Methane Conversion over C2N-Supported Fe2 Dimers


1
Physical School of Science and Technology, Inner Mongolia University, Hohhot 010021, China
2
Key Laboratory of Photonic and Electronic Bandgap Materials, College of Chemistry and Chemical Engineering, Ministry of Education, Harbin Normal University, Harbin 150025, China
*
Authors to whom correspondence should be addressed.
Catalysts 2020, 10(9), 973; https://0-doi-org.brum.beds.ac.uk/10.3390/catal10090973
Submission received: 30 June 2020
/
Revised: 13 August 2020
/
Accepted: 21 August 2020
/
Published: 29 August 2020
(This article belongs to the Special Issue Transition Metal Catalysis)
Abstract
:Methane is a vast hydrocarbon resource around the globe that has the potential to replace petroleum as a raw material and energy source. Therefore, the catalytic conversion of methane into high value-added chemicals is significantly important for the utilization of this hydrocarbon resource. However, this is a great challenge due to the high-energy input required to overcome the reaction barrier. Herein, a highly active catalytic conversion process of methane on an iron dimer anchored on a two-dimensional (2D) C2N monolayer (Fe2@C2N) is reported. Density functional theory calculations reveal that the superior properties of Fe2@C2N can be attributed to the formation of the Fe-O-Fe intermediate with H2O2 as the O-donor molecule, which facilitates the formation of methyl radicals and promotes the conversion of methane. This finding could pave the way toward highly efficient non-precious metal catalysts for methane oxidation reactions.
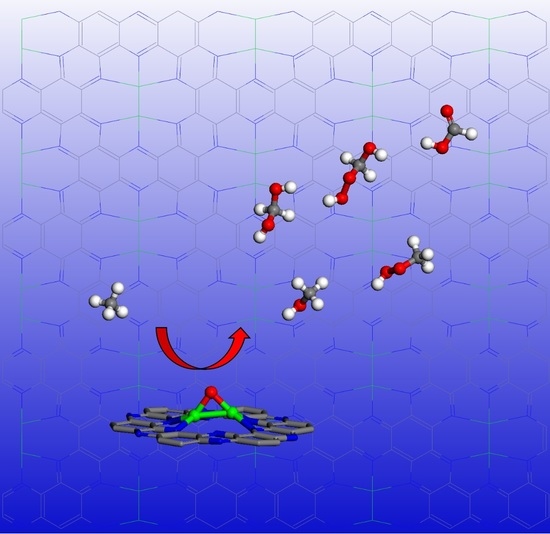
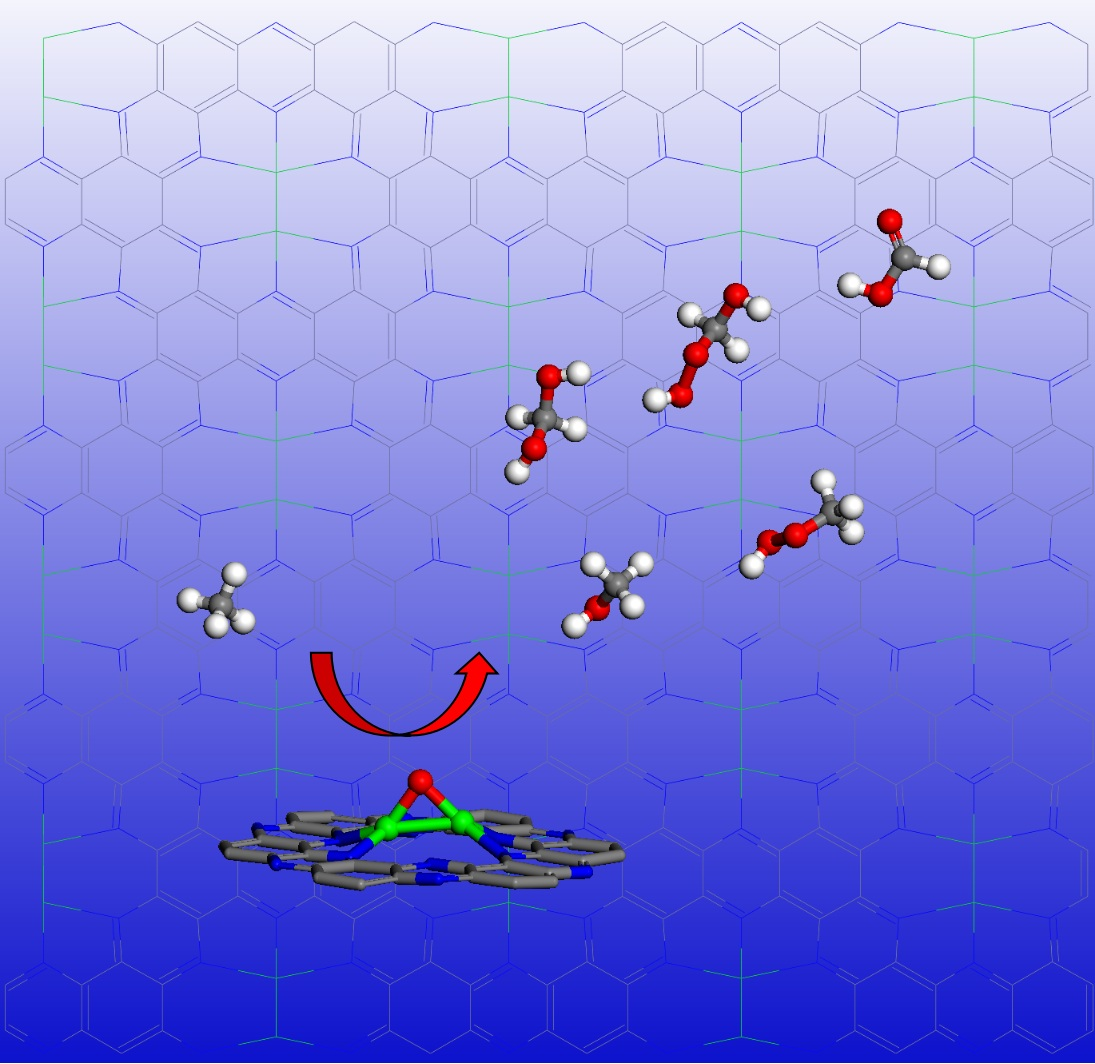{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Energy sources have become a major worldwide concern in recent years, as a result of global climate crisis and reduced fossil fuel reserves. China has relatively large reserves of natural gas and shale gas. The main component of natural gas is methane, which is a high-quality and clean energy source, abundant in nature, thus making natural gas an appealing feedstock for making valuable chemicals and fuels [1,2,3]. Today, with the depletion of petroleum resources, effective development and utilization of natural gas has attracted substantial interest from academia and industry. Unfortunately, most methane is distributed in remote and sparsely populated areas, which makes the transportation of methane over long distances energy-intensive and costly [4]. Therefore, there is a high demand for methods to convert methane into more transportable and high value-added chemicals.
Methane is the most stable hydrocarbon, with a tetrahedral geometry of four equivalent C–H bonds, where the carbon atom is located in the center of the regular tetrahedron in a sp3 hybridization with a C-H bond length of 1.09 Å and a H–C–H bond angle is 109.5°. The large bond dissociation energy of the methane molecule (435 kJ·mol−1) hinders the cleavage of the C–H bond [5], which means that methane conversion usually requires high temperatures to overcome the high reaction barrier. Such high temperatures are not conducive to industrial application. Although long-term efforts have been devoted to studying the “Holy Grail” of catalysis [6], i.e., the direct conversion of methane into methanol, formaldehyde, etc., the yield and selectivity of these reactions are low. Up till now, it is not able to compete with traditional petrochemical processes, making it difficult to achieve its industrialization in the short term. Therefore, over the past few decades many experimental and theoretical investigations have focused on developing catalysts to achieve high methane conversion rates under mild conditions [7,8,9]. It has been reported both experimentally and theoretically that [10,11,12,13] reducing the size of supported metal nanoparticles can enhance the catalytic efficiency. A heterogeneous catalytic process would be the preferred scheme for the low temperature methane oxidative conversion [14].
Due to the unique structure of two-dimensional (2D) materials, single atom embedded 2D materials offer an opportunity to significantly enhance catalytic activity. Sanjubala et al. [15] investigated the use of free and graphene-supported single transition metal Cr, Mn, Fe, Co and Cu atoms for the activation of methane, and found that Co atoms supported on graphene could be a very effective catalyst for methane activation. Yuan et al. [2] demonstrated a two-step reaction mechanism for the direct oxidation of methane to methanol over a single atom Co-embedded graphene catalyst by using first principle calculations, with N2O as the O-donor, and they predicted that the catalyst would be highly reactive and selective under mild conditions. Interestingly, the enzyme methane monooxygenase is capable of selectively oxidizing methane to methanol at room temperature, and the active site for this enzymatic system consists mostly of iron and copper [16,17,18,19]. Although such biocatalysts are difficult to scale-up, the nature of their active sites provides inspiration for the development of highly active catalysts. Some of the most efficient catalysts for activating methane under mild conditions are iron-exchanged zeolites, as they have active centers similar to that of methane monooxygenase [20,21]. In particular, iron-embedded graphene exhibits good catalytic activity for N2O decomposition, since the involved activation energy is only 8 kcal·mol−1 [22]. Based on this finding, Impeng et al. [7] recently showed that FeO/graphene provides excellent reactivity for the oxy-functional oxidation of methane to methanol. In addition to graphene, they also found that the methane C-H bond cleavage is favorable on the Fe/boron nitride (BN) system with a lower energy barrier of 10 kcal·mol−1 [14]. Cui et al. demonstrated that graphene-confined single Fe atoms can be used as an efficient metal catalyst to directly convert methane at room temperature, with a low rate-determining (C–H bond cleavage) reaction energy barrier of 0.79 eV [9]. That is, we can choose suitable supports to properly design catalysts with high activity. As an extension of these single atom catalysts, diatomic catalysts, which feature larger adsorption sites and modes, have been widely studied [23,24,25,26,27], and are thus conducive to improving the catalytic activity of metal atoms or providing other reaction pathways.
A novel carbon-nitrogen porous 2D C2N crystal compound, which was successfully prepared by a simple bottom-up wet chemical reaction method [28], has also attracted the interest of researchers [29,30,31]. With big pores, C2N can tightly anchor one or two metal atoms, whereby the lone pairs of electrons of the sp2 hybridized nitrogen atoms at the edge of the pores can strongly couple with the metal atoms, thereby preventing their mobility and providing a good coordination site for the metal single atom and clusters. A high surface-to-volume ratio ensures that the metal atoms can be sufficiently exposed to the substrate, which facilitates adsorption of reactant molecules and promotes catalytic reactions. For example, the diatomic catalysts Cu2@C2N and Fe1Cu1@C2N were found to possess better catalytic performance for CO oxidation than SAC Cu1@C2N does [22,24]. Compared with a single Fe atom, the Fe2 dimer provides larger adsorption sites, thus we expect that the Fe2 dimer supported on the 2D C2N monolayer (Fe2@C2N) will possesses good catalytic performance for methane conversion. A recent work by Zhao et al. found that Fe2@C2N has remarkable selectivity for C2H5OH against other C1 and C2 products on CO2 reduction [32]. In this work, we explore the potential of the Fe2@C2N as an efficient catalyst to catalyze the methane conversion reaction by means of periodic density functional theory (DFT) calculations. Our results indicate that the Fe2 dimer embedded in the C2N monolayer is a promising catalyst for methane conversion, whose activation barrier for the C-H breakage (0.66 eV) is estimated to be comparable to or lower than those of reported Fe-based catalysts (0.52 ~ 0.79 eV) [8,33,34]. This work should be helpful to provide guidelines for experimentalists to design low-cost and effective catalysts for methane conversion.
2. Results and Discussion
Methane activation on 2D C2N embedded Fe2 dimer was studied by means of DFT computations. We found that the methane first generates methyl radicals, which subsequently turn into value-added C1 oxygenated products. As a result, the methane C–H bond cleavage is both kinetically and thermodynamically favorable.
2.1. Geometric Structure and Electronic Properties of the Fe2@C2N Monolayer
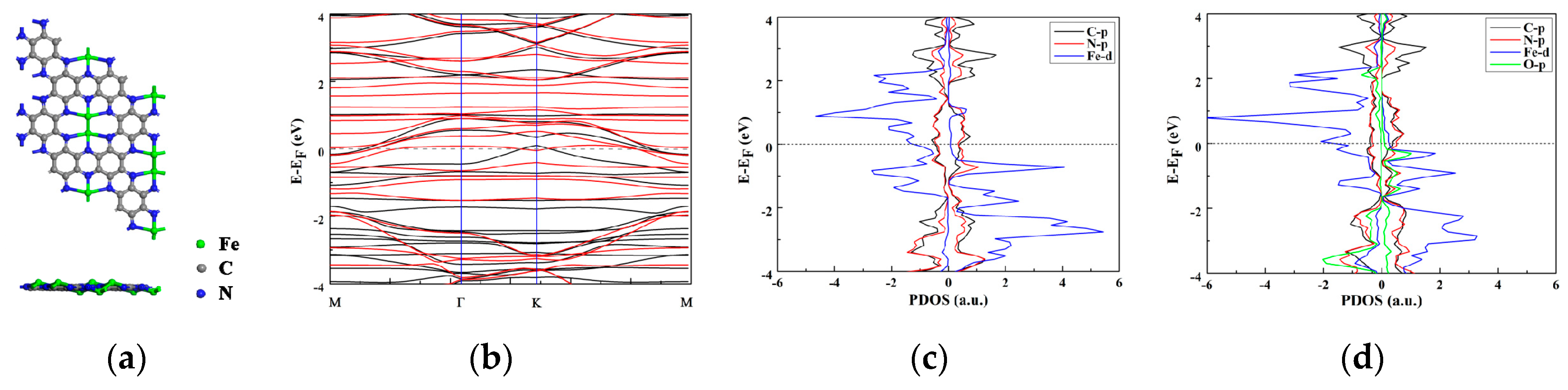
Firstly the geometric and electronic properties of Fe2 clusters on the C2N monolayer were investigated. According to a previous study [35], there are two configurations for the TM2@C2N, in which both two metal atoms coordinate with two N atoms or three N atoms of the C2N substrate, respectively. Herein, the coordination of Fe atoms with three N atoms is more stable (Figure 1a), the distances of Fe-N bonds are in the range of 1.93 ~ 2.43 Å, and the Fe2@C2N monolayer displays a slightly distorted plane, with the two Fe atoms above and below the plane. The large binding energy between the Fe atom and C2N (the binding energy of each Fe atom is 4.30 eV) implies that the Fe2 dimer could bond strongly with C2N, suggesting good stability. Previous ab initio molecular dynamics (AIMD) simulation results [35] have also shown that the Fe2@C2N structure has good thermodynamic stability. The electrical conductivity is an important indicator to evaluate the catalyst activity. Therefore, the band structures of Fe2@C2N, which exhibits metallic properties, were computed (Figure 1b). The electronic structures reflected by the partial density of states (PDOS) show that electronic coupling mainly originates from the Fe-3d and N-2p orbitals (Figure 1c). The Bader charge analysis showed that each Fe atom extracts 0.74 e from the C2N, indicating its high activity.
2.2. Methane Activation over the Fe2@C2N Monolayer
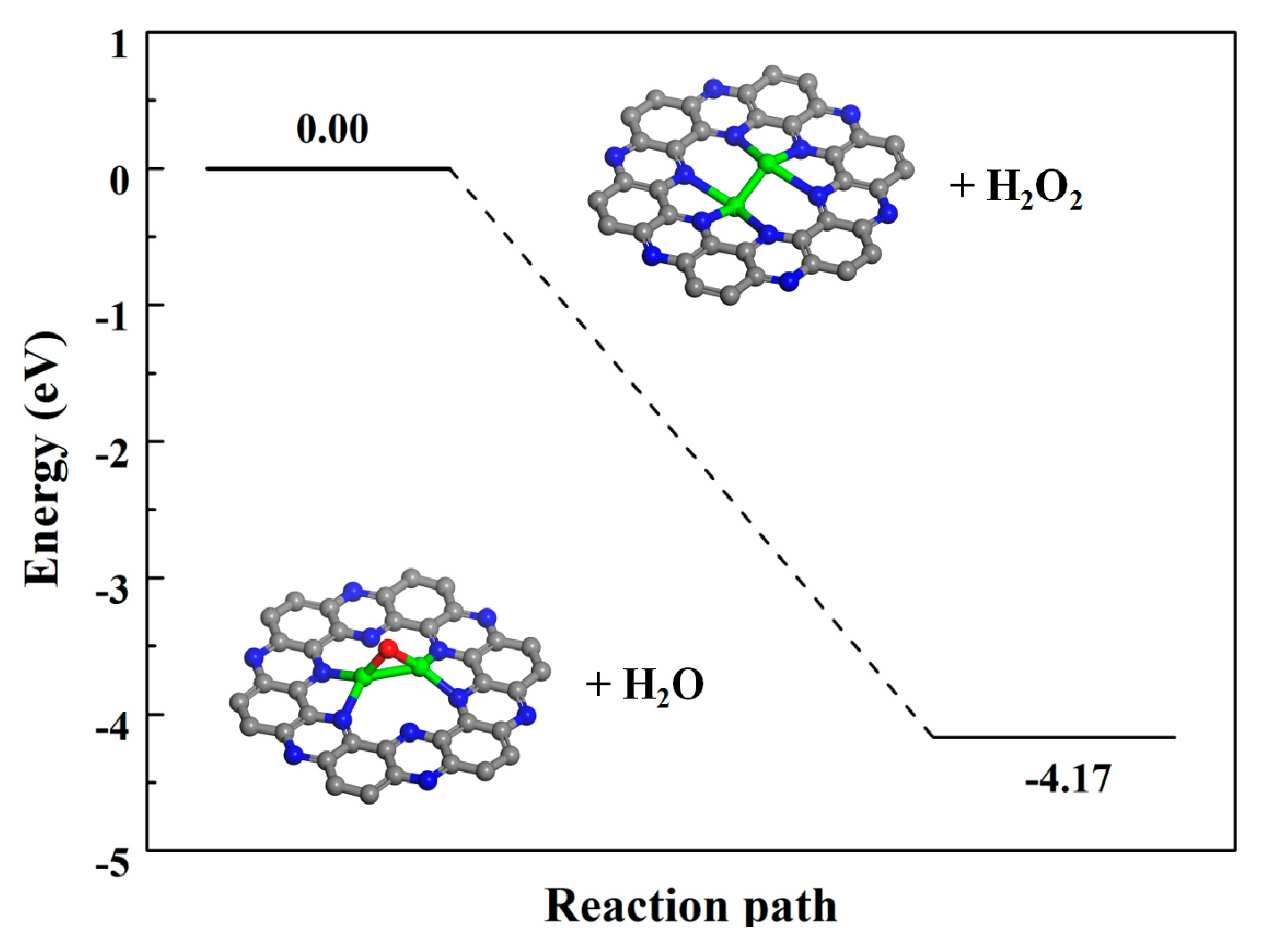
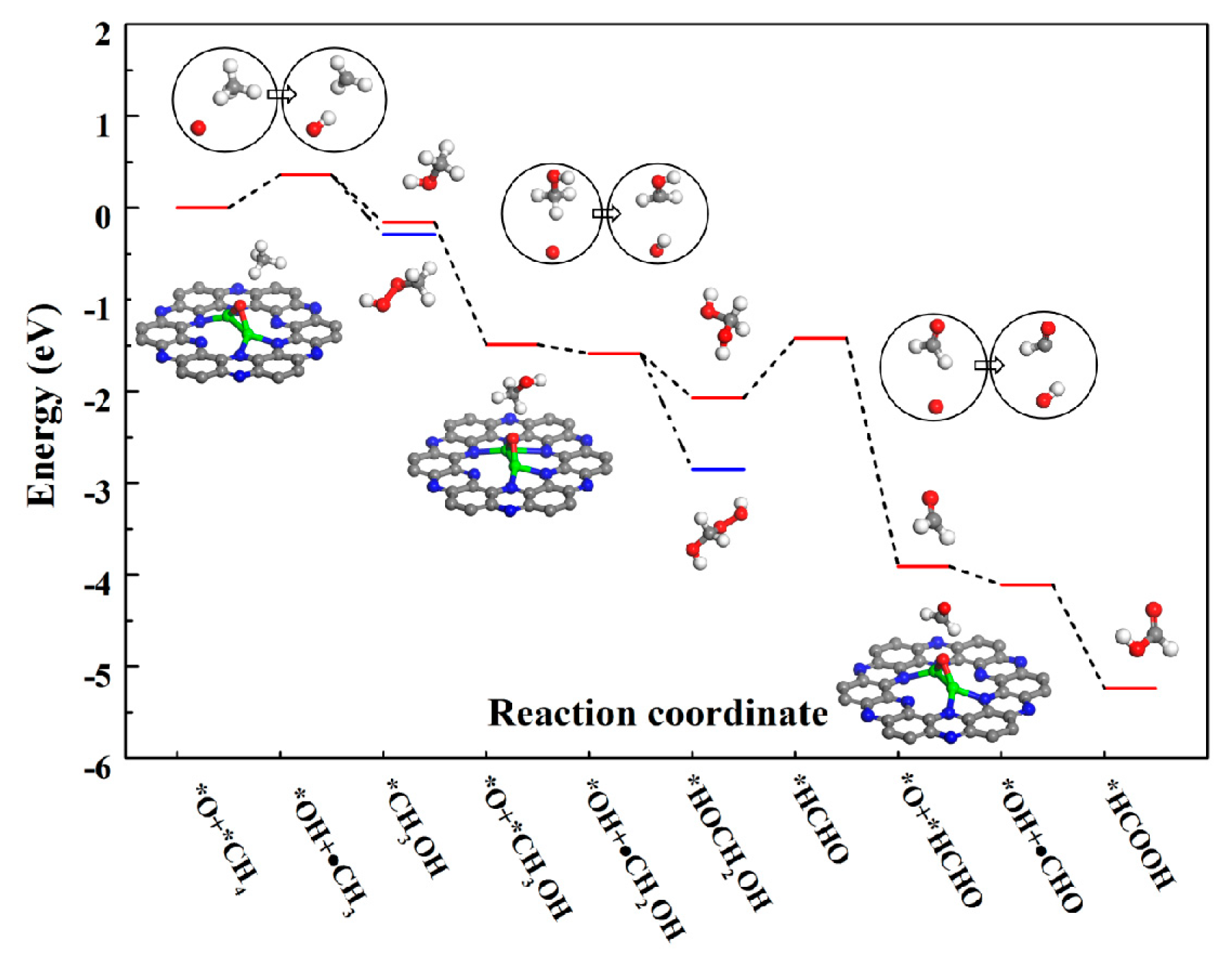
We first examined the adsorption of the oxidant H2O2 on the Fe2@C2N, and we found that the H2O2 dissociates (*H2O2 → *H2O + *O) spontaneously. The potential energy profile of H2O2 dissociation catalyzed by Fe2@C2N (Figure 2) is highly exothermic (−4.17 eV), revealing that the Fe2@C2N is an efficient catalyst for H2O2 decomposition. The H2O desorption from Fe2@C2N is very easily due to the very small adsorption energy of −0.23 eV. After the removal of the H2O molecule, the Fe-O-Fe intermediate was formed, where the two Fe-O bond lengths are both 1.80 Å, and the Fe-N bonds changed to 1.89 ~ 2.05 Å, while the C-N distances remain the same as those of the pristine Fe2@C2N. The electronic state of the Fe-O-Fe intermediate near the Fermi level is increased compared to the original Fe2@C2N (Figure 1c,d), leading to the Fe-O-Fe intermediate being more active, serving as the active site in all the subsequent oxidation reactions. The main reaction pathway of methane oxidation process as is shown in Figure 3. Below we will discuss the complete oxidation process.
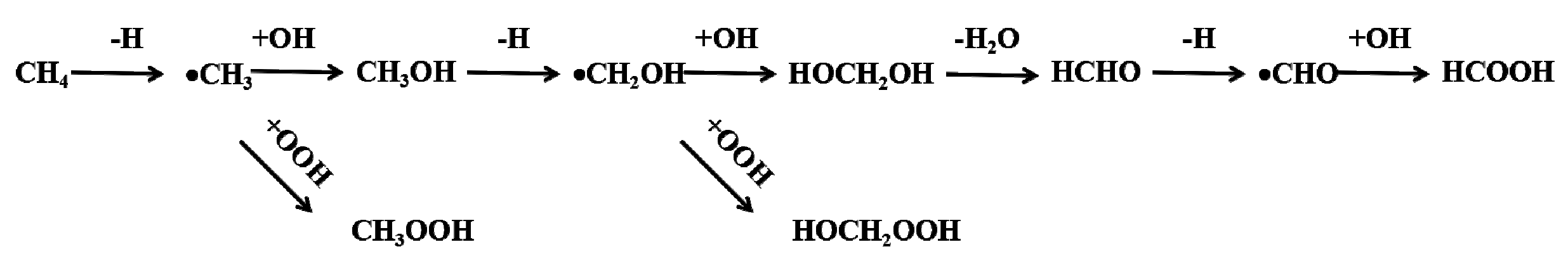
The calculated potential energy profile for methane conversion on the Fe-O-Fe active sites generated on Fe2@C2N by decomposition of H2O2 is illustrated in Figure 4 (the optimized geometries of intermediates in the reaction are given in the insets). The first elementary reaction starts with the adsorption of the methane on the Fe-O-Fe intermediate via van der Waals interactions, where one hydrogen atoms pointing towards the oxygen atom with a distance of about 2.29 Å and an adsorption energy of −0.58 eV. The C–H bond activation begins to transfer one H atom from C atom of methane to O atom coordinated to the Fe2 dimer, leading to the formation of the methyl species and the hydroxyl group, where the activated C–H bond of methane is elongated from 1.10 to 1.48 Å, and the formed O-H bond is 0.98 Å (the original distance is ~2.29 Å). This step is endothermic by 0.36 eV. After the first C-H bond of methane is broken, the Fe-O-Fe active site continues to sequentially activate other C-H bonds, generating a series of oxidation products. The formed methyl radical has two different ways to proceed: (1) It can combine with the *OH to form a C-O bond (the C-O bond length is 1.45 Å), leading to the formation of CH3OH adsorbed on the Fe2 active center, and the Fe-O distance in the CH3OH adsorption complex is 2.20 Å (the red line in Figure 4). (2) Or it can react with *OOH in the H2O2 solution to form CH3OOH, where the C-O bond length is 1.40 Å (the blue line in Figure 4). It is worth noting that whether the methyl radical forms a CH3OH or CH3OOH, both processes are exothermic, by −0.52 and −0.65 eV, respectively. The adsorption energies of *CH3OH or *CH3OOH are −0.45 and −0.18 eV, respectively. After the removal of *CH3OOH, the geometrical structure of the Fe2 dimer will recover to the original Fe2@C2N structure. Subsequently, the Fe2@C2N monolayer will react with further H2O2 in the solution to cycle back to the Fe-O-Fe active structure, and thus continue to convert CH3OH into other chemicals. The formed Fe-O-Fe intermediate will interact with the H atom bonded with the C atom in CH3OH to form *OH (the O-H bond length is 1.11 Å) and •CH2OH. Similar to the case of the methyl radical, CH2OH can also react either with *OH to form HOCH2OH (the red line in Figure 4), or with *OOH in the solution to form HOCH2OOH (the blue line in Figure 4), and the two steps are exothermic by −0.48 and −1.26 eV, respectively. The formed HOCH2OH will dehydrate to form HCHO with the reaction energy of 0.65 eV. The Fe-O-Fe intermediate will continue to extract one H in HCHO to form *OH and •CHO, a step that is exothermic by −0.20 eV. Finally, the •CHO and *OH combine to form HCOOH product, which releases an energy of −1.13 eV. As a result, the conversion of methane on the Fe2@C2N is exothermic by −5.24 eV for the overall steps, (the red lines in Figure 4), and the rate-determining step is the dehydration of HOCH2OH, which consumes the energy of 0.65 eV.
Similar to previous reports [34,36], the iron-oxo species is the most important form for methane activation. In this study the Fe-O-Fe intermediate, which is generated by the decomposition of H2O2 acts as a catalytic active center and as a source of oxygen. The interaction between Fe-O will increase the unoccupied state of Fe because of the electronegativity of O. The Fe–O bond with the length of 1.80 Å, is comparable to the reported Fe–O bond lengths in the zeolite and in the enzyme [37,38]. The calculated adsorption energy of methane (−0.58 eV) is comparable with the methane on Fe-GP (about −0.50 eV) and Fe-BN (about −0.62 eV) [14]. The most important point of the reaction pathway is the methane activation on the Fe-O-Fe species, and the reaction energy for this step is endothermic by 0.36 eV, which is much lower than the energy on traditional effective catalysts, such as CoO/Gr (0.56 eV), FeO/Gr (~0.70 eV) [2,7]. By calculating the formation energy of hydrogen, we estimate that the methane activation energy barrier is about 0.66 eV, which is comparable to that determined for Fe-exchanged zeolites (0.52 eV and 0.69 eV) [33,34]. More importantly, it is smaller than the energy barrier on O-FeN4-O in graphene (0.79 eV), which agrees with our conjecture that Fe2@C2N is a promising catalyst for CH4 conversion. Moreover, the recycling ability is major demand in developing suitable catalysts for the conversion of methane. The Fe2@C2N almost remains in the original structure throughout the reaction and the catalytically active center Fe-O-Fe can be regenerated by introduction of H2O2. From the computed free energies of the reaction pathways, we conclude that the reaction mechanism is that the methyl radical is first converted to CH3OH and CH3OOH, the CH3OH can be further converted to HOCH2OOH and HCOOH. The features of the energy diagrams are similar to those on the O-FeN4-O structure [9], and the overall reaction is exothermic by −5.24 eV. Furthermore, compared with the core steps of methane conversion in the graphene-confined single iron atom, the Fe2@C2N exhibits even higher catalytic activity: the key step of C–H cleavage is endothermic by 0.36 vs. ~0.45 eV [9]. Synder et al. [39] reported that the high O-H formation energy (ΔEO-H) will highly activate the abstraction of H-atom of methane. Therefore, we calculated ΔEO-H on the Fe-O-Fe intermediate (−4.81 eV), which is close to the value of the best represents active site [FeO]2+ reported in Fe-zeolites (−4.42 eV) [39], suggesting that the Fe-O-Fe intermediate shows good activity for the C-H activation.
3. Materials and Methods
Spin-polarized density functional theory (DFT) calculations were carried out with the Vienna ab initio simulation package (VASP) [40] using the projector augmented wave (PAW) method [41]. The generalized gradient approximation (GGA) with the Perdew-Burke-Ernzerhof (PBE) exchange-correlation functional was adopted [42]. The energy cutoff for the plane-wave basis set was chosen as 550 eV, the systemic energy tolerance and the remaining total force were set as 1 × 10−5 eV and 0.01 eV Å−1, respectively. The Brillouin zone was sampled with a 5 × 5 × 1 k-mesh grid of the Monkhorst-Pack scheme [43] for geometry optimization, and a denser k-mesh of 15 × 15 × 1 for electronic structure computations.
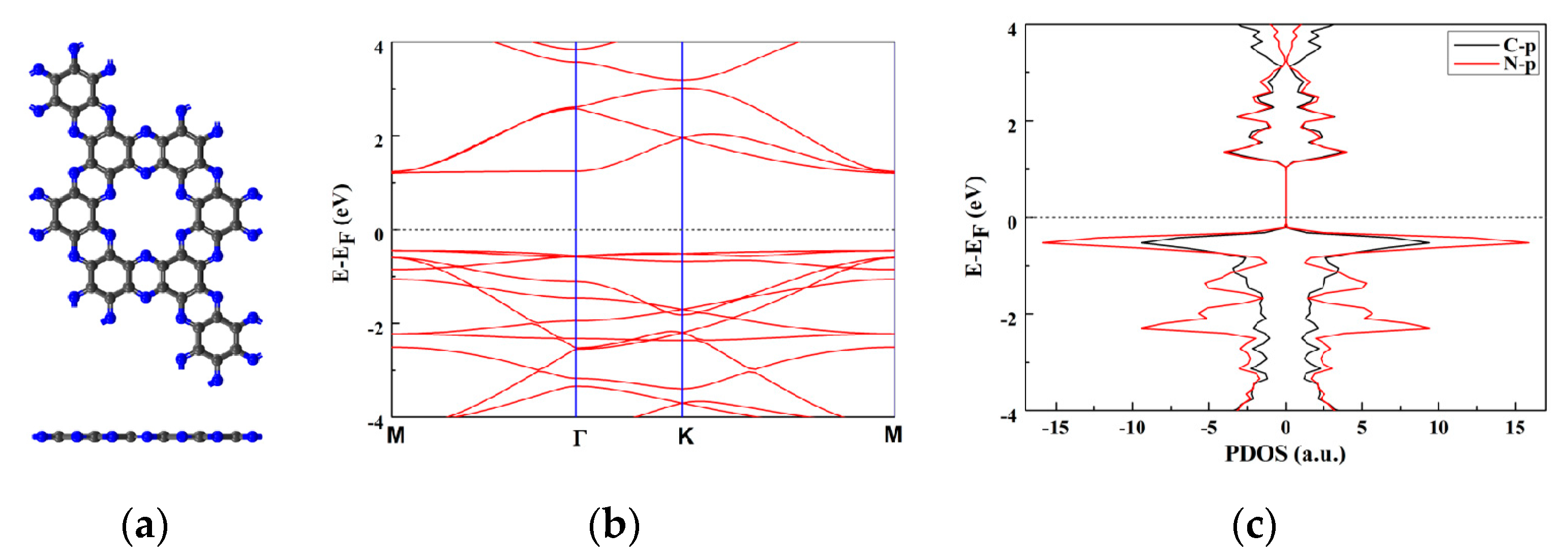
The lattice parameter of the 2D C2N monolayer was calculated to be a = b = 8.32 Å, which was consistent with the experiment reports [28]. A C2N supercell with 2 × 2 × 1 unit cells, containing 48 C atoms and 28 N atoms, was used in this work (Figure 5a). The computed energy band gap of C2N monolayer (Figure 5b,c) is 1.66 eV, which is consistent with previous reports [30]. The periodic boundary condition was set with a 20 Å vacuum region above the plane of one C2N unit cell, which was large enough to avoid interaction from the neighboring images. The reaction energy of elementary reaction was based on the expression ΔE = EFS − EIS, where EFS and EIS denote the energies of the final state and the initial state, respectively.
The binding energy (Eb) was computed to examine the structure stability:
where EFe2@C2N, EFe and EC2N represent the energy of the Fe2@C2N, the isolated Fe atom, and the clean C2N respectively. According to this definition, a more negative value of Eb means better thermodynamic stability.
Eb = EFe2@C2N − EC2N − 2EFe
The adsorption energy (Eads) of the adsorbates was computed according to the following equation:
where the Etot, EFe2@C2N and Eadsorbate represent the total energies of the system containing the Fe2@C2N catalyst and the adsorbate, the pristine Fe2@C2N, and the isolated adsorbate, respectively. According to this definition, a more negative value of Eads means better thermodynamic stability.
Eads = Etot − EFe2@C2N − Eadsorbate
The descriptor EH proposed by Latimer et al. [44,45] can accurately estimate the methane transition state (TS) energy, while avoiding the tedious transition state energy calculation, and the error of this descriptor is comparable to the typical accuracy of DFT adsorption energy. Therefore, in this work, we used this descriptor to quickly estimate the activation energy of the methane C–H bond:
where ETS and EH represent the energy of the methane TS energy and the affinity of hydrogen, respectively.
ETS = 0.75 EH + 1.09
The formation energy of O–H bond was computed according to the following equation:
where the EOH*, EH* are the total energies of O* and OH* adsorbed on the pristine Fe2@C2N, respectively. The calculation of H2 was to place it in a 15 × 15 × 15 cubic cell, using Gamma point only.
ΔEO-H = EOH* − EH* − 0.5 EH2
4. Conclusions
In this work, the direct conversion of methane over a Fe2@C2N monolayer, with H2O2 as the oxidant, was investigated by means of density functional theory calculations. Our calculations revealed that the Fe2@C2N is a highly efficient catalyst for methane activation, and the high activity is mainly due to the formation of Fe-O-Fe intermediates. Because of a strong formation energy of the O-H bond on the Fe-O-Fe intermediate, methane activation is driven to proceed with a low energy barrier. Based on the proposal of synthesizing Cu2@C2N [23], it is feasible to obtain Fe2@C2N by using FeCl2 and other proper metal precursors. Hopefully, our theoretical study may pave the way toward the design of highly efficient C2N-supported non-precious catalysts for the conversion of methane to high value-added chemicals. One should note that the support, coordination and electronic effects impact the activity and selectivity of the supported dimer catalysts [27], so other metal dimers may also have good catalytic performance for methane conversion [46].
Author Contributions
F.L. and J.Z. outline the work plan; H.M. and B.H. conducted the computations; H.M. draw the figures and drafted the manuscript. All authors participated in the reviewing and publication processes of the article. All authors have read and agreed to publish this version of the manuscript.
Funding
This work was funded by the National Natural Science Foundation of China (11704203), the Startup Project of Inner Mongolia University (21200-5175101), and the Natural Science Funds for Distinguished Young Scholar of Heilongjiang Province (JC2018004).
Acknowledgments
The authors acknowledge the support of this study by the National Natural Science Foundation of China, under Grant number (11704203), the Startup Project of Inner Mongolia University (21200-5175101), and the Natural Science Funds for Distinguished Young Scholar of Heilongjiang Province, under Grant number (JC2018004). Additionally, computational support from PARATEAR is appreciated.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Zhao, Y.; Li, S.; Sun, Y. Theoretical study on the dissociative adsorption of CH4 on Pd-doped Ni surface. Chin. J. Catal. 2013, 34, 911–922. [Google Scholar] [CrossRef]
- Yuan, J.; Zhang, W.; Li, X.; Yang, J. A high performance catalysts for methane conversion to methanol: Grapheme supported single atom Co. Chem. Commun. 2018, 54, 2284–2287. [Google Scholar] [CrossRef] [PubMed]
- Sushkevich, V.; Palagin, D.; Ranocchiari, M.; Van Bokhoven, J. Selective anaerobic oxidation of methane enables direct synthesis of methanol. Science 2017, 356, 523–527. [Google Scholar] [CrossRef] [PubMed]
- Schwach, P.; Pan, X.; Bao, X. Direct conversion of methane to value-added chemicals over heterogeneous catalysts: Challenges and prospects. Chem. Rev. 2017, 117, 8497–8520. [Google Scholar] [CrossRef]
- Narsimhan, K.; Iyoki, K.; Dinh, K.; Roman-Leshkov, Y. Catalytic oxidation of methane into methanol over copper exchanged zeolites with oxygen at low temperature. ACS Cent. Sci. 2016, 2, 424–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Research Grant Targeting “Holy Grail” of Catalysis. Available online: https://www.egr.uh.edu/news/201401/research-grant-targeting-holy-grail-catalysis (accessed on 13 January 2014).
- Impeng, S.; Khongpracha, P.; Warakulwit, C.; Jansang, B.; Sirijaraensre, J.; Masahiro Ehara, M.; Limtrakul, J. Direct oxidation of methane to methanol on Fe-O modified graphene. RSC Adv. 2014, 4, 12572–12578. [Google Scholar] [CrossRef]
- Tang, P.; Zhu, Q.; Wu, Z.; Ma, D. Methane activation: The past and future. Energy Environ. Sci. 2014, 7, 2580–2591. [Google Scholar] [CrossRef]
- Cui, X.; Li, H.; Wang, Y.; Hu, Y.; Hua, L.; Li, H.; Han, X.; Liu, Q.; Yang, F.; He, L.; et al. Room-temperature methane conversion by graphene-confined single iron atoms. Chem 2018, 4, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Bayatsarmadi, B.; Zheng, Y.; Vasileff, A.; Qiao, S. Recent advances in atomic metal doping of carbon-based nanomaterials for energy conversion. Small 2017, 13, 1700191. [Google Scholar] [CrossRef]
- Luo, Z.; Castleman, A.; Khanna, S. Reacitivy of metal clusters. Chem. Rev. 2016, 116, 14456–14492. [Google Scholar] [CrossRef]
- Tyo, E.; Vajda, S. Catalysis by clusters with precise numbers of atoms. Nat. Nanotechnol. 2015, 10, 577–588. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Wang, A.; Qiao, B.; Li, J.; Liu, J.; Zhang, T. Single-atom catalysts: A new frontier in heterogeneous catalysis. Acc. Chem. Res. 2013, 46, 1740–1748. [Google Scholar] [CrossRef] [PubMed]
- Impeng, S.; Khongpracha, P.; Sirijaraensre, J.; Jansang, A.; Ehara, M.; Limtrakul, J. Methane activation on Fe- and FeO-embedded graphene and boron nitride sheet: Role of atomic defects in catalytic activities. RSC Adv. 2015, 5, 97918–97927. [Google Scholar] [CrossRef]
- Sahoo, S.; Suib, S.; Pamir Alpay, S. Graphene supported single atom transition metal catalysts for methane activation. ChemCatChem 2018, 10, 3229–3235. [Google Scholar] [CrossRef]
- Solomon, E.; Heppner, D.; Johnston, E.; Ginsbach, J.; Cirera, J.; Qayyum, M.; Kieber-Emmons, M.; Kjaergaard, C.; Hadt, R.; Li, T. Copper active sites in biology. Chem. Rev. 2014, 114, 3659–3853. [Google Scholar] [CrossRef] [Green Version]
- Lieberman, R.; Rosenzweig, A. Crystal structure of a membrane-bound metalloenzyme that catalyses the biological oxidation of methane. Nature 2005, 434, 177–182. [Google Scholar] [CrossRef]
- Balasubramanian, R.; Smith, S.; Rawat, S.; Yatsunyk, L.; Stemmler, T.; Rosenzweig, A. Oxidation of methane by a biological dicopper centre. Nature 2010, 465, 115–119. [Google Scholar] [CrossRef] [Green Version]
- Panov, G.; Sobolev, V.; Dubkov, K.; Parmon, V.; Ovanesyan, N.; Shilov, A.; Shteinman, A. Iron complexes in zeolites as a new model of methane monooxygenase. React. Kinet. Catal. Lett. 1997, 61, 251–258. [Google Scholar] [CrossRef]
- Ovanesyan, N.; Shteinman, A.; Dubkov, K.; Sobolev, V.; Panov, G. The state of iron in the Fe-ZSM-5-N2O system for selective oxidation of methane to methanol from data of Mössbauer spectroscopy. Kinet. Catal. 1998, 39, 792–797. [Google Scholar]
- Narsimhan, K.; Michaelis, V.; Mathies, G.; Gunther, W.; Griffin, R.; Román-Leshkov, Y. Methane to acetic acid over Cu-exchanged zeolites: Mechanistic insights from a site-specific carbonylation reaction. J. Am. Chem. Soc. 2015, 137, 1825–1832. [Google Scholar] [CrossRef] [Green Version]
- Wannakao, S.; Nongnual, T.; Khongpracha, P.; Maihom, T.; Limtrakul, J. Reaction mechanisms for CO catalytic oxidation by N2O on Fe-embedded graphene. J. Phys. Chem. C 2012, 116, 16992–16998. [Google Scholar] [CrossRef]
- Li, F.; Chen, Z. Cu dimer anchored C2N Monolayer: Low-cost and efficient catalyst for CO oxidation. Nanoscale 2018, 10, 15696–15705. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Zhao, J.; Li, F.; Chen, Z. Copper dimer supported on C2N-layer as an efficient electrocatalyst for CO2 reduction reaction: A computational study. J. Phys. Chem. C 2018, 122, 19712–19721. [Google Scholar] [CrossRef]
- Li, F.; Liu, X.; Chen, Z. 1+1ʹ > 2: Heteronuclear bi-atom catalyst outperforms its homonuclear counterparts for CO oxidation. Small Methods 2019, 3, 1800480. [Google Scholar] [CrossRef]
- Chen, Z.; Chen, L.; Yang, C.; Jiang, Q. Atomic (single, double, and triple atoms) catalysis: Frontiers, opportunities, and challenges. J. Mater. Chem. A 2019, 7, 3492–3515. [Google Scholar] [CrossRef]
- Pan, Y.; Zhang, C.; Liu, Z.; Chen, C.; Li, Y. Structural regulation with atomic-level precision: From single-atomic site to diatomic and atomic interface catalysis. Matter 2020, 2, 78–110. [Google Scholar] [CrossRef]
- Mahmood, J.; Lee, E.; Jung, M.; Shin, D.; Jeon, I.; Jung, S.; Choi, H.; Seo, J.; Bae, S.; Sohn, S.; et al. Nitrogenated holey two-dimensional structure. Nat. Commun. 2015, 6, 6486–6492. [Google Scholar] [CrossRef] [Green Version]
- Su, Y.; Li, W.; Li, G.; Ao, Z.; An, T. Density functional theory investigation of the enhanced adsorption mechanism and potential catalytic activity for formaldehyde degradation on Al-decorated C2N monolayer. Chin. J. Catal. 2019, 40, 664–673. [Google Scholar] [CrossRef]
- He, B.; Shen, J.; Tian, Z. Iron-embedded C2N monolayer: A promising low-cost and high-activity single-atom catalyst for CO oxidation. Phys. Chem. Chem. Phys. 2016, 18, 24261–24269. [Google Scholar] [CrossRef]
- Li, X.; Zhong, W.; Cui, P.; Li, J.; Jiang, J. Design of efficient catalysts with double transition metal atoms on C2N layer. J. Phys. Chem. Lett. 2016, 7, 1750–1755. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhou, S.; Zhao, J. Selective C-C coupling by spatially confined dimeric metal centers. iScience 2020, 23, 101051. [Google Scholar] [CrossRef] [PubMed]
- Hammond, C.; Forde, M.; Rahim, M.; Thetford, A.; He, Q.; Jenkins, R.; Dimitratos, N.; Lopez-Sanchez, J.; Dummer, N.; Murphy, D.; et al. Direct catalytic conversion of methane to methanol in an aqueous medium by using copper-promoted Fe-ZSM-5. Angew. Chem. Int. Ed. 2012, 51, 5129–5133. [Google Scholar] [CrossRef] [PubMed]
- Yoshizawa, K.; Shiota, Y.; Yumura, T.; Yamabe, T. Direct methane-methanol and benzene-phenol conversions on Fe-ZSM-5 zeolite: Theoretical predictions on the reaction pathways and energetics. J. Phys. Chem. B 2000, 104, 734–740. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, A.; Zhang, Z.; Jiao, M.; Zhou, Z. Transition metal anchored C2N monolayers as efficient bifunctional electrocatalysts for hydrogen and oxygen evolution reactions. J. Mater. Chem. A 2018, 6, 11446–11452. [Google Scholar] [CrossRef]
- Pantu, P.; Pabchanda, S.; Limtrakul, J. Theoretical investigation of the selective oxidation of methane to methanol on nanostructured Fe-ZSM-5 by the ONIOM method. ChemPhysChem 2004, 5, 1901–1906. [Google Scholar] [CrossRef] [PubMed]
- Sirijaraensre, J.; Limtrakul, J. Structures and mechanisms of the dehydration of benzaldoxime over Fe-ZSM-5 zeolites: A DFT study. Struct. Chem. 2013, 24, 1307–1318. [Google Scholar] [CrossRef]
- Schlichting, I.; Berendzen, J.; Chu, K.; Stock, A.; Maves, S.; Benson, D.; Sweet, R.; Ringe, D.; Petsko, G.; Sligar, S. The catalytic pathway of cytochrome P450cam at atomic resolution. Science 2000, 287, 1615–1622. [Google Scholar] [CrossRef]
- Snydera, B.; Böttgera, L.; Bolsb, M.; Yana, J.; Rhodaa, H.; Jacobsa, A.; Huc, M.; Zhao, J.; Alpc, E.; Hedmand, B.; et al. Structural characterization of a non-heme iron active site in zeolites that hydroxylates methane. Proc. Natl. Acad. Sci. USA 2018, 115, 4565–4570. [Google Scholar] [CrossRef] [Green Version]
- Kresse, G.; Furthmüller, F. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef]
- Blöch, P. Projector augmented-wave method. Phy. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [Green Version]
- Perdew, J.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monkhorst, H.; Pack, J. Special points for brillouin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Latimer, A.; Aljama, H.; Kakekhani, A.; Yoo, J.; Kulkarni, A.; Tsai, C.; Melchor, M.; Abild-Pedersen, F.; Nørskov, J. Mechanistic insights into heterogeneous methane activation. Phys. Chem. Chem. Phys. 2017, 19, 3575–3581. [Google Scholar] [CrossRef] [PubMed]
- Latimer, A.; Kulkarni, A.; Aljama, H.; Montoya, J.; Yoo, J.; Tsai, C.; Abild-Pedersen, F.; Studt, F.; Nørskov, J. Understanding trends in C-H bond activation in heterogeneous catalysis. Nat. Mater. 2017, 16, 225–229. [Google Scholar] [CrossRef]
- Arvidsson, A.A.; Zhdanov, V.P.; Carlsson, P.-A.; Grönbecka, H.; Hellman, A. Metal dimer sites in ZSM-5 zeolite for methane-to-methanol conversion from first-principles kinetic modelling: Is the [Cu–O–Cu]2+ motif relevant for Ni, Co, Fe, Ag, and Au? Catal. Sci. Technol. 2017, 7, 1470–1477. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
(a) Top and side view of the structure of Fe2@C2N in a 2 × 2 × 1 supercell; (b) Band structure; (c,d) Projected density of state (PDOS) of the Fe2@C2N and Fe-O-Fe@C2N intermediate. The Fermi level is set to zero.
Figure 1.
(a) Top and side view of the structure of Fe2@C2N in a 2 × 2 × 1 supercell; (b) Band structure; (c,d) Projected density of state (PDOS) of the Fe2@C2N and Fe-O-Fe@C2N intermediate. The Fermi level is set to zero.

Figure 2.
Transformation process of a Fe2 dimer in H2O2 solution. The inset was the atomic structure model of each step.
Figure 2.
Transformation process of a Fe2 dimer in H2O2 solution. The inset was the atomic structure model of each step.

Figure 3.
The possible reaction pathways for methane oxidation processes.

Figure 4.
Potential energy diagram for methane oxidation on Fe-O-Fe active sites. The gray, blue, green, red and while balls represent C, N, Fe, O and H atoms.
Figure 4.
Potential energy diagram for methane oxidation on Fe-O-Fe active sites. The gray, blue, green, red and while balls represent C, N, Fe, O and H atoms.

Figure 5.
Top and side view of the structure of C2N in a 2 × 2 supercell (a) and band structure (b) and projected density of state (PDOS) (c) of the C2N, the Fermi level is set to zero.
Figure 5.
Top and side view of the structure of C2N in a 2 × 2 supercell (a) and band structure (b) and projected density of state (PDOS) (c) of the C2N, the Fermi level is set to zero.

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Meng, H.; Han, B.; Li, F.; Zhao, J. Methane Conversion over C2N-Supported Fe2 Dimers. Catalysts 2020, 10, 973. https://0-doi-org.brum.beds.ac.uk/10.3390/catal10090973
AMA Style
Meng H, Han B, Li F, Zhao J. Methane Conversion over C2N-Supported Fe2 Dimers. Catalysts. 2020; 10(9):973. https://0-doi-org.brum.beds.ac.uk/10.3390/catal10090973
Chicago/Turabian StyleMeng, Haihong, Bing Han, Fengyu Li, and Jingxiang Zhao. 2020. "Methane Conversion over C2N-Supported Fe2 Dimers" Catalysts 10, no. 9: 973. https://0-doi-org.brum.beds.ac.uk/10.3390/catal10090973
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.