
Optimizing Temperature Treatment of Copper Hollow Fibers for the Electrochemical Reduction of CO2 to CO

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. Morphology of the Fibers after Spinning
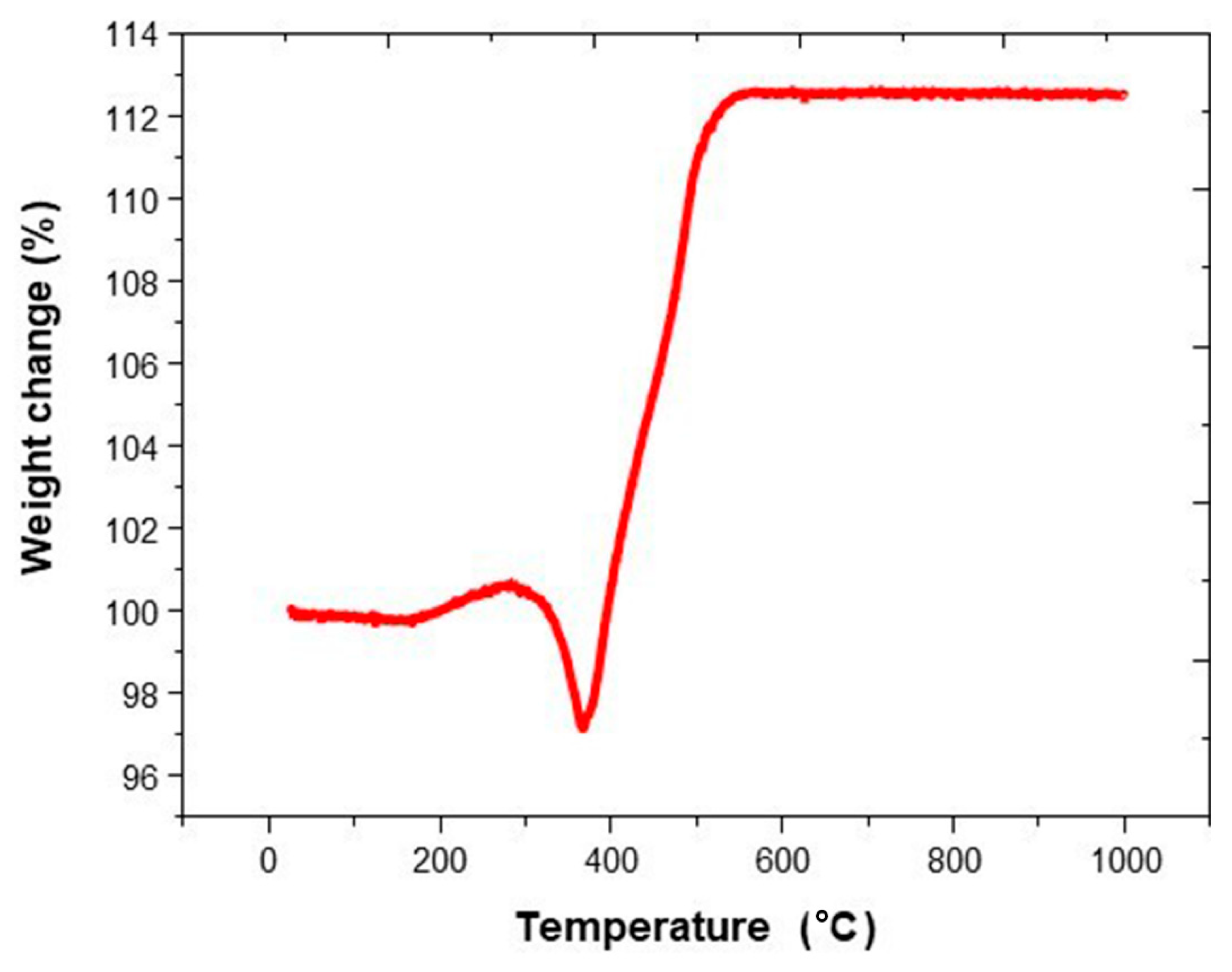
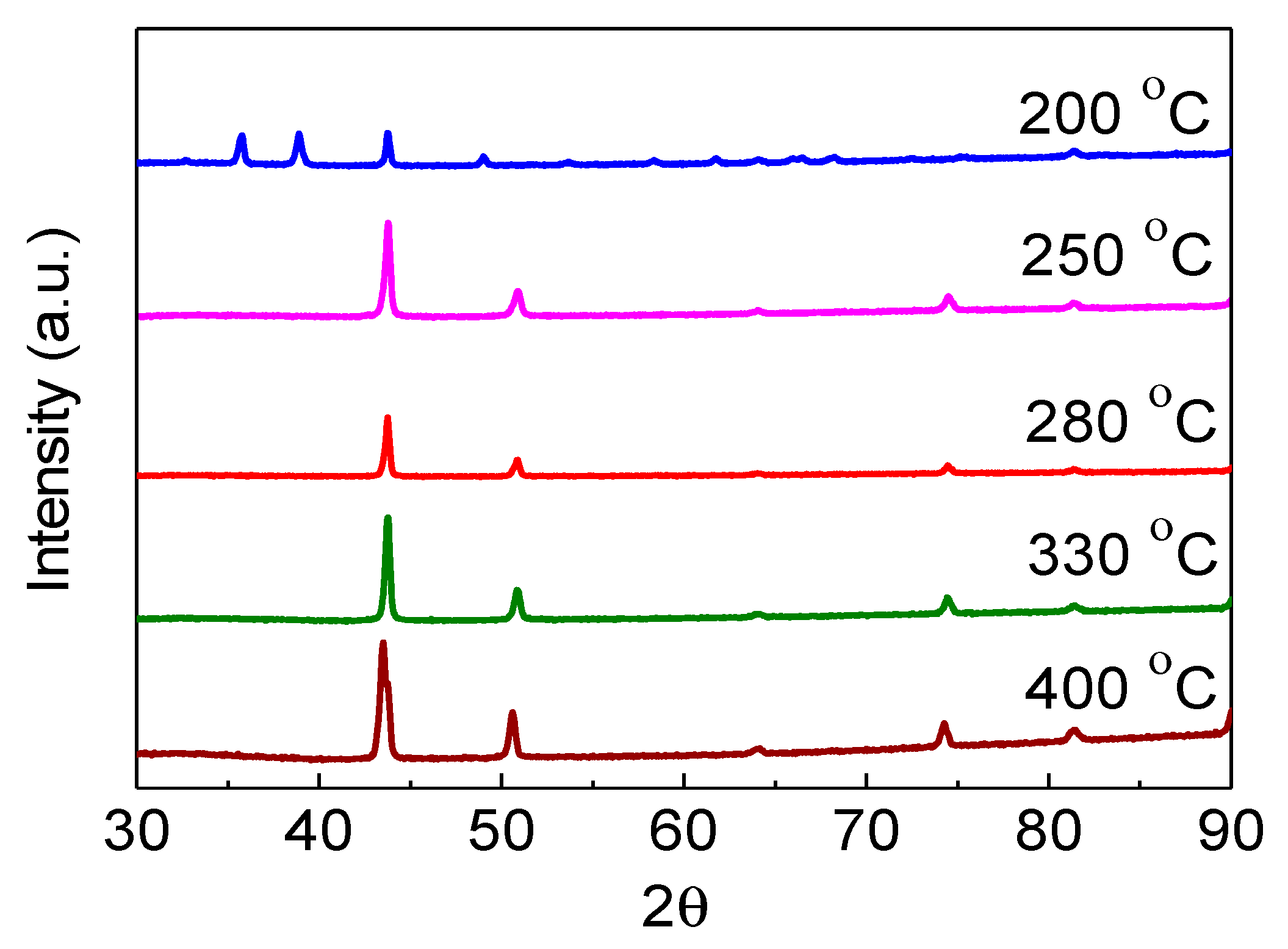
2.2. Calcination
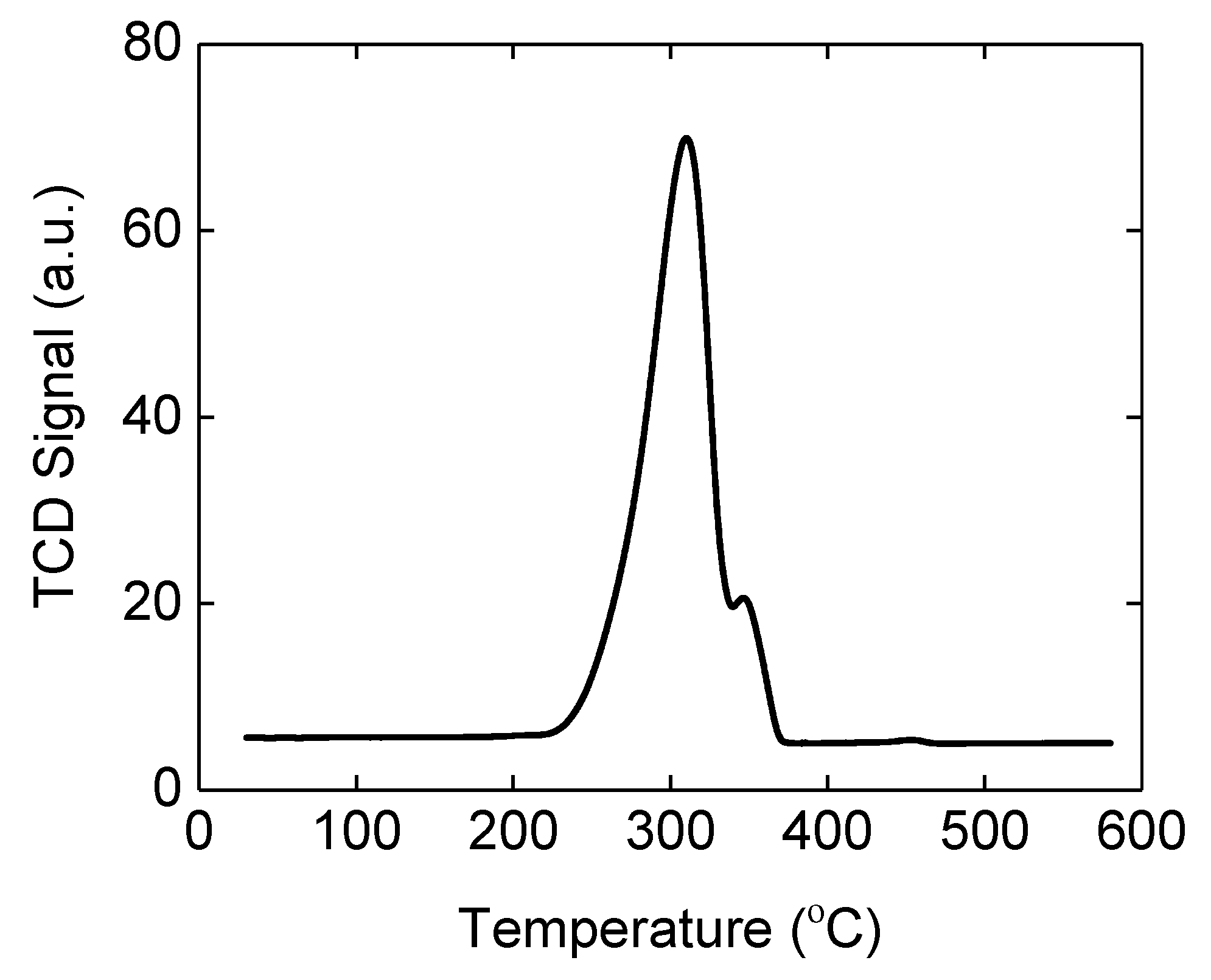
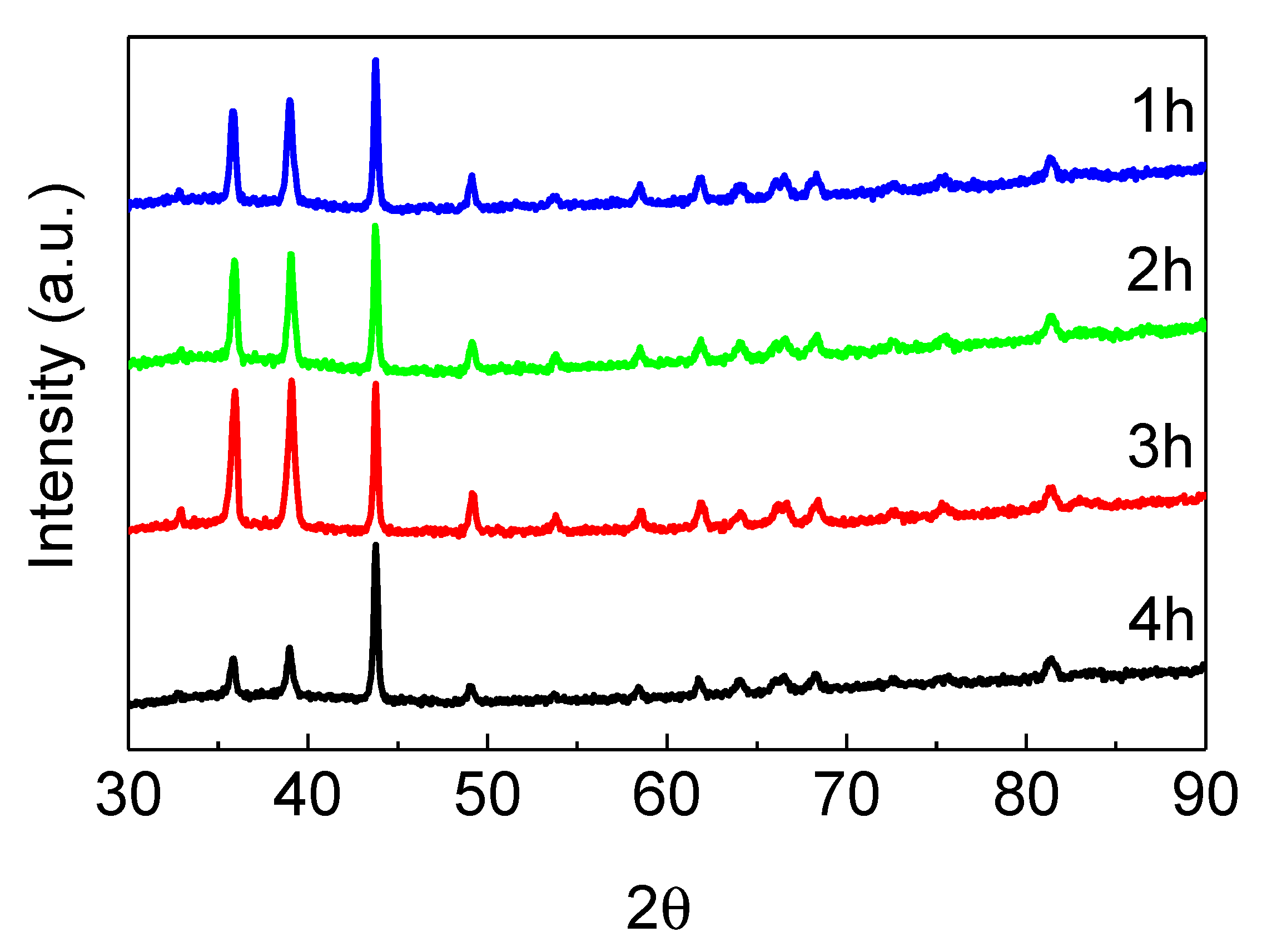
2.3. Reduction of Oxidized Fibers in 4% H2
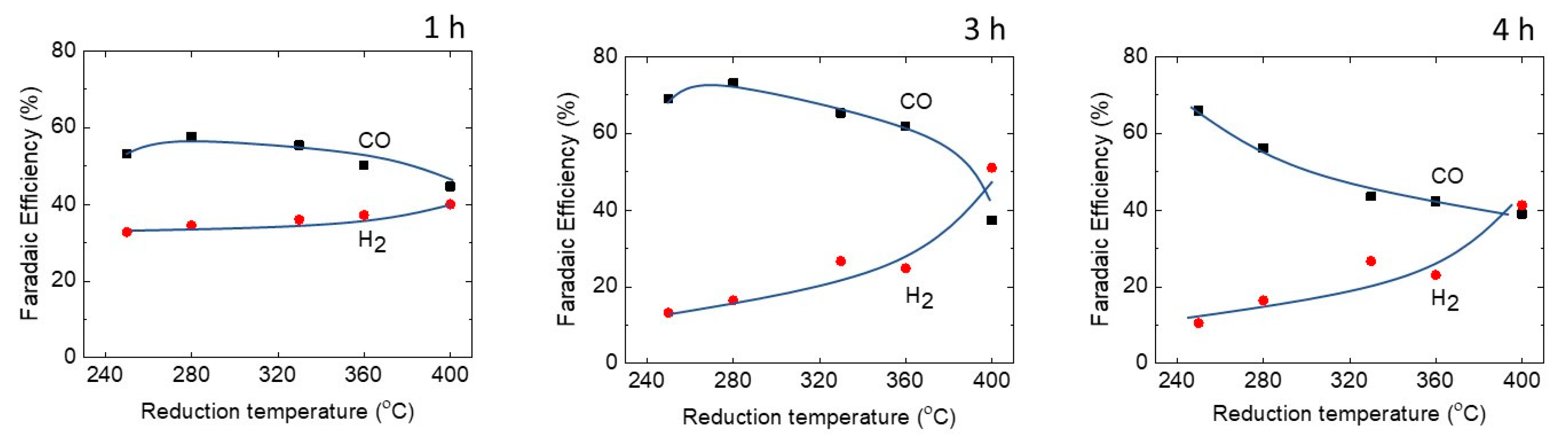
2.4. Performance of the Cu Hollow Fibers in CO2 Reduction
3. Discussion
4. Materials and Methods
4.1. Preparation of Copper-Polymer Spinning Mixtures
4.2. The Spinning Process
4.3. Treatment of the Fibers at Elevated Temperatures
4.4. Characterization
4.5. Determination of Electrochemical Performance
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Appendix A







References
- Kondratenko, E.V.; Mul, G.; Baltrusaitis, J.; Larrazábal, G.O.; Pérez-Ramírez, J. Status and perspectives of CO2 conversion into fuels and chemicals by catalytic, photocatalytic and electrocatalytic processes. Energy Environ. Sci. 2013, 6, 3112–3135. [Google Scholar] [CrossRef] [Green Version]
- Spurgeon, J.M.; Kumar, B. A comparative technoeconomic analysis of pathways for commercial electrochemical CO2 reduction to liquid products. Energy Environ. Sci. 2018, 11, 1536–1551. [Google Scholar] [CrossRef]
- Zhang, B.; Zhang, J.-T. Rational design of Cu-based electrocatalysts for electrochemical reduction of carbon dioxide. J. Energy Chem. 2017, 26, 1050–1066. [Google Scholar] [CrossRef] [Green Version]
- Lv, J.-J.; Jouny, M.; Luc, W.; Zhu, W.; Zhu, J.-J.; Jiao, F. A Highly Porous Copper Electrocatalyst for Carbon Dioxide Reduction. Adv. Mater. 2018, 30, 1803111. [Google Scholar] [CrossRef] [PubMed]
- Hori, Y.; Wakebe, H.; Tsukamoto, T.; Koga, O. Electrocatalytic process of CO selectivity in electrochemical reduction of CO2 at metal electrodes in aqueous media. Electrochim. Acta 1994, 39, 1833–1839. [Google Scholar] [CrossRef]
- Jhong, H.-R.M.; Ma, S.; Kenis, P.J.A. Electrochemical conversion of CO2 to useful chemicals: Current status, remaining challenges, and future opportunities. Curr. Opin. Chem. Eng. 2013, 2, 191–199. [Google Scholar] [CrossRef]
- Kas, R.; Kortlever, R.; Milbrat, A.; Koper, M.T.M.; Mul, G.; Baltrusaitis, J. Electrochemical CO2 reduction on Cu2O-derived copper nanoparticles: Controlling the catalytic selectivity of hydrocarbons. Phys. Chem. Chem. Phys. 2014, 16, 12194–12201. [Google Scholar] [CrossRef]
- Chen, C.; Khosrowabadi Kotyk, J.F.; Sheehan, S.W. Progress toward Commercial Application of Electrochemical Carbon Dioxide Reduction. Chem 2018, 4, 2571–2586. [Google Scholar] [CrossRef] [Green Version]
- Fang, Y.-X.; Flake, J.C. Electrochemical Reduction of CO2 at Functionalized Au Electrodes. J. Am. Chem. Soc. 2017, 139, 3399–3405. [Google Scholar] [CrossRef]
- Gattrell, M.; Gupta, N.; Co, A. A review of the aqueous electrochemical reduction of CO2 to hydrocarbons at copper. J. Electroanal. Chem. 2006, 594, 1–19. [Google Scholar] [CrossRef]
- Schreier, M.; Yoon, Y.; Jackson, M.N.; Surendranath, Y. Competition between H and CO for active sites governs Cu mediated electrosynthesis of hydrocarbon fuels. Angew. Chem. Int. Ed. 2018, 57, 10221–10225. [Google Scholar] [CrossRef]
- Dinh, C.-T.; Burdyny, T.; Kibria, M.G.; Seifitokaldani, A.; Gabardo, C.M.; García de Arquer, F.P.; Kiani, A.; Edwards, J.P.; De Luna, P.; Bushuyev, O.S.; et al. CO2 electroreduction to ethylene via hydroxide-mediated copper catalysis at an abrupt interface. Science 2018, 360, 783–787. [Google Scholar] [CrossRef] [Green Version]
- Kas, R.; Hummadi, K.H.K.H.; Kortlever, R.; Wit, P.D.; Milbrat, A.; Luiten-Olieman, M.W.J.; Benes, N.E.; Koper, M.T.M.; Mul, G. Three-dimensional porous hollow fibre copper electrodes for efficient and high-rate electrochemical carbon dioxide reduction. Nat. Commun. 2016, 7, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wickramasinghe, S.R.; Semmens, M.J.; Cussler, E.L. Mass transfer in various hollow fiber geometries. J. Mem. Sci. 1992, 69, 235–250. [Google Scholar] [CrossRef]
- Rabiee, H.; Zhang, X.; Ge, L.; Hu, S.; Li, M.; Smart, S.; Zhu, Z.; Yuan, Z. Tuning the Product Selectivity of the Cu Hollow Fiber Gas Diffusion Electrode for Efficient CO2 Reduction to Formate by Controlled Surface Sn Electrodeposition. ACS Appl. Mater. Interfaces 2020, 12, 21670–21681. [Google Scholar] [CrossRef] [PubMed]
- Jong, R.P.H.; Krzywda, P.M.; Benes, N.E.; Mul, G. Preparation of Ti, Ti/TiC or Ti/TiN based hollow fibres with extremely low electrical resistivity. RSC Adv. 2020, 10, 31901–31908. [Google Scholar] [CrossRef]
- Bell, D.; Rall, D.; Grosseheide, M.; Marx, L.; Hulsdunker, L.; Wessling, M. Tubular hollow fibre electrodes for CO2 reduction made from copper aluminum alloy with drastically increased intrinsic porosity. Electrochem. Commun. 2020, 111, 106645. [Google Scholar] [CrossRef]
- Rodriguez, J.A.; Kim, J.Y.; Hanson, J.C.; Perez, M.; Frenkel, A.I. Reduction of CuO in H2: In situ time-resolved XRD studies. Catal. Lett. 2003, 85, 247–254. [Google Scholar] [CrossRef]
- Zhu, Y.; Mimura, K.; Isshiki, M. Oxidation Mechansim of Cu2O to CuO at 600–1050 °C. Oxid. Met. 2004, 62, 207–222. [Google Scholar] [CrossRef]
- Wang, H.-P.; Yeh, C.-T. On the reduction of copper oxide. J. Chin. Chem. Soc. 1983, 30, 139–143. [Google Scholar] [CrossRef]
- Kim, J.Y.; Rodriguez, J.A.; Hanson, J.C.; Frenkel, A.I.; Lee, P.L. Reduction of CuO and Cu2O with H2: H Embedding and Kinetic Effects in the Formation of Suboxides. J. Am. Chem. Soc. 2003, 125, 10684–10692. [Google Scholar] [CrossRef] [PubMed]
- Li, C.W.; Kanan, M.W. CO2 Reduction at Low Overpotential on Cu Electrodes Resulting from the Reduction of Thick Cu2O Films. J. Am. Chem. Soc. 2012, 134, 7231–7234. [Google Scholar] [CrossRef]
- Qiao, J.; Liu, Y.; Hong, F.; Zhang, J. A review of catalysts for the electroreduction of carbon dioxide to produce low-carbon fuels. Chem. Soc. Rev. 2014, 43, 631–675. [Google Scholar] [CrossRef] [PubMed]
- Kuhl, K.P.; Cave, E.R.; Abramf, D.N.; Jaramillo, T.F. New insights into the electrochemical reduction of carbon dioxide on metallic copper surfaces. Energy Environ. Sci. 2012, 5, 7050–7059. [Google Scholar] [CrossRef]
- Hori, Y.; Konishi, H.; Futamura, T.; Murata, A.; Koga, O.; Sakurai, H.; Oguma, K. Deactivation of copper electrode, in electrochemical reduction of CO2. Electrochem. Acta 2005, 50, 5354–5369. [Google Scholar] [CrossRef]
- Kedzierzawski, P.; Augustynski, J. Poisoning and Activation of the Gold Cathode during Electroreduction of CO2. J. Electrochem. Soc. 1994, 141, L58–L60. [Google Scholar] [CrossRef]
- Velasco-Velez, J.-J.; Jones, T.; Gao, D.; Carbonio, E.; Arrigo, R.; Hsu, C.-J.; Huang, Y.-C.; Dong, C.-L.; Chen, J.-M.; Lee, J.-F.; et al. The Role of the Copper Oxidation State in the Electrocatalytic Reduction of CO2 into Valuable Hydrocarbons. ACS Sustain. Chem. Eng. 2019, 7, 1485. [Google Scholar] [CrossRef]
- Eilert, A.; Cavalca, F.; Roberts, F.S.; Osterwalder, J.; Liu, C.; Favaro, M.; Crumlin, E.J.; Ogasawara, H.; Friebel, D.; Pettersson, L.G.M.; et al. Subsurface Oxygen in Oxide-Derived Copper Electrocatalysts for Carbon Dioxide Reduction. J. Phys. Chem. Lett. 2017, 8, 285–290. [Google Scholar] [CrossRef] [Green Version]
- Chou, T.-C.; Chang, C.-C.; Yu, H.-L.; Yu, W.-Y.; Dong, C.-L.; Velasco-Vélez, J.-J.; Chuang, C.-H.; Chen, L.-C.; Lee, J.-F.; Chen, J.M.; et al. Controlling the Oxidation State of the Cu Electrode and Reaction Intermediates for Electrochemical CO2 Reduction to Ethylene. J. Am. Chem. Soc. 2020, 142, 2857–2867. [Google Scholar] [CrossRef]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hummadi, K.K.; Sustronk, A.; Kas, R.; Benes, N.; Mul, G. Optimizing Temperature Treatment of Copper Hollow Fibers for the Electrochemical Reduction of CO2 to CO. Catalysts 2021, 11, 571. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11050571
Hummadi KK, Sustronk A, Kas R, Benes N, Mul G. Optimizing Temperature Treatment of Copper Hollow Fibers for the Electrochemical Reduction of CO2 to CO. Catalysts. 2021; 11(5):571. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11050571
Chicago/Turabian StyleHummadi, Khalid Khazzal, Anne Sustronk, Recep Kas, Nieck Benes, and Guido Mul. 2021. "Optimizing Temperature Treatment of Copper Hollow Fibers for the Electrochemical Reduction of CO2 to CO" Catalysts 11, no. 5: 571. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11050571