | (2.19 ± 0.06) × 104 | |
| pH = 11, 0.5 M KNO3 + NaOH | (9.3 ± 2.0) × 104 [a] | |
| pH = 11, 0.5 M NaHCO3 + Na2CO3 | (1.7 ± 0.4) × 106 [a] | |
| NaOCl | pH = 9.3, NaOH | (4.1 ± 0.2) × 102 |
| pH = 9.3, 0.5 M KNO3 + NaOH | (1.42 ± 0.07) × 103 | |
| pH = 9.3, 0.5 M NaHCO3 + Na2CO3 | (1.46 ± 0.04) × 103 | |
| pH = 11, NaOH | (9.6 ± 0.4) × 102 | |
| pH = 11, 0.5 M KNO3 + NaOH | (3.65 ± 0.06) × 103 | |
| pH = 11, 0.5 M NaHCO3 + Na2CO3 | (1.19 ± 0.03) × 104 | |
| peroxomonosulfate | pH = 11, 0.5 M KNO3 + NaOH | (4.4 ± 1.1) × 104 [a] |
| pH = 11, 0.5 M NaHCO3 + Na2CO3 | (1.17 ± 0.02) × 105 | |
| perborate | pH = 11, 0.5 M KNO3 + NaOH | (8.8 ± 0.2) × 102 |
| pH = 11, 0.5 M NaHCO3 + Na2CO3 | (3.7 ± 0.2) × 104 |
[a] Taken from the initial slope of the plots in Figure S2b, Figure 3b and Figure S3b.
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Procner, M.; Orzel, Ł.; Stochel, G.; van Eldik, R. A Kinetic Study on the Efficient Formation of High-Valent Mn(TPPS)-oxo Complexes by Various Oxidants. Catalysts 2020, 10, 610. https://0-doi-org.brum.beds.ac.uk/10.3390/catal10060610
AMA Style
Procner M, Orzel Ł, Stochel G, van Eldik R. A Kinetic Study on the Efficient Formation of High-Valent Mn(TPPS)-oxo Complexes by Various Oxidants. Catalysts. 2020; 10(6):610. https://0-doi-org.brum.beds.ac.uk/10.3390/catal10060610
Chicago/Turabian StyleProcner, Magdalena, Łukasz Orzel, Grażyna Stochel, and Rudi van Eldik. 2020. "A Kinetic Study on the Efficient Formation of High-Valent Mn(TPPS)-oxo Complexes by Various Oxidants" Catalysts 10, no. 6: 610. https://0-doi-org.brum.beds.ac.uk/10.3390/catal10060610
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.