Fe3 Cluster Anchored on the C2N Monolayer for Efficient Electrochemical Nitrogen Fixation

1
Physical School of Science and Technology, Inner Mongolia University, Hohhot 010021, China
2
College of Chemistry and Chemical Engineering, Key Laboratory of Photonic and Electronic Bandgap Materials, Ministry of Education, Harbin Normal University, Harbin 150025, China
*
Authors to whom correspondence should be addressed.
Catalysts 2020, 10(9), 974; https://0-doi-org.brum.beds.ac.uk/10.3390/catal10090974
Submission received: 27 July 2020
/
Revised: 18 August 2020
/
Accepted: 22 August 2020
/
Published: 29 August 2020
(This article belongs to the Special Issue Transition Metal Catalysis)
Abstract
:Under the current double challenge of energy and the environment, an effective nitrogen reduction reaction (NRR) has become a very urgent need. However, the largest production of ammonia gas today is carried out by the Haber–Bosch process, which has many disadvantages, among which energy consumption and air pollution are typical. As the best alternative procedure, electrochemistry has received extensive attention. In this paper, a catalyst loaded with Fe3 clusters on the two-dimensional material C2N (Fe3@C2N) is proposed to achieve effective electrochemical NRR, and our first-principles calculations reveal that the stable Fe3@C2N exhibits excellent catalytic performance for electrochemical nitrogen fixation with a limiting potential of 0.57 eV, while also suppressing the major competing hydrogen evolution reaction. Our findings will open a new door for the development of non-precious single-cluster catalysts for effective nitrogen reduction reactions.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Ammonia (NH3) is an important chemical in agriculture and industry [1], and the direct reduction of nitrogen to ammonia is still considered to be one of the most important and challenging chemical transformations [2]. Dinitrogen (N2), as the component with the largest volume fraction (78.08%) in the Earth’s atmosphere, is the main source of nitrogen [3]. Therefore, ammonia can be directly synthesized from the Earth’s abundant nitrogen resources. Usually, the Haber–Bosch process (N2 + 3H2 = 2NH3) is used to synthesize ammonia on large scales. In this process, iron and ruthenium-based metal catalysts are used to convert atmospheric nitrogen (N2) into NH3 by reacting with hydrogen (H2) [4,5]; the adsorbed N2 molecule is firstly dissociated on specific active sites of the catalysts [6,7,8], and then the dissociated N species are hydrogenated, which is known as the dissociation mechanism [9]. However, extreme reaction conditions are usually required, such as high pressure (~100 bar) and high temperature (~700 K) [10]; as the reaction proceeds, a large amount of carbon dioxide is emitted, and the energy consumed each year is huge. On the other hand, the conditions of high temperature and pressure may not be necessary because the reaction is actually exothermic [10]. For this reason, it is of significance to search for a green and cost-effective method for the production of ammonia [11].
With continuous efforts, scientists have found that the electrochemical nitrogen reduction reaction (NRR, N2 + 6H+ + 6e− = 2NH3) [12] under environmental conditions is extremely attractive because of its greatly reduced energy input and good environmental compatibility. Here, the electrocatalyst is the core component for reducing the reaction limit potential and increasing the reaction speed and the selectivity of NH3 [13]. For NRR, one of the key challenges is the activation of nitrogen, which is due to the highly stable N≡N bond [14]. The capability of N2 adsorption strongly depends on the electronic structure of the elements in the catalysts, since only the elements possess unoccupied d orbitals; d orbitals with an appropriate symmetry can accept the electrons of N2, and the occupied d orbitals of these elements can back donate to the π orbitals of N2, thus weakening the N≡N bond [15]. Suitable and active catalysts can break the N≡N bond, and hence, the single nitrogen atoms react with hydrogen to form ammonia. In recent years, not only single-atom catalysts (SACs) [12,13,14,15,16,17,18,19,20,21] have been emerging as a novel strategy for designing effective electrocatalysts for NRR: the catalytic performance of double atomic catalysts (DACs) [21,22,23,24] and triple atomic catalysts (TACs) [25,26] for NRR has also been explored both experimentally and theoretically.
Many transition-metal (TM) species, such as Fe, Ru, Co and Mo-based complexes, have been used for nitrogen fixation, because the occupied d orbitals of these metals can donate electrons to the empty π*-orbital of N2 and accept electrons from its σ-orbital, thereby enhancing the adsorption of N2 [23]. Among the transition metals, Mo and Fe were considered to be the most promising metal species because, based on density functional theory (DFT) calculations, Mo and Fe were located at the top of the volcano map [27], and Fe has received the most attention because of its robustness and low cost [16]. For example, in 2018, Li’s group proposed that Fe3@Al2O3 as a catalyst for NRR has superior catalytic performance [25], and quite recently, Jiang and co-workers’ DFT studies showed that the Fe3 cluster loaded on a heterostructure of graphdiyne and graphene (GDY/Gra) also had excellent NRR catalytic activity [28]. Thus, we conceived that the Fe3 clusters should have capable catalytic performance when supported on a suitable substrate, such as the C2N monolayer. The C2N monolayer was first fabricated in 2015 [22] and has a unique porous structure originating from a 2D graphene layer, which provides an ideal support for metal atoms as the active center and has a high surface-to-volume ratio property that ensures sufficient exposure of TM atoms to interact with reactant molecules [29]. Thus, we explored the stability of Fe3 clusters anchored on the C2N monolayer (Fe3@C2N) and the electrocatalysis of NRR by means of density functional theory (DFT) computations. Our calculations show that Fe3@C2N is a stable metal and has excellent catalytic performance for the N2 reduction reaction with the supported Fe3 cluster serving as the active center.
2. Results and Discussion
2.1. The Geometry, Stability and Electronic Properties of the Fe3@C2N
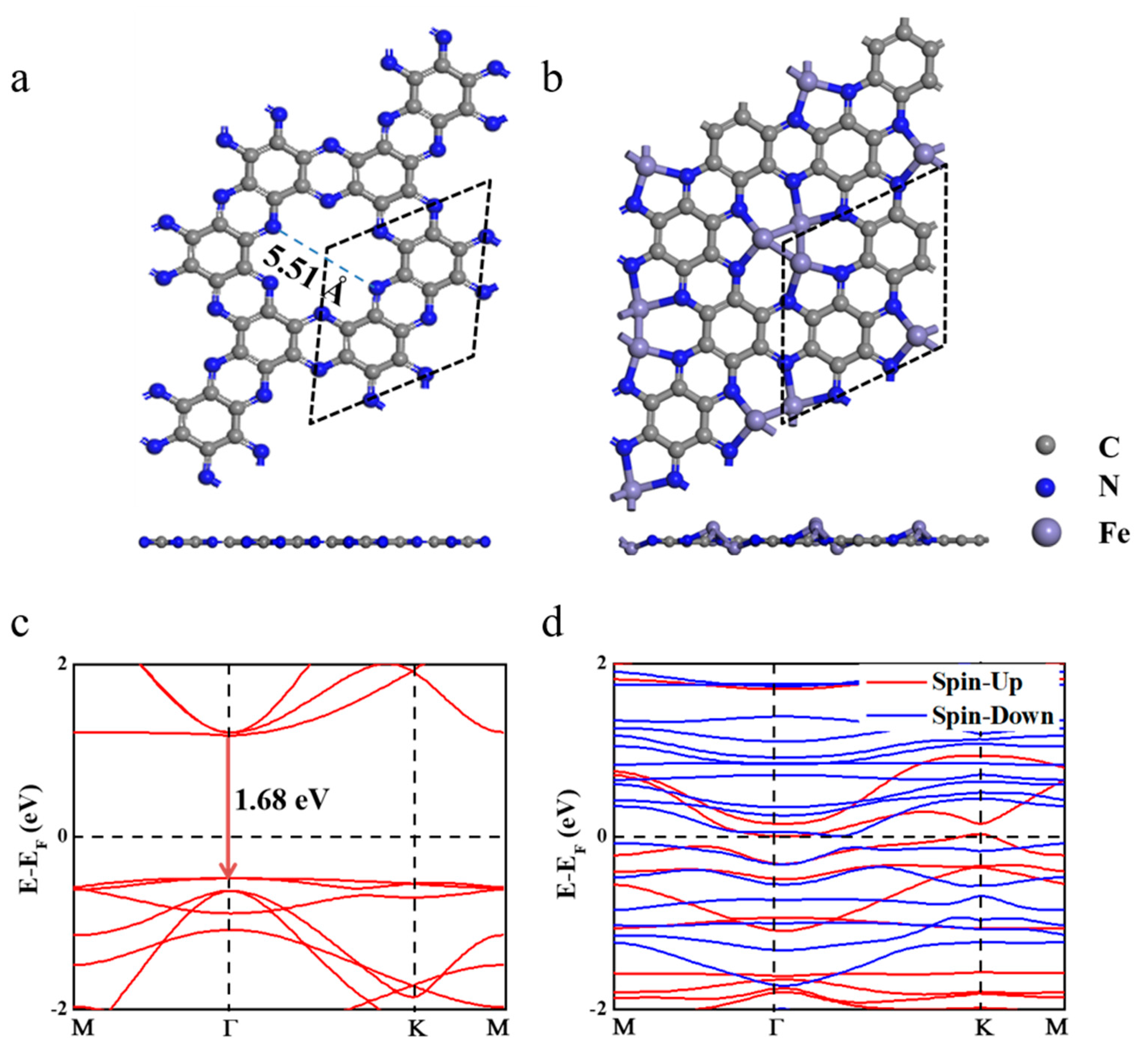
Two-dimensional (2D) C2N is a porous material similar to graphene, and the hexagonal lattice parameter of the C2N unit cell was calculated to be 8.32 Å, which matches well with previous studies [29]. The optimized geometric structure of C2N is given in Figure 1a. In the C2N monolayer, the C6 rings are connected by N atoms, resulting in a six-membered nitrogen pore with a para-N distance of 5.51 Å; the pores provide desired sites to anchor metal atoms or clusters. Previous theoretical studies [30,31] showed that adding the second Fe atom to Fe1@C2N is energetically favored: the binding energy of the second Fe atom is comparable to that of the first one, and Fe2@C2N maintains its original structure throughout a 10 ps molecular dynamics simulation at 800 K [30]. Based on the porous structural feature in the C2N monolayer and the stable Fe2@C2N complex [30,31], we conjectured that adding one more Fe atom to Fe2@C2N, i.e., Fe3@C2N, may also have good thermodynamic and thermal stability. According to our computations, the lowest-energy supported Fe3 cluster adopts a buckled triangle structure, with each Fe bonded to two N atoms, and the buckling height of the Fe3 structure is 1.29 Å, while the C2N support almost maintains its flat configuration (Figure 1b), similar to the model proposed by Pei et al. [32]. The Fe−Fe and Fe−N bond lengths are in the range of 2.28~2.32 Å (slightly shorter than the 2.48 Å in the bulk phase) and 1.97~2.01 Å, respectively. The average binding energy per Fe atom is −4.12 eV, which is slightly lower than the value of −4.84 eV per Fe atom in the bulk phase at the same level of theory, indicating the good thermodynamic stability of Fe3@C2N. Furthermore, a first-principles molecular dynamics (FPMD) simulation in an NVT ensemble with the temperature controlled by the Nosé–Hoover method [33] was performed, and the structure of Fe3@C2N was well kept through a 10 ps FPMD with a time step of 0.5 fs at 800 K (Figure S1). Thus, Fe3@C2N is extraordinarily stable.
Good electrical conductivity is required for fast charge transfer during efficient electrocatalytic processes. Compared to the semiconducting C2N monolayer, whose band gap was calculated to be 1.68 eV (Figure 1c), in line with a previous report [29], Fe3@C2N is a ferromagnetic (each Fe atom carries the magnetic moment of ~3 μB, Figure S2) metal (Figure 1d), and the metallic feature originates from the states that have crossed the Fermi level, which is dominatingly attributed to the Fe-d orbitals (Figure S3). The charge transfer between the Fe3 cluster and C2N is 2.12 |e|, indicating the high activity of the Fe3 cluster.
2.2. N2 Adsorption on Fe3@C2N
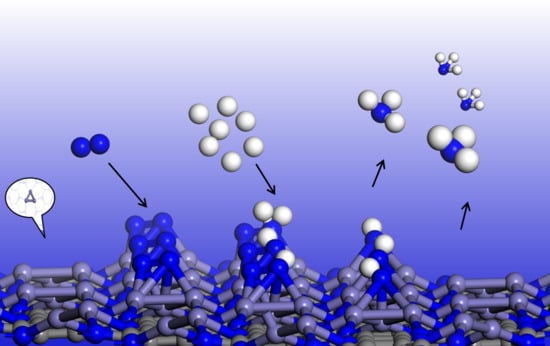
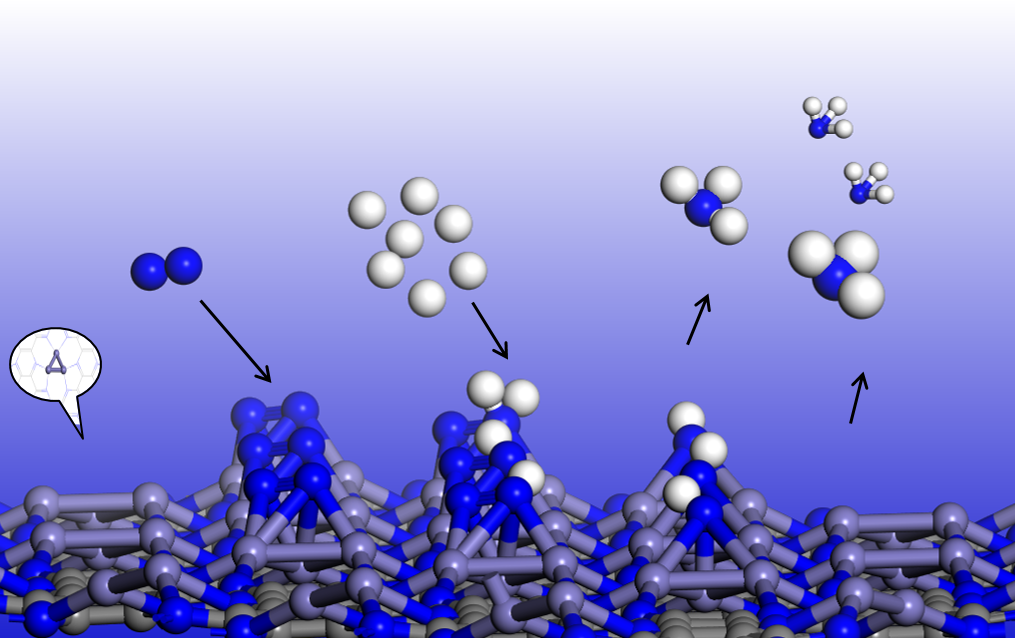
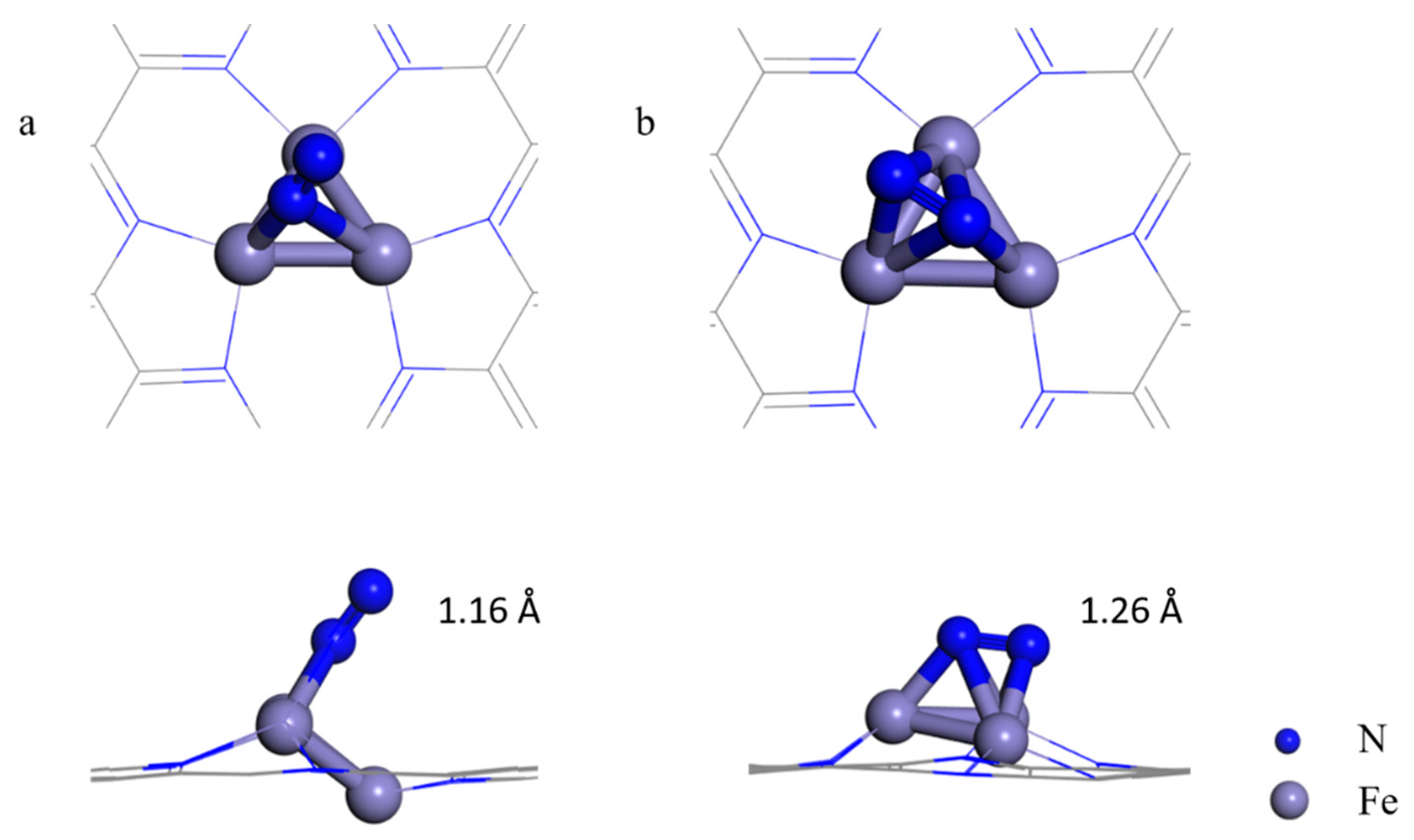
Previous investigations proposed the following criteria for an efficient electrocatalyst for NRR: (1) the catalyst can facilitate the chemisorption of N2 molecules to sufficiently activate the inert N≡N triple bonds, and (2) the catalyst can selectively stabilize N2H* and (3) destabilize NH2* species to lower the limiting potential [12]. In the electrochemical synthesis of ammonia, the adsorption of nitrogen is considered to be the first step, and the initial nitrogen adsorption configuration plays an important role in subsequent reactions [9]. On pristine C2N, the adsorption of N2 is very weak, and the adsorption energy (Eads) is only −0.01 eV, which indicates that the pristine C2N monolayer cannot effectively activate nitrogen. Zhao’s theoretical group also found that N2 adsorption on C and N atoms is rather weak (Eads < −0.20 eV), or the N2 molecule is spontaneously trapped by the central TM atoms after structural optimization on TM@C2N [34]. In our work, we considered two adsorption configurations, namely, end-on and side-on structures of N2 on the Fe3 cluster anchored on C2N (Figure 2), and the adsorption energies of the two configurations are −1.08 and −1.45 eV, respectively. The Eads values are sufficiently strong to capture and activate N2, as indicated by the elongated N≡N lengths of 1.16 and 1.26 Å (the isolated N≡N length was calculated to be 1.11 Å) and a charge transfer of −0.57 and −1.14 |e| for the end-on and side-on configurations, respectively. Compared to the maximum vibrational frequency of free N2 (2420 cm−1), the maximum vibrational mode of the adsorbed N-N in the side-on (end-on) configuration is 1380 (2011) cm−1. The remarkably reduced frequency in the side-on structure suggests an apparent elongation/weakening in the N-N bond upon N2 being adsorbed on Fe3@C2N.
2.3. N2 Reaction on Fe3@C2N
Considering the energetically preferred side-on configuration and the greater charge transfer between N2 and Fe3@C2N, as well as the much longer stretched N≡N length, we selected the N2 adsorption with the side-on structure as the starting state for NRR.
Previous studies revealed that the N2 molecule could dissociate directly on the specific iron surface, such as Fe(111) and Fe(211) surfaces [35,36]; we first examined the dissociative mechanism. However, our computations showed that the dissociated NN state on Fe3@C2N is not energetically favored, since it is 2.67 eV higher in energy than the N2 adsorbed state (Figure S4), suggesting a very high activation energy barrier (>2.67 eV) of N2 dissociation. For comparison, the calculated barrier of N2 dissociation is as high as 1.89 eV on Fe3/θ-Al2O3(010) [25]. Thus, the dissociative mechanism of NRR over Fe3@C2N was not further studied in our work. Quite recently, Wang’s theoretical group proposed a new mechanism for NRR, namely, a surface-hydrogenation mechanism [37], where the surface hydrogenation can drive the N2 reduction reaction. We also tested the surface-hydrogenation mechanism of NRR on Fe3@C2N; nevertheless, N2 adsorption on the hydrogenated Fe3 cluster is endothermic at 2.41 eV (Figure S5), indicating an unfavorable pathway for NRR. Therefore, we focused on the associative mechanism in the following experiment.
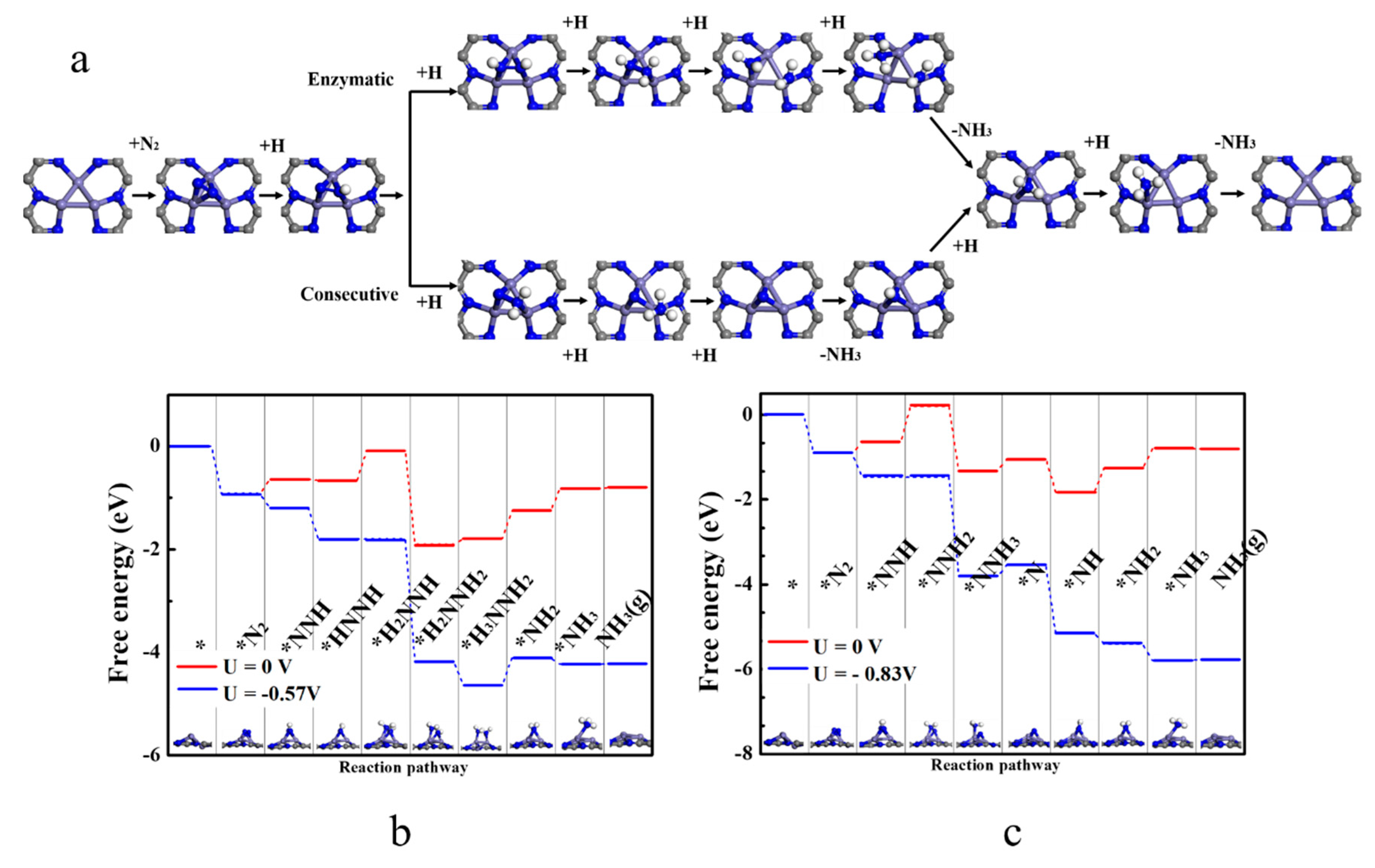
For the associative mechanism, we compared the enzymatic (* → *N2 → *NNH → *HNNH → *HNNH2 → *H2NNH2 → *H3NNH2 → *NH2 → *NH3 → NH3(g)) and consecutive (*→ *N2 → *NNH → *NNH2 → *NNH3 → *N → *NH → *NH2 → *NH3 → NH3(g)) pathways [38] for NRR (Figure 3a), and the free energy change for each step is illustrated in Figure 3b,c. The difference between the two mechanisms is that in the enzymatic mechanism, the proton–electron pair (H+ + e−) alternately attacks two N atoms, while in the consecutive mechanism, the proton–electron pair first continuously approaches one N atom to generate the first NH3 and then attacks the other N atom to generate the second NH3 [39]. The free energy difference for each elementary step in the enzymatic (consecutive) route is −0.92, 0.28, −0.02, 0.57, −1.82, 0.11, 0.54, 0.44 and 0.01 (−0.92, 0.28, 0.83, −1.51, 0.28, −0.78, 0.57, 0.44 and 0.01) eV, respectively, and accordingly, the potential limiting step *HNNH → *HNNH2 (*NNH → *NNH2) has a maximum free energy change of 0.57 (0.83) eV via the enzymatic (consecutive) mechanism. Thus, the limiting potential (η) of NRR on Fe3@C2N is as low as −0.57 V, lower than the η values on the Ru (0001) step surface [40] and Fe2@C2N [24] (0.98 eV and 1.23 eV, respectively). Typically, the potential limiting step on most metal surfaces (such as Re, Ru, Rh and Fe) or two-dimensional MBenes is either the first hydrogenation (forming *NNH) or the last protonation (*NH3 formation or NH3 desorption) [9,41]. However, on Fe3@C2N, the free energy change of *N2 → *NNH is only 0.28 eV. We also calculated the barriers of the potential limiting steps of the two mechanisms, and the barriers are both 1.11 eV based on the CI-NEB method (Figure S6), lower than the corresponding values (1.24 and 1.31 eV) on Fe3@θ-Al2O3(010) [25]. All of the above results suggest the high activity of Fe3@C2N for NRR.
In addition to the catalytic activity, another important aspect for the catalytic performance is the selectivity of NRR [22]. Due to the high content of protons in the acidic solution, the major competitive reaction of NRR is the hydrogen evolution reaction (HER). The adsorption energy of *H on Fe3@C2N is −0.94 eV, weaker than Eads of N2 (−1.45 eV), and expectedly, the charge transfer between *H and the catalyst is smaller (0.43 |e| vs. 1.14 |e|). By comparing the energies of two *H on Fe3@C2N (−1.69 eV) and the formation of H2 (1.79 eV, illustrated in Figure S7), we found that the reaction energy of HER is as high as 3.45 eV, indicating that Fe3@C2N has a high ability to suppress the competing HER during NRR under an acidic environment. The stable and metallic Fe3@C2N not only possesses low NRR limiting potential but also exhibits high selectivity against HER, and therefore, the low-cost Fe3@C2N is a highly promising electrocatalyst for NRR.
3. Materials and Methods
The spin-polarized density functional theory (DFT) calculations were carried out using the Vienna Ab initio Simulation Package (VASP 5.4.4) [42]. The exchange–correlation functional was described by the Perdew, Burke, and Ernzerhof (PBE) parameterization of the generalized gradient approximation (GGA) [43]. The projector augmented-wave (PAW) potential [44] was employed, and an electron configuration of 3d74s1 was adopted for Fe. According to our tests, we found that the system is ferromagnetic, with each Fe atom carrying a magnetic moment of ~3 μB (Figure S2), which is quite similar to the case of a Fe3 cluster on the θ-Al2O3(010) surface [25]. A cutoff energy of 550 eV was adopted. The van der Waals interactions were described using the empirical correction in the Grimme scheme (DFT-D2) [45], where the dispersion energy is corrected based on the pairwise atomic –C/R6 terms. The D2 scheme was widely used in these theoretical studies for the oxygen reduction reaction [29] and NRR [2]. During the structure relation, the maximum force and energy on each atom were less than 0.01 eV/Å and 10−5 eV, the width of smearing was chosen as 0.2 eV, and k-points were sampled using the 5 × 5 × 1 Monkhorst−Pack mesh [46]. To avoid the interaction between two neighboring surfaces, a vacuum space over 20 Å was used [2,40]. To simulate a C2N monolayer, a periodic 1 × 1 unit cell (with a lateral dimension of ~8.34 Å) containing 12 carbon atoms and 6 nitrogen atoms was constructed. The barriers of the potential limiting steps were identified by using the climbing image nudged elastic-band (CI-NEB) method [47].
The average binding energy (Eb) of each Fe atom on the C2N substrate in our work is given by
where EFe3@C2N, EC2N and EFe are the energies of the Fe3@C2N system, the C2N monolayer, and an isolated Fe atom, respectively.
Eb = (EFe3@C2N − EC2N − 3EFe)/3
The adsorption energy (Eads) of the single atom or NRR intermediates was determined according to the following equation:
where Etot, EFe3@C2N and Eadsorbate represent the total energies of the systems containing the Fe3@C2N catalyst and the adsorbate, Fe3@C2N, and the adsorbate, respectively.
Eads = Etot − EFe3@C2N − Eadsorbate
According to the calculated hydrogen electrode (CHE) model proposed by Nørskov and co-workers [48], the reaction free energy of each basic step ΔG is calculated by
ΔG = ΔE − ΔEZPE − TΔS + eU + ΔGpH
In the above equation, ΔE is the reaction energy difference between the products and reactants of the NRR occurring on the catalyst, which can be directly obtained from DFT computations, and ΔEZPE is the change in zero-point energies, which was calculated from the vibrational frequencies [12]. The vibrational frequencies were calculated based on the finite differences method, and only the adsorbed species were included. ΔS is the change in entropy at 298.15 K. Among them, the free energy correction of pH is expressed by ΔGpH, which is equal to kBT × ln10 × pH, where kB is the Boltzmann constant. In this study, it is assumed that pH = 0.
The limiting potential (η) of the entire reduction process is determined by the potential limiting step, which has the most positive ΔG (ΔGMax), as computed by η = −ΔGMax/e [49].
4. Conclusions
In summary, we designed a supported catalyst, i.e., Fe3 clusters anchored on a two-dimensional C2N monolayer (Fe3@C2N), to electrocatalyze the reduction of nitrogen to ammonia. Through our density functional theory computations, we found that Fe3@C2N is excellent as an electrocatalyst for NRR owing to the stability at high temperature (800 K), the metallic feature, the absence of precious metal, the low limiting potential (0.57 eV of the enzymatic mechanism) and the high selectivity against the competing side reaction, HER. Such superiority can be ascribed to the partially occupied d orbitals and largely negative charged Fe3 cluster, which is beneficial in activating the inert N≡N triple bond. Since it was feasible to reach Cu2@C2N using CuCl2 as a metal precursor in a previous theoretical protocol [50], Fe3@C2N could be synthesized using FeCl2 and other proper iron precursors. We hope that our work can inspire more experimental and theoretical studies to further explore the potential of non-precious metal clusters for NRR.
Supplementary Materials
The following are available online at https://0-www-mdpi-com.brum.beds.ac.uk/2073-4344/10/9/974/s1: Figure S1: Top and side views of the final structure of Fe3@C2N through a 10 ps FPMD simulation at 800 K; Figure S2: The magnesium distribution of Fe3@C2N; Figure S3: Density of states (DOS) of C2N and Fe3@C2N; Figure S4: The energy diagram of N2 dissociation on Fe3@C2N; Figure S5: The energy diagram of NRR via the surface-hydrogenation mechanism on Fe3@C2N; Figure S6: The reaction pathway of the potential limiting step of the enzymatic mechanism and the consecutive mechanism; Figure S7: The energy diagram of the hydrogen evolution reaction (HER0 on Fe3@C2N; Table S1: The calculated zero-point energy (ZPE) and entropy of different molecules (T = 298.15 K and P = 101.325 Pa, in eV); Table S2: The calculated E(DFT), ZPE and TS of the intermediates (T = 298.15 K and P = 101.325 Pa, in eV); Table S3: The estimated reaction rate of each step on different paths (k0 is the prefactor of the estimated reaction rate k = k0exp(−ΔG/kT)).
Author Contributions
F.L. and J.Z. outlined the work plan; B.H. and H.M. conducted the computations; B.H. drew the figures and drafted the manuscript. All authors participated in the reviewing and publication processes of the article. All authors have read and agreed to the published version of the manuscript.
Funding
This work was funded by the National Natural Science Foundation of China (11704203), the Startup Project of Inner Mongolia University (21200-5175101), and the Natural Science Funds for Distinguished Young Scholar of Heilongjiang Province (JC2018004).
Acknowledgments
The authors acknowledge the support of this study by the National Natural Science Foundation of China, under Grant number (11704203), the Startup Project of Inner Mongolia University (21200-5175101), and the Natural Science Funds for Distinguished Young Scholar of Heilongjiang Province, under Grant number (JC2018004). Additionally, computational support from PARATEAR is appreciated.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Guo, X.; Li, X.; Li, Y.; Yang, J.; Wan, X.; Chen, L.; Liu, J.; Liu, X.; Yu, R.; Zheng, L.; et al. Molecule template method for precise synthesis of Mo-based alloy clusters and electrocatalytic nitrogen reduction on partially reduced PtMo alloy oxide cluster. Nano Energy 2020, 78, 105211. [Google Scholar] [CrossRef]
- Guo, X.; Gu, J.; Lin, S.; Zhang, S.; Chen, Z.; Huang, S. Tackling the activity and selectivity challenges of electrocatalysts toward the nitrogen reduction reaction via atomically dispersed biatom catalysts. J. Am. Chem. Soc. 2020, 142, 5709–5721. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Li, N.; Kong, Z.; Ong, W.-J.; Zhao, X. Photocatalytic fixation of nitrogen to ammonia: State-of-the-art advancements and future prospects. Mater. Horiz. 2018, 5, 9–27. [Google Scholar] [CrossRef]
- Van der Ham, C.J.M.; Koper, M.T.; Hetterscheid, D.G. Challenges in reduction of dinitrogen by proton and electron transfer. Chem. Soc. Rev. 2014, 43, 5183–5191. [Google Scholar] [CrossRef]
- Shipman, M.A.; Symes, M.D. Recent progress towards the electrosynthesis of ammonia from sustainable resources. Catal. Today 2017, 286, 57–68. [Google Scholar] [CrossRef] [Green Version]
- Logadóttir, Á.; Nørskov, J.K. Ammonia synthesis over a Ru(0001) surface studied by density functional calculations. J. Catal. 2003, 220, 273–279. [Google Scholar] [CrossRef]
- Ertl, G.; Lee, S.B.; Weiss, M. Kinetics of nitrogen adsorption on Fe(111). Surf. Sci. 1982, 114, 515–526. [Google Scholar] [CrossRef]
- Montoya, J.H.; Tsai, C.; Vojvodic, A.; Nørskov, J.K. The challenge of electrochemical ammonia synthesis: A new perspective on the role of nitrogen scaling relations. ChemSusChem 2015, 8, 2180–2186. [Google Scholar] [CrossRef]
- Zhao, J.; Zhao, J.X.; Cai, Q.H. Single transition metal atom embedded into a MoS2 nanosheet as a promising catalyst for electrochemical ammonia synthesis. Phys. Chem. Chem. Phys. 2018, 20, 9248–9255. [Google Scholar] [CrossRef]
- Li, Q.Y.; He, L.Z.; Sun, C.H.; Zhang, X.W. Computational study of MoN2 monolayer as electrochemical catalysts for nitrogen reduction. J. Phys. Chem. C 2017, 121, 27563–27568. [Google Scholar] [CrossRef]
- Li, M.; Huang, H.; Low, J.; Gao, C.; Long, R.; Xiong, Y. Recent progress on electrocatalyst and photocatalyst design for nitrogen reduction. Small Methods 2019, 3, 1800388. [Google Scholar] [CrossRef]
- Zhao, J.; Chen, Z. Single Mo atom supported on defective boron nitride monolayer as an efficient electrocatalyst for nitrogen fixation: A computational study. J. Am. Chem. Soc. 2017, 139, 12480–12487. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Zhao, J.X.; Cabrera, C.R.; Chen, Z.F. Computational screening of efficient single-atom catalysts based on graphitic carbon nitride (g-C3N4) for nitrogen electroreduction. Small Methods 2018, 2, 1800368. [Google Scholar]
- Li, Q.; Liu, C.; Qiu, S.; Zhou, F.; He, L.; Zhang, X.; Sun, C. Exploration of iron borides as electrochemical catalysts for nitrogen reduction reaction. J. Mater. Chem. A 2019, 7, 21507–21513. [Google Scholar] [CrossRef]
- Cheng, H.; Ding, L.-X.; Chen, G.-F.; Zhang, L.; Xue, J.; Wang, H. Molybdenum carbide nanodots enable efficient electrocatalytic nitrogen fixation under ambient conditions. Adv. Mater. 2018, 30, 1803694. [Google Scholar] [CrossRef]
- Li, X.-F.; Li, Q.-K.; Cheng, J.; Liu, L.; Yan, Q.; Wu, Y.C.; Zhang, X.Z.; Wang, Z.-Y.; Qiu, Q.; Luo, Y. Conversion of dinitrogen to ammonia by FeN3 embedded graphene. J. Am. Chem. Soc. 2016, 138, 8706–8709. [Google Scholar] [CrossRef]
- Azofra, L.M.; Sun, C.; Cavallo, L.; MacFarlane, D.R. Feasibility of N2 binding and reduction to ammonia on Fe-deposited MoS2 2D sheets: A DFT study. Chem. Eur. J. 2017, 23, 8275–8279. [Google Scholar] [CrossRef] [Green Version]
- Ling, C.; Ouyang, Y.X.; Li, Q.; Bai, X.; Mao, X.; Du, A.; Wang, J.L. A general two-step strategy–based high-through put screening of single atom catalysts for nitrogen fixation. Small Methods 2018, 3, 1800376. [Google Scholar] [CrossRef]
- Li, Q.; Qiu, S.; Liu, C.; Liu, M.; He, L.; Zhang, X.; Sun, C. Computational design of single molybdenum catalysts for nitrogen reduction reaction. J. Phys. Chem. C 2019, 123, 2347–2352. [Google Scholar] [CrossRef]
- Wang, M.; Liu, S.; Qian, T.; Liu, J.; Zhou, J.; Ji, H.; Xiong, J.; Zhong, J.; Yan, C. Over 56.55% faradaic efficiency of ambient ammonia synthesis enabled by positively shifting the reaction potential. Nat. Commun. 2019, 10, 341. [Google Scholar] [CrossRef]
- Liu, C.; Li, Q.; Zhang, J.; Jin, Y.; MacFarlane, D.R.; Sun, C. Conversion of dinitrogen to ammonia on Ru atoms supported on boron sheets: A DFT study. J. Mater. Chem. A 2019, 7, 4771–4776. [Google Scholar] [CrossRef]
- Chen, Z.; Yan, J.; Jiang, Q. Single or double: Which is the altar of atomic catalysts for nitrogen reduction reaction? Small Methods 2018, 3, 1800291. [Google Scholar] [CrossRef]
- Li, F.; Chen, L.; Liu, H.; Wang, D.; Shi, C.; Pan, H. Enhanced N2-fixation by engineering the edges of two-dimensional transition-metal disulfides. J. Phys. Chem. C 2019, 123, 22221–22227. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, A.; Zhang, Z.H.; Zhou, Z. Double-atom catalysts: Transition metal dimer anchored C2N monolayers as N2 fixation electrocatalysts. J. Mater. Chem. A 2018, 6, 18599–18604. [Google Scholar] [CrossRef]
- Liu, J.; Ma, X.; Li, Y.; Wang, Y.; Xiao, H.; Li, J. Heterogeneous Fe3 single-cluster catalyst for ammonia synthesis via an associative mechanism. Nat. Commun. 2018, 9, 1610. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.W.; Zeng, Z.; Liu, L.; Huang, X.; Jia, Y. Computational evaluation of electrocatalytic nitrogen reduction on TM single-, double-, and triple-atom catalysts (TM = Mn, Fe, Co, Ni) based on graphdiyne monolayers. J. Phys. Chem. C 2019, 123, 19066–19076. [Google Scholar] [CrossRef]
- Li, Y.; Zhang, Q.; Fan, H.-N.; Luo, W.-B.; Liu, H.-K.; Dou, S.-X. Atomically dispersed metal dimer species with selective catalytic activity for nitrogen electrochemical reduction. J. Mater. Chem. A 2019, 7, 22242–22247. [Google Scholar] [CrossRef]
- Chen, Z.; Chen, L.; Jiang, M.; Chen, D.; Wang, Z.; Yao, X.; Singh, C.V.; Jiang, Q. Triple atom catalyst with ultrahigh loading potential for nitrogen electrochemical reduction. J. Mater. Chem. A 2020, 8, 15086–15093. [Google Scholar] [CrossRef]
- Li, X.; Zhong, W.; Cui, P.; Li, J.; Jiang, J. Design of efficient catalysts with double transition metal atoms on C2N layer. J. Phys. Chem. Lett. 2016, 7, 1750–1755. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, A.; Zhang, Z.H.; Jiao, M.G.; Zhou, Z. Transition metal anchored C2N monolayers as efficient bifunctional electrocatalysts for hydrogen and oxygen evolution reactions. J. Mater. Chem. A 2018, 6, 11446–11452. [Google Scholar] [CrossRef]
- Li, F.; Liu, X.; Chen, Z. 1 + 1′ > 2: Heteronuclear biatom aatalyst outperforms its homonuclear counterparts for CO oxidation. Small Methods 2019, 10, 1800480. [Google Scholar] [CrossRef]
- Pei, W.; Zhou, S.; Zhao, J.; Xu, X.; Du, Y.; Dou, S.X. Immobilized trimeric metal clusters: A family of the smallest catalysts for selective CO2 reduction toward multi-carbon products. Nano Energy 2020, 76, 105049. [Google Scholar] [CrossRef]
- Martyna, G.J.; Klein, M.L.; Tuckerman, M. Nosè-Hoover chains: The canonical ensemble via continuous dynamics. J. Chem. Phys. 1992, 97, 2635–2643. [Google Scholar] [CrossRef]
- Wang, Z.; Yu, Z.; Zhao, J. Computational screening of single transition metal atom supported on C2N monolayer for electrochemical ammonia synthesis. Phys. Chem. Chem. Phys. 2018, 20, 12835–12844. [Google Scholar] [CrossRef] [PubMed]
- Strongin, D.; Carrazza, J.; Bare, S.R.; Somorjai, G. The importance of C7 sites and surface roughness in the ammonia synthesis reaction over iron. J. Catal. 1987, 103, 213–215. [Google Scholar] [CrossRef] [Green Version]
- Somorjai, G.; Materer, N. Surface structures in ammonia synthesis. Top. Catal. 1994, 1, 215–231. [Google Scholar] [CrossRef]
- Ling, C.; Zhang, Y.; Li, Q.; Bai, X.; Shi, J.; Wang, J. New mechanism for N2 reduction: The essential role of surface hydrogenation. J. Am. Chem. Soc. 2019, 141, 18264–18270. [Google Scholar] [CrossRef]
- Gao, Z.; Huang, H.; Xu, S.; Li, L.; Yan, G.; Zhao, M.; Yang, W.; Zhao, X. Regulating the coordination environment through doping N atoms for single-atom Mn electrocatalyst of N2 reduction with high catalytic activity and selectivity: A theoretical study. Mol. Catal. 2020, 493, 111091. [Google Scholar] [CrossRef]
- Ling, C.; Bai, X.; Ouyang, Y.; Du, A.; Wang, J. Single molybdenum atom anchored on N-doped carbon as a promising electrocatalyst for nitrogen reduction into ammonia at ambient conditions. J. Phys. Chem. C 2018, 122, 16842–16847. [Google Scholar] [CrossRef]
- Choi, C.; Back, S.; Kim, N.Y.; Lim, J.; Kim, Y.H.; Jung, Y. Suppression of hydrogen evolution reaction in electrochemical N2 reduction using single-atom catalysts: A computational guideline. ACS Catal. 2018, 8, 7517–7525. [Google Scholar] [CrossRef]
- Yang, X.; Shang, C.; Zhou, S.; Zhao, J. MBenes: Emerging 2D materials as efficient electrocatalysts for the nitrogen reduction reaction. Nanoscale Horiz. 2020, 5, 1106–1115. [Google Scholar] [CrossRef] [PubMed]
- Kresse, G.; Furthmüller, F. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 1996, 54, 11169–11186. [Google Scholar] [CrossRef] [PubMed]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blöch, P.E. Projetor augmented-wave method. Phys. Rev. B 1994, 50, 17953–17979. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bucko, T.; Hafner, J.; Lebegue, S.; Aangyan, J.G. Improved description of the structure of molecular and layered crystals: Ab initio DFT calculations with van der Waals Corrections. J. Phys. Chem. A 2010, 114, 11814–11824. [Google Scholar] [CrossRef] [PubMed]
- Monkhorst, H.J.; Pack, D.J. Special points for Brillonin-zone integrations. Phys. Rev. B 1976, 13, 5188–5192. [Google Scholar] [CrossRef]
- Henkelman, G.; Uberuaga, B.P.; Jónsson, H. A climbing image nudged elastic band method for finding saddle points and minimum energy paths. J. Chem. Phys. 2000, 113, 9901. [Google Scholar] [CrossRef] [Green Version]
- Nørskov, J.K.; Rossmeisl, J.; Logadottir, A.; Lindqvist, L.; Kitchin, J.R.; Bligaard, T.; Jonsson, H. Origin of the overpotential for oxygen reduction at a fuel-cellcathode. J. Phys. Chem. B 2004, 108, 17886–17892. [Google Scholar] [CrossRef]
- Skulason, E.; Bligaard, T.; Gudmundsdottir, S.; Studt, F.; Rossmeisl, J.; Abild-Pedersen, F.; Vegge, T.; Jonsson, H.; Norskov, J.K. A theoretical evaluation of possible transition metal electro-catalysts for N2 reduction. Phys. Chem. Chem. Phys. 2012, 14, 1235–1245. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Chen, Z. Cu dimer anchored C2N monolayer: Low-cost and efficient catalyst for CO oxidation. Nanoscale 2018, 10, 15696–15705. [Google Scholar] [CrossRef]
Figure 1.
Top and side views of C2N (a) and Fe3@C2N (b), as well as their corresponding band structures (c,d).
Figure 1.
Top and side views of C2N (a) and Fe3@C2N (b), as well as their corresponding band structures (c,d).

Figure 2.
Top and side views of N2 adsorbed on the Fe3@C2N with the end-on (a) and side-on configuration (b).
Figure 2.
Top and side views of N2 adsorbed on the Fe3@C2N with the end-on (a) and side-on configuration (b).

Figure 3.
(a) Two nitrogen reduction reaction (NRR) pathways on Fe3@C2N, and the free energy diagram through enzymatic (b) and consecutive mechanisms (c) at different limiting potentials.
Figure 3.
(a) Two nitrogen reduction reaction (NRR) pathways on Fe3@C2N, and the free energy diagram through enzymatic (b) and consecutive mechanisms (c) at different limiting potentials.

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Han, B.; Meng, H.; Li, F.; Zhao, J. Fe3 Cluster Anchored on the C2N Monolayer for Efficient Electrochemical Nitrogen Fixation. Catalysts 2020, 10, 974. https://0-doi-org.brum.beds.ac.uk/10.3390/catal10090974
AMA Style
Han B, Meng H, Li F, Zhao J. Fe3 Cluster Anchored on the C2N Monolayer for Efficient Electrochemical Nitrogen Fixation. Catalysts. 2020; 10(9):974. https://0-doi-org.brum.beds.ac.uk/10.3390/catal10090974
Chicago/Turabian StyleHan, Bing, Haihong Meng, Fengyu Li, and Jingxiang Zhao. 2020. "Fe3 Cluster Anchored on the C2N Monolayer for Efficient Electrochemical Nitrogen Fixation" Catalysts 10, no. 9: 974. https://0-doi-org.brum.beds.ac.uk/10.3390/catal10090974
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.