Asymmetric Cyanation of Activated Olefins with Ethyl Cyanoformate Catalyzed by Ti(IV)-Catalyst: A Theoretical Study


1
Key Laboratory of Green Chemistry and Technology, Ministry of Education, College of Chemistry, Sichuan University, No. 29 Wangjiang Road, Chengdu 610064, China
2
Department of Chemistry and Chemical Engineering, Center for Design and Applications of Molecular Catalysts, Inha University, 100 Inha-ro, Michuhol-gu, Incheon 22212, Korea
*
Author to whom correspondence should be addressed.
Catalysts 2020, 10(9), 1079; https://0-doi-org.brum.beds.ac.uk/10.3390/catal10091079
Submission received: 30 July 2020
/
Revised: 14 September 2020
/
Accepted: 15 September 2020
/
Published: 18 September 2020
(This article belongs to the Special Issue Advanced Strategies for Catalyst Design)
Abstract
:The reaction mechanism and origin of asymmetric induction for conjugate addition of cyanide to the C=C bond of olefin were investigated at the B3LYP-D3(BJ)/6-31+G**//B3LYP-D3(BJ)/6-31G**(SMD, toluene) theoretical level. The release of HCN from the reaction of ethyl cyanoformate (CNCOOEt) and isopropanol (HOiPr) was catalyzed by cinchona alkaloid catalyst. The cyanation reaction of olefin proceeded through a two-step mechanism, in which the C-C bond construction was followed by H-transfer to generate a cyanide adduct. For non-catalytic reaction, the activation barrier for the rate-determining C-H bond construction step was 34.2 kcal mol−1, via a four-membered transition state. The self-assembly Ti(IV)-catalyst from tetraisopropyl titanate, (R)-3,3′-disubstituted biphenol, and cinchonidine accelerated the addition of cyanide to the C=C double bond by a dual activation process, in which titanium cation acted as a Lewis acid to activate the olefin and HNC was orientated by hydrogen bonding. The steric repulsion between the 9-phenanthryl at the 3,3′-position in the biphenol ligand and the Ph group in olefin raised the Pauli energy (ΔE≠Pauli) of reacting fragments at the re-face attack transition state, leading to the predominant R-product.
1. Introduction
The asymmetric catalytic cyanation of C=X bond (X = O, N or C) provides an outstanding method to obtain various optically active nitriles [1,2,3,4,5,6]. Compared to the intensively studied cyanation of aldehydes [1,2,5], ketones [1,2,5] and imine (Strecker reaction) [3], reports on the conjugate addition of cyanide to the C=C bond are limited [4,6]. Since the products from cyanide addition to C=C double bonds in the α,β-unsaturated carbonyl compounds could conveniently convert to the enantioenriched intermediates with great synthesizing value and pharmaceutical importance (e.g., γ-aminobutyric acids), developing straightforward synthetic procedures and exploring the relevant reaction mechanisms are in high demand.
Jacobsen’s group reported the first catalytic asymmetric cyanation of α,β-unsaturated imides, using Al(III) complex with chiral salen ligand as a catalyst. Based on the kinetic analyses, they proposed that the reaction involved a bimetallic, dual activation process. The salen–Al(III) complex-activated cyanide was delivered to the electrophile bound as an imidate complex for highly enantioenriched cyanide adducts [7]. In a heterobimetallic system with (salen)Al and (pybox)Er complexes (pybox = 2,6-bis(2-oxazolinyl) pyridine), two catalysts operated cooperatively in the rate-determining step, promoting the conjugate addition in a highly enantioselective manner [8]. The poly(norbornene)-supported (salen)AlCl catalyst could also realize this transformation. The proximity of catalytic sites in polymeric Al-catalyst facilitated the reaction to occur via a bimetallic pathway [9]. Shibasaki et al. developed chiral gadolinium complex catalysts for the cyanation of α,β-unsaturated N-acylpyrroles [10,11] and enones [12]. Mechanistic studies suggested that the reaction is carried out through an intramolecular cyanide transfer from the gadolinium cyanide to the activated N-acylpyrrole substrate. The protic additive (e.g., HCN) efficiently facilitated both catalyst activity and enantioselectivity. Other metal complexes containing Sr(II) [13], Ru(II)/Li(I) [14,15], Mg(II) [16,17] and Li(I) [18] were also active catalysts for this kind of reaction. The bifunctional catalysis model was proposed to interpret the activation mode as well as the stereochemical outcome [15,16]. Besides, the reaction could be realized by organocatalysis [19,20] or a phase-transfer process [21,22]. The spectroscopic studies by Khan et al. verified that the N-oxide additive participated in the cyanation of nitroalkenes as a ligand and activator of trimethylsilylcyanide (TMSCN) [23]. Experiments and density functional theory (DFT) calculations by Minakata and co-workers revealed that the cyanation of the boron enolates generated from α,β-unsaturated ketones with p-toluenesulfonyl cyanide (TsCN) proceeded through a six-membered ring transition state (TS) [24]. Based on the NMR spectroscopy results, Khan et al. proposed that imidazolium cations interacted with the substrate, facilitating the attack of cyanide ions generated by the activation of acetone cyanohydrin by the acetate counter ion in 1-butyl-3-methylimidazolium (BMIM)-based ionic-liquid-catalyzed conjugate cyanation of CF3-substituted alkylidenemalonates [25].
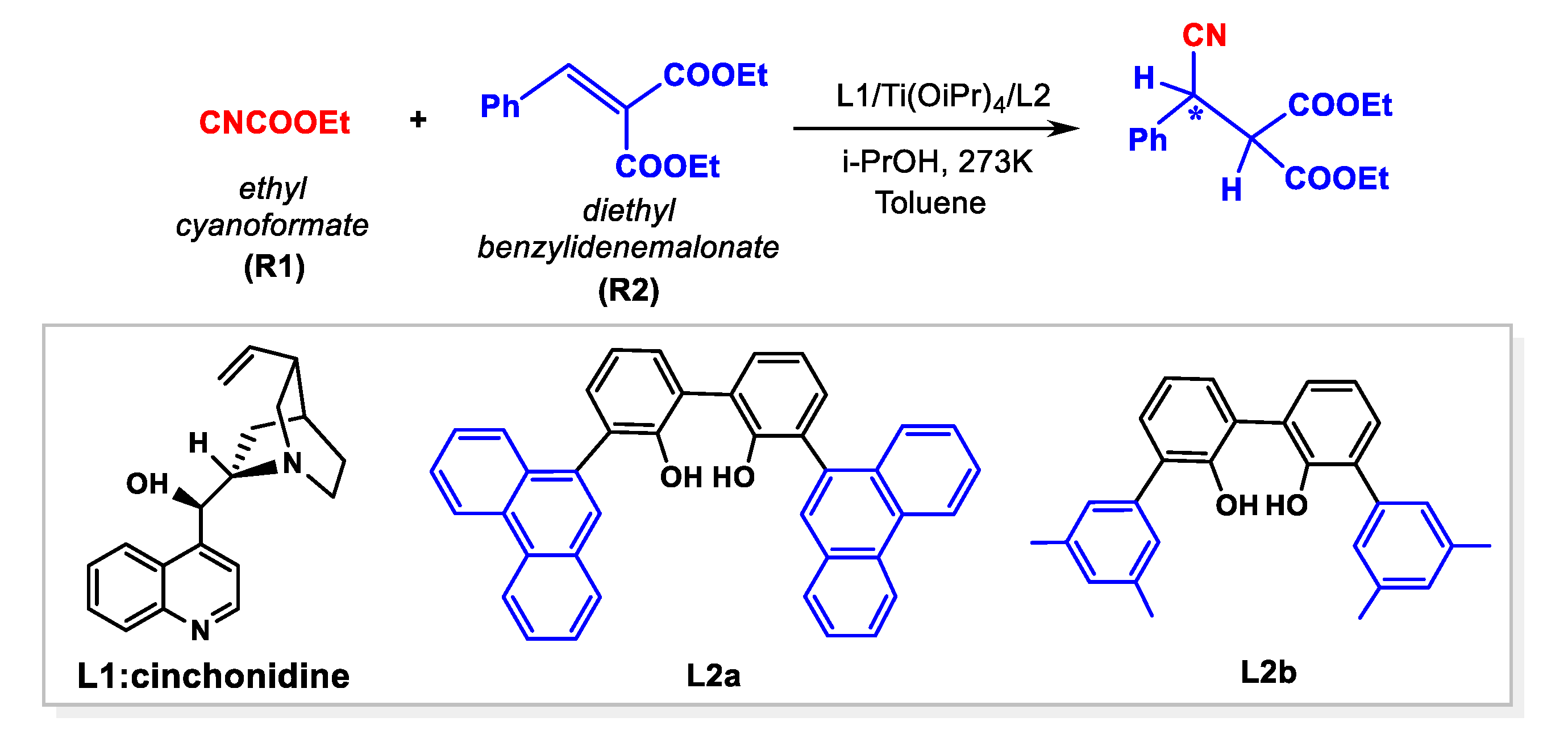
In 2010, Feng’s group developed a modular catalyst generated in situ from cinchona alkaloid, tetraisopropyl titanate (Ti(OiPr)4) and achiral 3,3′-disubstituted biphenol, achieving the efficient asymmetric cyanation of N-p-toluenesulfonyl aldimines and ketimines, as well as ketones and aldehydes [26,27]. Interestingly, this self-assembled catalyst system also exhibited excellent performance in the asymmetric cyanation of C=C bonds, using ethyl cyanoformate (CNCOOEt) as a cyanide source [28]. The enantioenriched cyanide adducts could be obtained with high yield (97%) and enantiomeric excess (ee) (up to 94%). In the reaction, the axial chirality of the biphenol ligand was induced in the formation of the complex to achieve asymmetric activation [29]. Based on previous work, they proposed that the catalytic species was (R)-biphenol, and chiral cinchonidine ligand coordinated to the Ti(IV) center simultaneously.
In this work, we employed DFT calculations to understand the mechanism for the asymmetric cyanation of activated olefin in detail (Scheme 1). The key factors controlling the enantioselectivity could be revealed to expedite the rational design of new Ti(IV)-complex catalysts.
2. Computational Details
DFT calculations were performed using the Gaussian 09 package [30] at the B3LYP-D3(BJ)/6-31G**(SMD, toluene) theoretical level. Geometries were optimized in toluene solvent and characterized by calculating the harmonic vibrational frequencies. The self-consistent reaction field (SCRF) method and SMD solvation model [31] were adopted to evaluate the effect of the solvent. The transition states were verified by the intrinsic reaction coordinate (IRC) calculation [32]. The optimized structures are summarized in the Supporting Information. Activation strain model (ASM) analysis [33,34,35], also known as distortion–interaction model calculation [36,37,38,39], was used to analyze the factors affecting the enantioselectivity of the catalytic reaction, in which the potential energy (ΔE) was decomposed into the distortion (ΔEstrain) and interaction (ΔEint) energies using the Gaussian 09 program. Besides, four energy contributors (i.e., electrostatic interaction (ΔVelstat), Pauli repulsion (ΔEPauli), dispersion effect (ΔEdisp) and orbital interaction (ΔEoi)) in ΔEint were partitioned by energy decomposition analysis (EDA) [40] using the Amsterdam Density Functional (ADF) program [41] at the B3LYP-D3(BJ)/TZ2P level. The energy of the optimized structure was re-evaluated by single-point calculations at the B3LYP-D3(BJ)/6-31+G**(SMD, toluene) level, in which dispersion correction was also included using Grimme’s D3(BJ) method [42,43]. Unless specified, the Gibbs free energies obtained at the B3LYP-D3(BJ)/6-31+G**//B3LYP-D3(BJ)/6-31G**(SMD, toluene) level at 273 K were used.
3. Results and Discussion
3.1. Release of HCN or HNC Species from CNCOOEt
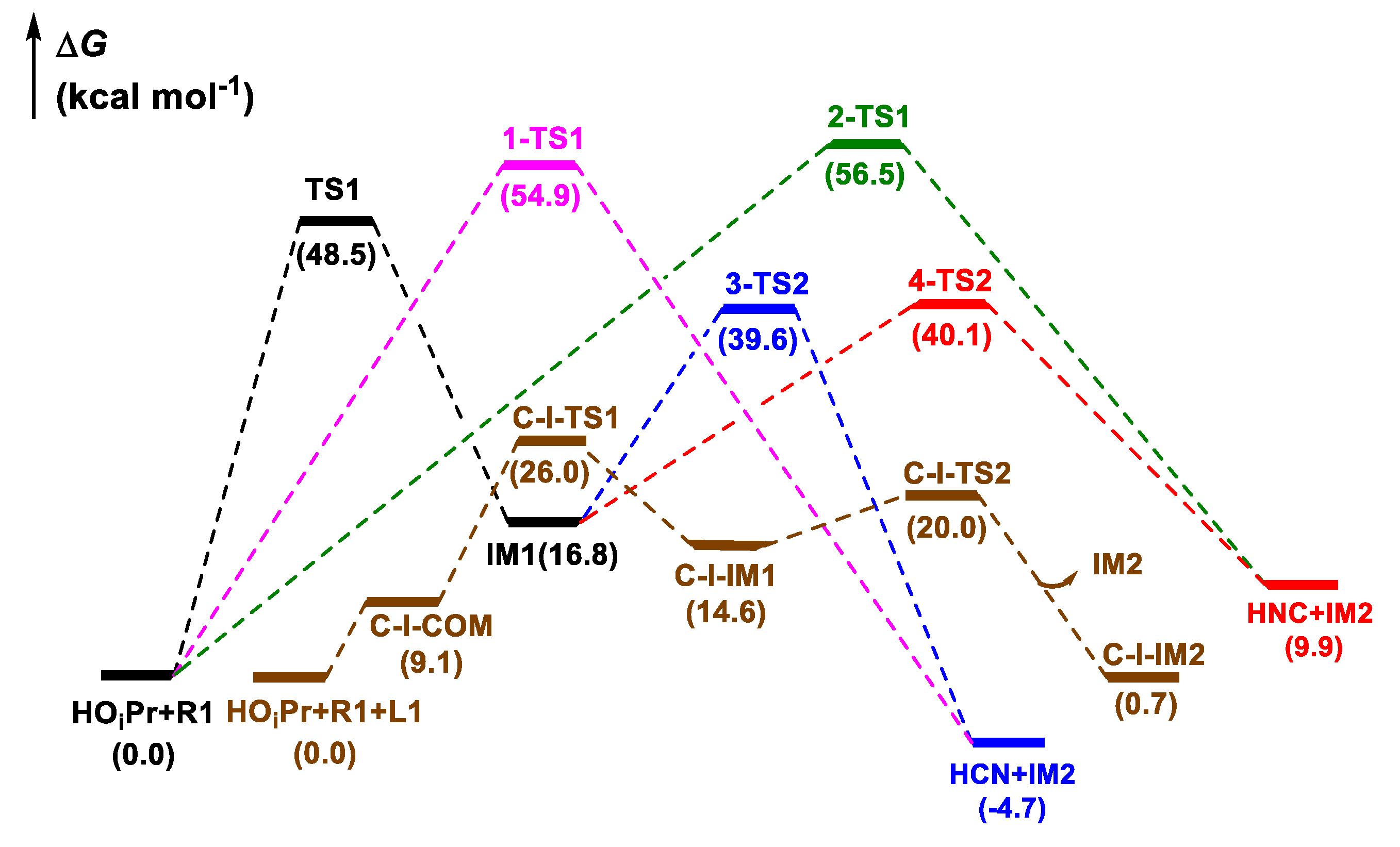
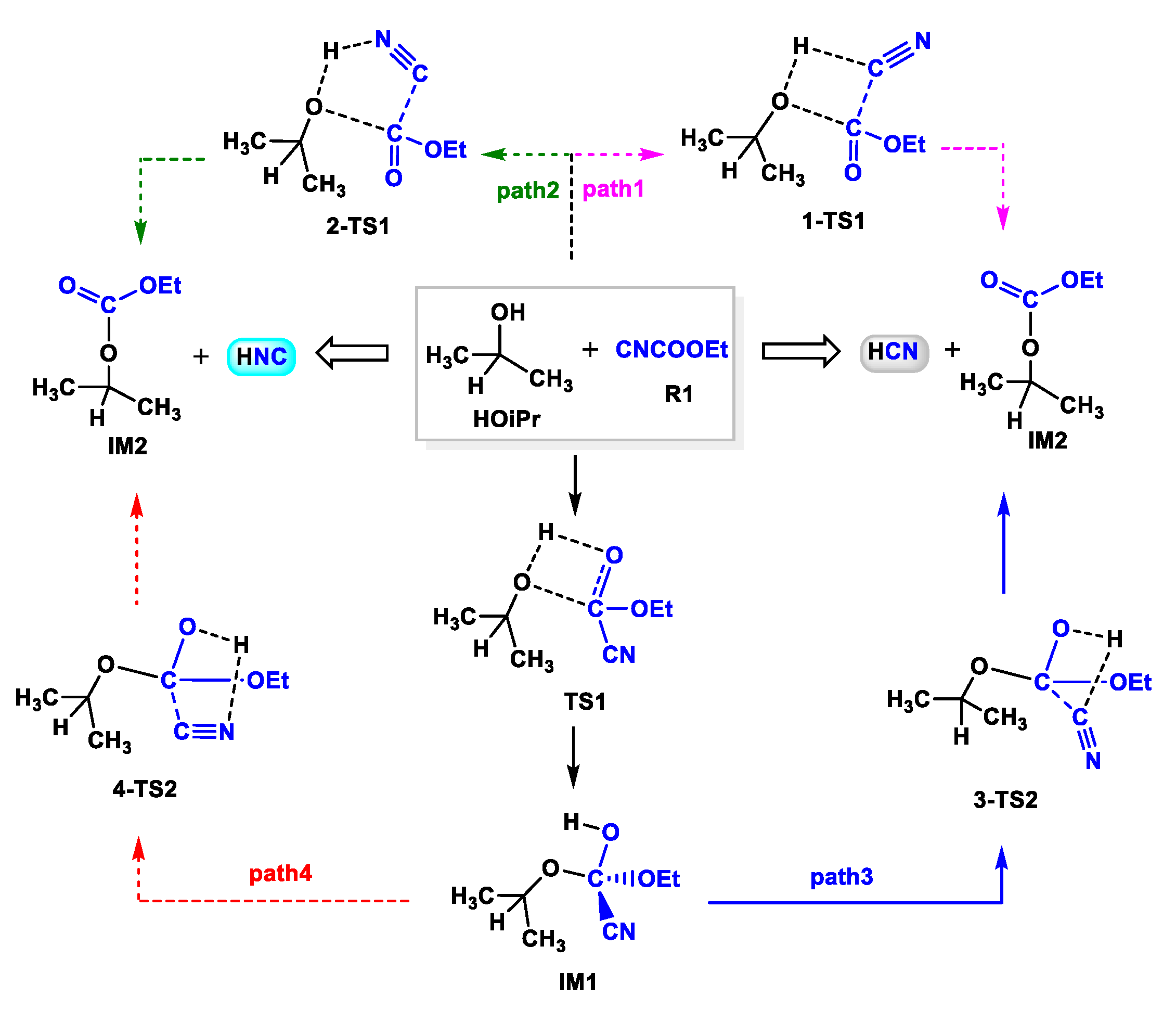
The previous experimental and theoretical investigations suggested that HCN was the real cyanation reagent in the cyanation of imines with TMSCN catalyzed by Ti(IV)-complex [29]. Moreover, isopropyl alcohol (HOiPr) had a positive effect on the release of HCN from TMSCN, consequently accelerating the reaction [26,27,29,44]. Based on these results, we first studied the formation of HCN or HNC species from the reaction between HOiPr and the cyanide source, ethyl cyanoformate (CNCOOEt). Four possible pathways were considered, in which HCN was formed along paths 1 and 3, while its isomer HNC was afforded along paths 2 and 4. As shown in Scheme 2, when CNCOOEt approached HOiPr, the H atom of HOiPr transferred to the C and N atoms of CNCOOEt, yielding HCN and HNC species, respectively. The potential energy surfaces for these pathways are shown in Figure 1. The activation energies were 54.9 and 56.5 kcal mol−1 via 1-TS1 and 2-TS1, respectively. Besides, the reaction could also occur via other stepwise mechanisms along paths 3 and 4, in which the H atom transferred initially to the O atom of the carbonyl group in CNCOOEt having a more negative charge (−0.548 at the O atom vs. −0.501 at the CN moiety) via a four-membered ring transition state TS1. Then, this H atom transferred to the C and N atoms of the CN group through transition states 3-TS2 and 4-TS2, producing HCN (path 3) and HNC (path 4), respectively. The calculations indicated that the ΔG associated with the generation of HCN along path 3 and HNC along path 4 were 39.6 and 40.1 kcal mol−1, respectively, which were lower than those along paths 1 and 2.
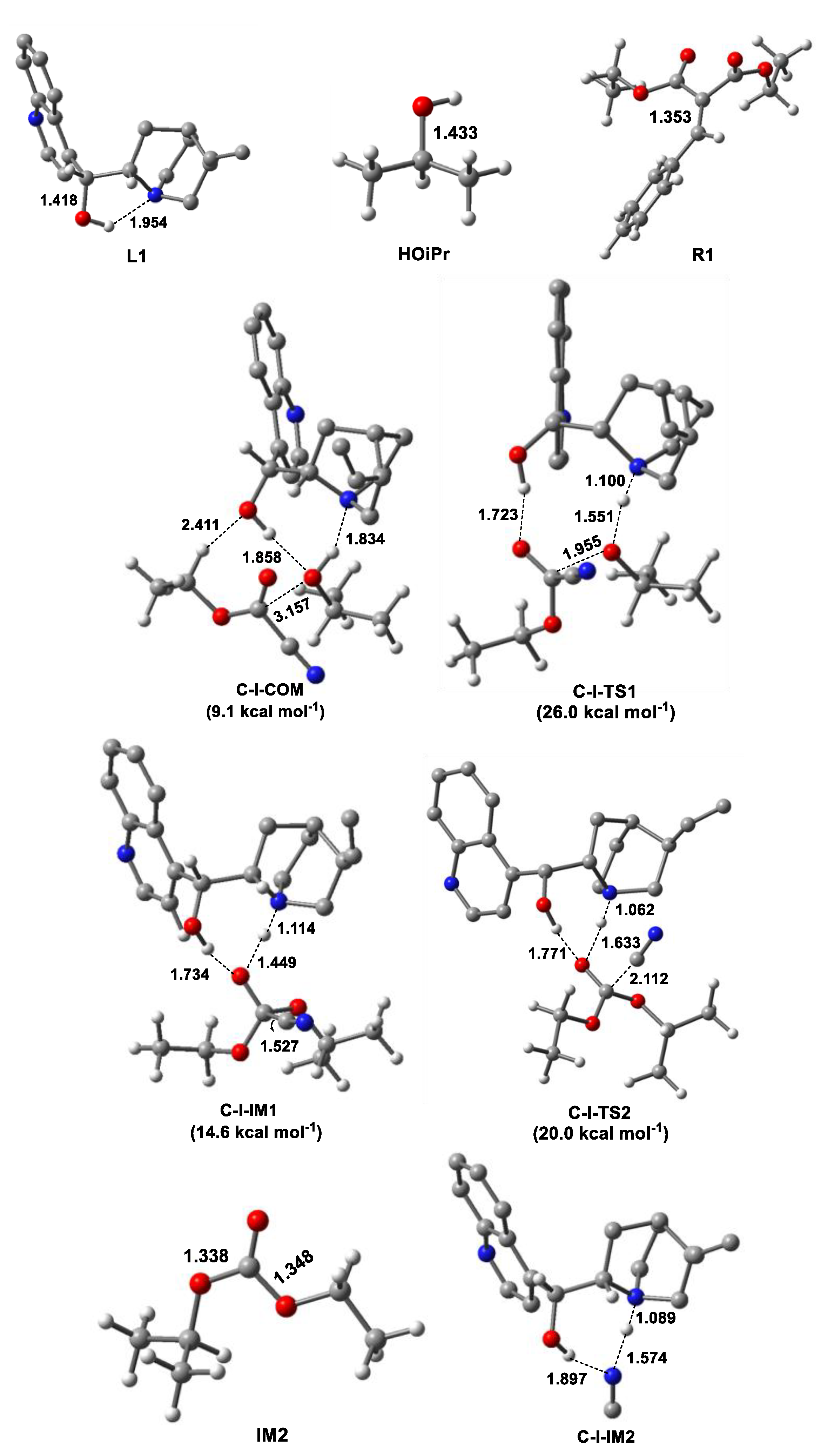
For comparison, the formation of HNC assisted by cinchona alkaloid (L1) was studied (Figure 1 and Figure 2). In the initial complex (C-I-COM), the cinchona alkaloid activated HOiPr by the N atom of the tertiary amine ring and OH group simultaneously with (O)H···O and N···H distances of 1.858 and 1.834 Å, respectively. These hydrogen bonds could be verified by atoms-in-molecules (AIM) analysis, with the positive Laplacian (∇2ρ) on (3, −1) bond critical points (BCPs) (Figure S1). In the next step, the O atom of HOiPr approached the C atom of CNCOOEt, accompanied with H transfer from the O atom to N atom via C-I-TS1. Then, HNC was formed by breaking the C-C bond via C-I-TS2, with a ΔG of 20.0 kcal mol−1. From the viewpoint of energy, cinchona alkaloid could promote the transformation of CNCOOEt to HNC by organocatalysis.
3.2. Reaction Mechanism
3.2.1. Noncatalytic Reaction
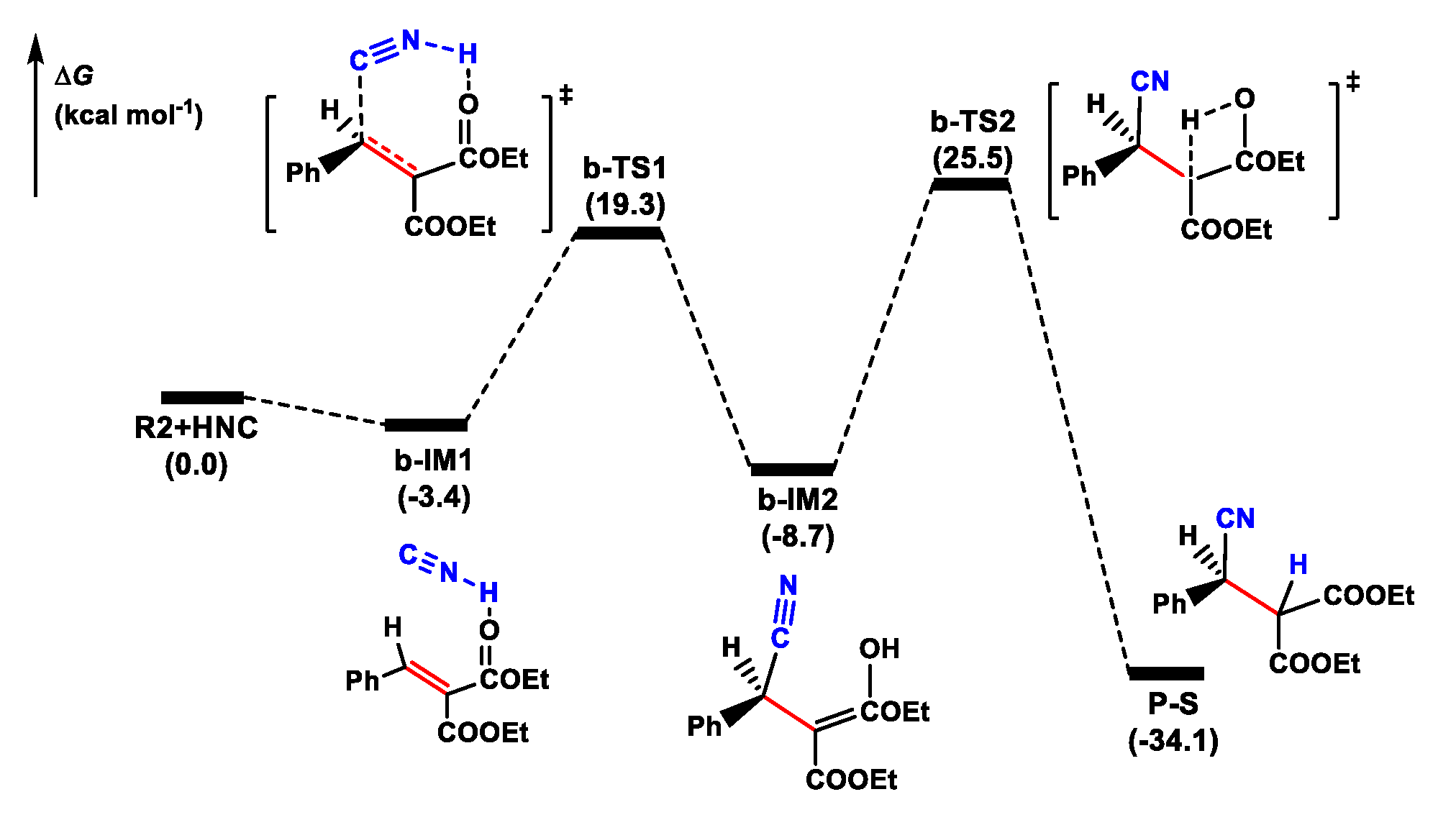
The isomerization between HCN and HNC can occur quickly in the presence of cinchona alkaloid [29], establishing rapid HCN–HNC equilibrium. Then, HNC can act as an active cyanide species to construct a C-C bond by interacting with an olefin in the cyanation reaction. Based on these results, we first studied the mechanism of the noncatalytic cyanation of olefins with HNC (Figure 3). The reaction occurred through a two-step process: C-C bond formation followed by C-H bond construction. In the initial step, HNC interacted with the COOEt moiety through hydrogen bonding, forming a molecular complex b-IM1. Then, the CN group attacked the olefin to form a C-C bond via a seven-membered ring transition state b-TS1. In the final step, the product P-S was formed by shifting an H atom to a C atom (tautomerization of enol to keto), with a ΔG≠ of 34.2 kcal mol−1. This H-transfer step was predicted to be the rate-determining step (RDS) in the background reaction. We also located the TS involving HCN as a proton donor (b-TS1-1, Figure S2). The relative Gibbs energy of b-TS1-1 was higher than that of b-TS2 (the highest point in the energy profile) by 8.4 kcal mol−1. These results indicated that the noncatalytic reaction was difficult to achieve owing to a higher barrier.
3.2.2. Catalytic Reaction
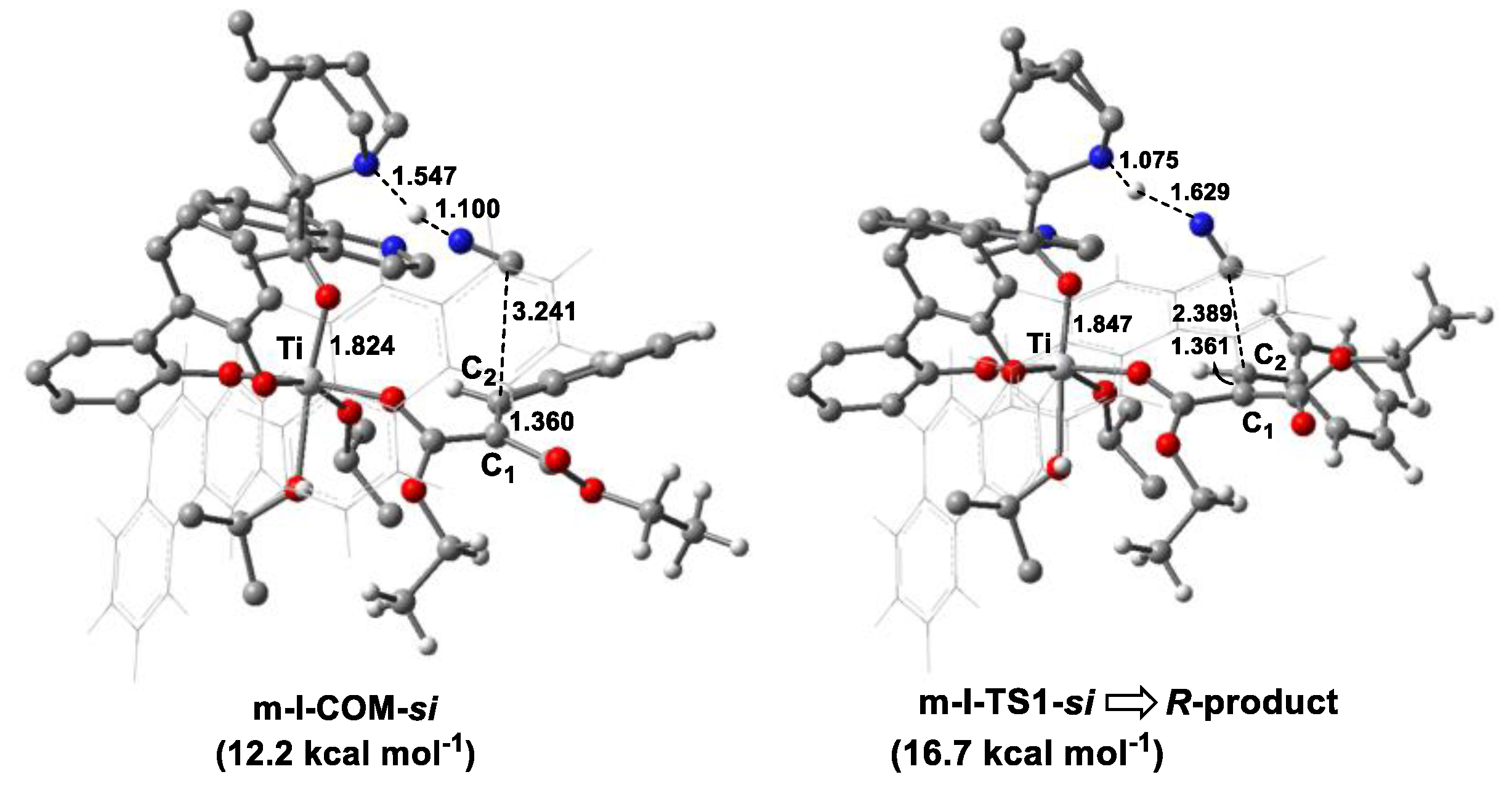
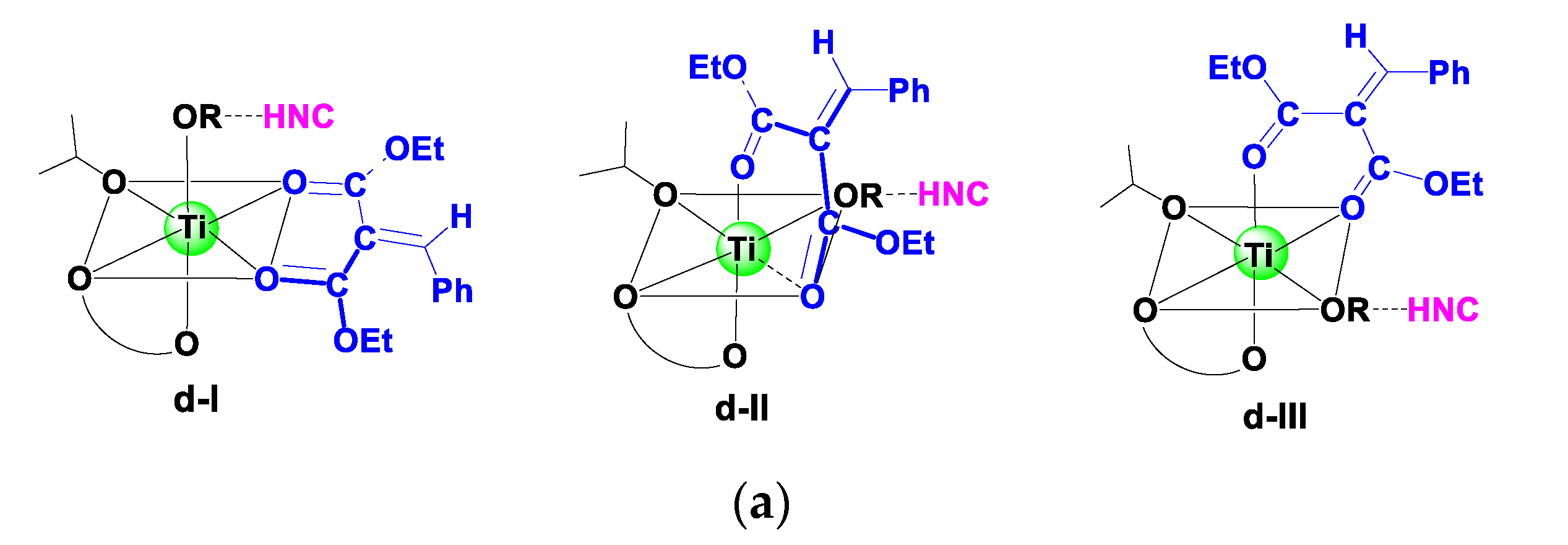
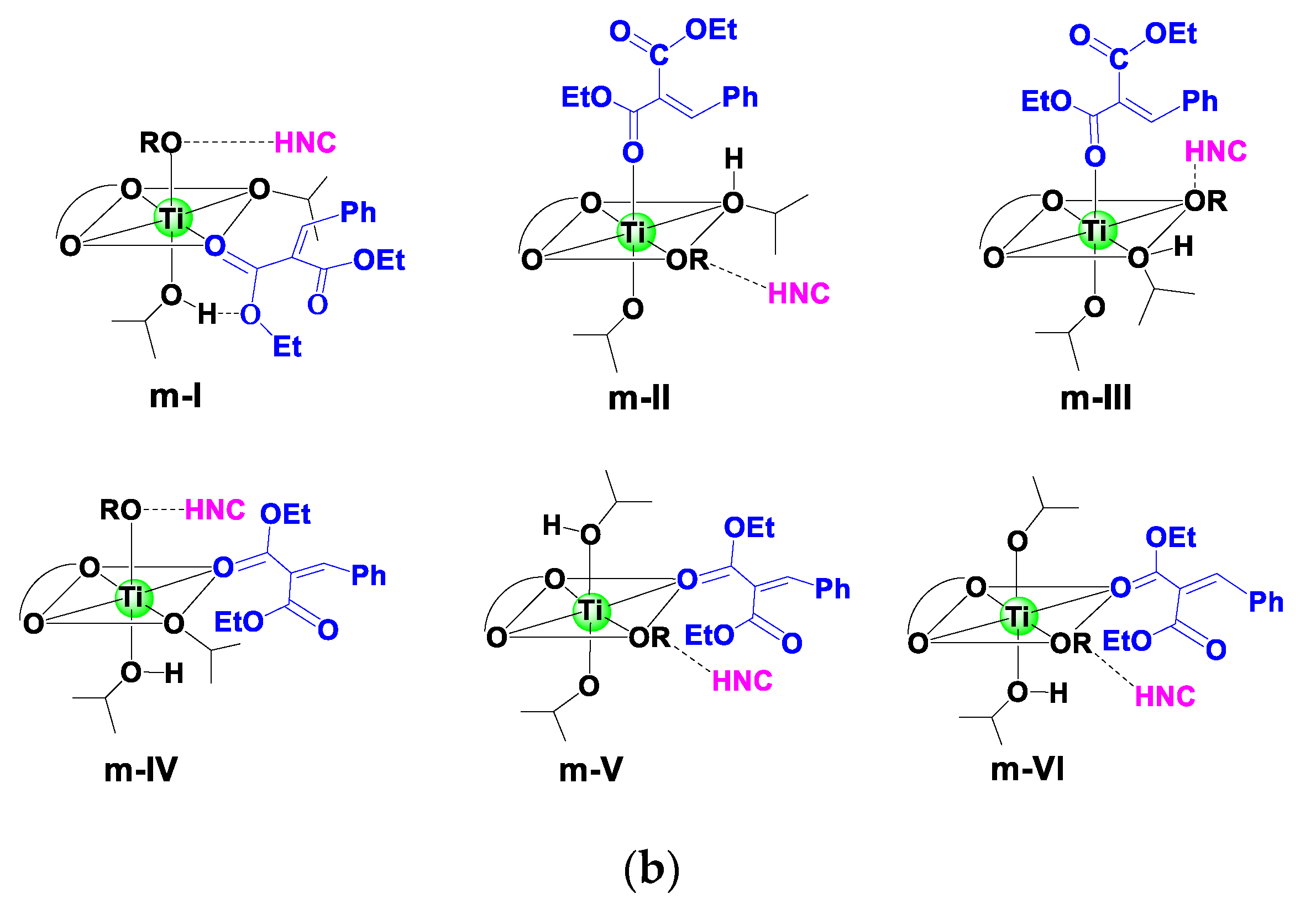
A previous study indicated that aldimine and HOiPr could coordinate to the Ti(IV), forming a reactive hexacoordinated Ti(IV)-complex in the Strecker reaction catalyzed by Ti(IV)-complex catalyst with cinchona alkaloid and achiral 3,3′-disubstituted 2,2′-biphenol ligands [29]. Based on experimental observation [28], a cinchonidine(L1)/Ti(IV)/(R)-biphenol (L2a) catalyst was employed as an active species in the reaction. Considering that there were two O-donors in the olefin substrate, a bidentate model was first studied in the present work, in which two carbonyl groups of olefin coordinated to the Ti(IV) center simultaneously, forming three possible hexacoordinated Ti(IV)-complexes (d-I–d-III). For comparison, the monocoordinated models (m-I–m-VI) were also investigated (Scheme 3). Nine low-energy Ti(IV)-complexes in mono- and bidentate models were located to allow the favorable si-face attack pathway observed in the experiment (see Table 1 and Figure S3). Table 1 shows that a bidentate Ti(IV)-complex (d-I-COM-si) had the lowest Gibbs free energy among nine models. In other words, this complex was the only starting species available in the reaction mixture.
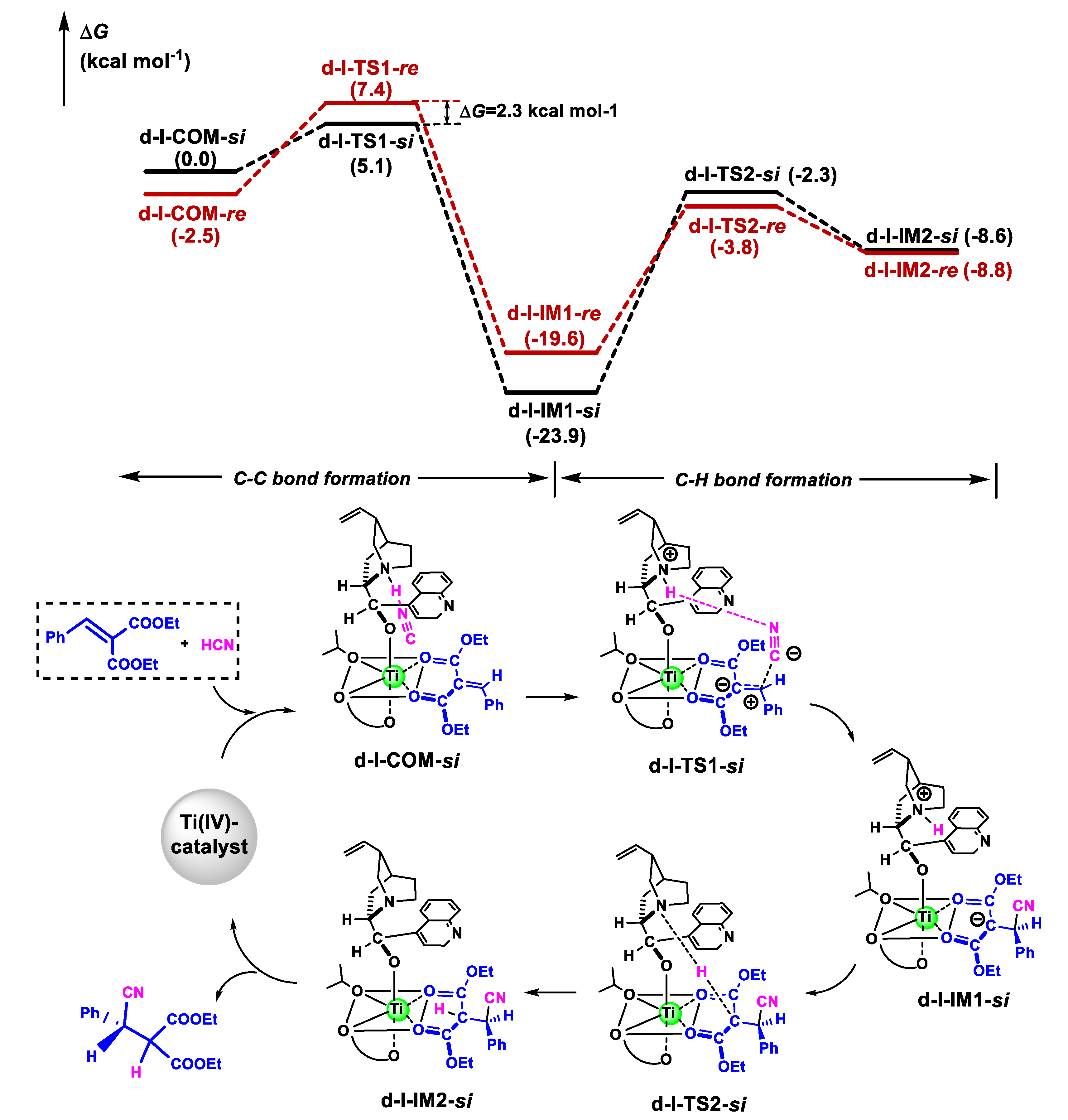
The reaction mechanisms of the cyanation of olefins in the presence of Ti(IV)-complex catalyst could be very similar for the nine coordination models, although the reaction might begin with different hexacoordinated Ti(IV)-complexes: C-C bond construction followed by H-shift. The potential energy surfaces for the si-face and re-face attack to produce R- and S-configuration enantiomers from d-I-COM-si and d-I-COM-re are shown in Figure 4.
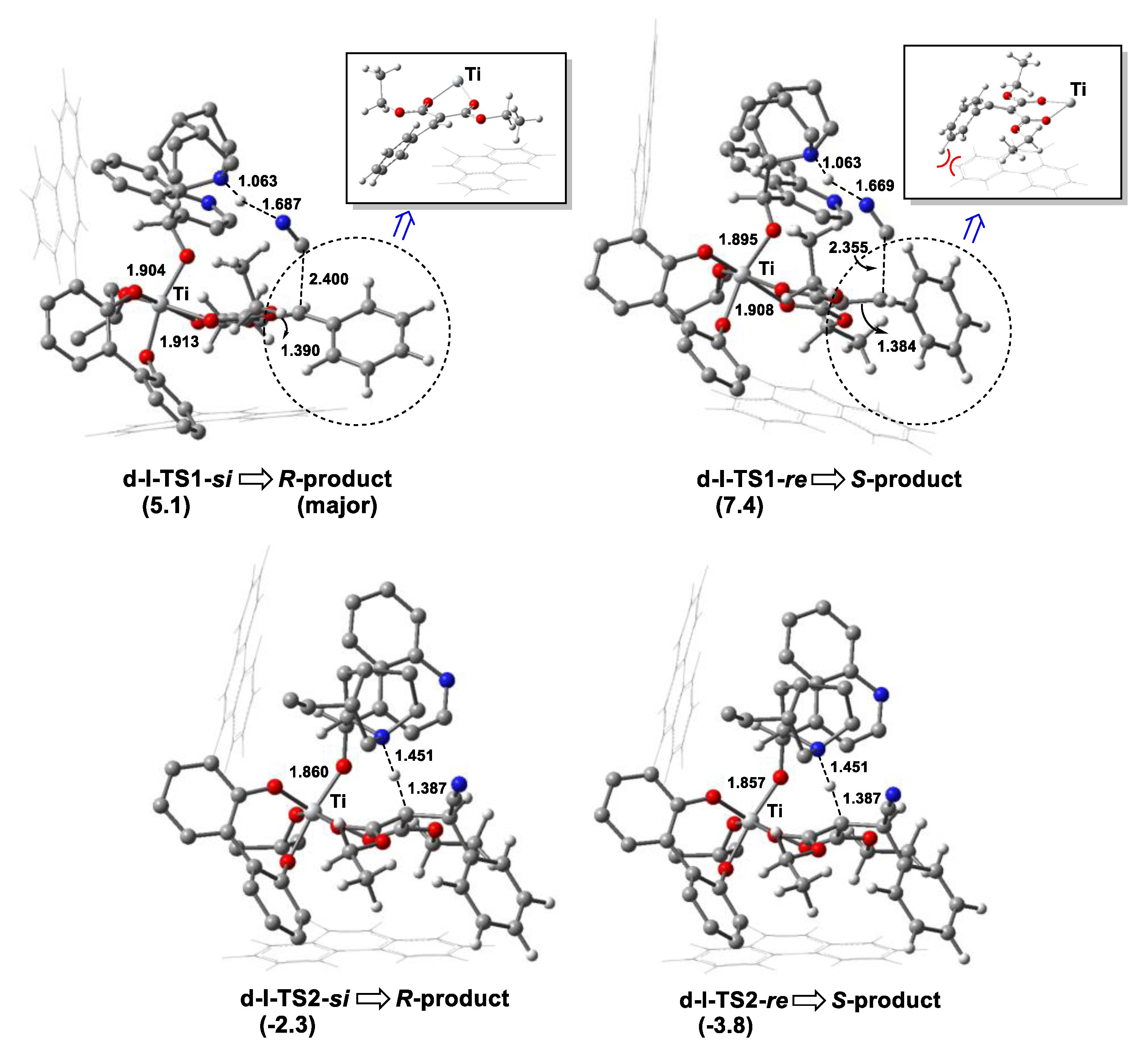
Like the noncatalytic reaction, the catalytic process occurred via a stepwise mechanism. Firstly, HNC was coordinated to the tertiary amine of cinchona alkaloid through hydrogen bonding, with an H···N distance of 1.612 Å. Then, the C-C bond was constructed by the attack of the CN group to olefin, with ΔG≠ of 5.1 kcal mol−1 via the TS d-I-TS1-si. Finally, the catalytic cycle was finished when the H atom was transferred from the N to C atom, via the TS d-I-TS2-si. A protonic reagent (e.g., HOiPr) could accelerate proton shift by hydrogen-bonding [29], and the H-shift barrier for HOiPr via the TS d-I-TS2-si-HOiPr was decreased by 3.4 kcal mol−1 (Figure S4). The activation barrier in the chiral-controlling step (C-C bond formation step) in the catalysis was lower than that for the noncatalytic process by 14.2 kcal mol−1. We also optimized the TSs in the chiral-controlling C-C bond construction along with the si-face attack in the d-II and d-III models (Figure S5). As expected, the activation free energies via d-II-TS1-si (ΔG≠ = 10.4 kcal mol−1) and d-III-TS1-si (ΔG≠ = 7.9 kcal mol−1) were higher than that via d-I-TS1-si (ΔG≠ = 5.1 kcal mol−1) in the d-I model. As shown in Figure 5, the ΔG of d-I-TS1-si was lower than that of d-I-TS1-re in a chiral-controlling step by 2.3 kcal mol−1 at 273 K, indicating the product with R-configuration was predominant. The theoretical enantioselectivity (ee %) was 96% using the Curtin–Hammett principle, which was close to that obtained experimentally (93% ee) [28].
We also studied the catalytic mechanism starting from the monodentate Ti(IV)-complex m-I-COM-si for comparison, and the corresponding C-C bond construction TS (m-I-TS1-si) in the chiral-controlling step was located (Figure 5). Compared with d-I-COM-si, the olefin substrate in m-I-COM-si was slightly weakened, with a large Wiberg bond index of 1.664 for the C1=C2 bond. Accordingly, m-I-TS1-si was less stable than d-I-TS1-si by 11.6 kcal mol−1. Thus, olefin tended to participate in the cyanation reaction in a bidentate fashion.
3.3. Origin of Stereoselectivity
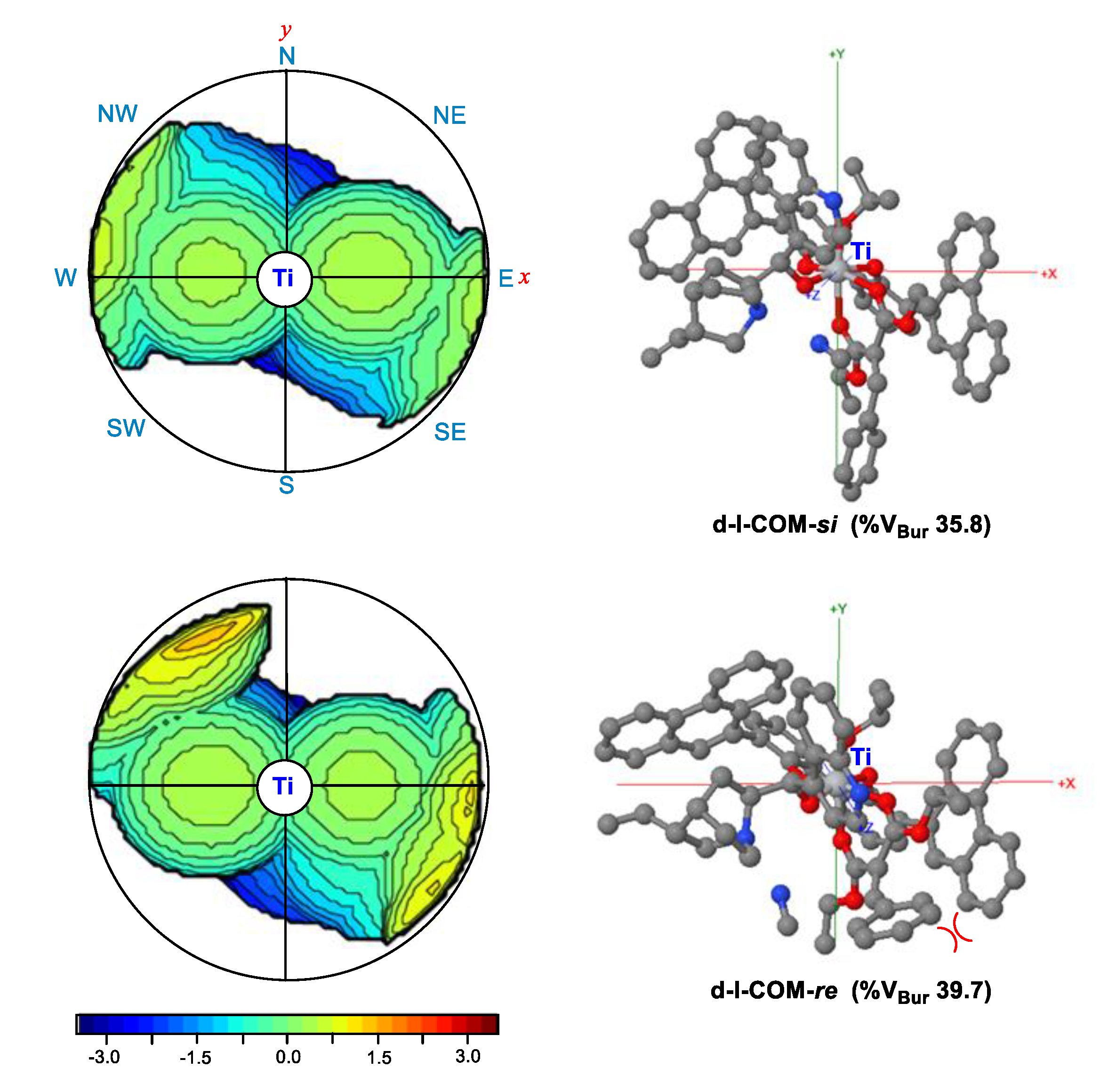
The hexacoordinated Ti(IV)-complexes d-I-COM-si and d-I-COM-re had a pocket-like chiral cavity, with the dihedral angles formed by the 9-phenanthryl groups at the 3,3′-position in L2a of 93.7° and 87.3°, respectively. The topographic steric maps [45,46,47] of d-I-COM-si and d-I-COM-re are shown in Scheme 4, which characterizes the surface that the ligand L2a offers to the olefin substrate. The percentage of buried volume (%VBur) quantified the first coordination sphere around the Ti center occupied by L2a ligand. For d-I-COM-re, %VBur was 39.7, which was larger than that of d-I-COM-si (35.8). Importantly, the 9-phenanthryl groups provided stronger hindrance in the north-western and south-eastern quadrants (yellow colored area). Consequently, the unfavorable steric repulsion between the Ph group of olefin and 9-phenanthryl group of ligand L2a (in the south-eastern quadrant) became more significant in d-I-COM-re as well as in the corresponding C-C bond formation TS.
Then, we further analyzed the structures of d-I-TS1-si and d-I-TS1-re (Figure 6). The Ph group of the olefin moiety in d-I-TS1-re was in proximity to the neighboring 3-substituted group in the biphenol ligand, with a Ph···Ph distance of about 2.5 Å. Accordingly, the steric repulsion raised the Pauli energy (∆E≠Pauli) of the reacting fragments at d-I-TS1-re (139.0 vs. 130.4 kcal mol−1) (Table 2). Consequently, the ΔG of d-I-TS1-re was higher than that of d-I-TS1-si (7.4 vs. 5.1 kcal mol−1). This steric repulsion was also visualized by noncovalent interaction analysis using Multiwfn software [48], in which the larger yellow area between the Ph of olefin and phenanthryl group of L2a (Figure S6) in d-I-TS1-re was observed. In contrast, this unfavorable steric interaction was avoided efficiently in d-I-TS1-si because the Ph groups were located far away. Consequently, two reacting fragments could interact easily, with a more stabilizing ΔE≠int (−5.6 kcal mol−1) and low reaction barrier of 5.1 kcal mol−1 in the C-C bond formation step.
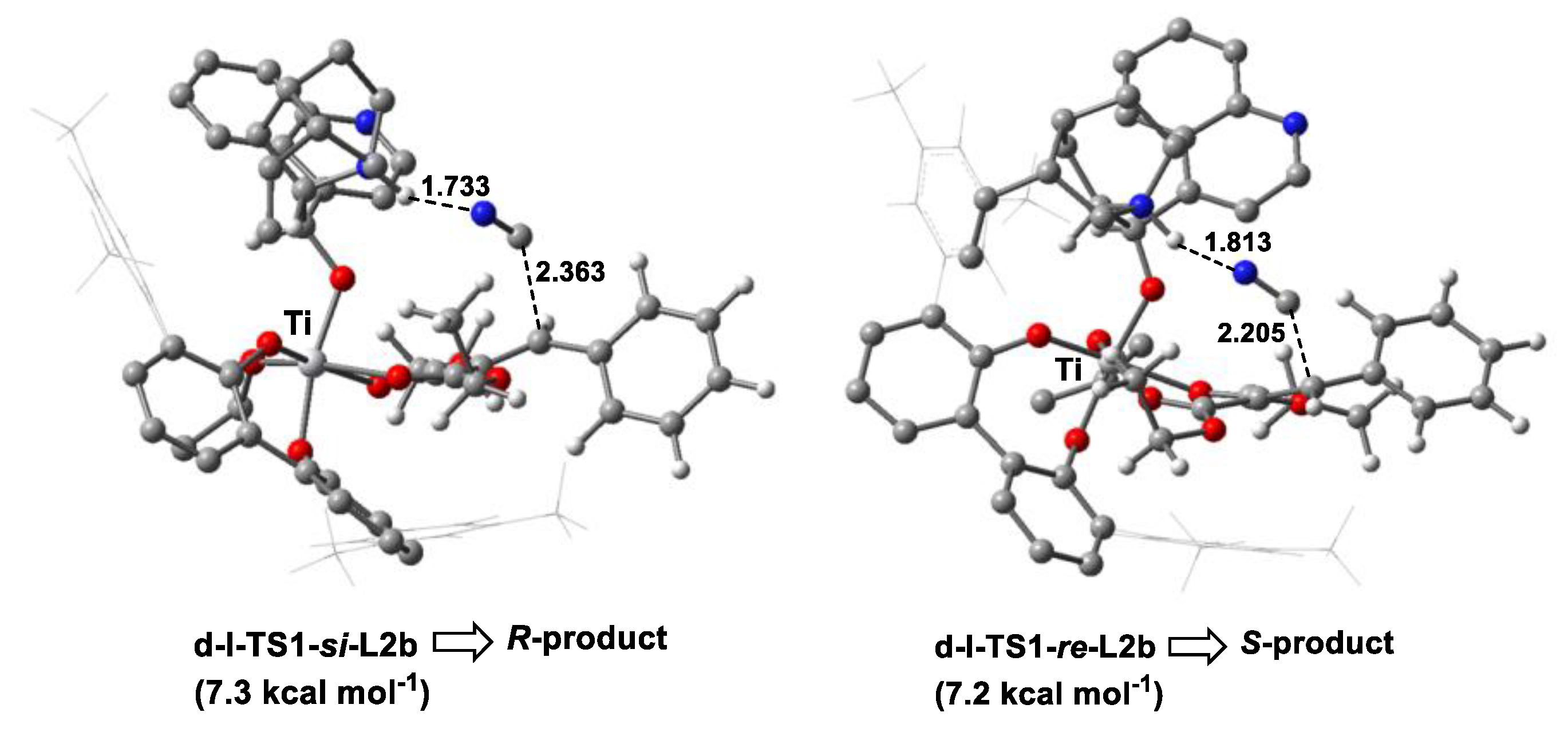
The influence of the 3,3′-substitute of biphenol on the stereoselectivity was further studied. When the 9-phenanthryl group in L2a was replaced by a 3,5-dimethyl phenyl group (L2b), an opening chiral pocket in d-I-COM-si-L2b was observed, with the dihedral angle of the two substituent groups at the 3,3′-position of 109.6°. The relative energy of the two competing TSs (d-I-TS1-si-L2b and d-I-TS1-re-L2b) in the chiral-controlling step (i.e., C-C bond construction step) was comparable (7.3 vs. 7.2 kcal mol−1), affording racemic products (Figure 7). These results indicated that the bulky substituent at the 3,3′-position of the biphenol ligand was essential for asymmetric induction in the Ti(IV)-complex-catalyzed cyanation of activated olefins.
4. Conclusions
DFT calculations on the reaction mechanism of asymmetric cyanation of activated olefin-catalyzed Ti(IV)-complexes revealed the following results:
- (i).
- Cinchona alkaloid facilitated the reaction between HOiPr and ethyl cyanoformate (CNCOOEt) to release the reacting species HCN (or HNC) by organocatalysis with free energy barrier of 26.0 kcal mol−1.
- (ii).
- The cyanation reaction of olefin proceeded via a two-step mechanism, in which the C-C bond construction was followed by H-transfer to generate a cyanide adduct. For noncatalytic reaction, the ∆G≠ for the rate-determining C-H bond construction step was up to 34.2 kcal mol−1, through a four-membered TS. In the catalytic reaction, the olefin coordinated to the self-assembly cinchonidine/Ti(IV)/(R)-3,3′-disubstituted biphenol catalyst in the bidentate model, forming a highly reactive hexacoordinated Ti(IV)-complex. The HNC activated by the quinuclidine tertiary amine moiety of cinchonidine ligand performed a nucleophilic attack towards the activated C=C bond of olefin, generating a cyanide adduct. The catalytic reaction required about 19.9 kcal mol−1 lower energy barrier compared to the noncatalytic reaction.
- (iii).
- EDA showed that the steric repulsion between the bulky group (e.g., 9-phenanthryl substituent) at the 3-position in the biphenol ligand and the phenyl group in olefin raised the Pauli energy (∆E≠Pauli) of the reacting fragments at the re-face attack TS, leading to the predominant R-product through the si-face attack, as observed in the experiment.
Supplementary Materials
The following are available online at https://0-www-mdpi-com.brum.beds.ac.uk/2073-4344/10/9/1079/s1, Figure S1: Laplacian (∇2ρ) and electronic density (ρ, in parentheses) of selected bond critical points (BCP) for molecular complex C-I-COM were obtained by AIM analysis, using Multiwfn software. Figure S2: Optimized geometry of transition state b-TS1-1. The intermolecular distance is in Angstroms (Å). The color definitions of atoms are Red = oxygen, blue = nitrogen, gray = carbon, and white = hydrogen. Figure S3: Optimized geometries of low-energy hexacoordinated Ti(IV)-complexes formed by coordinating olefin to metal center in (a) bidentate (b) monodentate fashion along si-face attack pathway. The intermolecular distance is in Angstroms (Å). The color definitions of atoms are Red = oxygen, blue = nitrogen, gray = carbon, and white = hydrogen. Figure S4: Optimized geometries of hexacoordinated Ti(IV)-complexes and H-shift transition state in the presence of HOiPr along si-face attack pathway. The intermolecular distance is in Angstroms (Å). The color definitions of atoms are Red = oxygen, blue = nitrogen, gray = carbon, and white = hydrogen. Figure S5: Optimized geometries of two competing transition states in C-C bond formation step in d-II and d-III models as well as their relative Gibbs free energies (in kcal mol−1). The intermolecular distance is in Angstroms (Å). The color definitions of atoms are Red = oxygen, blue = nitrogen, gray = carbon, and white = hydrogen. Figure S6: Visualization of the main noncovalent interaction described by contour plots of the reduced density gradient isosurfaces (density cutoff of 0.7 au) for transition states d-I-TS1-si and d-I-TS1-re. The surface color code is blue for strongly attractive, green for weakly attractive, and red for strongly repulsive interactions.
Author Contributions
Conceptualization, Z.S., C.H. and C.K.K.; methodology, Z.S., C.H. and C.K.K.; software, C.H.; validation, Z.S. and C.K.K.; formal analysis, Z.S. and N.S.; investigation, Z.S.; resources, C.H. and C.K.K.; data curation, N.S.; writing—original draft preparation, Z.S.; writing—review and editing, Z.S., C.H., N.S. and C.K.K.; visualization, Z.S.; supervision, C.H. and C.K.K.; project administration, C.K.K.; funding acquisition, Z.S. and C.K.K. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the Framework of International Cooperation Program managed by the National Research Foundation of Korea (2019K2A9A2A06023069, FY2019) and National Natural Science Foundation of China (21911540465).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Khan, N.H.; Kureshy, R.I.; Abdi, S.H.R.; Agrawal, S.; Jasra, R.V. Metal catalyzed asymmetric cyanation reactions. Coord. Chem. Rev. 2008, 252, 593–623. [Google Scholar] [CrossRef]
- North, M.; Usanov, D.L.; Young, C. Lewis acid catalyzed asymmetric cyanohydrin synthesis. Chem. Rev. 2008, 108, 5146–5226. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Liu, X.H.; Feng, X.M. Asymmetric strecker reactions. Chem. Rev. 2011, 111, 6947–6983. [Google Scholar] [CrossRef] [PubMed]
- Kurono, N.; Ohkuma, T. Catalytic Asymmetric Cyanation Reactions. ACS Catal. 2016, 6, 989–1023. [Google Scholar] [CrossRef]
- Zeng, X.P.; Sun, J.C.; Liu, C.; Ji, C.B.; Peng, Y.Y. Catalytic Asymmetric Cyanation Reactions of Aldehydes and Ketones in Total Synthesis. Adv. Synth. Catal. 2019, 361, 3281–3305. [Google Scholar] [CrossRef]
- Kouznetsov, V.V.; Galvis, C.E.P. Strecker reaction and α-amino nitriles: Recent advances in their chemistry, synthesis, and biological properties. Tetrahedron 2018, 74, 773–810. [Google Scholar] [CrossRef]
- Mazet, C.; Jacobsen, E.N. Dinuclear {(salen)Al} complexes display expanded scope in the conjugate cyanation of α,β-unsaturated imides. Angew. Chem. Int. Ed. 2008, 47, 1762–1765. [Google Scholar] [CrossRef]
- Sammis, G.M.; Danjo, H.; Jacobsen, E.N. Cooperative dual catalysis: Application to the highly enantioselective conjugate cyanation of unsaturated imides. J. Am. Chem. Soc. 2004, 126, 9928–9929. [Google Scholar] [CrossRef]
- Madhavan, N.; Weck, M. Highly Active Polymer-Supported (Salen)Al Catalysts for the Enantioselective Addition of Cyanide to α,β-Unsaturated Imides. Adv. Synth. Catal. 2008, 350, 419–425. [Google Scholar] [CrossRef]
- Mita, T.; Sasaki, K.; Kanai, M.; Shibasaki, M. Catalytic enantioselective conjugate addition of cyanide to α,β-unsaturated N-acylpyrroles. J. Am. Chem. Soc. 2005, 127, 514–515. [Google Scholar] [CrossRef]
- Fujimori, I.; Mita, T.; Maki, K.; Shiro, M.; Sato, A.; Furusho, S.; Kanai, M.; Shibasaki, M. Toward a rational design of the assembly structure of polymetallic asymmetric catalysts: Design, synthesis, and evaluation of new chiral ligands for catalytic asymmetric cyanation reactions. Tetrahedron 2007, 63, 5820–5831. [Google Scholar] [CrossRef]
- Tanaka, Y.; Kanai, M.; Shibasaki, M. A catalytic enantioselective conjugate addition of cyanide to enones. J. Am. Chem. Soc. 2008, 130, 6072–6073. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Kanai, M.; Shibasaki, M. Catalytic Enantioselective Construction of β-Quaternary Carbons via a Conjugate Addition of Cyanide to β,β-Disubstituted α,β-Unsaturated Carbonyl Compounds. J. Am. Chem. Soc. 2010, 132, 8862–8863. [Google Scholar] [CrossRef] [PubMed]
- Kurono, N.; Nii, N.; Sakaguchi, Y.; Uemura, M.; Ohkuma, T. Asymmetric hydrocyanation of alpha, beta-unsaturated ketones into beta-cyano ketones with the [Ru(phgly)2(binap)]/C6H5OLi catalyst system. Angew. Chem. Int. Ed. 2011, 50, 5541–5544. [Google Scholar] [CrossRef]
- Sakaguchi, Y.; Kurono, N.; Yamauchi, K.; Ohkuma, T. Asymmetric conjugate hydrocyanation of alpha, beta-unsaturated N-acylpyrroles with the Ru(phgly)2(binap)-CH3OLi catalyst system. Org. Lett. 2014, 16, 808–811. [Google Scholar] [CrossRef]
- Zhang, J.L.; Liu, X.H.; Wang, R. Magnesium complexes as highly effective catalysts for conjugate cyanation of α,β-unsaturated amides and ketones. Chem. Eur. J. 2014, 20, 4911–4915. [Google Scholar] [CrossRef]
- Dong, C.; Song, T.; Bai, X.F.; Cui, Y.M.; Xu, Z.; Xu, L.W. Enantioselective conjugate addition of cyanide to chalcones catalyzed by a magnesium-Py-BINMOL complex. Catal. Sci. Technol. 2015, 5, 4755–4759. [Google Scholar] [CrossRef]
- Hatano, M.; Yamakawa, K.; Ishihara, K. Enantioselective Conjugate Hydrocyanation of α,β-Unsaturated N-Acylpyrroles Catalyzed by Chiral Lithium(I) Phosphoryl Phenoxide. ACS Catal. 2017, 7, 6686–6690. [Google Scholar] [CrossRef]
- Kawai, H.; Okusu, S.; Tokunaga, E.; Sato, H.; Shiro, M.; Shibata, N. Organocatalytic asymmetric synthesis of trifluoromethyl-substituted diarylpyrrolines: Enantioselective conjugate cyanation of beta-aryl-beta-trifluoromethyl-disubstituted enones. Angew. Chem. Int. Ed. 2012, 51, 4959–4962. [Google Scholar] [CrossRef]
- Wang, Y.F.; Zeng, W.; Sohail, M.; Guo, J.; Wu, S.; Chen, F.X. Highly Efficient Asymmetric Conjugate Hydrocyanation of Aromatic Enones by an Anionic Chiral Phosphate Catalyst. Eur. J. Org. Chem. 2013, 2013, 4624–4633. [Google Scholar] [CrossRef]
- Liu, Y.; Shirakawa, S.; Maruoka, K. Phase-Transfer-Catalyzed Asymmetric Conjugate Cyanation of Alkylidenemalonates with KCN in the Presence of a Brønsted Acid Additive. Org. Lett. 2013, 15, 1230–1233. [Google Scholar] [CrossRef] [PubMed]
- Provencher, B.A.; Bartelson, K.J.; Liu, Y.; Foxman, B.M.; Deng, L. Structural study-guided development of versatile phase-transfer catalysts for asymmetric conjugate additions of cyanide. Angew. Chem. Int. Ed. 2011, 50, 10565–10569. [Google Scholar] [CrossRef] [PubMed]
- Jakhar, A.; Sadhukhan, A.; Khan, N.H.; Saravanan, S.; Kureshy, R.I.; Abdi, S.H.R.; Bajaj, H.C. Asymmetric Hydrocyanation of Nitroolefins Catalyzed by an Aluminum(III) Salen Complex. ChemCatChem 2014, 6, 2656–2661. [Google Scholar] [CrossRef]
- Nagata, T.; Tamaki, A.; Kiyokawa, K.; Tsutsumi, R.; Yamanaka, M.; Minakata, S. Enantioselective Electrophilic Cyanation of Boron Enolates: Scope and Mechanistic Studies. Chem. Eur. J. 2018, 24, 17027–17032. [Google Scholar] [CrossRef]
- Jakhar, A.; Ansari, A.; Nandi, S.; Gupta, N.; Khan, N.H.; Kureshy, R.I. Bronsted Basic Ionic Liquid as Catalytic and Reusable Media for Conjugate Cyanation of CF3-Substituted Alkylidenemalonates Using Acetone Cyanohydrin. ChemistrySelect 2017, 2, 11346–11351. [Google Scholar] [CrossRef]
- Wang, J.; Hu, X.L.; Jiang, J.; Gou, S.H.; Huang, X.; Liu, X.H.; Feng, X.M. Asymmetric Activation of Tropos 2,2′-Biphenol with Cinchonine Generates an Effective Catalyst for the Asymmetric Strecker Reaction of N-Tosyl-Protected Aldimines and Ketoimines. Angew. Chem. Int. Ed. 2007, 46, 8468–8470. [Google Scholar] [CrossRef]
- Wang, J.; Wang, W.T.; Li, W.; Hu, X.L.; Shen, K.; Tan, C.; Liu, X.H.; Feng, X.M. Asymmetric cyanation of aldehydes, ketones, aldimines, and ketimines catalyzed by a versatile catalyst generated from cinchona alkaloid, achiral substituted 2,2′-biphenol and tetraisopropyl titanate. Chem. Eur. J. 2009, 15, 11642–11659. [Google Scholar] [CrossRef]
- Wang, J.; Li, W.; Liu, Y.L.; Chu, Y.Y.; Lin, L.L.; Liu, X.H.; Feng, X.M. Asymmetric Cyanation of Activated Olefifins with Ethyl Cyanoformate Catalyzed by a Modular Titanium Catalyst. Org. Lett. 2010, 12, 1280–1283. [Google Scholar] [CrossRef]
- Su, Z.S.; Li, W.Y.; Wang, J.; Hu, C.W.; Feng, X.M. A theoretical investigation on the Strecker reaction catalyzed by a Ti(IV)-complex catalyst generated from a cinchona alkaloid, achiral substituted 2,2′-biphenol, and tetraisopropyl titanate. Chem. Eur. J. 2013, 19, 1637–1646. [Google Scholar] [CrossRef]
- Frisch, J.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Montgomery, J.A., Jr.; Vreven, T.; Kudin, K.N.; Burant, J.C.; Millam, J.M.; et al. Pople, Gaussian 09 (Revision D.01); Gaussian, Inc.: Wallingford, CT, USA, 2013. [Google Scholar]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Gonzalez, C.; Schlegel, H.B. An improved algorithm for reaction path following. J. Chem. Phys. 1989, 90, 2154. [Google Scholar] [CrossRef]
- Fernandez, I. Combined activation strain model and energy decomposition analysis methods: A new way to understand pericyclic reactions. Phys. Chem. Chem. Phys. 2014, 16, 7662–7671. [Google Scholar] [CrossRef]
- Zeist, W.J.; Bickelhaupt, F.M. The activation strain model of chemical reactivity. Org. Biomol. Chem. 2010, 8, 3118–3127. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, I.; Bickelhaupt, F.M. The activation strain model and molecular orbital theory: Understanding and designing chemical reactions. Chem. Soc. Rev. 2014, 43, 4953–4967. [Google Scholar] [CrossRef] [PubMed]
- Ess, D.H.; Houk, K.N. Distortion/Interaction Energy Control of 1,3-Dipolar Cycloaddition Reactivity. J. Am. Chem. Soc. 2007, 129, 10646–10647. [Google Scholar] [CrossRef]
- Thomas, B.E.; Loncharich, R.J.; Houk, K.N. Force Field Modeling of Transition Structures of Intramolecular Ene Reactions and ab Initio Transition Structures for an Activated Enophile. J. Org. Chem. 1992, 57, 1354–1362. [Google Scholar] [CrossRef]
- Hong, X.; Liang, Y.; Griffith, A.K.; Lambert, T.H.; Houk, K.N. Distortion-accelerated cycloadditions and strain-release-promoted cycloreversions in the organocatalytic carbonyl-olefin metathesis. Chem. Sci. 2014, 5, 471–475. [Google Scholar] [CrossRef]
- Houk, K.N.; Beno, B.R.; Nendel, M.; Black, K.; Yoo, H.Y.; Wilsey, S.; Lee, J.K. Exploration of pericyclic reaction transition structures by quantum mechanical methods: Competing concerted and stepwise mechanisms. J. Mol. Struct. Theochem. 1997, 398, 169–179. [Google Scholar] [CrossRef]
- Hopffgarten, M.; Frenking, G. Energy decomposition analysis. WIREs Comput. Mol. Sci. 2012, 2, 43–62. [Google Scholar] [CrossRef]
- Baerends, J.; Autschbach, J.; Bashford, D.; Bérces, A.; Bickelhaupt, F.M.; Bo, C.; Boerrigter, P.M.; Cavallo, L.; Chong, D.P.; Deng, L.; et al. ADF2016, Theoretical Chemistry; Vrije Universiteit: Amsterdam, The Netherlands, 2016; Available online: http://www.scm.com (accessed on 10 July 2020).
- Grimme, S.; Antony, J.; Ehrlich, S.; Krieg, H. A consistent and accurate ab initio parametrization of density functional dispersion correction (DFT-D) for the 94 elements H-Pu. J. Chem. Phys. 2010, 132, 154104. [Google Scholar] [CrossRef] [Green Version]
- Grimme, S.; Ehrlich, S.; Goerigk, L. Effect of the damping function in dispersion corrected density functional theory. J. Comput. Chem. 2011, 32, 1456–1465. [Google Scholar] [CrossRef] [PubMed]
- Sammis, G.M.; Jacobsen, E.N. Highly Enantioselective, Catalytic Conjugate Addition of Cyanide to α,β-Unsaturated Imides. J. Am. Chem. Soc. 2003, 125, 4442–4443. [Google Scholar] [CrossRef] [PubMed]
- Falivene, L.; Cao, Z.; Petta, A.; Serra, L.; Poater, A.; Oliva, R.; Scarano, V.; Cavallo, L. Towards the online computer-aided design of catalytic pockets. Nat. Chem. 2019, 11, 872–879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poater, A.; Ragone, F.; Giudice, S.; Costabile, C.; Dorta, R.; Nolan, S.P.; Cavallo, L. Thermodynamics of N-Heterocyclic Carbene Dimerization: The Balance of Sterics and Electronics. J. Am. Chem. Soc. 2008, 27, 2679–2681. [Google Scholar] [CrossRef]
- Poater, A.; Ragone, F.; Mariz, R.; Dorta, R.; Cavallo, L. Comparing the enantioselective power of steric and electrostatic effects in transition-metal-catalyzed asymmetric synthesis. Chem. Eur. J. 2010, 16, 14348–14353. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F.W. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
Scheme 1.
Asymmetric cyanation of activated olefin catalyzed by Ti(IV)-complex.

Scheme 2.
Formation of HCN or HNC from the reaction between HOiPr and CNCOOEt.

Figure 1.
Potential energy profiles for the formation of HCN or HNC by the reaction between HOiPr and CNCOOEt (R1) without a catalyst and in the presence of cinchona alkaloid (L1).
Figure 1.
Potential energy profiles for the formation of HCN or HNC by the reaction between HOiPr and CNCOOEt (R1) without a catalyst and in the presence of cinchona alkaloid (L1).

Figure 2.
Optimized geometries of reactants (R1 and HOiPr), cinchona alkaloid (L1), molecular complex (C-I-COM), transition states (TSs) (C-I-TS1 and C-I-TS2) and intermediates (C-I-IM1–C-I-IM3) for the HNC formation catalyzed by cinchona alkaloid (some H atoms in cinchona alkaloid are omitted for clarity) and their relative Gibbs free energies. The intermolecular distance is in Angstroms (Å). The color definitions of atoms are red = oxygen, blue = nitrogen, gray = carbon and white = hydrogen.
Figure 2.
Optimized geometries of reactants (R1 and HOiPr), cinchona alkaloid (L1), molecular complex (C-I-COM), transition states (TSs) (C-I-TS1 and C-I-TS2) and intermediates (C-I-IM1–C-I-IM3) for the HNC formation catalyzed by cinchona alkaloid (some H atoms in cinchona alkaloid are omitted for clarity) and their relative Gibbs free energies. The intermolecular distance is in Angstroms (Å). The color definitions of atoms are red = oxygen, blue = nitrogen, gray = carbon and white = hydrogen.

Figure 3.
Potential energy profile for the noncatalytic cyanation reaction of olefin.

Scheme 3.
Nine possible hexacoordinated Ti(IV)-complexes formed by coordinating olefin to Ti(IV) ion in a (a) bidentate and (b) monodentate fashion.
Scheme 3.
Nine possible hexacoordinated Ti(IV)-complexes formed by coordinating olefin to Ti(IV) ion in a (a) bidentate and (b) monodentate fashion.


Figure 4.
Gibbs free energy profiles for the catalytic cyanation of olefin mediated by Ti(IV)-complex along si- and re-face attack pathways and schematic catalytic cycle along si-face attack pathway as a representative.
Figure 4.
Gibbs free energy profiles for the catalytic cyanation of olefin mediated by Ti(IV)-complex along si- and re-face attack pathways and schematic catalytic cycle along si-face attack pathway as a representative.

Figure 5.
Optimized structures of the monodentate Ti(IV)-complex (m-I-COM-si) and TS (m-I-TS1-si) in the C-C bond construction step in the catalytic cyanation reaction of olefin along the si-face attack pathway and their relative Gibbs free energies. The intermolecular distance is in Angstroms (Å). The color definitions of atoms are red = oxygen, blue = nitrogen, gray = carbon and white = hydrogen.
Figure 5.
Optimized structures of the monodentate Ti(IV)-complex (m-I-COM-si) and TS (m-I-TS1-si) in the C-C bond construction step in the catalytic cyanation reaction of olefin along the si-face attack pathway and their relative Gibbs free energies. The intermolecular distance is in Angstroms (Å). The color definitions of atoms are red = oxygen, blue = nitrogen, gray = carbon and white = hydrogen.

Scheme 4.
Topographic steric maps for d-I-COM-si and d-I-COM-re. %VBur is the percentage of buried volume, obtained by SambVca 2.1 software [45,46,47].

Figure 6.
Optimized geometries of TSs in the C-C bond formation step (d-I-TS1-si and d-I-TS1-re) and H-transfer step (d-I-TS2-si and d-I-TS2-re) in the cyanation reaction of olefins catalyzed by Ti(IV)-complex along two competing pathways for R- and S-configuration products, respectively, associated with the relative Gibbs free energies (kcal mol−1). The intermolecular distance is in Angstroms (Å). The color definitions of atoms are red = oxygen, blue = nitrogen, gray = carbon and white = hydrogen.
Figure 6.
Optimized geometries of TSs in the C-C bond formation step (d-I-TS1-si and d-I-TS1-re) and H-transfer step (d-I-TS2-si and d-I-TS2-re) in the cyanation reaction of olefins catalyzed by Ti(IV)-complex along two competing pathways for R- and S-configuration products, respectively, associated with the relative Gibbs free energies (kcal mol−1). The intermolecular distance is in Angstroms (Å). The color definitions of atoms are red = oxygen, blue = nitrogen, gray = carbon and white = hydrogen.

Figure 7.
Optimized geometries of TSs in the C-C bond formation step (d-I-TS1-si-L2b and d-I-TS1-re-L2b) in the cyanation reaction of olefins catalyzed by Ti(IV)-complex with L2b along two competing pathways for R- and S-configuration products, respectively, associated with the relative Gibbs free energies. The intermolecular distance is in Angstroms (Å). The color definitions of atoms are red = oxygen, blue = nitrogen, gray = carbon and white = hydrogen.
Figure 7.
Optimized geometries of TSs in the C-C bond formation step (d-I-TS1-si-L2b and d-I-TS1-re-L2b) in the cyanation reaction of olefins catalyzed by Ti(IV)-complex with L2b along two competing pathways for R- and S-configuration products, respectively, associated with the relative Gibbs free energies. The intermolecular distance is in Angstroms (Å). The color definitions of atoms are red = oxygen, blue = nitrogen, gray = carbon and white = hydrogen.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Relative Gibbs free energy for the formation of hexacoordinated Ti(IV)-complexes.
| Model | Species | ΔG (kcal mol−1) 1 | |
|---|---|---|---|
| Monodentate | m-I | m-I-COM-si | 12.2 |
| m-I-COM-re | 7.5 | ||
| m-II | m-II-COM-si | 12.6 | |
| m-II-COM-re | 6.9 | ||
| m-III | m-III-COM-si | 12.6 | |
| m-III-COM-re | 16.0 | ||
| m-IV | m-IV-COM-si | 14.4 | |
| m-IV-COM-re | 13.5 | ||
| m-V | m-V-COM-si | 16.1 | |
| m-V-COM-re | 20.0 | ||
| m-VI | m-VI-COM-si | 16.2 | |
| m-VI-COM-re | 11.0 | ||
| Bidentate | d-I | d-I-COM-si | 0.0 |
| d-I-COM-re | −2.5 | ||
| d-II | d-II-COM-si | 5.5 | |
| d-II-COM-re | −0.6 | ||
| d-III | d-III-COM-si | 2.7 | |
| d-III-COM-re | 7.4 |
1 The energy of d-I-COM-si was set to zero.
Table 2.
Activation strain model (ASM) 1 analysis and energy decomposition analysis (EDA)2 for the catalytic cyanation reaction of olefins via d-I-TS1-si and d-I-TS1-re. The energies are given in kcal mol−1.
Table 2.
Activation strain model (ASM) 1 analysis and energy decomposition analysis (EDA)2 for the catalytic cyanation reaction of olefins via d-I-TS1-si and d-I-TS1-re. The energies are given in kcal mol−1.
| TS | ΔE≠strain | ΔE≠int | ΔE≠Pauli | ΔE≠oi | ΔV≠elstat | ΔE≠disp |
|---|---|---|---|---|---|---|
| d-I-TS1-si | 8.8 | −5.6 | 130.4 | −77.9 | −99.9 | −28.7 |
| d-I-TS1-re | 8.6 | −4.9 | 139.0 | −82.6 | −104.0 | −29.4 |
1 ASM calculations were done at the B3LYP-D3(BJ)/6-31+G**(SMD, toluene) level of theory; 2 EDA calculations were done at the B3LYP-D3(BJ)/TZ2P level of theory.
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Su, Z.; Hu, C.; Shahzad, N.; Kim, C.K. Asymmetric Cyanation of Activated Olefins with Ethyl Cyanoformate Catalyzed by Ti(IV)-Catalyst: A Theoretical Study. Catalysts 2020, 10, 1079. https://0-doi-org.brum.beds.ac.uk/10.3390/catal10091079
AMA Style
Su Z, Hu C, Shahzad N, Kim CK. Asymmetric Cyanation of Activated Olefins with Ethyl Cyanoformate Catalyzed by Ti(IV)-Catalyst: A Theoretical Study. Catalysts. 2020; 10(9):1079. https://0-doi-org.brum.beds.ac.uk/10.3390/catal10091079
Chicago/Turabian StyleSu, Zhishan, Changwei Hu, Nasir Shahzad, and Chan Kyung Kim. 2020. "Asymmetric Cyanation of Activated Olefins with Ethyl Cyanoformate Catalyzed by Ti(IV)-Catalyst: A Theoretical Study" Catalysts 10, no. 9: 1079. https://0-doi-org.brum.beds.ac.uk/10.3390/catal10091079
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.