
Metal-Organic Frameworks in Oxidation Catalysis with Hydrogen Peroxide


Boreskov Institute of Catalysis, Lavrentieva Ave. 5, 630090 Novosibirsk, Russia
*
Author to whom correspondence should be addressed.
Catalysts 2021, 11(2), 283; https://0-doi-org.brum.beds.ac.uk/10.3390/catal11020283
Submission received: 30 January 2021
/
Revised: 17 February 2021
/
Accepted: 19 February 2021
/
Published: 21 February 2021
(This article belongs to the Special Issue Heterogeneous Selective and Total Catalytic Oxidation)
Abstract
:In recent years, metal–organic frameworks (MOFs) have received increasing attention as selective oxidation catalysts and supports for their construction. In this short review paper, we survey recent findings concerning use of MOFs in heterogeneous liquid-phase selective oxidation catalysis with the green oxidant–aqueous hydrogen peroxide. MOFs having outstanding thermal and chemical stability, such as Cr(III)-based MIL-101, Ti(IV)-based MIL-125, Zr(IV)-based UiO-66(67), Zn(II)-based ZIF-8, and some others, will be in the main focus of this work. The effects of the metal nature and MOF structure on catalytic activity and oxidation selectivity are analyzed and the mechanisms of hydrogen peroxide activation are discussed. In some cases, we also make an attempt to analyze relationships between liquid-phase adsorption properties of MOFs and peculiarities of their catalytic performance. Attempts of using MOFs as supports for construction of single-site catalysts through their modification with heterometals will be also addressed in relation to the use of such catalysts for activation of H2O2. Special attention is given to the critical issues of catalyst stability and reusability. The scope and limitations of MOF catalysts in H2O2-based selective oxidation are discussed.
1. Introduction
In the quest for sustainable and green production of valuable chemicals, an important goal is the development of economic and ecologically sound oxidation processes using benign and readily available oxidizing agents [1,2,3]. Hydrogen peroxide is a clean and green oxidant because it contains 47% of potentially active oxygen and produces water as the sole byproduct [4,5]. New methodologies being developed for the direct synthesis of hydrogen peroxide from H2 and O2 are anticipated to expand significantly the scope of this oxidant in the near future [6,7,8].
Metal–organic frameworks (MOFs) are an emerging class of materials composed of metal ions or, more frequently, clusters connected by multidentate organic linkers into a regular porous structure. A unique combination of properties, including extraordinarily high surface areas, open nanoporosity, tunable functionality, and pore dimensions, together with a large fraction (20–40 wt.%) of accessible metal sites uniformly distributed over the framework, makes MOFs highly attractive materials for applications in gas storage and separation, molecular recognition, drug delivery, and heterogeneous catalysis [9,10,11,12,13,14,15,16,17,18,19,20,21,22]. Paz and coworkers in their review paper analyzed efforts of commercial companies to bring functional MOFs towards the industrial applications [18], while Choi and colleagues surveyed recent developments in process engineering and upcoming applications of MOFs [22].
Analysis of the recent review literature devoted to heterogeneous catalysis on MOFs showed that a considerable scientific effort was directed to evaluation of their potential for selective oxidation [23,24,25,26,27,28,29,30,31,32,33,34,35,36,37,38,39,40,41,42,43]. So far, the majority of works in this area deal with the use of molecular oxygen or anhydrous alkyl hydroperoxides as oxidants while not so many are devoted to oxidation catalysis with H2O2. This is not surprising if we remember that aqueous hydrogen peroxide possesses not only oxidizing but also hydrolyzing and strong complexing ability, which is a threat to the structure of most MOFs, especially at elevated temperatures. However, discoveries of MOFs having outstanding chemical and thermal stability, especially Zr-based ones, are currently changing the situation [44,45].
In this short review paper, we survey recent findings concerning use of MOFs as heterogeneous catalysts for selective oxidations using H2O2, with special attention drawn to the mechanistic aspects of the oxidation reactions and stability/reusability issues. Some attempts of using MOFs as supports for construction of single-site selective oxidation catalysts through their modification with heterometals will be also discussed in their relation to H2O2-based oxidations. We focus here on MOFs with high chemical and thermal stability, such as Cr(III)-based MIL-101, Ti(IV)-based MIL-125, Zn(II)-based ZIF-8, Zr(IV)-based UiO-66(67), and some others. Representative MOF structures are shown in Figure 1, while Table 1 summarizes typical physicochemical characteristics of these MOFs, including thermal stability. Since the incorporation of metal–oxygen clusters or polyoxometalates (POMs) into the structural nodes or cages of MOFs was recently surveyed [46,47], POM-MOF catalysts are beyond the scope of this work.
2. MOFs as Selective Oxidation Catalysts
2.1. Oxidation of S-Compounds
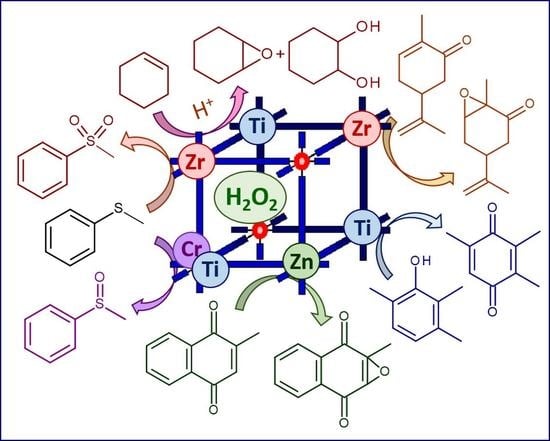
Organic sulfides are nucleophilic substrates which can be readily oxidized to corresponding sulfoxides with electrophilic oxidants (most peroxo complexes of d0 transition metals) whereas sulfoxides possess a biphilic nature, and therefore their oxidative transformation to sulfones can be accomplished using both electrophilic and nucleophilic oxidants [58,59]. Considering high reactivity of alkyl aryl sulfides that allows use of mild conditions for their oxidation, these substrates have been widely employed to evaluate catalytic activity of MOFs [26,42,60,61,62,63,64,65,66]. Below we discuss a few MOF-based catalyst systems which feature high selectivity to either sulfoxide or sulfone, thereby giving hints about the oxidation mechanism.
2.1.1. Electrophilic Oxidation of Thioethers to Sulfoxides over Zn- and Cr-MOFs
In 2006, Dybtsev et al. reported the synthesis of a robust homochiral microporous Zn-MOF, [Zn2(BDC)(L-lac)(DMF)], starting from Zn(NO3)2, L-lactic acid (L-H2lac), and 1,4-benzenedicarboxylic acid (H2BDC) and its application for enantioselective sorption of sulfoxides and catalytic oxidation of alkyl aryl sulfides with urea hydroperoxide or H2O2 [65]. Substrates with small substituents showed high conversions (92–100%) with 3 equiv. of H2O2 and excellent chemoselectivity toward sulfoxides (up to 100% for methyl phenyl sulfoxide), although no asymmetric induction was observed. The preferable formation of sulfoxides in this system allows suggestion about electrophilic activation of the oxidant [58]; however, the detailed mechanism of the thioether oxidation is not completely clear.
One of the first applications of MOFs in oxidation catalysis with H2O2 is related to the chromium(III) terephthalate Cr-MIL-101 ([Cr3X(H2O)2O(BDC)3]; X = F, OH; MIL stands for Matérial Institut Lavoisier) discovered by Férey and coworkers [48]. MIL-101 possesses a rigid zeotype (MTN) crystal structure, consisting of quasi-spherical cages of two modes (2.9 and 3.4 nm) accessible through windows of ca. 1.2 and 1.6 nm (Figure 1). Characteristic features of this MOF, which play a significant role in catalysis, are mesoporosity coupled to high chemical, thermal and hydrothermal stability. An additional advantage for catalysis arises from the possibility of creating coordinatively unsaturated sites (CUS) in the Cr(III) metal nodes by removal of terminal water molecules using thermal treatment upon evacuation (Figure 2) [23,66].
In contrast to oxidations with anhydrous alkyl hydroperoxides [24,67,68], the use of aqueous hydrogen peroxide as oxidant is generally detrimental for Cr-MIL-101 because a cooperative action of water and H2O2 causes MOF destruction and metal leaching. However, Chang, Férey and coworkers demonstrated that Cr-MIL-101 may preserve its structure in H2O2-based oxidation of highly reactive substrates such as aryl alkyl sulfides, where very mild conditions (room temperature) can be employed [66]. Moreover, they found that the catalytic activity can be improved by increasing the number of CUS in MIL-101 through its dehydration. The catalyst system produced exclusively sulfoxides (88–99 % yields) and no sulfones, pointing to electrophilic activation of hydrogen peroxide on the Cr(III) sites (Figure 2).
2.1.2. Oxidative Desulfurization
Since oxidative desulfurization (ODS) was considered as an alternative method to the currently existing hydrodesulfurization (HDS) to reduce the sulfur content in gasoline and diesel fuel to ultra-low levels [69], the oxidation of thiophene derivatives with hydrogen peroxide was the most frequently studied oxidative transformation catalyzed by various MOFs and MOF-derived materials.
The titanium terephthalate MIL-125 [49] (see Figure 1 for the structure) and MIL-125_NH2 [70] prepared using H2BDC and 2-amino benzene dicarboxylic acid (H2BDC-NH2), respectively, were employed for oxidative desulfurization using H2O2 [71,72]. In general, MIL-125 was more active than its NH2-modified analogue but stability of both under the ODS conditions was questionable [72]. Moreover, mesoporous carbons comprising TiO2 nanoparticles obtained by pyrolysis of MIL-125_NH2 appeared to be more efficient catalyst for the oxidation of dibenzothiophene than the original Ti-MOF [71]. A composite, ZIF-8@H2N-MIL-125, exhibited the highest activity among the catalysts studied toward the oxidative desulfurization of dibenzothiophene [71].
Since the pioneer work of Cavka et al. that reported the synthesis of UiO-66 (UiO stands for University of Oslo, see Figure 1 for the structure) [52], Zr-based MOFs constructed from very robust Zr6-oxo-hydroxo clusters and various carboxylate linkers, due to their outstanding chemical, thermal, hydrothermal, and mechanical stability [44,73,74,75,76,77], have become one of the most appealing classes of MOFs for applications in heterogeneous catalysis and, in particular, for ODS. Oxidation of thiophenes with UiO-66 as catalyst and H2O2 as oxidant was studied by several research groups [78,79,80,81,82,83,84]. Catalytic activity of UiO-66 and other Zr-MOFs was suggested to correlate with the number of missing-linker defects (accessible Zr-OH/-OH2 sites) [78,80,81,82,83,84,85]. Zheng et al. compared the catalytic performance of four Zr-MOFs, UiO-66, UiO-67, NU-1000, and MOF-808, and revealed superior activity of MOF-808 that correlated with its higher Lewis acidity [81]. The authors suggested that a higher concentration of readily accessible Zr-OH sites in the 6-connected MOF-808 compared to the 12-connected UiO-66/UiO-67 and the 8-connected NU-1000 may be crucial for the catalytic activity and that the active species are •OH and •O2− radicals derived from homolytic decomposition of H2O2. Subsequent studies revealed that engineering of defects in the structure of MOF-808 by acid treatment allows the ODS activity of MOF-808 to be increased significantly [85]. Other efforts to improve the catalytic performance of Zr-MOFs involved their modification with Ti(IV) [86,87] and other active species. Given that this research area was recently summarized by Piscopo and coworkers [87] we do not touch on this subject in more detail and refer the reader to that comprehensive review.
2.1.3. Unprecedented Selectivity of Zr-MOFs toward Sulfones
While studying catalytic activity of UiO-66 prepared by different methodologies and differing in their crystallinity for the oxidative desulfurization of fuel, Granadeiro and coworkers have found that all the samples reveal superior selectivity toward the formation of sulfones [78]. In line with these results, Nguyen and coworkers demonstrated a predomination of sulfone over sulfoxide in the oxidation of methyl phenyl sulfide (MPS) over UiO-66 in CH3CN [88]. However, if CH3OH was employed as solvent instead of CH3CN, sulfoxide became the principle oxidation product. A correlation between catalytic activity and the number of accessible “open” sites in the crystalline structure of UiO-66 was established [88]. On the basis of kinetic and computational data, the authors suggested that MPS oxidation involves a Zr-μ1-OOH active intermediate generated at the defect open sites of the MOF, which are supposed to be terminated with a couple of Zr-μ1-OH and Zr-μ1-OH2 [89,90]. The predomination of sulfone over sulfoxide was explained suggesting a reaction mechanism where the primary oxidation product, sulfoxide, binds to a Zr site adjacent to Zr-μ1-OOH, thereby favoring increased local concentration of sulfoxide and its overoxidation to sulfone.
More recently, Zalomaeva et al. [91] and Maksimchuk et al. [92] investigated thioether oxidation in aprotic solvents over a range of Zr-MOFs, including UiO-66, isoreticular UiO-67, MOF-801, and the recently discovered Zr-abtc [54] and MIP-200 [55]. They concluded that the unprecedentedly high selectivity toward sulfones attained with only one equivalent of H2O2 (90–99% at ca. 50% sulfide conversion) is an intrinsic feature of Zr-MOFs, regardless of their structural and morphological characteristics. Table 2 illustrates this finding, showing a comparison of the catalytic performances of various Zr-MOFs and the Ti-based MIL-125 under the same reaction conditions (the latter reveals predomination of sulfoxide, which is typical of electrophilic oxidation).
In contrast to selectivity, catalyst activity strongly depends on the specific Zr-MOF nature and sample characteristics. In general, it decreased in the order UiO-66/67 > Zr-abtc > MIP-200 (see Table 2). No direct correlation between activity and the average particle size, which decreased in the series UiO-67 > MIP-200 > Zr-abtc > UiO-66, or the size of pore apertures, that followed the order MIP-200 (1.3 nm hexagonal channels) > UiO-67 (0.8 nm windows) > Zr-abtc (0.7 nm channels) > UiO-66 (0.6 nm windows), could be found [92]. At the same time, for a specific Zr-MOF (e.g., UiO-66), activity may strongly depend on the size of MOF crystallites (see Table 2).
Interestingly, Zr-abtc and MIP-200 consisted of 8-connected Zr6-clusters and tetratopic linkers showed no advantages in terms of activity over the 12-coordinated UiO-66 and UiO-67. Moreover, termogravimetric analysis (TGA) showed that the amount of defects corresponding to the amount of terminal Zr-OH/-OH2 groups declined in the order Zr-abtc > MIP-200 > UiO-67 > UiO-66 and also did not correlate with the observed catalytic activity in the MPS oxidation. MIP-200 that possesses the largest pore entrance and high amount of Zr-OH groups showed the lowest activity among the Zr-MOFs studied, which was attributed to its specific hydrophilicity (high H2O uptake at low P/P0) [55] disfavoring adsorption of organic substrates and H2O2 in the presence of water.
This example shows that while MOF selectivity is mostly determined by the nature of transition metal that constitutes its nodes, activity is a complicated function of various factors, including structural, textural, and morphological characteristics, as well as specific sorption properties.
We should also mention here that the excellent selectivity toward sulfones casts doubt on the possibility of using Zr-MOFs for oxidative decontamination of mustard gas (bis(2-chloroethyl)sulfide) because the corresponding sulfoxide is much less toxic than the sulfone [94]. On the other hand, this feature may give benefits for ODS technologies where the formation of sulfones is often desirable.
Post-synthetic modification of Zr-MOFs may drastically change their activity and selectivity and lead to new opportunities in oxidation catalysis. For example, modification of an anionic Zr-MOF, NPF-201, with a photoactive cation, [Ru(bpy)3]2+, made possible photocatalytic oxidation of MPS with O2 as oxidant, producing sulfoxide as the major product [76].
2.1.4. Nucleophilic Activation of H2O2 and Mechanism of Thioether Oxidation
To understand the reasons for the unusually high sulfone selectivity over Zr-MOFs, the reaction mechanism was investigated using specific test substrates, kinetic, adsorption, 18O labeling, and spectroscopic methods [91]. Several lines of evidence pointed to a nucleophilic character of the peroxo species responsible for the superior formation of sulfones. The oxidation of the test substrate thianthrene 5-oxide (Scheme 1) gave the nucleophilic parameter XNu = 0.92, typical of nucleophilic oxidation [95,96]. The addition of 1 equiv. of acid relative to Zr reduced this parameter down to 0.47.
A complementary test [97], competitive oxidation of methyl phenyl sulfoxide and p-Br-methyl phenyl sulfide, gave the sulfone to sulfoxide ratio of 24, while kinetic studies showed significantly lower initial rates for MPS oxidation relative to methyl phenyl sulfoxide (kS/kSO = 0.05), which also points to a nucleophilic oxidation mechanism. Finally, a positive slope ρ = +0.42 of the Hammett plot for competitive oxidation of p-substituted aryl methyl sulfoxides also implies nucleophilic activation of H2O2 on Zr-MOF [91]. Kinetic modeling on methyl phenyl sulfoxide oxidation together with adsorption studies (the latter showed significant affinity of UiO-66 toward sulfoxide and not to sulfide and sulfone) implicated a mechanism that involves the interaction of H2O2 with Zr sites with the formation of a nucleophilic oxidizing species and release of water followed by oxygen atom transfer from the nucleophilic oxidant to sulfoxide that competes with water for Zr sites (Scheme 2).
The nucleophilic peroxo species co-exists with an electrophilic one, ZrOOH, which is responsible for the oxidation of nucleophilic sulfides (Scheme 2). This first step, most likely, occurs through electrophilic oxygen transfer from ZrOOH to sulfide rather than an alternative single electron transfer (SET) mechanism that involves the formation of an intermediate thioether radical cation followed by its rapid capture by water and further oxidation. This conclusion was drawn on the basis of isotopic (18OH2) labeling experiments, which revealed no incorporation of 18O into the sulfone and sulfoxide products, lack of products derived from radical cation fragmentation and/or dimerization (benzaldehyde, benzoic acid, and dibenzyl) in the oxidation of benzyl phenyl sulfide and finally, higher rates of diethyl sulfide oxidation relative to oxidation of MPS [91].
The prevalence of nucleophilic activation of H2O2 over electrophilic one in Zr-MOFs is, most likely, ensured by the presence of weak basic sites. Such sites have been identified in Zr-MOFs by FTIR spectroscopy of adsorbed CDCl3 as a probe molecule [91,92]. Measurements of the adsorption of isobutyric acid on Zr-MOFs revealed that the number of these basic sites is rather close to the amount of terminal Zr-OH groups present in the MOF defects that can be evaluated by TGA [91]. This finding agrees well with recent computational results, which implicated that basic sites in hydrated UiO-66 are, most likely, the terminal ZrOH groups [98,99].
2.2. Epoxidation of C=C Bonds
2.2.1. Nucleophilic Oxidation of Electron-Deficient C=C Bonds
The capability of Zr-MOFs to accomplish nucleophilic activation of H2O2 is also manifested in the epoxidation of electron-deficient C=C bonds in α,β-unsaturated ketones and quinones [91,92]. This process is accompanied by oxidation of acetonitrile solvent as evidenced by the formation of significant amounts of acetamide among the oxidation products. The latter fact implies that peroxycarboximidic acid H3CC(=NH)OOH resulted from the interaction of CH3CN and H2O2 under basic conditions could participate in the epoxidation process (the so-called Payne oxidation [100]). On the other hand, the epoxidation could also proceed if CH3CN was replaced with ethylacetate (see Table 3), suggesting that another oxidizing species, e.g., a nucleophilic peroxo intermediate derived from H2O2 and Zr-MOF, might be responsible for the oxygen transfer to the electron-deficient C=C bonds.
The 8-coordinated Zr-abtc demonstrated advantages over other Zr-MOFs in both substrate conversion and product selectivity for epoxidation of α,β-unsaturated ketones [92]. A significant predomination of 1,2-epoxide in the oxidation of carvone (i.e., epoxidation of the more electron poor C=C bond) strongly suggests a nucleophilic oxidation mechanism [101]. The superior catalytic performance of Zr-abtc correlated with a larger amount of weak basic sites found for this MOF relative to the other Zr-MOFs studied [92].
The imidazolate framework ZIF-8 consisting of zinc atoms linked through nitrogen atoms of 2-methylimidazole [56] (see Figure 1) turned out another MOF capable of epoxidizing electron-poor C=C bonds [102]. While in the oxidation of cyclohexenone catalytic performance of ZIF-8 was comparable with that of UiO-66/67, it revealed superior properties, in terms of attainable substrate conversion and epoxide selectivity, with 2-methyl-naphthoquinone (MNQ), also known as menadione or Vitamin K3 (see Table 3). Although the windows of ZIF-8 are able to increase their size up to 7.6 Å [103,104,105], the dimensions of the MNQ molecule are too large (ca. 6 × 12 Å) to envisage their penetration into the micropores, and the reaction, most likely, occurs on the external surface of the ZIF-8 crystallites. This is consistent with the fact that the MNQ epoxidation rate greatly increased with decreasing size of the catalyst particles (Figure 3).
Under optimized conditions, ZIF-8 having small crystallites (ca. 200 nm) produced MNQ epoxide with the yield as high as 90% [102]. The reasons for such superior performance of ZIF-8 in the MNQ epoxidation are not yet completely clear but, again, nucleophilic activation of H2O2 is, most likely, realized owing to the presence of weak basic sites on the MOF surface (N− moieties and OH groups located on the external surface and/or possibly in bulk defects [106]). We may suppose that such basic sites favor polarization of the H2O2 molecule via hydrogen-bonding and facilitate its dissociation, leading to the nucleophilic oxidant HOO−. In contrast to the epoxidation over Zr-MOFs, minor amounts of acetamide were found for the MNQ epoxidation with ZIF-8, indicating that oxidation of CH3CN and contribution of peroxycarboximidic acid into the ZIF-8-catalyzed oxidation process are insignificant.
2.2.2. Oxidation of Electron-Rich C=C Bonds: Effect of Protons on Heterolytic Pathway Selectivity
Epoxidation of C=C bonds in unfunctionalized alkenes is one of the most important reaction in the organic synthesis. To accomplish alkene epoxidation selectively, two-electron, heterolytic activation of H2O2 is required [1,2]. However, redox activity of transition metals which constitute MOF nodes favors homolytic activation of hydrogen peroxide (see Section 2.5), leading to the formation of one-electron oxidation products. Cyclohexene (CyH) is a well-known test substrate that enables an easy discrimination between one- and two-electron oxidation mechanisms (Scheme 3).
Heterolytic oxidation pathways give exclusively CyH epoxide and the epoxide ring opening product trans-cyclohexane-1,2-diol (diol), whereas homolytic routes produce, in addition to epoxide and diol, a significant amount of the allylic oxidation products, cyclohexenyl hydroperoxide (HP), 2-cyclohexene-1-ol (enol), and 2-cyclohexene-1-one (enone).
Nguyen et al. investigated CyH oxidation with H2O2 in the presence of UiO-66 and MIL-125-NH2 and found relatively low activity for both MOFs with predominant formation of the allylic oxidation products [107], which clearly indicated predomination of homolytic oxidation pathways. Attempts to modify the Zr-MOF by Ti(IV) using different techniques led to increasing catalytic activity but had very little impact on the product distribution: enol and enone remained the main products [107]. The formation of the allylic oxidation products could not be completely suppressed by grafting of Nb(V) oxide sites onto the nodes of Nu-1000 [108].
Maksimchuk et al., on the basis of their experience in polyoxometalate chemistry [109,110,111,112,113,114,115], suggested a simple tool that allowed heterolytic pathway selectivity and attainable CyH conversions to be considerably improved [116,117]. The addition of one equivalent of acid relative to Ti (or Zr) led to a great reduction of the yield of allylic oxidation products in the CyH oxidation (Table 4): from 60 to 22% for MIL-125 [116] and from 50 to only 2% for UiO-66 [117]. This phenomenon also took place for other Zr-MOFs, regardless their structural and morphological features (see Table 4) [92,117]. The addition of protons also markedly enhanced the oxidation rate (e.g., turnover frequencies (TOFs), increased from 0.1 to 1.3 min−1 for UiO-66 upon addition of 1 equiv. of acid [117]. In the absence of MOF, acid produced only minor changes in CyH conversion and oxidation product distribution (see Table 4). The addition of extra protons (>1 equiv.) to UiO-66 led to decreasing epoxide yield because of its transformation into diol [117].
Importantly, XRD, Raman and FTIR studies confirmed that the acid additives produced no changes in the crystalline structure of MIL-125 and UiO-66 [116,117]. By analogy with POMs [109,110,111,112,113,114,115] and metal-silicate catalysts [118,119,120,121], it was suggested that the addition of a source of protons favors the formation of active metal hydroperoxo intermediates, MOOH, responsible for oxygen atom transfer to alkenes to afford epoxides [116,117].
2.3. Oxidation of Alkylphenols
In contrast to the epoxidation of alkenes, the propensity of MOF to homolytic activation of H2O2 can provide benefits for oxidation reactions which can be realized effectively via one-electron mechanisms, for example, for the oxidation of alkyl-substituted phenols to corresponding benzoquinones. Previous studies on titanium-silicate catalysts implicated that this reaction involves two consecutive electron transfers from two adjacent Ti sites and, notwithstanding the homolytic oxidation mechanism, excellent quinone yields can be obtained due to a cooperative action of di(multi)nuclear Ti centers [122,123,124]. Bearing this idea in mind, Ivanchikova et al. investigated catalytic performance of MIL-125 and MIL-125_NH2 comprising Ti-oxohydroxo clusters in the oxidation of two representative alkylphenols, 2,3,6-trimethylphenol (TMP) and 2,6-di- tert-butylphenol (DTBP), with aqueous H2O2 [125]. Superior selectivity toward alkyl-p-benzoquinones (nearly 100%) was achieved with both Ti-MOFs (Table 5).
Samples with different size of crystallites (0.5, 1.5, and 5 μm) showed similar reaction rates, indicating the absence of internal diffusion limitations [125]. Even though the crystal structure of MIL-125 was destroyed under the turnover conditions, the MOF acted as a precursor for the highly active, selective, and recyclable catalyst (see Section 2.7). Comparison with other types of Ti catalysts revealed that catalytic performance of MIL-125 in TMP oxidation to trimethyl-p-benzoquinone (vitamin E precursor), in terms of the attainable product yield, is superior to amorphous and crystalline titania but inferior to mesoporous titanium-silicates containing di(oligo)meric Ti sites [126].
2.4. Oxidation of Alcohols and Diols
2.4.1. Alcohol Oxidation
Oxidation of alcohols is one of the most studied reactions in the MOF-based catalysis [23,30,32]. Modification of MOFs with noble metal nanoparticles is often employed to fulfill this type of oxidative transformations [17,20,23]. However, in the majority of works, molecular oxygen and/or alkyl hydroperoxides were employed as the oxidant, and only few works reported about the use of hydrogen peroxide.
A two-dimensional microporous dinuclear copper(II) trans-1,4-cyclohexanedicarboxylate [Cu2(OOCC6H10COO)2]·H2O was used as a heterogeneous catalyst for selective oxidations of various (primary, secondary, and benzylic) alcohols with hydrogen peroxide [127]. Although selectivity toward corresponding ketones and aldehydes was high (>99%), the attainable conversions were low even if a 113-fold excess of the oxidant was employed. The reaction was realized at room temperature, and no copper leaching was detected. An active peroxo copper(II) intermediate, H2[Cu2(OOCC6H10COO)2(O2)]·H2O was isolated and characterized by X-ray powder diffraction, spectroscopic and other techniques, which implicated a structure constructed by intramolecular bridging of µ-1,2-trans-Cu–OO–Cu species between two-dimensional [Cu2(O2CC6H10CO2)] layers. This peroxo complex revealed activity in the stoichiometric oxidation of 2-propanol to acetone, which proved its crucial role in the catalytic oxidation of alcohols.
Balu et al. prepared a Fe-containing MIL-101 using a microwave-assisted deposition methodology [128]. They reported that the incorporation of ca. 2 wt.% Fe in the form of hematite Fe2O3 led to Fe/MIL-101 with high catalytic activity in the oxidation of various alcohols, including benzylic and aliphatic ones, using hydrogen peroxide as oxidant and water as solvent. The reaction was accomplished under microwave irradiation. Feasible conversions, product yields and H2O2 efficiencies were obtained for benzylic alcohols and cyclohexanol while primary alcohols revealed relatively low conversions (<20%). Fe-free MIL-101 activated in vacuum at 150 °C during 12 h to produce CUS revealed poor activity (ca. 10% yield of benzaldehyde in the oxidation of benzylic alcohol vs. 90% yield acquired over 2.06 wt.% Fe/MIL-101, which clearly indicates the role of iron species in the observed catalysis. Surprisingly, no iron leaching and MOF destruction were observed, which could be due to the short reaction time (15 min).
Cohen and coworkers synthesized a robust Zr-based MOF bearing catechol functionality, UiO-66-CAT, and accomplished its metalation, resulting in a MOF decoration with coordinatively unsaturated Fe- and Cr-monocatecholato species [129]. The Cr-metalated UiO-66 turned out an efficient and recyclable catalyst for the selective oxidation of alcohols to ketones using both tert-butyl hydroperoxide (TBHP) and H2O2 as oxidant. TBHP gave fairly good yield of ketones for a wide range of secondary alcohols, including bulky substrates for which the reaction was suggested to occur at the external surface of the catalyst particles. The application of hydrogen peroxide was effective for oxidation of benzylic alcohols (95–99% yields with 2 equiv. of the oxidant) while aliphatic alcohols gave poor yields (e.g., ca. 39% for 2-heptanol). No chromium leaching into solution was found.
2.4.2. Propylene Glycol Oxidation
Another reaction where homolytic activation of H2O2 has led to the formation of useful products is oxidation of propylene glycol (PG) over UiO-66 [130]. The reaction proceeds with high regioselectivity, leading to the formation of hydroxyacetone (HA) as the main product. Additives of radical chain scavengers produce a rate-inhibiting effect, suggesting radical chain mechanism of the oxidation process. However, molecular oxygen does not participate in the oxidation process as indicated by close reaction rates in air and inert atmosphere.
The solvent nature caused a pronounced impact on HA yields and oxidant utilization efficiency. Adsorption studies revealed that PG adsorption on UiO-66 from pure CH3CN is significant (the adsorption constant evaluated using Langmuir model was 290 ± 60 M−1) while practically no adsorption occurred from a mixed solvent H2O/CH3CN 3/7 (v/v) (Figure 4) [130]. This allowed the authors to suggest that adsorbed PG molecules block Zr active sites and thereby prevent adsorption and activation of the oxidant. In agreement with this suggestion, the best catalytic performance (85% selectivity toward HA at ca. 10% PG conversion) was achieved in acetonitrile containing 30 vol.% of H2O.
Adsorption studies also allowed the authors to suggest that the low level of attainable PG conversions is, most likely, related to the catalyst deactivation caused by adsorption of the main reaction by-product, acetic acid (see Section 2.7).
2.5. Oxidation of Arenes and Alkanes
As can be inferred from the recent review literature [41,43], examples of C-H bond activation with H2O2 over MOFs where the catalyst stability toward leaching has been proved are still very rare. A survey of the early literature in this field can be found in the book chapter of Chang and coworkers [23].
The post-synthetic functionalization of the bridging OH-group between two metal centers of the secondary building units of the robust MOF MIL-53(Al) with 1,1′-ferrocenediyl-dimethylsilane has led to a functionalized material active in the liquid-phase H2O2-based oxidation of benzene to phenol [131]. Although the substrate conversion was rather low (ca. 15%) and some destruction of the catalyst could not be excluded, this work first demonstrated that MIL-53(Al) can be converted into a redox-active functional material. Wang et al. reported on a highly selective hydroxylation of benzene to phenol over Fe-based MOFs, MIL-100 and MIL-68, using hydrogen peroxide as oxidant under a visible light irradiation [132]. With MIL-100 and a 3:4 oxidant to benzene ratio, selectivity to phenol attained 97% at ca. 20% benzene conversion. On the basis of ESR and kinetic studies, the authors suggested a synergy between photocatalysis of Fe-O clusters in the MOF and a Fenton-like chemistry.
More recently, Gascon and coworkers synthesized a Fe-containing MOF, MIL-53(Al, Fe) which comprised a spatially isolated oxo-bridged Fe2 units in a coordination environment resembling that of the carboxylate-bridging diiron active site of methane monooxygenase (MMO) [133]. They demonstrated that the hybrid MIL-53(Al, Fe) material is able to catalyze methane oxidation with H2O2 in water, producing methanol as the principle oxidation product with selectivities toward oxygenates of ca. 80% and turnover number (TON) up to 350. The catalyst synthesized through an electrochemical route contained no extraframework iron oxide species and was rather stable toward aqueous H2O2 at temperatures below 60 °C. Pure phase MIL-53(Fe) revealed substantial iron leaching under the reaction conditions and behaved as a typical Fenton type catalyst rather than a MMO simulant, which proves that that the use of a nonredox scaffold, MIL-53(Al), is crucial for both the catalyst stability and catalytic performance [133]. Based on spectroscopic studies and DFT calculations, the authors have concluded that the observed MMO-like behavior is mostly due to antiferromagnetically coupled, high-spin Fe sites isolated within MOF. Computational studies also suggested that the catalytic cycle involves the replacement of one of the carboxylate linkers by H2O2 molecule followed by a sequence of redox steps leading to increasing the formal oxidation state of one of the Fe centers from III to V, subsequent homolytic dissociation of the C−H bond and recombination of CH3 radical with the terminal OH group to form methanol.
2.6. H2O2 Decomposition and Oxidant Utilization Efficiency
Most of H2O2-based oxidations over MOF catalysts suffer from relatively low substrate conversions. One of possible reasons for that is a high rate of the unproductive decomposition of H2O2 relative to the rates of the target oxidation reaction, i.e., rather low oxidant utilization efficiency (i.e., selectivity based on the oxidant). Figure 5 shows plots of H2O2 consumption over MIL-125 and UiO-66 in the absence of organic substrate. One can notice that activity of the Ti-MOF is higher than that of the Zr-MOF. A hot catalyst filtration test showed that the reaction occurs on the catalyst surface rather than in solution [117].
Various Zr-MOFs reveal very similar activity in H2O2 dismutation [92]. Interestingly, protons produce practically no effect on this reaction over MIL-125 while they strongly decrease the H2O2 decomposition rate in the presence of Zr-MOFs, including UiO-66 (Figure 5). Even small additives of HClO4 (e.g., 0.1 equiv. to Zr) resulted in a significant suppression of H2O2 decomposition [117]. Therefore, protons produce opposite effect on the alkene epoxidation rate (see Section 2.2.2) and the rate of H2O2 degradation over Zr-MOF, which suggests that different active sites are involved in these two reactions.
This finding gives us a tool of how we can increase the oxidant utilization efficiency in the MOF-catalyzed oxidations. Note that this parameter strongly depends on the substrate nature and the reaction conditions employed for its oxidation. For example, H2O2 efficiency is usually high for the oxidation of highly reactive alkyl aryl sulfides while it may significantly decrease for less reactive alkenes. Figure 6 shows how this important characteristic depends on the specific substrate and catalyst system. It is noteworthy that H2O2 utilization efficiency in the CyH oxidation increased from 45% to 75 and 63% with MIL-125 [116] and UiO-66 [117], respectively, due to the addition of a source of protons (Figure 6).
2.7. Catalyst Stability and Reusability
The catalyst instability toward leaching of active species under turnover conditions is often a critical issue of solid catalysts in the liquid-phase. Sheldon and coworkers suggested use of a hot filtration test to verify whether the observed catalysis is truly heterogeneous (occurs on the catalyst surface) or homogeneous (caused by active species leached into solution) [134]. Additionally, the absence of leaching should be confirmed by elemental analysis of the filtrate. In this short review, we cited almost exclusively works for which the nature of catalysis was validated.
Figure 7 shows examples of hot filtration tests for TMP oxidation over MIL-125 and CyH oxidation over UiO-66 in the presence of 1 equiv. HClO4. In both cases, the reaction stopped after separation of the catalyst, pointing to the truly heterogeneous nature of the catalytic reactions [117,125].
Nevertheless, the heterogeneous nature of catalysis does not necessarily mean that the structure of MOF remains intact. XRD patterns in Figure 8 demonstrate that the structure of both MIL-125 and UiO-66 was maintained after the CyH oxidation. Meanwhile, the crystalline structure of MIL-125 was completely destroyed under the conditions of TMP oxidation (Figure 8a, curve C).
Studies by high-resolution transmission electron microscopy (HRTEM) revealed that, during alkyl phenol oxidation, the structure of MIL-125 collapsed into disordered mesoporous amorphous particles composed of small (ca. 1 nm) dense Ti-based corpuscles connected into globules and surrounded with one or several concentric Ti-containing layers [126]. Bulk elemental analysis and EDX spectra showed reduction of carbon content in MIL-125 after the TMP oxidation, indicating that leaching of the organic linker rather than Ti occurs.
In spite of the structural changes, the MIL-125-derived catalyst could be recovered by simple filtration and reused without loss of the yield of the target quinone (Figure 9a). No catalyst regeneration was required before reuse. Catalytic activity expressed in TOF values decreased after the first use but then remained stable, which implies that the catalyst transformation occurs during the first operation cycle.
On the contrary, no decrease in the CyH oxidation rate was observed during recycling of the same MOF, MIL-125 (Figure 9b). This is consistent with the absence of metal or linker leaching and preservation of the MOF structure and morphology, which was confirmed by SEM, N2 adsorption, XRD and FTIR techniques [116]. Therefore, the difference in the reaction conditions employed for the alkylphenol oxidation (0.4 M H2O2, 80 °C) and cyclohexene oxidation (0.1 M H2O2, 50 °C) was critical for the catalyst stability.
Blockage of MOF micropores or active sites by the reaction products, including water that is introduced with H2O2 and additionally generated during the oxidation process, is most often the reason for fast catalyst deactivation in the liquid-phase oxidations, leading to incomplete substrate conversions. Nevertheless, the development of effective methods for catalyst regeneration may result in a good recycling performance, which in turn, partially compensates the relatively low conversions achieved during one operation cycle and increases the catalyst productivity. Kinetic studies with product additives and/or studies on adsorption of products/reagents help to elucidate the reasons for catalyst deactivation and suggest a regeneration procedure. Thus UiO-66 could be easily recycled without significant loss of catalytic properties, provided proper catalyst regeneration was employed before reuse [117,130].
Figure 10a shows how the addition of oxidation products, epoxide and water, retarded the overall CyH oxidation process over UiO-66, thereby indicating that adsorption of both products could be the reason for the catalyst deactivation. Indeed, washing of the spent UiO-66 with hot methanol and then acetone followed by drying at room temperature allowed the catalyst to restore its activity (Figure 10b).
In the case of PG oxidation over UiO-66, where the catalyst was deactivated by adsorption of the main by-product acetic acid, complete regeneration and recycling without loss of activity was possible if the catalyst was thoroughly washed with acetone before reuse [130].
3. Conclusions and Outlook
Less than 20 years have passed since the first demonstrations of the application of MOFs in liquid-phase oxidation catalysis. During these years various synthetic methodologies for the synthesis of chemically and thermally robust MOFs and engineering of catalytically active sites in their structure have been developed, which have opened up new opportunities for use of this class of materials in catalysis and, in particular, in selective oxidations with the green oxidant aqueous hydrogen peroxide. In this short review, we tried to survey recent developments in this field based, first of all, on our own research experience.
So far, oxidation of thioethers and thiophenes into corresponding sulfoxides and sulfones remains the most investigated oxidative transformation accomplished using various MOFs and H2O2. We demonstrated here that the choice of metal in the MOF nodes completely determines the selectivity of these oxidations, leading to either formation of sulfoxide or sulfone depending on the H2O2 activation mechanism. While Zn(II)-, Cr(III)- and Ti(IV)-based MOFs fulfill electrophilic activation of H2O2 and produce sulfoxides as the major oxidation products, Zr(IV)-based MOFs realize nucleophilic activation of this oxidant and afford sulfones even at low substrate conversions. This characteristic feature of Zr-MOFs is related to the presence of weak basic sites in their structure and also enables epoxidation of electron-deficient C=C bonds in α,β-unsaturated carbonyl compounds. Therefore, one might expect that other MOFs having weak basic centers would show this type of oxidative transformations. The superior catalytic performance of ZIF-8 in the epoxidation of menadione clearly confirmed this suggestion.
The selective H2O2-based epoxidation of unfunctionalized alkenes, especially those with highly reactive allylic H atoms, still remains a challenging goal for oxidation catalysis with MOFs because most of them possess significant activity in homolytic activation of hydrogen peroxide favoring the formation of allylic oxidation products rather than epoxides. The discovery of a simple tool that greatly improves heterolytic pathway selectivity without evident changes in the MOF crystalline structure, namely, the in situ addition of a source of protons, created a basis for the rational design and exploitation of MOF-based catalyst systems in selective oxidation. Importantly, this simple approach also allows one to control unproductive decomposition of the oxidant, thereby improving such important characteristic as oxidant utilization efficiency.
Homolytic oxidation routes may become useful for the selective oxidation of a range of organic substrates, e.g., phenols, alcohols (in some cases), alkanes, and some others. However, specific sorption properties favoring adsorption of both substrate and oxidant molecules in a close proximity to each other and the structure of active sites become crucial factors governing both catalyst activity and selectivity. The cluster-based structures of MOFs may provide advantages for realization of a sequence of one-electron transformations leading to the target oxidation products. Adsorption and mechanistic studies are indispensable in the way to designing new efficient MOF-based catalyst systems.
The development of new synthetic approaches for post-synthetic modification of MOFs may expand significantly the capabilities of catalyst selectivity control and create new opportunities for MOF applications in the field of oxidation catalysis, in particular, selective oxidations with H2O2.
One of the main problems that arise in the field of the oxidation catalysis with MOFs is catalyst deactivation during the catalytic process leading to incomplete substrate conversions. Most often, the reason for that is adsorption of the reaction products, including organic by-products and water, the amount of which always increases if we use H2O2 as the oxidant. Kinetic and adsorption studies help to understand the specific reason for catalyst deactivation and suggest regeneration procedure. Although good recycling performance may partially compensate incomplete substrate conversions attained during one operation cycle, the development of larger pores hydrophobic MOFs would, probably, help to increase catalyst productivity.
With the discovery of MOFs having high chemical and thermal stability a great progress has been achieved in solving the problem of MOF destruction and metal leaching during catalytic oxidations with H2O2. However, as we demonstrated here, some restrictions may exist with regards to the operation conditions (concentrations, temperature, and/or reaction time). Consequently, even robust MOFs should be used with a caution. At the moment, it is not always easy to decide which type of catalysts is superior in terms of activity and stability because direct comparative studies are still a rare case, while the reaction conditions employed may differ considerably. Moreover, samples of the same MOF quite often reveal very different activity and stability, depending on the specific method of their synthesis that may strongly affect the particle size, amount of defects, and other characteristics. Therefore, exchange of MOF samples between teams is very important for elucidation of structure/activity relationships.
So far, synthetic chemists working in the field of MOFs certainly outnumber researchers engaged in oxidation catalysis by MOFs, and the catalytic potential of many new MOFs still remains unexplored. Even fewer teams are immersed in a detailed elucidation of the mechanisms of oxidation reactions occurring on MOFs, although this is necessary for the rational design of catalytic centers and optimization of their operating conditions. Collaborative efforts of specialists from different fields would certainly lead to a rapid progress in this area.
Author Contributions
Writing-review and editing, O.K.; writing-review and editing, N.M. All authors have read and agreed to the published version of the manuscript.
Funding
This work was partially supported by the Ministry of Science and Higher Education of the Russian Federation within the governmental order for Boreskov Institute of Catalysis (project AAAA-A21-121011390008-4) and the Russian Foundation for Basic Research (grant № 18-29-04022).
Acknowledgments
The authors thank all co-authors of the joint papers published on oxidation catalysis by MOFs and cited in this Perspective. Our special thanks to Jong-San Chang (Korea Research Institute of Chemical Technology) for his fruitful collaboration in the field of MOFs.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Sheldon, R.; Arends, I.W.C.E.; Hanefeld, U. Green Chemistry and Catalysis; Wiley VCH: Weinheim, Germany, 2007. [Google Scholar]
- Liquid Phase Oxidation via Heterogeneous Catalysis: Organic Synthesis and Industrial Applications; Clerici, M.G.; Kholdeeva, O.A. (Eds.) Wiley: Hoboken, NJ, USA, 2013. [Google Scholar]
- Handbook of Advanced Methods and Processes in Oxidation Catalysis; Duprez, D.; Cavani, F. (Eds.) Imperial College Press: London, UK, 2014. [Google Scholar]
- Jones, C.W. Application of Hydrogen Peroxide and Derivatives; Royal Society of Chemistry: Cambridge, UK, 1999. [Google Scholar]
- Catalytic Oxidations with Hydrogen Peroxide as Oxidant; Strukul, G. (Ed.) Kluwer: Dordrecht, The Netherlands, 1992. [Google Scholar]
- Campos-Martin, J.M.; Blanco-Brieva, G.; Fierro, J.L.G. Hydrogen peroxide synthesis: An outlook beyond the anthraquinone process. Angew. Chem. Int. Ed. 2006, 45, 6962–6984. [Google Scholar] [CrossRef]
- Edwards, J.K.; Freakley, S.J.; Lewis, R.J.; Pritchard, J.C.; Hutchings, G.J. Advances in the direct synthesis of hydrogen peroxide from hydrogen and oxygen. Catal. Today 2015, 248, 3–9. [Google Scholar] [CrossRef]
- Menegazzo, F.; Signoretto, M.; Ghedini, E.; Strukul, G. Looking for the “dream catalyst” for hydrogen peroxide production from hydrogen and oxygen. Catalysts 2019, 9, 251. [Google Scholar] [CrossRef] [Green Version]
- Functional Metal-Organic Frameworks: Gas Storage, Separation and Catalysis; Schröder, M. (Ed.) Springer: Heidelberg, Germany, 2010. [Google Scholar]
- Metal–Organic Frameworks as Heterogeneous Catalysts; Xamena, F.L.I.; Gascon, J. (Eds.) RCS Publishing: Cambridge, UK, 2013. [Google Scholar]
- Metal-Organic Frameworks: Applications in Separations and Catalysis; García, H.; Navalón, S. (Eds.) Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2018. [Google Scholar]
- Elaboration and Applications of Metal-Organic Frameworks, Series on Chemistry, Energy and the Environment; Ma, S.; Perman, J.A. (Eds.) World Scientific Publishing Co. Pte. Ltd.: Singapore, 2018; Volume 2. [Google Scholar]
- Levason, B.; Bradshaw, D. Special Issue on Chemistry and Applications of Metal Organic Frameworks. Coord. Chem Rev. 2016, 307, 105–424. [Google Scholar] [CrossRef]
- Dietzel, P.; Kitagawa, H. Cluster Issue “Metal–Organic Frameworks Heading towards Application”. Eur. J. Inorg. Chem. 2016, 2016, 4265–4529. [Google Scholar]
- Batten, S.R.; Chen, B.; Vittal, J.J. Special Issue on Coordination Polymers/MOFs. ChemPlusChem 2016, 81, 666–898. [Google Scholar]
- Maurin, G.; Serre, C.; Cooper, A.; Férey, G. Themed issue Metal-organic frameworks and porous polymers—Current and future challenges. Chem. Soc. Rev. 2017, 46, 3104–3107. [Google Scholar] [CrossRef]
- Moon, H.R.; Lim, D.-W.; Suh, M.P. Fabrication of metal nanoparticles in metal–organic frameworks. Chem. Soc. Rev. 2013, 42, 1807–1824. [Google Scholar] [CrossRef]
- Silva, P.; Vilela, S.M.F.; Tomé, J.P.C.; Paz, F.A.A. Multifunctional metal–organic frameworks: From academia to industrial applications. Chem. Soc. Rev. 2015, 44, 6774–6803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, Y.; Li, B.; He, H.; Zhou, W.; Chen, B.; Qian, G. Metal–organic frameworks as platforms for functional materials. Acc. Chem. Res. 2016, 49, 483–493. [Google Scholar] [CrossRef]
- Yang, Q.; Xu, Q.; Jiang, H.-L. Metal–organic frameworks meet metal nanoparticles: Synergistic effect for enhanced catalysis. Chem. Soc. Rev. 2017, 46, 4774–4808. [Google Scholar] [CrossRef] [PubMed]
- Islamoglu, T.; Chen, Z.; Wasson, M.C.; Buru, C.T.; Kirlikovali, K.O.; Afrin, U.; Mian, M.R.; Farha, O.K. Metal−organic frameworks against toxic chemicals. Chem. Rev. 2020, 120, 8130–8160. [Google Scholar]
- Ryu, U.J.; Jee, S.; Rao, P.C.; Shin, J.; Ko, C.; Yoon, M.; Park, K.S.; Choi, K.M. Recent advances in process engineering and upcoming applications of metal–organic frameworks. Coord. Chem. Rev. 2021, 426, 213544. [Google Scholar] [CrossRef]
- Hwang, Y.K.; Férey, G.; Lee, U.-H.; Chang, J.-S. Liquid phase oxidation of organic compounds by metal-organic frameworks. In Liquid Phase Oxidation via Heterogeneous Catalysis: Organic Synthesis and Industrial Applications; Clerici, M.G., Kholdeeva, O.A., Eds.; Wiley: Hoboken, NJ, USA, 2013; pp. 371–409. [Google Scholar]
- Maksimchuk, N.V.; Zalomaeva, O.V.; Skobelev, I.Y.; Kovalenko, K.A.; Fedin, V.P.; Kholdeeva, O.A. Metal-organic frameworks of the MIL-101 family as heterogeneous single-site catalysts. Proc. R. Soc. A 2012, 468, 2017–2034. [Google Scholar] [CrossRef]
- Valvekens, P.; Vermoortele, F.; De Vos, D. Metal–organic frameworks as catalysts: The role of metal active sites. Catal. Sci. Technol. 2013, 3, 1435–1445. [Google Scholar] [CrossRef]
- Zhao, M.; Ou, S.; Wu, C.-D. Porous Metal–organic frameworks for heterogeneous biomimetic catalysis. Acc. Chem. Res. 2014, 47, 1199–1207. [Google Scholar] [CrossRef]
- Gascon, J.; Corma, A.; Kapteijn, F.; Llabrés i Xamena, F.X. Metal organic framework catalysis: Quo vadis? ACS Catal. 2014, 4, 361–378. [Google Scholar] [CrossRef]
- Liu, J.; Chen, L.; Cui, H.; Zhang, J.; Zhang, L.; Su, C.-Y. Applications of metal–organic frameworks in heterogeneous supramolecular catalysis. Chem. Soc. Rev. 2014, 43, 6011–6061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gu, Z.-Y.; Park, J.; Raiff, A.; Wei, Z.; Zhou, H.-C. Metal–organic frameworks as biomimetic catalysts. ChemCatChem 2014, 6, 67–75. [Google Scholar] [CrossRef]
- Leus, K.; Liu, Y.-Y.; Van Der Voort, P. Metal-organic frameworks as selective or chiral oxidation catalysts. Catal. Rev. 2014, 56, 1–56. [Google Scholar] [CrossRef] [Green Version]
- Chughtai, A.H.; Ahmad, N.; Younus, H.A.; Laypkov, A.; Verpoort, F. Metal–organic frameworks: Versatile heterogeneous catalysts for efficient catalytic organic transformations. Chem. Soc. Rev. 2015, 44, 6804–6849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhakshinamoorthy, A.; Asiri, A.M.; García, H. Metal–organic frameworks as catalysts for oxidation reactions. Chem. Eur. J. 2016, 22, 8012–8024. [Google Scholar] [CrossRef]
- Kholdeeva, O.A. Liquid-phase selective oxidation catalysis with metal-organic frameworks. Catal. Today 2016, 278, 22–29. [Google Scholar] [CrossRef]
- Hu, Z.; Zhao, D. Metal–organic frameworks with Lewis acidity: Synthesis, characterization, and catalytic applications. CrystEngComm 2017, 19, 4066–4081. [Google Scholar] [CrossRef]
- Rogge, S.M.J.; Bavykina, A.; Hajek, J.; Garcia, H.; Olivos-Suarez, A.I.; Sepúlveda-Escribano, A.; Vimont, A.; Clet, G.; Bazin, P.; Kapteijn, F.; et al. Metal–organic and covalent organic frameworks as single-site catalysts. Chem. Soc. Rev. 2017, 46, 3134–3184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, W.; Thapa, K.B.; Ju, Q.; Fang, Z.; Huang, W. Heterogeneous catalysts based on mesoporous metal–organic frameworks. Coord. Chem. Rev. 2018, 373, 199–232. [Google Scholar] [CrossRef]
- Dhakshinamoorthy, A.; Asiric, A.M.; Herance, J.R.; García, H. Metal organic frameworks as solid promoters for aerobic autoxidations. Catal. Today 2018, 306, 2–8. [Google Scholar] [CrossRef]
- Dhakshinamoorthy, A.; Li, Z.; García, H. Catalysis and photocatalysis by metal organic frameworks. Chem. Soc. Rev. 2018, 47, 8134–8172. [Google Scholar] [CrossRef] [PubMed]
- Qin, J.-S.; Yuan, S.; Lollar, C.; Pang, J.; Alsalme, A.; Zhou, H.-C. Stable metal–organic frameworks as a host platform for catalysis and biomimetics. Chem. Commun. 2018, 54, 4231–4249. [Google Scholar] [CrossRef] [PubMed]
- Yang, D.; Gates, B.C. Catalysis by Metal Organic Frameworks: Perspective and Suggestions for Future Research. ACS Catal. 2019, 9, 1779–1798. [Google Scholar] [CrossRef]
- Liu, M.; Wu, J.; Hou, H. Metal–organic framework (MOF)-based materials as heterogeneous catalysts for C-H Bond activation. Chem. Eur. J. 2019, 25, 2935–2948. [Google Scholar] [CrossRef]
- Kang, Y.-S.; Lu, Y.; Chen, K.; Zhao, Y.; Wang, P.; Sun, W.-Y. Metal–organic frameworks with catalytic centers: From synthesis to catalytic application. Coord. Chem. Rev. 2019, 378, 262–280. [Google Scholar] [CrossRef]
- Bavykina, A.; Kolobov, N.; Khan, I.S.; Bau, J.A.; Ramirez, A.; Gascon, J. Metal−organic frameworks in heterogeneous catalysis: Recent progress, new trends, and future perspectives. Chem. Rev. 2020, 120, 8468–8535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, Y.; Dou, Y.; Xie, L.-H.; Rutledge, W.; Li, J.-R.; Zhou, H.-C. Zr-Based metal–organic frameworks: Design, synthesis, structure, and applications. Chem. Soc. Rev. 2016, 45, 2327–2367. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Feng, L.; Wang, K.; Pang, J.; Bosch, M.; Lollar, C.; Sun, Y.; Qin, J.; Yang, X.; Zhang, P.; et al. Stable Metal–Organic Frameworks: Design, Synthesis, and Applications. Adv. Mater. 2018, 30, 1704303. [Google Scholar]
- Du, D.Y.; Qin, J.S.; Li, S.L.; Su, Z.M.; Lan, Y.Q. Recent advances in porous polyoxometalate based metal–organic framework materials. Chem. Soc. Rev. 2014, 43, 4615–4632. [Google Scholar] [CrossRef] [Green Version]
- Kholdeeva, O.A. Recent progress in selective oxidations with hydrogen peroxide catalyzed by polyoxometalates. In Frontiers of Green Catalytic Selective Oxidations; Bryliakov, K.P., Ed.; Springer Nature Singapore Pte Ltd.: Singapore, 2019; pp. 61–92. [Google Scholar]
- Ferey, G.; Mellot-Draznieks, C.; Serre, C.; Millange, F.; Dutour, J.; Surble, S.; Margiolaki, I. A Chromium terephthalate-based solid with unusually large pore volumes and surface area. Science 2005, 309, 2040–2042. [Google Scholar] [CrossRef]
- Dan-Hardi, M.; Serre, C.; Frot, T.; Rozes, L.; Maurin, G.; Sanchez, C.; Ferey, G. A new photoactive crystalline highly porous titanium (IV) dicarboxylate. J. Am. Chem. Soc. 2009, 131, 10857–10859. [Google Scholar] [CrossRef]
- Shearer, G.C.; Chavan, S.; Bordiga, S.; Svelle, S.; Olsbye, U.; Lillerud, K.P. Defect engineering: Tuning the porosity and composition of the metal−organic framework UiO-66 via modulated synthesis. Chem. Mater. 2016, 28, 3749–3761. [Google Scholar] [CrossRef]
- DeCoste, J.B.; Peterson, G.W.; Jasuja, H.; Glover, T.G.; Huang, Y.-g.; Walton, K.S. Stability and degradation mechanisms of metal–organic frameworks containing the Zr6O4(OH)4 secondary building unit. J. Mater. Chem. A 2013, 1, 5642–5650. [Google Scholar] [CrossRef]
- Cavka, J.H.; Jakobsen, S.; Olsbye, U.; Guillou, N.; Lamberti, C.; Bordiga, S.; Lillerud, K.P. A New zirconium inorganic building brick forming metal organic frameworks with exceptional stability. J. Am. Chem. Soc. 2008, 130, 13850–13851. [Google Scholar] [CrossRef]
- Wißmann, G.; Schaate, A.; Lilienthal, S.; Bremer, I.; Schneider, A.M.; Behrens, P. Modulated synthesis of Zr-fumarate MOF. Micropor. Mesopor. Mater. 2012, 152, 64–70. [Google Scholar] [CrossRef]
- Wang, H.; Dong, X.; Lin, J.; Teat, S.J.; Jensen, S.; Cure, J.; Alexandrov, E.V.; Xia, Q.; Tan, K.; Wang, Q.; et al. Topologically guided tuning of Zr-MOF pore structures for highly selective separation of C6 alkane isomers. Nat. Commun. 2018, 9, 1745. [Google Scholar] [CrossRef]
- Wang, S.; Lee, J.S.; Wahiduzzaman, M.; Park, J.; Muschi, M.; Martineau-Corcos, C.; Tissot, A.; Cho, K.H.; Marrot, J.; Shepard, W.; et al. A Robust large–pore zirconium carboxylate metal–organic framework for energy-efficient water–sorption–driven refrigeration. Nat. Energy 2018, 3, 985–993. [Google Scholar] [CrossRef]
- Huang, X.-C.; Lin, Y.-Y.; Zhang, J.-P.; Chen, X.-M. Ligand-directed strategy for zeolite-type metal–organic frameworks: Zinc(II) imidazolates with unusual zeolitic topologies. Angew. Chem. Int. Ed. 2006, 45, 1557–1559. [Google Scholar] [CrossRef] [PubMed]
- Park, K.S.; Ni, Z.; Côté, A.P.; Choi, J.Y.; Huang, R.; Uribe-Romo, F.J.; Chae, H.K.; O’Keeffe, M.; Yaghi, O.M. Exceptional chemical and thermal stability of zeolitic imidazolate frameworks. Proc. Natl. Acad. Sci. USA 2006, 103, 10186–10191. [Google Scholar] [CrossRef] [Green Version]
- Di Furia, F.; Modena, G. Mechanism of oxygen transfer from peroxo species. Pure Appl. Chem. 1982, 54, 1853–1866. [Google Scholar] [CrossRef]
- Bonchio, M.; Campestrini, S.; Conte, V.; Di Furia, F.; Moro, S. A Theoretical and experimental investigation of the electrophilic oxidation of thioethers and sulfoxides by peroxides. Tetrahedron 1995, 51, 12363–12372. [Google Scholar] [CrossRef]
- Gomez-Lor, B.; Gutiérrez-Puebla, E.; Iglesias, M.; Monge, M.A.; Ruiz-Valero, C.; Snejko, N. In2(OH)3(BDC)1.5 (BDC) = 1,4-Benzendicarboxylate): An In(III) supramolecular 3d framework with catalytic activity. Inorg. Chem. 2002, 41, 2428–2432. [Google Scholar] [CrossRef]
- Perles, J.; Iglesias, M.; Martin-Luengo, M.-A.; Monge, M.A.; Ruiz-Valero, C.; Snejko, N. Metal-organic scandium framework: Useful material for hydrogen storage and catalysis. Chem. Mater. 2005, 17, 5837–5842. [Google Scholar] [CrossRef]
- Gándara, F.; de Andrés, A.; Gómez-Lor, B.; Gutiérrez-Puebla, E.; Iglesias, M.; Monge, M.A.; Proserpio, D.M.; Snejko, N. Rare-earth MOF series: Fascinating structure, efficient light emitters, and promising catalyst. Cryst. Growth Des. 2008, 8, 378–380. [Google Scholar] [CrossRef]
- Gándara, F.; Gutierrez Puebla, E.; Iglesias, M.; Proserpio, D.M.; Snejko, N.; Angeles, M.M. Controlling the structure of arenedisulfonates toward catalytically active materials. Chem. Mater. 2009, 21, 655–661. [Google Scholar] [CrossRef]
- Perles, J.; Snejko, N.; Iglesias, M.; Monge, M.A. 3D scandium and yttrium arenedisulfonate MOF materials as highly thermally stable bifunctional heterogeneous catalysts. J. Mater. Chem. 2009, 19, 6504–6511. [Google Scholar] [CrossRef]
- Dybtsev, D.N.; Nuzhdin, A.L.; Chun, H.; Bryliakov, K.P.; Talsi, E.P.; Fedin, V.P.; Kim, K. A Homochiral metal–organic material with permanent porosity, enantioselective sorption properties, and catalytic activity. Angew. Chem. Int. Ed. 2006, 45, 916–920. [Google Scholar] [CrossRef]
- Hwang, Y.K.; Hong, D.-Y.; Chang, J.-S.; Seo, H.; Yoon, M.; Kim, J.; Jhung, S.H.; Serre, C.; Férey, G. Selective sulfoxidation of aryl sulfides by coordinatively unsaturated metal centers in chromium carboxylate MIL-101. Appl. Catal. A Gen. 2009, 358, 249–253. [Google Scholar] [CrossRef]
- Maksimchuk, N.V.; Kovalenko, K.A.; Fedin, V.P.; Kholdeeva, O.A. Heterogeneous selective oxidation of alkenes to α,β-unsaturated ketones over coordination polymer MIL-101. Adv. Synth. Catal. 2010, 352, 2943–2948. [Google Scholar] [CrossRef]
- Maksimchuk, N.V.; Kovalenko, K.A.; Fedin, V.P.; Kholdeeva, O.A. Cyclohexane selective oxidation over metal–organic frameworks of MIL-101 family: Superior catalytic activity and selectivity. Chem. Commun. 2012, 48, 6812–6814. [Google Scholar] [CrossRef] [PubMed]
- Campos-Martin, J.M.; Capel-Sanchez, M.C.; Perez-Presas, P.; Fierro, J.L.G. Oxidative processes of desulfurization of liquid fuels. J. Chem. Technol. Biotechnol. 2010, 85, 879–890. [Google Scholar] [CrossRef] [Green Version]
- Zlotea, C.; Phanon, D.; Mazaj, M.; Heurtaux, D.; Guillerm, V.; Serre, C.; Horcajada, P.; Devic, T.; Magnier, E.; Cuevas, F.; et al. Effect of NH2 and CF3 functionalization on the hydrogen sorption properties of MOFs. Dalton Trans. 2011, 40, 4879–4881. [Google Scholar] [CrossRef]
- Bhadra, B.N.; Song, J.Y.; Khan, N.A.; Jhung, S.H. TiO2-containing carbon derived from a metal–organic framework composite: A highly active catalyst for oxidative desulfurization. ACS Appl. Mater. Interfaces 2017, 9, 31192–31202. [Google Scholar] [CrossRef]
- Zhang, Y.; Lia, G.; Kong, L.; Lu, H. Deep oxidative desulfurization catalyzed by Ti-based metal-organic frameworks. Fuel 2018, 219, 103–110. [Google Scholar] [CrossRef]
- Mondloch, J.E.; Bury, W.; Fairen-Jimenez, D.; Kwon, S.; DeMarco, E.J.; Weston, M.H.; Sarjeant, A.A.; Nguyen, S.T.; Stair, P.C.; Snurr, R.Q.; et al. Vapor-phase metalation by atomic layer deposition in a metal–organic framework. J. Am. Chem. Soc. 2013, 135, 10294–10297. [Google Scholar] [CrossRef] [PubMed]
- Devic, T.; Serre, C. High Valence 3p and transition metal based MOFs. Chem. Soc. Rev. 2014, 43, 6097–6115. [Google Scholar] [CrossRef]
- Howarth, A.J.; Liu, Y.; Li, P.; Li, Z.; Wang, T.C.; Hupp, J.T.; Farha, O.K. Chemical, thermal and mechanical stabilities of metal–organic frameworks. Nat. Rev. Mater. 2016, 1, 15018. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, X.; Johnson, J.A.; Chen, Y.-S.; Zhang, J. Highly porous zirconium metal−organic frameworks with β-UH3-like topology based on elongated tetrahedral linkers. J. Am. Chem. Soc. 2016, 138, 8380–8383. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhakshinamoorthy, A.; Santiago-Portillo, A.; Asiri, A.M.; Garcia, H. Engineering UiO-66 metal organic framework for heterogeneous catalysis. ChemCatChem 2019, 11, 899–923. [Google Scholar] [CrossRef]
- Granadeiro, C.M.; Ribeiro, S.O.; Karmaoui, M.; Valenca, R.; Ribeiro, J.C.; de Castro, B.; Cunha-Silva, L.; Balula, S.S. Production of ultra–deep sulfur–free diesels using a sustainable catalytic system based on UiO-66(Zr). Chem. Commun. 2015, 51, 13818–13821. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Huang, P.; Liu, A.; Zhu, M. A Metal–Organic Framework for Oxidative Desulfurization: UIO-66(Zr) as a Catalyst. Fuel 2017, 209, 417–423. [Google Scholar] [CrossRef]
- Ye, G.; Zhang, D.; Li, X.; Leng, K.; Zhang, W.; Ma, J.; Sun, Y.; Xu, W.; Ma, S. Boosting catalytic performance of metal–organic framework by increasing the defects via a facile and green approach. ACS Appl. Mater. Interfaces 2017, 9, 34937–34943. [Google Scholar] [CrossRef]
- Xiao, W.; Dong, Q.; Wang, Y.; Li, Y.; Deng, S.; Zhang, N. Time modulation of defects in UiO-66 and application in oxidative desulfurization. CrystEngComm 2018, 20, 5658–5662. [Google Scholar] [CrossRef]
- Ye, G.; Qi, H.; Zhou, W.; Xu, W.; Sun, Y. Green and scalable synthesis of nitro- and amino-functionalized UiO-66(Zr) and the effect of functional groups on the oxidative desulfurization performance. Inorg. Chem. Front. 2019, 6, 1267–1274. [Google Scholar] [CrossRef]
- Viana, A.M.; Ribeiro, S.O.; de Castro, B.; Balula, S.S.; Cunha-Silva, L. Influence of UiO-66(Zr) Preparation strategies in its catalytic efficiency for desulfurization process. Materials 2019, 12, 3009. [Google Scholar] [CrossRef] [Green Version]
- Zheng, H.-Q.; Zeng, Y.-N.; Chen, J.; Lin, R.-G.; Zhuang, W.-E.; Cao, R.; Lin, Z.-J. Zr-Based metal–organic frameworks with intrinsic peroxidase-like activity for ultradeep oxidative desulfurization: Mechanism of H2O2 decomposition. Inorg. Chem. 2019, 58, 6983–6992. [Google Scholar] [CrossRef]
- Gu, Y.; Xu, W.; Sun, Y. Enhancement of catalytic performance over MOF-808(Zr) by acid treatment for oxidative desulfurization of dibenzothiophene. Catal. Today 2020. [Google Scholar] [CrossRef]
- Ye, G.; Qi, H.; Li, X.; Leng, K.; Sun, Y.; Xu, W. Enhancement of oxidative desulfurization performance over UiO-66(Zr) by titanium ion exchange. ChemPhysChem 2017, 18, 1903–1908. [Google Scholar] [CrossRef]
- Piscopo, C.G.; Voellinger, L.; Schwarzer, M.; Polyzoidis, A.; Bošković, D.; Loebbecke, S. Continuous flow desulfurization of a model fuel catalysed by titanium functionalized UiO-66ю. Chem. Sel. 2019, 4, 2806–2809. [Google Scholar]
- Limvorapitux, R.; Chen, H.; Mendonca, M.L.; Liu, M.; Snurr, R.Q.; Nguyen, S.B.T. Elucidating the mechanism of the UiO-66−catalyzed sulfide oxidation: Activity and selectivity enhancements through changes in the node coordination environment and solvent. Catal. Sci. Technol. 2019, 9, 327–335. [Google Scholar] [CrossRef]
- Trickett, C.A.; Gagnon, K.J.; Lee, S.; Gándara, F.; Bürgi, H.-B.; Yaghi, O.M. Definitive molecular level characterization of defects in UiO-66 crystals. Angew. Chem. Int. Ed. 2015, 54, 11162–11167. [Google Scholar] [CrossRef] [PubMed]
- Ling, S.; Slater, B. Dynamic acidity in defective UiO-66. Chem. Sci. 2016, 7, 4706–4712. [Google Scholar] [CrossRef] [Green Version]
- Zalomaeva, O.V.; Evtushok, V.Y.; Ivanchikova, I.D.; Glazneva, T.S.; Chesalov, Y.A.; Larionov, K.P.; Kholdeeva, O.A. Nucleophilic vs. electrophilic activation of hydrogen peroxide over Zr-based metal-organic frameworks. Inorg. Chem. 2020, 59, 10634–10649. [Google Scholar] [CrossRef]
- Maksimchuk, N.V.; Ivanchikova, I.D.; Cho, K.H.; Zalomaeva, O.V.; Evtushok, V.Y.; Larionov, K.P.; Glazneva, T.S.; Chang, J.-S.; Kholdeeva, O.A. Catalytic performance of Zr-based metal-organic frameworks Zr-abtc and MIP-200 in selective oxidations with H2O2. Chem. Eur. J. 2021. [Google Scholar] [CrossRef]
- Zalomaeeva, O.V. Boreskov Institute of Catalysis, Novosibirsk, Russia, Unpublished Results.
- Gall, R.D.; Faraj, M.; Hill, C.L. Role of water in polyoxometalate-catalyzed oxidations in nonaqueous media. Scope, kinetics, and mechanism of oxidation of thioether mustard (HD) analogs by tert-butyl hydroperoxide catalyzed by H5PV2Mo10040. Inorg. Chem. 1994, 33, 5015–5021. [Google Scholar] [CrossRef]
- Adam, W.; Haas, W.; Sieker, G. Thianthrene 5-oxide as mechanistic probe in oxygen-transfer reactions: The case of carbonyl oxides vs. dioxiranes. J. Am. Chem. Soc. 1984, 106, 5020–5022. [Google Scholar] [CrossRef]
- Herrmann, W.A.; Fischer, R.W.; Correia, J.D.G. Multiple bonds between main-group elements and transition-metals. Part 133. Methyltrioxorhenium as a catalyst of the Baeyer-Villiger oxidation. J. Mol. Catal. 1994, 94, 213–223. [Google Scholar] [CrossRef]
- Ballistreri, F.P.; Tomaselli, G.A.; Toscano, R.M.; Conte, V.; Di Furia, F. Application of the thianthrene 5-oxide mechanistic probe to peroxometal complexes. J. Am. Chem. Soc. 1991, 113, 6209–6212. [Google Scholar] [CrossRef]
- Caratelli, C.; Hajek, J.; Cirujano, F.G.; Waroquier, M.; Llabrés i Xamena, F.X.; Van Speybroeck, V. Nature of active sites on UiO-66 and beneficial influence of water in the catalysis of Fischer esterification. J. Catal. 2017, 352, 401–414. [Google Scholar] [CrossRef] [Green Version]
- Caratelli, C.; Hajek, J.; Rogge, S.M.J.; Vandenbrande, S.; Meijer, E.J.; Waroquier, M.; Van Speybroeck, V. Influence of a confined methanol solvent on the reactivity of active sites in UiO-66. ChemPhysChem 2018, 19, 420–429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Payne, G.B.; Deming, P.H.; Williams, P.H. Reactions of hydrogen peroxide. VII. Alkali-Catalyzed epoxidation and oxidation using a nitrile as co-reactant. J. Org. Chem. 1961, 26, 659–663. [Google Scholar] [CrossRef]
- Mak, K.K.W.; Lai, Y.M.; Siu, Y.-H. Regiospecific epoxidation of carvone: A Discovery-oriented experiment for understanding the selectivity and mechanism of epoxidation reactions. J. Chem. Educ. 2006, 83, 1058–1061. [Google Scholar] [CrossRef]
- Ivanchikova, I.D.; Evtushok, V.Y.; Zalomaeva, O.V.; Kolokolov, D.I.; Stepanov, A.G.; Kholdeeva, O.A. Heterogeneous epoxidation of menadione with hydrogen peroxide over zeolite imidazolate framework ZIF-8. Dalton Trans. 2020, 49, 12546–12549. [Google Scholar] [CrossRef]
- Peralta, D.; Chaplais, G.; Paillaud, J.L.; Simon-Masseron, A.; Barthelet, K.; Pirngruber, G.D. The separation of xylene isomers by ZIF-8: A demonstration of the extraordinary flexibility of the ZIF-8 framework. Micropor. Mesopor. Mater. 2013, 173, 1–5. [Google Scholar] [CrossRef]
- Kolokolov, D.I.; Diestel, L.; Caro, J.; Freude, D.; Stepanov, A.G. Rotational and translational motion of benzene in ZIF-8 studied by 2H NMR: Estimation of microscopic self-diffusivity and its Comparison with macroscopic measurements. J. Phys. Chem. C 2014, 118, 12873–12879. [Google Scholar] [CrossRef]
- Khudozhitkov, A.E.; Arzumanov, S.S.; Kolokolov, D.I.; Stepanov, A.G. Mobility of aromatic guests and isobutane in ZIF-8 metal–organic framework studied by 2H solid state NMR spectroscopy. J. Phys. Chem. C 2019, 123, 13765–13774. [Google Scholar] [CrossRef]
- Chizallet, C.; Lazare, S.; Bazer-Bachi, D.; Bonnier, F.; Lecocq, V.; Soyer, E.; Quoineaud, A.-A.; Bats, N. Catalysis of transesterification by a nonfunctionalized metal−organic Framework: Acido-basicity at the external surface of ZIF-8 probed by FTIR and ab initio calculations. J. Am. Chem. Soc. 2010, 132, 12365–12377. [Google Scholar] [CrossRef]
- Nguyen, H.G.T.; Mao, L.; Peters, A.W.; Audu, C.O.; Brown, Z.J.O.; Farha, K.; Hupp, J.T.; Nguyen, S.B.T. Comparative study of titanium-functionalized UiO-66: Support effect on the oxidation of cyclohexene using hydrogen peroxide. Catal. Sci. Technol. 2015, 5, 4444–4451. [Google Scholar] [CrossRef]
- Ahn, S.; Thornburg, N.E.; Li, Z.; Wang, T.C.; Gallington, L.C.; Chapman, K.W.; Notestein, J.M.; Hupp, J.T.; Farha, O.K. Stable metal−organic framework-supported niobium catalysts. Inorg. Chem. 2016, 55, 11954–11961. [Google Scholar] [CrossRef] [PubMed]
- Kholdeeva, O.A.; Maksimov, G.M.; Maksimovskaya, R.I.; Kovaleva, L.A.; Fedotov, M.A. Role of protons in methyl phenyl sulfide oxidation with hydrogen peroxide catalyzed by Ti (IV)-monosubstituted heteropolytungstates. React. Kinet. Catal. Lett. 1999, 66, 311–317. [Google Scholar] [CrossRef]
- Kholdeeva, O.A.; Trubitsina, T.A.; Timofeeva, M.N.; Maksimov, G.M.; Maksimovskaya, R.I.; Rogov, V.A. The role of protons in cyclohexene oxidation with H2O2 catalysed by Ti (IV)-monosubstituted Keggin polyoxometalate. J. Mol. Catal. A Chem. 2005, 232, 173–178. [Google Scholar] [CrossRef]
- Kholdeeva, O.A.; Maksimovskaya, R.I. Titanium- and zirconium-monosubstituted polyoxometalates as molecular models for studying mechanisms of oxidation catalysis. J. Mol. Catal. A Chem. 2007, 262, 7–24. [Google Scholar] [CrossRef]
- Jiménez-Lozano, P.; Ivanchikova, I.D.; Kholdeeva, O.A.; Poblet, J.M.; Carbó, J.J. Alkene oxidation by Ti-containing polyoxometalates. Unambiguous characterization of the role of the protonation state. Chem. Commun. 2012, 48, 9266–9268. [Google Scholar] [CrossRef]
- Kholdeeva, O.A. Hydrogen peroxide activation over TiIV: What have we learned from studies on Ti-containing polyoxometalates? Eur. J. Inorg. Chem. 2013, 2013, 1595–1605. [Google Scholar] [CrossRef]
- Maksimchuk, N.V.; Maksimov, G.M.; Evtushok, V.Y.; Ivanchikova, I.D.; Chesalov, Y.A.; Maksimovskaya, R.I.; Kholdeeva, O.A.; Solé-Daura, A.; Poblet, J.M.; Carbó, J.J. Relevance of protons in heterolytic activation of H2O2 over Nb(V): Insights from model studies on nb-substituted polyoxometalates. ACS Catal. 2018, 8, 9722–9737. [Google Scholar] [CrossRef]
- Maksimchuk, N.V.; Ivanchikova, I.D.; Maksimov, G.M.; Eltsov, I.V.; Evtushok, V.Y.; Kholdeeva, O.A.; Lebbie, D.; Errington, R.J.; Solé-Daura, A.; Poblet, J.M.; et al. Why does Nb(V) show higher heterolytic pathway selectivity than Ti(IV) in epoxidation with H2O2? Answers from model studies on Nb- and Ti-substituted Lindqvist tungstates. ACS Catal. 2019, 9, 6262–6275. [Google Scholar] [CrossRef]
- Maksimchuk, N.; Lee, J.S.; Ayupov, A.; Chang, J.-S.; Kholdeeva, O. Cyclohexene oxidation with H2O2 over metal-organic framework MIL-125(Ti): The effect of protons on the reactivity. Catalysts 2019, 9, 324. [Google Scholar] [CrossRef] [Green Version]
- Maksimchuk, N.V.; Lee, J.S.; Solovyeva, M.V.; Cho, K.H.; Shmakov, A.N.; Chesalov, Y.A.; Chang, J.-S.; Kholdeeva, O.A. Protons make possible heterolytic activation of hydrogen peroxide over Zr-based metal-organic frameworks. ACS Catal. 2019, 9, 9699–9704. [Google Scholar] [CrossRef]
- Clerici, M.G.; Domine, M.E. Oxidation reactions catalyzed by transitionmetal-substituted zeolites. In Liquid Phase Oxidation via Heterogeneous Catalysis: Organic Synthesis and Industrial Applications; Clerici, M.G., Kholdeeva, O.A., Eds.; Wiley: Hoboke, NJ, USA, 2013; pp. 21–93. [Google Scholar]
- Bordiga, S.; Groppo, E.; Agostini, G.; van Bokhoven, J.A.; Lamberti, C. Reactivity of surface species in heterogeneous catalysts probed by in situ X-ray absorption techniques. Chem. Rev. 2013, 113, 1736–1850. [Google Scholar] [CrossRef] [Green Version]
- Clerici, M.G. The activity of titanium silicalite-1 (TS-1): Some considerations on its origin. Kinet. Catal. 2015, 56, 450–455. [Google Scholar] [CrossRef]
- Kholdeeva, O.A.; Ivanchikova, I.D.; Maksimchuk, N.V.; Skobelev, I.Y. H2O2-based selective epoxidations: Nb-silicates versus Ti-silicates. Catal. Today 2019, 333, 63–70. [Google Scholar] [CrossRef]
- Kholdeeva, O.A.; Ivanchikova, I.D.; Guidotti, M.; Ravasio, N.; Sgobba, M.; Barmatova, M. How to reach 100% selectivity in H2O2-based oxidation of 2,3,6-trimethylphenol to trimethyl-p-benzoquinone over Ti,Si-catalysts. Catal. Today 2009, 141, 330–336. [Google Scholar] [CrossRef]
- Kholdeeva, O.A.; Ivanchikova, I.D.; Guidotti, M.; Pirovano, C.; Ravasio, N.; Barmatova, M.V.; Chesalov, Y.A. Highly selective H2O2-based oxidation of alkylphenols to benzoquinones over silica-supported titanium catalysts: Ti cluster site versus Ti single site. Adv. Synth. Catal. 2009, 351, 1877–1889. [Google Scholar] [CrossRef]
- Kholdeeva, O.A. Recent developments in liquid-phase selective oxidation using environmentally benign oxidants and mesoporous metal silicates. Catal. Sci. Technol. 2014, 4, 1869–1889. [Google Scholar] [CrossRef]
- Ivanchikova, I.D.; Lee, J.S.; Maksimchuk, N.V.; Shmakov, A.N.; Chesalov, Y.A.; Ayupov, A.B.; Hwang, Y.K.; Jun, C.-H.; Chang, J.-S.; Kholdeeva, O.A. Highly selective H2O2-based oxidation of alkylphenols to p-benzoquinones over MIL-125 metal–organic frameworks. Eur. J. Inorg. Chem. 2014, 2014, 132–139. [Google Scholar] [CrossRef]
- Kholdeeva, O.A.; Ivanchikova, I.D.; Maksimchuk, N.V.; Mel’gunov, M.S.; Chang, J.-S.; Guidotti, M.; Shutilov, A.A.; Zaikovskii, V.I. Environmentally benign oxidation of alkylphenols to p-benzoquinones: A comparative study of various Ti-containing catalysts. Top. Catal. 2014, 57, 1377–1384. [Google Scholar] [CrossRef]
- Kato, N.; Hasegawa, M.; Sato, T.; Yoshizawa, A.; Inoue, T.; Mori, W. Microporous dinuclear copper(II) trans-1,4-cyclohexanedicarboxylate: Heterogeneous oxidation catalysis with hydrogen peroxide and X-ray powder structure of peroxo copper(II) intermediate. J. Catal. 2005, 230, 226–236. [Google Scholar] [CrossRef]
- Balu, A.M.; Lin, C.S.K.; Liu, H.; Li, Y.; Vargas, C.; Luque, R. Iron oxide functionalised MIL-101 materials in aqueous phase selective oxidations. Appl. Catal. A Gen. 2013, 455, 261–266. [Google Scholar] [CrossRef]
- Fei, H.; Shin, J.W.; Meng, Y.S.; Adelhardt, M.; Sutter, J.; Meyer, K.; Cohen, S.M. Reusable oxidation catalysis using metal-monocatecholato species in a robust metal−organic framework. J. Am. Chem. Soc. 2014, 136, 4965–4973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Torbina, V.V.; Nedoseykina, N.S.; Ivanchikova, I.D.; Kholdeeva, O.A.; Vodyankina, O.V. Propylene glycol oxidation with hydrogen peroxide over Zr-containing metal–organic framework UiO-66. Catal. Today 2019, 333, 47–53. [Google Scholar] [CrossRef]
- Meilikhov, M.; Yusenko, K.; Fischer, R.A. Turning MIL-53(Al) redox-active by functionalization of the bridging OH-group with 1,1′-ferrocenediyl-dimethylsilane. J. Am. Chem. Soc. 2009, 131, 9644–9645. [Google Scholar] [CrossRef]
- Wang, D.; Wang, M.; Li, Z. Fe-Based metal–organic frameworks for highly selective photocatalytic benzene hydroxylation to phenol. ACS Catal. 2015, 5, 6852–6857. [Google Scholar] [CrossRef]
- Osadchii, D.Y.; Olivos-Suarez, A.I.; Szécsényi, L.G.; Nasalevich, M.A.; Dugulan, I.A.; Crespo, P.S.; Hensen, E.J.M.; Veber, S.L.; Fedin, M.V.; Sankar, G.; et al. Isolated Fe sites in metal organic frameworks catalyze the direct conversion of methane to methanol. ACS Catal. 2018, 8, 5542–5548. [Google Scholar] [CrossRef] [Green Version]
- Sheldon, R.A.; Wallau, M.; Arends, I.W.C.E.; Schuchardt, U. Heterogeneous catalysts for liquid-phase oxidations: Philosophers’ stones or Trojan horses? Acc. Chem. Res. 1998, 31, 485–493. [Google Scholar] [CrossRef]
Figure 1.
Representative structures of metal–organic frameworks (MOFs) with high thermal and chemical stability.
Figure 1.
Representative structures of metal–organic frameworks (MOFs) with high thermal and chemical stability.

Figure 2.
Activation of MIL-101 and thioether oxidation over coordinatively unsaturated sites (CUS).
Figure 2.
Activation of MIL-101 and thioether oxidation over coordinatively unsaturated sites (CUS).

Scheme 1.
Oxidation of thianthrene 5-oxide over UiO-66.

Scheme 2.
Proposed catalytic cycle for thioether oxidation over Zr-MOF defects. Reprinted with permission from [91]. Copyright 2020 American Chemical Society.
Scheme 2.
Proposed catalytic cycle for thioether oxidation over Zr-MOF defects. Reprinted with permission from [91]. Copyright 2020 American Chemical Society.

Figure 3.
Plots of 2-methyl-naphthoquinone (MNQ) conversion and epoxide yield vs. time over ZIF-8 with different particle size. Reaction conditions: MNQ 0.1 mmol, H2O2 0.8 mmol, catalyst 3.3 mg, CH3CN 1 mL, and 70 °C.
Figure 3.
Plots of 2-methyl-naphthoquinone (MNQ) conversion and epoxide yield vs. time over ZIF-8 with different particle size. Reaction conditions: MNQ 0.1 mmol, H2O2 0.8 mmol, catalyst 3.3 mg, CH3CN 1 mL, and 70 °C.

Scheme 3.
CyH oxidation products typical of homolytic and heterolytic oxidation pathways.

Figure 4.
Propylene glycol (PG) adsorption on UiO-66 from CH3CN and CH3CN/H2O (7/3 (v/v)).

Figure 5.
Effect of acid on H2O2 decomposition over UiO-66 and MIL-125. Reaction conditions: H2O2 0.4 mmol, catalyst 0.04 mmol of Zr or Ti, HClO4 0.04 mmol (if any), CH3CN 2 mL, 50 °C.
Figure 5.
Effect of acid on H2O2 decomposition over UiO-66 and MIL-125. Reaction conditions: H2O2 0.4 mmol, catalyst 0.04 mmol of Zr or Ti, HClO4 0.04 mmol (if any), CH3CN 2 mL, 50 °C.

Figure 6.
Oxidant utilization efficiency in selective oxidations with 30% H2O2 over MOF catalysts.

Figure 7.
Hot catalyst filtration tests for (a) 2,3,6-trimethylphenol (TMP) oxidation over MIL-125 and (b) CyH oxidation over UiO-66 in the presence of 1 equiv. HClO4.
Figure 7.
Hot catalyst filtration tests for (a) 2,3,6-trimethylphenol (TMP) oxidation over MIL-125 and (b) CyH oxidation over UiO-66 in the presence of 1 equiv. HClO4.

Figure 8.
XRD patterns for (a) MIL-125 and (b) UiO-66: (A) initial, (B) after CyH oxidation (H2O2 0.1 M, 50 °C, 1 equiv. of HClO4) and (C) after TMP oxidation (H2O2 0.4 M, 80 °C).
Figure 8.
XRD patterns for (a) MIL-125 and (b) UiO-66: (A) initial, (B) after CyH oxidation (H2O2 0.1 M, 50 °C, 1 equiv. of HClO4) and (C) after TMP oxidation (H2O2 0.4 M, 80 °C).

Figure 9.
Reuse of MIL-125 in (a) TMP oxidation with H2O2 and (b) CyH oxidation with H2O2 in the presence of 1 equiv. of HClO4.
Figure 9.
Reuse of MIL-125 in (a) TMP oxidation with H2O2 and (b) CyH oxidation with H2O2 in the presence of 1 equiv. of HClO4.

Figure 10.
(a) Effect of reaction products on CyH oxidation over UiO-66 and (b) recycling of UiO-66. Reaction conditions: CyH 0.1 mmol, H2O2 0.1 mmol, 2 mg UiO-66 (7 µmol Zr), HClO4 7 µmol, CH3CN 1 mL, 50 °C.
Figure 10.
(a) Effect of reaction products on CyH oxidation over UiO-66 and (b) recycling of UiO-66. Reaction conditions: CyH 0.1 mmol, H2O2 0.1 mmol, 2 mg UiO-66 (7 µmol Zr), HClO4 7 µmol, CH3CN 1 mL, 50 °C.


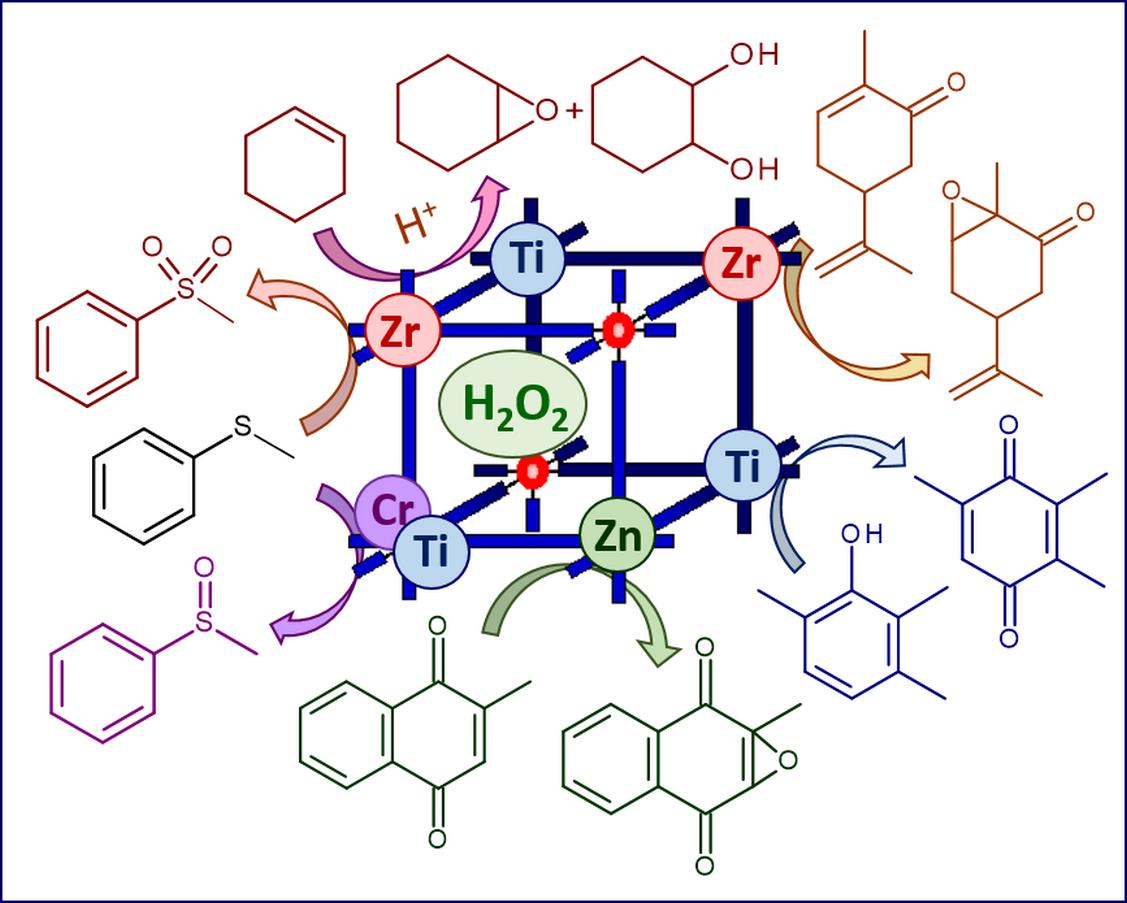{kind=link}
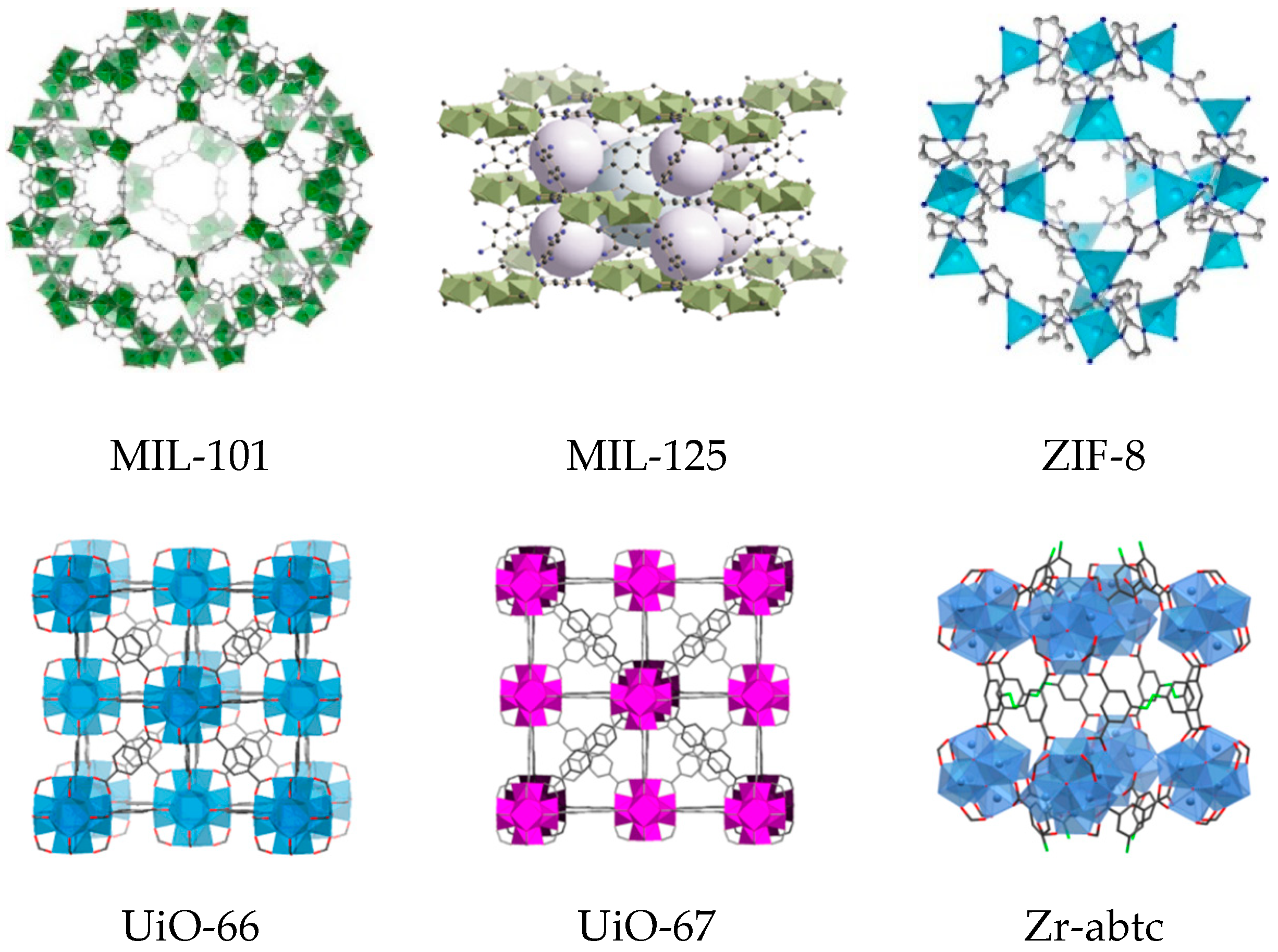{kind=link}
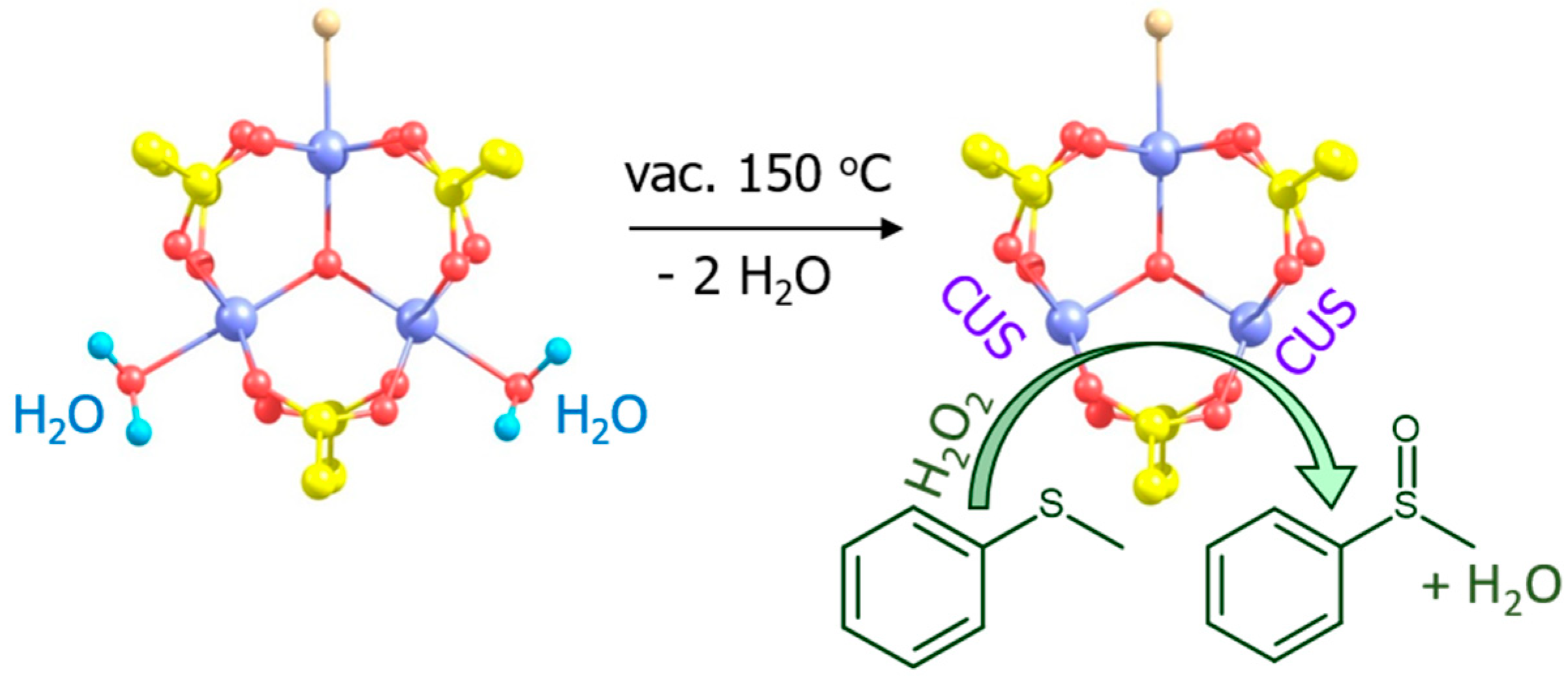{kind=link}
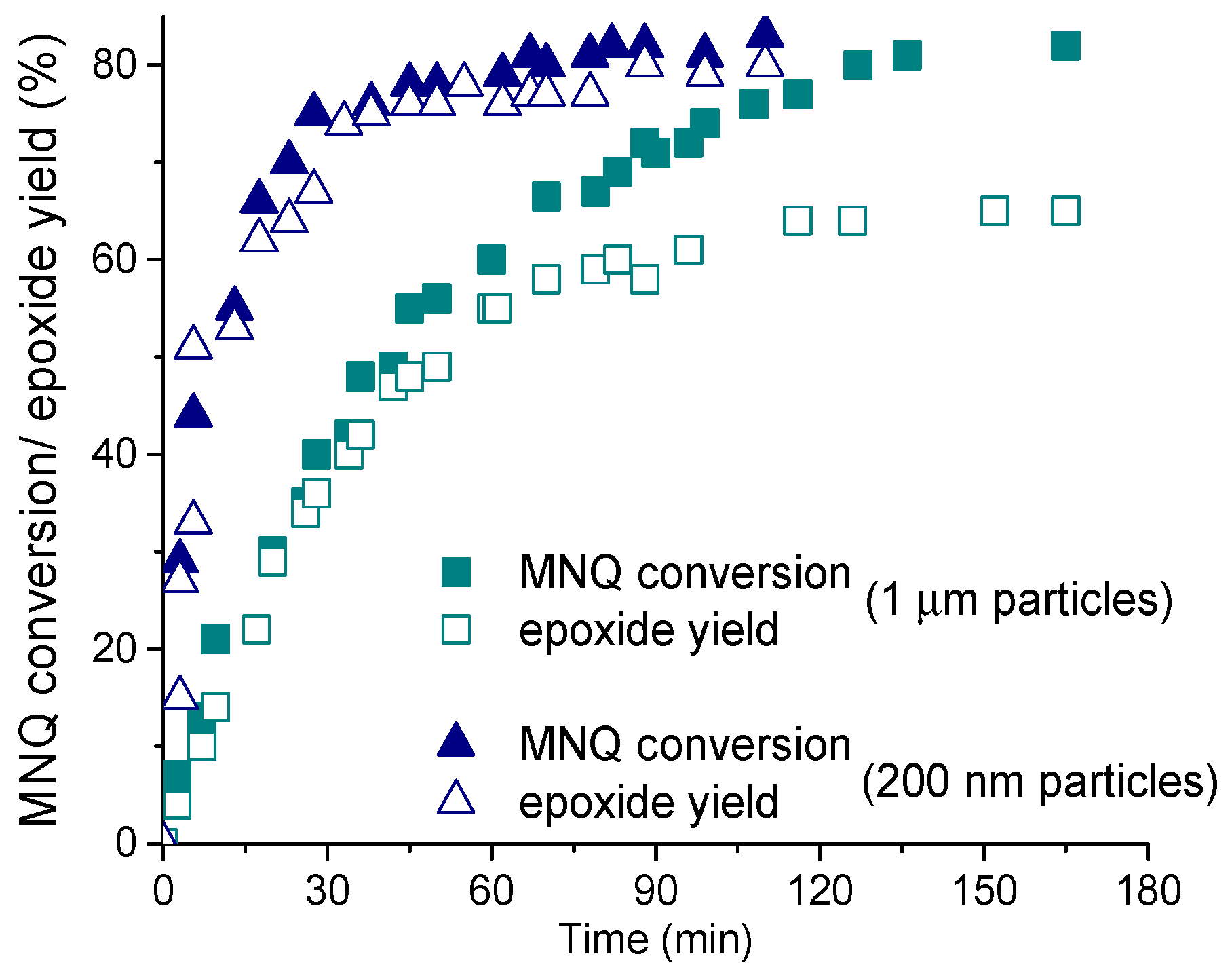{kind=link}
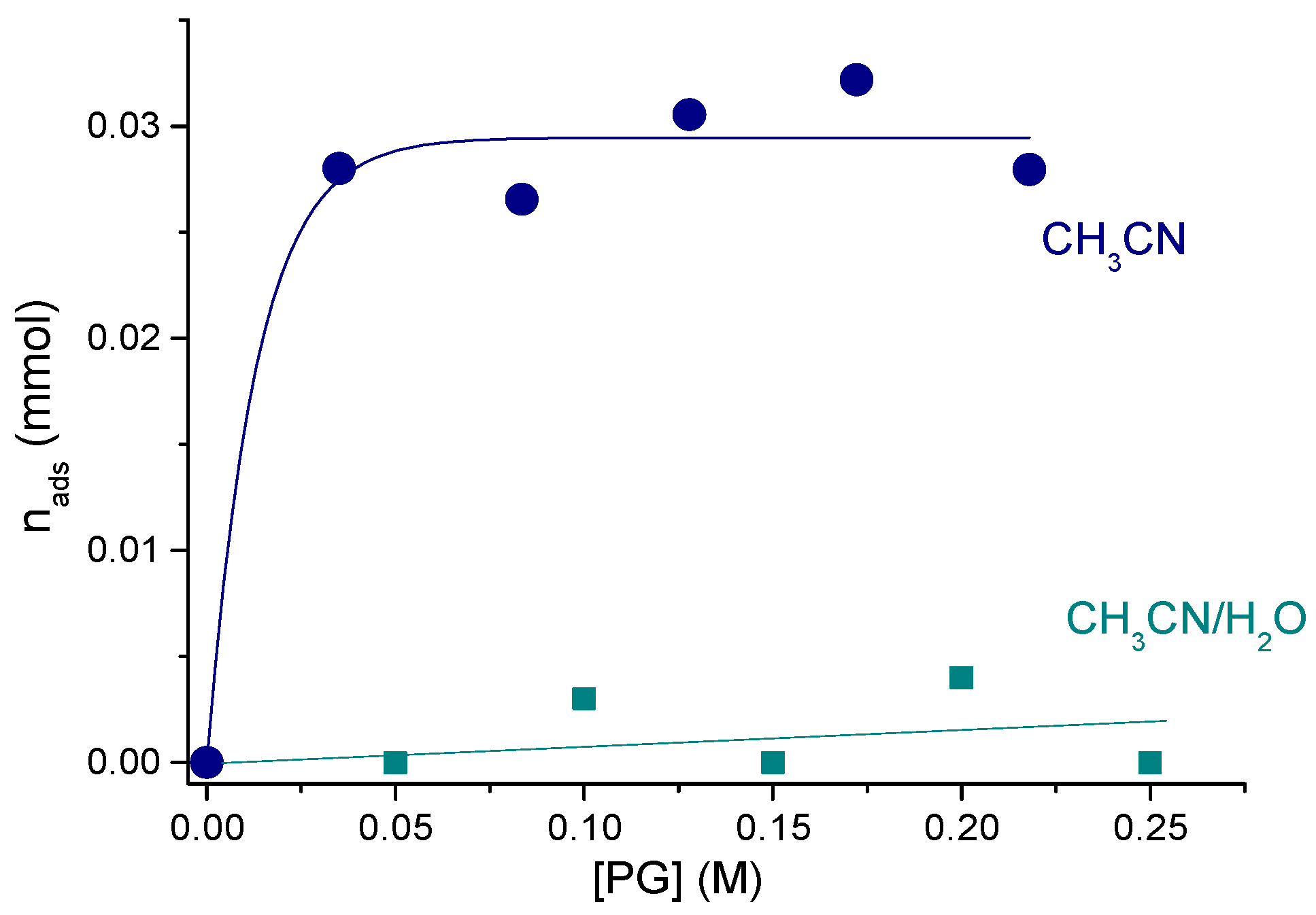{kind=link}
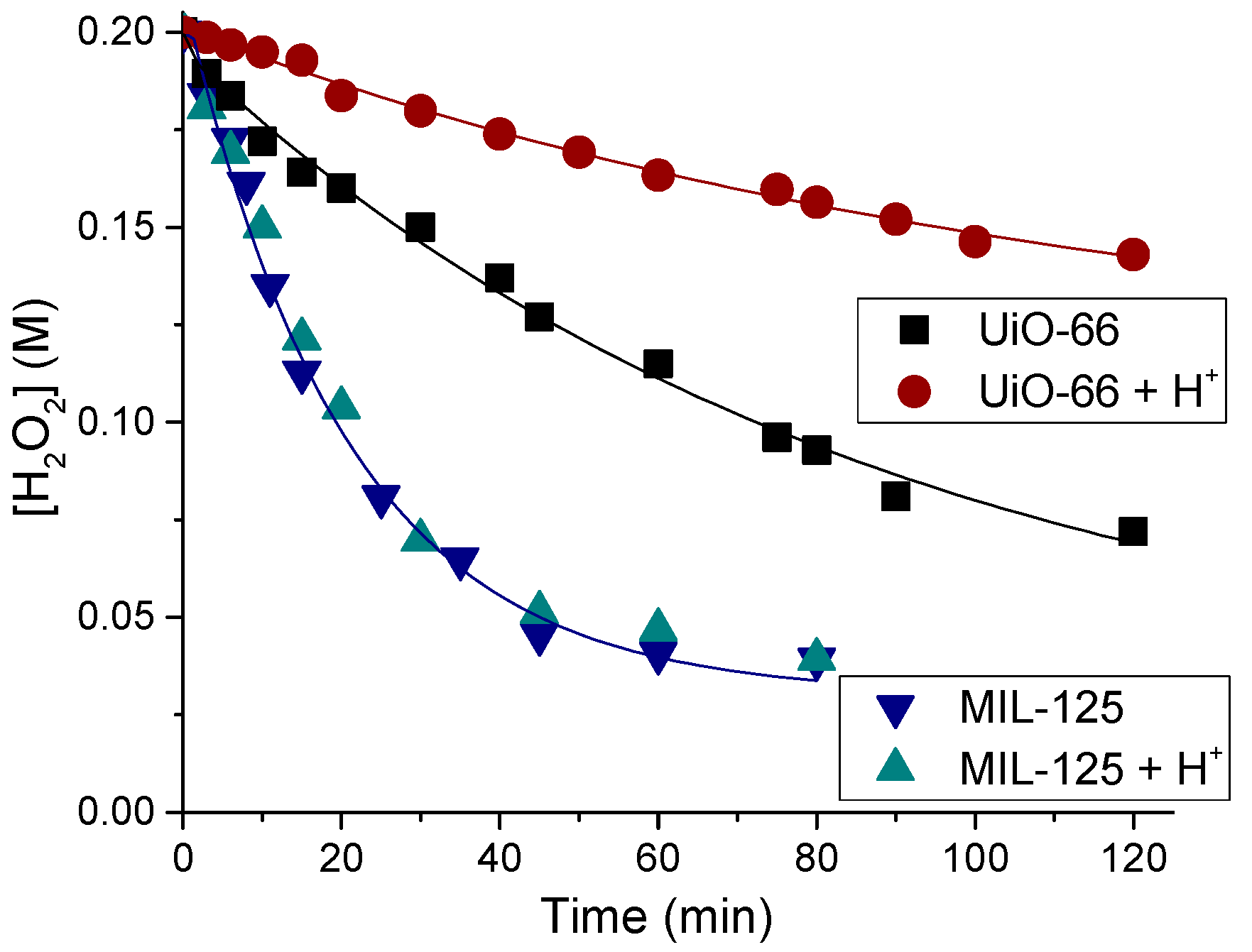{kind=link}
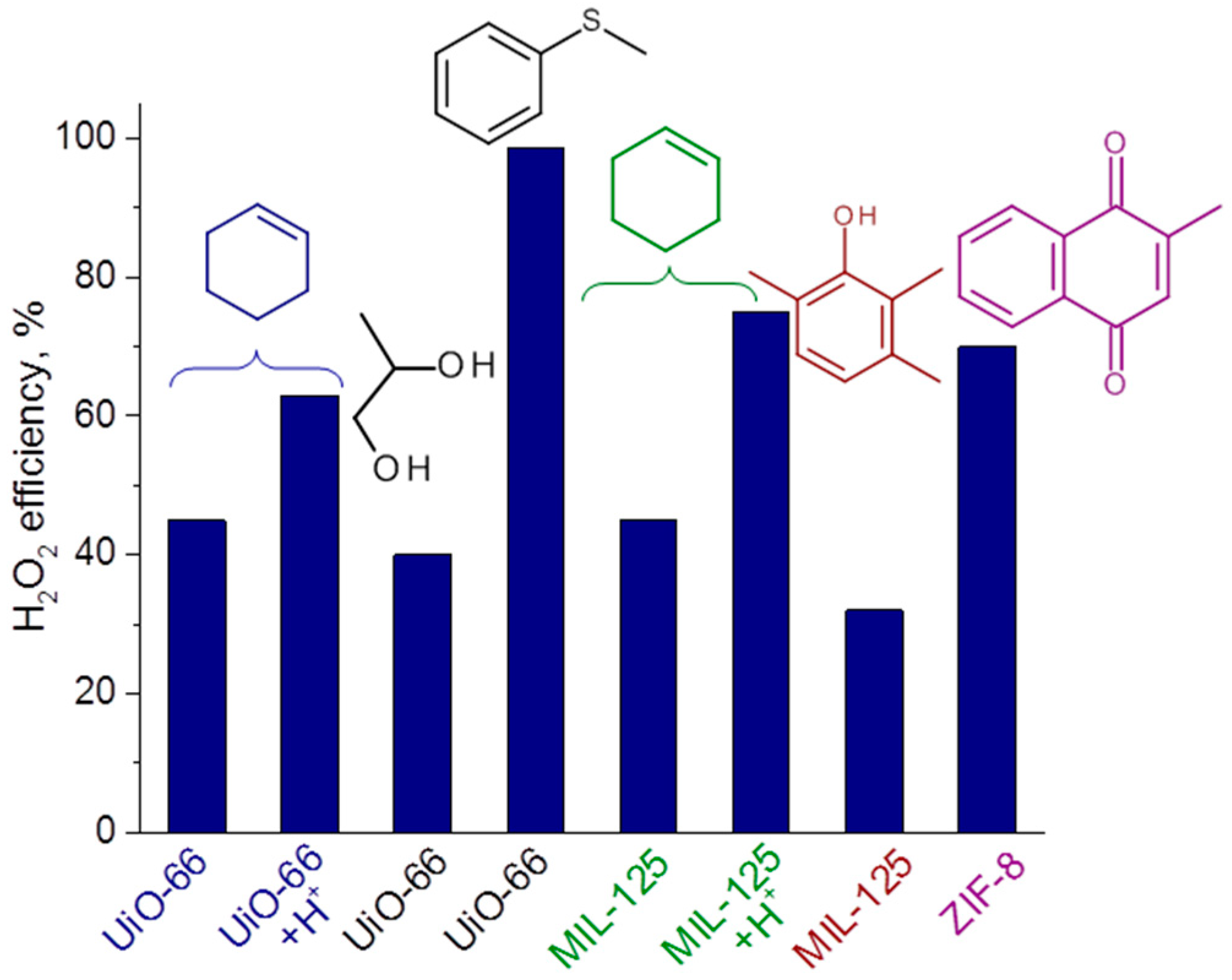{kind=link}
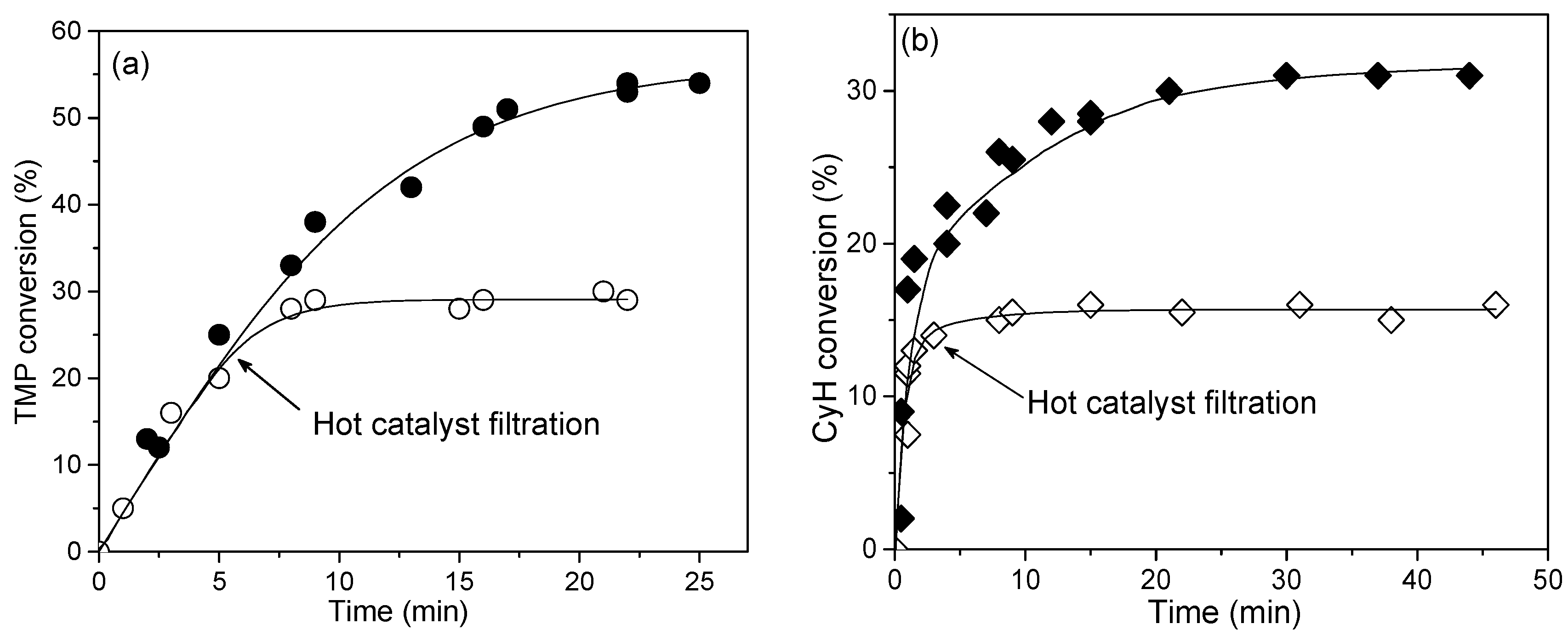{kind=link}
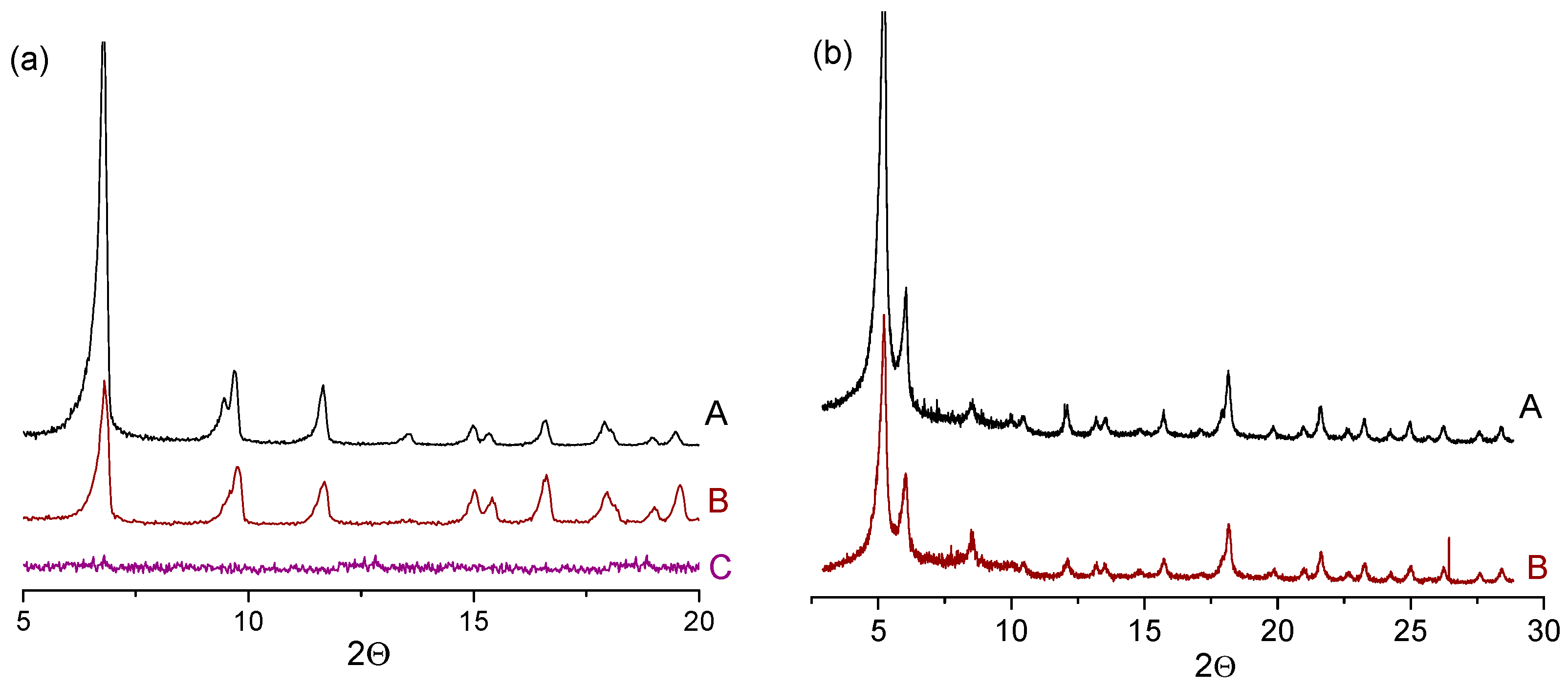{kind=link}
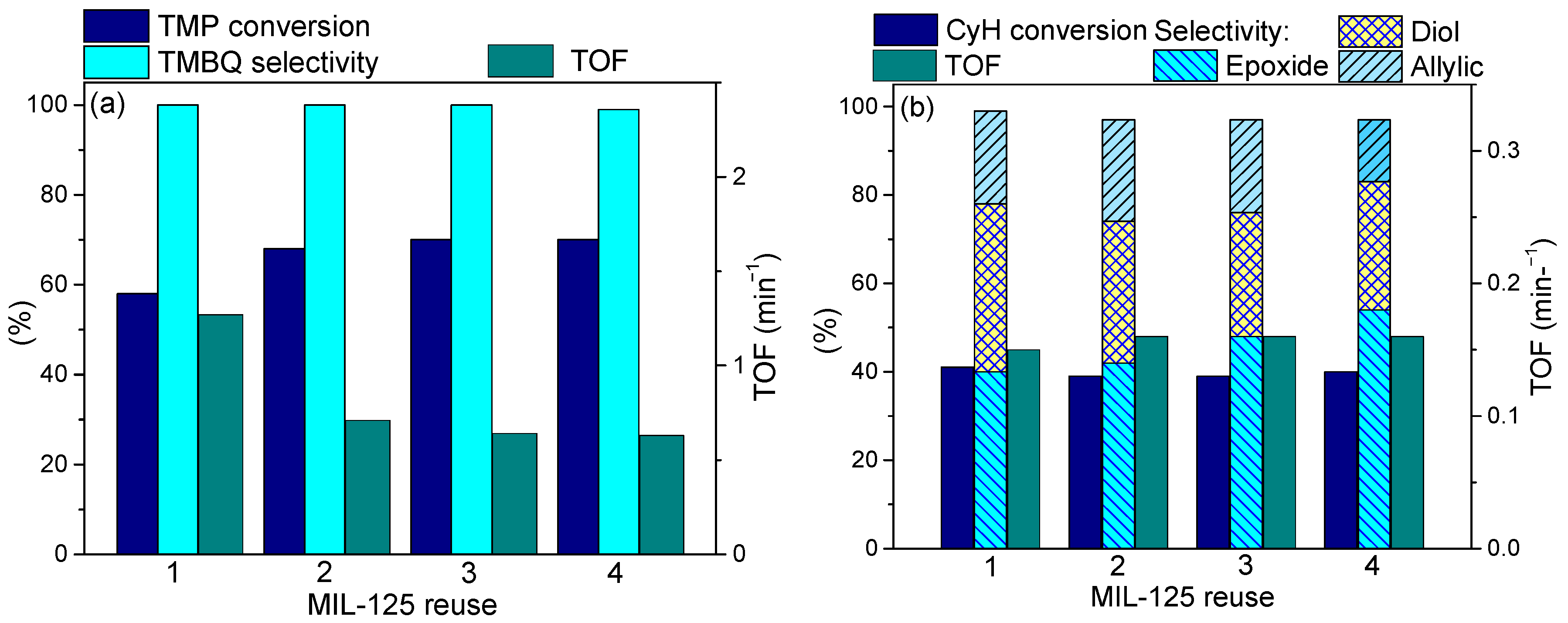{kind=link}
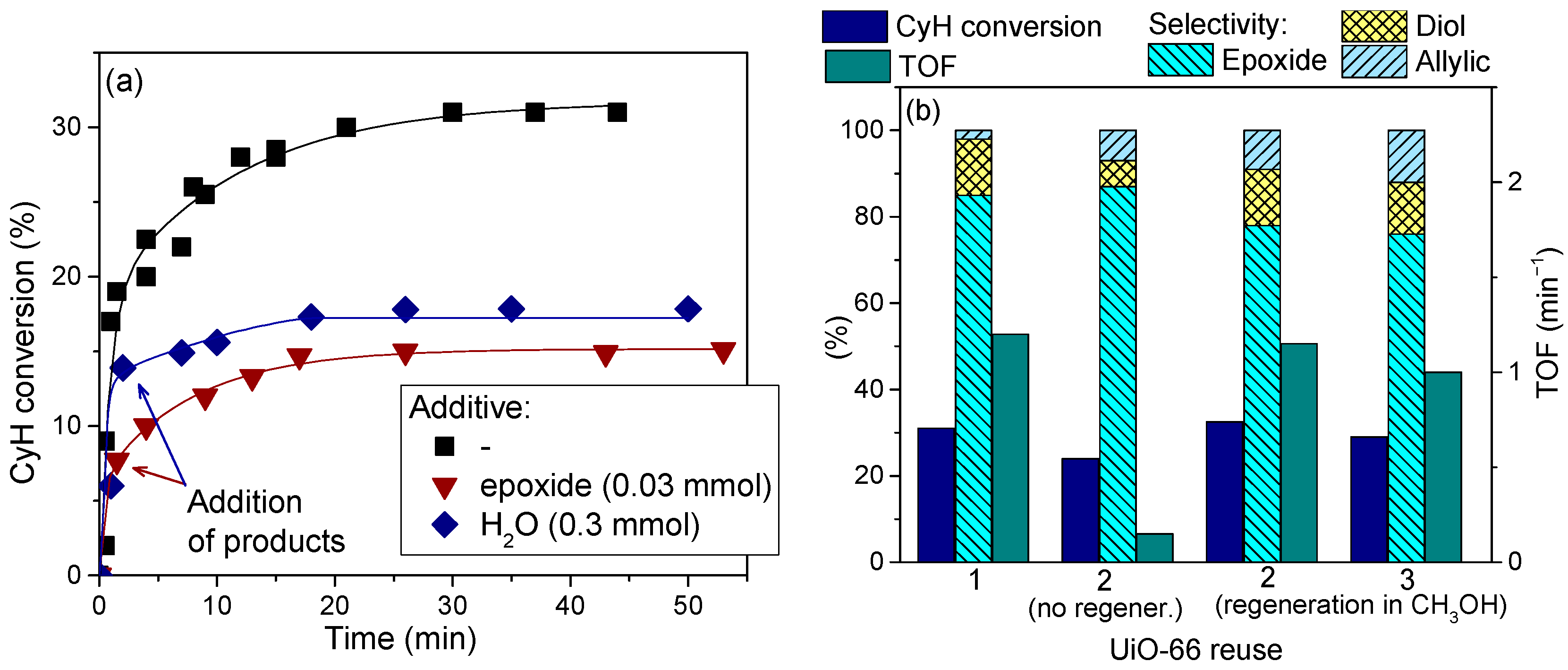{kind=link}
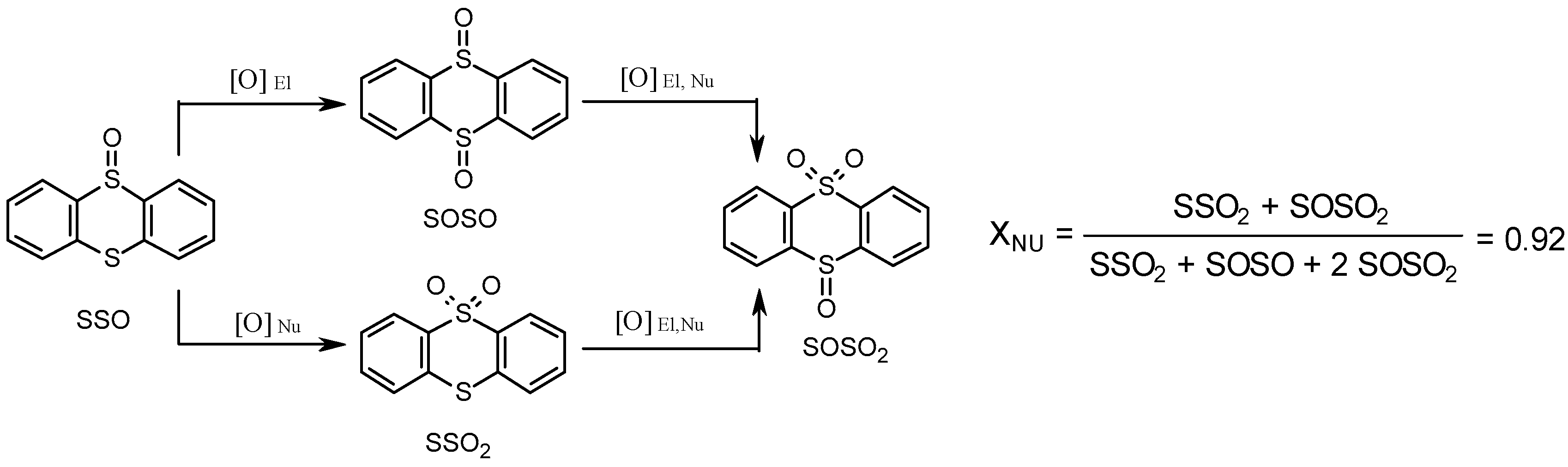{kind=link}
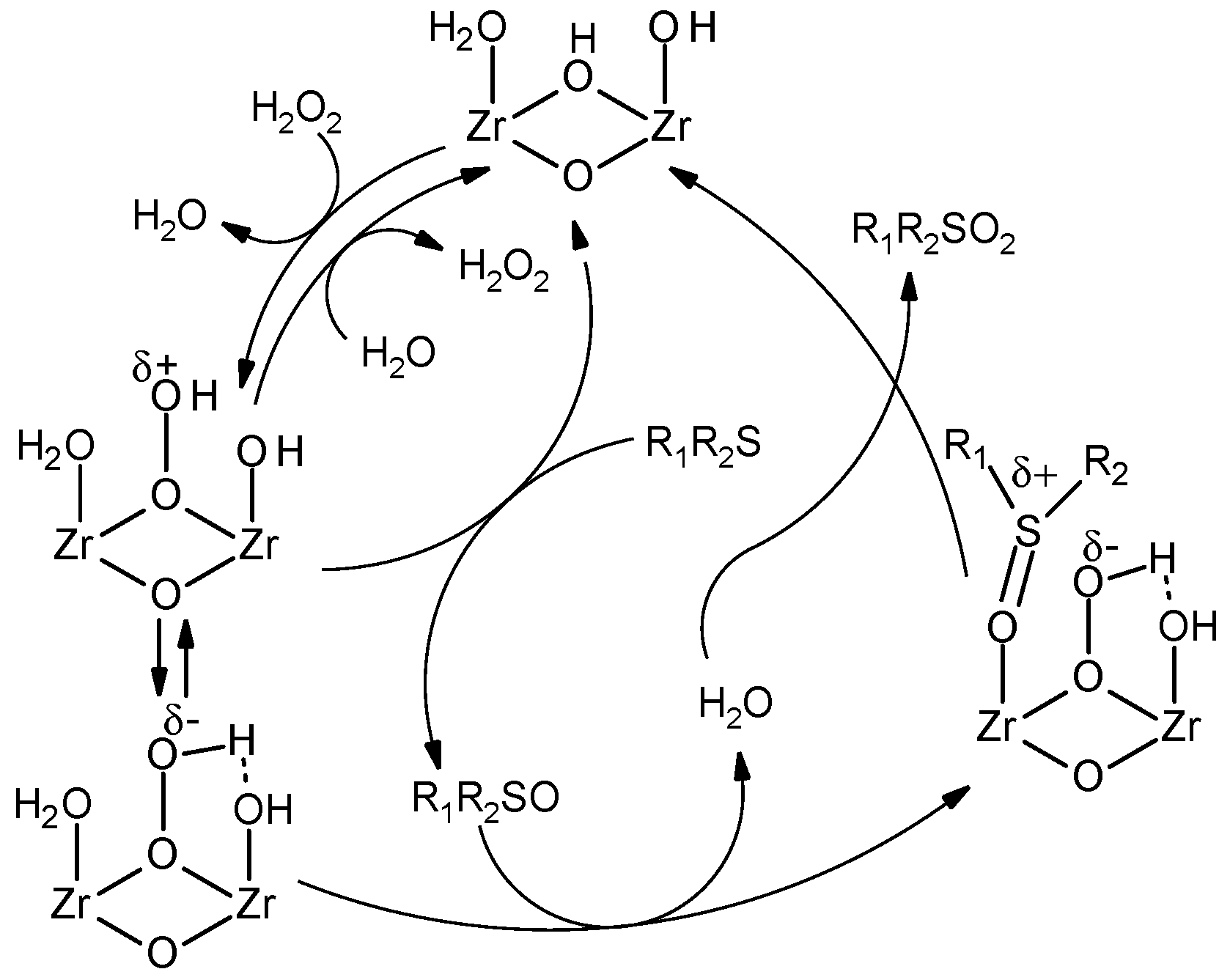{kind=link}
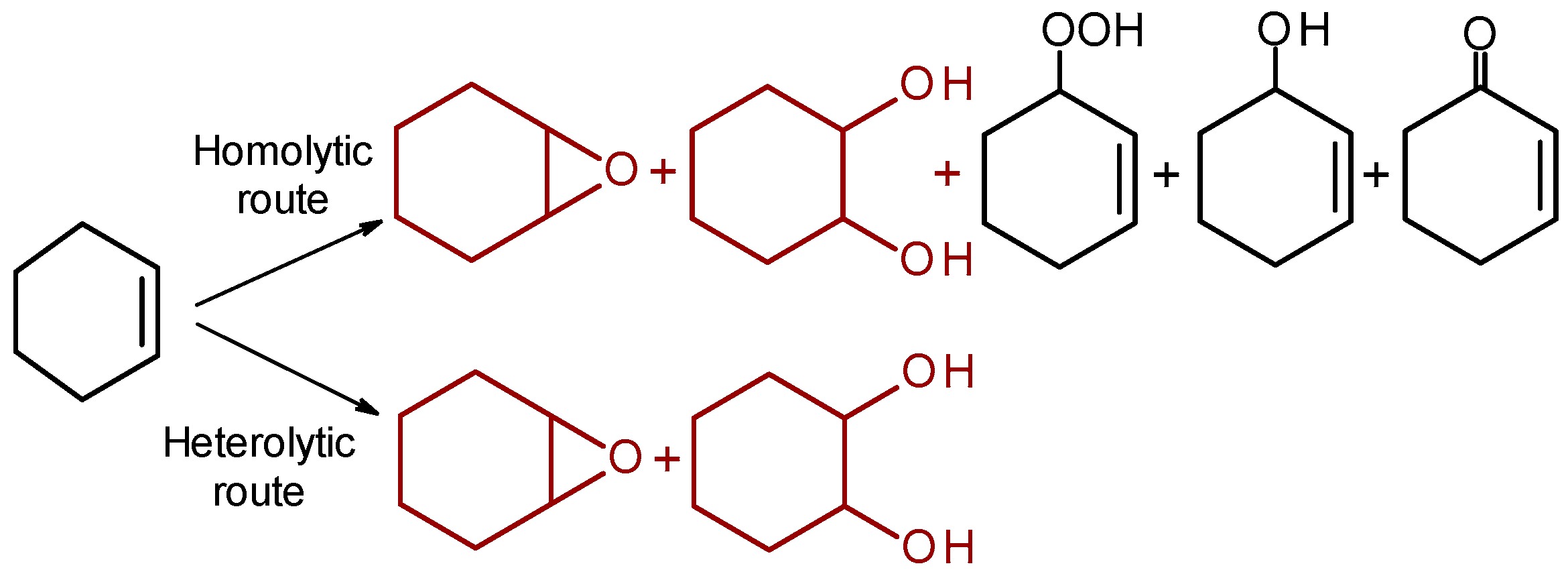{kind=link}
Table 1.
Physicochemical characteristics of some MOFs with high chemical and thermal stability.
| MOF | Molecular Formula a | SBET | dp | Metal Content | Thermal Stability |
|---|---|---|---|---|---|
| (m2/g) | (Å) | (%) a | (oC) b | ||
| MIL-101 | Cr3OX(BDC)3(H2O) | 3200–3900 | 12 and 16 (windows) | 21.7 | 275 [48] |
| (X = F, OH) | 29 and 34 (cages) | ||||
| MIL-125 | Ti8O8(OH)4(BDC)6 | 1500 | 5–7 (windows) | 24.5 | 360 [49] |
| 6 and 12.5 (cages) | |||||
| UiO-66 | Zr6O4(OH)4(BDC)6 | 1200 | 6 (windows) | 32.9 | 520 [50,51] |
| 8 and 11 (cages) | 540 c [52] | ||||
| UiO-67 | Zr6O4(BPDC)6 | 2300 | 8 (windows) | 25.8 | 520 [51] |
| 11.5 and 18 (cages) | 540 c [52] | ||||
| MOF-801 | Zr6O4(OH)4(fum)6 | 900 | 5 (windows) | 40.1 | 400 [53] |
| 5.5 and 7 (cages) | |||||
| Zr-abtc | Zr6O4(OH)4(abtc)2(OH)4(H2O)4 | 1300 | 7 (1D channels) | 39.5 | 450 c [54] |
| MIP-200 | Zr6O4(OH)4(mdip)2(formate)4 | 1000 | 13 and 6.8 (hexagonal and triangular channels) | 35.5 | 500 [55] |
| ZIF-8 | Zn(MeIM)2 | 1300–1800 | 3.4 (windows) | 28.7 | 400 [56] |
| 11.6 (cages) | 550 c [57] |
a For ideal, non-defective MOF; b Under air (O2) atmosphere; c Under N2 atmosphere; BDC = 1,4-benzene-dicarboxylate; BPDC = 4,4’-biphenyl-dicarboxylate; fum = fumarate; abtc = 3,3’,5,5-azobenzene-tetracarboxylate; mdip = 5,5′-methylenediisophthalate; MeIM = 2-methylimidazolate.
Table 2.
Oxidation of methyl phenyl sulfide (MPS) with H2O2 over various Zr-MOFs (adapted from refs [91,92]) and comparison with MIL-125(Ti) [93] a.
| MOF | Time, h | MPS Conv. (%) | Product Selectivity (%) | |
|---|---|---|---|---|
| MPSO | MPSO2 | |||
| - | 24 | 14 | 79 | 21 |
| UiO-66 b | 0.25 | 49 | 1 | 99 |
| UiO-66 c | 0.5 | 51 | 8 | 92 |
| UiO-67 | 0.5 | 51 | 4 | 96 |
| MOF-801 | 2.5 | 47 | 4 | 96 |
| Zr-abtc c | 2 | 52 | 8 | 92 |
| Zr-abtc d | 2 | 46 | 9 | 91 |
| MIP-200 | 4 | 51 | 10 | 90 |
| MIL-125 | 2 | 69 | 80 | 20 |
a Reaction conditions: MPS 0.1 mmol, H2O2 0.1 mmol, MOF 1.6–2.0 mg, 27 °C, CH3CN 1 mL; b Average particle size 20 nm; c Average particle size 200 nm; d Average particle size 50 nm.
Table 3.
Epoxidation of α,β-unsaturated carbonyl compounds over Zr-MOFs and ZIF-8 (adapted from references [91,92,102]) a.
| Substrate | Catalyst | Time | Substrate Conv. | Epoxide Selectivity | |
|---|---|---|---|---|---|
| (h) | (%) | (%) b | |||
 | - | 4 | 7 |  | 22 |
| Zr-abtc | 1 | 26 | 62 | ||
| Zr-abtc c | 2 | 20 | 55 | ||
| MIP-200 | 2 | 15 | 10 | ||
| UiO-66 | 1 | 20 | 60 | ||
| UiO-67 | 1 | 20 | 55 | ||
| ZIF-8 | 5 | 20 | 50 | ||
 | - | 2 | 4 |  | 75 |
| Zr-abtc | 1.5 | 12 | 92 | ||
| MIP-200 | 2 | 7 | 57 | ||
| UiO-66 | 4 | 10 | 80 | ||
| UiO-67 | 4 | 8 | 88 | ||
| ZIF-8 | 1.5 | 81 | 97 | ||
 | - | 1 | 4 |  | 50 (1:1) d |
| Zr-abtc | 1 | 15 | 87 (6:1) | ||
| MIP-200 | 1 | 5 | 40 (1:1) | ||
| UiO-66 | 0.5 | 8 | 38 (1:2) | ||
| UiO-67 | 1 | 11 | 64 (3:4) | ||
 | UiO-66 | 0.5 | 30 |  | 50 |
a Reaction conditions: substrate 0.1 mmol, H2O2 0.8 mmol, MOF 3–5 mg, CH3CN 1 mL, 70 °C; b Epoxide yield based on the substrate consumed; c EtOAc was used as solvent; d In parentheses, the ratio of 1,2- to 7,8-epoxide for carvone oxidation.
Table 4.
Effect of protons on CyH oxidation with H2O2 over Ti-MIL-125 and Zr-MOFs (adapted from references [92,116,117]) a.
| Catalyst | Time, min | CyH conv. b (%) | Product Selectivity c (%) | ||
|---|---|---|---|---|---|
| Epoxide | Diol | Allylic d | |||
| – e | 60 | 6 | 16 | 16 | 65 |
| H+ f | 60 | 8 | 25 | 26 | 47 |
| MIL-125 | 60 | 26 | 25 | 10 | 60 |
| MIL-125 + 1 eq. H+ | 45 | 42 | 48 | 27 | 22 |
| UiO-66 | 60 | 16 | 35 | 10 | 50 |
| UiO-66 + 1 eq. H+ | 20 | 30 | 85 | 13 | 2 |
| UiO-67 | 30 | 17 | 28 | 25 | 43 |
| UiO-67 + 1 eq. H+ | 20 | 33 | 45 | 43 | 11 |
| Zr-abtc | 60 | 10 | 33 | 33 | 30 |
| Zr-abtc + 1 eq. H+ | 20 | 26 | 61 | 25 g | 7 |
| MIP-200 | 60 | 10 | 29 | 43 | 29 |
| MIP-200 + 1 eq. H+ | 20 | 27 | 70 | 19 h | 6 |
| MOF-801 | 60 | 10 | 35 | 20 | 45 |
| MOF-801 + 1 eq. H+ | 20 | 24 | 55 | 35 | 8 |
a Reaction conditions: CyH 0.1 mmol, H2O2 0.1 mmol, catalyst 0.01 mmol Ti or Zr, HClO4 0.01 mmol (if any), CH3CN 1 mL, 50 °C; b Maximum achievable conversion. c GC yield based on CyH consumed; d Total amount of allylic oxidation products (HP + enol + enone); e No catalyst was present; f No MOF was present; g The product of diol overoxidation, 2-hydroxycyclohexanone (ketol) was also found (7% selectivity); h Ketol (5% selectivity) was also formed.
Table 5.
Oxidation of alkylphenols with H2O2 over MIL-125 and MIL-125_NH2 (adapted from [125]) a.
Table 5.
Oxidation of alkylphenols with H2O2 over MIL-125 and MIL-125_NH2 (adapted from [125]) a.
| Catalyst | Phenol Conversion (%) | Quinone Selectivity (%) | ||
|---|---|---|---|---|
| MIL-125 |  | 56 |  | 100 |
| MIL-125_NH2 | 50 | 100 | ||
| MIL-125 |  | 20 |  | 100 |
a Reaction conditions: phenol 0.1 m, MOF 3 mg, H2O2 0.4 m, CH3CN 2 mL, 80 °C, 15 min.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Kholdeeva, O.; Maksimchuk, N. Metal-Organic Frameworks in Oxidation Catalysis with Hydrogen Peroxide. Catalysts 2021, 11, 283. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11020283
AMA Style
Kholdeeva O, Maksimchuk N. Metal-Organic Frameworks in Oxidation Catalysis with Hydrogen Peroxide. Catalysts. 2021; 11(2):283. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11020283
Chicago/Turabian StyleKholdeeva, Oxana, and Nataliya Maksimchuk. 2021. "Metal-Organic Frameworks in Oxidation Catalysis with Hydrogen Peroxide" Catalysts 11, no. 2: 283. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11020283
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.