
Electrocatalysis by Heme Enzymes—Applications in Biosensing


Instituto de Tecnologia Química e Biológica António Xavier, Universidade NOVA de Lisboa, av. da República, 2780-157 Oeiras, Portugal
*
Author to whom correspondence should be addressed.
Catalysts 2021, 11(2), 218; https://0-doi-org.brum.beds.ac.uk/10.3390/catal11020218
Submission received: 18 January 2021
/
Revised: 2 February 2021
/
Accepted: 3 February 2021
/
Published: 6 February 2021
(This article belongs to the Special Issue Perspectives in Bioinorganic Catalysis)
Abstract
:Heme proteins take part in a number of fundamental biological processes, including oxygen transport and storage, electron transfer, catalysis and signal transduction. The redox chemistry of the heme iron and the biochemical diversity of heme proteins have led to the development of a plethora of biotechnological applications. This work focuses on biosensing devices based on heme proteins, in which they are electronically coupled to an electrode and their activity is determined through the measurement of catalytic currents in the presence of substrate, i.e., the target analyte of the biosensor. After an overview of the main concepts of amperometric biosensors, we address transduction schemes, protein immobilization strategies, and the performance of devices that explore reactions of heme biocatalysts, including peroxidase, cytochrome P450, catalase, nitrite reductase, cytochrome c oxidase, cytochrome c and derived microperoxidases, hemoglobin, and myoglobin. We further discuss how structural information about immobilized heme proteins can lead to rational design of biosensing devices, ensuring insights into their efficiency and long-term stability.
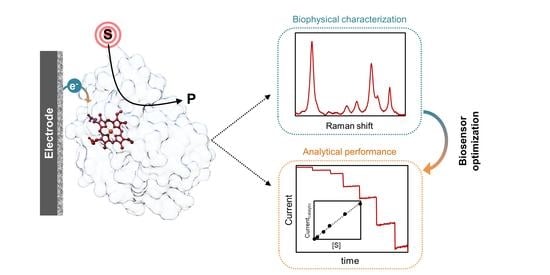
1. Introduction
Heme proteins perform a plethora of distinct cellular functions. The interactions of heme (iron protoporphyrin IX) with the surrounding protein matrix fine-tune its reactivity and the protein function. The heme moiety shuttles electrons between proteins in mitochondrial respiration, transports and stores O2 in hemoglobin and myoglobin, performs enzymatic chemical transformation by incorporating oxygen atoms into organic substrates in oxygenases, reduces O2 to water in oxygen reductases, and reduces H2O2 to water with concomitant oxidation of a variety of structurally different molecules in peroxidases, among other functions [1,2]. Due to unique electronic, magnetic, and thermodynamic properties of the heme, spectroscopic and electrochemical methods can provide a wealth of fine molecular details on heme proteins. The diversity of physiological roles, abundance of heme among the naturally occurring cofactors in metalloproteins, and the facilitated monitoring, isolation, and characterization, have furthermore placed heme proteins into a spotlight for development of biotechnological applications. They are engineered and optimized by Nature to sense and bind small diatomic molecules like O2, CO, and NO, as well as H2O2, the detection of which is highly relevant in biomedical, environmental, food safety, and water quality fields, among others. In biosensors, the heme enzyme plays a role of biological recognition element that sensitively and selectively (and sometimes even region- and stereo-specifically) detects substrates, which is subsequently communicated to a signal transducer that provides a measurable response proportional to the concentration of the analyte. The development of biosensors lies at interface of chemistry, biochemistry, physics, biophysics, and nanotechnology.
In this review, we focus on the biophysical aspects of electrochemical, and in particular amperometric biosensors, which owing to the high sensitivity, accuracy, and relatively low price, are currently among the most explored bioanalytical tools [3,4,5]. First, we present an overview of the basic concepts of enzyme biosensing and define analytical and kinetic parameters that are crucial for understanding and relative comparison of biosensor performance (Section 2). Then we describe different transducing schemes and distinct strategies for immobilization of heme enzymes on biocompatible electrode supports. The latter is crucial for biosensor development, as the immobilization needs to ensure efficient electrical communication between the enzyme and electrode and the preservation of the enzyme native structure. A rational design of the devices offers obvious advantages over empirical trial and error approaches, but it can be achieved only via in situ monitoring and optimization of individual steps in the development of these devices; we describe several specific biophysical methods that exclusively provide these insights. In Section 3, we explore the universe of heme proteins in biosensing. Emphasis is given to heme enzymes, which have been employed either in development of proof-of-concept biosensor or an actual commercial device. Special attention is given to peroxidases that are the main protagonists in the currently developed devices. We furthermore dwell on biosensors that employ microperoxidases, cytochromes P450, oxygen, and nitrite reductases and other heme proteins (cytochrome c [cyt c], hemoglobin and myoglobin), which can acquire catalytic properties under specific conditions. We conclude with an outlook (Section 4) that describes future perspectives and challenges to wider implementation of heme protein biosensors.
2. Amperometric Enzyme-Based Biosensors
A biosensor is an analytical device that detects a target analyte in complex matrices, in situ and without sample preparation, by exploiting the high specificity of biological recognition reactions. It is composed of a biological moiety capable of recognizing the analyte (e.g., antibody, cell, DNA, enzyme, tissue), which is in direct contact with a physico-chemical transducer (e.g., electrochemical, magnetic, optical, piezoelectric, thermal) that converts the bio-recognition event into a detectable signal. Owing to the huge diversity of potential targets, biosensors are highly sought after in a wide range of areas from research, medicine, industry and agriculture to food quality, environment, and public health [3,6].
The combination of enzymes as bio-components and electrochemical transducers constitutes the most common and widespread type of biosensors [4,5,7]. These devices combine the advantages of electroanalytical methods, such as high sensitivity, simplicity, low cost, potential for miniaturization, and portability, with the selectivity and high turnover rates of biological catalysts. Electrochemical biosensors are classified according to the physico-chemical signal that is generated by the bio-recognition reaction as (i) potentiometric, which detect charge accumulation or cell potential; (ii) conductometric, which monitor conductivity changes; (iii) impedimetric, which probe resistance and capacitance variations; and (iv) amperometric, which measure current [4,5,7,8]. The amperometric biosensors are the most explored option and the focus of this review.
The typical amperometric biosensor consists of a thin layer of enzyme immobilized on the surface of a conductive material, such as gold, platinum, silver, or carbon, including graphite, glassy carbon (GC), carbon nanotubes (CNTs), and boron-doped diamond, which acts as working electrode [7]. The attachment of the enzyme to the electrode can be achieved through a variety of immobilization methods (cf. Section 2.2). When placed in the sample solution, the enzyme interacts with the target analyte, i.e., the enzymatic substrate, and the ensuing catalytic reaction consumes or produces an electroactive species (e.g., (co-)substrate, product, redox mediator, or enzyme redox cofactor) that is detected by the transducer. The catalytic reaction can also be monitored indirectly via enzyme inhibitors or activators [7,9].
2.1. Electron Transfer in Enzyme Biosensors
Amperometric enzyme biosensors are divided into three classes based on the electron transfer (ET) mode between the enzyme and the electrode. In first-generation biosensors, the substrate or product of the enzymatic reaction is directly measured electrochemically; second-generation biosensors make use of a biological enzyme redox partner or an artificial redox mediator, which is responsible for the interfacial ET and electrical communication with the enzyme; and third-generation biosensors are based on direct electrochemical reduction or oxidation of the redox center of the enzyme in the presence of substrate (Figure 1) [10,11].
Development of first-generation biosensors was led by the work of Clark and Lyons on glucose oxidase (GOx) [12]. The enzyme was immobilized on a Pt electrode poised at a negative working potential to promote O2 reduction at −0.4 V vs. NHE (henceforward all potentials are quoted vs. normal hydrogen electrode, NHE, unless stated otherwise). The decrease of the co-substrate concentration (O2) during the enzyme catalyzed glucose oxidation (Equation (1)) was monitored at the electrode, and directly correlated to the concentration of glucose in the sample [13,14].
β-D-glucose + O2 → δ-D-gluconolactone + H2O2
First-generation biosensors are mainly based on O2-dependent enzymes, in particular oxygenases and oxidases. In the latter case, the H2O2 that is generated in the enzymatic reaction, can also be used to monitor the response of the biosensor. Its direct oxidation at electrode surfaces is typically measured at highly positive working potentials (e.g., +0.5 V) [14]. Although the detection mode in first-generation biosensors is quite simple, these devices have several drawbacks, such as the need for abundant amount of O2 to ensure that enzyme activity is not limited by the lack of the co-substrate or by its variation in the sample. Since high operating potentials are employed, the oxidation of interfering species present in the samples is also likely to occur, resulting in a low selectivity [13,14,15].
In second-generation biosensors, the electrochemical response is based on mediated electron transfer (MET). The use of mediators eliminates the difficulties that limit first-generation devices, i.e., direct measurement of O2 and H2O2, expanding the list of possible enzymes and target analytes. The electron shuttle molecules should have a facile and reversible electrochemistry at the working electrode and react easily with the active site of the enzyme, thus efficiently exchanging the electrons between the two; the generated current is proportional to the concentration of analyte. Importantly, mediator molecules should be oxidized/reduced at relatively low potentials (close to 0 V) to avoid the interference of other electroactive species with the response of the biosensor (e.g., sample matrix interferences) [5,7,10]. Artificial redox mediators include quinones derivatives, metal complexes like ferrocene, ferricyanide, ruthenium, and osmium complexes and organic dyes, such as methylene blue, methyl viologen and phenazines [16]. Low molecular weight ET proteins, like cytochromes and azurines, can also be used as redox mediators [17]. When the mediator protein is the physiological partner of the enzyme, the selectivity of the biosensor can be further increased due to the specific interaction between the two [18,19]. A common problem in second-generation/MET biosensors is the difficulty to secure the redox mediators on the electrode surface, which due to leakage into the sample solution limits the applications in continuous measurements and the re-usability of the devices. This can be overcome by (i) using redox polymers that have ET groups attached to the backbone (e.g., polypyrrole (PPY) viologen) [20,21], (ii) employing modified enzymes with chemically attached mediating groups (e.g., ferrocene modified GOx) [22], or (iii) reconstituting apoenzymes on cofactor-redox mediator functionalized electrodes (e.g., apo-GOx reconstitution on a pyrroloquinoline quinone-flavinadenine dinucleotide, PQQ-FAD, modified electrode) [23,24]. Although these approaches can reduce or even eliminate mediator leakage, they can also lead to loss of enzyme activity due to structural changes or unfavorable interactions with the redox matrices.
Third-generation biosensors have a simplified design that relies on direct electron transfer (DET) between the enzyme and the electrochemical transducer. Redox mediators or co-substrates are therefore not required [11,25], since the enzyme is in direct electrical contact with the electrode transducer. These devices offer superior selectivity, particularly in comparison with biosensors based on artificial mediators [11,15,25]. Furthermore, the working potential (cf. Section 2.3.1) is close to the redox potential of the employed enzyme, which in most of the cases falls within a physiological range that excludes extreme values (usually ≥−0.5 V at pH 7, an extreme limit exemplified by some ferredoxins) [26,27], thus decreasing the possibility for detection of interfering redox reactions. Still, establishing DET is not always an easy task, since the redox centers can be buried into the protein matrix, which can act like an isolation barrier hindering the ET process with the electrode. Additionally, direct contact with the electrodes can disturb the enzyme conformation and cause denaturation and activity loss [28]. Despite these concerns, several oxidoreductases display efficient DET upon immobilization on electrode surfaces, including cytochromes, laccases, peroxidases, and alcohol and cellobiose dehydrogenases [5,25,26,29]. The use of these enzymes in third-generation/DET biosensors has been gaining momentum in recent years [15,30]. This is in part due to the advances in protein engineering, which led to the production of tailored biocatalysts with improved characteristics, and alongside in materials sciences, which led to the development of new conductive nanomaterials that can facilitate interfacial ET and provide high surface area on electrodes for enhanced loading of enzymes [31,32,33,34].
2.2. Enzyme Immobilization
Enzyme attachment to the electrode surface is the most critical part of biosensor design. The main types of immobilization account for adsorption, encapsulation, and chemical binding, namely covalent coupling and cross-linking (Figure 2) [7,35,36]. The strategy of choice often results from the combination of several of these methods [37,38,39,40]. In general terms, enzyme immobilization should guarantee (i) biocompatibility, (ii) efficient electrical contact between enzyme and transducer that also ensures exposed substrate binding sites, and (iii) preservation of catalytic activity [7,35,36,41]. For the chemical modification of electrodes, we highlight the use of self-assembled monolayers (SAMs) of alkanethiols, which represent one of the most common electrode coatings [42,43]. Due to the high affinity of the thiol groups for metals, bifunctional SAMs form covalent bonds with metallic electrodes, while the head groups (e.g., thiols, disulphides, amines, acids, and silanes) offer a variety of possibilities and promote interactions that can anchor the enzymes to the modified electrode [42,44]. The electrostatic interaction, for example, is driven by surface charge distribution of the enzyme and the corresponding SAM.
Physical adsorption of the enzyme to a bare or modified electrode is one of the simplest immobilization methods (Figure 2a). The enzyme solution is typically deposited onto the electrode surface and allowed to incubate for a determined period of time; the weakly adsorbed molecules are afterwards removed by washing. Adsorption is governed by electrostatic and hydrophobic interactions and van der Waals forces, which are considered to be less disruptive for enzyme structure and activity than e.g., covalent binding [35,36]. Nonetheless, the direct contact with bare metal or carbon electrodes can cause enzyme denaturation [28]. Functionalization of the electrodes with biocompatible materials (e.g., SAMs) can overcome this issue [43]. Furthermore, due to the weak binding, enzyme desorption can occur as a result of varying experimental conditions, such as temperature, pH and ionic strength [35]. Thus, biosensors based on adsorbed enzymes often have poor operational and storage stability (cf. Section 2.3.1).
Encapsulation/entrapment into porous matrices (Figure 2b) fixes the protein molecules on the electrode support, while allowing the access of substrate molecules [35,36]. Commonly used materials include carbon pastes, conducting and non-conducting polymers, such as polyaniline (PANI), PPY, Nafion, and silica sol-gels [5]. Electrodes can be prepared by simple co-deposition of the enzyme, mediators, and the encapsulation matrix or by more complex processes, such as electropolymerization in the presence of the enzyme and the monomers of the entrapment film [35]. In this manner, the stability and lifetime of the biosensor can be increased due to reduced leakage of enzyme and prevention of non-specific adsorption of fouling agents. In addition, the access of interfering species to the enzyme layer can be selectively blocked by e.g., size exclusion and charge repulsion. The main drawbacks of encapsulation/entrapment are (i) the potentially limited access of substrate to the active site due to the diffusion barrier created by the matrix film, (ii) the reduced enzyme loading capacity, and (iii) the possible alterations to the enzyme structure [7,35,36].
Covalent binding enables the formation of stable complexes between the enzyme and the electrode support (Figure 2c). The coupling involves surface functional groups of the enzyme, such as amino (Lys), thiol (Cys), or carboxylic acid (Asp, Glu) side chains [35,45,46]. It is usually carried out after an initial activation of the immobilization surface using glutaraldehyde or carbodiimides (e.g., 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide, EDC, coupled to N-hydroxysuccinimide, NHS). This method can facilitate the control of enzyme orientation on the interface, particularly if the enzyme possesses only one tethering functional group [31,47]. Despite these advantages, covalent binding may cause alterations of enzyme conformation and consequently decreased activity and stability. In addition, electrode reproducibility is often poor and the process may involve several, sometimes toxic reagents [35].
Enzyme immobilization by cross-linking relies on bifunctional agents (usually glutaraldehyde) to generate a three-dimensional enzyme network on the electrode surface (Figure 2d) [35,46]. The enzyme molecules can be linked to each other or to a functionally inert protein, such as bovine serum albumin (BSA) [9,48]. The immobilization is highly stable and allows for increased enzyme loading in comparison to covalent binding. Still, like covalent binding, it can lead to loss of activity due to the distortion of enzyme structure and chemical alterations during the cross-linking reaction [35,46].
Taken together, each immobilization strategy has its advantages and drawbacks. The most adequate approach is case dependent, since it must take into account enzyme characteristics and envisioned application.
2.3. Biophysical Characterization of Amperometric Enzyme Biosensors
2.3.1. Biosensor Performance
Amperometric biosensors detect the substrate of the enzyme, the analyte, upon applying a potential that drives the redox reaction to the working electrode. Measurements are performed by chrono-amperometry or potential scanning techniques including cyclic, differential pulse and square-wave voltammetries [7,28]. Herein, we review the basic concepts of amperometric methods for detection of biosensor responses and define the main analytical and kinetic parameters that are used to evaluate its performance. Detailed reviews of the subject can be found elsewhere [5,8,49].
The working potential of the biosensor is the value at which the enzyme activity is determined as a current variation. It is related to the reduction potential of the electroactive species that is monitored (e.g., (co-)substrate or product, redox mediator, and enzyme redox cofactor in first-, second-, and third-generation biosensors, respectively). The working potential is usually set at a higher value than the current peak for the oxidation of the electroactive species (or at a lower value if the catalytic reaction is monitored via reduction of the electroactive species). Note that by increasing the overpotential, i.e., deviation from the equilibrium potential, the rate of the ET reaction can be considerably increased, and thus the response of the biosensor (Figure 3A). However, care must be taken not to compromise the biosensor selectivity by applying too high (or too low) potentials at which interfering species can also be oxidized (or reduced) (Figure 3B) [5].
In chrono-amperometric detection, the electrode is poised at a constant potential (the working potential of the biosensor) and the current is monitored over time. The current change observed upon the injection of the analyte is proportional to its concentration in solution. It is the most commonly used technique for practical applications owing to its simplicity, low background signals and detection limits [7]. In voltammetric detection the potential is scanned over a specific range while the current is measured as a function of the applied potential. The catalytic current response is usually a peak or a plateau that is proportional to the concentration of the analyte. These techniques, and cyclic voltammetry (CV) in particular, are useful in development stages of the biosensor, e.g., for selecting the working potential. If enzyme DET response is observed in the absence of analyte (i.e., non-catalytic signal) its reduction potential can also be determined. Noteworthily, the reduction potential of the same enzyme immobilized on different modified electrodes can vary significantly [51,52]. Furthermore, it can be different from the reduction potential of the enzyme in solution, suggesting that the electrocatalytic response may arise from denatured enzyme species. The reduction potential is thus a reliable indicator of possible immobilization-induced alterations of the enzyme. Nonetheless, structural characterization of the immobilized enzymes using sensitive spectroscopic methods (cf. Section 2.3.2) enables a more accurate identification of non-native states.
After defining the operational conditions, i.e., the detection technique, working potential, and other experimental variables (e.g., pH, ionic strength, temperature) the calibration of the biosensor is performed by measuring the current after adding a standard solution of the analyte. The calibration plot is obtained by representing the steady-state current response corrected for the background as a function of analyte concentration or its logarithm [8].
The performance of the biosensor is evaluated by a series of analytical parameters: sensitivity, linear concentration range, limit of detection (LOD), reproducibility, selectivity/response to interferences, stability, and response time [8].
The sensitivity corresponds to the response of the biosensor per unit of analyte concentration. It is determined from the slope of the linear calibration curve and is typically represented in units of current or current density per concentration (e.g., A M−1 or A M−1 cm−2, respectively). It is the most commonly used parameter to evaluate and compare biosensor performance.
Linear range is the range of analyte concentrations for which the biosensor response varies linearly. It is obtained from the linear section of the calibration curve for a set of measurements with different concentrations of analyte and is usually represented as I = m C, where I is the current response, m is the sensitivity of the biosensor, and C is the concentration of the analyte. Ideally, the biosensor should have a wide linear working range to avoid sample dilution or pre-concentration steps.
The limit of detection can be defined as the minimum analyte concentration that is detected, but not necessarily quantified, by the biosensor. According to IUPAC definitions, the LOD of an analytical method is related to the background signal and its fluctuations [53]. In the case of biosensors, it is most commonly estimated from the calibration curve parameters, typically as the analyte concentration at a signal-to-noise ratio of 3.
Reproducibility is the ability of a biosensor to generate the same response in repeated experiments. It is generally determined as the relative standard deviation of the current response at a specific analyte concentration (within the linear range) or of the sensitivity of multiple biosensor preparations.
The selectivity is related to the ability of the biosensor to identify the target analyte in a complex sample. It is usually expressed as the percentage of response variation upon addition of a potential interfering species to the solution. The selectivity depends on both the bio-recognition element and the transducer. Many enzymes used in biosensing are highly specific for their substrates and the selectivity is usually high. Still, non-selective enzymes can be useful for simultaneous monitoring of a group of analytes, such as phenolic compounds or catecholamines that can be found in clinical, environmental, or food samples [54,55,56,57].
Response time is the time required to reach 95% of the maximum current signal after adding the analyte to solution. It depends on the diffusion rates of the compounds involved in the detection reaction (e.g., analytes, products, and redox mediators) through the biosensor film and can be affected by the speed at which the solution is homogenized. The response time is usually proportional to the turnover rate of the enzyme, a fast turnover resulting in short response times.
The stability is usually estimated from activity measurements for one analyte concentration over time, in comparison with the initial value. The biosensors can also be calibrated multiple times to monitor variations in sensitivity, linear range, and LOD, providing a more complete description of their performance over time. The stability of the biosensor is often evaluated in terms of operational lifetime (repeated measurements using the same electrode) and storage stability or shelf-life (analysis of electrode batches at different times).
The kinetic parameters of the biosensor can be estimated if the enzyme reaction at the electrode surface follows a Michaelis-Menten type kinetic behavior [58]. The catalytic currents are fitted by a modified Michaelis-Menten equation:
where Icat is the catalytic current, Imax is the maximum Icat observed at enzyme saturating conditions, C is the analyte concentration, and is the apparent Michaelis-Menten constant. The refers to the immobilized enzyme and frequently differs from the solution value, sometimes over orders of magnitude. An increased indicates that there is a significant analyte diffusion barrier between the sample and the biosensing layer [8]. It may reflect restricted access of the analyte to the active site on the enzyme, due to e.g., a low permeability of the biosensor film and/or unfavorable orientation of the enzyme on the transducer surface. Furthermore, enzyme interaction with the immobilization matrix can induce conformational changes that decrease the affinity for the substrate (increase of ) or cause partial or total inactivation (decrease of Imax) [5].
2.3.2. Structural Characterization of Immobilized Proteins
Structural features of the immobilized enzyme govern the analytic and kinetic performance of the biosensor. Coupling of electrochemistry with microscopy and spectroscopy can, in particular, provide information about enzyme amount, structure, and conformation upon immobilization, which cannot be assessed by electrochemical methods [59,60,61,62]. Ideally, the structural/morphologic characterization should be obtained simultaneously with the electrochemical measurements. However, this is not always possible, and the immobilized enzymes are often analyzed by spectroscopy and microscopy prior to (or after) the electrochemical experiment and not necessarily on the working electrodes [59].
Atomic force (AFM) and scanning electron (SEM) microscopies are the most widely used to characterize biosensor surface morphology, providing information on enzyme distribution on the electrode and homogeneity of the films [59]. Spectroscopic techniques reveal insights into the enzyme-electrode interfaces on a molecular level. In particular, the structure, orientation, and cofactor redox states of the enzyme can be probed using UV-Visible, fluorescence, circular dichroism (CD), Fourier-transform infrared spectroscopy (FTIR), resonance Raman (RR), and in some cases surface-enhanced IR (SEIRA) and RR (SERR) spectroscopies [60,61,63,64,65,66]. In the case of heme enzymes, UV-Visible, RR, and SERR spectroscopy typically probe heme active site environment, whereas CD and FTIR spectroscopies report on subtle changes of the protein secondary structure upon immobilization (Figure 4) [67,68,69,70].
Owing to its simplicity and wide availability, UV-Visible spectroscopy is useful for characterization of biosensors prepared on transparent conductive oxide electrodes, such as indium tin oxide (ITO) films deposited on glass. The analysis can also be performed by reproducing the enzyme films on glass and quartz slides or, alternatively, in solution, after enzyme incubation with the composite materials that are used to prepare electrodes [71]. In these cases, identical experimental conditions cannot be guaranteed, such as enzyme orientation on the electrode and effects of electric fields upon application of potential. The Soret absorption band of the heme group, at around 410 nm, is typically monitored due to the high extinction coefficient and the sensitivity to the redox state of the heme; the weaker Q and charge-transfer (CT) bands, which can probe heme coordination state, are analyzed less frequently [72]. Band shifting in comparison with the solution spectra is indicative of possible denaturation of the immobilized enzyme [71,73,74] (Figure 3B).
The high frequency bands (1300–1700 cm−1) in RR and SERR spectra of heme proteins obtained under Soret band excitation are sensitive to the redox and spin state and coordination pattern of the iron. SERR spectroscopy selectively enhances the signal of molecules situated in close proximity to plasmonic metal surfaces, which can serve as working electrodes [60,62,75]. In this manner, it can be coupled with electrochemical methods, enabling in situ probing of the enzyme structure in the immobilized state under biosensor working conditions. SERR spectroelectrochemistry is, nonetheless, limited mainly to nanostructured Ag electrodes. On the other hand, RR can in principle be used to evaluate the immobilized enzyme on any type of electrode, but the concentration must be much higher. The RR and SERR spectra of the enzyme in the biosensor are usually compared to reference RR spectra of the native enzyme in solution to evaluate the effects of immobilization on structural features [67,70,76]. They can provide fine details on eventual exchange of heme ligands upon immobilization, which can be correlated with the catalytic and thermodynamic properties of the biosensor [70,77,78].
FTIR reveals detailed information about the secondary structure of enzymes in biosensors [79,80,81]. Analysis is based on protein amide I and amide II bands at approximately 1650 and 1550 cm−1, respectively. The FTIR spectra of the native enzyme in solution and immobilized state are compared [68,82] and eventual alterations of the relative intensities, positions and shapes of the amide I and amide II bands indicate enzyme structural changes upon immobilization (Figure 3A). In addition, the orientation of enzymes attached onto Au electrodes can be probed by SEIRA spectroscopy [60,61,76].
Similarly, CD can probe structural properties of immobilized enzymes revealing changes in the conformation of the peptide backbone and interactions with other molecules [68,83].
Fluorescence spectroscopy can also be used to evaluate immobilized heme enzymes in biosensors, since the intrinsic fluorescence of Trp residues, if present, is sensitive to the polarity of its environment and to the vicinity of the heme group [64,65]. Its modulation is a marker for the accessibility of the substrate to the heme and thus of the folding of the immobilized enzyme [84,85].
3. Biosensors Based on Heme Proteins and Enzymes
3.1. Peroxidases
Peroxidases constitute a large group of enzymes that catalyze the oxidation of a broad range of organic and inorganic substrates using H2O2 or organic hydroperoxides as electron acceptors [86,87,88]. The typical oxidation reaction of aromatic amine and phenolic substrates (RH) to free radicals (R°) is represented by Equation (3).
2RH + H2O2 → 2R° + 2H2O
The majority of known peroxidases contain a single heme b cofactor. The iron is coordinated by a His residue on the proximal heme face, while the sixth axial site is either vacant (penta-coordinated, 5c) or it carries a weakly bound water molecule (hexa-coordinated, 6c) to enable binding of the peroxide substrate [51,86]. The reduction potentials of the Fe(III)/Fe(III) couple are generally negative, consistent with the stability of the Fe(III) state that is required to initiate the catalytic reaction, and display a high variability (e.g., ca. −0.160 V for soybean ascorbate peroxidase and −0.300 V for horseradish peroxidase-C, HRP-C) [51].
Heme peroxidases share a common catalytic cycle involving the formation of high valence oxy-ferryl heme intermediate species, compound I ([Fe(IV)=O]+•) and compound II ([Fe(IV)=O]+), which are responsible for substrate oxidation (Figure 5, dashed line). The reaction is initiated by the two-electron oxidation of the resting Fe(III) state enzyme by H2O2, leading to the formation of compound I (Equation (4)), which contains the oxy-ferryl center and an organic cation radical located on the heme (porphyrin π-cation radical) or, less commonly, on a tryptophan or tyrosine residue [89]. The cation radical is reduced by the first substrate molecule, generating compound II (Equation (5)), which reacts with a second substrate molecule and regenerates the resting state peroxidase (Equation (6)) [51,87,88]. In the case of immobilized peroxidases, the electrode can mimic the substrate molecules (RH) providing electrons for H2O2 reduction (Figure 5, solid line).
Fe(III) + H2O2 → Compound I + H2O
Compound I + RH → Compound II + R°
Compound II + RH → Fe(III) + R° + H2O
3.1.1. Detection Modes in Peroxidase-Based Biosensors
Peroxidase modified electrodes can be used for the detection of H2O2 and organic hydroperoxides (oxidizing substrates), enzyme reducing substrates, including numerous phenolic compounds and aromatic amines, as well as non-typical substrates, such as uric acid, trichloroacetic acid (TCA), and nitrite [55,56,90]. The main biosensor response mechanisms are detailed bellow. Although they refer to heme peroxidases, the detection modes are similar for other heme proteins. In most cases, the electrocatalytic activity is monitored through MET or DET (in second- or third-generation biosensors, respectively) via an increase of cathodic current, which is related to the concentration of the substrate (i.e., the analyte) in solution [91].
In DET-based biosensors for detection of H2O2, the ferric [Fe(III)] resting state peroxidase is first oxidized by H2O2, forming compound I (Figure 5, solid line). The intermediate is then reduced to compound II and the ferric enzyme is restored, via heterogeneous ET directly from the electrode. The catalytic currents are typically measured by amperometric methods at working potentials between +0.15 and +0.5 V [37,92,93], owing to the high reduction potentials of the catalytic redox couples Fe(III)/compound I and compound I/compound II (e.g., HRP-C E0’CI/CII +0.898 V) [51,87]. It is noteworthy that cathodic currents are often observed at much lower potentials, consistent with the Fe(III)/Fe(II) redox transition (e.g., −0.35 V for HRP immobilized on modified GC electrodes [94]). In this case, it is likely that the H2O2 detection follows an alternative non-native route, which proceeds via formation of the ferrous [Fe(II)] enzyme. This low potential redox cycle of peroxidase electrocatalysis has been associated with non-native enzyme states and transient formation of inactive catalytic intermediates (e.g., Fe(II)-O2), which can also be generated in the presence of high concentrations of H2O2 [51,91,95,96]. Among the DET mechanisms proposed to rationalize these experimental findings (cf. reference [96] for details), we highlight the one that postulates that the alternative electrocatalytic reaction follows a catalase-like mechanism, in which H2O2 acts as an oxidant and as a reductant. Consequently, the generated compound I (Equation (4)) can be reduced by H2O2, returning the peroxidase to the Fe(III) state and producing O2 (Equation (7)). After reduction of the Fe(III) enzyme at the electrode (Equation (8)), O2 reacts with the ferrous peroxidase (Equation (9)) to form Fe(II)–O2 oxy-peroxidase, which in turn undergoes electrochemical reduction at the electrode (Equation (10)), at the potential of the Fe(III)/Fe(II) reduction.
Compound I + H2O2 → Fe(III) + O2 + H2O
Fe(III) + H+ + e− → Fe(II), at electrode
Fe(II) + O2 → Fe(II)−O2
Fe(II)–O2 + 2H+ + 2e− → Fe(II) + H2O2, at electrode
This reaction produces H2O2 and regenerates the Fe(II) enzyme for a new cycle (Equations (8)–(10)) [73]. Due to the exceptionally efficient redox chemistry of the heme, it is likely that this non-physiological process contributes to the electrocatalytic response of different heme enzyme-based electrodes, including peroxidases, catalases, cyt c, globins, and cytochrome P450 [68,94,97]. The extent of this contribution is likely to be related to the rates of the chemical and electrochemical reactions and the relative stability of the catalytic intermediates [98].
DET-based peroxidase biosensors for detection of non-classical peroxidase substrates, such as nitrite, NO, and TCA have also been developed [81,99,100,101]. In these systems, the peroxidase acts as a reductase, and the Fe(II) state is responsible for the direct reduction of the substrates at negative working potentials.
Mediated peroxidase biosensors rely on the reduction of compound I and compound II by a redox mediator or enzyme substrate (Figure 5). The oxidized mediator is then reduced at the electrode surface generating the catalytic current. These biosensors operate at the reduction potential of the mediator [91].
MET-based biosensors for detection of H2O2 have taken advantage of numerous redox mediators, including aromatic amines and phenolic substrates, thionine, methylene blue, methylene green, ferrocenes, hydroquinone, osmium, and ruthenium complexes, which can be co-immobilized with the enzyme on the electrode or added to the solution [95,102,103,104]. Overall, MET is considered to be more efficient than DET, since it does not critically depend on the enzyme orientation and since the small mediator molecules can have better access to the heme active site. It has nevertheless been shown that in some cases, MET and DET can occur simultaneously [11,105].
MET-based biosensors for detection of peroxidase reducing substrates have been developed for phenol, cathecol, dopamine, norepinephrine, epinephrine, or serotonin, among others [56,57,90,106]. In these cases, the enzyme oxidized product is reduced electrochemically, enabling monitoring of the catalytic reaction. The generated cathodic current is proportional to the concentration of the peroxidase substrate, provided that concentration of H2O2 is non-limiting. Importantly, no direct reduction of the substrate can occur at the electrode surface in the absence of H2O2 [56,106].
While the reaction mechanism in MET is usually straightforward, this is not the case for DET-based devices, particularly those employing heme proteins that at the same time can act as peroxidases and reductases, including heme peroxidases, catalases, cyt c, myoglobin, and hemoglobin. Note that a catalytic reduction current is typically observed in all cases, but that the heme species involved in the respective reactions are different. The Fe(II) heme state is present in catalytic reduction, while peroxidase activity involves high valence oxy-ferryl intermediates and/or Fe(II)-O2 species (cf. the low potential redox cycle).
3.1.2. Plant Peroxidases
Plant peroxidases belong to class III of the plant, fungal and bacterial peroxidase family. They typically display monomeric structures with a conserved helical fold and a high level of glycosylation. Despite a low amino acid sequence identity between members of this family, all peroxidases contain invariant amino acid residues in the heme b-containing active site that are important for enzyme activity, including the conserved catalytic His residue on the distal heme face [86,87,88]. These enzymes take part in fundamental processes in the plant life cycle, such as cell wall metabolism, defense against pathogens, lignification, and reactive oxygen (ROS) and reactive nitrogen species (RNS) metabolism. Owing to their catalytic versatility, plant peroxidases have a well-recognized potential for application in bioremediation, paper and pulp industry, polymer synthesis, biosensor development, and other fields [87,107]. In particular, tobacco peroxidase (TOP), barley peroxidase (BaP), peanut peroxidase (PNP), guinea grass peroxidase (GGP), soybean peroxidase (SBP), and HRP have been employed in biosensor development. In fact, HRP is one of the most explored enzymes for development of amperometric biosensors due to (i) commercial availability, (ii) low production costs, (iii) high stability under standard conditions, (iv) activity towards H2O2, a broad range of phenolic compounds and aromatic amines, and (v) the well understood catalytic and structural properties [88,106,108]. The main shortcomings of HRP use in amperometric biosensors are the substrate inhibition (H2O2), and the high level of surface glycosylation. The carbohydrate residues can constitute a considerable fraction of the molecular weight of plant peroxidases (around 25% in HRP) and affect binding and electronic wiring of the enzymes to electrodes, as well as DET [87,105,109].
Biosensors based on plant peroxidases have been developed for detection of H2O2, phenolics and other classical peroxidase substrates [90,95,106], as well as nitrite, NO, uric acid, and TCA, among others [81,99,100,101]. In the simplest configuration, the peroxidase is directly adsorbed on the electrode interface and used for quantification of H2O2. Electrocatalytic activity has been observed in materials like graphite [105,106,109,110], graphene thick films [111], and gold [112,113]. This strategy is, nonetheless, commonly associated with slow ET rates because of (i) the deeply buried heme cofactor in the enzyme matrix; (ii) the presence of a high percentage of non-active enzyme population, owing to poor orientation and/or altered conformational structure; (iii) the high glycosylation levels [114]. Besides forming an insulating barrier, the surface carbohydrates increase the distance between the electrode and the heme site, hindering ET [105]. The use of recombinant non-glycosylated HRP and TOP favors the adsorption and uniform orientation on the interface, improving the ET rates and overall bioelectrode performance [105,109,112,113].
Electron exchange between plant peroxidases and electrodes can be facilitated employing conducting and non-conducting polymers, detergents, SAMs, ionic liquids (ILs), and nanomaterials, among others [68,74,92,115,116]. We highlight immobilization on polymeric films and nanostructured electrodes as the most common strategies. Plant peroxidases have been physically entrapped [115] or chemically attached [95] to polymer films, which can mimic the interactions in the physiological environment, thus promoting the native conformation of the enzyme. Sun et al. described the incorporation of HRP in a non-conducting silica sol-gel film of poly (N-isopropylacyamide-co-3-methacryloxy-propyltrimethoxysilane) (PNM) deposited on a GC electrode [115]. The PNM film showed good compatibility with HRP, which retained its native structure in the polymer, as demonstrated by FTIR spectroscopy. The resulting biosensor was used for sensitive amperometric detection of H2O2 in a limited range of concentrations, 0.19 to 1.35 µM, with high sensitivity and low detection limit of 0.620 A M−1 cm−2 and 0.0475 µM, respectively (Table 1) [115].
Incorporation of plant peroxidases into redox polymer matrices has a twofold role, as it physically and electrically connects the enzyme to the electrode [38,95,110,139]. Both single polymeric materials [140] and composite matrices that incorporate CNTs or redox mediators (e.g., osmium doped polymers) have been explored, demonstrating that favorable electrostatic interactions between the enzymes and the polymer films are important for the performance of the biosensor [38,92,95,110,139]. Comparison of the electrocatalytic behavior of the cationic HRP and the anionic TOP upon immobilization on a cationic osmium polymer revealed improved catalytic response towards H2O2, sensitivity, and long-term stability for TOP (cf. Table 1), which was attributed to the electrostatic attraction between the polymer matrix and the enzyme that increases coupling between the two [95].
Immobilization of plant peroxidases on electrodes modified with nanomaterials is a particularly common strategy in the design of third-generation biosensors. A vast number of nanostructures, including Au nanoparticles (NPs) [71,74,93,141], multi-walled CNTs (MWCNTs) [39,79,83], graphene [82,142], Co3O2, and MoS2 nanosheets [99,143] has been tested. Owing to the small size of the nanomaterials and high surface area/volume ratio, the resulting biosensors typically show increased enzyme surface coverage and sensitivity. Nanomaterials like CNTs and AuNPs can also form tailored scaffolds that provide favorable orientation of the enzymes, which is determinant for enhanced electrochemical response [94]. Wang and co-workers described a H2O2 biosensor based on the incorporation of HRP into a composite MWCNT/thionine/AuNP film, via electrostatic interactions between the enzyme and the negatively charged NPs (Figure 2a). The synergistic effect of the three components played a key role in accelerating the ET between the heme active site and the GC electrode, resulting in the reversible electrochemical response of HRP and high electrocatalytic currents in the presence of H2O2 [37]. In another example, HRP was covalently attached to γ-aminobutyric acid (GABA) functionalized MWCNTs, drop-casted on a GC electrode, and used for the direct electrochemical quantification of H2O2. Docking and molecular dynamics calculations suggested that GABA interacts with the Lys residues of HRP, inducing a specific and uniform enzyme orientation on the nanostructured interface [39]. The importance of enzyme arrangement on the electrode surface was further demonstrated employing HRP attachment on AuNPs/4-aminothiophenol polymer (Figure 6). The electrochemical response of randomly deposited AuNPs was poorer, despite the higher enzyme loading, than in the case of well-dispersed deposits. SEM images of the modified electrodes revealed that the latter configuration provided larger spacing between AuNPs and between the attached HRP molecules, which contributed to the preservation of enzyme folding, diminished steric hindrance, and improved substrate accessibility [94].
3.1.3. Dye-Decolorizing Peroxidases
Dye-decolorizing peroxidases (DyPs) constitute a relatively recently discovered family of heme peroxidases that are mostly found in fungi and bacteria [144,145,146,147]. Although they have been associated with a range of processes, including lignin degradation and deferrochelation, thus far their physiological role remains to be identified [146]. DyPs have unique properties that set them apart from other heme peroxidases, notably: (i) a broad catalytic versatility, which includes poor peroxidase substrates, such as carotenoids and anthraquinone-based and azo dyes, and (ii) distinct structural and consequently mechanistic properties [145,146,147,148]. These enzymes have a ferredoxin-like fold, composed of α-helices and β-sheets, instead of the α-helix rich arrangement found in other peroxidases. Furthermore, DyPs lack the highly conserved catalytic distal His residue. In turn, the distal side of the heme pocket typically contains an Asp (or Glu) and an Arg, which are thought to be involved in the catalytic reaction, although their exact roles are not well understood yet [145,146,147,149].
DyPs have a widely recognized biotechnological potential, particularly for lignin degradation and valorization, and dye decolorization of wastewater [145,146,148]. Nevertheless, only one biosensing application has been reported so far. A DET-based H2O2 biosensor was prepared by attaching a bacterial DyP onto biocompatible SAM coated Ag electrodes [67]. SERR spectroscopy demonstrated that the enzyme maintained its native structure upon adsorption, in the absence and presence of H2O2 [67,150]. The biosensor exhibited high sensitivity (1.31 A M−1 cm−2) and a linear response range of 1–200 µM H2O2 (Table 1, Figure 7). Optimization of the device was attempted employing enzyme variants obtained by directed evolution, which displayed increased resistance to H2O2 inactivation in solution [151]. The biosensor based on a variant harboring mutations at the second shell of the heme cavity showed an improved storage stability in comparison with the wild-type enzyme (Figure 7b) [67].
Development of DyP-based biosensors is likely to increase in the future, particularly considering that the bacterial enzymes are devoid of glycosylation and can be easily genetically modified to attain variant forms with improved catalytic and stability properties [151], which is highly advantageous in comparison to the more commonly explored plant peroxidases.
3.1.4. Cytochrome c Peroxidases
Cytochrome c peroxidases (CcPs) take part in peroxide stress management in prokaryotic and eukaryotic organisms and are possibly involved in bacterial H2O2 respiration in anoxic environments [89,152]. The enzymes from prokaryotes and eukaryotes display different structures and catalytic mechanisms; the former CcPs contain two (or three) covalently bound c-type hemes, while the latter have a single heme b [86,87,89,152].
The electrochemical response of CcPs has been demonstrated at pyrolytic graphite (PG) and Au surfaces, using DET and MET [153,154,155,156]. In the latter case, physiological redox partners, including c-type cytochromes and small blue copper proteins, typically act as mediators [155,156,157,158]. However, the applications of CcPs in biosensor construction are very limited. In one report, CcP was co-immobilized with horse heart cyt c in a gelatin B-based matrix deposited on a SAM modified Au electrode and tested as a biosensor for H2O2 monitoring [158]. The catalytic response towards H2O2 was linear up to 300 µM and the LOD was 10 µM. The hydrophilic nature of the gelatin was proposed to contribute to the biocompatibility of the encapsulation matrix and maintenance of catalytic activity [158]. CcPs, and in particular bacterial ones, are less sought after for biosensing applications owing to the complex activation mechanisms and the limited substrate range in comparison with other peroxidases.
3.2. Cytochromes P450
Cytochromes P450 (P450s) constitute a large family of heme b-containing monooxygenases that can be found in all living organisms. P450s are involved in a wide variety of biosynthetic processes, such as the production of fatty acids and endogenous steroids, as well as in the metabolism of xenobiotics, including drugs, pesticides, and carcinogens [159]. They catalyze diverse types of reactions, including hydroxylations, sulfoxidations, epoxidations, deaminations, and N-oxide reduction [159]. In the typical monooxygenase reaction (Equation (11)), P450s use O2, electrons, and protons to oxidize organic substrates (RH) in a stereo- and region-selective manner [159,160]. The electrons derive from NAD(P)H via iron-sulfur- and/or flavin-containing redox partners, i.e., P450 reductases, depending on the type of organism or the cellular compartment [161].
RH + O2 + NAD(P)H + H+ → ROH + H2O + NAD(P)+
P450s display monomeric structures with a similar overall fold [1,162]. The ferric heme is coordinated by a proximal Cys residue and a distal water molecule, giving origin to a 6c resting state. Upon initiating the reaction, binding of the organic substrate molecule in the vicinity of the active site displaces the axial water and converts the heme iron into a 5c state. Similarly to heme peroxidases, the reduction potentials of P450s fall into a negative window (ca. −0.220 to −0.370 V) and vary among different enzymes [163,164,165,166].
The reaction cycle starts with binding of the organic molecule, as described above, followed by enzyme reduction to the Fe(II) state, enabling binding of O2. Subsequent reduction and protonation steps generate heme iron-oxo intermediates including compound I (cf. Section 3.1). This species then readily oxygenates the bound substrate molecule (RH) to ROH [1,159,162,167,168,169]. Interestingly, some P450s can use H2O2 and other peroxides to generate compound I, thus bypassing the electron delivery by redox partners and leading to product formation (peroxide shunt pathway) [159,168,169]. This peroxide-dependent catalysis is also observed in P450 peroxygenases, a subgroup of the P450s family that has been identified in some microorganisms [169].
Owing to the broad substrate range, P450s are highly sought after for biocatalysis, bioremediation and drug development [170,171]. In biosensing, P450s have been employed for the quantification of metabolites, e.g., testosterone, progesterone; drugs, e.g., codeine, omeprazole, paracetamol; and pesticides [172]. Despite the great interest in exploring P450s in the construction of amperometric biosensors, so far the success has been limited [173,174]. This is mainly due to (i) the inherent instability of the enzyme, as it is particularly prone to be converted to the inactive P420 state, which is characterized by an altered heme ligation pattern and redox potential, and is easily induced by immobilization and (ii) uncoupling (branching) reactions in which the heme iron-oxo intermediates lead to the formation of superoxide anion, H2O2 or water, thereby consuming electrons and generating a current response that is not related to substrate conversion [170,174]. For these reasons, identification of the reaction product is often required to confirm if the substrate was consumed in the electrochemical reaction [172]. SERR and UV-Visible spectroscopies can provide fine details about the active site structure upon enzyme immobilization in biosensor devices, including formation of the catalytically inactive P420 state (Figure 8) [70,175]. Shifts in the reduction potential may also point out to structural changes caused by immobilization, however this criterion alone is not sufficient to distinguishing between P450 and P420 [70].
Several methods have been employed for detection of P450 electrocatalytic activity in biosensors, including consumption of the electron donor NAD(P)H or of the co-substrate O2 [167,172,176], which correspond to first-generation biosensing strategies. Likewise, if substrate conversion is driven by H2O2, a decrease of its concentration can be correlated with P450 catalytic activity. In MET approaches (second-generation biosensors), the reduction of the electron shuttles, like viologens, cobalt sepulchrate, and phenosafranine, have been used to activate P450s [172,176,177]. In turn, the DET driven devices (third-generation) can be prepared with the P450 enzyme alone, via co-immobilization with a natural redox partner or with genetically engineered multi-domain systems in which the P450 is fused to the redox partner [170,178]. The interpretation of electrochemical signals in DET biosensors is complex because the reduction of the ferric P450 requires a potential at which the co-substrate O2 is also reduced. It is generally accepted that the current for the O2 catalytic reduction is further increased in the presence of P450 substrates [179]. However, this increase may simply be due to a variation of O2 concentration upon addition of the organic substrate. The difficulties in separating O2 reduction currents from the monooxygenase reaction highlight the importance of monitoring formation of the product in P450 electrochemical systems. Alternatively, biosensor performance can be assessed by measuring enzyme activity in presence of known P450 inhibitors [170,176].
P450s used in biosensor development are mainly of human origin, due to their commercial availability and the capacity to metabolize around 95% of known drugs [180]. P450s have been encapsulated into polymers and lipid membranes or attached to SAM, surfactant, and nanomaterial coated electrodes [52]. To that end, P450-1A2 in complex with its redox partners (i.e., P450 reductase and cytochrome b5) was adsorbed on graphite screen-printed electrodes (SPEs) coated with MWCNTs and used for quantification of the anti-inflammatory drug naproxen [181]. A similar P450-3A4/P450 reductase complex was adsorbed on 3D-nanoporous graphene foams (NCFs) and employed in detection of testosterone, estrone, and progesterone [182]. The highest activity and thermal stability of the device was achieved when the dimensions of the foam pore matched the size of the enzyme. Several biosensors for prescription drugs have been developed via chemical binding of the enzyme to functionalized surfaces using EDC/NHS [48], glutaraldehyde [117,118], or NN-carbonylimidazole [118]. Omeprazole was detected on GC electrodes modified with a nanocomposite made of chitosan, reduced graphene, and ceria NPs onto which P450-2C19 was cross-linked in the presence of glutaraldehyde [117]. The intrinsic capability of the ceria NPs to accommodate O2, required for P450s catalytic activity, resulted in a biosensor capable of detecting omeprazole concentrations in a 2 to 50 µM range (Table 1). A multi-enzyme complex of P450-1A2 and P450-3A4 was assembled on top of a nanocomposite of AuNPs, chitosan, and graphene oxide nanosheets by covalent binding using glutaraldehyde and NN-carbonylimidazole. The two enzymes had a coupled action, allowing detection of clopidogrel with a good sensitivity and a low limit of detection (Table 1) [118]. Asturia-Arribas et al. developed a biosensor for codeine, based on P450-2D6 immobilized on a carbon SPE. From the three explored strategies (simple adsorption, cross-linking with glutaraldehyde and BSA, and covalent binding using EDC/NHS) covalent immobilization resulted in the best amperometric response towards codeine. The biosensor showed extended linear range and was functional up to three weeks [48].
Reports on P450 peroxygenase-based biosensors are scarcer, owing to more recent discovery of these enzymes. They include devices for quantification of e.g., 4-nitrophenol [183], naphthalene [184], aniline, and paracetamol [185], constructed employing AuNPs and chitosan nanocomposites deposited on GC electrodes. The devices succeeded in detecting substrate concentrations as low as 0.1 µM.
It is noteworthy that despite of promising examples, real applications of P450s and related enzymes still do not match their potential. Due to their inherent instability, interactions with a vast majority of electrode supports results in structural changes at the level of the active site that alter catalytic properties of these enzymes in the immobilized state impeding their broader utilization (Figure 8).
3.3. Catalases
Catalases are ubiquitous enzymes that are found in nearly all aerobic organisms. They are involved in ROS detoxification and catalyze the decomposition of H2O2 into water and O2, Equation (12) [89,186,187].
2H2O2 → 2H2O + O2
The mono-functional catalases have been identified in fungi, bacteria and archaebacteria and display significant peroxidase activity in addition to the H2O2 dismutation reaction. They are phylogenetically related to the superfamily of plant, fungal and bacterial peroxidases. The mono-functional catalase family is the largest and most widespread (from bacteria to eukaryotic organisms) [89,187] and the most explored in biosensor development, in particular the commercially available bovine liver catalase [187,188,189]. They are tetrameric proteins containing one 5c heme b or heme d in each of the four identical subunits [186,187]. Like peroxidases and P450s, catalase reduction potentials are typically negative (e.g., −0.260 V for the heme b-containing bovine liver catalase), which is consistent with a stable Fe(III) resting state [190].
The catalytic dismutation reaction occurs in two steps and requires two molecules of H2O2. In the first step, the resting Fe(III) enzyme is oxidized by H2O2, forming compound I and water (Section 3.1, Equation (4)). The catalytic intermediate is reduced by the second H2O2 molecule in the second step, regenerating the resting state enzyme and releasing a water molecule and O2 (Section 3.1.1, Equation (7)).
Catalases are, in general, highly thermo-stable, robust enzymes; they also have one of the highest turnover rates among known enzymes, which together with their wide availability, have prompted a variety of applications, including amperometric biosensors [188]. A limitation to catalase use in comparison to peroxidases is the narrower substrate range. Catalase biosensors are mostly used in H2O2 detection, although some devices have been employed for quantification of e.g., nitrite [119] and for indirect measurement of activators and inhibitors of enzyme activity, such as calcium [191] and mercury [9].
Both first- and third-generation biosensing schemes have been exploited in catalase devices. In the former, detection is based on direct measurements of the electroactive substrate (H2O2) or product (O2). In the latter, DET between catalase and the electrode in the presence of H2O2 gives origin to the increase of cathodic currents resulting from reduction of compound I at the electrode surface, as described for peroxidase biosensors (cf. Section 3.1.1) [91,106]. However, it has been proposed that this reaction may not be the main contributor to the catalytic current [98]. The observed catalytic reduction peak for H2O2 is usually close to the reduction potential of the Fe(III)/Fe(II) redox pair, which indicates that an alternative reaction involving direct reduction of Fe(II)-O2 species at the electrode, equivalent to the low potential redox cycle described for peroxidases (cf. Section 3.1.1) probably takes place [96]. Further evidence for this hypothesis is the catalytic response to O2, observed at similar potential values [98].
Catalase-based amperometric biosensors employ a variety of materials, including cross-linking agents, polymers, sol-gels, lipid films, surfactants, and diverse nanomaterials, and most commonly carbon electrodes (GC, graphite, carbon paste) as transducing surfaces. An inhibition biosensor for quantification of Hg2+ was prepared by cross-linking catalase with glutaraldehyde and BSA on a GC surface. The inhibitory effects on the enzyme reaction induced a decrease of the current response, which was dependent on Hg2+ concentration. The device displayed high selectivity for Hg2+ in the presence of other heavy metal ions and organic pesticides [9]. Combination of catalase cross-linking with gelatin encapsulation was explored for the development of a first-generation biosensor for detection of Ca2+ in milk and water samples, taking advantage of the increased enzyme activity in the presence of the cation. A dissolved O2 electrode was used to measure the concentration of the reaction product in the presence of constant amount of H2O2 [191]. A first-generation biosensor employing catalase adsorbed on Pt electrodes coated with Nafion and poly-o-phenylenediamine (PPD) layers and subsequently cross-linked with glutaraldehyde was used for following the direct oxidation of H2O2. The current was simultaneously measured at an enzyme-modified and a control electrode without catalase. The dual electrode device performed well in the presence of interfering species, since background currents were subtracted using the control electrode. The biosensor was envisioned for in vivo monitoring of H2O2 [192].
For the last 10 to 15 years, nanostructured electrodes have been recurrently used in the construction of third-generation catalase biosensors. This is in line with development of electrochemical biosensor in general and is justified by the advantages that carbon and metal nanomaterials provide in terms of biocompatibility, high conductivity, and large surface area [119,120,122,193,194,195]. Quantification of H2O2 and nitrite has been achieved with a catalase/NiO NPs biosensor prepared by electrodeposition of enzyme and the NPs on GC and ITO electrodes. UV-Visible studies of the latter showed no apparent enzyme denaturation upon interactions with the NiO nanostructures [119]. Composite materials based on surfactants, polymers, and ILs are often used to facilitate the dispersion of the nanomaterials, improve enzyme retention on the electrode, and help preserve the native conformation of the catalyst. To that end, catalase has been (i) adsorbed on GC surfaces modified with poly-L-lysine/MWCNT and 1-butyl-3-methylimidazolium tetrafluoroborate ([BMIM]BF4)-IL/MWCNT-NH2 films [121,122], (ii) co-deposited with mixtures of dodecyltrimethyl ammonium bromide (DTAB)/MWCNT [195], Nafion/MWCNT-COOH [196], or chitosan/SnO2 nanofibers/PANI on GC electrodes [194], and (iii) incorporated into multilayered assemblies with amine terminated IL (NH2-IL) prepared on titanium nitride (TiN) modified GC electrodes [120]. FTIR and UV-Visible spectra of the catalase/poly-L-lysine/MWCNT and catalase/NH2-IL/TiN films indicated that the enzyme was intact upon immobilization, suggesting suitable microenvironments for catalase activity [120,122]. The electrocatalytic response of catalase towards H2O2 reduction was improved on the nanocomposite electrodes. In particular, the IL/MWCNT-NH2 bioelectrode displayed one of the lowest reported LODs (3.7 × 10−3 µM) in a limited linear concentration range (8.6 × 10−3–0.140 µM) [121]. The poly-L-lysine/MWCNT and the NH2-IL/TiN biosensors had much broader dynamic ranges, up to 2.1 mM H2O2 in the latter (cf. Table 1) [120,122].
3.4. Nitrite Reductases
Nitrite reductases (NiRs) constitute a structurally diverse group of enzymes that are responsible for nitrite reduction in bacteria [197]. The heme containing cytochrome cd1 NiRs (cd1NiRs) and cytochrome c NiRs (ccNiRs) have been explored for the construction of electrochemical biosensors [198]. cd1NiRs reduce nitrite to NO in a one-electron reaction (Equation (13)), as part of the bacterial denitrification pathway, an anaerobic respiratory process in which nitrate is reduced to N2 for energy production. cd1NiRs have homodimeric structures with a heme d1 in the active site and an electron acceptor heme c per subunit [197,199,200]. The reduction potentials of the heme groups have been reported in the +0.2 to +0.3 V range [201,202,203]. Upon reduction of the enzyme, the catalytic heme d1 adopts a 5c configuration capable of binding the nitrite substrate [204,205]. ccNiRs catalyze the six-electron reduction of nitrite to ammonium (Equation (14)). They are periplasmic multi-heme complexes, composed of ET (NrfB or NrfH) and catalytic subunits (NrfA), involved in dissimilatory nitrate reduction that provides reduced nitrogen for cell growth [197,200]. The active site is a 5c heme c with an unusual Lys residue as the proximal ligand [200,206,207]. The ET heme groups have reduction potentials between +0.15 and −0.48 V, while the value for the catalytic heme has been reported between −0.05 and −0.2 V [208,209,210].
NO2− + 2H+ + e− → NO + H2O
NO2− + 8H+ + 6e− → NH4+ + 2H2O
cd1NiRs and ccNiRs have been employed in amperometric biosensors for detection of nitrite, which is a relevant analyte for the medical research, clinical diagnosis, and environmental fields [211,212,213]. Both enzymes have been used in devices relying on MET, while only ccNiR has been explored in DET-based designs [19,124,198,214]. In the former, a reduced mediator converts the enzyme to the Fe(II) catalytically active state, which reduces nitrite. The oxidized mediator is then re-reduced at the electrode surface generating the electrocatalytic current and subsequently reducing the enzyme back to the Fe(II) state [19,214]. In DET-based biosensors, the current signal derives from the direct regeneration of the reduced enzyme cofactors (i.e., Fe(III)→Fe(II)), following the reduction of nitrite [215].
In ccNiR biosensors, the enzyme has been incorporated into polyacrylamide, Nafion and poly(carbamoyl sulfonate) non-conducting layers and into redox active [ZnCr-AQS] layered double hydroxides (LDHs) containing anthraquinone disulfonate (AQS), and PPY films prepared on graphite or GC electrodes. Enzyme activity could be detected via MET using methyl viologen, phenosafranine, AQS, and viologen functionalized PPY [20,123,198,214]. The analytical performance of the resulting devices included some of the highest and lowest LODs reported to date (60 µM vs. 4.0 × 10−3 µM, for Nafion- and [ZnCr-AQS] LDH-based biosensors, respectively) [123,214]. Electrodes prepared with non-conducting polymers generally displayed relatively low sensitivities (e.g., 0.445 A M−1 cm−2 for the Nafion biosensor) and broad linear ranges. The redox active matrices provide improved the electrical wiring of the enzyme to the electrode, resulting in higher sensitivities (1.7–1.8 A M−1 cm−2), low detections limits, but more limited dynamic ranges (0.015–2.35 µM for the [ZnCr-AQS] LDH biosensor, Table 1) [20,123].
The ability of ccNiRs to directly exchange electrons with electrode interfaces [209,216,217] has prompted the development of third-generation nitrite biosensors employing mostly PG and carbon ink/paste electrodes [124,215,218]. ccNiR electrocatalytic activity has also been observed upon deposition onto ITO [125] and encapsulation into non-conducting materials, such as a sol-gel matrix based on an alkoxysilane precursor [124]. The protective sol-gel film ensured catalytic activity up to six months. Characterization of the ccNiR/sol-gel films by UV-Visible and RR spectroscopies revealed no indications of denaturation or conformational changes of the heme groups upon enzyme encapsulation [124].
Further improvement of DET-based electrocatalysis by ccNiR was achieved with the introduction of nanomaterials for electrode modification [125,215,219]. The highest current response to nitrite so far, has been obtained with ccNiR adsorbed on multilayered single-walled CNT (SWCNT) deposits (sensitivity 2.4 A M−1 cm−2) [215]. Likewise, WO3NPs enhanced DET between ccNiR and ITO electrodes (Figure 9). The RR spectra demonstrated that the native state of the ccNiR was preserved in the construct. Confocal Raman spectroscopy measurements of the enzyme/NP construct revealed that the WO3NPs underneath the deposited ccNiR film were also not affected by the presence of the enzyme. The biosensor had a high sensitivity (2.1 A M−1 cm−2) and a relatively narrow linear range (Table 1) [125].
Thus far, cd1NiRs have had limited application in biosensing in comparison to ccNiRs. This is mainly due to the necessity to activate cd1NiRs. The mechanism includes reduction of the hemes, conformational rearrangement, and heme ligand exchange, processes that are triggered by specific interactions with electron donor partners and cannot be easily mimicked at electrode surfaces [220,221]. SERR spectroelectrochemical studies of cd1NiR attached to SAM coated Ag electrodes showed that about half of the immobilized enzyme retained redox activity and native structure at the level of the heme c, but no electrochemical catalysis was observed in the presence of nitrite via DET [201]. MET-based approaches include early trials in which cd1NiR was retained by dialysis membranes on the surface of graphite electrodes, while the mediating species were added to solution [222,223]. These were followed by the development of a functional biosensor based on the co-immobilization of cd1NiR and a putative electron donor, cytochrome c552, on carbon SPEs, employing a cross-linked poly(vinyl alcohol) matrix. The resulting biosensor was able to detect nitrite within a 10 to 200 µM range [19].
3.5. Cytochrome c Oxidases
Cytochrome c oxidase (CcO) is a cytochrome c: oxygen oxidoreductase that catalyzes the four-electron reduction of O2 to water. The catalytic subunit of CcO houses a 6c ET heme and a binuclear 5c heme a3-CuB catalytic center. Although it could theoretically be explored for construction of both O2 biosensors and cyt c immunosensors, applications of CcO are still limited, due to its hydrophobic nature and a complex multi-subunit structure prone to degradation. To that end, the few existing reports have mainly focused on detection of cyt c in biological samples, since its presence in blood can be indicative of several pathologies related to damaged mitochondria, such as apoptotic cell death, myocardial infarctions, and a number of neurological diseases [224]. The reported cyt c sensing applications include those that rely on (i) immobilization of CcO onto Au electrode-supported lipid bilayer membranes; the oxidase-modified electrode operates as an amperometric biosensor capable of detection of reduced cyt c with detection limit of 0.1 µM [127], (ii) casting of CcO-didodecyldimethylammonium bromide (DDAB) vesicle films onto Au electrodes for detection of ferric cyt c in human blood serum [126], and (iii) CcO deposited over Au electrodes modified with NiONPs, CNTs and a conducting PANI polymer (NiONPs/cMWCNT/PANI hybrid) [225]. The amperometric biosensors for cyt c based on these hybrid films were investigated by CV, electrochemical impedance spectroscopy (EIS), SEM, and FTIR [225], revealing a linear range between 1 × 10−6 and 0.1 µM, a superior detection limit in different serum samples (5 × 10−6 µM) and sensitivity of 3.7 × 106 A M−1 cm−2 [225]. Bioelectrocatalytic reduction of O2, employing cyt c functionalized SAM coated surfaces, in which the CcO complex was stabilized by cross-linking with glutaraldehyde, was studied in an attempt to develop a biofuel cell in which CcO plays the role of a cathode [226].
3.6. Cytochrome c and Microperoxidases
Cytochrome c is a small heme c-containing protein that, under normal physiological conditions, shuttles electrons from Complex III to Complex IV of the respiratory chain in bacteria and in the mitochondria of eukaryotic organisms. Nowadays, it is fully recognized that upon changes of the active site configuration, cyt c acquires peroxidase activity and becomes involved in the early stages of apoptosis [227]. The peroxidase activity of cyt c is induced by a transition of the heme iron from the native 6c conformation, with Met and His as axial iron ligands, to a 5c state [228,229]. The conformational exchange occurs via the detachment of the distal Met and is accompanied by a remarkable lowering of the reduction potential from +0.250 to −0.090 V [230,231]. The plastic nature of the heme coordination in cyt c is therefore very important for its exploitation in biosensors. Frequently, cyt c immobilization on electrode surfaces leads to partially unfolded configurations that display catalytic activity towards H2O2 and O2 [232]. For instance, non-native cyt c states can be generated upon adsorption on metal electrodes coated with SAMs [78]. In this context, SERRs and SEIRA spectroscopies have played a particularly important role in providing structural details about the immobilized cyt c and the parameters that control its structure and function [233,234]. In the absence of structural information, the reduction potential has also been used to evaluate the nature (native vs. non-native) of the immobilized cyt c species. As alternative to non-native protein states, the easy design and production of engineered variants can offer another broad range of possibilities for development of cyt c biosensors with more efficient catalytic activity. For example, variants that favor 5c species often display enhanced peroxidase activity [235,236,237], some of which lead to enhanced formation of compound I [237,238].
Biosensors based on cyt c are frequently employed in H2O2 monitoring and follow DET detection schemes similar to those described for plant peroxidases (cf. Section 3.1.1) [239]. Other cyt c biosensors have been designed for detection of nitrite [128], NO [130], and rebaudioside A [240], among others. Device construction has relied on diverse immobilization strategies, such as adsorption, entrapment in polymeric matrices, layer-by-layer assemblies, and others [239]. Herein, we highlight recent works focusing on covalent immobilization and nanomaterial modified electrodes.
Detection of H2O2 has been achieved with cyt c adsorbed or entrapped on different nanostructured electrodes, including (i) Au/Fe2O3/N-doped CNT composites [241]), (ii) three-dimensional graphene aerogels in the presence of AuNPs [242], (iii) mixtures of MWCNTs and conductive polymers, namely PANI [239] or PPY [243], and (iv) AuNPs grown on electrochemically reduced graphene oxide/Nafion composites [244]. In a recent work, cyt c was adsorbed on a l-cysteine layer deposited onto a hybrid conducting polymer/MWCNT modified GC electrode [128]. The protein had a reduction potential of +0.275 V in the construct, pointing out that its native structure was preserved. The device had a low sensitivity (0.0178 A M−1 cm−2) in a broad linear range (Table 1) [128]. In the absence of polymers, the sensitivity was increased up to 0.043 A M–1 cm−2 [245]. However, in this case, the interaction of cyt c with the immobilization components induced a red-shift of the Soret band and a lower formal potential (−0.128 V), which indicated the presence of a non-native state [245].
Cyt c contains two positively charged Lys patches on the surface that govern its binding to negatively charged surfaces and enable immobilization via covalent attachment using EDC/NHS (cf. Section 2.2). The possibility to introduce carboxylic functions in a broad variety of support materials has driven to a remarkable diversity of cyt c biosensors that explore this type of covalent attachment [129,130,246]. One example explores covalent binding of the protein onto AuNPs, and subsequent entrapment with chitosan, for the development of a NO biosensor [130]. UV-Visible spectroscopy indicated that the native structure of cyt c was preserved in the construct. The NO biosensor had a good sensitivity and a linear range from 10 to 215 μM (Table 1). In another approach, carboxylated boron-doped diamond and ILs covalently bound to cyt c resulted in a H2O2 biosensor with a broad linear range (1–450 μM) but low sensitivity (cf. Table 1) [129]. Alternatively, cyt c was chemically modified with carboxylated alkanethiols before adsorption onto Au electrodes (Figure 10).
The redox activity of the protein was determined as a function of the orientation and distance from the interface, which were governed by the length of the alkanethiol chain. in situ generated AuNPs were shown to greatly improve the ET efficiency (Figure 10). The optimized device was able to detect H2O2 overproduced in vitro by living cells, despite a very low sensitivity (2.00 × 10−4 A M−1 cm−2) [246].
Other c-type cytochromes have shown catalytic activity under specific conditions, similarly to what is observed with cyt c [229,247]. A detailed CV, RR, and SERR spectroscopic characterization of cytochrome PccH, immobilized on SAM-modified Au and Ag electrodes, revealed that the protein had residual peroxidase activity. Cytochrome PccH derived microperoxidase displayed an increased activity toward H2O2 [229]. Likewise, CytD, showed peroxidase activity upon immobilization on Au-SAM surfaces, which was attributed to a minor 5c population generated upon protein adsorption [247].
Microperoxidases (MPs) are heme-peptide fragments obtained by the proteolytic digestion of horse heart cyt c. They contain the CXXCH structural motif and 8 or 11 amino acid residues (MP8-11). MPs have lower reduction potential than cyt c, i.e., −0.160 V [248], and retain only the proximal axial His ligand that gives origin to a 5c heme state. MPs efficiently reduce H2O2 via compound I formation. They also exhibit typical P450 activities [249] and can catalyze specific reactions of organic chemistry, such as aniline p-hydroxylation [250], S-oxidation [251], O- and N- dealkylation, or nitration [252]. The heme is highly exposed to the solvent, which leads to instability in solution and easy auto-aggregation [253,254].
Biosensors based on MPs have been developed for similar target analytes as cyt c devices. A H2O2 biosensor was prepared by immobilization of MP11 on mesoporous antimony doped ITO electrodes, coated with the polycation poly-diallyldimethylammonium chloride (PDADMAC) [131]. The presence of PDADMAC improved biosensor performance due to increased protein surface coverage and slower desorption of MP11 from the electrode. UV-Visible spectroelectrochemistry revealed that the direct reduction of H2O2 proceeded via a compound I-type intermediate, as deduced by the high potential at which H2O2 reduction was detected (>+0.660 V). Despite the low affinity to H2O2 ( = 1.8 μM), probably due to decreased access to the active site, the biosensor had a wide linear range of 10 to 750 μM, although a quite low sensitivity (3.87 × 10−3 A M−1 cm−2) [131]. A biosensor with similar linear range but almost an order of magnitude higher sensitivity was obtained by adsorption of MP11 onto a PG electrode in the presence of ZnONPs, thus demonstrating the advantages of using nanostructured electrodes [255]. High sensitivities were also observed for the encapsulation of MP11 on mesoporous SnO2 electrodes coated with poly-L-lysine [256] and within metal-organic frameworks (MOF) of porous coordination Al-based network, PCN-333 (Al), grown on three-dimensional kenaf stem-derived porous carbon (3D-KSC), Table 1 [132]. The latter device also showed an extended linear range of 0.4 to 1725 μM [132]. Recently, a MP11-based biosensor was developed for detection of the antimalaria drug artemisinin [257]. The protein was adsorbed onto DDAB and further entrapped into mesoporous SnO2NPs. The ITO electrode employed as primary support allowed the spectroelectrochemical investigation of MP11/artemisinin interaction, revealing a shift of the Soret band of MP11 and possible structural alterations in the presence of artemisinin.
In the last decades, the tendency in employing MPs in biosensing devices shifted towards their coupling with other enzymes. To that end, MPs have been employed as mediators in glucose sensing [258,259,260] and together with catalase and superoxide dismutase to increase the sensitivity towards NO detection [133].
3.7. Globins
Hemoglobin (Hb) and myoglobin (Mb) are the most well-known members of the globins superfamily; they are responsible for carrying/storing O2 in living organisms. Both house a 5c heme b as prosthetic group with His as the proximal ligand. Hb and Mb have reduction potentials of ca. 0.050 and 0.150 V, respectively [270]. The ferric states of both proteins, i.e., methemoglobin (metHb) and metmyoglobin (metMb), are physiologically inactive for O2 transport and storage, however their redox chemistry can lead to pathologic conditions due to free-radical production and/or oxidation of fatty acids [271]. In this context, MetHb and metMb show peroxidase-like catalytic activity, which involves the formation of high valence oxy-ferryl heme intermediates, such as compound I [272,273,274]. MetHb and metMb can also be reactive toward other compounds, including NO and sulfide [271].
Mb and Hb are readily available and therefore have often been explored in biosensing. Besides typical analytes, such as H2O2, nitrite, and TCA, globin biosensors have been developed for detection of artemisinin [136], nitromethane [275], and chloropropandiol [276]. Recently, Mb-based nitrite and TCA biosensors, relying on DET transduction schemes, have been characterized in acidic pH conditions [138,277,278]. A Mg-based MOF/AuNPs biosensor employing Mb was able to detect up to 18 mM nitrite (Table 1) [138]. A broad linear range has also been reported for Hb adsorbed on a composite of reduced graphene oxide and TiO2 nanosheets [134]. In both cases, entrapment of the proteins inside Nafion polymer improved the biosensor performance. A similar approach, based on Hb encapsulated onto TiO2 hollow microspheres, led to a H2O2 biosensor with increased affinity ( 141.4 μM vs. 81 μM) and a good sensitivity (0.417 A M−1 cm−2) [99]. Much higher sensitivity (14.1 A M−1 cm−2) was attained using a broccoli-like Bi2S3 and chitosan nanocomposite (Table 1) [135]. A very low LOD (7 × 10−3 μM) was obtained by entrapment of Hb inside a nanocomposite of Nafion and Mn3(PO4)2 nanoflowers [65]. The good performance of the device was rationalized in terms of partial unfolding of Hb interacting with the inorganic matrix, as indicated by fluorescence spectroscopy. Enhanced catalytic activity was also observed for urea treated partially unfolded Hb adsorbed on a DDAB film deposited on GC electrodes. The device was used for H2O2 and nitrite biosensing, revealing improved sensitivities in comparison to the immobilized native protein [64].
4. Outlook
Heme proteins and enzymes have been widely exploited in the development of amperometric biosensors. Among different applications reported herein, H2O2 monitoring noticeably stands out, as it can be carried out by diverse heme proteins like peroxidases, catalases, cyt c, hemoglobin, and myoglobin. Biosensors for other relevant analytes, such as nitrite, NO, phenolic compounds, as well as several drugs, hormones, and pesticides have also been constructed. Notably, the possibility to develop simple and highly selective third-generation biosensors for various analytes is an advantage over e.g., glucose oxidases and dehydrogenases, which often rely on mediator-based transduction schemes.
Taken together, we witness a continuous growth of developed heme enzyme-based biosensing devices, some of which display very good performance, including in the analysis of real samples [95,195,218,225,246]. In parallel, the theoretical knowledge of the processes occurring at the electrode interface, and in particular the mechanisms of heterogeneous ET between electrodes and heme enzymes, together with general aspects of efficient DET have been well understood.
Still, transferring laboratory developed methodologies into practical applications remains challenging and the majority of biosensor reports do not go beyond the proof-of-concept stage. Perhaps the main current problems are the short lifetime of the devices (cf. Table 1), which is related to the stability of the heme biocatalyst, and the low efficiency of the heterogeneous ET reactions, due to e.g., unfavorable immobilization and/or poor orientation of the heme redox centers towards the electrode and the substrate. Use of nanomaterials represents a promising solution to tackle these issues, however, it has become evident that simple adsorption of the enzymes onto nanostructured electrodes is not sufficient. Instead, trends have been shifting towards construction of multicomponent architectures with carefully controlled deposition and immobilization chemistry, usually employing tailored made, functionalized nanomaterials. In parallel, rational modification of the enzymes through genetic engineering has also been explored to improve the efficiency and control the electrocatalytic properties of heme enzymes. This includes changes on the surface of the molecules to facilitate anchoring to functionalized electrodes, as well as development of variants with enhanced catalytic and stability properties, which are crucial for the overall performance of the biosensors. We highlight the potential of directed evolution approaches, which constitute the fastest and most efficient methods for tailoring enzymes for desired properties, such as a higher thermal stability, better substrate specificity, or lower inhibition by substrate/product [151,279,280]. The production of artificial heme-inspired catalysts, such as mimochromes and synthetic porphyrins, displaying increased robustness and activity, is also likely to gain more protagonism. However, care must be taken to ensure that resulting devices can deliver improved sensitivity and selectivity in comparison with those based on biological heme catalysts. Another important factor is the availability of sophisticated biophysical methods for in situ monitoring of the immobilized biocatalyst, which sensitively report on potential structural alterations or desorption from the electrode. We emphasize the role of SERR and SEIRA spectroscopies in providing molecular details of the immobilized heme enzymes that can guide biosensor construction, which therefore no longer depends on empirical trial and error approaches.
The final barriers to the implementation of biosensors are market interest and efficient passage to production on a large industrial scale. On the first aspect, heme enzyme biosensors can have an impact, since they have a broad substrate range that includes analytes that are relevant for the healthcare field, which offers the largest opportunities for commercialization [3,4]. As for the scaling-up of proof-of-concept devices, stability and reproducibility issues can be technically solved, although this often entails time and resource consuming development processes. Therefore, the future efforts towards development of heme enzyme biosensors need to ensure that the emerging advancements in electronics, material sciences and protein engineering are efficiently combined to produce functional and robust systems in a cost effective manner.
Author Contributions
Conceptualization, S.T. and C.M.S.; writing—original draft preparation, L.Z., C.B., S.T. and C.M.S.; writing—review and editing, S.T. and C.M.S. All authors have read and agreed to the published version of the manuscript.
Funding
The APC was funded by TIMB3 project, European Union’s Horizon 2020 Research and Innovation Program grant agreement No 810856.
Data Availability Statement
No new data were created or analyzed in this study. Data sharing is not applicable to this article.
Acknowledgments
We acknowledge the support from Project LISBOA-01-0145-FEDER-007660 (Microbiologia Molecular, Estrutural e Celular) funded by FEDER funds through COMPETE 2020-Programa Operacional Competitividade e Internacionalização (POCI); from FCT—Fundação para a Ciência e a Tecnologia (PTDC/BIA-BFS/31026/2017 and 2020.05017.BD) and from the European Union’s Horizon 2020 Research and Innovation Program, through TIMB3 and B-LigZymes projects (grant agreements No 810856 and 824017, respectively). We thank Edilson Galdino for critical reading of the manuscript and helpful discussions.
Conflicts of Interest
The authors declare no conflict of interest.
Abbreviations
| 5c | penta-coordinated |
| 6c | hexa-coordinated |
| AFM | atomic force microscopy |
| AQS | anthraquinone disulfonate |
| BSA | bovine serum albumin |
| ccNiR | cytochrome c nitrite reductase |
| CcO | cytochrome c oxidase |
| CcP | cytochrome c peroxidase |
| CD | circular dichroism |
| cd1NiR | cytochrome cd1 nitrite reductase |
| CNT | carbon nanotube |
| CT | charge-transfer |
| CV | cyclic voltammetry |
| cyt c | cytochrome c |
| DDAB | didodecyldimethylammonium bromide |
| DET | direct electron transfer |
| DyP | dye-decolorizing peroxidase |
| EDC | 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide |
| ET | electron transfer |
| FTIR | Fourier-transform infrared |
| GC | glassy carbon |
| GOx | glucose oxidase |
| Hb | hemoglobin |
| HRP | horseradish peroxidase |
| IL | ionic liquid |
| ITO | indium tin oxide |
| LDH | layered double hydroxide |
| LOD | limit of detection |
| Mb | myoglobin |
| MET | mediated electron transfer |
| MP | microperoxidase |
| MWCNTs | multi-walled carbon nanotubes |
| NHS | N-hydroxysuccinimide |
| NiR | nitrite reductase |
| NPs | nanoparticles |
| P420 | cytochrome P420 |
| P450 | cytochrome P450 |
| PANI | polyaniline |
| PG | pyrolytic graphite |
| PPY | polypyrrole |
| ROS | reactive oxygen species |
| RR | resonance Raman |
| SAM | self-assembled monolayer |
| SEIRA | surface-enhanced infrared absorption |
| SEM | scanning electron microscopy |
| SERR | surface-enhanced resonance Raman |
| SPEs | screen-printed electrodes |
| SWCNTs | single-walled carbon nanotubes |
| TCA | trichloroacetic acid |
| TOP | tobacco peroxidase |
References
- Poulos, T.L. Heme Enzyme Structure and Function. Chem. Rev. 2014, 114, 3919–3962. [Google Scholar] [CrossRef] [Green Version]
- Spiro, T.G.; Jarzecki, A.A. Heme-based sensors: Theoretical modeling of heme-ligand-protein interactions. Curr. Opin. Chem. Biol. 2001, 5, 715–723. [Google Scholar] [CrossRef]
- Turner, A.P.F. Biosensors: Sense and sensibility. Chem. Soc. Rev. 2013, 42, 3184–3196. [Google Scholar] [CrossRef] [Green Version]
- Labib, M.; Sargent, E.H.; Kelley, S.O. Electrochemical Methods for the Analysis of Clinically Relevant Biomolecules. Chem. Rev. 2016, 116, 9001–9090. [Google Scholar] [CrossRef]
- Monteiro, T.; Almeida, M.G. Electrochemical Enzyme Biosensors Revisited: Old Solutions for New Problems. Crit. Rev. Anal. Chem. 2019, 49, 44–66. [Google Scholar] [CrossRef]
- Sekretaryova, A.N.; Eriksson, M.; Turner, A.P. Bioelectrocatalytic systems for health applications. Biotechnol. Adv. 2016, 34, 177–197. [Google Scholar] [CrossRef]
- Ronkainen, N.J.; Halsall, H.B.; Heineman, W.R. Electrochemical biosensors. Chem. Soc. Rev. 2010, 39, 1747–1763. [Google Scholar] [CrossRef]
- Thevenot, D.R.; Toth, K.; Durst, R.A.; Wilson, G.S. Electrochemical Biosensors: Recommended Definitions and Classification. Biosens. Bioelectron. 2001, 16, 121–131. [Google Scholar] [CrossRef] [Green Version]
- Elsebai, B.; Ghica, M.E.; Abbas, M.N.; Brett, C.M.A. Catalase Based Hydrogen Peroxide Biosensor for Mercury Determination by Inhibition Measurements. J. Hazard. Mater. 2017, 340, 344–350. [Google Scholar] [CrossRef] [PubMed]
- Habermuller, K.; Mosbach, M.; Schuhmann, W. Electron-Transfer Mechanisms in Amperometric Biosensors. Fresenius J. Anal. Chem. 2000, 366, 560–568. [Google Scholar] [CrossRef]
- Gorton, L.; Lindgren, A.; Larsson, T.; Munteanu, F.D.; Ruzgas, T.; Gazaryan, I. Direct Electron Transfer between Heme-Containing Enzymes and Electrodes as Basis for Third Generation Biosensors. Anal. Chim. Acta 1999, 400, 91–108. [Google Scholar] [CrossRef]
- Clark, L.C., Jr.; Lyons, C. Electrode Systems for Continuous Monitoring In Cardiovascular Surgery. Ann. N. Y. Acad. Sci. 1962, 102, 29–45. [Google Scholar] [CrossRef]
- Wang, J. Electrochemical Glucose Biosensors. Chem. Rev. 2008, 108, 814–825. [Google Scholar] [CrossRef] [PubMed]
- Heller, A.; Feldman, B. Electrochemical Glucose Sensors and Their Applications in Diabetes Management. Chem. Rev. 2008, 108, 2482–2505. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, P.; Das, M.; Chinnadayyala, S.R.; Singha, I.M.; Goswami, P. Recent Advances on Developing 3rd Generation Enzyme Electrode for Biosensor Applications. Biosens. Bioelectron. 2016, 79, 386–397. [Google Scholar] [CrossRef]
- Chaubey, A.; Malhotra, B.D. Mediated Biosensors. Biosens. Bioelectron. 2002, 17, 441–456. [Google Scholar] [CrossRef]
- Silveira, C.M.; Almeida, M.G. Small Electron-Transfer Proteins as Mediators in Enzymatic Electrochemical Biosensors. Anal. Bioanal. Chem. 2013, 405, 3619–3635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Astier, Y.; Canters, G.W.; Davis, J.J.; Hill, H.A.O.; Verbeet, M.P.; Wijma, H.J. Sensing Nitrite through a Pseudoazurin–Nitrite Reductase Electron Transfer Relay. ChemPhysChem 2005, 6, 1114–1120. [Google Scholar] [CrossRef]
- Serra, A.S.; Jorge, S.R.; Silveira, C.M.; Moura, J.J.G.; Jubete, E.; Ochoteco, E.; Cabañero, G.; Grande, H.; Almeida, M.G. Cooperative Use of Cytochrome Cd1 Nitrite Reductase and Its Redox Partner Cytochrome C552 to Improve the Selectivity of Nitrite Biosensing. Anal. Chim. Acta 2011, 693, 41–46. [Google Scholar] [CrossRef]
- Da Silva, S.; Cosnier, S.; Almeida, M.G.; Moura, J.J.G. An Efficient Poly(Pyrrole–Viologen)-Nitrite Reductase Biosensor for the Mediated Detection of Nitrite. Electrochem. Commun. 2004, 6, 404–408. [Google Scholar] [CrossRef]
- Cosnier, S.; Da Silva, S.; Shan, D.; Gorgy, K. Electrochemical Nitrate Biosensor Based on Poly(Pyrrole–Viologen) Film–Nitrate Reductase–Clay Composite. Bioelectrochemistry 2008, 74, 47–51. [Google Scholar] [CrossRef] [PubMed]
- Schuhmann, W. Electron-Transfer Pathways in Amperometric Biosensors. Ferrocene-Modified Enzymes Entrapped in Conducting-Polymer Layers. Biosens. Bioelectron. 1995, 10, 181–193. [Google Scholar] [CrossRef]
- Willner, I.; Willner, B.; Katz, E. Functional Biosensor Systems via Surface-Nanoengineering of Electronic Elements. Rev. Mol. Biotechnol. 2002, 82, 325–355. [Google Scholar] [CrossRef]
- Fruk, L.; Kuo, C.-H.; Torres, E.; Niemeyer, C.M. Apoenzyme Reconstitution as a Chemical Tool for Structural Enzymology and Biotechnology. Angew. Chem. Int. Ed. 2009, 48, 1550–1574. [Google Scholar] [CrossRef]
- Nöll, T.; Nöll, G. Strategies for “Wiring” Redox-Active Proteins to Electrodes and Applications in Biosensors, Biofuel Cells, and Nanotechnology. Curr. Soc. Rev. 2011, 40, 3564–3576. [Google Scholar] [CrossRef]
- Léger, C.; Bertrand, P. Direct Electrochemistry of Redox Enzymes as a Tool for Mechanistic Studies. Chem. Rev. 2008, 108, 2379–2438. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Chakraborty, S.; Hosseinzadeh, P.; Yu, Y.; Tian, S.; Petrik, I.; Bhagi, A.; Lu, Y. Metalloproteins Containing Cytochrome, Iron–Sulfur, or Copper Redox Centers. Chem. Rev. 2014, 114, 4366–4469. [Google Scholar] [CrossRef]
- Borgmann, S.; Hartwich, G.; Schulte, A.; Schuhmann, W. Amperometric Enzyme Sensors based on Direct and Mediated Electron Transfer. In Perspectives in Bioanalysis; Paleček, E., Scheller, F., Wang, J., Eds.; Elsevier: Amsterdam, The Netherlands, 2005; Volume 1, pp. 599–655. ISBN 1871-0069. [Google Scholar]
- Bollella, P.; Gorton, L. Enzyme Based Amperometric Biosensors. Curr. Opin. Electrochem. 2018, 10, 157–173. [Google Scholar] [CrossRef]
- Taurino, I.; Sanzò, G.; Antiochia, R.; Tortolini, C.; Mazzei, F.; Favero, G.; De Micheli, G.; Carrara, S. Recent Advances in Third Generation Biosensors Based on Au and Pt Nanostructured Electrodes. TrAC Trends Anal. Chem. 2016, 79, 151–159. [Google Scholar] [CrossRef]
- Redeker, E.S.; Ta, D.T.; Cortens, D.; Billen, B.; Guedens, W.; Adriaensens, P. Protein Engineering for Directed Immobilization. Bioconjug. Chem. 2013, 24, 1761–1777. [Google Scholar] [CrossRef] [PubMed]
- Bollella, P.; Katz, E. Chapter Ten—Bioelectrocatalysis at carbon nanotubes. In Methods Enzymology; Kumar, C.V., Ed.; Academic Press: Cambridge, MA, USA, 2020; Volume 630, pp. 215–247. ISBN 0076-6879. [Google Scholar]
- Mazurenko, I.; Hitaishi, V.P.; Lojou, E. Recent Advances in Surface Chemistry of Electrodes to Promote Direct Enzymatic Bioelectrocatalysis. Curr. Opin. Electrochem. 2020, 19, 113–121. [Google Scholar] [CrossRef]
- Ju, H. Functional Nanomaterials and Nanoprobes for Amplified Biosensing. Appl. Mater. Today 2018, 10, 51–71. [Google Scholar] [CrossRef]
- Sassolas, A.; Blum, L.J.; Leca-Bouvier, B.D. Immobilization Strategies to Develop Enzymatic Biosensors. Biotechnol. Adv. 2012, 30, 489–511. [Google Scholar] [CrossRef] [PubMed]
- Kaur, J.; Choudhary, S.; Chaudhari, R.; Jayant, R.D.; Joshi, A. 9-Enzyme-based biosensors. In Bioelectronics and Medical Devices; Pal, K., Kraatz, H.-B., Khasnobish, A., Bag, S., Banerjee, I., Kuruganti, U., Eds.; Woodhead Publishing: Sawston, UK, 2019; pp. 211–240. ISBN 978-0-08-102420-1. [Google Scholar]
- Wang, Z.; Li, M.; Su, P.; Zhang, Y.; Shen, Y.; Han, D.; Ivaska, A.; Niu, L. Direct Electron Transfer of Horseradish Peroxidase and Its Electrocatalysis Based on Carbon Nanotube/Thionine/Gold Composites. Electrochem. Commun. 2008, 10, 306–310. [Google Scholar] [CrossRef]
- Rosca, V.; Popescu, I.C. Kinetic Analysis of Horseradish Peroxidase “Wiring” in Redox Polyelectrolyte–Peroxidase Multilayer Assemblies. Electrochem. Commun. 2002, 4, 904–911. [Google Scholar] [CrossRef]
- Feizabadi, M.; Soleymanpour, A.; Faridnouri, H.; Ajloo, D. Improving Stability of Biosensor Based on Covalent Immobilization of Horseradish Peroxidase by Gamma-Aminobutyric Acid and Application in Detection of H2O2. Int. J. Biol. Macromol. 2019, 136, 597–606. [Google Scholar] [CrossRef] [PubMed]
- Satvekar, R.K.; Rohiwal, S.S.; Raut, A.V.; Karande, V.A.; Tiwale, B.M.; Pawar, S.H. A Silica-Dextran Nanocomposite as a Novel Matrix for Immobilization of Horseradish Peroxidase, and Its Application to Sensing Hydrogen Peroxide. Microchim. Acta 2014, 181, 71–77. [Google Scholar] [CrossRef]
- Datta, S.; Christena, L.R.; Rajaram, Y.R.S. Enzyme Immobilization: An Overview on Techniques and Support Materials. 3 Biotech 2013, 3, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arya, S.K.; Solanki, P.R.; Datta, M.; Malhotra, B.D. Recent Advances in Self-Assembled Monolayers Based Biomolecular Electronic Devices. Biosens. Bioelectron. 2009, 24, 2810–2817. [Google Scholar] [CrossRef]
- Samanta, D.; Sarkar, A. Immobilization of Bio-Macromolecules on Self-Assembled Monolayers: Methods and Sensor Applications. Curr. Soc. Rev. 2011, 40, 2567–2592. [Google Scholar] [CrossRef]
- Hitaishi, V.P.; Clement, R.; Bourassin, N.; Baaden, M.; De Poulpiquet, A.; Sacquin-Mora, S.; Ciaccafava, A.; Lojou, E. Controlling Redox Enzyme Orientation at Planar Electrodes. Catalysts 2018, 8, 192. [Google Scholar] [CrossRef] [Green Version]
- Liébana, S.; Drago, G.A. Bioconjugation and Stabilisation of Biomolecules in Biosensors. Essays Biochem. 2016, 60, 59–68. [Google Scholar] [CrossRef] [Green Version]
- Yates, N.D.J.; Fascione, M.A.; Parkin, A. Methodologies for “Wiring” Redox Proteins/Enzymes to Electrode Surfaces. Chem. Eur. J. 2018, 24, 12164–12182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, B.; Guo, C.; Zhang, M.; Park, B.; Xu, B. High-Resolution Single-Molecule Recognition Imaging of the Molecular Details of Ricin–Aptamer Interaction. J. Phys. Chem. B 2012, 116, 5316–5322. [Google Scholar] [CrossRef] [PubMed]
- Asturias-Arribas, L.; Alonso-Lomillo, M.A.; Domínguez-Renedo, O.; Arcos-Martínez, M.J. Cytochrome P450 2D6 Based Electrochemical Sensor for the Determination of Codeine. Talanta 2014, 129, 315–319. [Google Scholar] [CrossRef] [PubMed]
- Karunakaran, C.; Rajkumar, R.; Bhargava, K. (Eds.) Introduction to Biosensors. In Biosens. Bioelectronics; Elsevier: Amsterdam, The Netherlands, 2015; Chapter 1; pp. 1–68. ISBN 978-0-12-803100-1. [Google Scholar]
- Zhou, J.; Liao, C.; Zhang, L.; Wang, Q.; Tian, Y. Molecular Hydrogel-Stabilized Enzyme with Facilitated Electron Transfer for Determination of H 2 O 2 Released from Live Cells. Anal. Chem. 2014, 86, 4395–4401. [Google Scholar] [CrossRef]
- Battistuzzi, G.; Bellei, M.; Bortolotti, C.A.; Sola, M. Redox Properties of Heme Peroxidases. Arch. Biochem. Biophys. 2010, 500, 21–36. [Google Scholar] [CrossRef]
- Schneider, E.; Clark, D.S. Cytochrome P450 (CYP) Enzymes and the Development of CYP Biosensors. Biosens. Bioelectron. 2013, 39, 1–13. [Google Scholar] [CrossRef]
- McNaught, A.D.; Wilkinson, A. IUPAC Compendium of Chemical Terminology, 2nd ed.; Blackwell Scientific Publications: Oxford, UK, 1997. [Google Scholar]
- Ruzgas, T.; Csöregi, E.; Emnéus, J.; Gorton, L.; Marko-Varga, G. Peroxidase-Modified Electrodes: Fundamentals and Application. Anal. Chim. Acta 1996, 330, 123–138. [Google Scholar] [CrossRef]
- Gazaryan, I.G.; Gorton, L.; Ruzgas, T.; Csoregi, E.; Schuhmann, W.; Lagrimini, L.M.; Khushpul’yan, D.M.; Tishkov, V.I. Tobacco Peroxidase as a New Reagent for Amperometric Biosensors. J. Anal. Chem. 2005, 60, 558–566. [Google Scholar] [CrossRef] [Green Version]
- Castilho, T.J.; Sotomayor, M.P.T.; Kubota, L.T. Amperometric Biosensor Based on Horseradish Peroxidase for Biogenic Amine Determinations in Biological Samples. J. Pharm. Biomed. Anal. 2005, 37, 785–791. [Google Scholar] [CrossRef] [PubMed]
- Kermad, A.; Sam, S.; Ghellai, N.; Khaldi, K.; Gabouze, N. Horseradish Peroxidase-Modified Porous Silicon for Phenol Monitoring. MSEB 2013, 178, 1159–1164. [Google Scholar] [CrossRef]
- Rusling, J.F. Electrochemistry of Redox Enzymes. In Bioelectrochemistry; Bartlett, P.N., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2008; pp. 39–85. [Google Scholar]
- Zigah, D.; Lojou, E.; de Poulpiquet, A. Micro- and Nanoscopic Imaging of Enzymatic Electrodes: A Review. ChemElectroChem 2019, 6, 5524–5546. [Google Scholar] [CrossRef]
- Kornienko, N.; Ly, K.H.; Robinson, W.E.; Heidary, N.; Zhang, J.Z.; Reisner, E. Advancing Techniques for Investigating the Enzyme–Electrode Interface. Acc. Chem. Res. 2019, 52, 1439–1448. [Google Scholar] [CrossRef] [Green Version]
- López-Lorente, Á.I.; Kranz, C. Recent Advances in Biomolecular Vibrational Spectroelectrochemistry. Curr. Opin. Electrochem. 2017, 5, 106–113. [Google Scholar] [CrossRef]
- Sezer, M.; Millo, D.; Weidinger, I.M.; Zebger, I.; Hildebrandt, P. Analyzing the Catalytic Processes of Immobilized Redox Enzymes by Vibrational Spectroscopies. IUBMB Life 2012, 64, 455–464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenfield, N.J. Using Circular Dichroism Spectra to Estimate Protein Secondary Structure. Nat. Protoc. 2006, 1, 2876–2890. [Google Scholar] [CrossRef]
- Wu, H.; Wang, X.; Qiao, M.; Zhang, H.; Jin, X.; Fan, S. Enhancing Sensitivity of Hemoglobin-Based Electrochemical Biosensor by Using Protein Conformational Intermediate. Sens. Actuators B 2015, 221, 694–699. [Google Scholar] [CrossRef]
- Gao, J.; Liu, H.; Tong, C.; Pang, L.; Feng, Y.; Zuo, M.; Wei, Z.; Li, J. Hemoglobin-Mn3(PO4)2 Hybrid Nanoflower with Opulent Electroactive Centers for High-Performance Hydrogen Peroxide Electrochemical Biosensor. Sens. Actuators B 2020, 307, 127628. [Google Scholar] [CrossRef]
- Murphy, M.; Theyagarajan, K.; Ganesan, P.; Senthilkumar, S.; Thenmozhi, K. Electrochemical Biosensor for the Detection of Hydrogen Peroxide Using Cytochrome c Covalently Immobilized on Carboxyl Functionalized Ionic Liquid/Multiwalled Carbon Nanotube Hybrid. Appl. Surf. Sci. 2019, 492, 718–725. [Google Scholar] [CrossRef]
- Barbosa, C.; Silveira, C.M.; Silva, D.; Brissos, V.; Hildebrandt, P.; Martins, L.O.; Todorovic, S. Immobilized Dye-Decolorizing Peroxidase (DyP) and Directed Evolution Variants for Hydrogen Peroxide Biosensing. Biosens. Bioelectron. 2020, 153, 112055. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Peng, X.; Kong, J.; Deng, J. Facilitated Electron Transfer from an Electrode to Horseradish Peroxidase in a Biomembrane-like Surfactant Film. J. Electroanal. Chem. 2000, 480, 26–33. [Google Scholar] [CrossRef]
- Chen, X.; Xie, H.; Kong, J.; Deng, J. Characterization for Didodecyldimethylammonium Bromide Liquid Crystal Film Entrapping Catalase with Enhanced Direct Electron Transfer Rate. Biosens. Bioelectron. 2001, 16, 115–120. [Google Scholar] [CrossRef]
- Todorovic, S.; Jung, C.; Hildebrandt, P.; Murgida, D.H. Conformational Transitions and Redox Potential Shifts of Cytochrome P450 Induced by Immobilization. J. Biol. Inorg. Chem. 2006, 11, 119–127. [Google Scholar] [CrossRef]
- Liu, X.; Feng, H.; Zhao, R.; Wang, Y.; Liu, X. A Novel Approach to Construct a Horseradish Peroxidase|hydrophilic Ionic Liquids|Au Nanoparticles Dotted Titanate Nanotubes Biosensor for Amperometric Sensing of Hydrogen Peroxide. Biosens. Bioelectron. 2012, 31, 101–104. [Google Scholar] [CrossRef]
- Dong, J.; Wen, Y.; Miao, Y.; Xie, Z.; Zhang, Z.; Yang, H. A Nanoporous Zirconium Phytate Film for Immobilization of Redox Protein and the Direct Electrochemical Biosensor. Sens. Actuators B 2010, 150, 141–147. [Google Scholar] [CrossRef]
- Zhang, Y.; He, P.; Hu, N. Horseradish Peroxidase Immobilized in TiO2 Nanoparticle Films on Pyrolytic Graphite Electrodes: Direct Electrochemistry and Bioelectrocatalysis. Electrochim. Acta 2004, 49, 1981–1988. [Google Scholar] [CrossRef]
- Wang, Y.; Ma, X.; Wen, Y.; Xing, Y.; Zhang, Z.; Yang, H. Direct Electrochemistry and Bioelectrocatalysis of Horseradish Peroxidase Based on Gold Nano-Seeds Dotted TiO2 Nanocomposite. Biosens. Bioelectron. 2010, 25, 2442–2446. [Google Scholar] [CrossRef] [PubMed]
- Siebert, F.; Hildebrandt, P. Heme Proteins. In Vibrational Spectroscopy in Life Science; Wiley-VCH Verlag GmbH & Co. KGaA: Hoboken, NJ, USA, 2008; pp. 227–282. ISBN 978-3-527-62134-7. [Google Scholar]
- Todorovic, S.; Verissimo, A.; Wisitruangsakul, N.; Zebger, I.; Hildebrandt, P.; Pereira, M.M.; Teixeira, M.; Murgida, D.H. SERR-Spectroelectrochemical Study of a Cbb3 Oxygen Reductase in a Biomimetic Construct. J. Phys. Chem. B 2008, 112, 16952–16959. [Google Scholar] [CrossRef]
- Murgida, D.H.; Hildebrandt, P. Heterogeneous Electron Transfer of Cytochrome c on Coated Silver Electrodes. Electric Field Effects on Structure and Redox Potential. J. Phys. Chem. B 2001, 105, 1578–1586. [Google Scholar] [CrossRef]
- Murgida, D.H.; Hildebrandt, P. Electron-Transfer Processes of Cytochrome c at Interfaces. New Insights by Surface-Enhanced Resonance Raman Spectroscopy. Acc. Chem. Res. 2004, 37, 854–861. [Google Scholar] [CrossRef]
- Moyo, M.; Okonkwo, J.O.; Agyei, N.M. A Novel Hydrogen Peroxide Biosensor Based on Adsorption of Horseradish Peroxidase onto a Nanobiomaterial Composite Modified Glassy Carbon Electrode. Electroanalysis 2013, 25, 1946–1954. [Google Scholar] [CrossRef]
- Tulli, F.; Gulotta, F.A.; Martino, D.M.; Zanini, V.I.P.; Borsarelli, C.D. Ultrasensitive Amperometric Biosensing of Polyphenols Using Horseradish Peroxidase Immobilized in a Laponite/Au/DNA-Bioinspired Polycation Nanocomposite. J. Electrochem. Soc. 2018, 165, B452–B457. [Google Scholar] [CrossRef]
- Zhu, Z.; Li, X.; Wang, Y.; Zeng, Y.; Sun, W.; Huang, X. Direct Electrochemistry and Electrocatalysis of Horseradish Peroxidase with Hyaluronic Acid–Ionic Liquid–Cadmium Sulfide Nanorod Composite Material. Anal. Chim. Acta 2010, 670, 51–56. [Google Scholar] [CrossRef]
- Song, H.; Ni, Y.; Kokot, S. Investigations of an Electrochemical Platform Based on the Layered MoS2–Graphene and Horseradish Peroxidase Nanocomposite for Direct Electrochemistry and Electrocatalysis. Biosens. Bioelectron. 2014, 56, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Cahuantzi-Muñoz, S.L.; González-Fuentes, M.A.; Ortiz-Frade, L.A.; Torres, E.; Ţălu, Ş.; Trejo, G.; Méndez-Albores, A. Electrochemical Biosensor for Sensitive Quantification of Glyphosate in Maize Kernels. Electroanalysis 2019, 31, 927–935. [Google Scholar] [CrossRef] [Green Version]
- Mai, Z.; Zhao, X.; Dai, Z.; Zou, X. Contributions of Components in Guanidine Hydrochloride to Hemoglobin Unfolding Investigated by Protein Film Electrochemistry. J. Phys. Chem. B 2010, 114, 7090–7097. [Google Scholar] [CrossRef]
- Li, X.; Zheng, W.; Zhang, L.; Yu, P.; Lin, Y.; Su, L.; Mao, L. Effective Electrochemical Method for Investigation of Hemoglobin Unfolding Based on the Redox Property of Heme Groups at Glassy Carbon Electrodes. Anal. Chem. 2009, 81, 8557–8563. [Google Scholar] [CrossRef]
- Smulevich, G.; Feis, A.; Howes, B.D.; Ivancich, A. Structure-Function Relationships among Heme Peroxidases: New Insights from Electronic Absorption, Resonance Raman and Multifrequency Electron Paramagnetic Resonance Spectroscopies. In Handbook of Porphyrin Science; World Scientific Publishing Co.: Singapore, 2010; pp. 367–453. ISBN 978-981-4307-19-2. [Google Scholar]
- Torres, E.; Ayala, M. Biocatalysis Based on Heme Peroxidases; Springer: Berlin/Heidelberg, Germany, 2010. [Google Scholar]
- Poulos, T.L. Thirty Years of Heme Peroxidase Structural Biology. Arch. Biochem. Biophys. 2010, 500, 3–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nóbrega, C.S.; Pauleta, S.R. Reduction of hydrogen peroxide in gram-negative bacteria—bacterial peroxidases. In Advances in Microbial Physiology; Poole, R.K., Ed.; Academic Press: Cambridge, MA, USA, 2019; Volume 74, pp. 415–464. ISBN 0065-2911. [Google Scholar]
- Mossanha, R.; Erdmann, C.A.; Santos, C.S.; Wohnrath, K.; Fujiwara, S.T.; Pessoa, C.A. Construction of a Biosensor Based on SAM of Thiolactic Acid on Gold Nanoparticles Stabilized by Silsesquioxane Polyelectrolyte for Cathecol Determination. Sens. Actuators B 2017, 252, 747–756. [Google Scholar] [CrossRef]
- Ferapontova, E.E. Direct Peroxidase Bioelectrocatalysis on a Variety of Electrode Materials. Electroanalysis 2004, 16, 1101–1112. [Google Scholar] [CrossRef]
- Narvaez, A.; Suarez, G.; Popescu, I.C.; Katakis, I.; Dominguez, E. Reagentless Biosensors Based on Self-Deposited Redox Polyelectrolyte-Oxidoreductases Architectures. Biosens. Bioelectron. 2000, 15, 43–52. [Google Scholar] [CrossRef]
- Okawa, Y.; Yokoyama, N.; Sakai, Y.; Shiba, F. Direct Electron Transfer Biosensor for Hydrogen Peroxide Carrying Nanocomplex Composed of Horseradish Peroxidase and Au-Nanoparticle—Characterization and Application to Bienzyme Systems. Anal. Chem. Res. 2015, 5, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Huerta-Miranda, G.A.; Arrocha-Arcos, A.A.; Miranda-Hernandez, M. Gold Nanoparticles/4-Aminothiophenol Interfaces for Direct Electron Transfer of Horseradish Peroxidase: Enzymatic Orientation and Modulation of Sensitivity towards Hydrogen Peroxide Detection. Bioelectrochemistry 2018, 122, 77–83. [Google Scholar] [CrossRef]
- Bollella, P.; Medici, L.; Tessema, M.; Poloznikov, A.A.; Hushpulian, D.M.; Tishkov, V.I.; Andreu, R.; Leech, D.; Megersa, N.; Marcaccio, M.; et al. Highly Sensitive, Stable and Selective Hydrogen Peroxide Amperometric Biosensors Based on Peroxidases from Different Sources Wired by Os-Polymer: A Comparative Study. Solid State Ion. 2018, 314, 178–186. [Google Scholar] [CrossRef]
- Olloqui-Sariego, J.L.; Zakharova, G.S.; Poloznikov, A.A.; Calvente, J.J.; Hushpulian, D.M.; Gorton, L.; Andreu, R. Fenton-like Inactivation of Tobacco Peroxidase Electrocatalysis at Negative Potentials. ACS Catal. 2016, 6, 7452–7457. [Google Scholar] [CrossRef]
- Rusling, J.F. Enzyme Bioelectrochemistry in Cast Biomembrane-Like Films. Acc. Chem. Res. 1998, 31, 363–369. [Google Scholar] [CrossRef]
- Zhang, Z.; Chouchane, S.; Magliozzo, R.S.; Rusling, J.F. Direct Voltammetry and Catalysis with Mycobacterium Tuberculosis Catalase−Peroxidase, Peroxidases, and Catalase in Lipid Films. Anal. Chem. 2002, 74, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Guo, K.; Lv, J.; Gao, Y.; Duan, C.; Deng, L.; Zhu, Z. A Novel Nitrite Biosensor Based on the Direct Electrochemistry of Horseradish Peroxidase Immobilized on Porous Co3O4 Nanosheets and Reduced Graphene Oxide Composite Modified Electrode. Sens. Actuators B 2017, 238, 249–256. [Google Scholar] [CrossRef] [Green Version]
- Niu, Y.; Liu, J.; Chen, W.; Yin, C.; Weng, W.; Li, X.; Wang, X.; Li, G.; Sun, W. A Direct Electron Transfer Biosensor Based on a Horseradish Peroxidase and Gold Nanotriangle Modified Electrode and Electrocatalysis. Anal. Methods 2018, 10, 5297–5304. [Google Scholar] [CrossRef]
- Sun, W.; Guo, Y.; Li, T.; Ju, X.; Lou, J.; Ruan, C. Electrochemistry of Horseradish Peroxidase Entrapped in Graphene and DsDNA Composite Modified Carbon Ionic Liquid Electrode. Electrochim. Acta 2012, 75, 381–386. [Google Scholar] [CrossRef]
- Şenel, M.; Çevik, E.; Abasıyanık, M.F. Amperometric Hydrogen Peroxide Biosensor Based on Covalent Immobilization of Horseradish Peroxidase on Ferrocene Containing Polymeric Mediator. Sens. Actuators B 2010, 145, 444–450. [Google Scholar] [CrossRef]
- Tatsuma, T.; Okawa, Y.; Watanabe, T. Enzyme Monolayer- and Bilayer-Modified Tin Oxide Electrodes for the Determination of Hydrogen Peroxide and Glucose. Anal. Chem. 1989, 61, 2352–2355. [Google Scholar] [CrossRef]
- Wang, Y.; Hasebe, Y. Carbon-Felt-Based Bioelectrocatalytic Flow-Detectors: Optimization of the Adsorption Conditions of Horseradish Peroxidase and Thionine onto Carbon-Felt for Highly Sensitive Amperometric Determination of H2O2. Anal. Sci. 2011, 27, 401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindgren, A.; Tanaka, M.; Ruzgas, T.; Gorton, L.; Gazaryan, I.; Ishimori, K.; Morishima, I. Direct Electron Transfer Catalysed by Recombinant Forms of Horseradish Peroxidase: Insight into the Mechanism. Electrochem. Commun. 1999, 1, 171–175. [Google Scholar] [CrossRef]
- Ruzgas, T.; Emnéus, J.; Gorton, L.; Marko-Varga, G. The Development of a Peroxidase Biosensor for Monitoring Phenol and Related Aromatic Compounds. Anal. Chim. Acta 1995, 311, 245–253. [Google Scholar] [CrossRef]
- Pandey, V.P.; Awasthi, M.; Singh, S.; Tiwari, S.; Dwivedi, U.N. A Comprehensive Review on Function and Application of Plant Peroxidases. Biochem. Anal. Biochem. 2017, 6, 308. [Google Scholar] [CrossRef]
- Krainer, F.W.; Glieder, A. An Updated View on Horseradish Peroxidases: Recombinant Production and Biotechnological Applications. Appl. Microbiol. Biotechnol. 2015, 99, 1611–1625. [Google Scholar] [CrossRef] [Green Version]
- Castillo, J.; Ferapontova, E.; Hushpulian, D.; Tasca, F.; Tishkov, V.; Chubar, T.; Gazaryan, I.; Gorton, L. Direct Electrochemistry and Bioelectrocatalysis of H2O2 Reduction of Recombinant Tobacco Peroxidase on Graphite. Effect of Peroxidase Single-Point Mutation on Ca2+-Modulated Catalytic Activity. J. Electroanal. Chem. 2006, 588, 112–121. [Google Scholar] [CrossRef]
- Gaspar, S.; Popescu, I.C.; Gazaryan, I.G.; Gerardo Bautista, A.; Sakharov, I.Y.; Mattiasson, B.; Csöregi, E. Biosensors Based on Novel Plant Peroxidases: A Comparative Study. Electrochim. Acta 2000, 46, 255–264. [Google Scholar] [CrossRef]
- Centeno, D.A.; Solano, X.H.; Castillo, J.J. A New Peroxidase from Leaves of Guinea Grass (Panicum Maximum): A Potential Biocatalyst to Build Amperometric Biosensors. Bioelectrochemistry 2017, 116, 33–38. [Google Scholar] [CrossRef] [PubMed]
- Ferapontova, E.E.; Grigorenko, V.G.; Egorov, A.M.; Börchers, T.; Ruzgas, T.; Gorton, L. Mediatorless Biosensor for H2O2 Based on Recombinant Forms of Horseradish Peroxidase Directly Adsorbed on Polycrystalline Gold. Biosens. Bioelectron. 2001, 16, 147–157. [Google Scholar] [CrossRef]
- Ferapontova, E.; Gorton, L. Effect of PH on Direct Electron Transfer in the System Gold Electrode–Recombinant Horseradish Peroxidase. Bioelectrochemistry 2002, 55, 83–87. [Google Scholar] [CrossRef]
- Andreu, R.; Ferapontova, E.E.; Gorton, L.; Calvente, J.J. Direct Electron Transfer Kinetics in Horseradish Peroxidase Electrocatalysis. J. Phys. Chem. B 2007, 111, 469–477. [Google Scholar] [CrossRef]
- Sun, Y.-X.; Zhang, J.-T.; Huang, S.-W.; Wang, S.-F. Hydrogen Peroxide Biosensor Based on the Bioelectrocatalysis of Horseradish Peroxidase Incorporated in a New Hydrogel Film. Sens. Actuators B 2007, 124, 494–500. [Google Scholar] [CrossRef]
- Guo, C.; Song, Y.; Wei, H.; Li, P.; Wang, L.; Sun, L.; Sun, Y.; Li, Z. Room Temperature Ionic Liquid Doped DNA Network Immobilized Horseradish Peroxidase Biosensor for Amperometric Determination of Hydrogen Peroxide. Anal. Bioanal. Chem. 2007, 389, 527–532. [Google Scholar] [CrossRef]
- Tian, J.; Wang, J.; Li, Y.; Huang, M.; Lu, J. Electrochemically Driven Omeprazole Metabolism via Cytochrome P450 Assembled on the Nanocomposites of Ceria Nanoparticles and Graphene. J. Electrochem. Soc. 2017, 164, H470–H476. [Google Scholar] [CrossRef]
- Lu, J.; Cui, D.; Li, H.; Zhang, Y.; Liu, S. Cytochrome P450 Bienzymes Assembled on Au/Chitosan/Reduced Graphene Oxide Nanosheets for Electrochemically-Driven Drug Cascade Metabolism. Electrochim. Acta 2015, 165, 36–44. [Google Scholar] [CrossRef]
- Salimi, A.; Sharifi, E.; Noorbakhsh, A.; Soltanian, S. Direct Electrochemistry and Electrocatalytic Activity of Catalase Immobilized onto Electrodeposited Nano-Scale Islands of Nickel Oxide. Biophys. Chem. 2007, 125, 540–548. [Google Scholar] [CrossRef] [PubMed]
- Saadati, S.; Salimi, A.; Hallaj, R.; Rostami, A. Layer by Layer Assembly of Catalase and Amine-Terminated Ionic Liquid onto Titanium Nitride Nanoparticles Modified Glassy Carbon Electrode: Study of Direct Voltammetry and Bioelectrocatalytic Activity. Anal. Chim. Acta 2012, 753, 32–41. [Google Scholar] [CrossRef]
- Rahimi, P.; Rafiee-Pour, H.-A.; Ghourchian, H.; Norouzi, P.; Ganjali, M.R. Ionic-Liquid/NH2-MWCNTs as a Highly Sensitive Nano-Composite for Catalase Direct Electrochemistry. Biosens. Bioelectron. 2010, 25, 1301–1306. [Google Scholar] [CrossRef] [PubMed]
- Vilian, A.T.E.; Chen, S.-M.; Lou, B.-S. A Simple Strategy for the Immobilization of Catalase on Multi-Walled Carbon Nanotube/Poly (l-Lysine) Biocomposite for the Detection of H2O2 and Iodate. Biosens. Bioelectron. 2014, 61, 639–647. [Google Scholar] [CrossRef]
- Chen, H.; Mousty, C.; Cosnier, S.; Silveira, C.; Moura, J.J.G.; Almeida, M.G. Highly Sensitive Nitrite Biosensor Based on the Electrical Wiring of Nitrite Reductase by [ZnCr-AQS] LDH. Electrochem. Commun. 2007, 9, 2240–2245. [Google Scholar] [CrossRef]
- Silveira, C.M.; Gomes, S.P.; Araújo, A.N.; Montenegro, M.C.B.S.M.; Todorovic, S.; Viana, A.S.; Silva, R.J.C.; Moura, J.J.G.; Almeida, M.G. An Efficient Non-Mediated Amperometric Biosensor for Nitrite Determination. Biosens. Bioelectron. 2010, 25, 2026–2032. [Google Scholar] [CrossRef]
- Santos, L.; Silveira, C.M.; Elangovan, E.; Neto, J.P.; Nunes, D.; Pereira, L.; Martins, R.; Viegas, J.; Moura, J.J.G.; Todorovic, S.; et al. Synthesis of WO3 Nanoparticles for Biosensing Applications. Sens. Actuators B 2016, 223, 186–194. [Google Scholar] [CrossRef]
- Ashe, D.; Alleyne, T.; Iwuoha, E. Serum Cytochrome c Detection Using a Cytochrome c Oxidase Biosensor. Biotechnol. Appl. Biochem. 2007, 46, 185. [Google Scholar] [CrossRef] [PubMed]
- Lewis, K.L.; Lianyong, S.; Hawkridge, F.M.; Ward, K.R.; Rhoten, M.C. Immobilization of Cytochrome c Oxidase into Electrode-Supported Lipid Bilayer Membranes for in Vitro Cytochrome c Sensing. IEEE Sens. J. 2006, 6, 420–427. [Google Scholar] [CrossRef]
- Eguílaz, M.; Agüí, L.; Yáñez-Sedeño, P.; Pingarrón, J.M. A Biosensor Based on Cytochrome c Immobilization on a Poly-3-Methylthiophene/Multi-Walled Carbon Nanotubes Hybrid-Modified Electrode. Application to the Electrochemical Determination of Nitrite. J. Electroanal. Chem. 2010, 644, 30–35. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhi, J.; Zou, Y.; Zhang, W.; Lee, S.-T. Direct Electrochemistry and Electrocatalytic Activity of Cytochrome c Covalently Immobilized on a Boron-Doped Nanocrystalline Diamond Electrode. Anal. Chem. 2008, 80, 4141–4146. [Google Scholar] [CrossRef] [PubMed]
- Pashai, E.; Najafpour Darzi, G.; Jahanshahi, M.; Yazdian, F.; Rahimnejad, M. An Electrochemical Nitric Oxide Biosensor Based on Immobilized Cytochrome c on a Chitosan-Gold Nanocomposite Modified Gold Electrode. Int. J. Biol. Macromol. 2018, 108, 250–258. [Google Scholar] [CrossRef]
- Neumann, B.; Kielb, P.; Rustam, L.; Fischer, A.; Weidinger, I.M.; Wollenberger, U. Bioelectrocatalytic Reduction of Hydrogen Peroxide by Microperoxidase-11 Immobilized on Mesoporous Antimony-Doped Tin Oxide. ChemElectroChem 2017, 4, 913–919. [Google Scholar] [CrossRef]
- Gong, C.; Shen, Y.; Chen, J.; Song, Y.; Chen, S.; Song, Y.; Wang, L. Microperoxidase-11@PCN-333 (Al)/Three-Dimensional Macroporous Carbon Electrode for Sensing Hydrogen Peroxide. Sens. Actuators B 2017, 239, 890–897. [Google Scholar] [CrossRef]
- Abdelwahab, A.A.; Koh, W.C.A.; Noh, H.-B.; Shim, Y.-B. A Selective Nitric Oxide Nanocomposite Biosensor Based on Direct Electron Transfer of Microperoxidase: Removal of Interferences by Co-Immobilized Enzymes. Biosens. Bioelectron. 2010, 26, 1080–1086. [Google Scholar] [CrossRef]
- Liu, H.; Duan, C.; Su, X.; Dong, X.; Huang, Z.; Shen, W.; Zhu, Z. A Hemoglobin Encapsulated Titania Nanosheet Modified Reduced Graphene Oxide Nanocomposite as a Mediator-Free Biosensor. Sens. Actuators B 2014, 203, 303–310. [Google Scholar] [CrossRef]
- Chen, X.; Wang, Q.; Wang, L.; Gao, F.; Wang, W.; Hu, Z. Imidazoline Derivative Templated Synthesis of Broccoli-like Bi2S3 and Its Electrocatalysis towards the Direct Electrochemistry of Hemoglobin. Biosens. Bioelectron. 2015, 66, 216–223. [Google Scholar] [CrossRef]
- Mazzochette, Z.; Newton, E.; Mugweru, A. Electrochemical Catalysis of Artemisinin on Hemoglobin Functionalized Carbon Nanofibers. Anal. Methods 2017, 9, 2997–3002. [Google Scholar] [CrossRef]
- Vilian, A.T.E.; Veeramani, V.; Chen, S.-M.; Madhu, R.; Kwak, C.H.; Huh, Y.S.; Han, Y.-K. Immobilization of Myoglobin on Au Nanoparticle-Decorated Carbon Nanotube/Polytyramine Composite as a Mediator-Free H2O2 and Nitrite Biosensor. Sci. Rep. 2015, 5, 18390. [Google Scholar] [CrossRef] [Green Version]
- Luo, G.; Xie, H.; Niu, Y.; Liu, J.; Huang, Y.; Li, B.; Li, G.; Sun, W. Electrochemical Myoglobin Biosensor Based on Magnesium Metal-Organic Frameworks and Gold Nanoparticles Composite Modified Electrode. Int. J. Electrochem. Sci. 2019, 2405–2413. [Google Scholar] [CrossRef]
- Ozoner, S.K.; Yilmaz, F.; Celik, A.; Keskinler, B.; Erhan, E. A Novel Poly(Glycine Methacrylate-Co-3-Thienylmethyl Methacrylate)-Polypyrrole-Carbon Nanotube-Horseradish Peroxidase Composite Film Electrode for the Detection of Phenolic Compounds. Curr. Appl. Phys. 2011, 11, 402–408. [Google Scholar] [CrossRef]
- Cao, S.; Yuan, R.; Chai, Y.; Zhang, L.; Li, X.; Gao, F. A Mediator-Free Amperometric Hydrogen Peroxide Biosensor Based on HRP Immobilized on a Nano-Au/Poly 2,6-Pyridinediamine-Coated Electrode. Bioprocess Biosyst. Eng. 2007, 30, 71–78. [Google Scholar] [CrossRef]
- Ahirwal, G.K.; Mitra, C.K. Direct Electrochemistry of Horseradish Peroxidase-Gold Nanoparticles Conjugate. Sensors 2009, 9, 881–894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komori, K.; Terse-Thakoor, T.; Mulchandani, A. Electrochemical Properties of Seamless Three-Dimensional Carbon Nanotubes-Grown Graphene Modified with Horseradish Peroxidase. Bioelectrochemistry 2016, 111, 57–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, G.-X.; Bao, W.-J.; Wang, J.; Lu, Q.-Q.; Xia, X.-H. Immobilization and Catalytic Activity of Horseradish Peroxidase on Molybdenum Disulfide Nanosheets Modified Electrode. Electrochem. Commun. 2013, 35, 146–148. [Google Scholar] [CrossRef]
- Sugano, Y. DyP-Type Peroxidases Comprise a Novel Heme Peroxidase Family. Cell. Mol. Life Sci. 2009, 66, 1387–1403. [Google Scholar] [CrossRef] [PubMed]
- Colpa, D.I.; Fraaije, M.W.; van Bloois, E. DyP-Type Peroxidases: A Promising and Versatile Class of Enzymes. J. Ind. Microbiol. Biotechnol. 2014, 41, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Eltis, L.D. The Multihued Palette of Dye-Decolorizing Peroxidases. Arch. Biochem. Biophys. 2015, 574, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, T.; Sugano, Y. A Structural and Functional Perspective of DyP-Type Peroxidase Family. Arch. Biochem. Biophys. 2015, 574, 49–55. [Google Scholar] [CrossRef]
- Mendes, S.; Robalo, M.P.; Martins, L.O. Bacterial Enzymes and Multi-enzymatic Systems for Cleaning-up Dyes from the Environment. In Microbial Degradation of Synthetic Dyes in Wastewaters; Singh, S.N., Ed.; Springer International Publishing: Cham, Switzerland, 2015; pp. 27–55. ISBN 978-3-319-10941-1. [Google Scholar]
- Silveira, C.M.; Moe, E.; Fraaije, M.; Martins, L.O.; Todorovic, S. Resonance Raman View of the Active Site Architecture in Bacterial DyP-Type Peroxidases. RSC Adv. 2020, 10, 11095–11104. [Google Scholar] [CrossRef]
- Sezer, M.; Genebra, T.; Mendes, S.; Martins, L.O.; Todorovic, S. A DyP-Type Peroxidase at a Bio-Compatible Interface: Structural and Mechanistic Insights. Soft Matter 2012, 8, 10314–10321. [Google Scholar] [CrossRef]
- Brissos, V.; Tavares, D.; Sousa, A.C.; Robalo, M.P.; Martins, L.O. Engineering a Bacterial DyP-Type Peroxidase for Enhanced Oxidation of Lignin-Related Phenolics at Alkaline PH. ACS Catal. 2017, 7, 3454–3465. [Google Scholar] [CrossRef]
- Atack, J.M.; Kelly, D.J. Structure, Mechanism and Physiological Roles of Bacterial Cytochrome c Peroxidases. In Advances in Microbial Physiology; Poole, R.K., Ed.; Academic Press: Cambridge, MA, USA, 2006; Volume 52, pp. 73–106. ISBN 0065-2911. [Google Scholar]
- Ellis, K.E.; Seidel, J.; Einsle, O.; Elliott, S.J. Geobacter Sulfurreducens Cytochrome c Peroxidases: Electrochemical Classification of Catalytic Mechanisms. Biochemistry 2011, 50, 4513–4520. [Google Scholar] [CrossRef] [Green Version]
- Becker, C.F.; Watmough, N.J.; Elliott, S.J. Electrochemical Evidence for Multiple Peroxidatic Heme States of the Diheme Cytochrome c Peroxidase of Pseudomonas Aeruginosa. Biochemistry 2009, 48, 87–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Wael, K.; Buschop, H.; Heering, H.A.; De Smet, L.; Van Beeumen, J.; Devreese, B.; Adriaens, A. Electrochemical Determination of Hydrogen Peroxide Using Rhodobacter Capsulatus Cytochrome c Peroxidase at a Gold Electrode. Microchim. Acta 2008, 162, 65–71. [Google Scholar] [CrossRef] [Green Version]
- Sousa, P.M.P.; Pauleta, S.R.; Simões Gonçalves, M.L.; Pettigrew, G.W.; Moura, I.; Correia dos Santos, M.M.; Moura, J.J.G. Mediated Catalysis of Paracoccus Pantotrophus Cytochrome c Peroxidase by P. Pantotrophus Pseudoazurin: Kinetics of Intermolecular Electron Transfer. JBIC J. Biol. Inorg. Chem. 2007, 12, 691–698. [Google Scholar] [CrossRef] [Green Version]
- Sousa, P.M.P.; Pauleta, S.R.; Rodrigues, D.; Simões Gonçalves, M.L.; Pettigrew, G.W.; Moura, I.; Moura, J.J.G.; Correia dos Santos, M.M. Benefits of Membrane Electrodes in the Electrochemistry of Metalloproteins: Mediated Catalysis of Paracoccus Pantotrophus Cytochrome c Peroxidase by Horse Cytochrome c: A Case Study. JBIC J. Biol. Inorg. Chem. 2008, 13, 779–787. [Google Scholar] [CrossRef] [PubMed]
- De Wael, K.; Bashir, Q.; Van Vlierberghe, S.; Dubruel, P.; Heering, H.A.; Adriaens, A. Electrochemical Determination of Hydrogen Peroxide with Cytochrome c Peroxidase and Horse Heart Cytochrome c Entrapped in a Gelatin Hydrogel. Bioelectrochemistry 2012, 83, 15–18. [Google Scholar] [CrossRef] [PubMed]
- Bernhardt, R. Cytochrome P-450. In Encyclopedia of Biological Chemistry; Elsevier: Amsterdam, The Netherlands, 2013; pp. 607–612. ISBN 978-0-12-378631-9. [Google Scholar]
- Guengerich, F.P. Mechanisms of Cytochrome P450-Catalyzed Oxidations. ACS Catal. 2018, 8, 10964–10976. [Google Scholar] [CrossRef] [PubMed]
- Hannemann, F.; Bichet, A.; Ewen, K.M.; Bernhardt, R. Cytochrome P450 Systems—Biological Variations of Electron Transport Chains. Biochim. Biophys. Acta Gen. Subj. 2007, 1770, 330–344. [Google Scholar] [CrossRef]
- Denisov, I.G.; Makris, T.M.; Sligar, S.G.; Schlichting, I. Structure and Chemistry of Cytochrome P450. Chem. Rev. 2005, 105, 2253–2278. [Google Scholar] [CrossRef]
- Reipa, V.; Mayhew, M.P.; Holden, M.J.; Vilker, V.L. Redox Control of the P450cam Catalytic Cycle: Effects of Y96F Active Site Mutation and Binding of a Non-Natural Substrate. Chem. Comm. 2002, 318–319. [Google Scholar] [CrossRef]
- Daff, S.N.; Chapman, S.K.; Holt, R.A.; Govindaraj, S.; Poulos, T.L.; Munro, A.W. Redox Control of the Catalytic Cycle of Flavocytochrome P-450 BM3. Biochemistry 1997, 36, 13816–13823. [Google Scholar] [CrossRef]
- Martinis, S.A.; Blanke, S.R.; Hager, L.P.; Sligar, S.G.; Hui Bon Hoa, G.; Rux, J.J.; Dawson, J.H. Probing the Heme Iron Coordination Structure of Pressure-Induced Cytochrome P420cam. Biochemistry 1996, 35, 14530–14536. [Google Scholar] [CrossRef]
- Das, A.; Grinkova, Y.V.; Sligar, S.G. Redox Potential Control by Drug Binding to Cytochrome P450 3A4. J Am. Chem. Soc. 2007, 129, 13778–13779. [Google Scholar] [CrossRef] [Green Version]
- Krishnan, S.; Rusling, J.F. Electrochemically Activated Catalytic Pathways of Human Metabolic Cytochrome P450s in Ultrathin Films. In Electrochemistry of N4 Macrocyclic Metal Complexes; Zagal, J.H., Bedioui, F., Eds.; Springer International Publishing: Cham, Switzerland, 2016; pp. 83–105. ISBN 978-3-319-31330-6. [Google Scholar]
- Munro, A.W.; Girvan, H.M.; Mason, A.E.; Dunford, A.J.; McLean, K.J. What Makes a P450 Tick? Trends Biochem. Sci. 2013, 38, 140–150. [Google Scholar] [CrossRef] [PubMed]
- Munro, A.W.; McLean, K.J.; Grant, J.L.; Makris, T.M. Structure and Function of the Cytochrome P450 Peroxygenase Enzymes. Biochem. Soc. Trans. 2018, 46, 183–196. [Google Scholar] [CrossRef] [Green Version]
- Gilardi, G. Cytochromes P450 Redox Activity. In Encyclopedia of Interfacial Chemistry; Elsevier: Amsterdam, The Netherlands, 2018; pp. 90–109. ISBN 978-0-12-809894-3. [Google Scholar]
- Krishnan, S.; Schenkman, J.B.; Rusling, J.F. Bioelectronic Delivery of Electrons to Cytochrome P450 Enzymes. J. Phys. Chem. B 2011, 115, 8371–8380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheller, F.W.; Yarmana, A.; Wollenberger, U. Cytochrome P450: Electron Transfer and Sensors. In Cytochrome P450 Enzymes: Biochemistry Pharmacology and Health Implications; Jian Wu, Ed.; Nova Science Publishers: New York, NY, USA, 2014; pp. 1–12. ISBN 978-1-61942-209-4. [Google Scholar]
- Hagen, K.D.; Gillan, J.M.; Im, S.-C.; Landefeld, S.; Mead, G.; Hiley, M.; Waskell, L.A.; Hill, M.G.; Udit, A.K. Electrochemistry of Mammalian Cytochrome P450 2B4 Indicates Tunable Thermodynamic Parameters in Surfactant Films. J. Inorg. Biochem. 2013, 129, 30–34. [Google Scholar] [CrossRef] [Green Version]
- Sadeghi, S.J.; Fantuzzi, A.; Gilardi, G. Breakthrough in P450 Bioelectrochemistry and Future Perspectives. Biochim. Biophys. Acta Proteins Proteom. 2011, 1814, 237–248. [Google Scholar] [CrossRef]
- Udit, A.K.; Hagen, K.D.; Goldman, P.J.; Star, A.; Gillan, J.M.; Gray, H.B.; Hill, M.G. Spectroscopy and Electrochemistry of Cytochrome P450 BM3-Surfactant Film Assemblies. J. Am. Chem. Soc. 2006, 128, 10320–10325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yarman, A.; Wollenberger, U.; Scheller, F.W. Sensors Based on Cytochrome P450 and CYP Mimicking Systems. Electrochim. Acta 2013, 110, 63–72. [Google Scholar] [CrossRef]
- Çekiç, S.Z.; Holtmann, D.; Güven, G.; Mangold, K.-M.; Schwaneberg, U.; Schrader, J. Mediated Electron Transfer with P450cin. Electrochem. Commun. 2010, 12, 1547–1550. [Google Scholar] [CrossRef]
- Dodhia, V.R.; Sassone, C.; Fantuzzi, A.; Nardo, G.D.; Sadeghi, S.J.; Gilardi, G. Modulating the Coupling Efficiency of Human Cytochrome P450 CYP3A4 at Electrode Surfaces through Protein Engineering. Electrochem. Commun. 2008, 10, 1744–1747. [Google Scholar] [CrossRef]
- Shumyantseva, V.V.; Kuzikov, A.V.; Masamrekh, R.A.; Bulko, T.V.; Archakov, A.I. From Electrochemistry to Enzyme Kinetics of Cytochrome P450. Biosens. Bioelectron. 2018, 121, 192–204. [Google Scholar] [CrossRef]
- Ducharme, J.; Auclair, K. Use of Bioconjugation with Cytochrome P450 Enzymes. Biochim. Biophys. Acta Proteins Proteom. 2018, 1866, 32–51. [Google Scholar] [CrossRef] [Green Version]
- Baj-Rossi, C.; Rezzonico Jost, T.; Cavallini, A.; Grassi, F.; De Micheli, G.; Carrara, S. Continuous Monitoring of Naproxen by a Cytochrome P450-Based Electrochemical Sensor. Biosens. Bioelectron. 2014, 53, 283–287. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Zheng, Q.; Bai, G.; Dai, Q.; Cao, X.; Yao, Y.; Liu, S.; Yao, C. Polydopamine Functionalized Nanoporous Graphene Foam as Nanoreactor for Efficient Electrode-Driven Metabolism of Steroid Hormones. Biosens. Bioelectron. 2018, 119, 182–190. [Google Scholar] [CrossRef]
- Peng, L.; Wollenberger, U.; Hofrichter, M.; Ullrich, R.; Scheibner, K.; Scheller, F.W. Bioelectrocatalytic Properties of Agrocybe Aegerita Peroxygenase. Electrochim. Acta 2010, 55, 7809–7813. [Google Scholar] [CrossRef]
- Peng, L.; Wollenberger, U.; Kinne, M.; Hofrichter, M.; Ullrich, R.; Scheibner, K.; Fischer, A.; Scheller, F.W. Peroxygenase Based Sensor for Aromatic Compounds. Biosens. Bioelectron. 2010, 26, 1432–1436. [Google Scholar] [CrossRef]
- Yarman, A.; Gröbe, G.; Neumann, B.; Kinne, M.; Gajovic-Eichelmann, N.; Wollenberger, U.; Hofrichter, M.; Ullrich, R.; Scheibner, K.; Scheller, F.W. The Aromatic Peroxygenase from Marasmius Rutola—a New Enzyme for Biosensor Applications. Anal. Bioanal. Chem. 2012, 402, 405–412. [Google Scholar] [CrossRef]
- Díaz, A.; Loewen, P.C.; Fita, I.; Carpena, X. Thirty Years of Heme Catalases Structural Biology. Arch. Biochem. Biophys. 2012, 525, 102–110. [Google Scholar] [CrossRef]
- Karakus, Y.Y. Typical Catalases: Function and Structure. In Glutathione System and Oxidative Stress in Health and Disease; Bagatini, M.D., Ed.; IntechOpen: London, UK, 2020. [Google Scholar]
- Lončar, N.; Fraaije, M.W. Catalases as Biocatalysts in Technical Applications: Current State and Perspectives. Appl. Microbiol. Biotechnol. 2015, 99, 3351–3357. [Google Scholar] [CrossRef]
- Prakash, P.A.; Yogeswaran, U.; Chen, S.-M. A Review on Direct Electrochemistry of Catalase for Electrochemical Sensors. Sensors 2009, 9, 1821–1844. [Google Scholar] [CrossRef] [PubMed]
- Tehrani, H.S.; Moosavi-Movahedi, A.A.; Ghourchian, H. Correlation between Biological Activity and Electron Transferring of Bovine Liver Catalase: Osmolytes Effects. Electrochim. Acta 2013, 113, 591–602. [Google Scholar] [CrossRef]
- Akyilmaz, E.; Kozgus, O. Determination of Calcium in Milk and Water Samples by Using Catalase Enzyme Electrode. Food Chem. 2009, 115, 347–351. [Google Scholar] [CrossRef]
- O’Brien, K.B.; Killoran, S.J.; O’Neill, R.D.; Lowry, J.P. Development and Characterization in Vitro of a Catalase-Based Biosensor for Hydrogen Peroxide Monitoring. Biosens. Bioelectron. 2007, 22, 2994–3000. [Google Scholar] [CrossRef] [Green Version]
- Huang, K.-J.; Niu, D.-J.; Liu, X.; Wu, Z.-W.; Fan, Y.; Chang, Y.-F.; Wu, Y.-Y. Direct Electrochemistry of Catalase at Amine-Functionalized Graphene/Gold Nanoparticles Composite Film for Hydrogen Peroxide Sensor. Electrochim. Acta 2011, 56, 2947–2953. [Google Scholar] [CrossRef]
- Alim, S.; Vejayan, J.; Kafi, A.K.M. Direct Electrochemistry of Catalase Immobilized at Polymerized-SnO2 Multiporous Modified Electrode for an Amperometric H2O2 Biosensor. Biomed. J. Sci. Tech. Res. 2018, 3, 3488–3493. [Google Scholar] [CrossRef]
- Fusco, G.; Bollella, P.; Mazzei, F.; Favero, G.; Antiochia, R.; Tortolini, C. Catalase-Based Modified Graphite Electrode for Hydrogen Peroxide Detection in Different Beverages. J. Anal. Methods Chem. 2016, 2016, 8174913. [Google Scholar] [CrossRef] [PubMed]
- Hashemnia, S.; Khayatzadeh, S.; Moosavi-Movahedi, A.A.; Ghourchian, H. Direct Electrochemistry of Catalase in Multiwall Carbon Nanotube/Dodecyl Trimethylammonium Bromide Film Covered With a Layer of Nafion on a Glassy Carbon Electrode. Int. J. Electrochem. Sci. 2011, 6, 581–595. [Google Scholar]
- Castiglione, N.; Rinaldo, S.; Giardina, G.; Stelitano, V.; Cutruzzola, F. Nitrite and Nitrite Reductases: From Molecular Mechanisms to Significance in Human Health and Disease. Antioxid. Redox Signal. 2012, 17, 684–716. [Google Scholar] [CrossRef] [Green Version]
- Almeida, M.G.; Serra, A.; Silveira, C.M.; Moura, J.J. Nitrite Biosensing via Selective Enzymes—A Long but Promising Route. Sensors 2010, 10, 11530–11555. [Google Scholar] [CrossRef] [Green Version]
- Brown, K.; Roig-Zamboni, V.; Cutruzzola’, F.; Arese, M.; Sun, W.; Brunori, M.; Cambillau, C.; Tegoni, M. Domain Swing upon His to Ala Mutation in Nitrite Reductase of Pseudomonas Aeruginosa. J. Mol. Biol. 2001, 312, 541–554. [Google Scholar] [CrossRef]
- Maia, L.B.; Moura, J.J.G. How Biology Handles Nitrite. Chem. Rev. 2014, 114, 5273–5357. [Google Scholar] [CrossRef] [PubMed]
- Silveira, C.M.; Quintas, P.O.; Moura, I.; Moura, J.J.G.; Hildebrandt, P.; Almeida, M.G.; Todorovic, S. SERR Spectroelectrochemical Study of Cytochrome Cd1 Nitrite Reductase Co-Immobilized with Physiological Redox Partner Cytochrome C552 on Biocompatible Metal Electrodes. PLoS ONE 2015, 10, e0129940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Besson, S.; Carneiro, C.; Moura, J.J.G.; Moura, I.; Fauque, G. A Cytochrome Cd1-Type Nitrite Reductase Isolated from the Marine Denitrifier Pseudomonas Nautica 617: Purification and Characterization. Anaerobe 1995, 1, 219–226. [Google Scholar] [CrossRef]
- Koppenhöfer, A.; Turner, K.L.; Allen, J.W.A.; Chapman, S.K.; Ferguson, S.J. Cytochrome Cd1 from Paracoccus Pantotrophus Exhibits Kinetically Gated, Conformationally Dependent, Highly Cooperative Two-Electron Redox Behavior. Biochemistry 2000, 39, 4243–4249. [Google Scholar] [CrossRef] [PubMed]
- Williams, P.A.; Fülöp, V.; Garman, E.F.; Saunders, N.F.W.; Ferguson, S.J.; Hajdu, J. Haem-Ligand Switching during Catalysis in Crystals of a Nitrogen-Cycle Enzyme. Nature 1997, 389, 406–412. [Google Scholar] [CrossRef]
- Moura, I.; Pauleta, S.R.; Moura, J.J.G. Enzymatic Activity Mastered by Altering Metal Coordination Spheres. JBIC J. Biol. Inorg. Chem. 2008, 13, 1185–1195. [Google Scholar] [CrossRef] [Green Version]
- Cunha, C.A.; Macieira, S.; Dias, J.M.; Almeida, G.; Gonçalves, L.L.; Costa, C.; Lampreia, J.; Huber, R.; Moura, J.J.G.; Moura, I.; et al. Cytochrome c Nitrite Reductase from Desulfovibrio Desulfuricans ATCC 27774: The Relevance of The Two Calcium Sites In The Structure Of The Catalytic Subunit (NrfA). J. Biol. Chem. 2003, 278, 17455–17465. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, M.L.; Oliveira, T.F.; Pereira, I.A.C.; Archer, M. X-Ray Structure of the Membrane-Bound Cytochrome c Quinol Dehydrogenase NrfH Reveals Novel Haem Coordination. EMBO J. 2006, 25, 5951–5960. [Google Scholar] [CrossRef] [Green Version]
- Costa, C.; Moura, J.J.G.; Moura, I.; Wang, Y.; Huynh, B.H. Redox Properties of Cytochrome c Nitrite Reductase from Desulfovibrio Desulfuricans ATCC 27774. J. Biol. Chem. 1996, 271, 23191–23196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almeida, M.G.; Silveira, C.M.; Guigliarelli, B.; Bertrand, P.; Moura, J.J.G.; Moura, I.; Léger, C. A Needle in a Haystack: The Active Site of the Membrane-Bound Complex Cytochrome c Nitrite Reductase. FEBS Lett. 2007, 581, 284–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todorovic, S.; Rodrigues, M.L.; Matos, D.; Pereira, I.A.C. Redox Properties of Lysine- and Methionine-Coordinated Hemes Ensure Downhill Electron Transfer in NrfH2A4 Nitrite Reductase. J. Phys. Chem. B 2012, 116, 5637–5643. [Google Scholar] [CrossRef]
- Dutt, J.; Davis, J. Current Strategies in Nitrite Detection and Their Application to Field Analysis. J. Environ. Monit. 2002, 4, 465–471. [Google Scholar] [CrossRef]
- Delgado, J.A.; Follett, R.F. Advances in Nitrogen Management for Water Quality. J. Soil Water Conserv. 2011, 66, 25A. [Google Scholar] [CrossRef]
- Lundberg, J.O.; Gladwin, M.T.; Ahluwalia, A.; Benjamin, N.; Bryan, N.S.; Butler, A.; Cabrales, P.; Fago, A.; Feelisch, M.; Ford, P.C.; et al. Nitrate and Nitrite in Biology, Nutrition and Therapeutics. Nat. Chem. Biol. 2009, 5, 865–869. [Google Scholar] [CrossRef] [PubMed]
- Almeida, M.G.; Silveira, C.M.; Moura, J.J.G. Biosensing Nitrite Using the System Nitrite Redutase/Nafion/Methyl Viologen—A Voltammetric Study. Biosens. Bioelectron. 2007, 22, 2485–2492. [Google Scholar] [CrossRef] [PubMed]
- Silveira, C.M.; Baur, J.; Holzinger, M.; Moura, J.J.G.; Cosnier, S.; Almeida, M.G. Enhanced Direct Electron Transfer of a Multihemic Nitrite Reductase on Single-Walled Carbon Nanotube Modified Electrodes. Electroanalysis 2010, 22, 2973–2978. [Google Scholar] [CrossRef]
- Gwyer, J.D.; Richardson, D.J.; Butt, J.N. Diode or Tunnel-Diode Characteristics? Resolving the Catalytic Consequences of Proton Coupled Electron Transfer in a Multi-Centered Oxidoreductase. J. Am. Chem. Soc. 2005, 127, 14964–14965. [Google Scholar] [CrossRef] [PubMed]
- Angove, H.C.; Cole, J.A.; Richardson, D.J.; Butt, J.N. Protein Film Voltammetry Reveals Distinctive Fingerprints of Nitrite and Hydroxylamine Reduction by a Cytochrome c Nitrite Reductase. J. Biol. Chem. 2002, 277, 23374–23381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monteiro, T.; Rodrigues, P.R.; Goncalves, A.L.; Moura, J.J.; Jubete, E.; Anorga, L.; Piknova, B.; Schechter, A.N.; Silveira, C.M.; Almeida, M.G. Construction of Effective Disposable Biosensors for Point of Care Testing of Nitrite. Talanta 2015, 142, 246–251. [Google Scholar] [CrossRef] [Green Version]
- Silveira, C.M.; Pimpão, M.; Pedroso, H.A.; Rodrigues, P.R.S.; Moura, J.J.G.; Pereira, M.F.R.; Almeida, M.G. Probing the Surface Chemistry of Different Oxidized MWCNT for the Improved Electrical Wiring of Cytochrome c Nitrite Reductase. Electrochem. Commun. 2013, 35, 17–21. [Google Scholar] [CrossRef] [Green Version]
- Lojou, E.; Cutruzzolà, F.; Tegoni, M.; Bianco, P. Electrochemical Study of the Intermolecular Electron Transfer to Pseudomonas Aeruginosa Cytochrome Cd1 Nitrite Reductase. Electrochim. Acta 2003, 48, 1055–1064. [Google Scholar] [CrossRef]
- Pedroso, H.A.; Silveira, C.M.; Almeida, R.M.; Almeida, A.; Besson, S.; Moura, I.; Moura, J.J.G.; Almeida, M.G. Electron Transfer and Docking between Cytochrome Cd1 Nitrite Reductase and Different Redox Partners—A Comparative Study. Biochim. Biophys. Acta Bioenerg. 2016, 1857, 1412–1421. [Google Scholar] [CrossRef]
- Strehlitz, B.; Gründig, B.; Schumacher, W.; Kroneck, P.M.H.; Vorlop, K.-D.; Kotte, H. A Nitrite Sensor Based on a Highly Sensitive Nitrite Reductase Mediator-Coupled Amperometric Detection. Anal. Chem. 1996, 68, 807–816. [Google Scholar] [CrossRef] [PubMed]
- Strehlitz, B.; Gründig, B.; Vorlop, K.D.; Bartholmes, P.; Kotte, H.; Stottmeister, U. Artificial Electron Donors for Nitrate and Nitrite Reductases Usable as Mediators in Amperometric Biosensors. Fresenius J. Anal. Chem. 1994, 349, 676–678. [Google Scholar] [CrossRef]
- Karunakaran, C.; Pandiaraj, M.; Santharaman, P. Immunosensors. In Biosensors and Bioelectronics; Karunakaran, C., Bhargava, K., Benjamin, R., Eds.; Elsevier: Amsterdam, The Netherlands, 2015; Chapter 4; pp. 205–245. ISBN 978-0-12-803100-1. [Google Scholar]
- Batra, B.; Lata, S.; Rani, S.; Pundir, C.S. Fabrication of a Cytochrome c Biosensor Based on Cytochrome Oxidase/NiO-NPs/CMWCNT/PANI Modified Au Electrode. J. Biomed. Nanotechnol. 2013, 9, 409–416. [Google Scholar] [CrossRef]
- Katz, E.; Lioubashevski, O.; Willner, I. Magnetic Field Effects on Bioelectrocatalytic Reactions of Surface-Confined Enzyme Systems: Enhanced Performance of Biofuel Cells. J. Am. Chem. Soc. 2005, 127, 3979–3988. [Google Scholar] [CrossRef]
- Santucci, R.; Sinibaldi, F.; Cozza, P.; Polticelli, F.; Fiorucci, L. Cytochrome c: An Extreme Multifunctional Protein with a Key Role in Cell Fate. Int. J. Biol. Macromol. 2019, 136, 1237–1246. [Google Scholar] [CrossRef]
- Hannibal, L.; Tomasina, F.; Capdevila, D.A.; Demicheli, V.; Tórtora, V.; Alvarez-Paggi, D.; Jemmerson, R.; Murgida, D.H.; Radi, R. Alternative Conformations of Cytochrome c: Structure, Function, and Detection. Biochemistry 2016, 55, 407–428. [Google Scholar] [CrossRef]
- Silveira, C.M.; Castro, M.A.; Dantas, J.M.; Salgueiro, C.; Murgida, D.H.; Todorovic, S. Structure, Electrocatalysis and Dynamics of Immobilized Cytochrome PccH and Its Microperoxidase. Phys. Chem. Chem. Phys. 2017, 19, 8908–8918. [Google Scholar] [CrossRef] [PubMed]
- Eddowes, M.J.; Hill, H.A.O. Electrochemistry of horse heart cytochrome c. J. Am. Chem. Soc. 1979, 101, 4461–4464. [Google Scholar] [CrossRef]
- Murgida, D.H.; Hildebrandt, P. Disentangling Interfacial Redox Processes of Proteins by SERR Spectroscopy. Curr. Soc. Rev. 2008, 37, 937–945. [Google Scholar] [CrossRef]
- Tavagnacco, C.; Monari, S.; Ranieri, A.; Bortolotti, C.A.; Peressini, S.; Borsari, M. Immobilized Unfolded Cytochrome c Acts as a Catalyst for Dioxygen Reduction. Chem. Commun. 2011, 47, 11122. [Google Scholar] [CrossRef]
- Wisitruangsakul, N.; Zebger, I.; Ly, K.H.; Murgida, D.H.; Ekgasit, S.; Hildebrandt, P. Redox-Linked Protein Dynamics of Cytochrome c Probed by Time-Resolved Surface Enhanced Infrared Absorption Spectroscopy. Phys. Chem. Chem. Phys. 2008, 10, 5276–5286. [Google Scholar] [CrossRef]
- Ly, H.K.; Sezer, M.; Wisitruangsakul, N.; Feng, J.-J.; Kranich, A.; Millo, D.; Weidinger, I.M.; Zebger, I.; Murgida, D.H.; Hildebrandt, P. Surface-Enhanced Vibrational Spectroscopy for Probing Transient Interactions of Proteins with Biomimetic Interfaces: Electric Field Effects on Structure, Dynamics and Function of Cytochrome c. FEBS J. 2011, 1382–1390. [Google Scholar] [CrossRef]
- Ranieri, A.; Bernini, F.; Bortolotti, C.A.; Castellini, E. The Met80Ala Point Mutation Enhances the Peroxidase Activity of Immobilized Cytochrome c. Catal. Sci. Technol. 2012, 2, 2206. [Google Scholar] [CrossRef]
- Casalini, S.; Battistuzzi, G.; Borsari, M.; Bortolotti, C.A.; Ranieri, A.; Sola, M. Electron Transfer and Electrocatalytic Properties of the Immobilized Methionine80Alanine Cytochrome c Variant. J. Phys. Chem. B 2008, 112, 1555–1563. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.-H.; Lin, Y.-W.; Rosell, F.I.; Ni, F.-Y.; Lu, H.-J.; Yang, P.-Y.; Tan, X.-S.; Li, X.-Y.; Huang, Z.-X.; Mauk, A.G. Converting Cytochrome c into a Peroxidase-Like Metalloenzyme by Molecular Design. ChemBioChem 2007, 8, 607–609. [Google Scholar] [CrossRef]
- Casalini, S.; Battistuzzi, G.; Borsari, M.; Bortolotti, C.A.; Di Rocco, G.; Ranieri, A.; Sola, M. Electron Transfer Properties and Hydrogen Peroxide Electrocatalysis of Cytochrome c Variants at Positions 67 and 80. J. Phys. Chem. B 2010, 114, 1698–1706. [Google Scholar] [CrossRef]
- Aghamiri, Z.S.; Mohsennia, M.; Rafiee-Pour, H.-A. Immobilization of Cytochrome c and Its Application as Electrochemical Biosensors. Talanta 2018, 176, 195–207. [Google Scholar] [CrossRef]
- Bathinapatla, A.; Kanchi, S.; Singh, P.; Sabela, M.I.; Bisetty, K. An Ultrasensitive Performance Enhanced Novel Cytochrome c Biosensor for the Detection of Rebaudioside A. Biosens. Bioelectron. 2016, 77, 116–123. [Google Scholar] [CrossRef] [PubMed]
- Zhang, M.; Zheng, J.; Wang, J.; Xu, J.; Hayat, T.; Alharbi, N.S. Direct Electrochemistry of Cytochrome c Immobilized on One Dimensional Au Nanoparticles Functionalized Magnetic N-Doped Carbon Nanotubes and Its Application for the Detection of H2O2. Sens. Actuators B 2019, 282, 85–95. [Google Scholar] [CrossRef]
- Zhao, Y.; Hu, Y.; Hou, J.; Jia, Z.; Zhong, D.; Zhou, S.; Huo, D.; Yang, M.; Hou, C. Electrochemical Biointerface Based on Electrodeposition AuNPs on 3D Graphene Aerogel: Direct Electron Transfer of Cytochrome c and Hydrogen Peroxide Sensing. J. Electroanal. Chem. 2019, 842, 16–23. [Google Scholar] [CrossRef]
- Aghamiri, Z.S.; Mohsennia, M.; Rafiee-Pour, H.-A. Immobilization of Cytochrome c on Polyaniline/Polypyrrole/Carboxylated Multi-Walled Carbon Nanotube/Glassy Carbon Electrode: Biosensor Fabrication. J. Solid State Electrochem. 2019, 23, 2233–2242. [Google Scholar] [CrossRef]
- Guo, C.; Wang, J.; Chen, X.; Li, Y.; Wu, L.; Zhang, J.; Tao, C. Construction of a Biosensor Based on a Combination of Cytochrome c, Graphene, and Gold Nanoparticles. Sensors 2019, 19, 40. [Google Scholar] [CrossRef] [Green Version]
- Eguílaz, M.; Gutiérrez, A.; Rivas, G. Non-Covalent Functionalization of Multi-Walled Carbon Nanotubes with Cytochrome c: Enhanced Direct Electron Transfer and Analytical Applications. Sens. Actuators B 2016, 225, 74–80. [Google Scholar] [CrossRef]
- Suárez, G.; Santschi, C.; Martin, O.J.F.; Slaveykova, V.I. Biosensor Based on Chemically-Designed Anchorable Cytochrome c for the Detection of H2O2 Released by Aquaticcells. Biosens. Bioelectron. 2013, 42, 385–390. [Google Scholar] [CrossRef] [Green Version]
- Molinas, M.F.; Benavides, L.; Castro, M.A.; Murgida, D.H. Stability, Redox Parameters and Electrocatalytic Activity of a Cytochrome Domain from a New Subfamily. Bioelectrochemistry 2015, 105, 25–33. [Google Scholar] [CrossRef]
- Harbury, H.A.; Cronin, J.R.; Fanger, M.W.; Hettinger, T.P.; Murphy, A.J.; Myer, Y.P.; Vinogradov, S.N. Complex Formation between Methionine and a Heme Peptide from Cytochrome c. Proc. Natl. Acad. Sci. USA 1965, 54, 1658–1664. [Google Scholar] [CrossRef] [Green Version]
- Osman, A.M.; Koerts, J.; Boersma, M.G.; Boeren, S.; Veeger, C.; Rietjens, I.M.C.M. Microperoxidase/H2O2-Catalyzed Aromatic Hydroxylation Proceeds by a Cytochrome-P-450-Type Oxygen-Transfer Reaction Mechanism. Eur. J. Biochem. 1996, 240, 232–238. [Google Scholar] [CrossRef]
- Rusvai, E.; Végh, M.; Kramer, M.; Horváth, I. Hydroxylation of Aniline Mediated by Heme-Bound Oxy-Radicals in a Heme Peptide Model System. Biochem. Pharmacol. 1988, 37, 4574–4577. [Google Scholar] [CrossRef]
- Colonna, S.; Gaggereo, N.; Carrea, G.; Pasta, P. The Microperoxidase-11 Catalyzed Oxidation of Sulfides Is Enantioselective. Tetrahedron Lett. 1994, 35, 9103–9104. [Google Scholar] [CrossRef]
- Boersma, M.G.; Primus, J.-L.; Koerts, J.; Veeger, C.; Rietjens, I.M.C.M. Heme-(Hydro)Peroxide Mediated O- and N-Dealkylation. Eur. J. Biochem. 2000, 267, 6673–6678. [Google Scholar] [CrossRef] [PubMed]
- Verbaro, D.; Hagarman, A.; Soffer, J.; Schweitzer-Stenner, R. The PH Dependence of the 695 Nm Charge Transfer Band Reveals the Population of an Intermediate State of the Alkaline Transition of Ferricytochrome c at Low Ion Concentrations. Biochemistry 2009, 48, 2990–2996. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.S.; Van Wart, H.E. Resonance Raman Characterization of the Heme c Group in N-Acetyl Microperoxidase-8: A Thermal Intermediate Spin-High Spin State Mixture. J. Phys. Chem. 1989, 93, 7925–7931. [Google Scholar] [CrossRef]
- Zhu, X.; Yuri, I.; Gan, X.; Suzuki, I.; Li, G. Electrochemical Study of the Effect of Nano-Zinc Oxide on Microperoxidase and Its Application to More Sensitive Hydrogen Peroxide Biosensor Preparation. Biosens. Bioelectron. 2007, 22, 1600–1604. [Google Scholar] [CrossRef]
- Astuti, Y.; Topoglidis, E.; Durrant, J.R. Use of Microperoxidase-11 to Functionalize Tin Dioxide Electrodes for the Optical and Electrochemical Sensing of Hydrogen Peroxide. Anal. Chim. Acta 2011, 686, 126–132. [Google Scholar] [CrossRef]
- Ioannidis, L.A.; Nikolaou, P.; Panagiotopoulos, A.; Vassi, A.; Topoglidis, E. Microperoxidase-11 Modified Mesoporous SnO 2 Film Electrodes for the Detection of Antimalarial Drug Artemisinin. Anal. Methods 2019, 11, 3117–3125. [Google Scholar] [CrossRef]
- Graça, J.S.; Oliveira, R.F.; Moraes, M.L.; Ferreira, M. Amperometric Glucose Biosensor Based on Layer-by-Layer Films of Microperoxidase-11 and Liposome-Encapsulated Glucose Oxidase. Bioelectrochemistry 2014, 96, 37–42. [Google Scholar] [CrossRef]
- Kogikoski, S.; Sousa, C.P.; Liberato, M.S.; Andrade-Filho, T.; Prieto, T.; Ferreira, F.F.; Rocha, A.R.; Guha, S.; Alves, W.A. Multifunctional Biosensors Based on Peptide–Polyelectrolyte Conjugates. Phys. Chem. Chem. Phys. 2016, 18, 3223–3233. [Google Scholar] [CrossRef]
- Miyazaki, C.M.; Shimizu, F.M.; Mejía-Salazar, J.R.; Oliveira, O.N.; Ferreira, M. Surface Plasmon Resonance Biosensor for Enzymatic Detection of Small Analytes. Nanotechnology 2017, 28, 145501. [Google Scholar] [CrossRef]
- Nastri, F.; Lista, L.; Ringhieri, P.; Vitale, R.; Faiella, M.; Andreozzi, C.; Travascio, P.; Maglio, O.; Lombardi, A.; Pavone, V. A Heme–Peptide Metalloenzyme Mimetic with Natural Peroxidase-Like Activity. Chem. Eur. J. 2011, 17, 4444–4453. [Google Scholar] [CrossRef]
- Vitale, R.; Lista, L.; Cerrone, C.; Caserta, G.; Chino, M.; Maglio, O.; Nastri, F.; Pavone, V.; Lombardi, A. An Artificial Heme-Enzyme with Enhanced Catalytic Activity: Evolution, Functional Screening and Structural Characterization. Org. Biomol. Chem. 2015, 13, 4859–4868. [Google Scholar] [CrossRef] [PubMed]
- Caserta, G.; Chino, M.; Firpo, V.; Zambrano, G.; Leone, L.; D’Alonzo, D.; Nastri, F.; Maglio, O.; Pavone, V.; Lombardi, A. Enhancement of Peroxidase Activity in Artificial Mimochrome VI Catalysts through Rational Design. ChemBioChem 2018, 19, 1823–1826. [Google Scholar] [CrossRef] [PubMed]
- Zambrano, G.; Nastri, F.; Pavone, V.; Lombardi, A.; Chino, M. Use of an Artificial Miniaturized Enzyme in Hydrogen Peroxide Detection by Chemiluminescence. Sensors 2020, 20, 3793. [Google Scholar] [CrossRef] [PubMed]
- Neumann, B.; Götz, R.; Wrzolek, P.; Scheller, F.W.; Weidinger, I.M.; Schwalbe, M.; Wollenberger, U. Enhancement of the Electrocatalytic Activity of Thienyl-Substituted Iron Porphyrin Electropolymers by a Hangman Effect. ChemCatChem 2018, 10, 4353–4361. [Google Scholar] [CrossRef]
- Li, T.; Hu, P.; Li, J.; Huang, P.; Tong, W.; Gao, C. Enhanced Peroxidase-like Activity of Fe@PCN-224 Nanoparticles and Their Applications for Detection of H2O2and Glucose. Colloids Surf. A 2019, 577, 456–463. [Google Scholar] [CrossRef]
- Palanisamy, S.; Velusamy, V.; Chen, S.-W.; Yang, T.C.K.; Balu, S.; Banks, C.E. Enhanced Reversible Redox Activity of Hemin on Cellulose Microfiber Integrated Reduced Graphene Oxide for H2O2 Biosensor Applications. Carbohydr. Polym. 2019, 204, 152–160. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.; Tang, H.; Wu, Y.; Zhang, Y.; Li, Z. Highly Electrocatalytic Biosensor Based on Hemin@AuNPs/Reduced Graphene Oxide/Chitosan Nanohybrids for Non-Enzymatic Ultrasensitive Detection of Hydrogen Peroxide in Living Cells. Biosens. Bioelectron. 2019, 132, 217–223. [Google Scholar] [CrossRef]
- Zhao, P.; Chen, S.; Zhou, J.; Zhang, S.; Huo, D.; Hou, C. A Novel Fe-Hemin-Metal Organic Frameworks Supported on Chitosan-Reduced Graphene Oxide for Real-Time Monitoring of H2O2 Released from Living Cells. Anal. Chim. Acta 2020, 1128, 90–98. [Google Scholar] [CrossRef]
- Ye, J.; Baldwin, R.P. Catalytic Reduction of Myoglobin and Hemoglobin at Chemically Modified Electrodes Containing Methylene Blue. Anal. Chem. 1988, 60, 2263–2268. [Google Scholar] [CrossRef]
- Reeder, B.J. The Redox Activity of Hemoglobins: From Physiologic Functions to Pathologic Mechanisms. Antioxid. Redox Signal. 2010, 13, 1087–1123. [Google Scholar] [CrossRef] [PubMed]
- Zu, X.; Lu, Z.; Zhang, Z.; Schenkman, J.B.; Rusling, J.F. Electroenzyme-Catalyzed Oxidation of Styrene and Cis -β-Methylstyrene Using Thin Films of Cytochrome P450cam and Myoglobin. Langmuir 1999, 15, 7372–7377. [Google Scholar] [CrossRef]
- Keilin, D.; Hartree, E.F. Reaction of Methæmoglobin with Hydrogen Peroxide. Nature 1950, 166, 513–514. [Google Scholar] [CrossRef]
- Onuoha, A.C.; Zu, X.; Rusling, J.F. Electrochemical Generation and Reactions of Ferrylmyoglobins in Water and Microemulsions. J. Am. Chem. Soc. 1997, 119, 3979–3986. [Google Scholar] [CrossRef]
- Wen, Y.; Wen, W.; Zhang, X.; Wang, S. Highly Sensitive Amperometric Biosensor Based on Electrochemically-Reduced Graphene Oxide-Chitosan/Hemoglobin Nanocomposite for Nitromethane Determination. Biosens. Bioelectron. 2016, 79, 894–900. [Google Scholar] [CrossRef] [PubMed]
- Yuan, Y.; Wang, J.; Ni, X.; Cao, Y. A Biosensor Based on Hemoglobin Immobilized with Magnetic Molecularly Imprinted Nanoparticles and Modified on a Magnetic Electrode for Direct Electrochemical Determination of 3-Chloro-1,2-Propandiol. J. Electroanal. Chem. 2019, 834, 233–240. [Google Scholar] [CrossRef]
- Niu, Y.; Xie, H.; Luo, G.; Weng, W.; Ruan, C.; Li, G.; Sun, W. Electrochemical Performance of Myoglobin Based on TiO 2 -Doped Carbon Nanofiber Decorated Electrode and Its Applications in Biosensing. RSC Adv. 2019, 9, 4480–4487. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Weng, W.; Xie, H.; Luo, G.; Li, G.; Sun, W.; Ruan, C.; Wang, X. Myoglobin- and Hydroxyapatite-Doped Carbon Nanofiber-Modified Electrodes for Electrochemistry and Electrocatalysis. ACS Omega 2019, 4, 15653–15659. [Google Scholar] [CrossRef] [Green Version]
- Brissos, V.; Ferreira, M.; Grass, G.; Martins, L.O. Turning a Hyperthermostable Metallo-Oxidase into a Laccase by Directed Evolution. ACS Catal. 2015, 5, 4932–4941. [Google Scholar] [CrossRef]
- Packer, M.S.; Liu, D.R. Methods for the Directed Evolution of Proteins. Nat. Rev. Genet. 2015, 16, 379–394. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Schematic representation of different electron transfer (ET) modes in amperometric enzyme biosensors. The depicted examples show the reduction process of the enzyme. In first-generation biosensors (yellow), the co-substrate (O2) or product (H2O2) of the enzymatic reaction are detected directly at the electrode. In second-generation biosensors (green), the amperometric signal arises from the reduction of the mediator following ET to the enzyme. Third-generation biosensors (blue) measure the direct reduction of the enzyme redox center by the electrode. Med—mediator; Red—reduced; Ox—oxidized; DET—direct ET.
Figure 1.
Schematic representation of different electron transfer (ET) modes in amperometric enzyme biosensors. The depicted examples show the reduction process of the enzyme. In first-generation biosensors (yellow), the co-substrate (O2) or product (H2O2) of the enzymatic reaction are detected directly at the electrode. In second-generation biosensors (green), the amperometric signal arises from the reduction of the mediator following ET to the enzyme. Third-generation biosensors (blue) measure the direct reduction of the enzyme redox center by the electrode. Med—mediator; Red—reduced; Ox—oxidized; DET—direct ET.

Figure 2.
Enzyme immobilization methods. (a) Adsorption onto a multi-walled carbon nanotubes (MWCNTs)/thionine/Au nanoparticles (NPs) composite film on glassy carbon (GC) [37]; (b) entrapment of chemically modified enzyme within a positively charged redox hydrogel deposited on Au coated with a negatively charged self-assembled monolayer (SAM) [38]; (c) covalent binding onto functionalized MWCNTs deposited on GC [39]; (d) glutaraldehyde cross-linking of enzyme and bovine serum albumin (BSA) on a silica-dextran nanocomposite sol-gel deposited on GC [40]. HRP—horseradish peroxidase.
Figure 2.
Enzyme immobilization methods. (a) Adsorption onto a multi-walled carbon nanotubes (MWCNTs)/thionine/Au nanoparticles (NPs) composite film on glassy carbon (GC) [37]; (b) entrapment of chemically modified enzyme within a positively charged redox hydrogel deposited on Au coated with a negatively charged self-assembled monolayer (SAM) [38]; (c) covalent binding onto functionalized MWCNTs deposited on GC [39]; (d) glutaraldehyde cross-linking of enzyme and bovine serum albumin (BSA) on a silica-dextran nanocomposite sol-gel deposited on GC [40]. HRP—horseradish peroxidase.

Figure 3.
Effect of the working potential on the response of a cytochrome c-based H2O2 biosensor. (A) Biosensor calibration plots obtained at (a) 22120.10, (b) −0.05, and (c) 0.00 V vs. Ag/AgCl. (B) Response to interfering compounds at the different working potentials. Upon lowering the potential, the sensitivity of the device is increased and the selectivity is decreased (i.e., interference from AA, DA, and DOPAC is increased). AA—ascorbic acid, DA—dopamine, DOPAC—dihydroxyphenylacetic acid, UA—uric acid. Adapted with permission from Zhou et al. [50]. Copyright (2014) American Chemical Society.
Figure 3.
Effect of the working potential on the response of a cytochrome c-based H2O2 biosensor. (A) Biosensor calibration plots obtained at (a) 22120.10, (b) −0.05, and (c) 0.00 V vs. Ag/AgCl. (B) Response to interfering compounds at the different working potentials. Upon lowering the potential, the sensitivity of the device is increased and the selectivity is decreased (i.e., interference from AA, DA, and DOPAC is increased). AA—ascorbic acid, DA—dopamine, DOPAC—dihydroxyphenylacetic acid, UA—uric acid. Adapted with permission from Zhou et al. [50]. Copyright (2014) American Chemical Society.

Figure 4.
Comparison of (A) FTIR and (B) UV-Visible spectra of cytochrome c (b) in solution and (c) covalently attached onto a carboxylated ionic liquid (triphenylphosphonium bis ((trifluoromethyl)sulfonyl) amide, TPP-HA[TFSI]). The spectroscopic characterization of the ionic liquid (a) is also shown. Reprinted from Murphy et al. [66], Copyright (2019), with permission from Elsevier.
Figure 4.
Comparison of (A) FTIR and (B) UV-Visible spectra of cytochrome c (b) in solution and (c) covalently attached onto a carboxylated ionic liquid (triphenylphosphonium bis ((trifluoromethyl)sulfonyl) amide, TPP-HA[TFSI]). The spectroscopic characterization of the ionic liquid (a) is also shown. Reprinted from Murphy et al. [66], Copyright (2019), with permission from Elsevier.

Figure 5.
Reaction scheme of peroxidase-based electrochemical biosensors (solid lines) and of the native catalytic cycle (dashed lines). RH—substrate; R°—product; Medox—oxidized mediator; Medred—reduced mediator; DET—direct electron transfer; MET—mediated electron transfer.
Figure 5.
Reaction scheme of peroxidase-based electrochemical biosensors (solid lines) and of the native catalytic cycle (dashed lines). RH—substrate; R°—product; Medox—oxidized mediator; Medred—reduced mediator; DET—direct electron transfer; MET—mediated electron transfer.

Figure 6.
Immobilization of horseradish peroxidase (HRP) on gold nanoparticle (GNP) deposits modified with 4-aminothiophenol (4-ATP). Two different GNP arrangements on the glassy carbon (GC) electrode are represented: (a) random and (b) well-dispersed GNP electrodeposits. Reprinted from Huerta-Miranda et al. [94], Copyright (2018), with permission from Elsevier.
Figure 6.
Immobilization of horseradish peroxidase (HRP) on gold nanoparticle (GNP) deposits modified with 4-aminothiophenol (4-ATP). Two different GNP arrangements on the glassy carbon (GC) electrode are represented: (a) random and (b) well-dispersed GNP electrodeposits. Reprinted from Huerta-Miranda et al. [94], Copyright (2018), with permission from Elsevier.

Figure 7.
Catalytic response and stability of a dye-decolorizing peroxidase (DyP)/SAM/Ag biosensor used for H2O2 quantification. (a) Amperometric response of wild-type (wt) and variant DyP-based biosensors to increasing H2O2. Inset: Calibration plots showing the linear dependence of the catalytic current on H2O2 concentration. (b) Relative catalytic current response to 50 μM H2O2 during storage for one month, measured on a batch of electrodes prepared on the same day (shelf-life). Reprinted from Barbosa et al. [67], Copyright (2020), with permission from Elsevier.
Figure 7.
Catalytic response and stability of a dye-decolorizing peroxidase (DyP)/SAM/Ag biosensor used for H2O2 quantification. (a) Amperometric response of wild-type (wt) and variant DyP-based biosensors to increasing H2O2. Inset: Calibration plots showing the linear dependence of the catalytic current on H2O2 concentration. (b) Relative catalytic current response to 50 μM H2O2 during storage for one month, measured on a batch of electrodes prepared on the same day (shelf-life). Reprinted from Barbosa et al. [67], Copyright (2020), with permission from Elsevier.

Figure 8.
Comparison of the surface-enhanced resonance Raman (SERR) (b and c) and RR (a and d) spectra of cytochrome P450 adsorbed on a self-assembled monolayer (SAM) coated Ag electrode and in solution, respectively. (a) RR of dithionite reduced form; (b) SERR of reduced form at −0.40 V poised potential; (c) SERR of oxidized form at 0.34 V poised potential; (d) RR of oxidized form. Reprinted from Todorovic et al. [70], Copyright (2005), with permission from Springer.
Figure 8.
Comparison of the surface-enhanced resonance Raman (SERR) (b and c) and RR (a and d) spectra of cytochrome P450 adsorbed on a self-assembled monolayer (SAM) coated Ag electrode and in solution, respectively. (a) RR of dithionite reduced form; (b) SERR of reduced form at −0.40 V poised potential; (c) SERR of oxidized form at 0.34 V poised potential; (d) RR of oxidized form. Reprinted from Todorovic et al. [70], Copyright (2005), with permission from Springer.

Figure 9.
Electrochemical and spectroscopic characterization of a cytochrome c nitrite reductase (ccNiR)/WO3 nanoparticles (NPs)/indium tin oxide (ITO) biosensor. (a) Schematic representation of the enzyme/electrode construct showing the catalytic conversion of nitrite into ammonium; (b) scanning electron microscopy (SEM) images of three different hydrothermally synthesized WO3NP samples (W1, W2, and W3); (c) cyclic voltammetry response obtained upon addition of increasing nitrite concentration; (d) Raman spectra of WO3NPs samples; (e) resonance Raman (RR) spectra of ccNiR immobilized on the WO3NPs/ITO electrodes. Reprinted from Santos et al. [125], Copyright (2016), with permission from Elsevier.
Figure 9.
Electrochemical and spectroscopic characterization of a cytochrome c nitrite reductase (ccNiR)/WO3 nanoparticles (NPs)/indium tin oxide (ITO) biosensor. (a) Schematic representation of the enzyme/electrode construct showing the catalytic conversion of nitrite into ammonium; (b) scanning electron microscopy (SEM) images of three different hydrothermally synthesized WO3NP samples (W1, W2, and W3); (c) cyclic voltammetry response obtained upon addition of increasing nitrite concentration; (d) Raman spectra of WO3NPs samples; (e) resonance Raman (RR) spectra of ccNiR immobilized on the WO3NPs/ITO electrodes. Reprinted from Santos et al. [125], Copyright (2016), with permission from Elsevier.

Figure 10.
Schematic representation and electrochemical response of native and chemically modified cytochrome c (cyt c) adsorbed on Au electrodes. Cyclic voltammograms obtained with (A) native, (B) mercapto-propionic acid (MPA) modified, and (C) mercapto-undecanoic acid (MUA) modified cyt c in the (1) absence or (2) presence of AuNPs. MPA and MUA anchors are represented in purple; AuNPs are shown as yellow spheres; black arrows represent the ET between heme group and electrode. Reprinted from Suárez et al. [246], Copyright (2013), with permission from Elsevier.
Figure 10.
Schematic representation and electrochemical response of native and chemically modified cytochrome c (cyt c) adsorbed on Au electrodes. Cyclic voltammograms obtained with (A) native, (B) mercapto-propionic acid (MPA) modified, and (C) mercapto-undecanoic acid (MUA) modified cyt c in the (1) absence or (2) presence of AuNPs. MPA and MUA anchors are represented in purple; AuNPs are shown as yellow spheres; black arrows represent the ET between heme group and electrode. Reprinted from Suárez et al. [246], Copyright (2013), with permission from Elsevier.


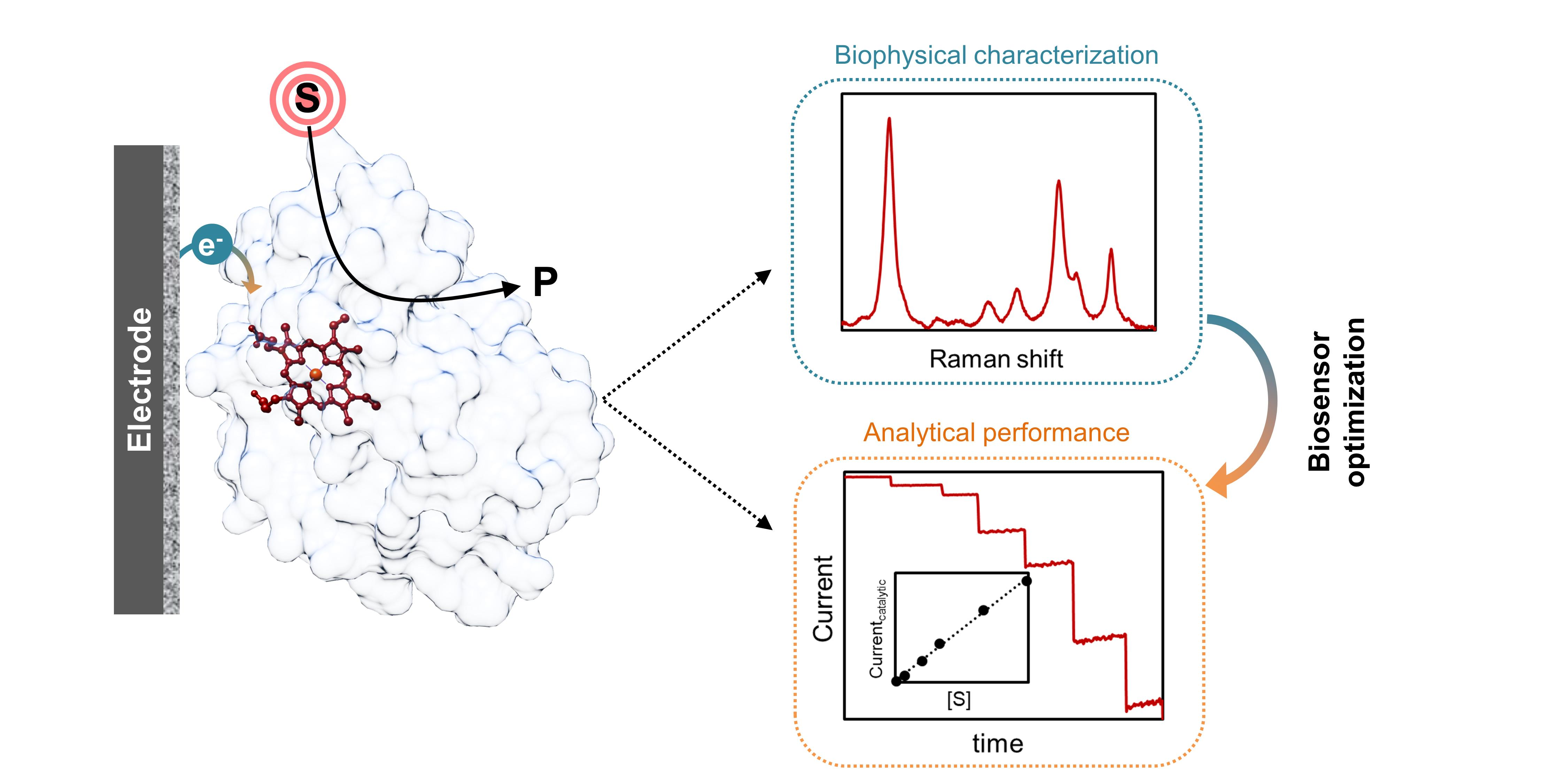{kind=link}
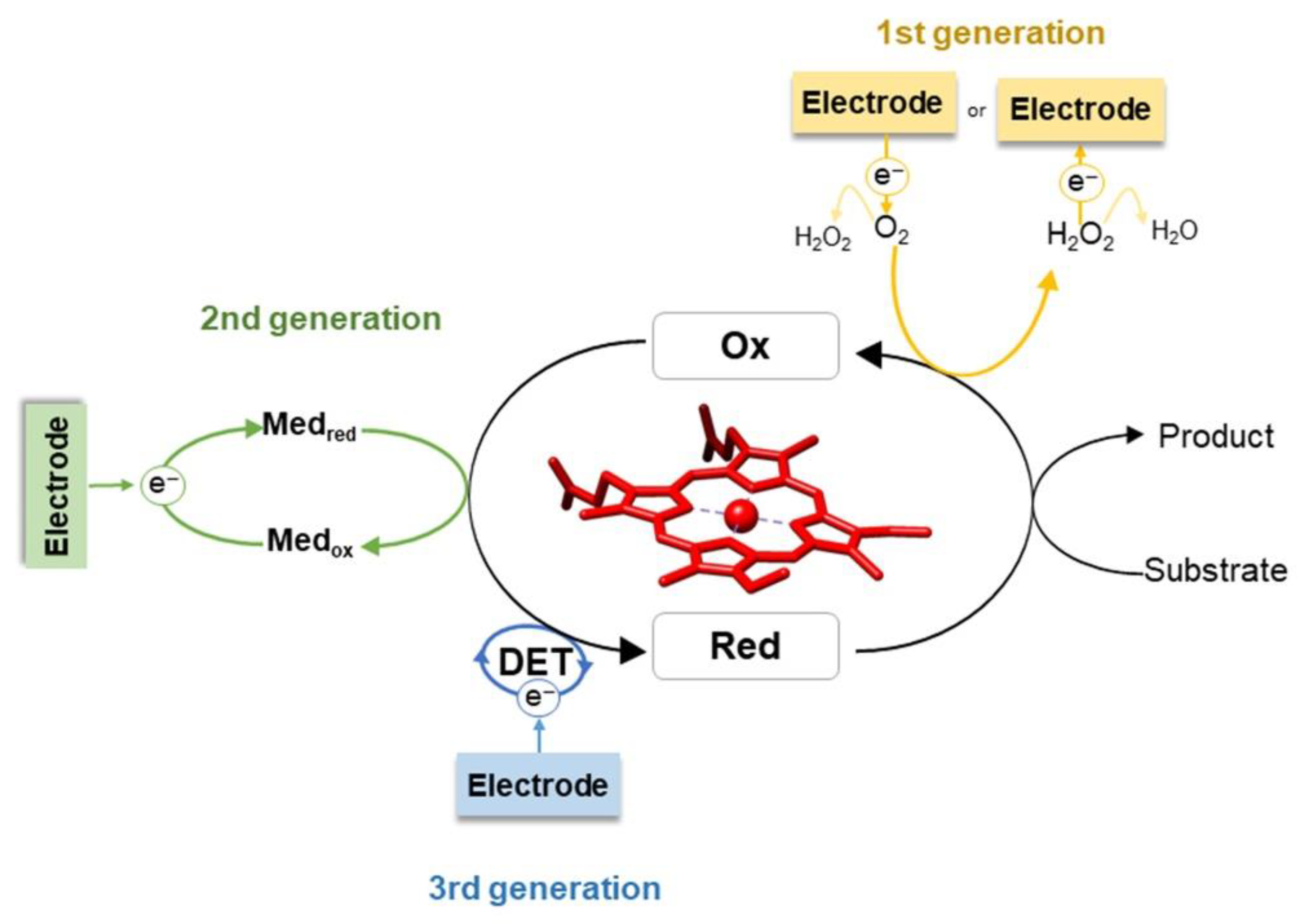{kind=link}
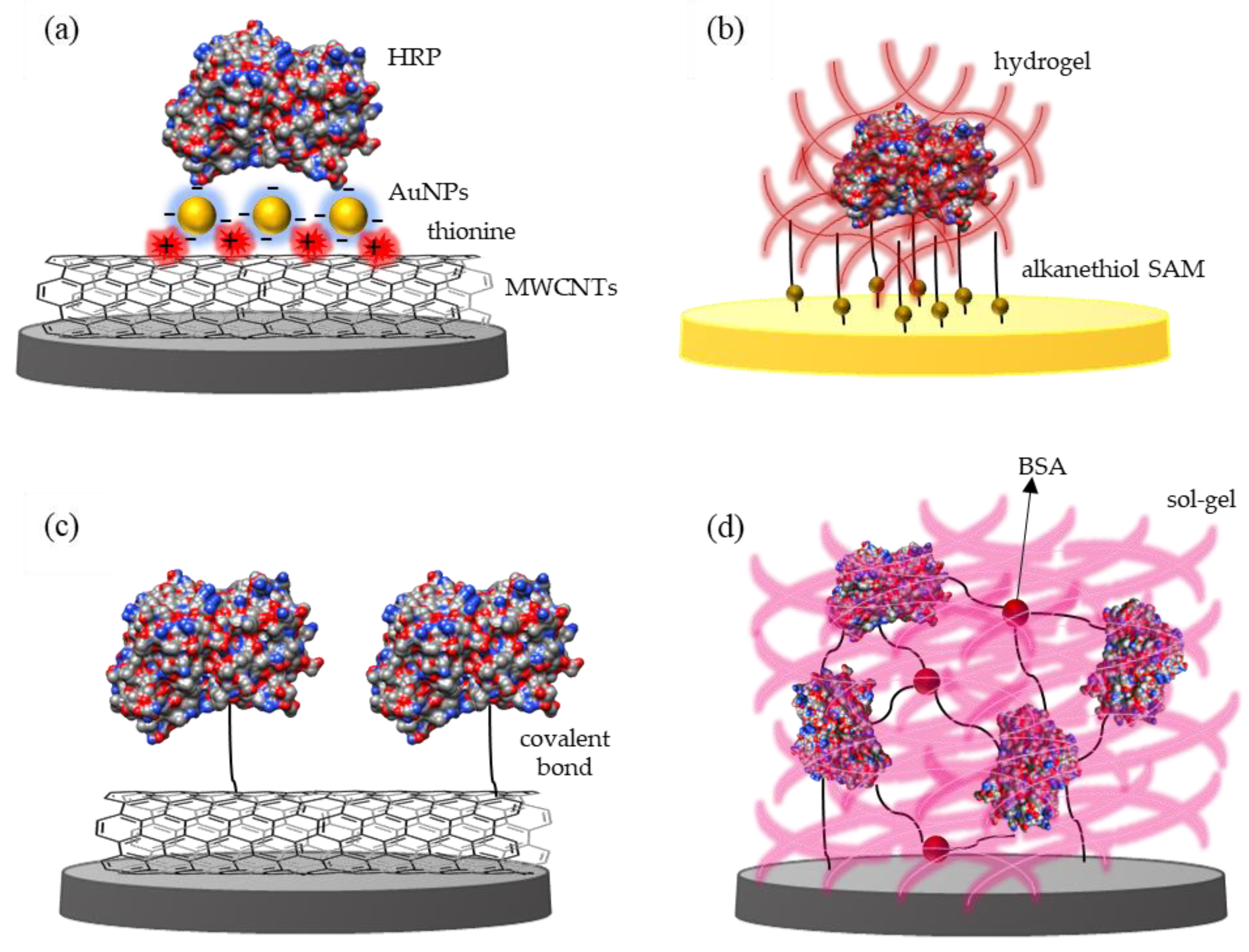{kind=link}
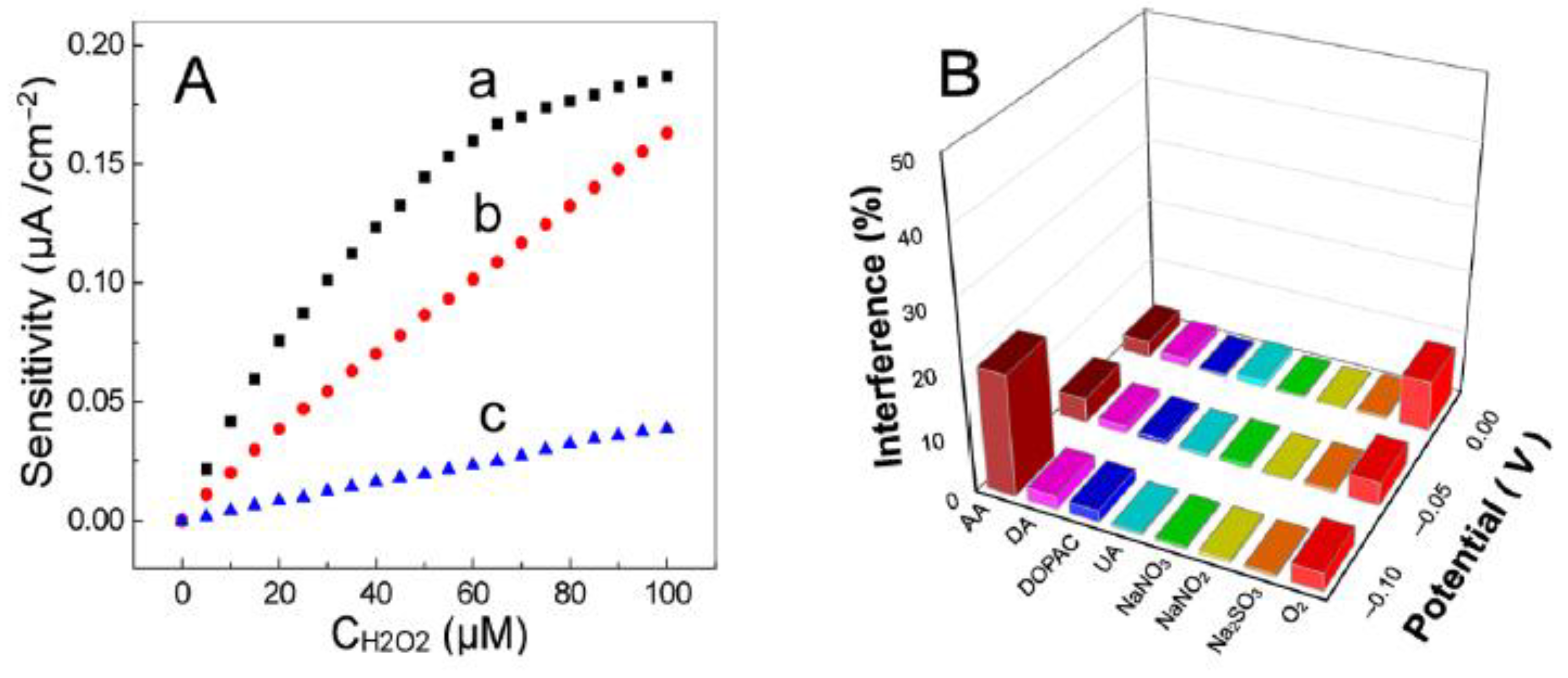{kind=link}
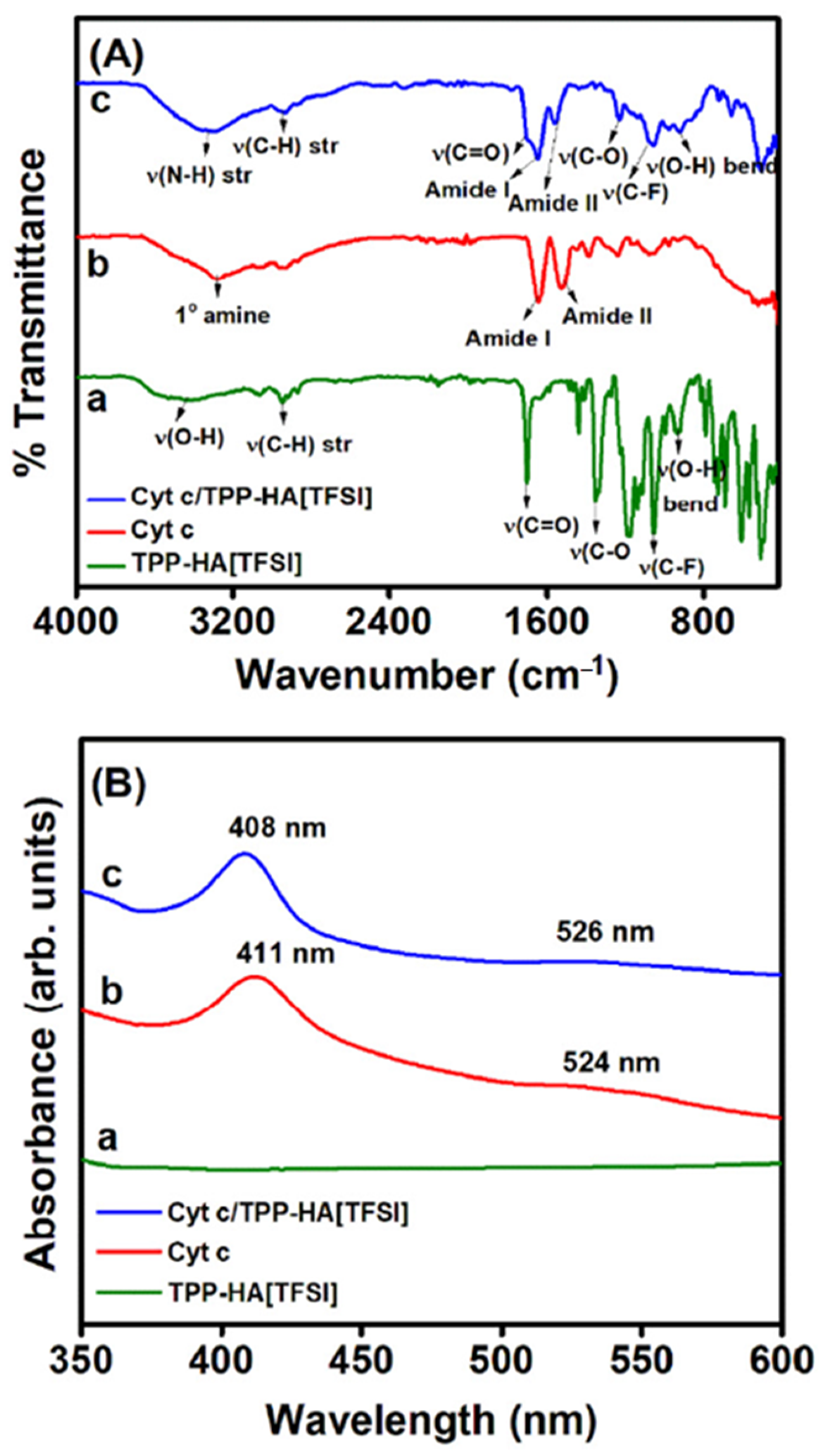{kind=link}
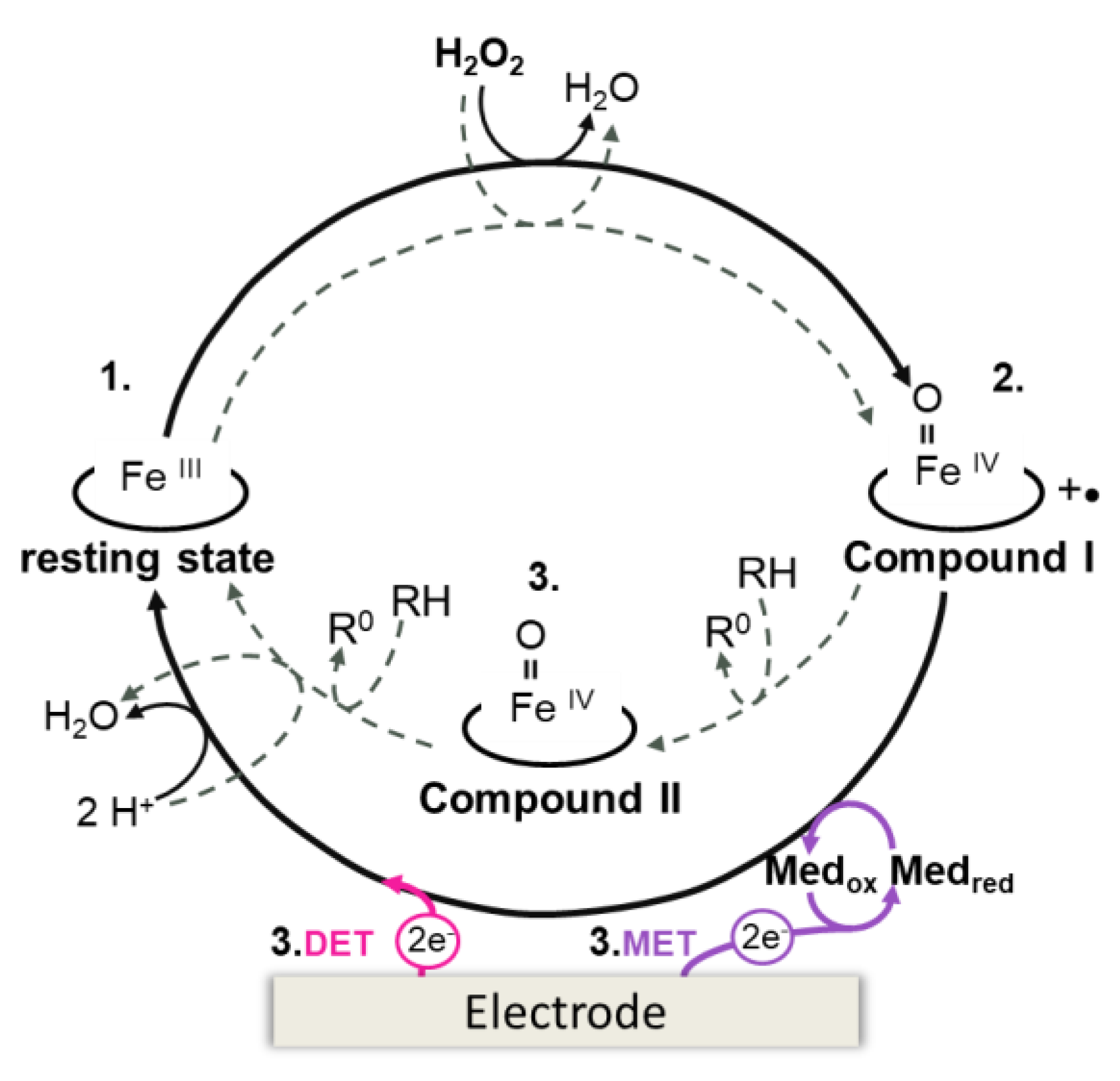{kind=link}
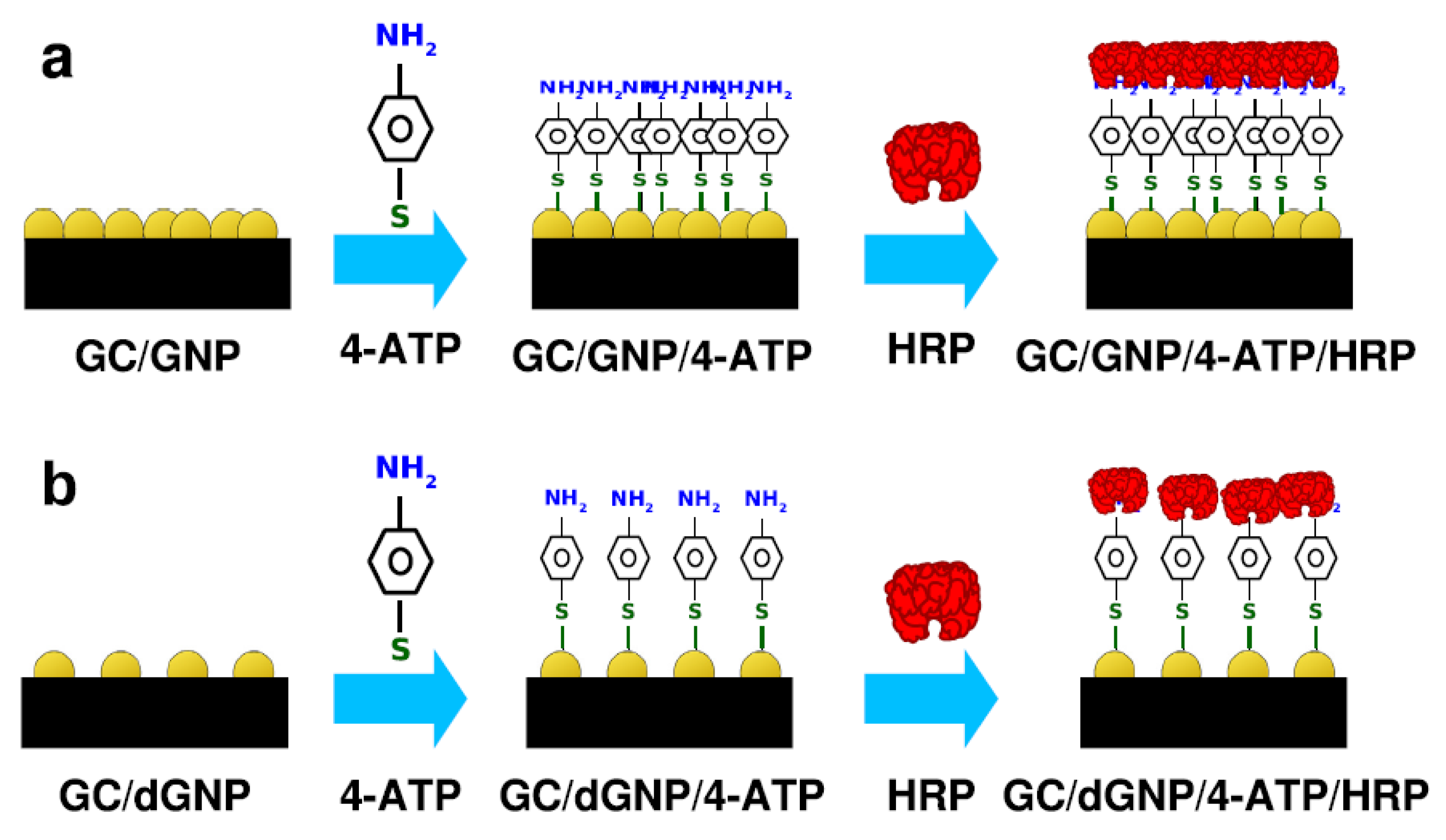{kind=link}
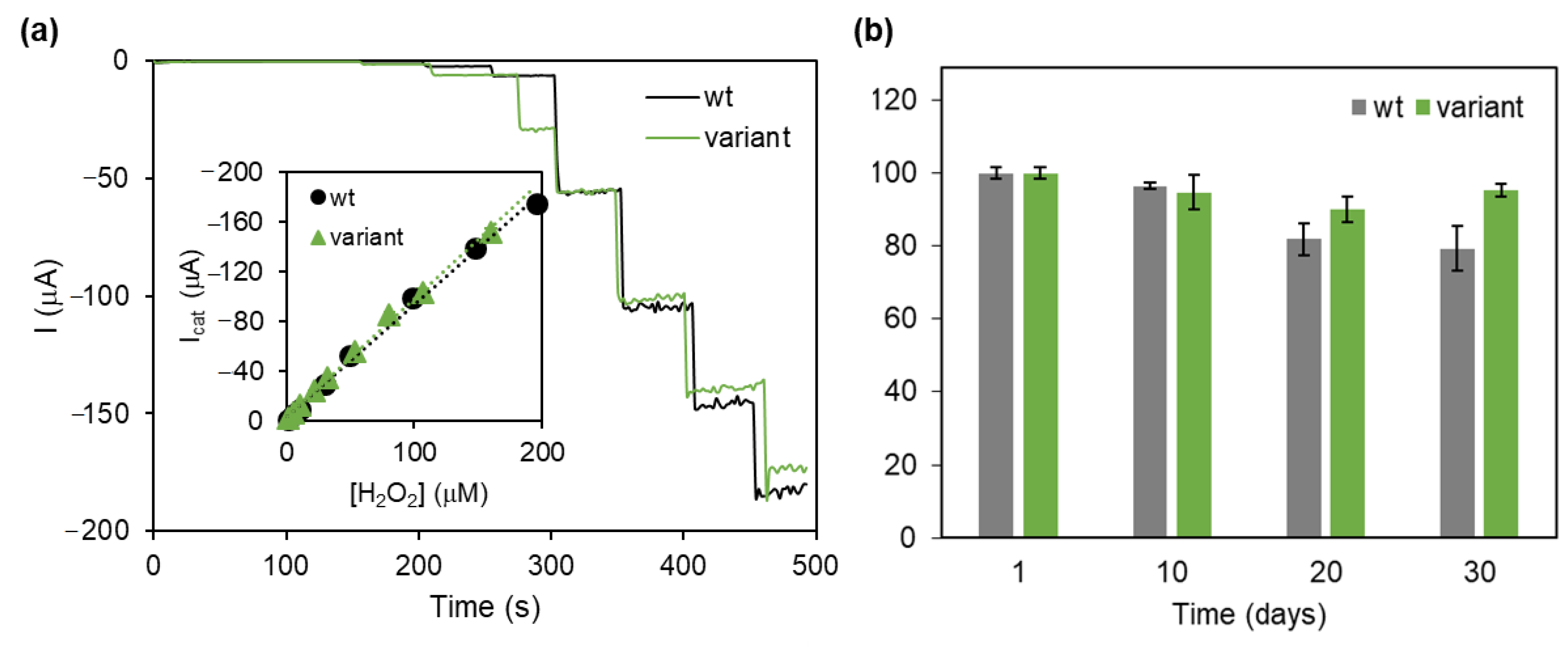{kind=link}
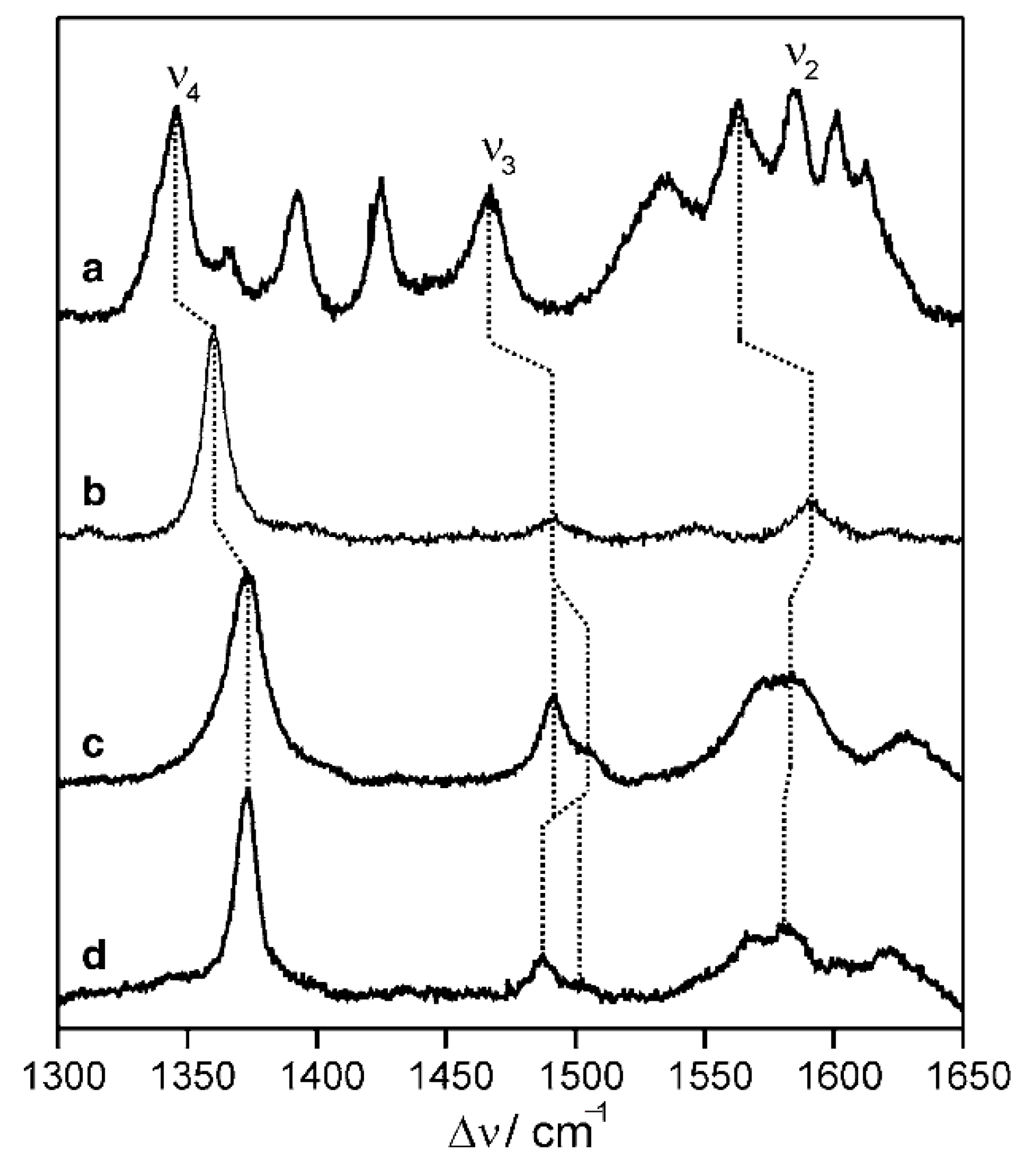{kind=link}
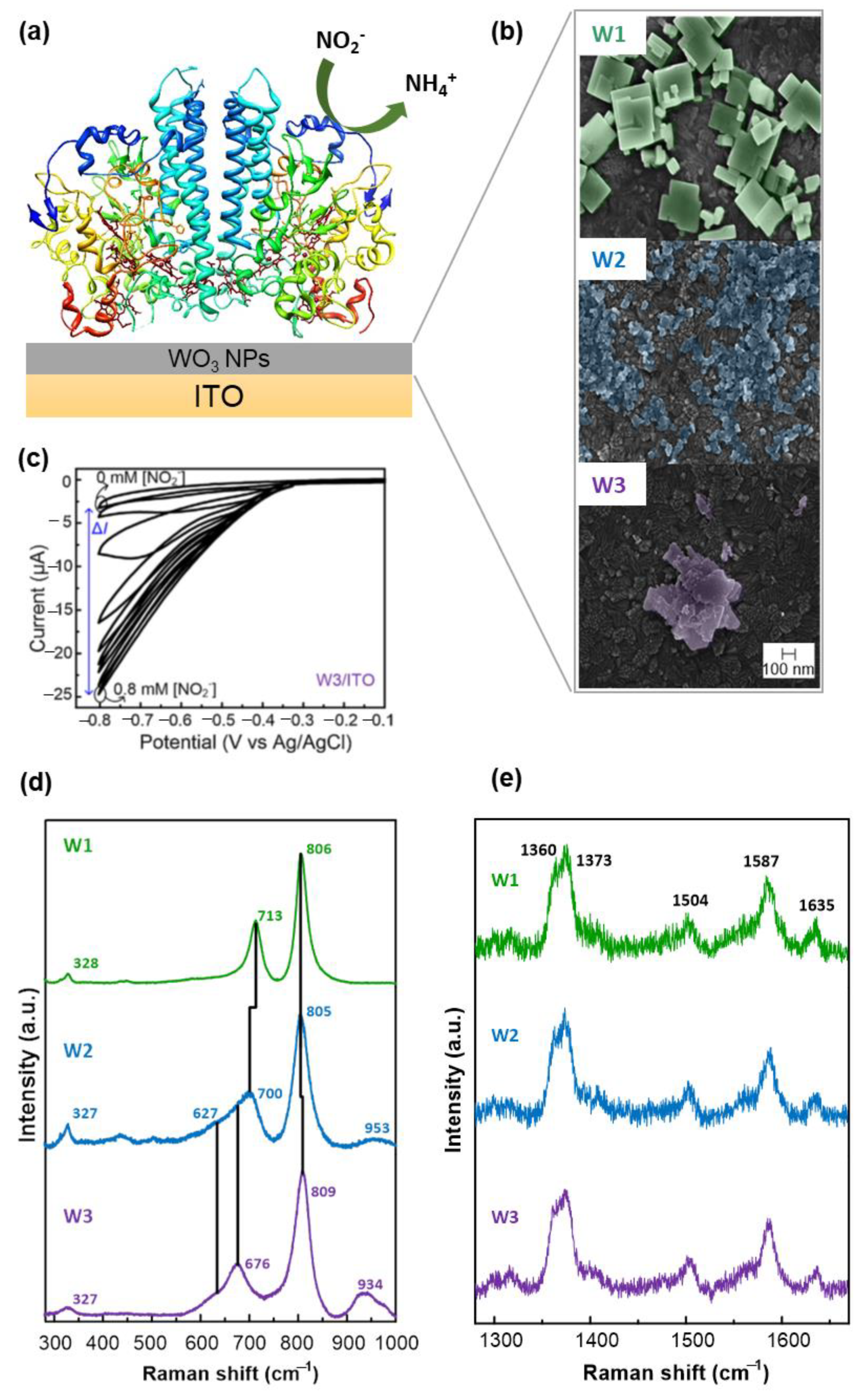{kind=link}
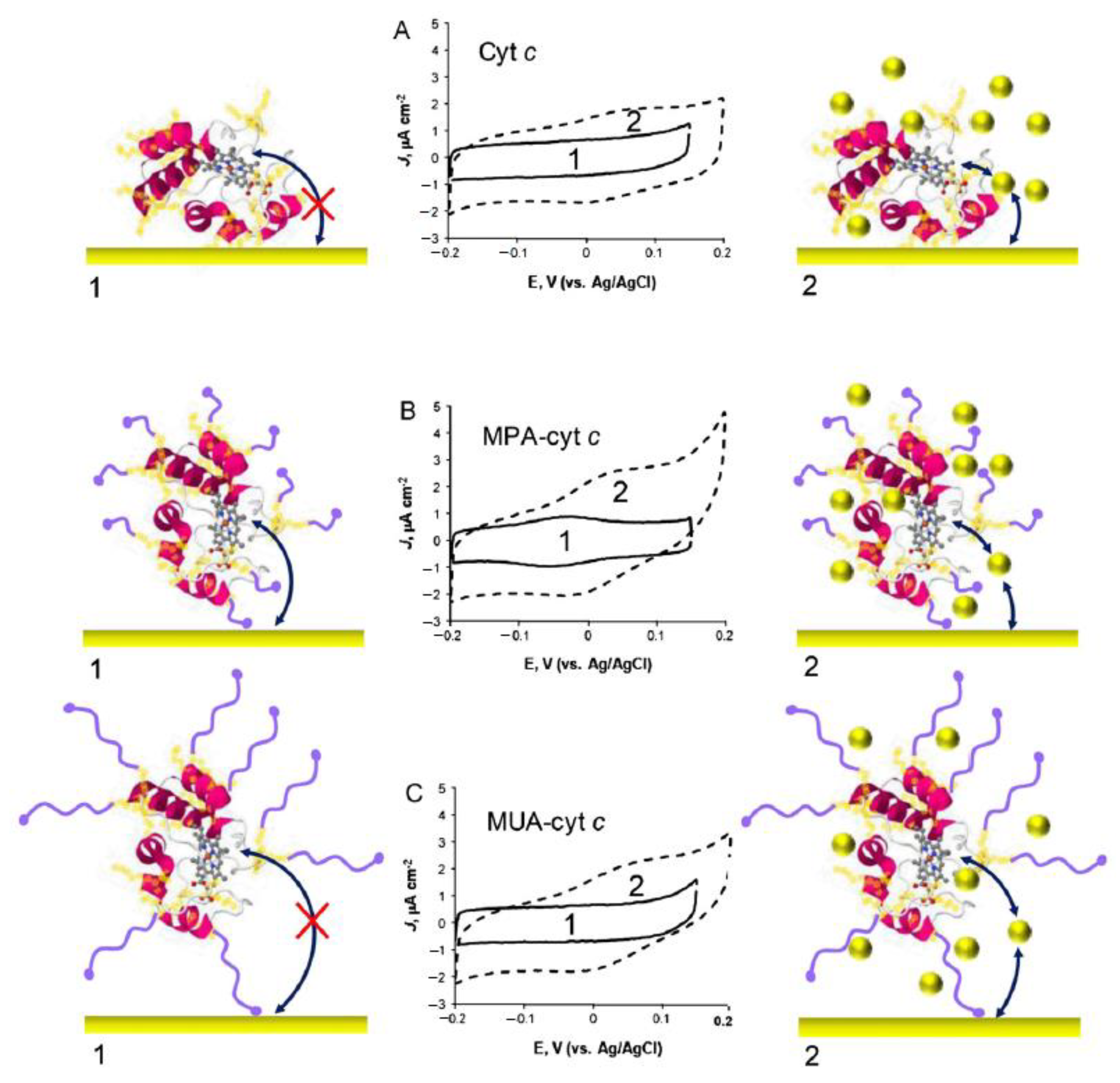{kind=link}
Table 1.
Analytical parameters of heme protein-based biosensors. EW—Working potential; a/b—normalized using the electrode geometric/active area; c—estimated from electrochemical data shown in the reference; n.d.—not determined.
Table 1.
Analytical parameters of heme protein-based biosensors. EW—Working potential; a/b—normalized using the electrode geometric/active area; c—estimated from electrochemical data shown in the reference; n.d.—not determined.
| Biosensor Configuration/Electrode | Target Analyte | Detection Mode (Mediator) | Ew (V) vs. NHE | Sensitivity (A M−1 cm−2) | Linear Range (µM) | Limit of Detection (µM) | Stability (% Initial Activity/Time) | [Ref] |
|---|---|---|---|---|---|---|---|---|
| HRP + PMN/GC | H2O2 | DET | Amp −0.006 | 0.620 | 0.19–1.35 | 0.0475 | 92%/1 month | [115] |
| HRP/PEGDGE/[Os(dmp)PVI]+/2+/G | MET ([Os(dmp)PVI]+/2) | Amp +0.188 | 0.300 | 1–500 | 0.3 | 50%/1 month | [95] | |
| HRP/MWCNT/thionine/AuNP/GC | DET | Amp +0.199 | 6.87 × 10−5 a | 640–7000 | 0.1 | n.d. | [37] | |
| HRP + HA + CdS-IL/CILE | DET | CV n.d. | 0.040 a | 10–170 | 3.30 | 95.6%/2 weeks | [81] | |
| TCA | DET | CV ~−0.060 c | 0.00208 a | 1600–18,000 | 530 | 95.6%/2 weeks | ||
| TOP/PEGDGE/[Os(dmp)PVI]+/2+/G | H2O2 | MET ([Os(dmp)PVI]+/2) | Amp +0.188 | 0.470 | 1–500 | 0.3 | 83%/1 month | [95] |
| DyP/SAM/Ag | H2O2 | DET | Amp +0.100 | 1.31 | 1–200 | 3.60 | 85%/1 month | [67] |
| P450-2C19/CS/CeNP/RGO/GC | Omeprazole | DET | Amp −0.276 | 1.440 a | 2–50 | 0.42 | n.d. | [117] |
| P450-3A4/P450-1A2/GA/AuNP/CS/RGO /GC | Clopidogel | DET | CV ~−0.360 c | 0.730 a | 2–50 | 0.63 | n.d. | [118] |
| Cat + NiO/GC | H2O2 | DET | Amp −0.059 | 0.478 a | 1–10 | 0.60 | 93%/2 weeks | [119] |
| [(Cat + NH2-IL)]7 + TiN NP/GC | DET | Amp −0.150 | 0.380 | 1–2100 | 0.100 | n.d. | [120] | |
| Cat/[BMIM]BF4-IL/NH2-MWCNT/GC | DET | Amp −0.345 | 69.3 a | 0.0086–0.140 | 0.0037 | n.d. | [121] | |
| Cat/PLL/f-MWCNT/GC | DET | Amp −0.250 | 0.392 | 0.001–3.6 | 0.008 | 89%/14 days | [122] | |
| ccNiR/[ZnCr-AQS] LDH/GC | NO2− | MET (ZnCr-AQS) | Amp −0.400 | 1.8 | 0.015–2.35 | 0.004 | 60%/32 days | [123] |
| ccNiR/sol-gel/PG | DET | Amp −0.700 | 0.430 | 0.25–50 | 0.120 | >90%/2 weeks | [124] | |
| ccNiR + WO3NP/ITO | DET | CV −0.600 | 2.143 | 5–50 | 5 | n.d. | [125] | |
| CcO/DDAB/Au | Cyt c | DET | SQW ~+0.550 c | n.d. | 0.2–4 | 0.2 | n.d. | [126] |
| CcO/DOPE-DOPC/Au | DET | Amp +0.485 | n.d. | 0–10 | 0.1 | n.d. | [127] | |
| Cyt c/l-Cys/P3MT/MWCNT/GC | H2O | DET | Amp +0.210 | 0.0178 a | 0.7–400 | 0.23 | n.d. | [128] |
| Cyt c-BDND | DET | Amp +0.149 | 0.0756 | 1–450 | 0.7 | 85%/6 weeks | [129] | |
| Nafion/Cyt c-MPA-AuNP-CS/SAM/Au | NO | DET | Amp −0.500 | 0.443 b | 10–215 | 4.5 | 96.3%/1 month | [130] |
| MP-11/PDADMAC-mpATO/ITO | H2O2 | DET | Amp +0.210 | 0.00387 | 10–750 | n.d. | n.d. | [131] |
| MP-11 @ PCN-333 (Al)/3D-KSC | H2O2 | DET | Amp −0.056 | 0.168 | 0.4–1725 | 0.127 | 89.5%/1 month | [132] |
| MP/Cat/SOD/MWCNT-PTTCA/AuNP/GC | NO | DET | Amp −0.600 | 15.7 a | 1.0–40 | 0.0043 | 97%/1 month | [133] |
| Nafion/Hb/H-TiO2-rGOMS/GC | H2O2 | DET | Amp −0.140 | n.d. | 0.1–145 | 0.01 | 95%/21 days | [134] |
| Nafion/Hb+CS+bBi2S3/GC | DET | Amp −0.203 | 14.1 a | 0.4–4.8 | 0.096 | 95.7%/15 days | [135] | |
| Hb/CNF/GC | ART | DET | CV ~−0.200 c | 4.70 a | 0–200 | n.d. | n.d. | [136] |
| Mb/AuNP-PTy-f-MWCNT/GC | H2O2 | DET | Amp −0.100 | 0.140 | 2–5000 | 0.01 | n.d. | [137] |
| Nafion/Mb/AuNP/Mg-MOF-74/CILE | NO2− | DET | CV −0.335 c | 0.1157 AM−1 | 800–18,000 | 267 | n.d. | [138] |
| TCA | DET | CV −0.098 c | 6.942 × 10−3 AM−1 | 1000–200,000 | 333 | n.d. |
3D-KSC—three-dimensional kenaf stem-derived porous carbon; Amp—amperometry; AQS—anthraquinone disulfonate; ART—artemisinin; AuNP—gold nanoparticle; bBi2S3—broccoli-like bismuth sulfide; BDND—boron-doped nanocrystalline diamond; [BMIM]BF4-IL—1-butyl-3-methylimidazolium tetrafluoroborate ionic liquid; Cat—catalase; Cyt c—cytochrome c; ccNiR—cytochrome c nitrite reductase; CcO—cytochrome c oxidase; CdS-IL—cadmium sulfide ionic liquid; CeNP—ceria nanoparticle; CILE—carbon ionic liquid electrode; CNF—carbon nanofiber; CS—chitosan; CV—cyclic voltammetry; DDAB—didodecyldimethylammonium bromide; DET—direct electron transfer; DOPC—dioleoyl phosphatidylcholine; DOPE—dioleoyl phosphatidylethanolamine; DyP—dye-decolorizing peroxidase; G—graphite rod; GA—glutaraldehyde; GC—glassy carbon; HA—hyaluronic acid; Hb—hemoglobin; HRP—horseradish peroxidase; ITO—indium tin oxide; LDH—layered double hydroxide; Mb—myoglobin; MET—mediated electron transfer; Mg-MOF-74—magnesium metal-organic framework; MP—microperoxidase; MPA—3-mercaptopropionic acid; mpATO—mesoporous antimony doped tin oxide thin film; MWCNT—multi-walled carbon nanotube; NH2-IL—amine terminated ionic liquid, 1-(3-Aminopropyl)-3-methylimidazolium bromide; NHE—normal hydrogen electrode; NiO—nickel oxide film; [Os(dmp)PVI]+/2+—osmium redox polymer; P3MT—poly-(3-methylthiophene); P450—cytochrome P450; PCN-333 (Al)—porous coordination Al-based network; PDADMAC—polydiallyldimethylammonium chloride; PEGDGE—poly(ethyleneglycol) diglycidyl ether; PG—pyrolytic graphite; PLL/f-MWCNT—poly-L-lysine/multi-walled carbon nanotube; PNM—N-isopropylacyamide-co-3-methacryloxy-propyltrimethoxysilane; PTTCA—poly-5,2:5,2-terthiophene-3-carboxylic acid; Pty—poly tyramine; RGO—reduced graphene oxide; rGOMs—reduced graphene oxide microspheres; SAM—self-assembled monolayer; SOD—super oxide dismutase; SQW—square wave voltammetry; TCA—trichloroacetic acid; TiN NP—titanium nitride nanoparticle; TOP—tobacco peroxidase; WO3NP—tungsten oxide nanoparticle.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Zuccarello, L.; Barbosa, C.; Todorovic, S.; Silveira, C.M. Electrocatalysis by Heme Enzymes—Applications in Biosensing. Catalysts 2021, 11, 218. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11020218
AMA Style
Zuccarello L, Barbosa C, Todorovic S, Silveira CM. Electrocatalysis by Heme Enzymes—Applications in Biosensing. Catalysts. 2021; 11(2):218. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11020218
Chicago/Turabian StyleZuccarello, Lidia, Catarina Barbosa, Smilja Todorovic, and Célia M. Silveira. 2021. "Electrocatalysis by Heme Enzymes—Applications in Biosensing" Catalysts 11, no. 2: 218. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11020218
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.