
Synthesis of Xylyl-Linked Bis-Benzimidazolium Salts and Their Application in the Palladium-Catalyzed Suzuki–Miyaura Cross-Coupling Reaction of Aryl Chlorides


Abstract
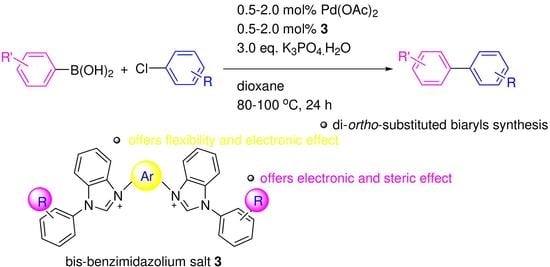
:
1. Introduction
2. Results
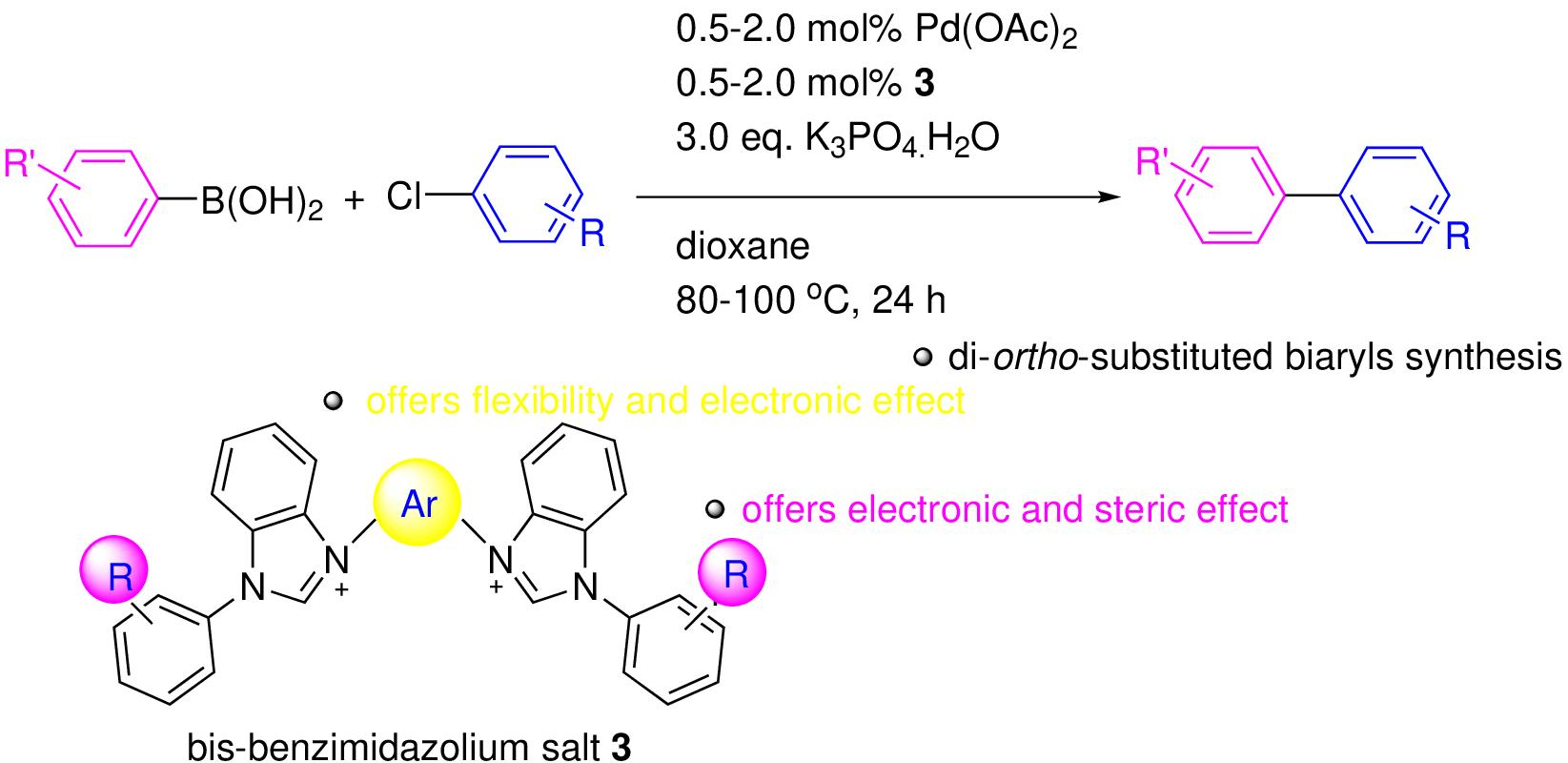
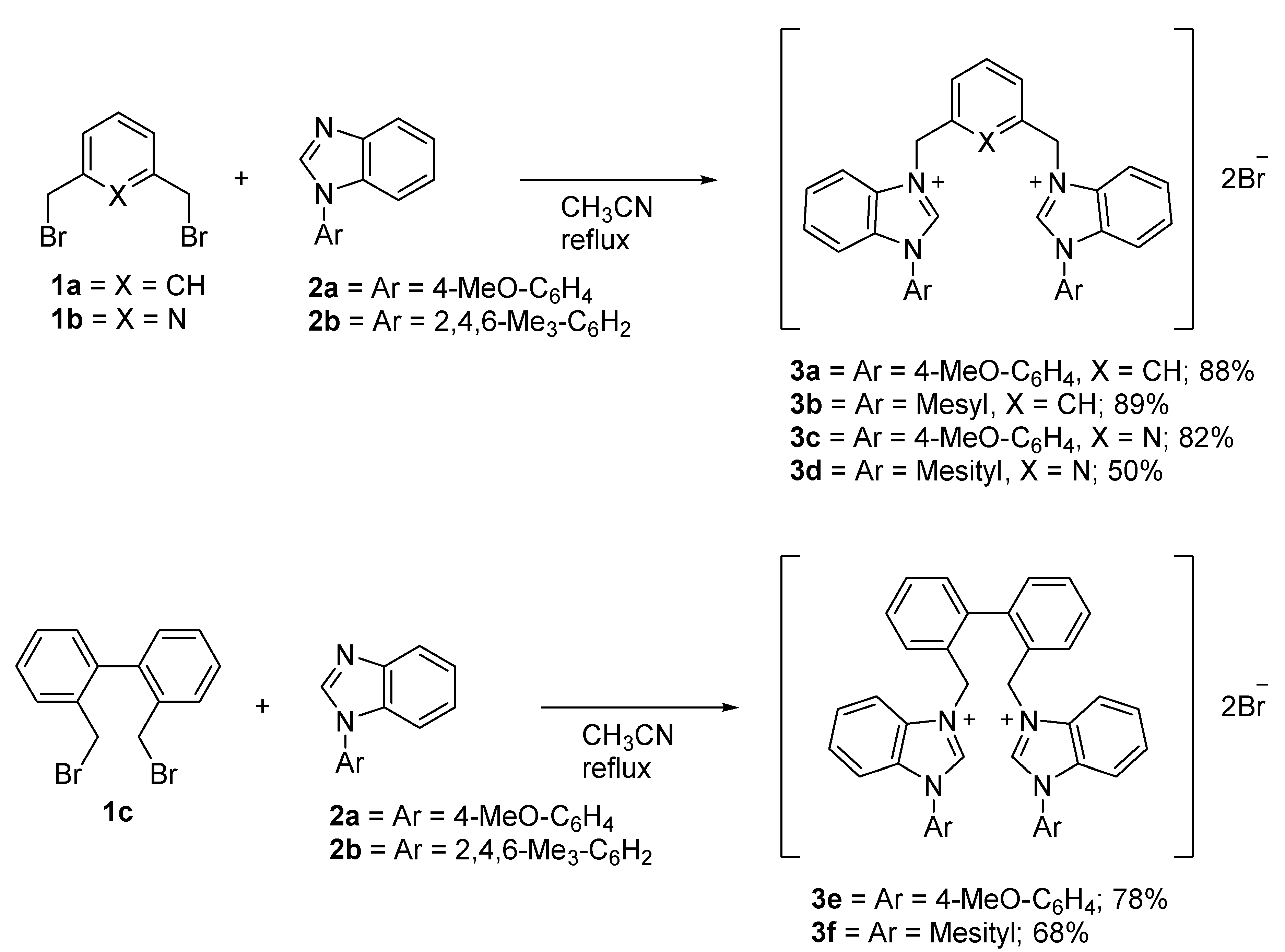
2.1. Synthesis and Characterization of the Bis-Benzimidazolium Salts 3
2.2. The Suzuki–Miyaura Cross-Coupling Reaction
3. Materials and Methods
3.1. General Methods
3.2. Experimental Procedures and Spectral Data
3.2.1. Synthesis of 1-(2,4,6-Trimethylphenyl)-1H-benzimidazole 2b
3.2.2. General Procedures for the Synthesis of Bis-Benzimidazolium Salts 3
3.2.3. General Procedures for Suzuki–Miyaura Cross-Coupling Reactions under N2
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Miyaura, N.; Suzuki, A. Palladium-Catalyzed Cross-Coupling Reactions of Organoboron Compounds. Chem. Rev. 1995, 95, 2457–2483. [Google Scholar] [CrossRef] [Green Version]
- Stanforth, S.P. Catalytic Cross-Coupling Reactions in Biaryl Synthesis. Tetrahedron 1998, 54, 263–303. [Google Scholar] [CrossRef]
- Suzuki, A. Recent advances in the cross-coupling reactions of organoboron derivatives with organic electrophiles, 1995–1998. J. Organomet. Chem. 1999, 576, 147–168. [Google Scholar] [CrossRef]
- Mingji, D.; Liang, B.; Wang, C.; You, Z.; Xiang, J.; Dong, G.; Chen, J.; Yang, Z. A Novel Thiourea Ligand Applied in the Pd-Catalyzed Heck, Suzuki and Suzuki Carbonylative Reactions. Adv. Synth. Catal. 2004, 346, 1669–1673. [Google Scholar] [CrossRef]
- Suzuki, A.; Yamamoto, Y. Cross-coupling Reactions of Organoboranes: An Easy Method for C–C Bonding. Chem. Lett. 2011, 40, 894–901. [Google Scholar] [CrossRef]
- Grushin, V.V.; Alper, H. Transformations of Chloroarenes, Catalyzed by Transition-Metal Complexes. Chem. Rev. 1994, 94, 1047–1062. [Google Scholar] [CrossRef]
- Littke, A.F.; Fu, G.C. Palladium-Catalyzed Coupling Reactions of Aryl Chlorides. Angew. Chem. Int. Ed. 2002, 41, 4176–4211. [Google Scholar] [CrossRef]
- Corbet, J.-P.; Mignani, G. Selected Patented Cross-Coupling Reaction Technologies. Chem. Rev. 2006, 106, 2651–2710. [Google Scholar] [CrossRef]
- Wong, S.M.; Yuen, O.Y.; Choy, P.Y.; Kwong, F.Y. When Cross-Coupling Partners Meet Indolylphosphines. Coord. Chem. Rev. 2015, 293–294, 158–186. [Google Scholar] [CrossRef]
- Schwarz, J.B.; Böhm, V.P.W.; Gardiner, M.G.; Grosche, M.; Hermann, W.A.; Hieringer, W.; Raudaschl-Sieber, G. Polymer-Supported Carbene Complexes of Palladium: Well-Defined, Air-Stable, Recyclable Catalysts for the Heck Reaction. Chem. Eur. J. 2000, 6, 1773–1780. [Google Scholar] [CrossRef]
- Hermann, W.A.; Öfele, K.; Preysing, D.; Herdtweck, E. Metal complexes of acyclic diaminocarbenes: Links between N-heterocyclic carbene (NHC)- and Fischer-carbene complexes. J. Organomet. Chem. 2003, 684, 235–248. [Google Scholar] [CrossRef]
- Herrmann, W.A.; Reisinger, C.-P.; Spiegler, M. Chelating N-heterocyclic carbene ligands in palladium-catalyzed heck-type reactions. J. Organomet. Chem. 1998, 557, 93–96. [Google Scholar] [CrossRef]
- Zhang, C.; Trudell, M.L. Palladium-bisimidazol-2-ylidene complexes as catalysts for general and efficient Suzuki cross-coupling reactions of aryl chlorides with arylboronic acids. Tetrahedron Lett. 2000, 41, 595–598. [Google Scholar] [CrossRef]
- Özdemir, İ.; Çetinkaya, B.; Demir, S.; Gürbüz, N. Palladium-catalyzed Suzuki-Miyaura reaction using saturated N-heterocyclic ligands. Catal. Lett. 2004, 97, 37–40. [Google Scholar] [CrossRef]
- Özdemir, İ.; Gök, Y.; Gürbüz, N.; Çetinkaya, E.; Çetinkaya, B. Suzuki-Miyaura Reaction of Unactivated Aryl Chlorides Using Beznimidazol-2-Ylidene Ligands. Synth. Commun. 2004, 34, 4135–4144. [Google Scholar] [CrossRef]
- Özdemir, İ.; Gök, Y.; Gürbüz, N.; Çetinkaya, E.; Çetinkaya, B. Palladium-Catalyzed Suzuki Reaction Using 1,3-Dialkylbenzimidazol-2-ylidene Ligands in Aqueous Media. Heteroat. Chem. 2004, 15, 419–423. [Google Scholar] [CrossRef]
- Shi, M.; Qian, H.-X. A new dimeric bidentated NHC–Pd(II) complex from trans-cyclohexane-1,2-diamine for Suzuki reaction and Heck reaction. Tetrahedron 2005, 61, 4949–4955. [Google Scholar] [CrossRef]
- Xu, Q.; Duan, W.-L.; Lei, Z.-Y.; Zhu, Z.-B.; Shi, M. A novel cis-chelated Pd(II)–NHC complex for catalyzing Suzuki and Heck-type cross-coupling reactions. Tetrahedron 2005, 61, 11225–11229. [Google Scholar] [CrossRef]
- Demir, S.; Özdemir, İ.; Çetinkaya, B. Use of bis(benzimidazolium)–palladium system as a convenient catalyst for Heck and Suzuki coupling reactions of aryl bromides and chlorides. Appl. Organometal. Chem. 2006, 20, 254–259. [Google Scholar] [CrossRef]
- Özdemir, İ.; Demir, S.; Çetinkaya, B. Novel tetrahydropyrimidinium/palladium system as a convenient catalysts: Suzuki coupling reactions of aryl chlorides. Arkivoc 2007, 13, 71–78. [Google Scholar] [CrossRef]
- Liu, Z.; Zhang, T.; Shi, M. Cyclometalated cis-Chelated Bidentate N-Heterocyclic Carbene Palladium Complexes: Synthetic, Structural, and Catalytic Studies. Organometallics 2008, 27, 2668–2671. [Google Scholar] [CrossRef]
- Avery, K.B.; Devine, W.G.; Kormos, C.M.; Leadbeater, N.E. Use of a silicon carbide multi-well plate in conjunction with microwave heating for rapid ligand synthesis, formation of palladium complexes, and catalyst screening in a Suzuki coupling. Tetrahedron Lett. 2009, 50, 2851–2853. [Google Scholar] [CrossRef]
- Yilmaz, U.; Şireci, N.; Deniz, S.; Küçükbay, H. Synthesis and microwave-assisted catalytic activity of novel bis-benzimidazoles salts bearing furfuryl and thenyl moieties in Heck and Suzuki cross-coupling reactions. Appl. Organometal. Chem. 2010, 24, 414–420. [Google Scholar]
- Micksch, M.; Strassner, T. Palladium(II) Complexes with Chelating Biscarbene Ligands in the Catalytic Suzuki–Miyaura Cross-Coupling Reaction. Eur. J. Org. Chem. 2012, 5872–5880. [Google Scholar] [CrossRef]
- Li, Y.; Tang, J.; Gu, J.; Wang, Q.; Sun, P.; Zhang, D. Chiral 1,2-Cyclohexane-Bridged Bis-NHC Palladium Catalysts for Asymmetric Suzuki–Miyaura Coupling: Synthesis, Characterization, and Steric Effects on Enantiocontrol. Organometallics 2014, 33, 876–884. [Google Scholar] [CrossRef]
- Charbonneau, M.; Addoumieh, G.; Oguadinma, P.; Schmitzer, A.R. Support-Free Palladium–NHC Catalyst for High Recyclable Heterogeneous Suzuki–Miyaura Coupling in Neat Water. Organometallics 2014, 33, 6544–6549. [Google Scholar] [CrossRef]
- Thapa, R.; Kilyanek, S.M. Synthesis and structural characterization of 20-membered macrocyclic rings bearing trans-chelating bis(N-heterocyclic carbene) ligands and the catalytic activity of their palladium(II) complexes. Dalton Trans. 2019, 48, 12577–12590. [Google Scholar] [CrossRef]
- Lin, Y.-R.; Chiu, C.-C.; Chiu, H.-T.; Lee, D.-S.; Lu, T.-J. Bis-benzimidazolium-palladium system catalyzed Suzuki–Miyaura coupling reaction of aryl bromides under mild conditions. Appl. Organometal. Chem. 2017, 32, e3896. [Google Scholar] [CrossRef]
- Chiu, C.-C.; Chiu, H.-T.; Lee, D.-S.; Lu, T.-J. An efficient class of bis-NHC salts: Applications in Pd-catalyzed reactions under mild reaction conditions. RSC Adv. 2018, 8, 26407–26415. [Google Scholar] [CrossRef] [Green Version]
- Tran, V.M.; Nguyen, T.K.N.; Sorna, V.; Loganathan, D.; Kuberan, B. Synthesis and Assessment of Glycosaminoglycan Priming Activity of Cluster-xylosides for Potential Use as Proteoglycan Mimetics. ACS Chem. Biol. 2013, 8, 949–957. [Google Scholar] [CrossRef]
- Yu, K.-K.; Li, K.; Hou, J.-T.; Yu, X.-Q. Coumarin-TPA Derivative: A reaction-based ratiometric fluorescent probe for Cu(I). Tetrahedron Lett. 2013, 54, 5771–5774. [Google Scholar] [CrossRef]
- Cervantes-Reyes, A.; Rominger, F.; Rudolph, M.; Hashmi, A.S.K. Gold(I) Complexes Stabilized by Nine- and Ten-Membered N-Heterocyclic Carbene Ligands. Chem. Eur. J. 2019, 25, 11745–11757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.; Fridman, N.; Tamm, M.; Eisten, M.S. Addition of E–H (E = N, P, C, O, S) Bonds to Heterocumulenes Catalyzed by Benzimidazolin-2-iminato Actinide Complexes. Organometallics 2017, 36, 3896–3903. [Google Scholar] [CrossRef]
- Chan, A.; Scheidt, K.A. Highly Stereoselective Formal [3+3] Cycloaddition of Enals and Azomethine Imines Catalyzed by N-Heterocyclic Carbenes. J. Am. Che. Soc. 2007, 129, 5334–5335. [Google Scholar] [CrossRef] [PubMed]
- Glannerini, M.; Vila, C.; Hornillos, V.; Feringa, B.L. One-Pot, Fast and Modular Approach to Alkyl- and Aryl Ketones via Sequential 1,2-Addition,Pd-Catalyzed Cross-Coupling of Organolithium Reagents with Weinreb Amides. Chem. Commun. 2016, 52, 1206–1209. [Google Scholar] [CrossRef]
- Liu, Z.; Tan, H.; Wang, L.; Fu, T.; Xia, Y.; Zhang, Y.; Wang, J. Transition-Metal-Free Intramolecular Carbene Aromatic Substitution/Büchner Reaction: Synthesis of Fluorenes and [6,5,7]Benzo-fused Rings. Angew. Chem. Int. Ed. 2015, 54, 3056–3060. [Google Scholar] [CrossRef]
- Mao, P.; Yang, L.; Xiao, Y.; Yuan, J.; Liu, X.; Song, M. Suzuki cross-coupling catalyzed by palladium(II) complexes bearing 1-aryl-3,4,5,6-tetrahydropyrimidine ligands. J. Organomet. Chem. 2012, 705, 39–43. [Google Scholar] [CrossRef]
- Chen, L.; Lang, H.; Fang, L.; Yu, J.; Wang, L. Nickel-Catalyzed Desulfitative Suzuki–Miyaura Cross-Coupling of N,N-Disulfinylmethylamines and Arylboromic Acids. Eur. J. Org. Chem. 2014, 2014, 6385–6389. [Google Scholar] [CrossRef]
- Han, C.; Zhang, Z.; Xu, S.; Wang, K.; Chen, K.; Zhao, J. Palladium-Catalyzed Hiyama Coupling of Benzylic Ammonium Salts via C–N Bond Cleavage. J. Org. Chem. 2019, 84, 16308–16313. [Google Scholar] [CrossRef]
- Piontek, A.; Ochędzan-Siodłak, W.; Bisz, E.; Szostak, M. Nickel-Catalyzed C(sp2)-C(sp3) Kumada Cross-Coupling of Aryl Tosylates with Alkyl Grignard Reagents. Adv. Synth. Catal. 2019, 361, 2329–2335. [Google Scholar] [CrossRef]
- Liu, H.; Yin, B.; Gao, Z.; Li, Y.; Jiang, H. Transition-metal-free highly chemo- and regioselective arylation of unactivated arenes with aryl halides over recyclable heterogeneous catalysts. Chem. Commun. 2012, 48, 2033–2035. [Google Scholar] [CrossRef] [PubMed]
- Lücke, A.-L.; Wlechmann, S.; Freese, T.; Schmidt, A. Suzuki–Miyaura Cross-Coupling Reactions in Acetic Acid Employing Sydnone-Derived Catalyst Systems. Synlett 2017, 28, 1990–1993. [Google Scholar]
- Song, B.; Knauber, T.; Gooßen, L.J. Decarboxylative Cross-Coupling of Mesylates Catalyzed by Copper/Palladium Systems with Customized Imidazolyl Phosphine Ligands. Angew. Chem. Int. Ed. 2013, 52, 2954–2958. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}

| Entry | Ligand | Temperature (°C) | 4a GC Yield (%) 2 | 6aa GC Yield (%) 2 |
|---|---|---|---|---|
| 1 | L | 30 | 88 | 7 |
| 2 | 3b | 30 | 78 | 21 |
| 3 | 3b | 80 | 5 | 88 |
| 4 | 3a | 80 | 5 | 87 |
| 5 | 3c | 80 | 62 | 31 |
| 6 | 3d | 80 | 25 | 70 |
| 7 | 3e | 80 | 71 | 25 |
| 8 | 3f | 80 | 37 | 58 |
| Entry | 3b/Pd(OAc)2 (mol%) | Solvent | Base | Time (h) | 4a GC Yield (%) 2 | 6aa GC Yield (%) 2 |
|---|---|---|---|---|---|---|
| 1 | 1.0 | Toluene | K3PO4·H2O | 24 | 0 | 91 (91) 3 |
| 2 | 1.0 | 1,4-Dioxane | K3PO4·H2O | 24 | 0 | 94 (94) 3 |
| 3 | 1.0 4 | 1,4-Dioxane | K3PO4·H2O | 24 | – | (20) 3 |
| 4 | 1.0 | CH3CN | K3PO4·H2O | 24 | 7 | 87 |
| 5 | 1.0 | t-BuOH | K3PO4·H2O | 24 | 5 | 88 |
| 6 | 1.0 | t-BuOH/H2O 5 | K3PO4·H2O | 24 | 31 | 55 |
| 7 | 1.0 | 1,4-Dioxane | KOtBu | 24 | 1 | 4 |
| 8 | 1.0 | 1,4-Dioxane | K2CO3 | 24 | 13 | 82 |
| 9 | 1.0 | 1,4-Dioxane | Cs2CO3 | 24 | − | (93) 3 |
| 10 | 1.0 | 1,4-Dioxane | KOAc | 24 | 86 | 8 |
| 11 | 1.0 | 1,4-Dioxane | K3PO4·H2O | 12 | 0 | 96 |
| 12 | 1.0 | 1,4-Dioxane | K3PO4·H2O | 6 | 0 | 99 (98) 3 |
| 13 | 1.0 | 1,4-Dioxane | K3PO4·H2O | 3 | 60 | 35 |
| 14 | 0.5 | 1,4-Dioxane | K3PO4·H2O | 6 | 0 | 99 (98) 3 |
| 15 | 0.1 | 1,4-Dioxane | K3PO4·H2O | 6 | 65 | 30 |
| 16 | 0.05 | 1,4-Dioxane | K3PO4·H2O | 6 | 90 | 0.4 |

| Entry | 3b/Pd(OAc)2 (mol%) | Temp. (°C) | 6 | Yield (%) | Entry | 3b/Pd(OAc)2 (mol%) | Temp. (°C) | 6 | Yield (%) |
|---|---|---|---|---|---|---|---|---|---|
| 1 | 0.5 | 80 |  | 99 | 12 | 1.5 | 100 |  | 92 |
| 2 | 1.0 | 80 |  | 99 | 13 | 1.5 | 100 |  | 92 |
| 3 | 1.0 | 100 |  | 98 | 14 | 1.5 | 100 |  | 90 |
| 4 | 1.0 | 80 |  | 99 | 15 | 1.0 | 80 |  | 95 |
| 5 | 1.0 | 80 |  | 99 | 16 | 1.5 | 100 |  | 99 |
| 6 | 1.0 | 80 |  | 96 | 17 | 1.0 | 80 |  | 17 2 |
| 7 | 1.0 | 100 |  | 81 | 18 | 1.0 | 80 |  | 27 2 |
| 8 | 1.0 | 100 |  | 89 | 19 | 1.0 | 80 |  | 95 |
| 9 | 1.0 | 80 |  | 90 | 20 | 2.0 | 100 |  | 93 |
| 10 | 1.5 | 100 |  | 99 | 21 | 2.0 | 100 |  | 92 |
| 11 | 1.5 | 100 |  | 97 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, T.; Wei, T.-R.; Huang, S.-J.; Lai, Y.-T.; Lee, D.-S.; Lu, T.-J. Synthesis of Xylyl-Linked Bis-Benzimidazolium Salts and Their Application in the Palladium-Catalyzed Suzuki–Miyaura Cross-Coupling Reaction of Aryl Chlorides. Catalysts 2021, 11, 817. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11070817
Wang T, Wei T-R, Huang S-J, Lai Y-T, Lee D-S, Lu T-J. Synthesis of Xylyl-Linked Bis-Benzimidazolium Salts and Their Application in the Palladium-Catalyzed Suzuki–Miyaura Cross-Coupling Reaction of Aryl Chlorides. Catalysts. 2021; 11(7):817. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11070817
Chicago/Turabian StyleWang, Tsui, Ting-Rong Wei, Shu-Jyun Huang, Yu-Ting Lai, Dong-Sheng Lee, and Ta-Jung Lu. 2021. "Synthesis of Xylyl-Linked Bis-Benzimidazolium Salts and Their Application in the Palladium-Catalyzed Suzuki–Miyaura Cross-Coupling Reaction of Aryl Chlorides" Catalysts 11, no. 7: 817. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11070817