
Recent Advances in Homogeneous/Heterogeneous Catalytic Hydrogenation and Dehydrogenation for Potential Liquid Organic Hydrogen Carrier (LOHC) Systems

Abstract
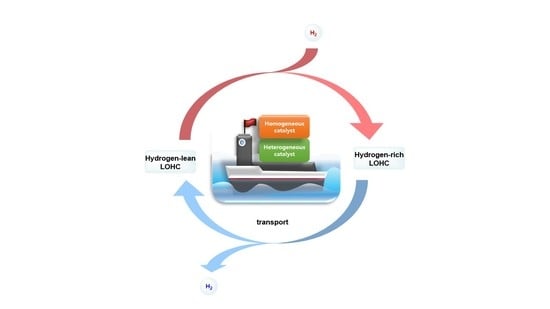
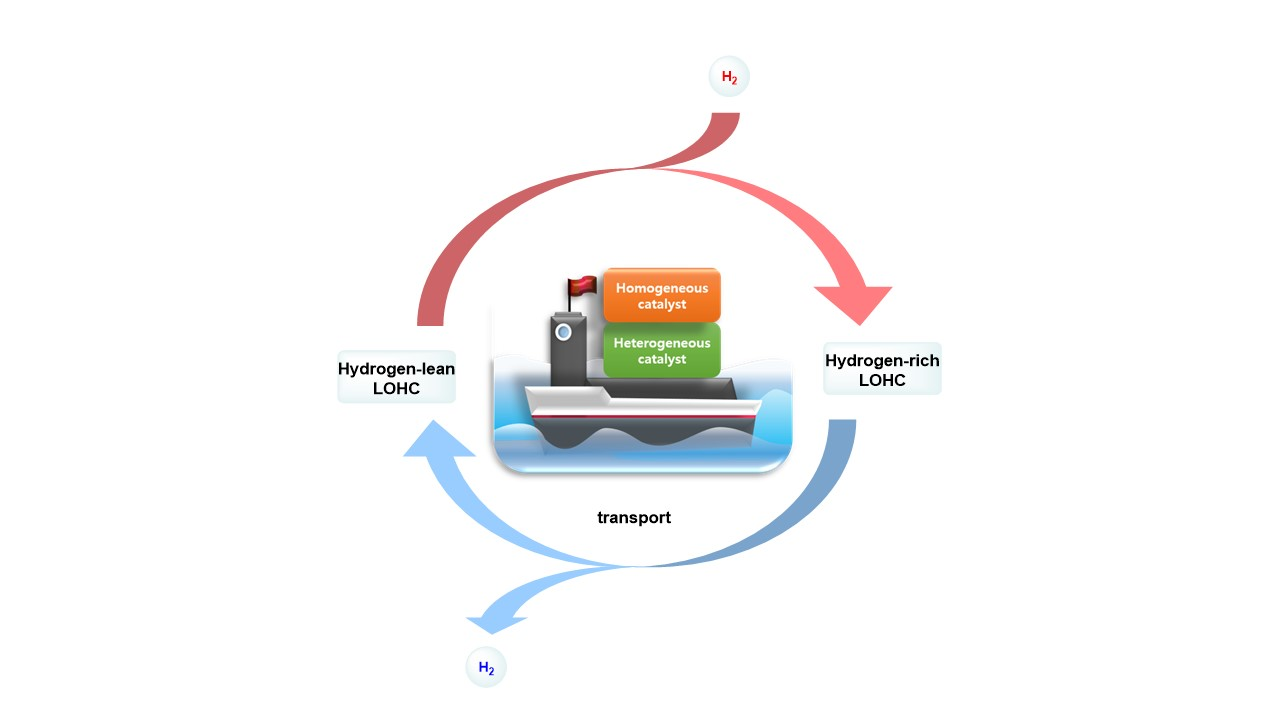
:
1. Introduction
- (1)
- Physical characteristic: Various physical properties, as given below, should be investigated to discover promising LOHC systems.
- Melting point: Liquids and low-melting solid organic compounds are considered LOHCs, but hydrogen-rich LOHCs, hydrogen-lean LOHCs, or a mixture of hydrogen-rich/hydrogen-lean LOHCs should exist in a liquid state for easy storage and transportation in an existing gasoline infrastructure at ambient temperature.
- Boiling point: LOHCs with a high boiling point are more effective in easily separating pure hydrogen gas from hydrogen-lean LOHCs after completion of dehydrogenation.
- Thermal stability: Decomposed LOHCs, through hydrogenation and dehydrogenation at high temperatures, could affect the ability of catalysts. Thus, the high thermal stability of LOHCs is an important factor for sustainable catalytic cycles.
- Viscosity: From a transportation standpoint, the lower viscosity of LOHCs makes them useful for pumping into existing tanks and pipelines.
- (2)
- Reversibility: Most LOHC systems can be recycled because hydrogen-rich and hydrogen-lean LOHCs are reversible in the presence of suitable catalysts via hydrogenation (exothermic reaction) and dehydrogenation (endothermic reaction). Irreversible circular hydrogen carriers, such as formic acid and ammonia, can also be used in LOHC systems, but re-use is inefficient as gaseous carbon dioxide or nitrogen is released into the atmosphere during dehydrogenation and must be injected into a new batch for hydrogenation with each cycle.
- (3)
- Hydrogen storage capacity: In 2020, the hydrogen and fuel cell technologies office (HFTO) in the U.S. Department of Energy suggested research and development goals for advanced hydrogen storage system, shown below. Thus, promising LOHC systems need to meet these conditions
- Hydrogen gravimetric storage density—6.0 wt% hydrogen (1.5 kWh/kg system).
- Hydrogen volumetric storage density—0.030 kg hydrogen/L (1.0 kWh/L).
- Cost—$333/kg stored hydrogen capacity ($10/kWh).
- (4)
- Eco-friendly characteristic: To minimize environmental damage, it is necessary to develop LOHC systems that produce only benign byproducts, such as water, during hydrogenation and dehydrogenation. Moreover, the toxicity of LOHCs should be investigated to prevent its effects on human health.
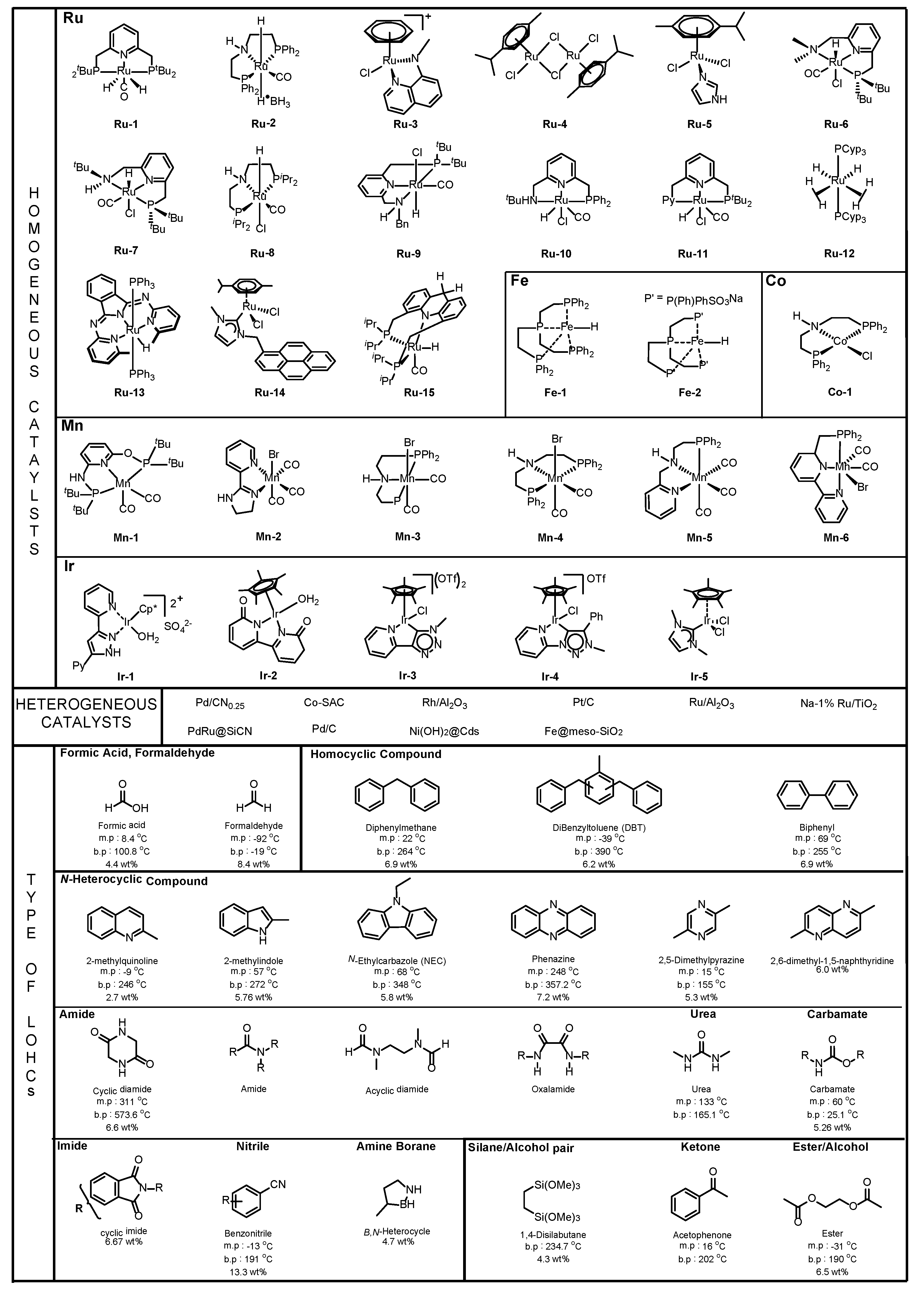
- Homogeneous catalysts: The performance of homogeneous catalysis at lower temperatures is generally better than heterogeneous catalysis because it is more useful to modify the catalyst with various ligands to enhance the catalyst’s ability [14]. As a result, homogeneous catalysis is also applicable to LOHCs. However, recyclability is not good, since it is difficult to recover the catalyst after the reaction.
- Heterogeneous catalysts: Heterogeneous catalysts are typically effective for application in potential LOHC systems in terms of thermal stability and recyclability because it is easier to separate the catalyst from LOHCs by filtering. Furthermore, heterogeneous catalysts can be used for continuous flow processes to scale up for hydrogenation and dehydrogenation to take and release hydrogen in industry.
2. Circular Hydrogen Carriers
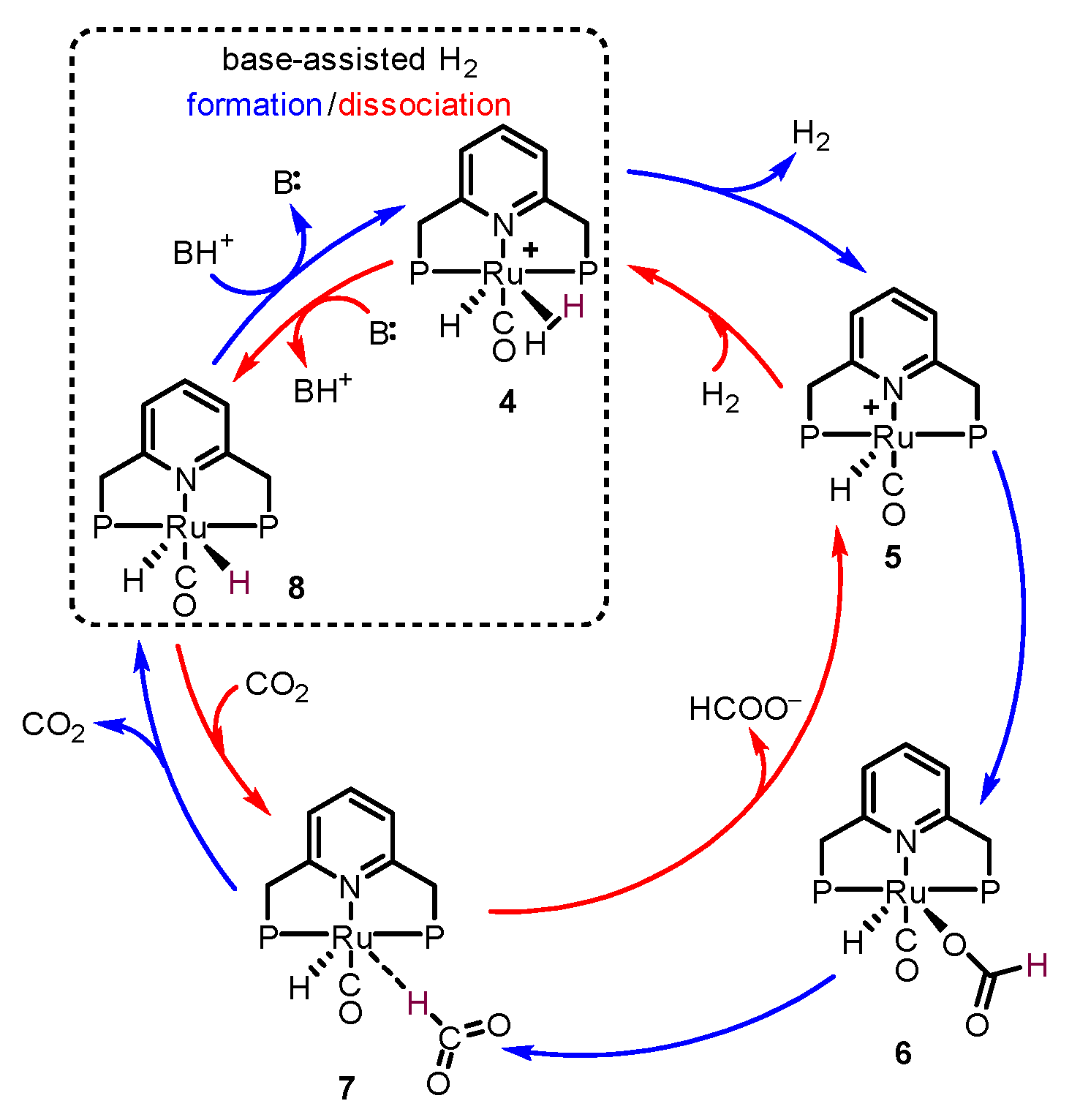
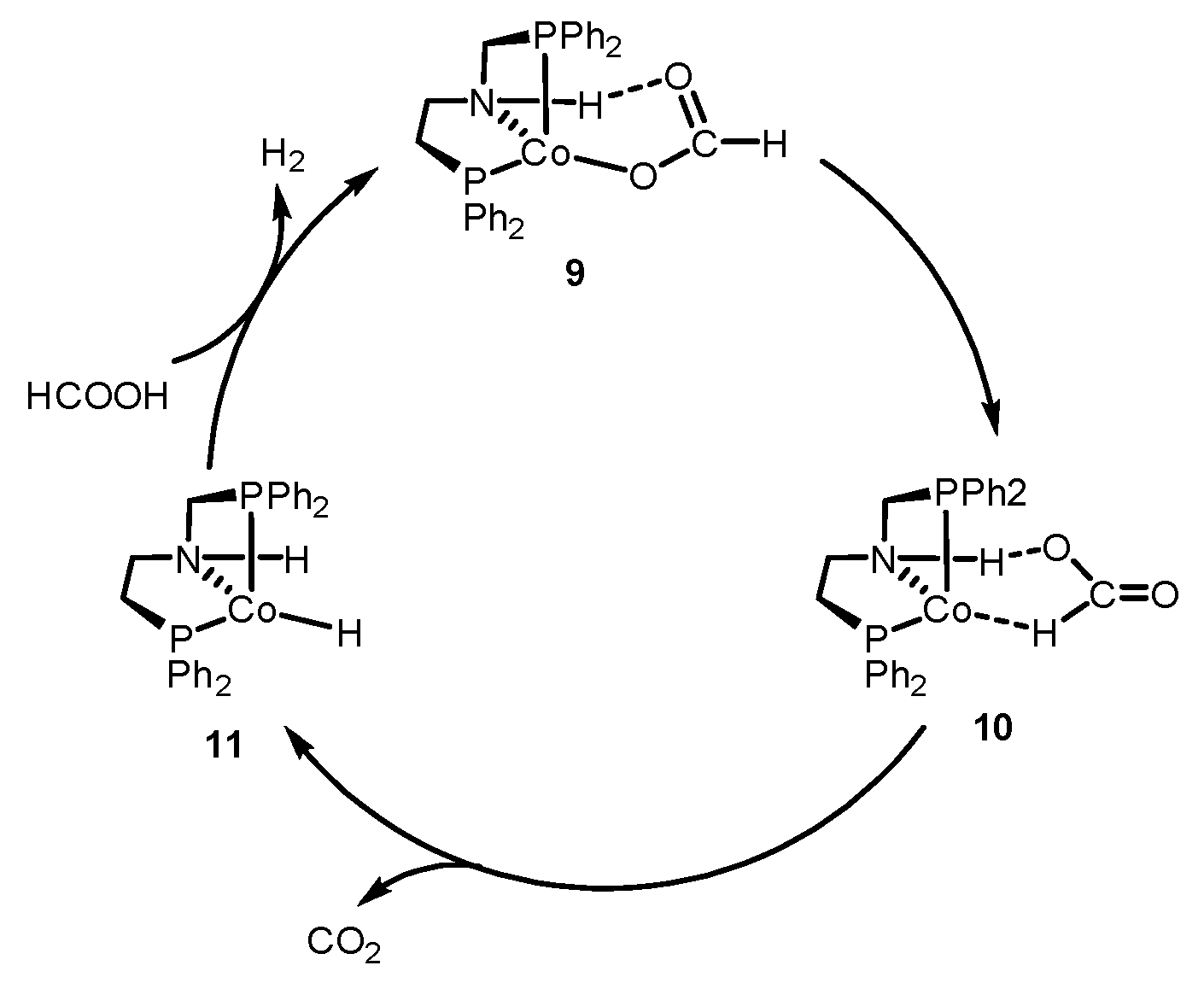
2.1. Formic Acid
2.2. Formaldehyde


 | ||||||
|---|---|---|---|---|---|---|
| Entry | Starting Material | Catalyst | Dehydrogenation Condition | TON | TOF | Ref. |
| A | 3 | Fe-1 | Propylene carbonate, 80 °C | 92,417 | 9425 | [27] |
| B | 3 | Ru-1 | DMF, NEt3, 90 °C | - | 257,000 | [28] |
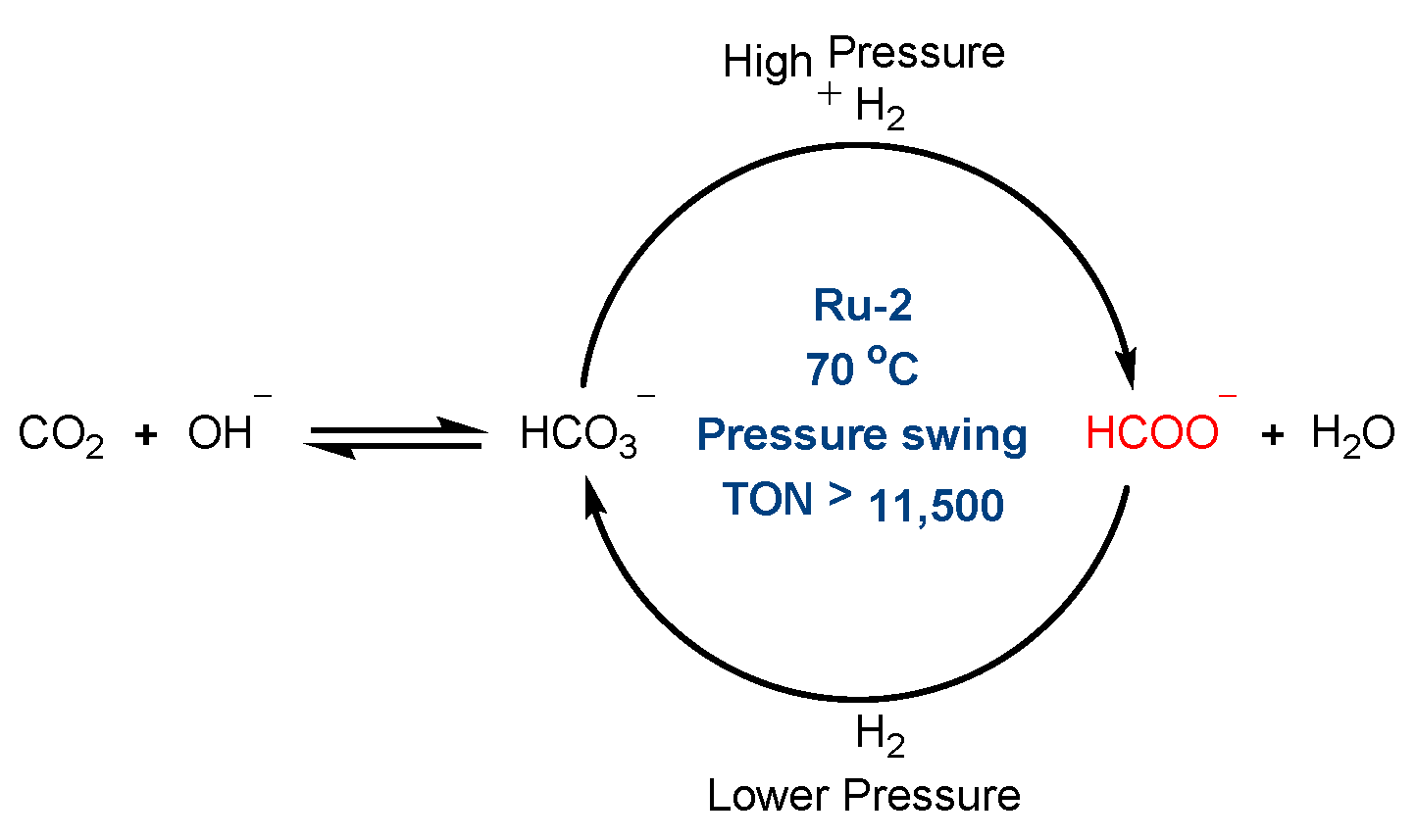
| C | 3 | Ru-2 | 70 °C | - | >11,500 | [29] |
| D | 3 | Pd/CN0.25 | H2O, 25 °C | 50,040 | 5530 | [30] |
| E | 3 | Fe-2 | H2O, 25 °C | - | - | [31] |
| F | 3 | Ru-3 | H2O, HCOONa, 90 °C | 2248 | 940 | [32] |
| G | 3 | Co-1 | H2O, HCOOK, NaBEt3H 80 °C | 2260 | - | [33] |
| H | 3 | Mn-1 | Chlorobenzene, NEt3, 80 °C | - | 8500 | [34] |
| I | 3 | Mn-2 | H2O, Triglyme, HCOOK, 92.5 °C | 2637 | - | [35] |
| J | 3 | Ir-1 | - | - | 8250 | [36] |
| K | 1 | Ru-4 | - | 700 | 3142 | [44] |
| L | 1 | Ru-5 | - | 515 | 850 | [45] |
| M | 1 | Ru-5 | - | 12,905 | - | [45] |
2.3. Ammonia
3. Reversible Liquid Organic Hydrogen Carriers
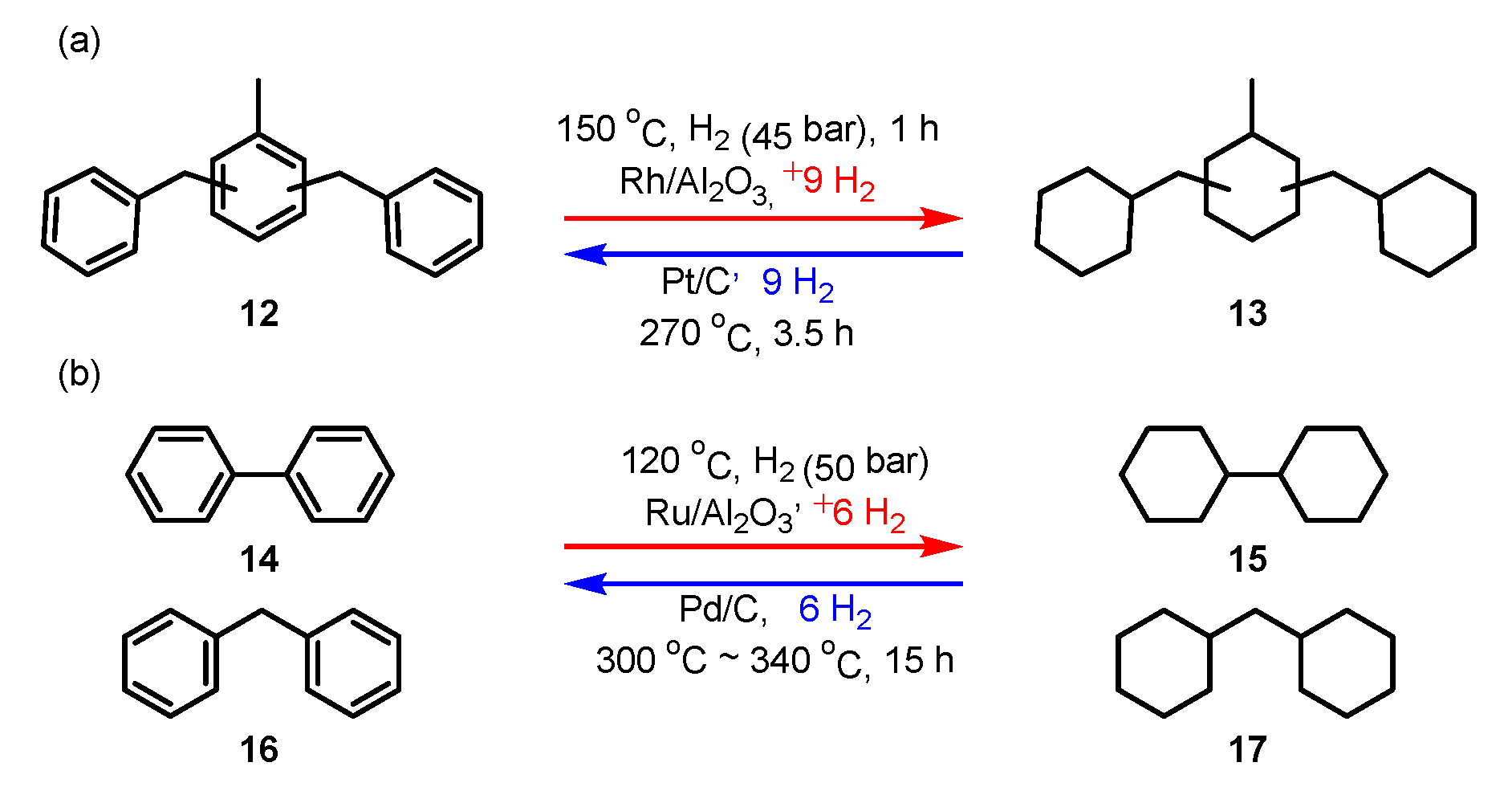
3.1. Homocyclic Compounds

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
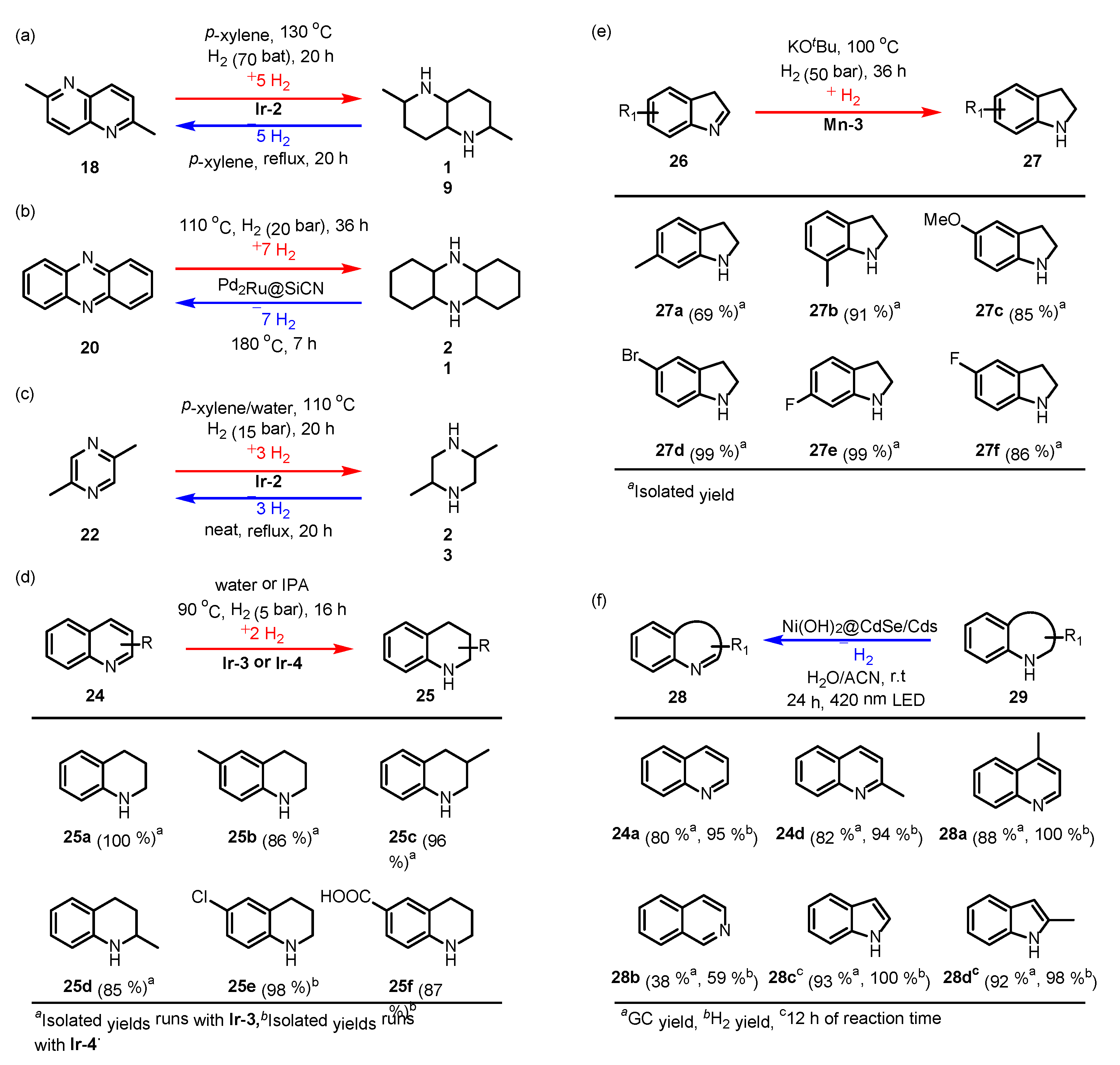
3.2. Nitrogen-Containing Compounds
3.2.1. N-Heterocyclic Compounds
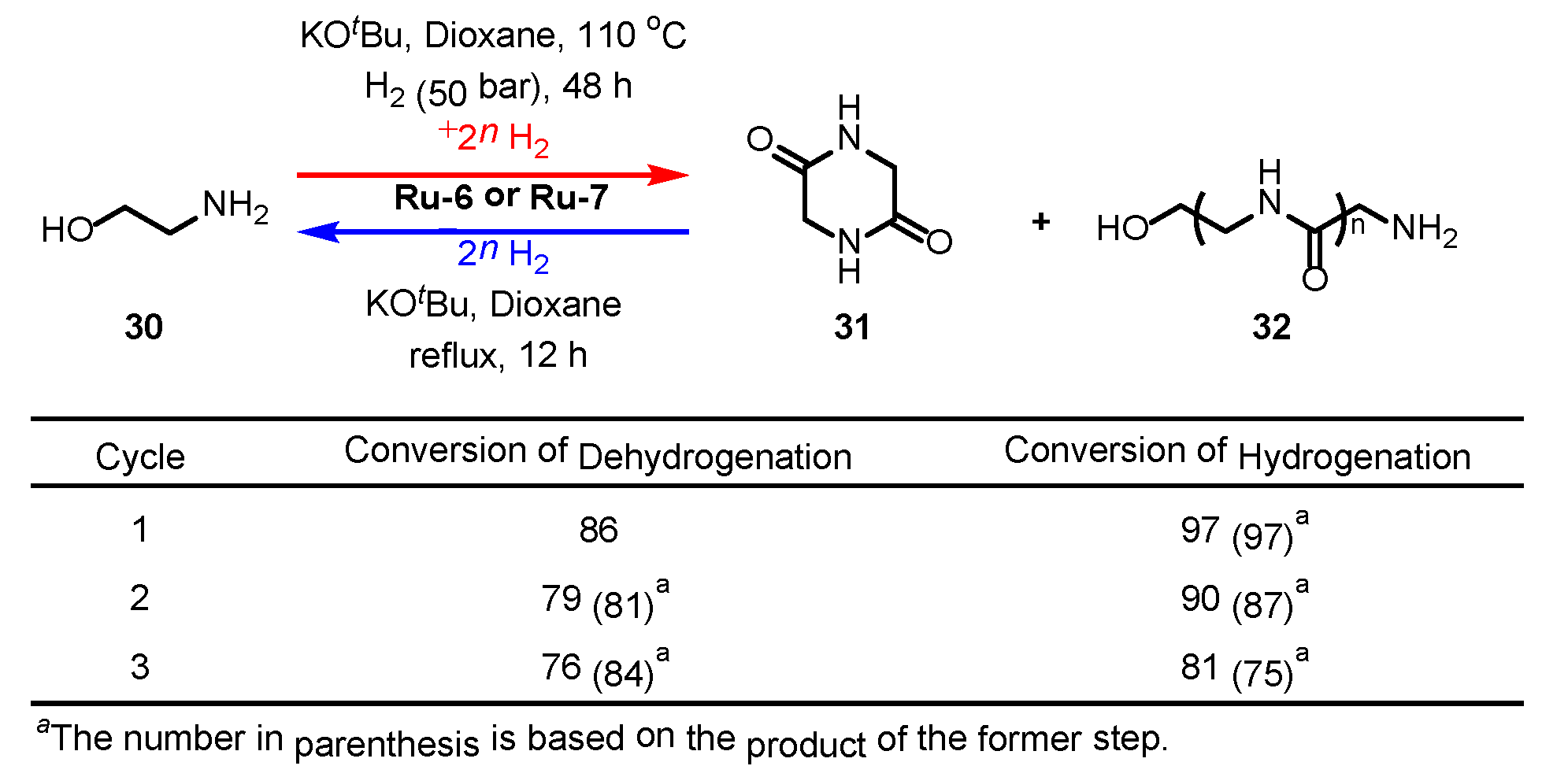
3.2.2. Amide
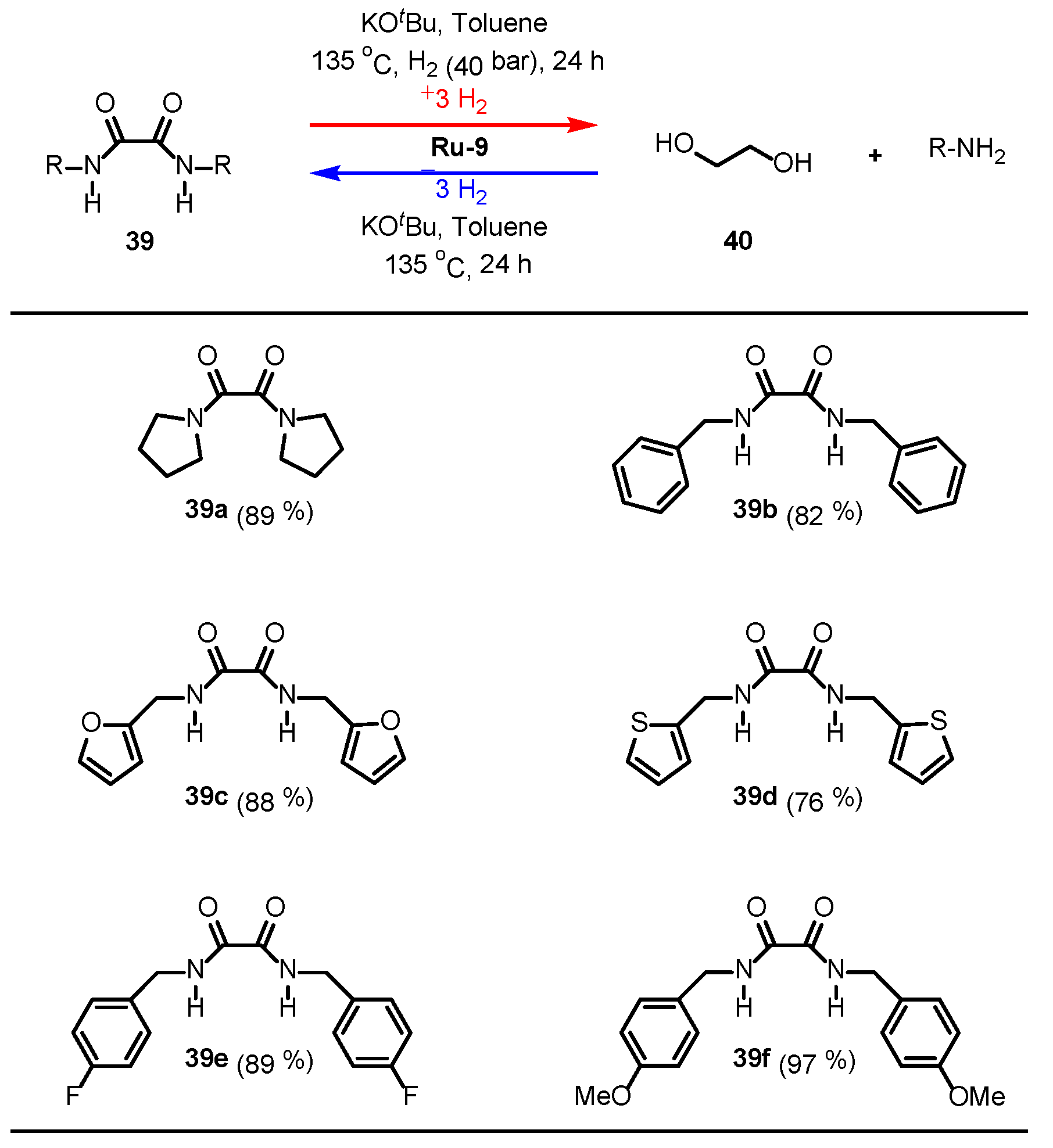
3.2.3. Imide
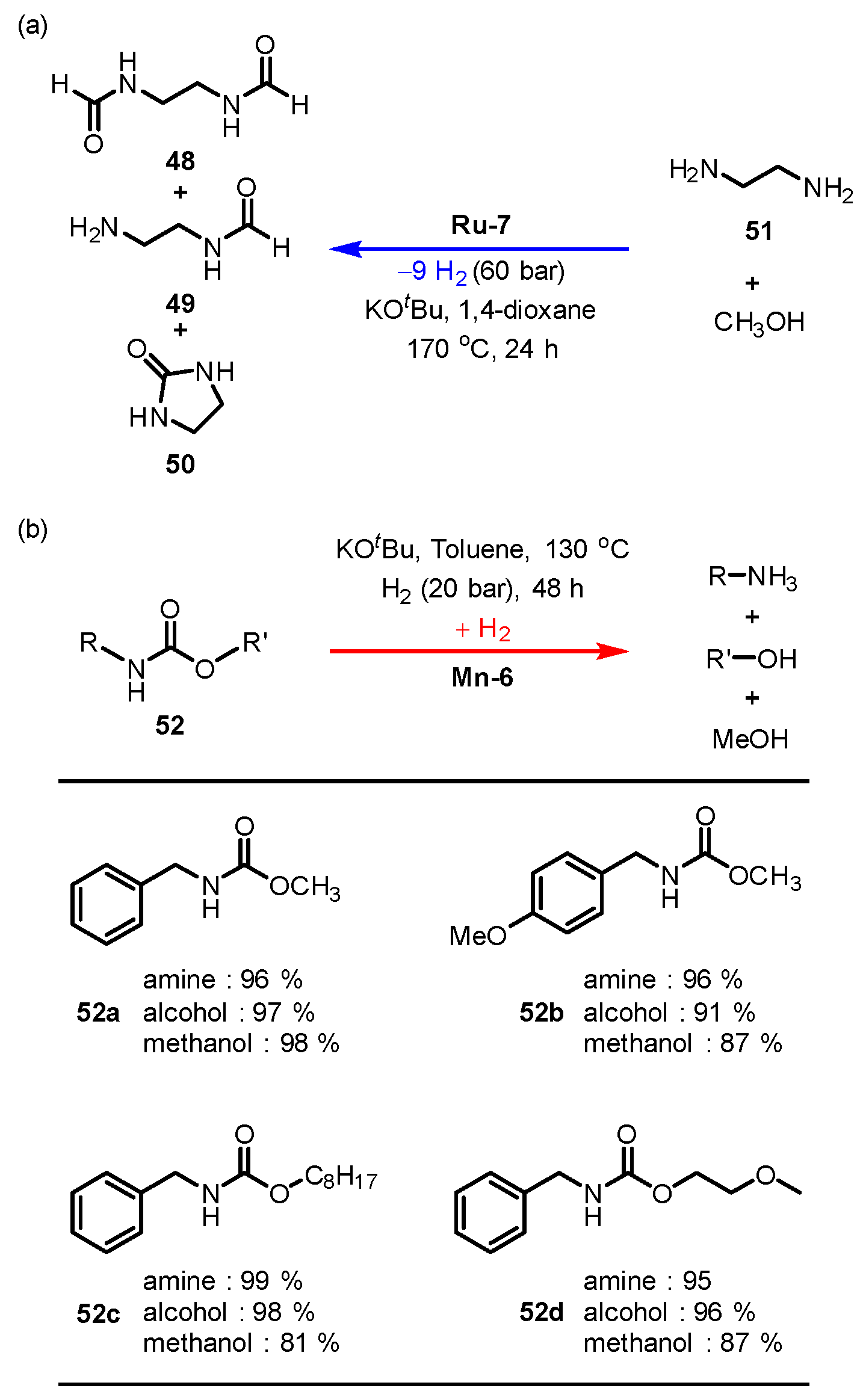
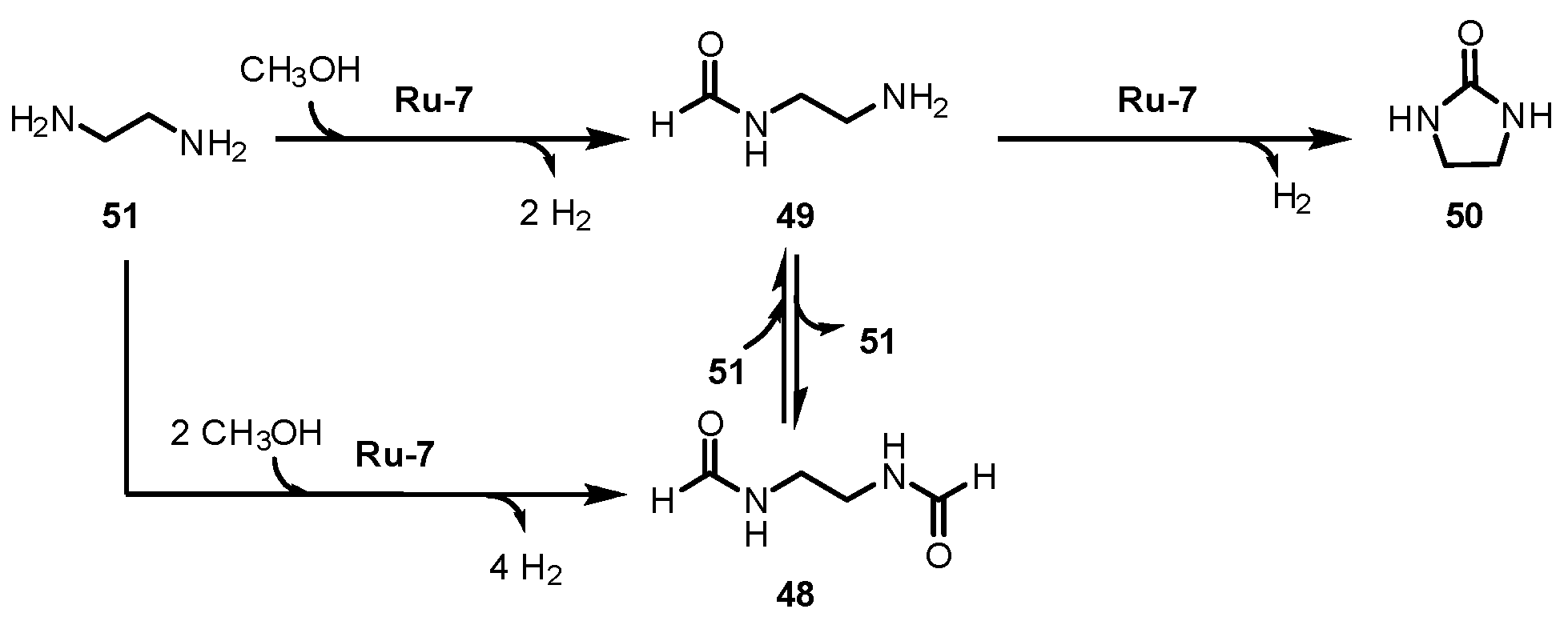
3.2.4. Urea/Carbamate
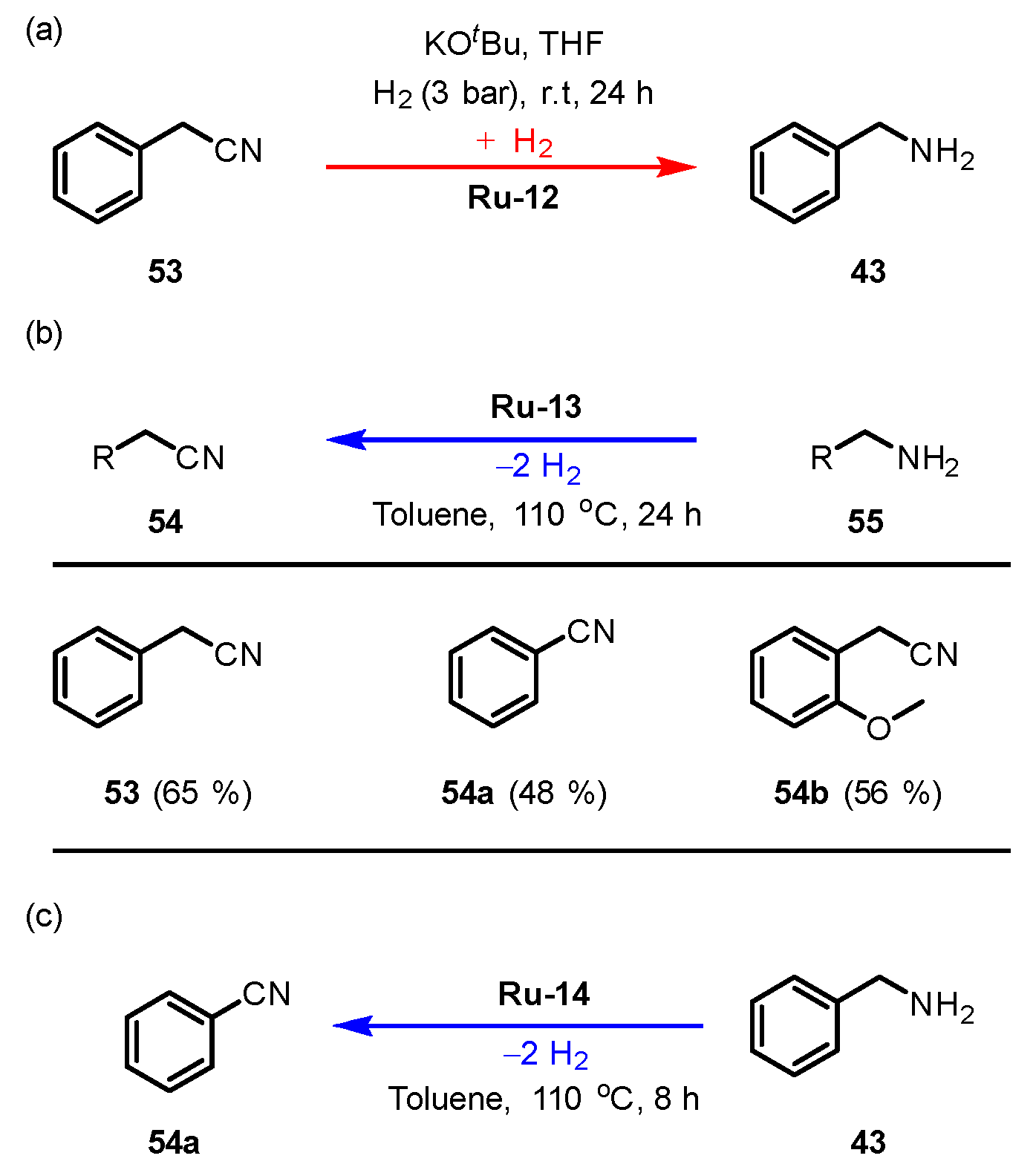
3.2.5. Nitrile
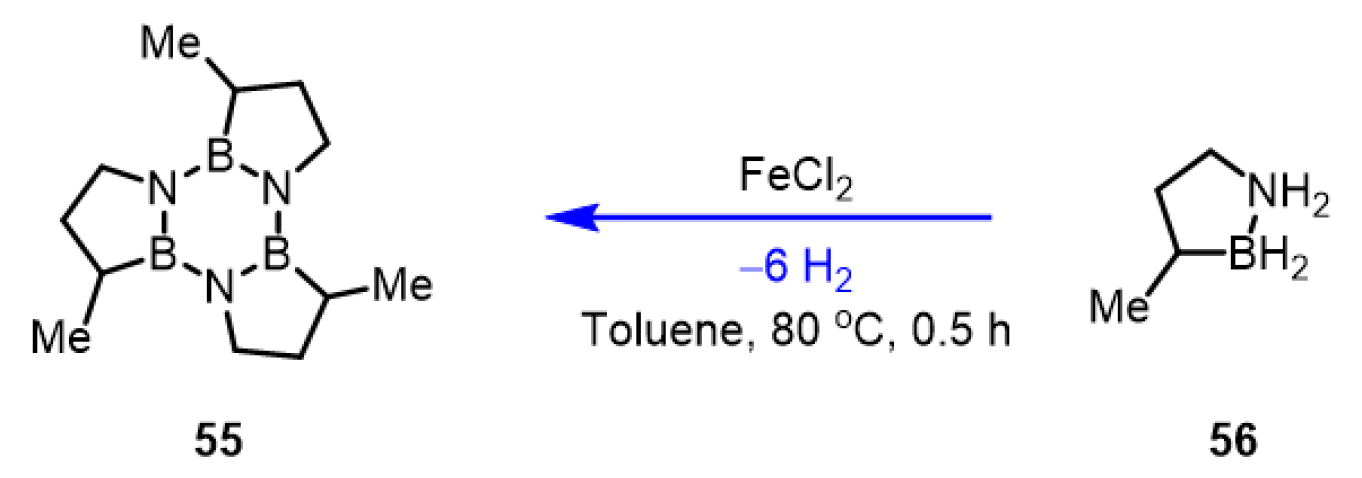
3.2.6. Amine-Borane
3.3. Oxygen-Containing Compounds
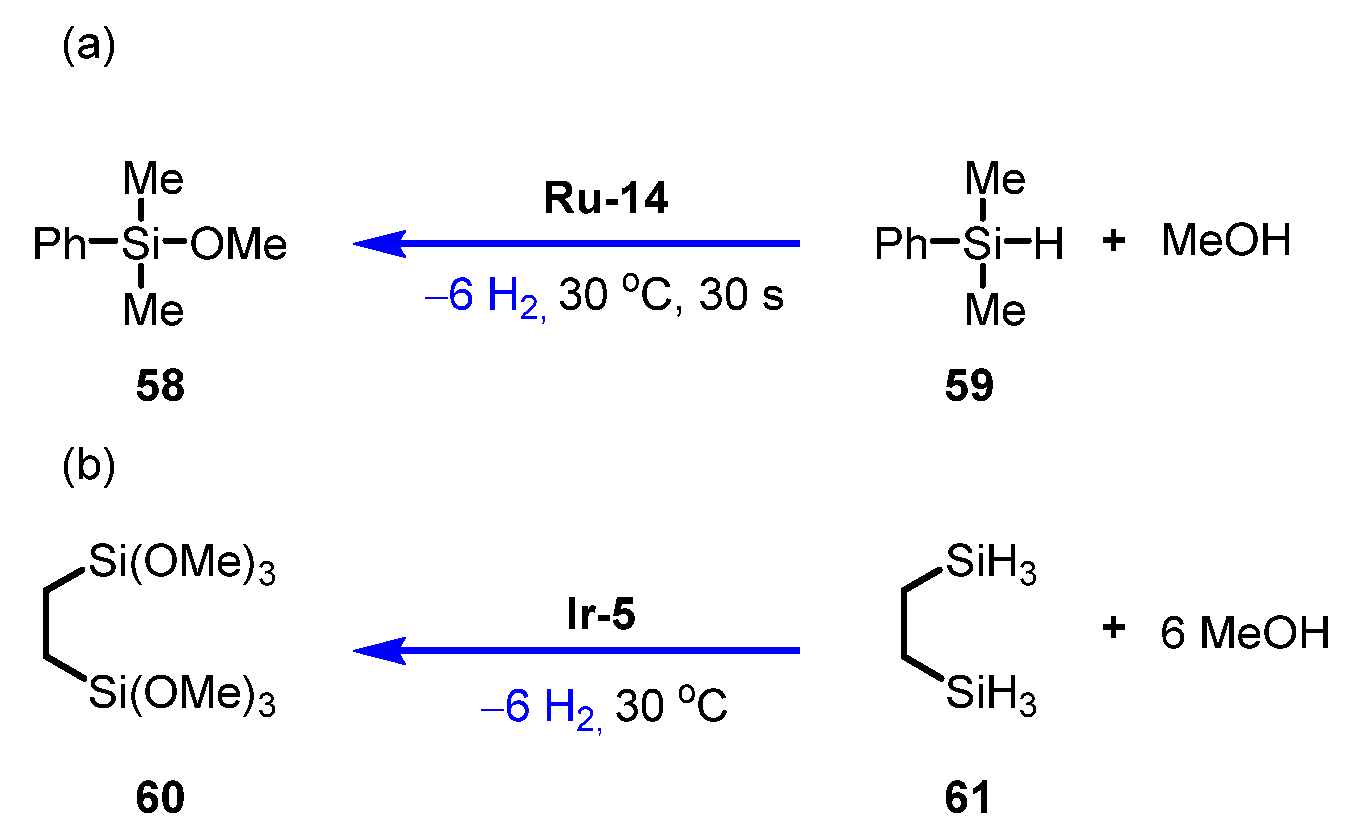
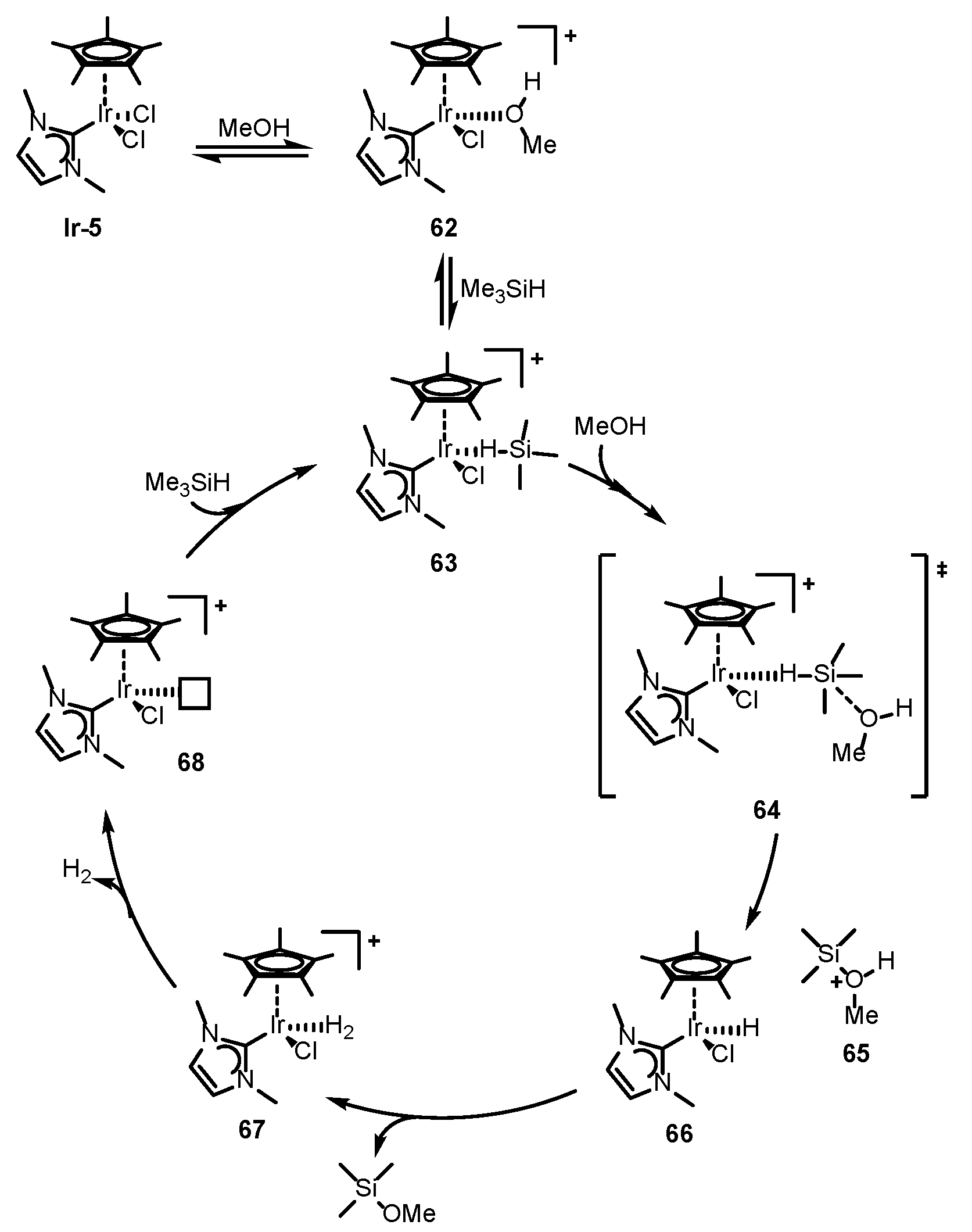
3.3.1. Silane /Alcohol Pair
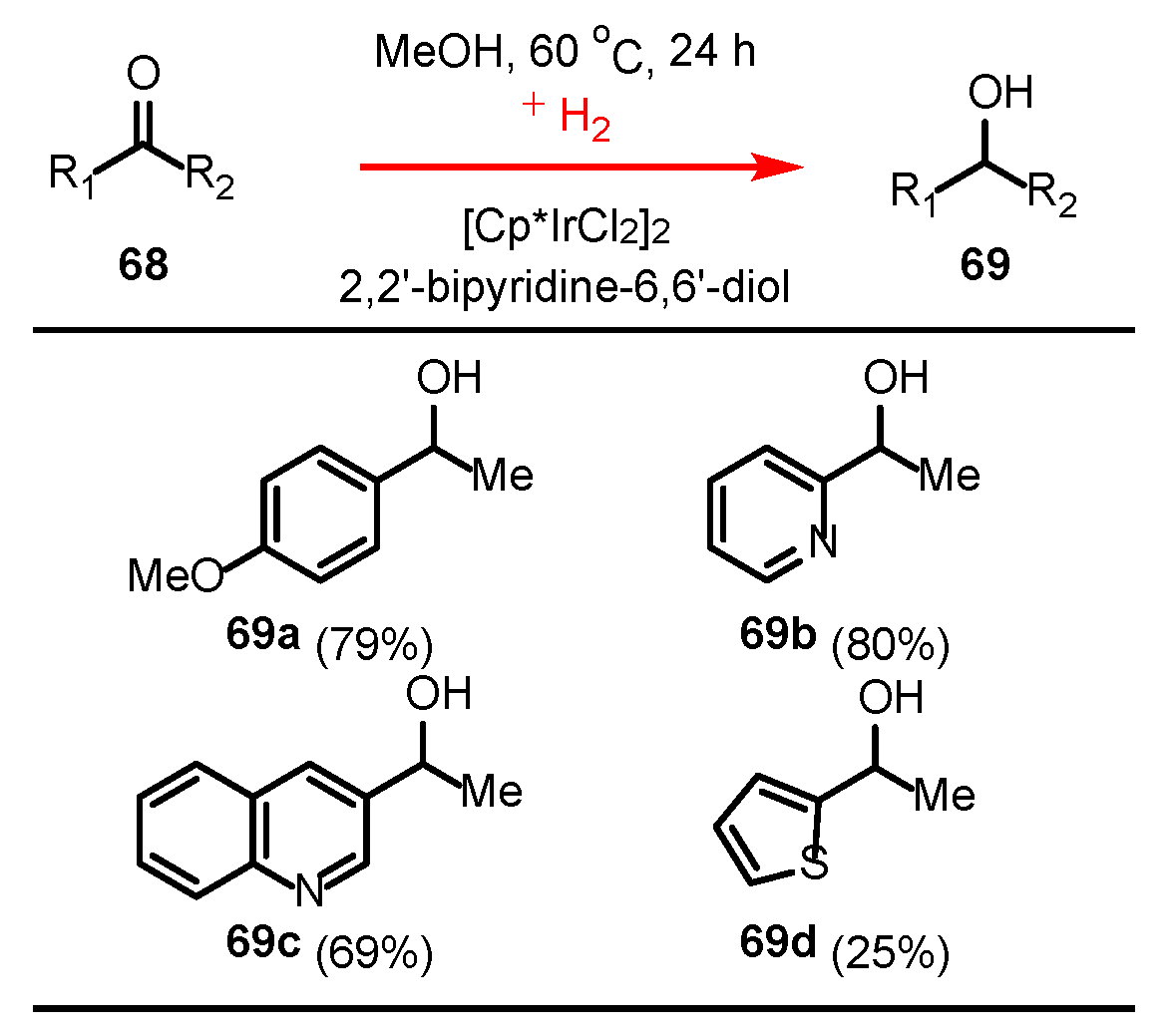
3.3.2. Ketone
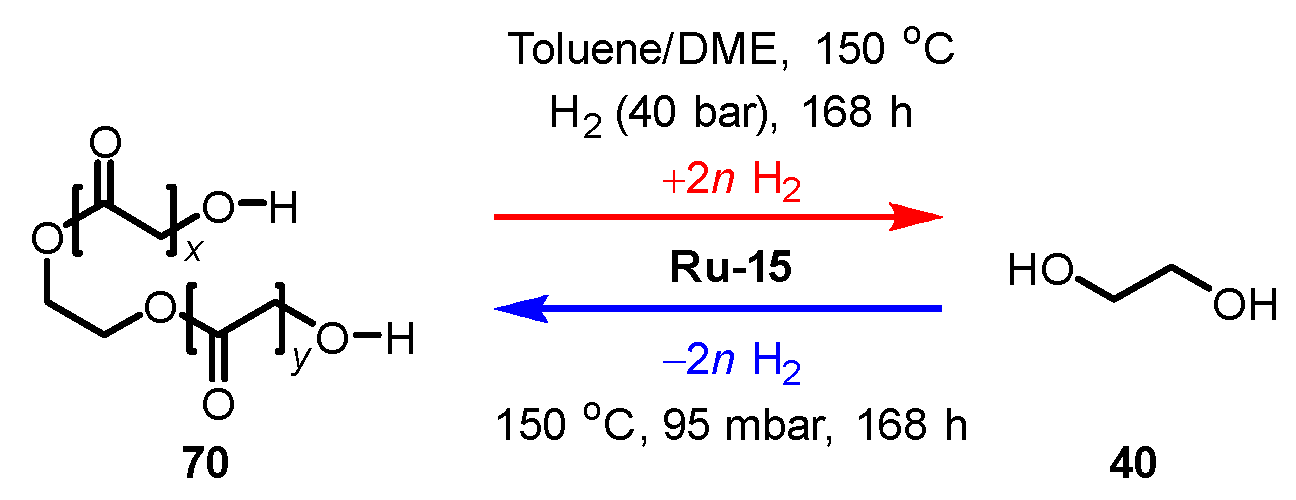
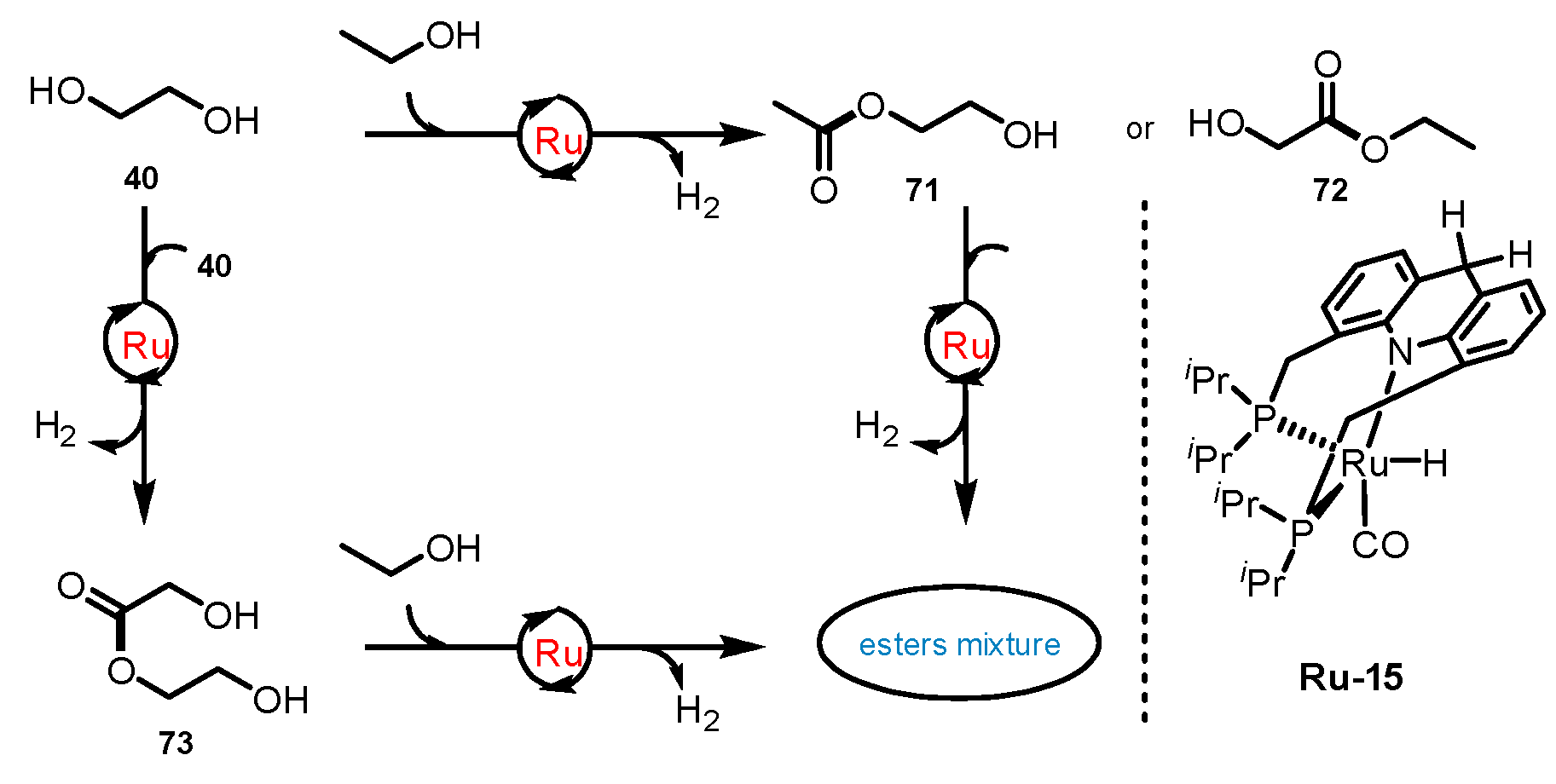
3.3.3. Ester/Alcohol Pair
4. Reactor Type for LOHC System
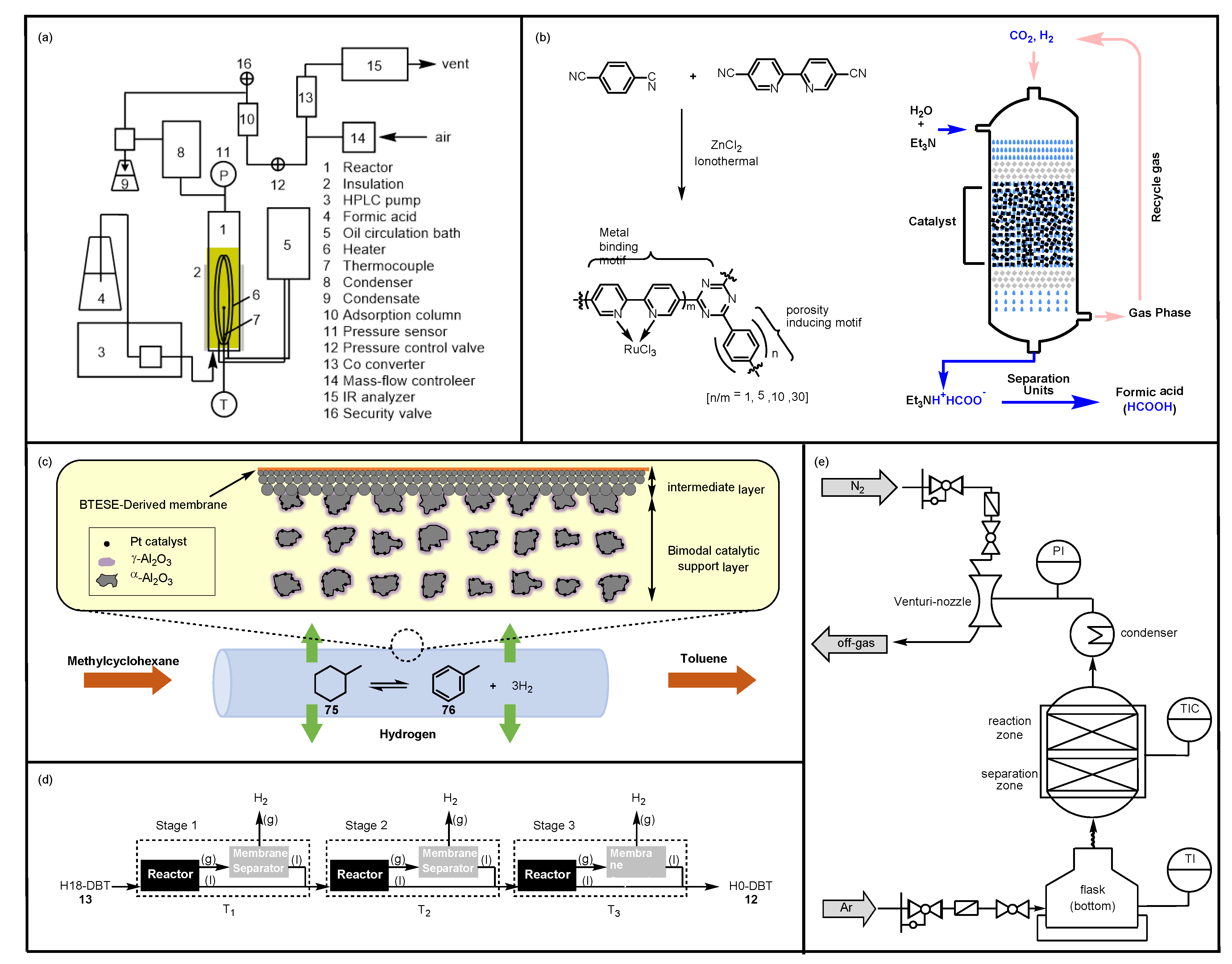
4.1. Heterogeneous Catalytic Reactor
4.2. Membrane Reactor
4.3. Hot Pressure-Swing Reactor
4.4. Reactive Distillation System
5. Conclusions
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Balzani, V.; Armaroli, N. Energy for a Sustainable World: From the Oil Age to a Sun-Powered Future; Wiley-VCH: Hoboken, NJ, USA, 2011; p. 50. ISBN 978-3-527-32540-5. [Google Scholar]
- Bockris, J.O.M. The hydrogen economy: Its history. Int. J. Hydrog. Energy 2013, 38, 2579–2588. [Google Scholar] [CrossRef]
- Singh, S.; Jain, S.; Venkateswaran, P.S.; Tiwari, A.K.; Nouni, M.R.; Pandey, J.K.; Goel, S. Hydrogen: A sustainable fuel for future of the transport sector. Renew. Sust. Energ. Rev. 2015, 51, 623–633. [Google Scholar] [CrossRef]
- Teichmann, D.; Arlt, W.; Schlücker, E.; Wasserscheid, P. Transport and Storage of Hydrogen via Liquid Organic Hydrogen Carrier (LOHC) Systems; Wiley-VCH: Hoboken, NJ, USA, 2016. [Google Scholar]
- Satyapal, S.; Petrovic, J.; Read, C.; Thomas, G.; Ordaz, G. The U.S. Department of Energy’s National Hydrogen Storage Project: Progress towards meeting hydrogen-powered vehicle requirements. Catal. Today 2007, 120, 246–256. [Google Scholar] [CrossRef] [Green Version]
- He, T.; Pachfule, P.; Wu, H.; Xu, Q.; Chen, P. Hydrogen carriers. Nat. Rev. Mater. 2016, 1, 16059–16075. [Google Scholar] [CrossRef]
- Gianotti, E.; Taillades-Jacquin, M.; Rozière, J.; Jones, D.J. High-Purity Hydrogen Generation via Dehydrogenation of Organic Carriers: A Review on the Catalytic Process. ACS Catal. 2018, 8, 4660–4680. [Google Scholar] [CrossRef]
- Dalebrook, A.F.; Gan, W.; Grasemann, M.; Moret, S.; Laurenczy, G. Hydrogen storage: Beyond conventional methods. Chem. Commun. 2013, 49, 8735–8751. [Google Scholar] [CrossRef]
- Sordakis, K.; Tang, C.; Vogt, L.K.; Junge, H.; Dyson, P.J.; Beller, M.; Laurenczy, G. Homogeneous Catalysis for Sustainable Hydrogen Storage in Formic Acid and Alcohols. Chem. Rev. 2018, 118, 372–433. [Google Scholar] [CrossRef] [PubMed]
- Clot, E.; Eisenstein, O.; Crabtree, R.H. Computational structure—Activity relationships in H2 storage: How placement of N atoms affects release temperatures in organic liquid storage materials. Chem. Commun. 2007, 22, 2231–2233. [Google Scholar] [CrossRef]
- Cooper, A.C.; Campbell, K.M.; Pez, G.P. An integrated hydrogen storage and delivery approach using organic liquid-phase carriers. Energy Conf. 2006, 16, 1–12. [Google Scholar]
- Dobereiner, G.E.; Crabtree, R.H. Dehydrogenation as a Substrate-Activating Strategy in Homogeneous Transition-Metal Catalysis. Chem. Rev. 2010, 110, 681–703. [Google Scholar] [CrossRef]
- Crabtree, R.H. Nitrogen-Containing Liquid Organic Hydrogen Carriers: Progress and Prospects. ACS Sustain. Chem. Eng. 2017, 5, 4491–4498. [Google Scholar] [CrossRef]
- Crabtree, R.H. Hydrogen storage in liquid organic heterocycles. Energy Environ. Sci. 2008, 1, 134–138. [Google Scholar] [CrossRef]
- He, T.; Pei, Q.; Chen, P. Liquid Organic Hydrogen Carriers. J. Energy Chem. 2015, 24, 587–594. [Google Scholar] [CrossRef] [Green Version]
- Preuster, P.; Papp, C.; Wasserscheid, P. Liquid Organic Hydrogen Carriers (LOHCs): Toward a Hydrogen-free Hydrogen Economy. Acc. Chem. Res. 2017, 50, 74–85. [Google Scholar] [CrossRef]
- Niermann, M.; Beckendorff, A.; Kaltschmitt, M.; Bonhoff, K. Liquid Organic Hydrogen Carrier (LOHC)—Assessment based on chemical and economic properties. Int. J. Hydrog. 2019, 44, 6631–6654. [Google Scholar] [CrossRef]
- Zhong, H.; Iguchi, M.; Chatterjee, M.; Himeda, Y.; Xu, Q.; Kawanami, H. Formic Acid-Based Liquid Organic Hydrogen Carrier System with Heterogeneous Catalysts. Adv. Sustain. Syst. 2018, 2, 1700161–1700178. [Google Scholar] [CrossRef]
- Aakko-Saksa, P.T.; Cook, C.; Kiviaho, J.; Repo, T. Liquid organic hydrogen carriers for transportation and storing of renewable energy—Review and discussion. J. Power Sources 2018, 396, 803–823. [Google Scholar] [CrossRef]
- Niermann, M.; Timmerberg, S.; Drünert, S.; Kaltschmitt, M. Liquid Organic Hydrogen Carriers and alternatives for international transport of renewable hydrogen. Renew. Sust. Energ. Rev. 2021, 135, 110171–110185. [Google Scholar] [CrossRef]
- Uhrig, F.; Kadar, J.; Müller, K. Reliability of liquid organic hydrogen carrier-based energy storage in a mobility application. Energy Sci. Eng. 2020, 8, 2044–2053. [Google Scholar] [CrossRef] [Green Version]
- Mondisha, P.M.; Ouma, C.N.M.; Garidziral, R.; Wasserscheid, P.; Bessarabov, D. The Prospect of Hydrogen Storage Using Liquid Organic Hydrogen Carriers. Energy Fuels 2019, 33, 2778–2796. [Google Scholar] [CrossRef]
- Rüde, T.; Bösmann, A.; Preuster, P.; Wasserscheid, P.; Arlt, W.; Müller, K. Resilience of Liquid Organic Hydrogen Carrier Based Energy-Storage Systems. Energy Technol. 2018, 6, 529–539. [Google Scholar] [CrossRef] [Green Version]
- Turunen, H. CO2-Balance in the Atmosphere and CO2-Utilisation: An. Engineering Approach; Acta University Oulu: Oulu, Finland, 2011; p. 386. ISBN 978-951-42-9487-7. [Google Scholar]
- Markiewicz, M.; Zhang, Y.Q.; Bösmann, A.; Brückner, N.; Thöming, J.; Wasserscheid, P.; Stolte, S. Environmental and health impact assessment of Liquid Organic Hydrogen Carrier (LOHC) systems—Challenges and preliminary results. Energy Environ. Sci. 2015, 8, 1035–1045. [Google Scholar] [CrossRef] [Green Version]
- Drury, D.J. Formic acid. In Kirk-Othmer Encyclopedia of Chemical Technology; John Wiley & Sons: New York, NY, USA, 2013; Volume 11, pp. 951–958. [Google Scholar]
- Langer, R.; Diskin-Posner, Y.; Leitus, G.; Shimon, L.J.W.; Ben-David, Y.; Milstein, D. Low-Pressure Hydrogenation of Carbon Dioxide Catalyzed by an Iron Pincer Complex Exhibiting Noble Metal Activity. Angew. Chem. Int. Ed. 2011, 50, 9948–9952. [Google Scholar] [CrossRef]
- Filonenko, G.A.; van Putten, R.; Schulpen, E.N.; Hensen, E.J.M.; Pidko, E.A. Highly Efficient Reversible Hydrogenation of Carbon Dioxide to Formates Using a Ruthenium PNP-Pincer Catalyst. ChemCatChem 2014, 6, 1526–1530. [Google Scholar] [CrossRef]
- Kothandaraman, J.; Czaun, M.; Goeppert, A.; Haiges, R.; Jones, J.P.; May, R.B.; Prakash, G.K.S.; Olah, G.A. Amine-Free Reversible Hydrogen Storage in Formate Salts Catalyzed by Ruthenium Pincer Complex without pH Control or Solvent Change. ChemSusChem 2015, 8, 1442–1451. [Google Scholar] [CrossRef]
- Bi, Q.Y.; Lin, J.D.; Liu, Y.M.; He, H.Y.; Huang, F.Q.; Cao, Y. Dehydrogenation of Formic Acid at Room Temperature: Boosting Palladium Nanoparticle Efficiency by Coupling with Pyridinic-Nitrogen-Doped Carbon. Angew. Chem. Int. Ed. 2016, 55, 11849–11853. [Google Scholar] [CrossRef]
- Montandon-Clerc, M.; Laurenczy, G. Additive free, room temperature direct homogeneous catalytic carbon dioxide hydrogenation in aqueous solution using an iron(II) phosphine catalyst. J. Catal. 2018, 362, 76–84. [Google Scholar] [CrossRef]
- Patra, S.; Awasthi, M.K.; Rai, R.K.; Deka, H.; Mobin, S.M.; Singh, S.K. Dehydrogenation of Formic Acid Catalyzed by Water-Soluble Ruthenium Complexes: X-ray Crystal Structure of a Diruthenium Complex. Eur. J. Inorg. Chem. 2019, 2019, 1046–1053. [Google Scholar] [CrossRef]
- Zhou, W.; Wei, Z.; Spannenberg, A.; Jiao, H.; Junge, K.; Junge, H.; Beller, M. Cobalt-Catalyzed Aqueous Dehydrogenation of Formic Acid. Chem. Eur. J. 2019, 25, 8459–8464. [Google Scholar] [CrossRef] [PubMed]
- Anderson, N.H.; Boncella, J.; Tondreau, A.M. Manganese-Mediated Formic Acid Dehydrogenation. Chem. Eur. J. 2019, 25, 10557–10560. [Google Scholar] [CrossRef]
- Léval, A.; Agapova, A.; Steinlechner, C.; Alberico, E.; Junge, H.; Beller, M. Hydrogen production from formic acid catalyzed by a phosphine free manganese complex: Investigation and mechanistic insights. Green Chem. 2020, 22, 913–920. [Google Scholar] [CrossRef]
- Wang, W.H.; Wang, H.; Yang, Y.; Lai, X.; Li, Y.; Wang, J.; Himeda, Y.; Bao, M. Synergistic Effect of Pendant N Moieties for Proton Shuttling in the Dehydrogenation of Formic Acid Catalyzed by Biomimetic IrIII Complexes. ChemSusChem 2020, 13, 5015–5022. [Google Scholar] [CrossRef]
- Li, X.; Surkus, A.E.; Rabeah, J.; Anwar, M.; Dastigir, S.; Junge, H.; Brückner, A.; Beller, M. Cobalt Single-Atom Catalysts with High Stability for Selective Dehydrogenation of Formic Acid. Angew. Chem. Int. Ed. 2020, 59, 15849–15854. [Google Scholar] [CrossRef]
- Trincado, M.; Grützmacher, H.; Prechtl, M.H.G. CO2-based hydrogen storage—Hydrogen generation from formaldehyde/water. Phys. Sci. Rev. 2018, 3, 20170013. [Google Scholar]
- Fujita, K.I.; Kawahara, R.; Aikawa, T.; Yamaguchi, R. Hydrogen Production from a Methanol–Water Solution Catalyzed by an Anionic Iridium Complex Bearing a Functional Bipyridonate Ligand under Weakly Basic Conditions. Angew. Chem. Int. Ed. 2015, 54, 9057–9060. [Google Scholar] [CrossRef]
- Heim, L.E.; Schlörer, N.E.; Choi, J.H.; Prechtl, M.H.G. Selective and mild hydrogen production using water and formaldehyde. Nat. Commun. 2014, 5, 3621–3628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suenobu, T.; Isaka, Y.; Shibata, S.; Fukuzumi, S. Catalytic hydrogen production from paraformaldehyde and water using an organoiridium complex. Chem. Commun. 2015, 51, 1670–1672. [Google Scholar] [CrossRef] [Green Version]
- Trincado, M.; Sinha, V.; Rodriguez-Lugo, R.E.; Pribanic, B.; Bruin, B.D.; Grützmacher, H. Homogeneously catalysed conversion of aqueous formaldehyde to H2 and carbonate. Nat. Commun. 2017, 8, 14990–15001. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Ertem, M.Z.; Murata, K.; Muckerman, J.T.; Fujita, E.; Himeda, Y. Highly Efficient and Selective Methanol Production from Paraformaldehyde and Water at Room Temperature. ACS Catal. 2018, 8, 5233–5239. [Google Scholar] [CrossRef]
- Heim, L.E.; Vallazza, S.; van der Waalsa, D.; Prechtl, M.H.G. Water decontamination with hydrogen production using microwave-formed minute-made ruthenium catalysts. Green Chem. 2016, 18, 1469–1474. [Google Scholar] [CrossRef] [Green Version]
- Awasthi, M.K.; Singh, S.K. Ruthenium catalyzed hydrogen production from formaldehyde—Water solution. Sustain. Energy Fuels 2021, 5, 549–555. [Google Scholar] [CrossRef]
- Li, Y.; Liu, S.; Yao, L.; Ji, W.; Au, C.T. Core-shell structured iron nanoparticles for the generation of COx-free hydrogen via ammonia decomposition. Catal. Commun. 2010, 11, 368–372. [Google Scholar] [CrossRef]
- Okura, K.; Okanishi, T.; Muroyama, H.; Matsui, T.; Eguchi, K. Promotion effect of rare-earth elements on the catalytic decomposition of ammonia over Ni/Al2O3 catalyst. APPL 2015, 505, 77–85. [Google Scholar] [CrossRef] [Green Version]
- Yin, S.F.; Zhang, Q.H.; Xu, B.Q.; Zhu, W.X.; Ng, C.F.; Au, C.T. Investigation on the catalysis of COx-free hydrogen generation from ammonia. J. Catal. 2004, 224, 384–396. [Google Scholar] [CrossRef]
- Yin, S.F.; Xu, B.Q.; Zhou, X.P.; Au, C.T. A mini-review on ammonia decomposition catalysts for on-site generation of hydrogen for fuel cell applications. APPL 2004, 277, 1–9. [Google Scholar] [CrossRef]
- Pelka, R.; Moszyńska, I.; Arabczyk, W. Catalytic Ammonia Decomposition Over Fe/Fe4N. Catal. Lett. 2009, 128, 72–76. [Google Scholar] [CrossRef]
- Yao, L.; Shi, T.; Li, Y.; Zhao, J.; Ji, W.; Au, C.T. Core–shell structured nickel and ruthenium nanoparticles: Very active and stable catalysts for the generation of COx-free hydrogen via ammonia decomposition. Catal. Today 2011, 164, 112–118. [Google Scholar] [CrossRef]
- Zheng, W.; Cotter, T.P.; Kaghazchi, P.; Jacob, T.; Frank, B.; Schlichte, K.; Zhang, W.; Su, D.S.; Schuth, F.; Schlogl, R. Experimental and Theoretical Investigation of Molybdenum Carbide and Nitride as Catalysts for Ammonia Decomposition. J. Am. Chem. Soc. 2013, 135, 3458–3464. [Google Scholar] [CrossRef] [PubMed]
- Tuuzani, A.; Belanger, G.; Klvana, D. Dehydrogenation reactor for a vehicle equipped with a hydrogen engine: A simulation study. Int. J. Hydrog. Energy 1984, 9, 929–936. [Google Scholar] [CrossRef]
- Cacciola, G.; Giordano, N.; Restuccia, G. Cyclohexane as a liquid phase carrier in hydrogen storage and transport. Int. J. Hydrog. Energy 1984, 9, 411–419. [Google Scholar] [CrossRef]
- Taube, M.; Rippin, D.W.T.; Cresswell, D.L.; Knecht, W. A system of hydrogen-powered vehicles with liquid organic hydrides. Int. J. Hydrog. Energy 1983, 8, 213–225. [Google Scholar] [CrossRef]
- Taube, M.; Rippin, D.; Knecht, W.; Hakimifard, D.; Milisavljevic, B.; Gruenefelder, N. A prototype truck powered by hydrogen from organic liquid hydrides. Int. J. Hydrog. Energy 1985, 10, 595–599. [Google Scholar] [CrossRef]
- Jorschick, H.; Bösmann, A.; Preuster, P.; Wasserscheid, P. Charging a Liquid Organic Hydrogen Carrier System with H2/CO2 Gas Mixtures. ChemCatChem 2018, 10, 4329–4337. [Google Scholar] [CrossRef]
- Bruckner, N.; Obesser, K.; Bosmann, A.; Teichmann, D.; Arlt, W.; Dungs, J.; Wasserscheid, P. Evaluation of Industrially Applied Heat-Transfer Fluids as Liquid Organic Hydrogen Carrier Systems. ChemSusChem 2014, 7, 229–235. [Google Scholar] [CrossRef]
- Kalenchuk, A.N.; Bogdan, V.I.; Dunaev, S.; Kustov, L.M. Effect of Isomerization on the Reversible Reaction of Hydrogenation-Dehydrogenation of ortho-Terphenyl on a Pt/C Catalyst. Chem. Eng. Technol. 2018, 41, 1842–1846. [Google Scholar] [CrossRef]
- Jang, M.; Jo, Y.S.; Lee, W.J.; Shin, B.S.; Sohn, H.; Jeong, H.; Jang, S.C.; Kwak, S.K.; Kang, J.W.; Yoon, C.W. A High-Capacity, Reversible Liquid Organic Hydrogen Carrier: H2-Release Properties and an Application to a Fuel Cell. ACS Sustain. Chem. Eng. 2019, 7, 1185–1194. [Google Scholar] [CrossRef]
- Dean, D.; Davis, B.; Jessop, P.G. The effect of temperature, catalyst and sterics on the rate of N-heterocycledehydrogenation for hydrogenstorage. New J. Chem. 2011, 35, 417–422. [Google Scholar] [CrossRef]
- Wechsler, D.; Davis, B.; Jessop, P.G. The dehydrogenation of combined organic and inorganic hydrogen-storage carriers. Can. J. Chem. 2010, 88, 548–555. [Google Scholar] [CrossRef]
- Smith, A.M.; Whyman, R. Review of Methods for the Catalytic Hydrogenation of Carboxamides. Chem. Rev. 2014, 114, 5477–5510. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, J.; Filonenko, G.A.; van Putten, R.; Hensen, E.J.M.; Pidko, E.A. Heterogeneous and homogeneous catalysis for the hydrogenation of carboxylic acid derivatives: History, advances and future directions. Chem. Soc. Rev. 2015, 44, 3808–3833. [Google Scholar] [CrossRef] [Green Version]
- Yamaguchi, R.; Ikeda, C.; Takahashi, Y.; Fujita, K. Homogeneous Catalytic System for Reversible Dehydrogenation−Hydrogenation Reactions of Nitrogen Heterocycles with Reversible Interconversion of Catalytic Species. J. Am. Chem. Soc. 2009, 131, 8410–8412. [Google Scholar] [CrossRef] [PubMed]
- Forberg, D.; Schwob, T.; Zaheer, M.; Friedrich, M.; Miyajima, N.; Kempe, R. Single-catalyst high-weight% hydrogen storage in an N-heterocycle synthesized from lignin hydrogenolysis products and ammonia. Nat. Commun. 2016, 7, 13201. [Google Scholar] [CrossRef]
- Fujita, K.; Wada, T.; Shiraishi, T. Reversible Interconversion between 2,5-Dimethylpyrazine and 2,5-Dimethylpiperazine by Iridium-Catalyzed Hydrogenation/Dehydrogenation for Efficient Hydrogen Storage. Angew. Chem. Int. Ed. 2017, 56, 10886–10889. [Google Scholar] [CrossRef]
- Vivancos, Á.; Beller, M.; Albrecht, M. NHC-Based Iridium Catalysts for Hydrogenation and Dehydrogenation of N-Heteroarenes in Water under Mild Conditions. ACS Catal. 2018, 8, 9945–9957. [Google Scholar] [CrossRef]
- Søgaard, A.; Scheuermeyer, M.; Bösmann, A.; Wasserscheid, P.; Riisager, A. Homogeneously-catalysed hydrogen release/storage using the 2-methylindole/2-methylindoline LOHC system in molten salt-organic biphasic reaction systems. Chem. Commun. 2019, 55, 2046–2049. [Google Scholar] [CrossRef]
- Liu, Y.; Yu, T.; Zeng, Y.; Chen, J.; Yang, G.; Li, Y. Efficient acceptorless dehydrogenation of hydrogen-rich N-heterocycles photocatalyzed by Ni(OH)2@CdSe/CdS quantum dots. Catal. Sci. Technol. 2021, 11, 3810–3817. [Google Scholar] [CrossRef]
- Xie, Y.; Milstein, D. Pd Catalyzed, Acid Accelerated, Rechargeable, Liquid Organic Hydrogen Carrier System Based on Methylpyridines/Methylpiperidines. ACS Appl. Energy Mater. 2019, 2, 4302–4308. [Google Scholar] [CrossRef]
- Zubar, V.; Borghs, J.C.; Rueping, M. Hydrogenation or Dehydrogenation of N-Containing Heterocycles Catalyzed by a Single Manganese Complex. Org. Lett. 2020, 22, 3974–3978. [Google Scholar] [CrossRef]
- Hu, P.; Fogler, E.; Diskin-Posner, Y.; Iron, M.A.; Milstein, D. A novel liquid organic hydrogen carrier system based on catalytic peptide formation and hydrogenation. Nat. Commun. 2015, 6, 6859–6865. [Google Scholar] [CrossRef]
- Kothandaraman, J.; Kar, S.; Sen, R.; Goeppert, A.; Olah, G.A.; Prakash, G.K.S. Efficient Reversible Hydrogen Carrier System Based on Amine Reforming of Methanol. J. Am. Chem. Soc. 2017, 139, 2549–2552. [Google Scholar] [CrossRef]
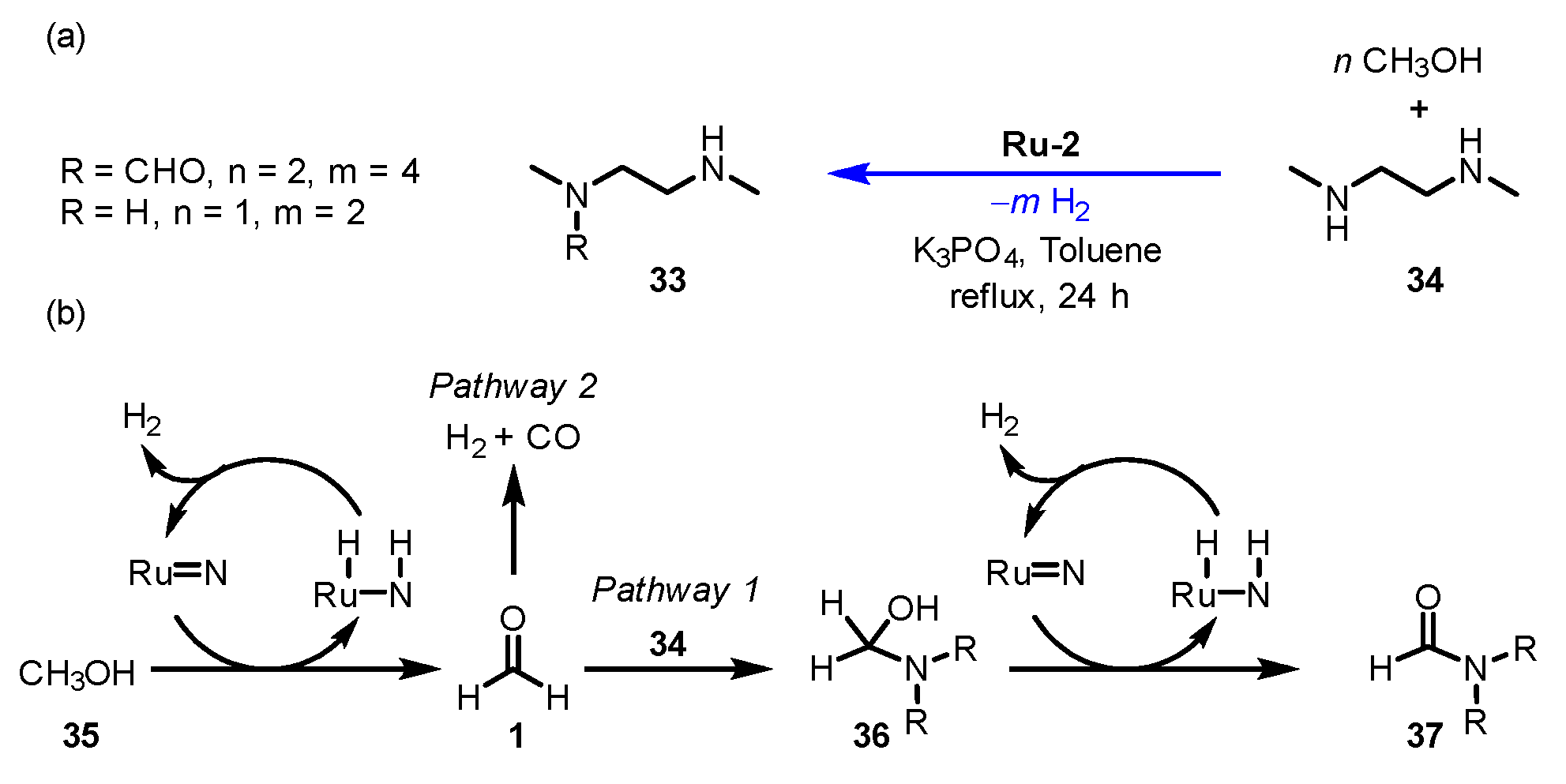
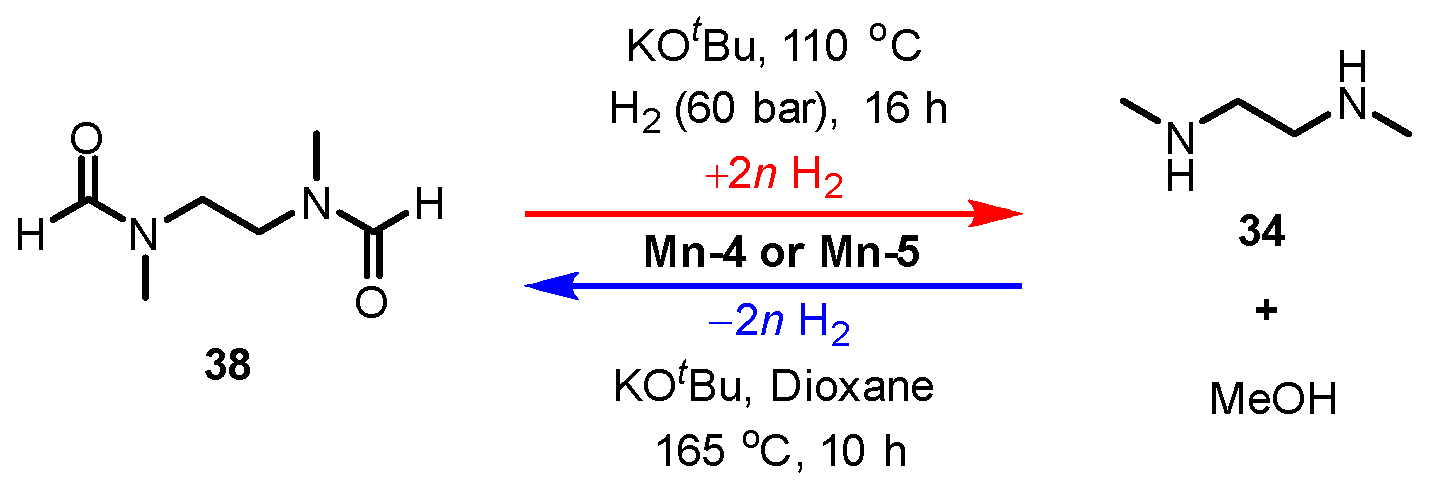
- Shao, Z.; Li, Y.; Liu, C.; Ai, W.; Luo, S.P.; Liu, Q. Reversible interconversion between methanoldiamineand diamide for hydrogen storage based on manganese catalyzed (de)hydrogenation. Nat. Commun. 2020, 11, 591–597. [Google Scholar] [CrossRef] [Green Version]
- Zou, Y.Q.; Zhou, Q.Q.; Diskin-Posner, Y.; Ben-David, Y.; Milstein, D. Synthesis of oxalamides by acceptorless dehydrogenative coupling of ethylene glycol and amines and the reverse hydrogenation catalyzed by ruthenium. Chem. Sci. 2020, 11, 7188–7193. [Google Scholar] [CrossRef]
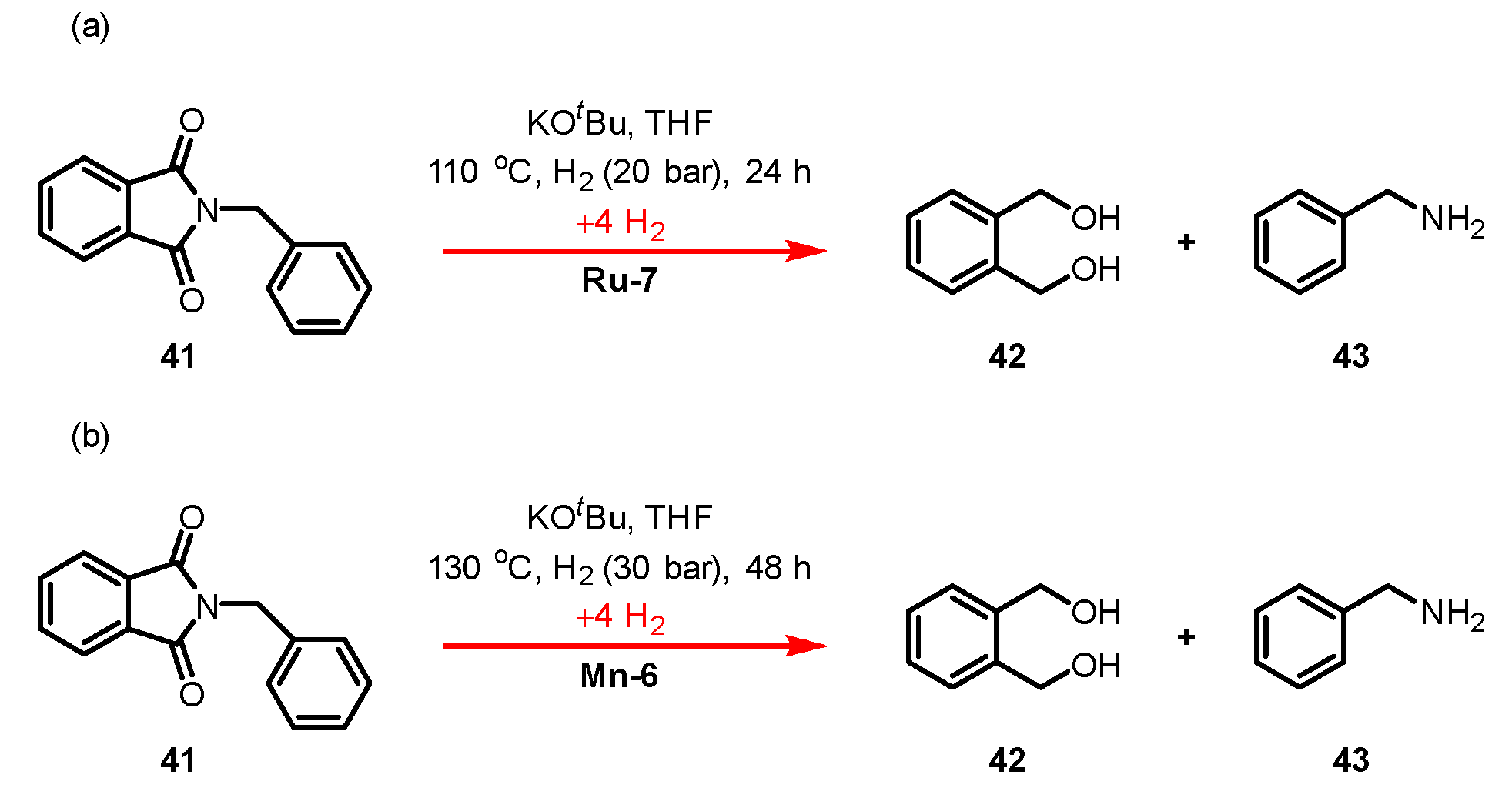
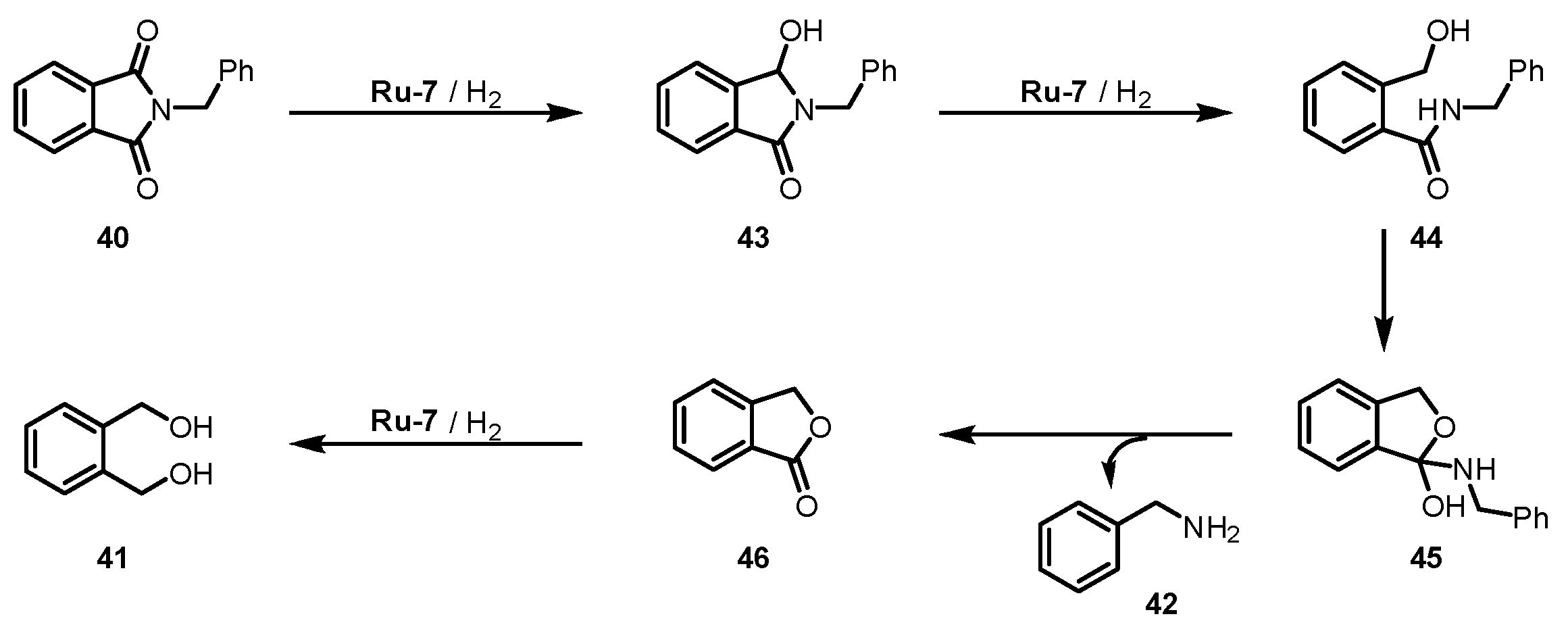
- Kumar, A.; Janes, T.; Espinosa-Jalapa, N.A.; Milstein, D. Selective Hydrogenation of Cyclic Imides to Diols and Amines and Its Application in the Development of a Liquid Organic Hydrogen Carrier. J. Am. Chem. Soc. 2018, 140, 7453–7457. [Google Scholar] [CrossRef]
- Das, U.K.; Jane, T.; Kumar, A.; Milstein, D. Manganese catalyzed selective hydrogenation of cyclic imides to diols and amines. Green Chem. 2020, 22, 3079–3082. [Google Scholar] [CrossRef]
- Xie, Y.; Hu, P.; Ben-David, Y.; Milstein, D. A Reversible Liquid Organic Hydrogen Carrier System Based on Methanol-Ethylenediamine and Ethylene Urea. Angew. Chem. Int. Ed. 2019, 58, 5105–5109. [Google Scholar] [CrossRef]
- Das, U.K.; Kumar, A.; Ben-David, Y.; Iron, M.A.; Milstein, D. Manganese Catalyzed Hydrogenation of Carbamates and Urea Derivatives. J. Am. Chem. Soc. 2019, 141, 12962–12966. [Google Scholar] [CrossRef]
- Reguillo, R.; Grellier, M.; Vautravers, N.; Vendier, L.; Sabo-Etienne, S. Ruthenium-Catalyzed Hydrogenation of Nitriles: Insights into the Mechanism. J. Am. Chem. Soc. 2010, 132, 7854–7855. [Google Scholar] [CrossRef] [PubMed]
- Tseng, K.N.T.; Rizzi, A.M.; Szymczak, N.K. Oxidant-Free Conversion of Primary Amines to Nitriles. J. Am. Chem. Soc. 2013, 135, 16352–16355. [Google Scholar] [CrossRef] [PubMed]
- Ventura-Espinosa, D.; Marzá-Beltrán, A.; Mata, J.A. Catalytic Hydrogen Production by Ruthenium Complexes from the Conversion of Primary Amines to Nitriles: Potential Application as a Liquid Organic Hydrogen Carrier. Chem. Eur. J. 2016, 22, 17758–17766. [Google Scholar] [CrossRef]
- Luo, W.; Campbell, P.G.; Zakharov, L.N.; Liu, S.Y. A Single-Component Liquid-Phase Hydrogen Storage Material. J. Am. Chem. Soc. 2011, 133, 19326–19329. [Google Scholar] [CrossRef]
- Stöcker, M. Methanol-to-hydrocarbons: Catalytic materials and their behavior. Microporous Mesoporous Mater. 1999, 29, 3–48. [Google Scholar] [CrossRef]
- Haw, J.F.; Song, W.; Marcus, D.M.; Nicholas, J.B. The mechanism of methanol to hydrocarbon catalysis. Acc. Chem. Res. 2003, 36, 317–326. [Google Scholar] [CrossRef] [PubMed]
- Olsbye, U.; Bjørgen, M.; Svelle, S.; Lillerud, K.P.; Kolboe, S. Mechanistic insight into the methanol-to-hydrocarbons reaction. Catal. Today 2005, 106, 108–111. [Google Scholar] [CrossRef]
- Palo, D.R.; Dagle, R.A.; Holladay, J.D. Methanol Steam Reforming for Hydrogen Production. Chem. Rev. 2007, 107, 3992–4021. [Google Scholar] [CrossRef] [PubMed]
- Olah, G.A.; Goeppert, A.; Prakash, G.K.S. Beyond Oil and Gas: The Methanol Economy; Wiley-VCH: Hoboken, NJ, USA, 2009; ISBN 9783527324224. [Google Scholar]
- Olah, G.A. Towards Oil Independence Through Renewable Methanol Chemistry. Angew. Chem. Int. Ed. 2013, 52, 104–107. [Google Scholar] [CrossRef]
- Shen, Y.; Zhan, Y.; Li, S.; Ning, F.; Du, Y.; Huang, Y.; He, T.; Zhou, X. Hydrogen generation from methanol at near-room temperature. Chem. Sci. 2017, 8, 7498–7504. [Google Scholar] [CrossRef] [Green Version]
- Ventura-Espinosa, D.; Carretero-Cerdán, A.; Baya, M.; García, H.; Mata, J.A. Catalytic Dehydrogenative Coupling of Hydrosilanes with Alcohols for the Production of Hydrogen On-demand: Application of a Silane/Alcohol Pair as a Liquid Organic Hydrogen Carrier. Chem. Eur. J. 2017, 23, 10815–10821. [Google Scholar] [CrossRef]
- Ventura-Espinosa, D.; Sabater, S.; Carretero-Cerdan, A.; Baya, M.; Mata, J.A. High Production of Hydrogen on Demand from Silanes Catalyzed by Iridium Complexes as a Versatile Hydrogen Storage System. ACS Catal. 2018, 8, 2558–2566. [Google Scholar] [CrossRef] [Green Version]
- Garg, N.; Paira, S.; Sundararaju, B. Efficient Transfer Hydrogenation of Ketones using Methanol as Liquid Organic Hydrogen Carrier. ChemCatChem 2020, 12, 3472–3476. [Google Scholar] [CrossRef]
- Zou, Y.Q.; von Wolff, N.; Anaby, A.; Xie, Y.; Milstein, D. Ethylene Glycol as an Efficient and Reversible Liquid Organic Hydrogen Carrier. Nat. Catal. 2019, 2, 415–422. [Google Scholar] [CrossRef]
- Zhou, Q.Q.; Zou, Y.Q.; Ben-David, Y.; Milstein, D. A Reversible Liquid-to-Liquid Organic Hydrogen Carrier System Based on Ethylene Glycol and Ethanol. Chem. Eur. J. 2020, 26, 15487–15490. [Google Scholar] [CrossRef]
- Fellay, C.; Yan, N.; Dyson, P.J.; Laurenczy, G. Selective formic acid decomposition for high-pressure hydrogen generation: A mechanistic study. Chem. Eur. J. 2009, 15, 3752–3760. [Google Scholar] [CrossRef] [PubMed]
- Orava, V.; Souček, O.; Cendula, P. Multi-phase modeling of non-isothermal reactive flow in fluidized bed reactors. J. Comput. Appl. Math. 2015, 289, 282–295. [Google Scholar] [CrossRef]
- Mellmann, D.; Sponholz, P.; Junge, H.; Beller, M. Formic acid as a hydrogen storage material—Development of homogeneous catalysts for selective hydrogen release. Chem. Soc. Rev. 2016, 45, 3954–3988. [Google Scholar] [CrossRef] [PubMed]
- van Putten, R.; Wissink, T.; Swinkels, T.; Padko, E.A. Fuelling the hydrogen economy: Scale-up of an integrated formic acid-to-power system. Int. J. Hydrog. Energy 2019, 44, 28533–28541. [Google Scholar] [CrossRef]
- Peters, R.; Deja, R.; Fang, Q.; Nguyen, V.N.; Preuster, P.; Blum, L.; Wasserscheid, P.; Stolten, D. A solid oxide fuel cell operating on liquid organic hydrogen carrier-based hydrogen—A kinetic model of the hydrogen release unit and system performance. Int. J. Hydrog. Energy 2019, 44, 13794–13806. [Google Scholar] [CrossRef]
- Yuranov, I.; Autissier, N.; Sordakis, K.; Dalebrook, A.F.; Grasemann, M.; Orava, V.; Cendula, P.; Gubler, L.; Laurenczy, G. Heterogeneous Catalytic Reactor for Hydrogen Production from Formic Acid and Its Use in Polymer Electrolyte Fuel Cells. ACS Sustain. Chem. Eng. 2018, 6, 6635–6643. [Google Scholar] [CrossRef]
- Hwang, S.; Smith, R. Heterogeneous catalytic reactor design with optimum temperature profile I: Application of catalyst dilution and side-stream distribution. Chem. Eng. Sci. 2004, 59, 4229–4243. [Google Scholar] [CrossRef]
- Javaid, R.; Kawasaki, S.; Suzuki, A.; Suzuki, T.M. Simple and rapid hydrogenation of p-nitrophenol with aqueous formic acid in catalytic flow reactors. Beilstein. J. Org. Chem. 2013, 9, 1156–1163. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Javaid, R.; Qzai, U.Y.; Kawasaki, S. Efficient and Continuous Decomposition of Hydrogen Peroxide Using a Silica Capillary Coated with a Thin Palladium or Platinum Layer. Bull. Chem. Soc. Jpn. 2015, 88, 976–980. [Google Scholar] [CrossRef] [Green Version]
- Park, K.; Gunasekar, G.H.; Kim, S.H.; Park, H.; Kim, S.; Park, K.; Jung, K.D.; Yoon, S. CO2 hydrogenation to formic acid over heterogenized ruthenium catalysts using a fixed bed reactor with separation units. Green Chem. 2020, 22, 1639–1649. [Google Scholar] [CrossRef]
- Meng, L.; Yu, X.; Niimi, T.; Nagasawa, H.; Kanezashi, M.; Yoshioka, T.; Tsuru, T. Methylcyclohexane Dehydrogenation for Hydrogen Production via a Bimodal Catalytic Membrane Reactor. AIChE J. 2015, 61, 1628–1638. [Google Scholar] [CrossRef]
- Wunsch, A.; Mohr, M.; Pfeifer, P. Intensified LOHC-Dehydrogenation Using Multi-Stage Microstructures and Pd-Based Membranes. Membranes 2018, 8, 112. [Google Scholar] [CrossRef] [Green Version]
- Jorschick, H.; Preuster, P.; Durr, S.; Seidel, A.; Muller, K.; Bosmann, A.; Wasserscheid, P. Hydrogen storage using a hot pressure swing reactor. Energy Environ. Sci. 2017, 10, 1652–1659. [Google Scholar] [CrossRef] [Green Version]
- Geißelbrecht, M.; Mrusek, S.; Muller, K.; Preuster, P.; Bosmann, A.; Wasserscheid, P. Highly efficient, low-temperature hydrogen release from perhydro-benzyltoluene using reactive distillation. Energy Environ. Sci. 2020, 13, 3119–3128. [Google Scholar] [CrossRef]





















Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Cho, J.-Y.; Kim, H.; Oh, J.-E.; Park, B.Y. Recent Advances in Homogeneous/Heterogeneous Catalytic Hydrogenation and Dehydrogenation for Potential Liquid Organic Hydrogen Carrier (LOHC) Systems. Catalysts 2021, 11, 1497. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11121497
Cho J-Y, Kim H, Oh J-E, Park BY. Recent Advances in Homogeneous/Heterogeneous Catalytic Hydrogenation and Dehydrogenation for Potential Liquid Organic Hydrogen Carrier (LOHC) Systems. Catalysts. 2021; 11(12):1497. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11121497
Chicago/Turabian StyleCho, Jun-Young, Hahyeon Kim, Jeong-Eun Oh, and Boyoung Y. Park. 2021. "Recent Advances in Homogeneous/Heterogeneous Catalytic Hydrogenation and Dehydrogenation for Potential Liquid Organic Hydrogen Carrier (LOHC) Systems" Catalysts 11, no. 12: 1497. https://0-doi-org.brum.beds.ac.uk/10.3390/catal11121497