
Catalytic Hydrofunctionalization Reactions of 1,3-Diynes


Departamento de Química Orgánica e Inorgánica, Facultad de Química, Universidad de Oviedo, Julián Clavería 8, E-33006 Oviedo, Spain
Catalysts 2022, 12(1), 89; https://0-doi-org.brum.beds.ac.uk/10.3390/catal12010089
Submission received: 16 December 2021
/
Revised: 10 January 2022
/
Accepted: 10 January 2022
/
Published: 13 January 2022
(This article belongs to the Special Issue 10th Anniversary of Catalysts: Molecular Catalysis)
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:Metal-catalyzed hydrofunctionalization reactions of alkynes, i.e., the addition of Y–H units (Y = heteroatom or carbon) across the carbon–carbon triple bond, have attracted enormous attention for decades since they allow the straightforward and atom-economic access to a wide variety of functionalized olefins and, in its intramolecular version, to relevant heterocyclic and carbocyclic compounds. Despite conjugated 1,3-diynes being considered key building blocks in synthetic organic chemistry, this particular class of alkynes has been much less employed in hydrofunctionalization reactions when compared to terminal or internal monoynes. The presence of two C≡C bonds in conjugated 1,3-diynes adds to the classical regio- and stereocontrol issues associated with the alkyne hydrofunctionalization processes’ other problems, such as the possibility to undergo 1,2-, 3,4-, or 1,4-monoadditions as well as double addition reactions, thus increasing the number of potential products that can be formed. In this review article, metal-catalyzed hydrofunctionalization reactions of these challenging substrates are comprehensively discussed.
1. Introduction
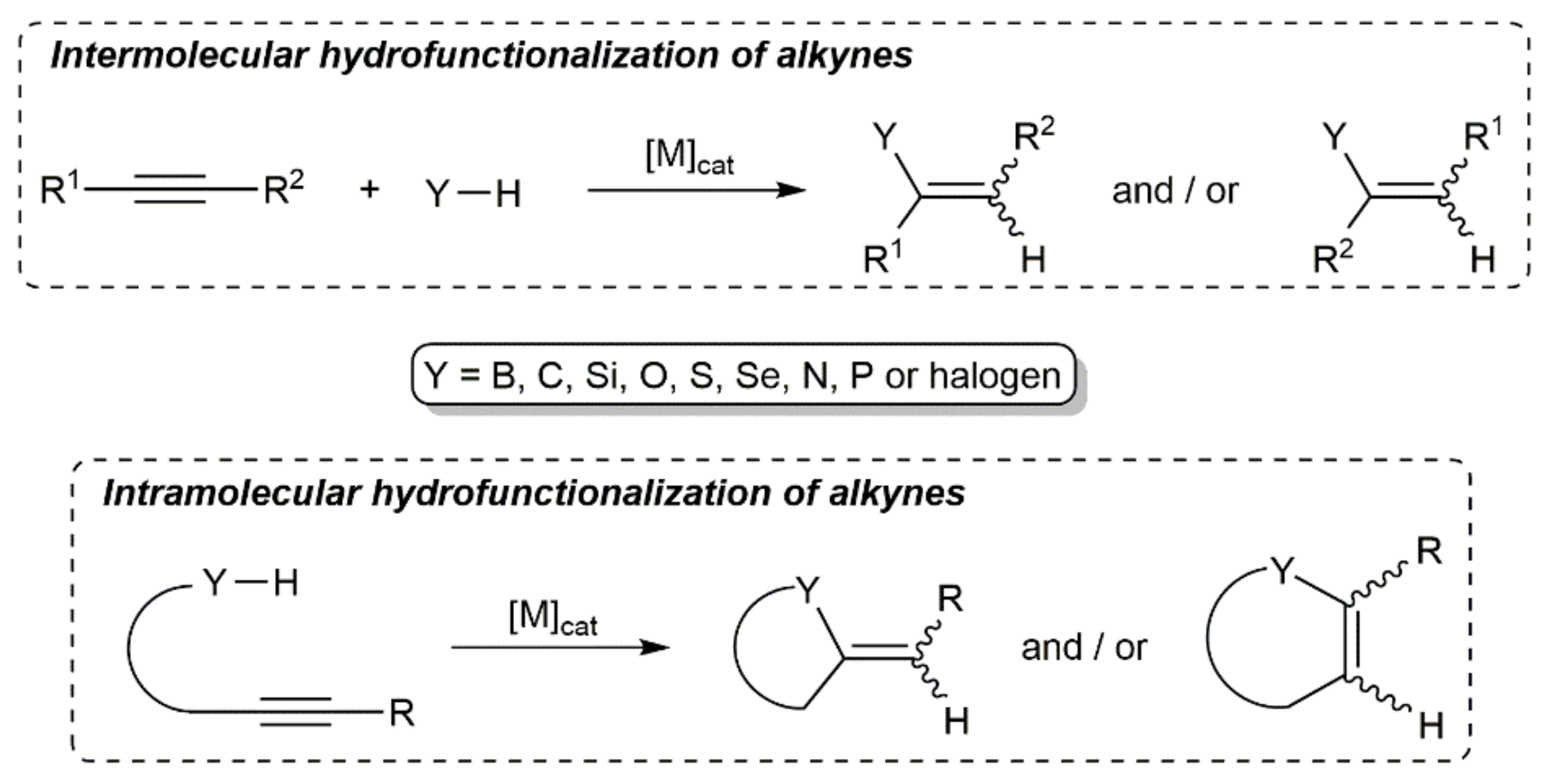
Alkynes are a pivotal class of compounds in organic chemistry and their electrophilic activation by transition metals has become a mainstay in the toolbox of synthetic chemists, biochemists, and materials scientists [1,2,3]. In this context, the metal-catalyzed hydrofunctionalization of alkynes, i.e., the addition of Y–H units (Y = heteroatom- or carbon-centered nucleophile) across the C≡C bond, has emerged in the last decades as one of the most powerful tools to access functionalized olefins in an atom-economical manner, its intramolecular version allowing also the rapid assembly of a wide range of heterocyclic and carbocyclic compounds (see Scheme 1) [4,5,6,7,8,9].
A wide variety of transition metal compounds (complexes, salts, or nanoparticles) are known to promote the addition of Y–H units to alkynes by π-activation of C≡C bond, the regio- and stereoselectivity (syn- or anti-type addition) of the process being strongly dependent on the nature of the reactants, catalyst, and reaction conditions employed, and a priori difficult to predict, particularly in the case of non-symmetrically substituted alkynes (up to four isomeric products can be potentially formed) [4,5,6,7,8,9].
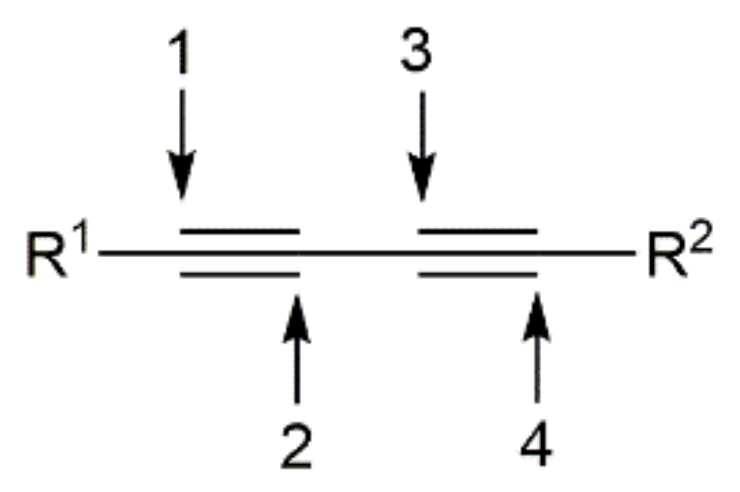
On the other hand, although their unique structure, relative stability, and unsaturated nature have turned conjugated 1,3-diynes into useful and versatile building blocks in organic synthesis and materials science [10,11,12,13,14,15], this particular class of alkynes has been much less employed in hydrofunctionalization reactions when compared to terminal or internal monoynes. Obviously, the hydrofunctionalization of 1,3-diynes having two triple bonds and four active positions (see Figure 1) is much more challenging, since they can undergo 1,2-, 3,4-, or 1,4-monoadditions, as well as double Y–H addition processes, thus expanding the range of regioisomeric and stereoisomeric products that can be formed.
The aim of the present review article is to provide a comprehensive overview on the metal-catalyzed hydrofunctionalization of conjugated 1,3-diynes. Although reactions not involving metal-based catalysts are considered out of the scope of this review, some of them will be mentioned in order to give the reader a complete prospect of the current state of the art.
2. Hydroboration Processes
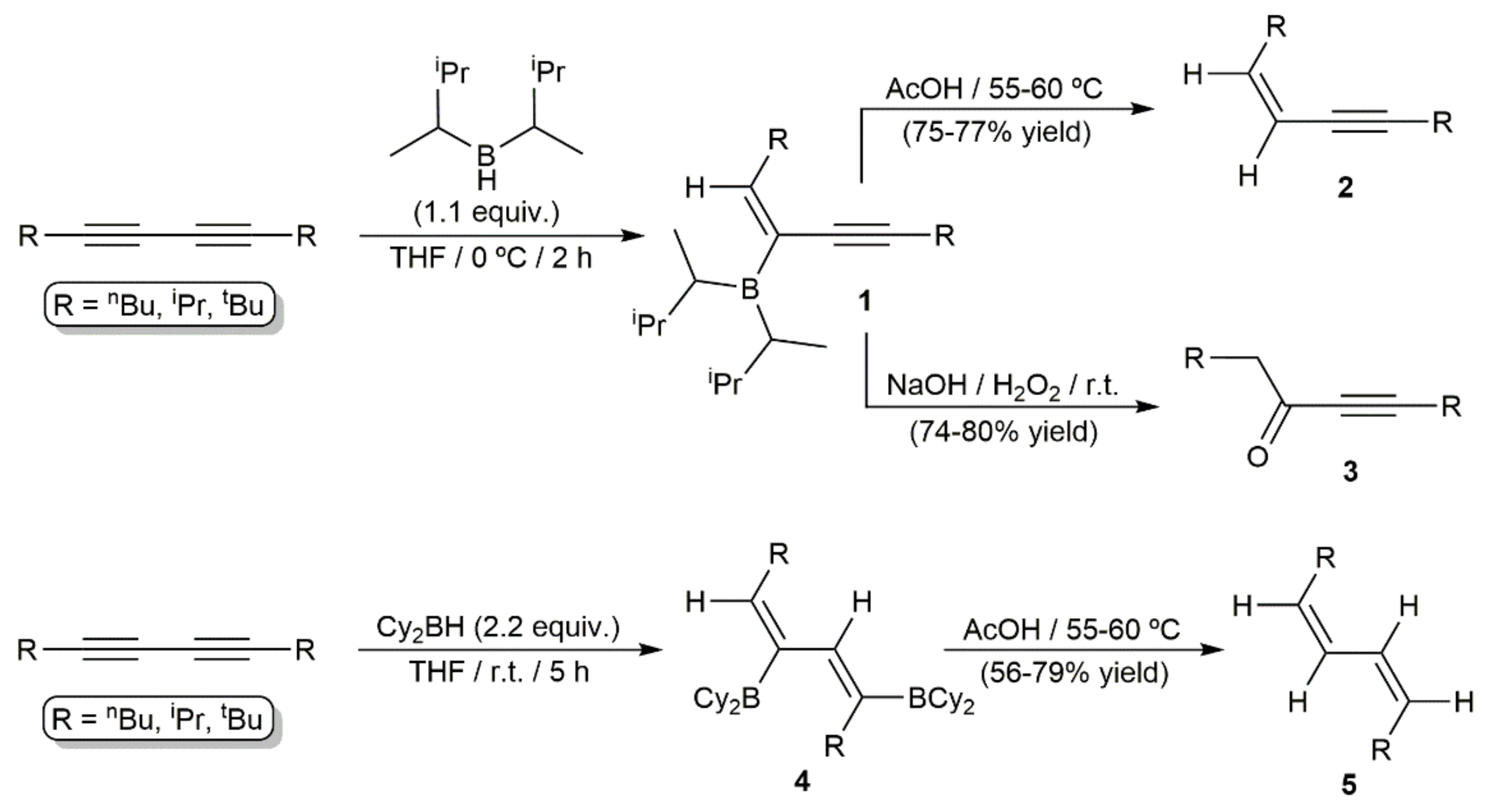
Organoboron compounds are important building blocks in organic chemistry because of their low toxicity, stability, and unique reactivity in various synthetic processes, as, for example, in the well-known Suzuki–Miyaura-type cross-couplings or the Petasis borono–Mannich reactions. In this context, the hydroboration of alkynes, including both metal-free and transition-metal-catalyzed reactions, has been widely studied in the last decades, as it provides a simple and straightforward access to valuable vinylborane synthetic intermediate compounds [16]. However, as a special class of alkynes, 1,3-diynes have been barely investigated in hydroboration processes. The first example quoted in the literature was described by Zweifel and Polston in 1970, who described the metal-free monohydroboration of symmetrically alkyl-substituted 1,3-diynes with disiamylborane [17]. The reactions led to the functionalized enynes 1 resulting from a syn-type addition of the B–H bond to one of the C≡C units, with the boron atom attacking the less sterically hindered internal position of the diyne system (Scheme 2). Compounds 1 were not isolated and transformed immediately into the corresponding cis-1,3-enynes 2 or α,β-acetylenic ketones 3 upon protonolysis with acetic acid or oxidation with alkaline hydrogen peroxide, respectively. In the same work, dihydroboration of the same alkyl-substituted diynes was successfully achieved employing a twofold excess of dicyclohexylborane, thus allowing the stereoselective access to the cis,cis-dienes 5 by protonolysis of the organoborane intermediates 4 (see Scheme 2). Monohydroboration of non-symmetrically alkyl-substituted 1,3-diynes was also attempted by the authors, but no discrimination between the triple bonds was observed [17]. Only when diynes of type Alk-C≡C-C≡C-SiR3 substituted with a bulky trialkylsilyl group (SiMe2tBu or SiMe2(CMe2nPr)) were employed as substrates, hydroboration of the less sterically hindered alkyl-substituted C≡C bond was preferentially observed [18].
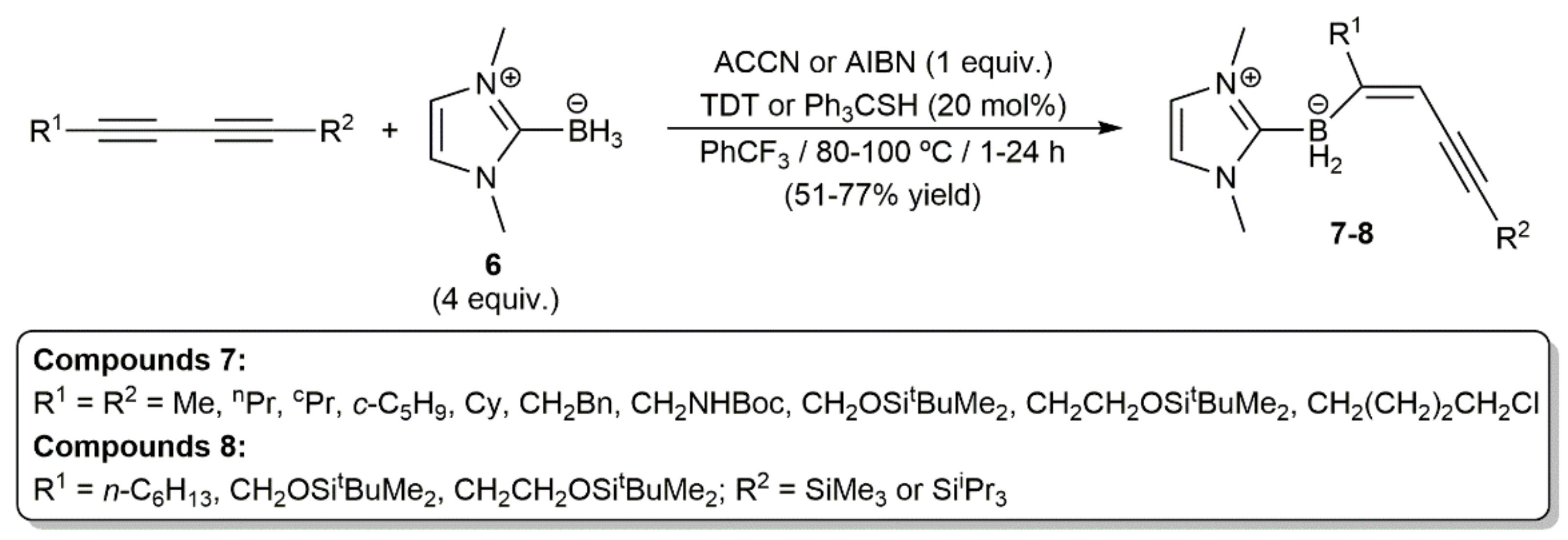
Following previous studies on the anti-hydroboration of internal alkynes with N-heterocyclic carbene (NHC) boranes under radical conditions [19], Curran, Taniguchi, and colleagues reported very recently the highly regio- and stereoselective access to the NHC-(E)-enynylboranates 7 and 8 by monohydroboration of symmetrical and unsymmetrical 1,3-dynes, respectively, with 1,3-dimethylimidazol-2-ylidene borane 6 (Scheme 3) [20]. In the reactions, an azo compound, i.e., ACCN (1,1′-azobis(cyclohexane-1-carbonitrile)) or AIBN (2,2′-azobis(2-methylpropionitrile)), was employed as the free radical initiator, and a thiol, i.e., TDT (tert-dodecanethiol) or Ph3CSH, as a polarity-reversal catalyst.
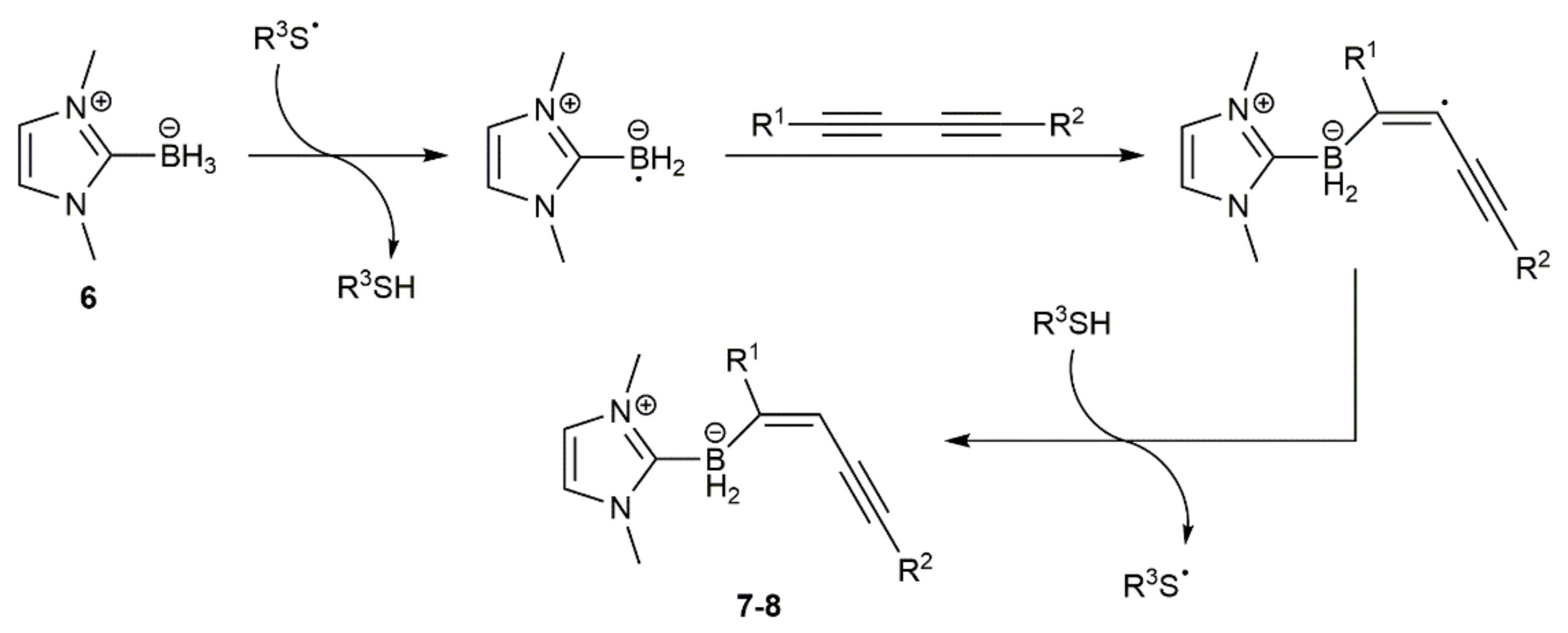
As shown in Scheme 4, a thiyl radical formed from the thiol by thermal decomposition of ACCN or AIBN initiates the reaction by abstracting a hydrogen atom from the NHC-borane 6 to generate an NHC-boryl radical, which adds to the C≡C bond of the diyne. The chemoselectivity observed in the reactions of the unsymmetrical silyl-substituted 1,3-diynes was rationalized in terms of the steric hindrance associated to the bulky SiR3 groups. The usefulness in organic synthesis of the NHC-(E)-enynylboranates 7–8 was additionally demonstrated by the authors, who successfully conducted palladium-catalyzed Suzuki–Miyaura C–C coupling reactions with some representatives [20].
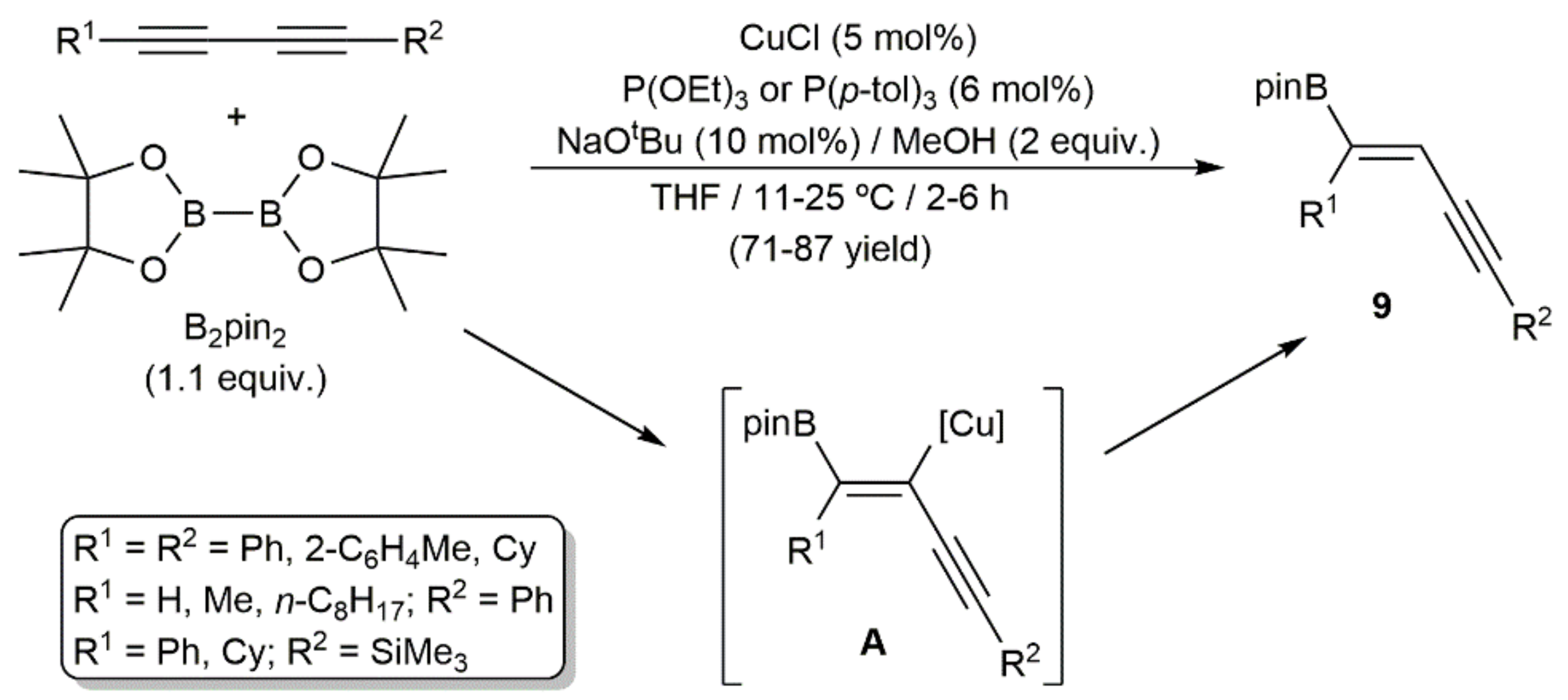
Regarding the use of metal-based catalysts, Yun and colleagues reported in 2015 the formal copper(I)-catalyzed monohydroboration of different symmetrical and unsymmetrical 1,3-diynes with bis(pinacolato)diboron (B2pin2), which allowed the regio- and stereoselective access to 1-boryl-substituted 1,3-enynes 9 (Scheme 5) [21]. According to the authors, the process proceeds through the initial borylcupration of the C≡C bond of the diynes by an in situ generated copper–boryl complex, followed by protonolysis of the metallated intermediate A by the methanol present in the medium.
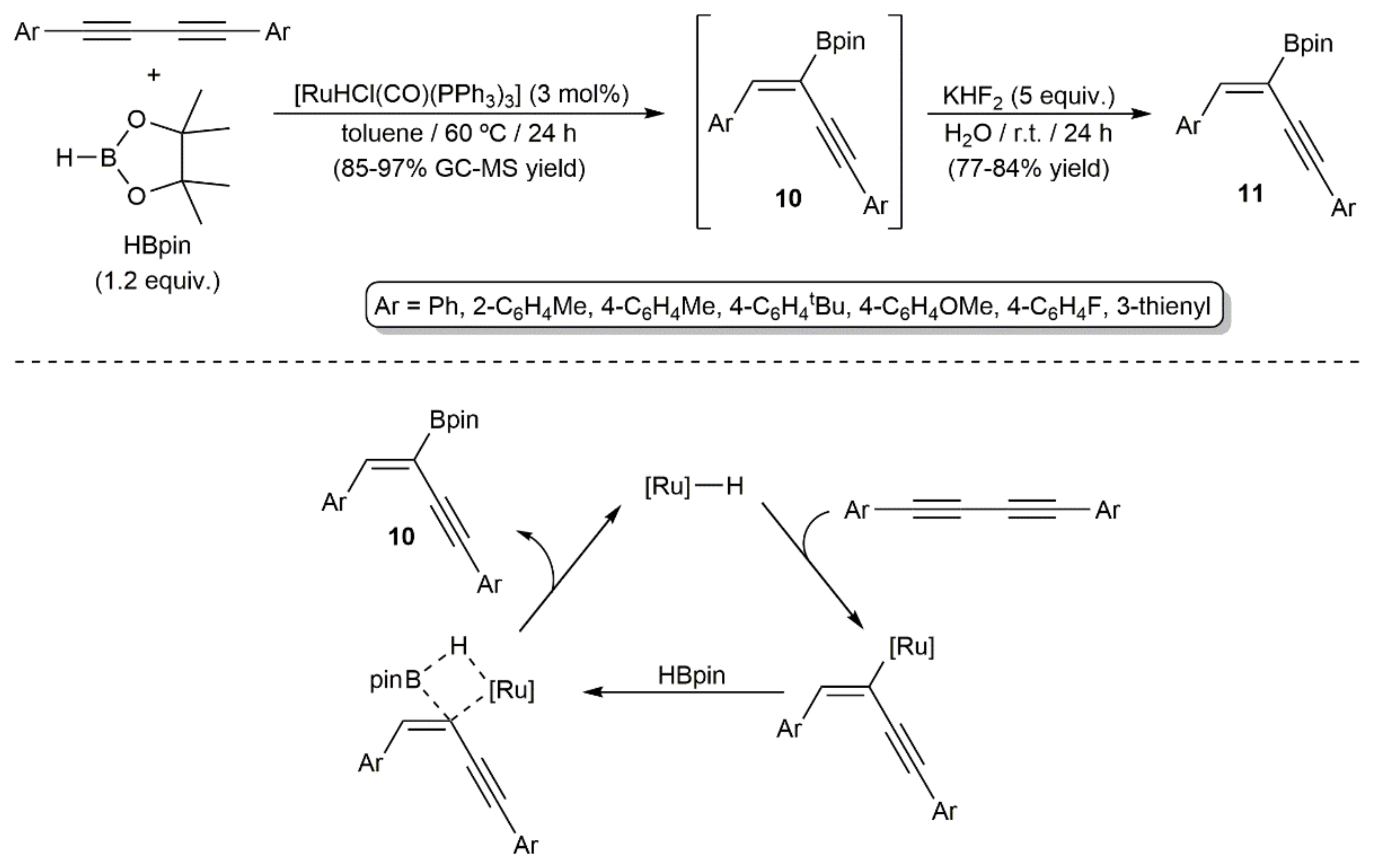
Hydroboration of a series of symmetrical 1,4-diaryl-substituted 1,3-diynes with pinacolborane (HBpin) was more recently described by Walkowiak and colleagues employing the ruthenium(II) hydride complex [RuHCl(CO)(PPh3)3] as a catalyst (Scheme 6) [22]. The reactions, carried out in toluene at 60 °C, led to the 2-boryl-substituted 1,3-enynes 10, which were rapidly transformed into the more stable trifluoroborate salts 11, with complete regio- and stereocontrol (syn-addition). This process nicely complements the previous one, since the borane attaches now to the less hindered internal carbon atoms of the diyne. Nonetheless, it should be noted that its scope is restricted to 1,4-diaryl-substituted 1,3-diynes, with the use of alkyl- or silyl-substituted ones leading to mixtures of the corresponding mono- and dihydroborated products, along with other unidentified species. Regarding the mechanism, a reaction pathway involving the initial insertion of the diyne into the Ru–H bond to generate a ruthenium(II)-enynyl intermediate, which subsequently reacts with pinacolborane through a σ-bond metathesis process, was proposed by the authors.
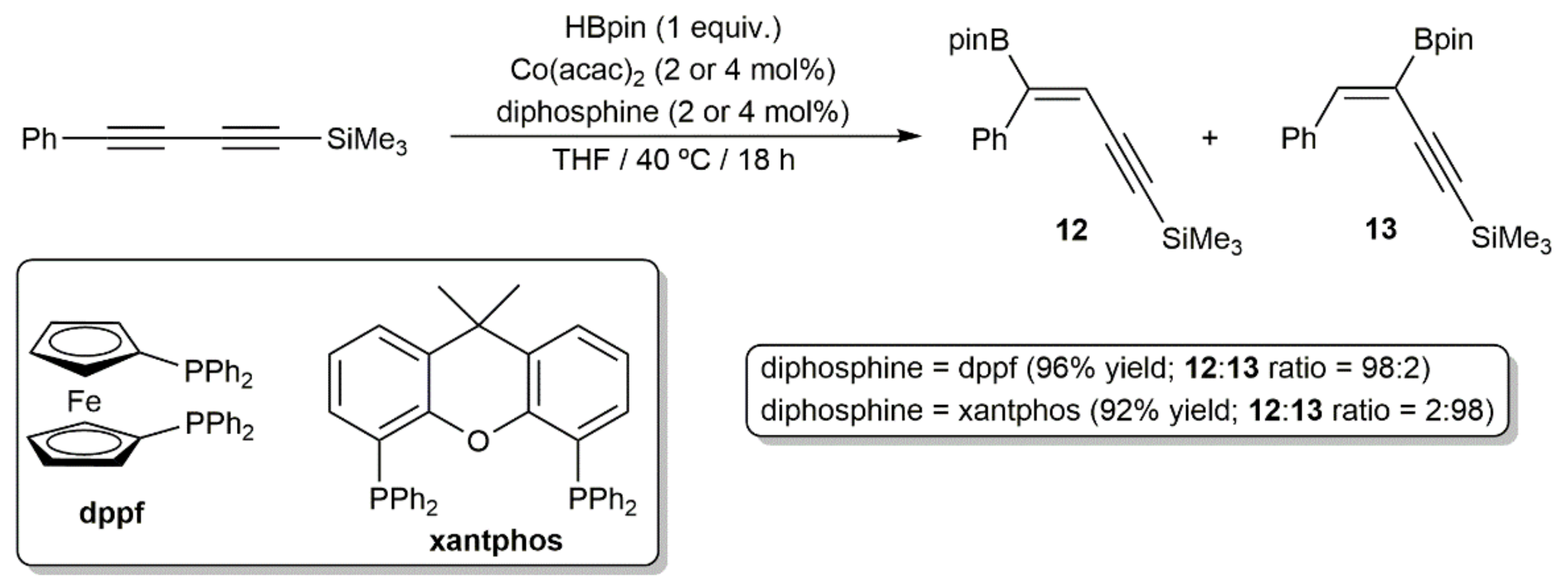
Finally, it merits to be particularly highlighted the work reported by Ge and colleagues, who were able to develop a regio-divergent stereoselective syn-monohydroboration protocol of symmetrical and unsymmetrical 1,3-diynes with HBpin, catalyzed by the acetylacetonate Co(II) complex Co(acac)2 in combination with a diphosphine ligand (dppf or xantphos) [23]. Thus, as shown in the example given in Scheme 7, while the use of the ferrocenyl-diphosphine dppf led to the major formation of the enynylborane product 12 with boron addition to the external carbon of the 1,3-diyne unit, an opposite regioselectivity was observed when xantphos was employed as the auxiliary ligand (product 13). The scope of this regio-divergent process was very high (more than 40 enynylboranes were synthesized), showed a good functional group compatibility, and could be performed in gram-scale.
3. Hydroarylation and Hydroalkylation Processes
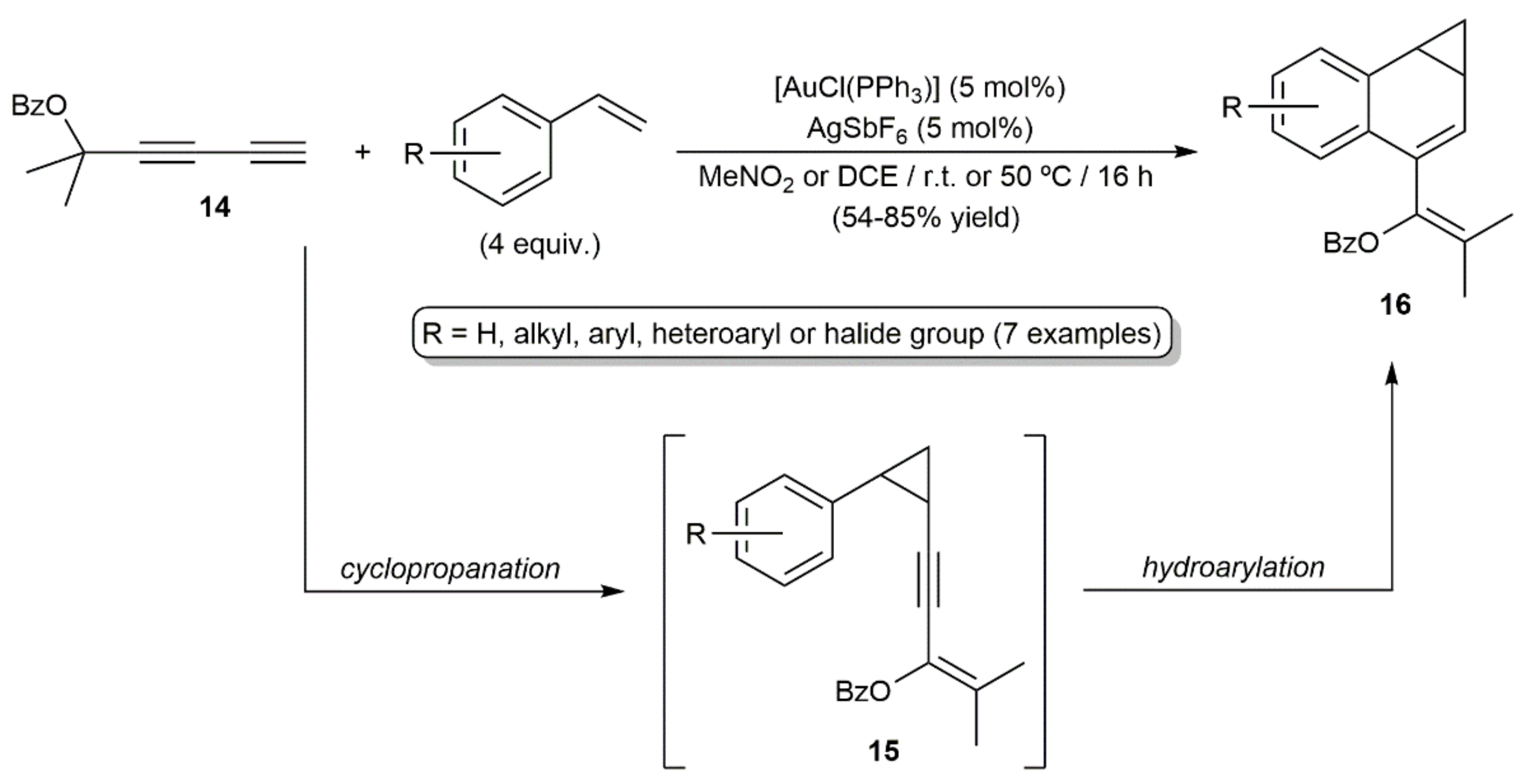
Transition-metal-catalyzed hydroarylation reactions of alkynes with substituted aromatic and heteroaromatic compounds allow the rapid construction of complex molecules from simple precursors without the need for prefunctionalization steps [24,25,26]. Involvement of 1,3-diynes in such processes was documented for the first time by Toste and colleagues in 2006, with the gold(I)-catalyzed [4+3] annulation reactions of the ester containing diyne 14 with styrenes depicted in Scheme 9 [27]. The benzonorcaradienes 16 were generated as the major reaction products through a cyclopropanation/hydroarylation sequence, along with minor amounts of the intermediate enynyl-cyclopropanes 15.
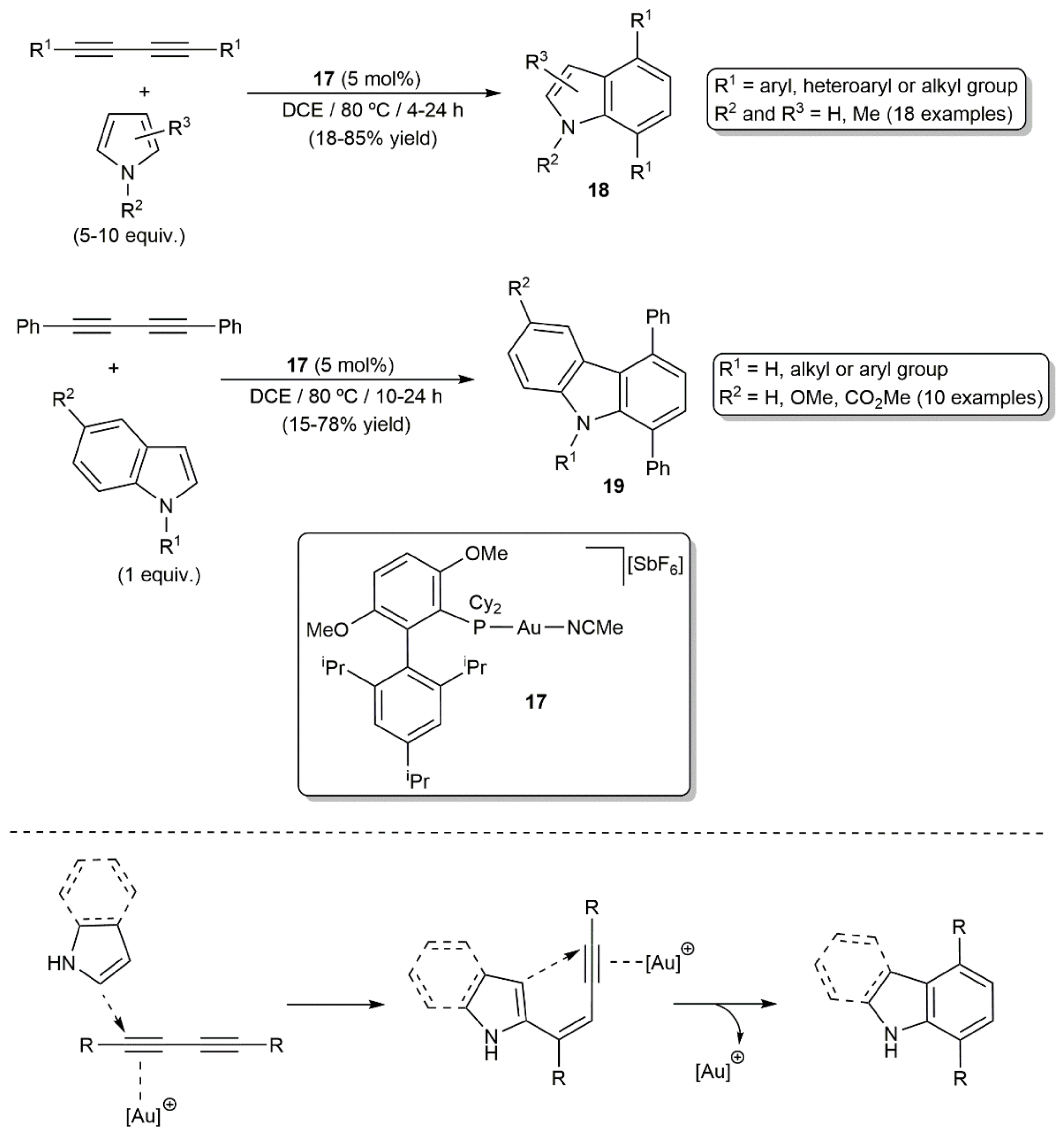
A few years later, Ohno´s group reported the coupling of pyrrole derivatives with different 1,3-diynes in the presence of catalytic amounts of the cationic gold(I) complex [Au(MeCN)(BrettPhos)][SbF6] (17) (Scheme 10) [28]. The reactions, performed in 1,2-dichloroethane (DCE) at 80 °C, provided access to substituted indoles 18 in moderate to high yields. As shown in Scheme 10, the use of indoles instead of pyrroles as the coupling partners allowed also the direct synthesis of carbazoles 19. Although symmetrically substituted 1,3-diynes were mainly employed in this study, the reactivity of a couple of unsymmetrical 1,3-diynes towards pyrrole was also explored, the reactions leading to the isomeric mixtures of the corresponding indole products due to the difficulty associated with controlling the regioselectivity of the first hydroarylation step. Indeed, these gold-catalyzed [4+2] cycloaddition reactions involve a stepwise double hydroarylation pathway, with participation of the pyrrole-C2 and the indole-C3 in the first intermolecular hydroarylation of the diyne, and a 6-endo-dig cyclization in the second one. Density functional theory (DFT) calculations have recently corroborated this mechanistic proposal [29].
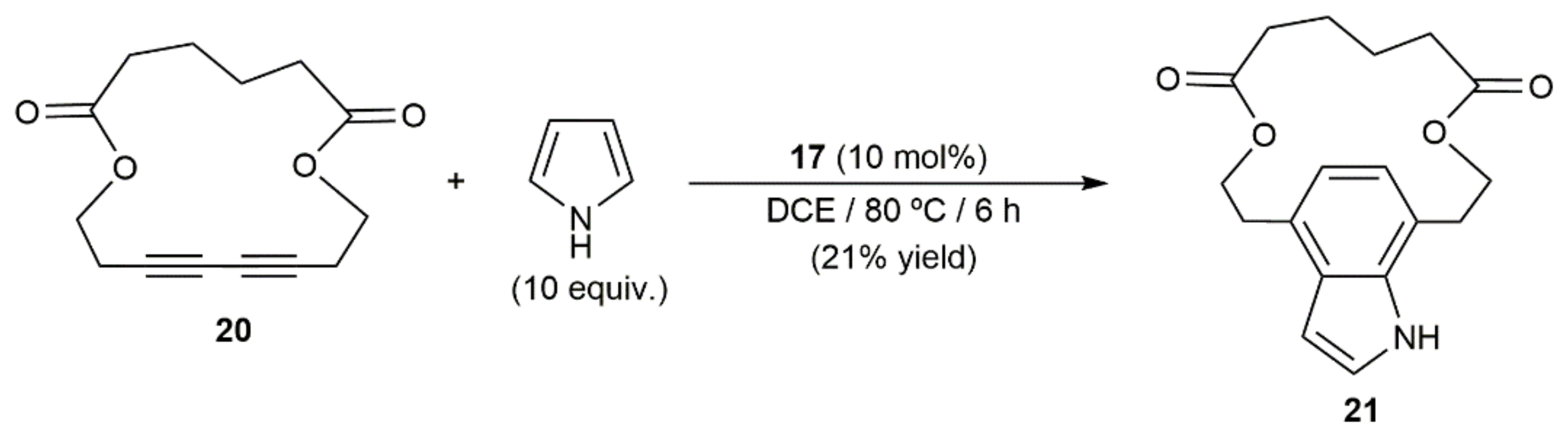
The synthetic utility of the process was fully demonstrated by Ohno and colleagues with the total synthesis of the naturally occurring dictyodendrins B, C, E, and F (see Figure 2), in which the central pyrrolo[2,3-c]carbazole cores were generated by intermolecular annulation of a conjugated diyne with a pyrrole derivative [30]. Moreover, of note is the fact that, employing Ohno’s protocol, Ungeheuer and Fürstner were able to synthesize the para-indolophane 21 by double hydroarylation of the macrocyclic 1,3-diyne 20 with pyrrole (Scheme 11), ratifying again the broad scope and synthetic relevance of the process [31].
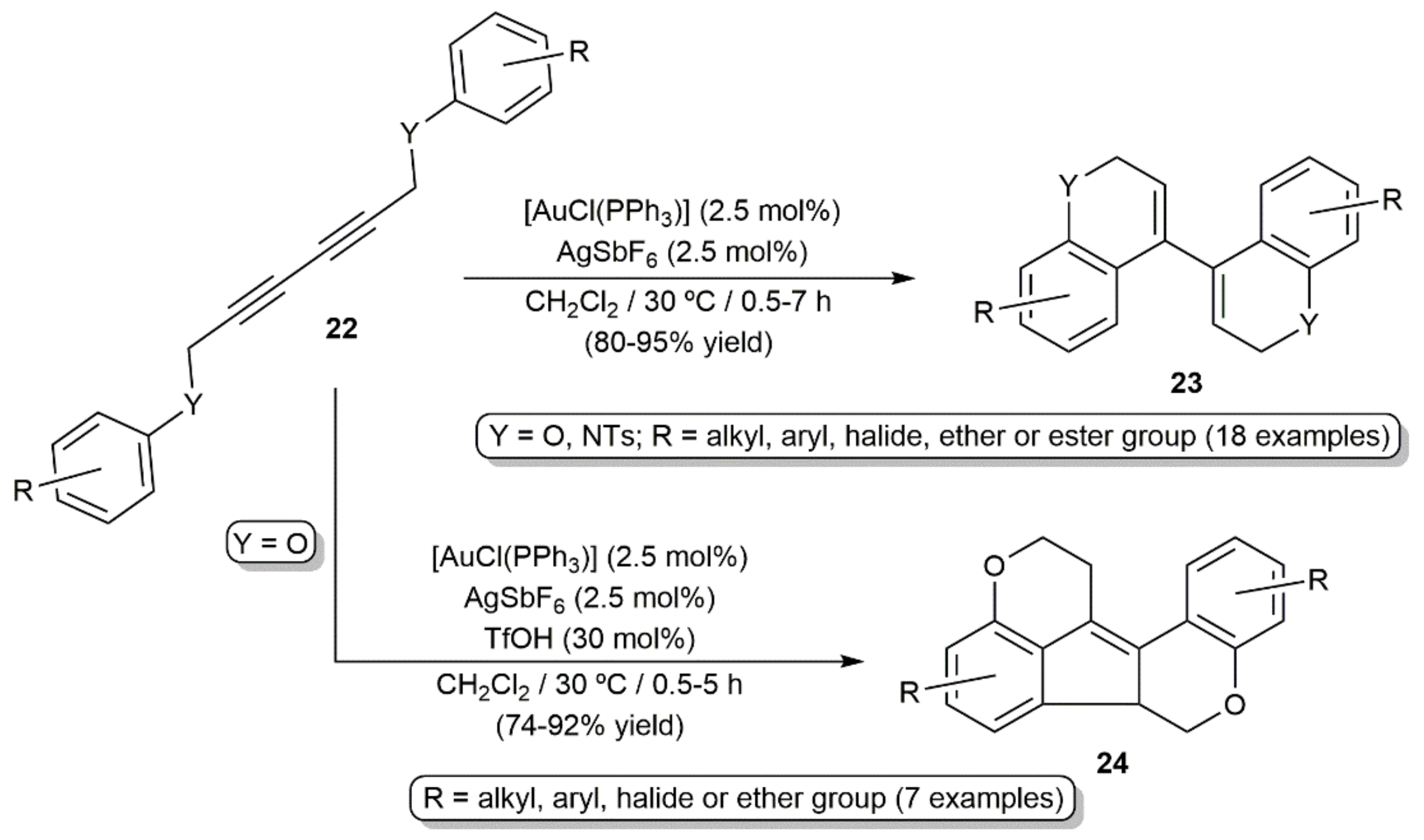
Intramolecular double hydroarylation of the 2,4-diyne-1,6-diethers and diamines 22 catalyzed by the in situ generated gold(I) cation [Au(PPh3)]+ was additionally described by Lee and colleagues, the reactions providing a straightforward access to 4,4′-bi(2H-chromene) and bi(2H-quinoline) derivatives 23 (Scheme 12) [32]. Combining the gold catalyst with triflic acid allowed also the synthesis of different dioxafluoranthenes 24 from the 2,4-diyne-1,6-diethers by acid-promoted cyclization of the initially generated 4,4′-bi(2H-chromenes) 23 [32].
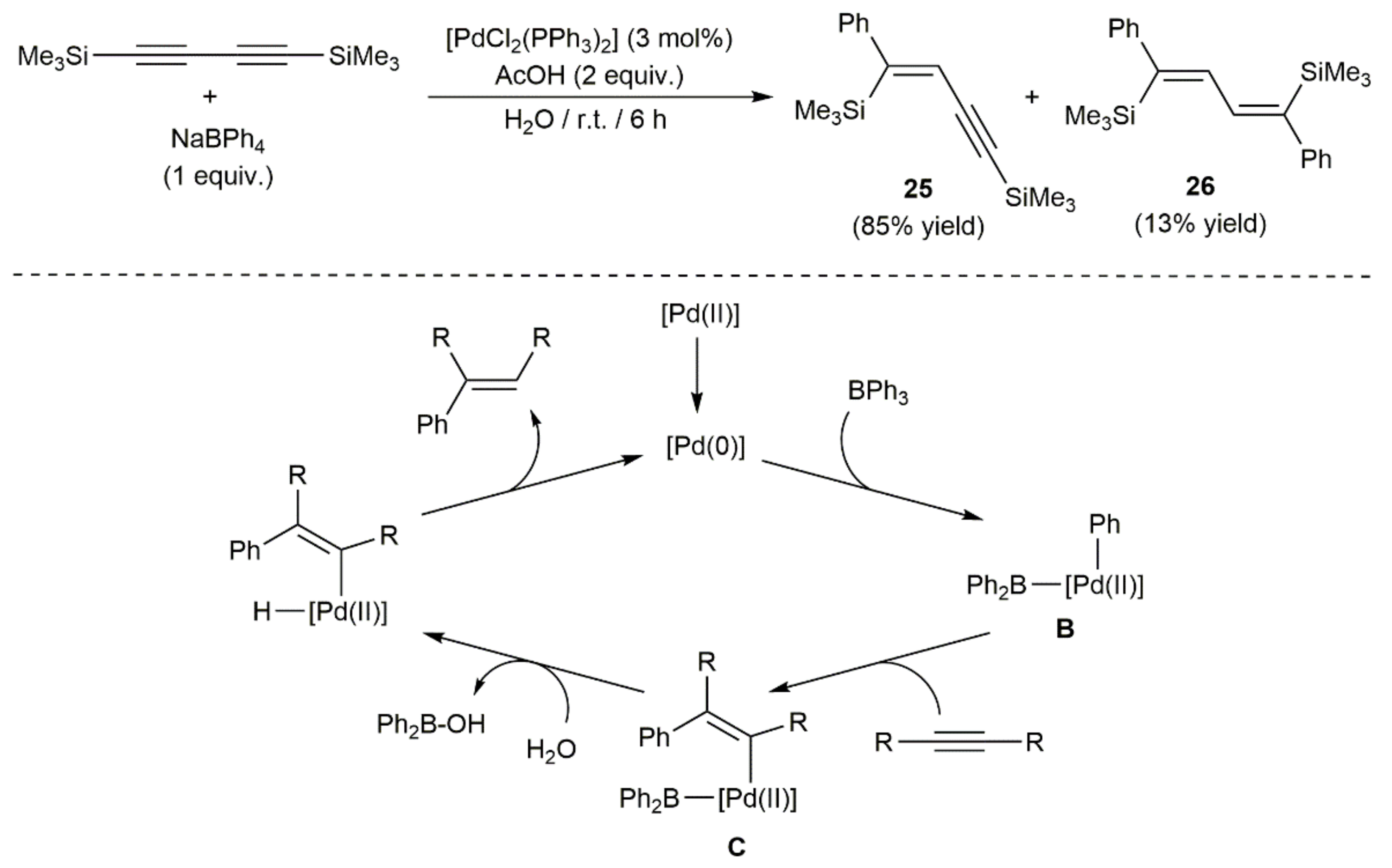
A couple of examples of palladium-catalyzed arylation of 1,3-diynes can be found in the literature. Thus, in the context of a broad study on the hydrophenylation of alkynes with sodium tetraphenylborate mediated by [PdCl2(PPh3)2] in water, Zeng and Hua tested the behavior of 1,4-bis(trimethylsilyl)-1,3-butadiyne [33]. As shown in Scheme 13, the conversion of the diyne was almost quantitative, the reaction affording both singly and doubly phenylated products 25 and 26 in 85 and 13% yields, respectively. As exemplified with a simple alkyne, the hydrophenylation process is believed to proceed through the oxidative addition of the B–C bond of BPh3, resulting from the reaction of NaBPh4 with the acetic acid present in the medium, to in situ generated Pd(0) species to give intermediate B, which evolves into C by syn-insertion of the alkyne. Hydrolysis of C, followed by reductive elimination, leads to the final phenylated product.
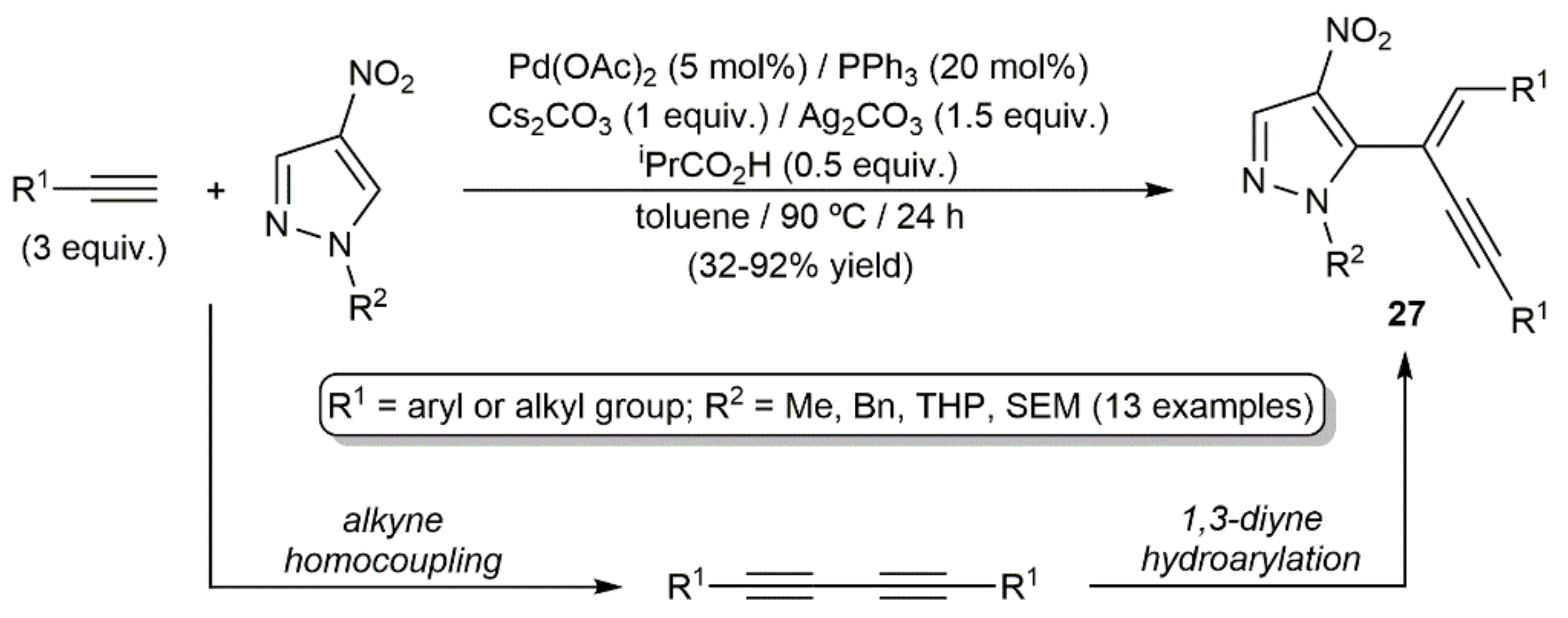
The second example, consisting in a tandem homocoupling/hydroarylation of terminal alkynes with nitropyrazoles to afford the functionalized enynes 27, is depicted in Scheme 14 [34].
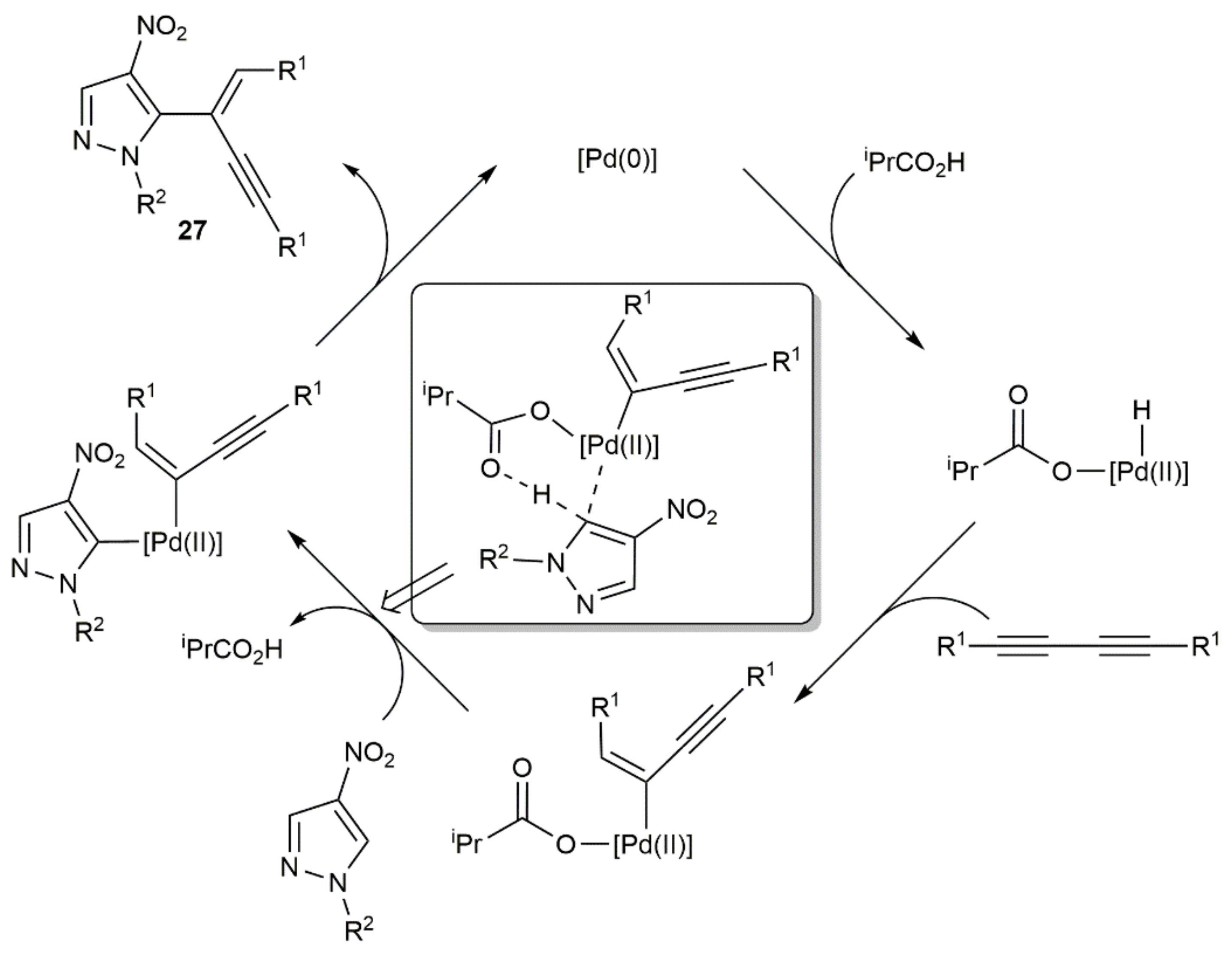
As in the precedent case, the hydroarylation of the 1,3-diyne intermediates proceeded in a syn manner, with the internal carbon of the diyne participating now in the formation of the new C–C bond. A reaction pathway for the hydroarylation step involving, again, in situ generated Pd(0) species was proposed by the authors, with the co-ordinated isobutyrate anion assisting the activation of the pyrazole C–H bond (see Scheme 15).
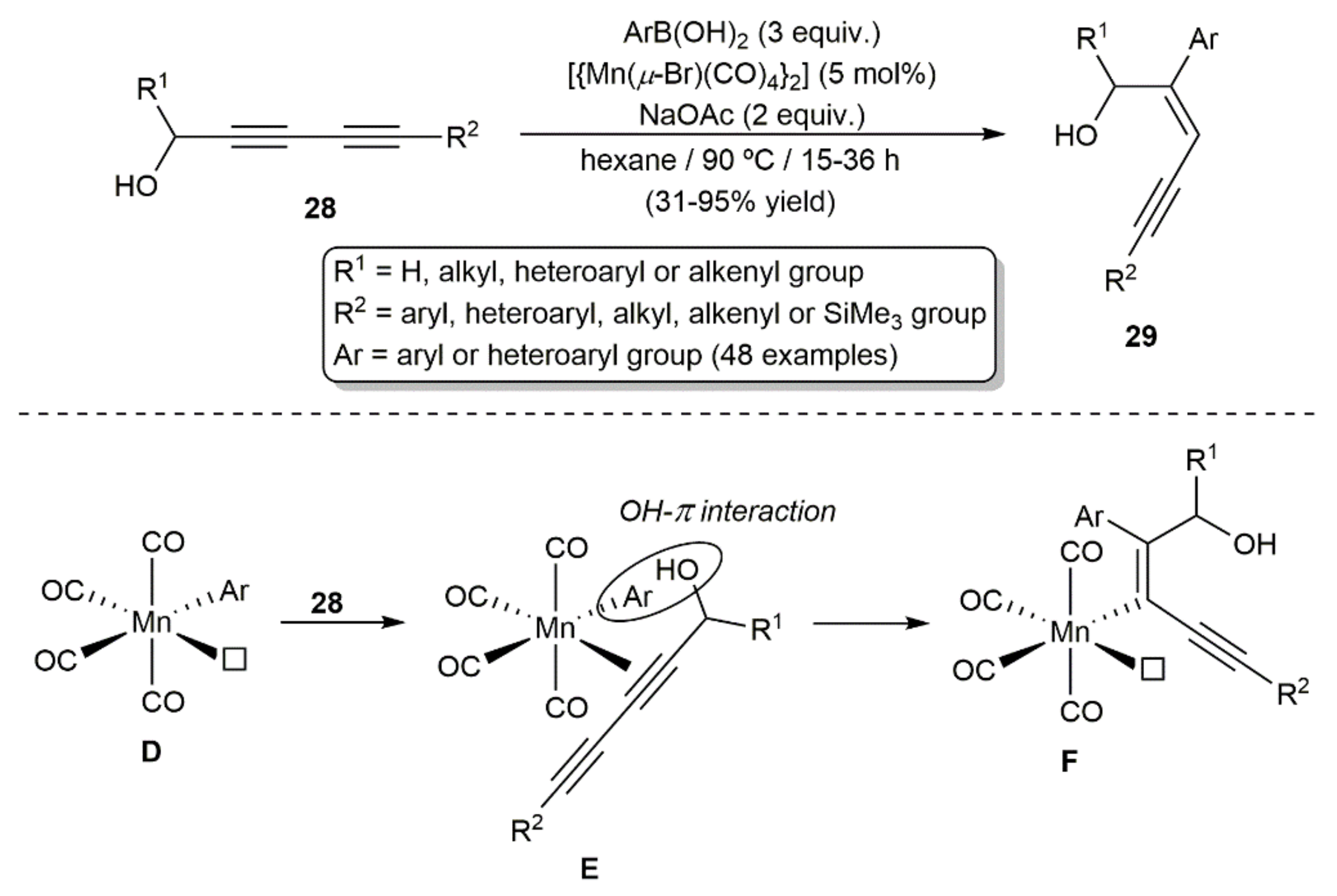
Hydroarylation reactions of diynes employing less expensive earth-abundant metals can also be found in the literature. For example, Xie and colleagues developed a wide-scope procedure for the regio-, stereo-, and chemoselective synthesis of enynes 29 by hydroarylation of alcohol-substituted diynes 28 with (hetero)aryl boronic acids ArB(OH)2 catalyzed by the Mn(I) dimer [{Mn(µBr)(CO)4}2] in conjunction with sodium acetate (Scheme 16) [35,36]. Mechanistic studies suggested the in situ generation in the reaction medium of the mononuclear 16-electron species [Mn(OAc)(CO)4] by cleavage of the dimer with NaOAc, which rapidly evolves into the arylated intermediate [MnAr(CO)4] (D) by transmetallation with the boronic acid. Selective co-ordination of the diyne on the vacant co-ordination site of D would lead to intermediate E, which would evolve into F through a syn-1,2-migratory insertion process. A weak π OH–aryl interaction in intermediate E, found by DFT calculations, would explain the regioselectivity observed. The final enyne product is liberated from F by protodemetallation with water, generated by partial decomposition of the aryl boronic acid employed, with concomitant regeneration of the active species [Mn(OAc)(CO)4] by the acetate present in the medium. It is worth mentioning that, despite the involvement of mononuclear Mn(I) species in the reaction, dimer [{Mn(µ-Br)(CO)4}2] proved to be a much more efficient catalyst than [MnBr(CO)5].
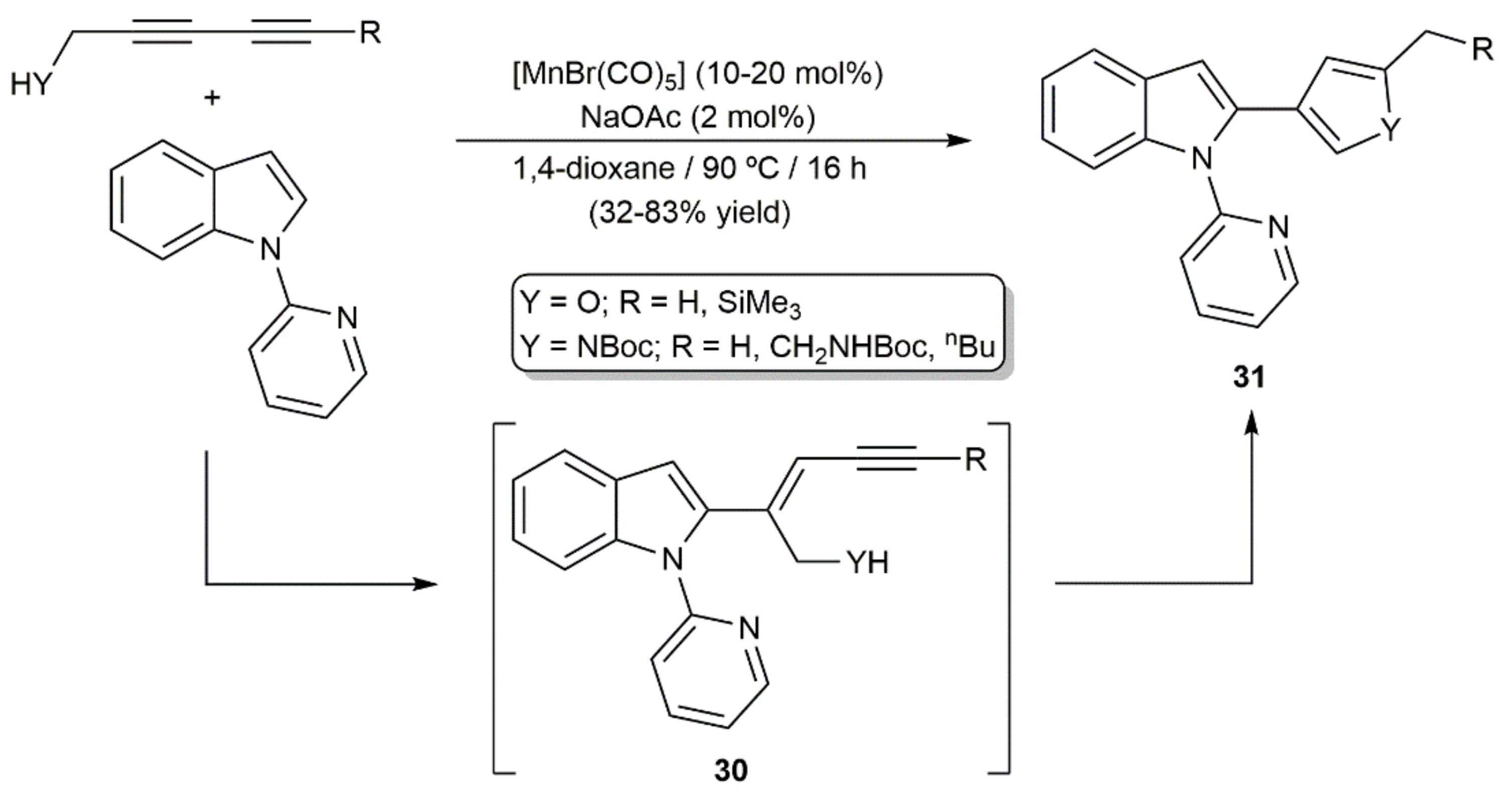
More recently, Glorius and colleagues reported the use of [MnBr(CO)5] to promote the regio- and stereoselective alkenylation of different 2-picolyl-substituted arenes and heteroarenes with 1,3-diynes [37]. The process featured a wide functional-group tolerance and single isomers of the 1,3-enyne products were, in most cases, obtained, even when unsymmetrically substituted diynes were employed as substrates (some representative examples are shown in Figure 3). Furthermore, starting from diynes bearing alcohol or protected amine substituents, furans and pyrroles 31 were accessed as single products via catalytic cyclization of the initially formed 1,3-enynes 30 (Scheme 17) [37].
As illustrated in Scheme 18, a reaction pathway involving the initial directing group-assisted C–H activation of the (hetero)arene, followed by co-ordination and regioselective insertion of the diyne into the Mn–C bond of intermediate G, and a final protodemetallation step was proposed by Glorius and colleagues for these [MnBr(CO)5]-catalyzed hydroarylation processes [37].
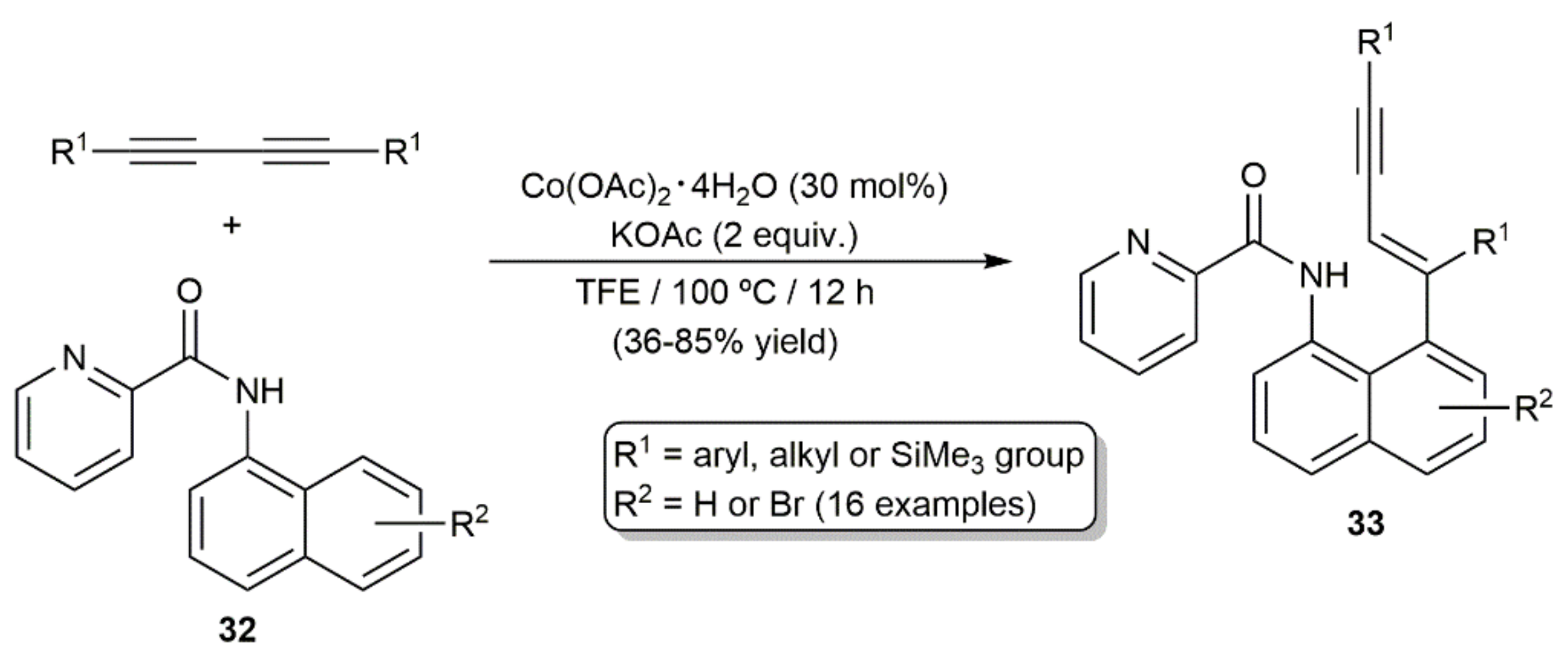
On the other hand, cobalt(II) acetate, in conjunction with KOAc, was additionally employed by Feng´s group to promote the hydroarylation of a variety of disubstituted diynes with picolinamides 32 (Scheme 19) [38]. The reactions, conducted in 2,2,2-trifluoroethanol (TFE) at 100 °C, afforded regioselectively the conjugated enynes 33 in moderate to good yields, and with complete E-selectivity (syn addition). As in the precedent case, a pycolyl-group-directed C–H activation/diyne insertion pathway was proposed.
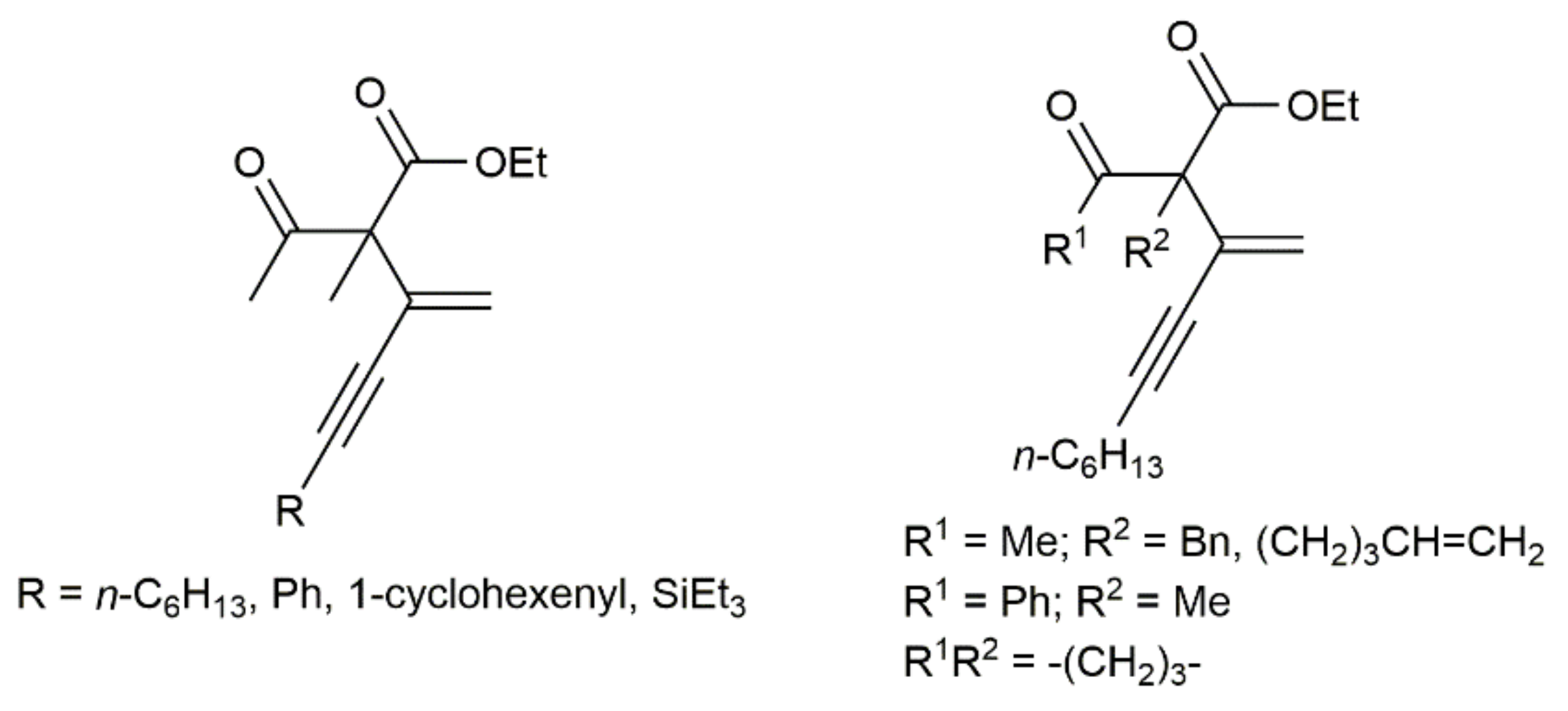
Concerning hydroalkylation processes, extending previous studies on the indium-catalyzed addition of active methylene compounds to terminal alkynes [39], Nakamura and colleagues reported the efficient synthesis (70–96% yield) of different α-ynenylated keto esters (see Figure 4) by hydroalkylation of monosubstituted 1,3-diynes HC≡CC≡CR with β-keto esters [40]. The reactions, which were performed at 60 °C under solvent-free conditions with 5 mol% of In(NTf2)3 (NTf2 = bis(trifluoromethane)sulfonamide) as the catalyst and equimolar amounts of the reactants, were completely regioselective (only the terminal C≡C bond of the diyne was found to participate in the addition reaction).
4. Hydrocyanation Processes
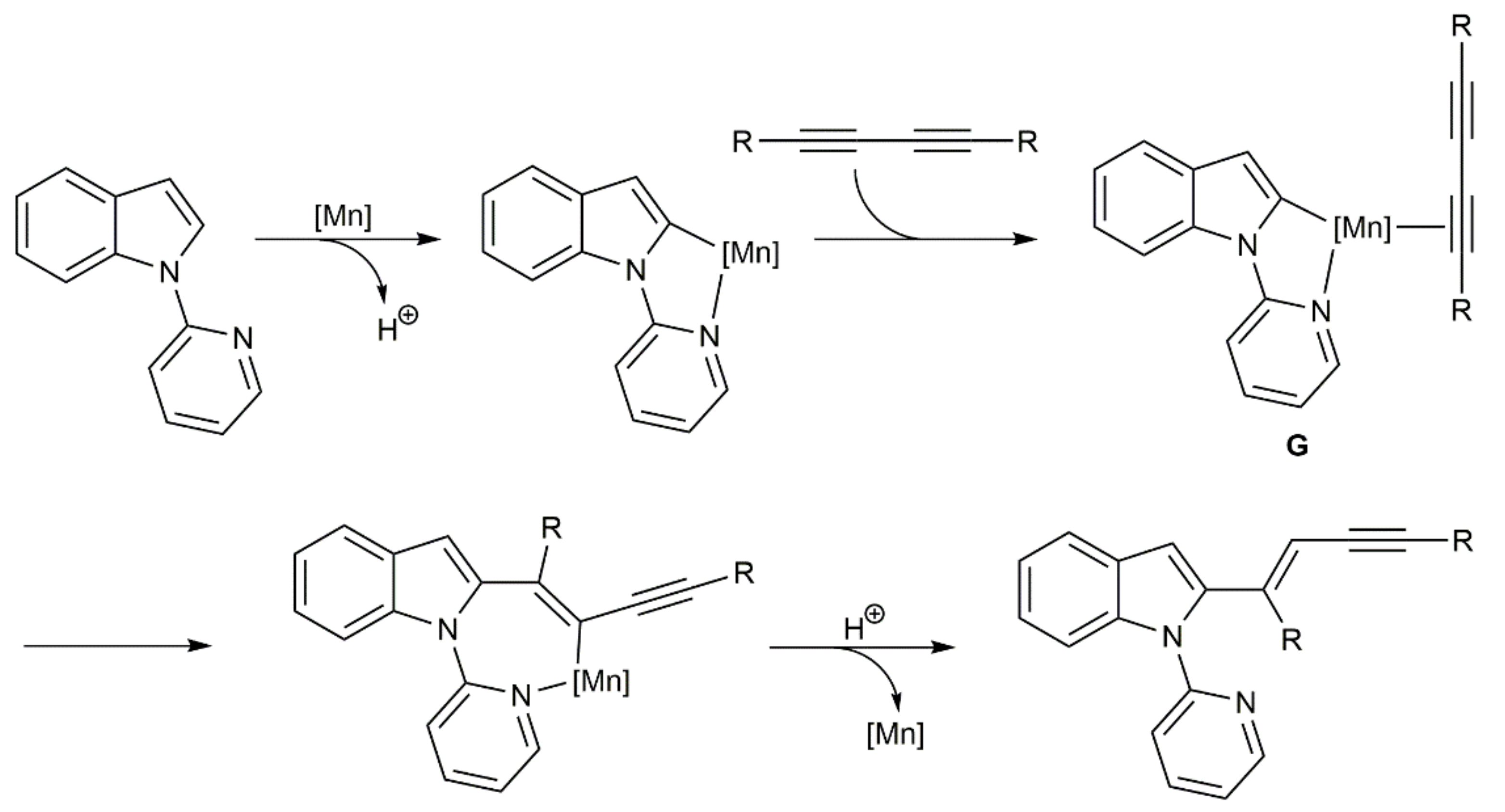
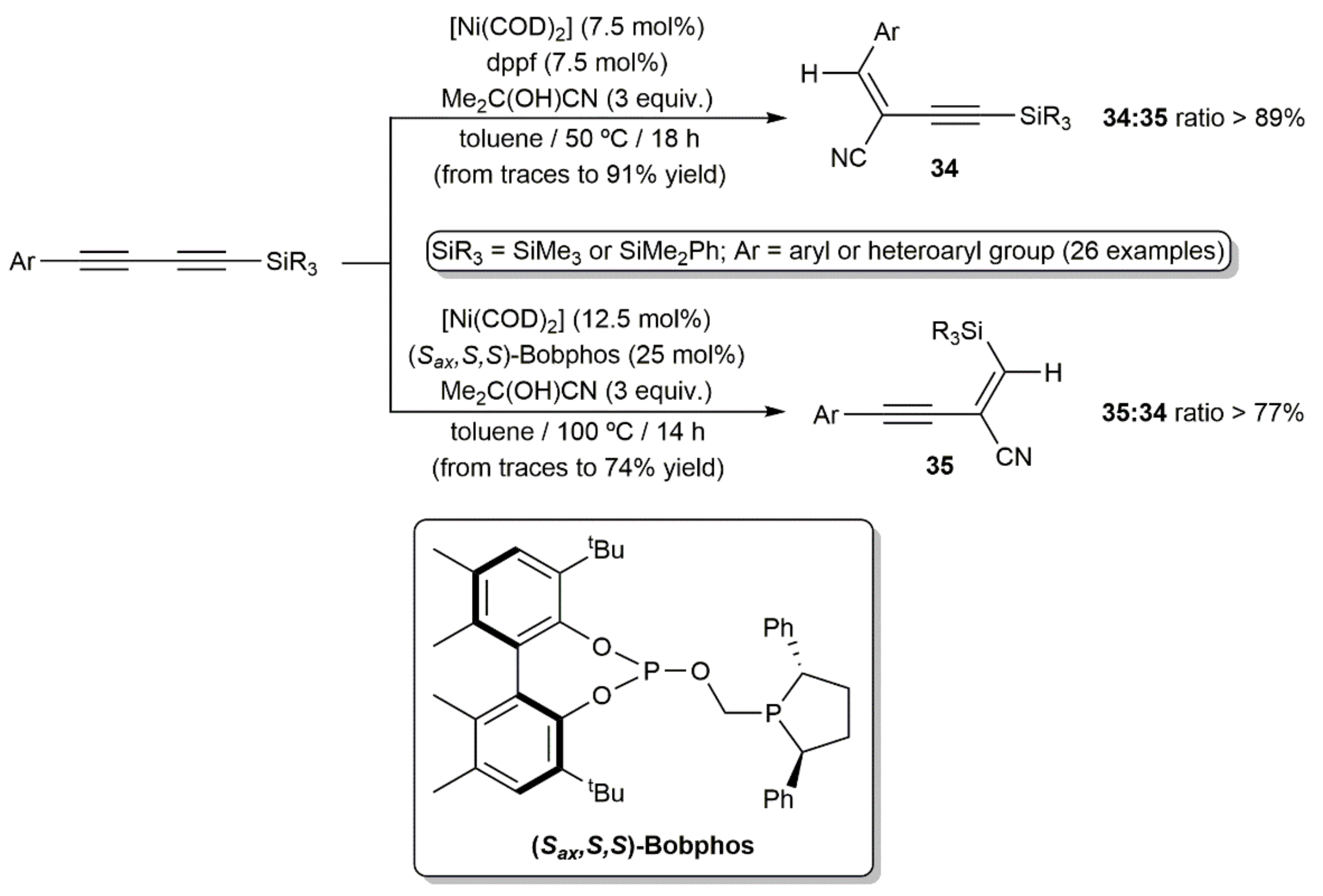
Hydrocyanation of unsaturated carbon–carbon bonds is a powerful method for the preparation of functionalized nitriles. Such reactions are commonly performed employing Ni-, Pd-, and Co-based catalysts and acetone cyanohydrin, Me3SiCN, or Zn(CN)2, the latter avoiding the direct use of harmful HCN [41,42,43]. In sharp contrast to simple alkynes, hydrocyanation of 1,3-diynes remains a neglected area of research. Indeed, just one very recent work can be found in the literature describing the Ni-catalyzed regio-divergent hydrocyanation of unsymmetrical 1-aryl-4-silyl-1,3-butadiynes (Scheme 20) [44]. The reactions, performed in toluene with catalytic amounts of [Ni(COD)2] (COD = 1,5-cyclooctadiene) and acetone cyanohydrin as the HCN surrogate, afforded with high regioselectivity and complete stereoselectivity (syn addition) two kinds of enynyl nitriles, i.e., compounds 34 and 35, depending on the conditions and, capitally, the ligand employed. Thus, while the ferrocenyl-diphosphine dppf oriented the hydrocyanation process towards the formation of enynes 34, their regioisomers 35 were preferentially generated when the chiral phosphine–phosphite (Sax,S,S)-Bobphos was used as the ligand. The process featured a broad scope and tolerated the presence of common functional groups on the aromatic or heteroaromatic substituent of the starting 1,3-diynes.
5. Other Formal C–H Bond Addition Processes
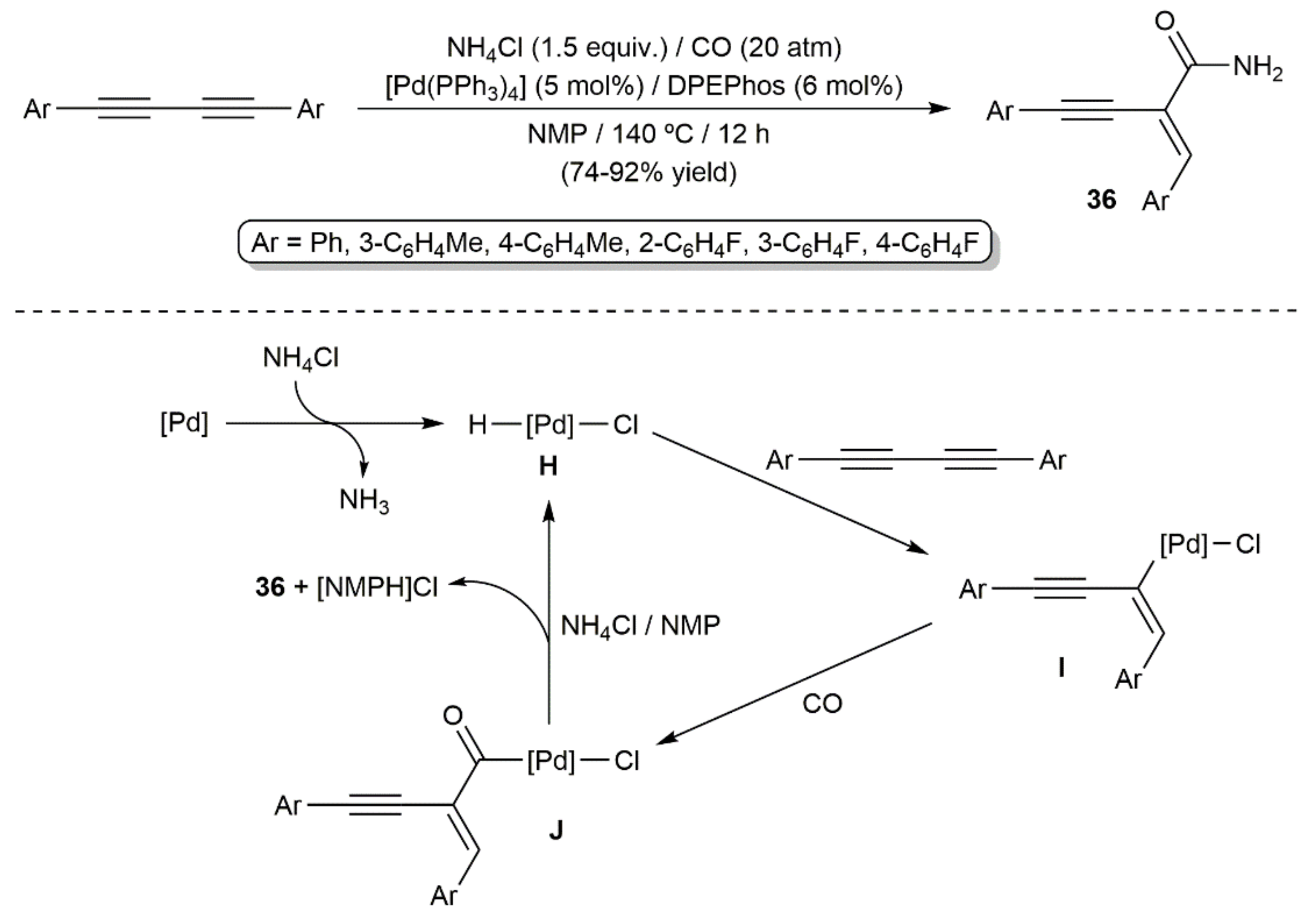
Transition-metal-catalyzed hydroaminocarbonylation reactions of alkenes and alkynes have emerged in recent years as powerful tools to access amides [45]. Within the topic covered in the present review article, only two works can be commented on. In the first one, published by Huang and colleagues in 2018, a small family of symmetrically substituted aromatic 1,3-diynes was smoothly converted into the primary amides 36 under 20 atm CO pressure, employing NH4Cl as the amine source (Scheme 21) [46]. The reactions, catalyzed by [Pd(PPh3)4] in combination with the diphosphine DPEPhos (bis[(2-diphenylphosphino)phenyl]ether) and carried out in N-methyl-2-pyrrolidone (NMP) at 140 °C, led to amides 36 in high yields and with complete regio- and stereocontrol. A mechanism involving the initial oxidative addition of NH4Cl to Pd(0) to furnish the catalytically active Pd(II)-hydride species H was proposed by the authors. Insertion of one of the C≡C bonds of the starting diyne into the Pd–H one affords the alkenyl-palladium intermediates I, which immediately react with CO to generate the acyl derivative J. The final amide products 36 result from the reaction of J with NH4Cl.
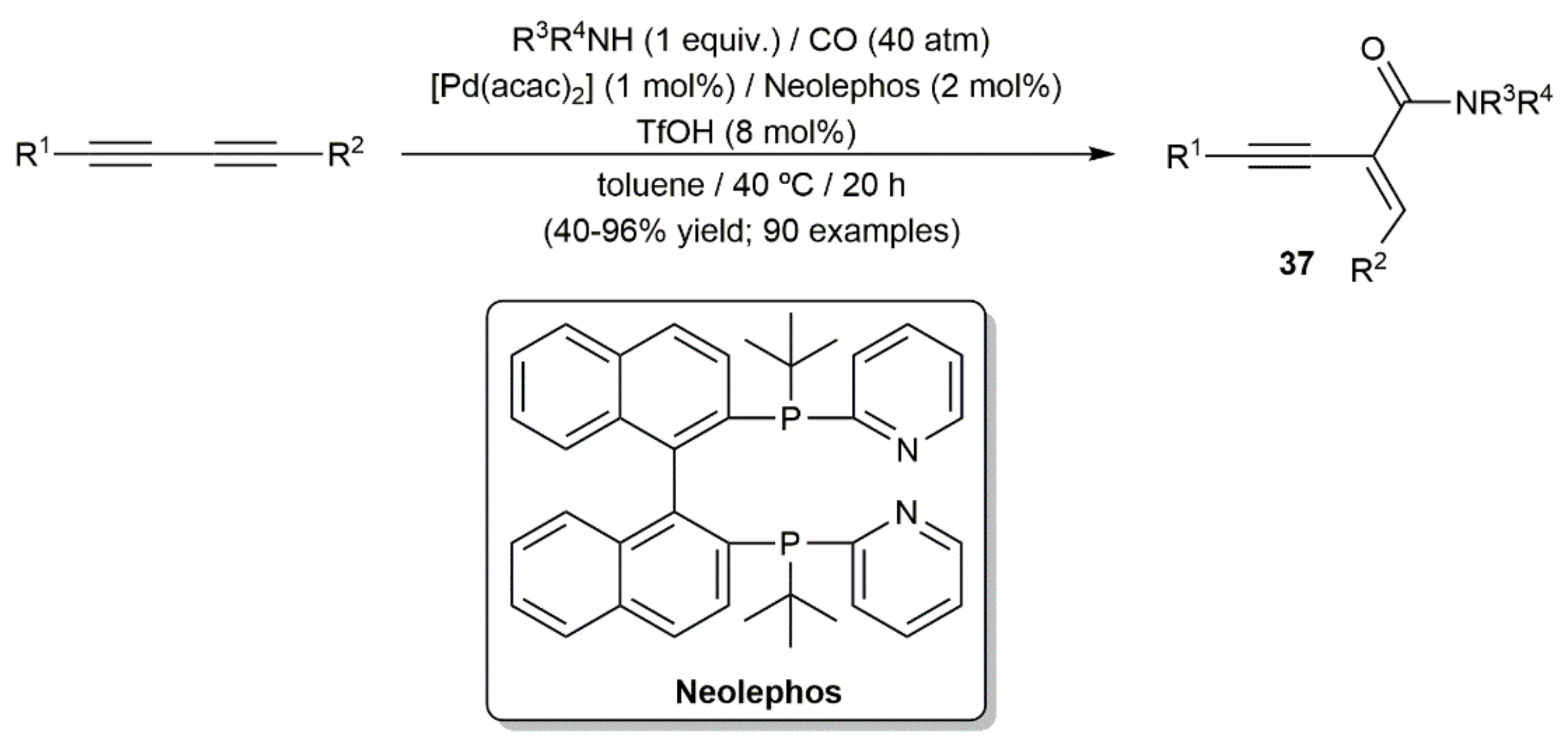
In the second one, Beller and colleagues reported the hydroaminocarbonylation of both symmetrically and unsymmetrically substituted 1,3-diynes with primary and secondary amines and CO, employing a catalytic system composed of the palladium(II) complex [Pd(acac)2] (acac = acetylacetonate), the diphosphine ligand Neolephos, and triflic acid (Scheme 22) [47]. The reactions proceeded in this case under milder conditions (toluene/40 °C), with mechanistic studies revealing that the pyridyl groups of Neolephos act as proton shuttles, facilitating the nucleophilic attack of the amine on the corresponding acyl-palladium intermediates. The scope of the process was very high and common functional groups on both the diyne and amine partners were tolerated. In addition, in the case of unsymmetrical diynes the regioselectivity of the process was exquisite, indicating a subtle role of the substituents in controlling the outcome of the initial diyne insertion step. In all the cases, the final amide products 37 were obtained with complete stereocontrol on the C=C bond formed.
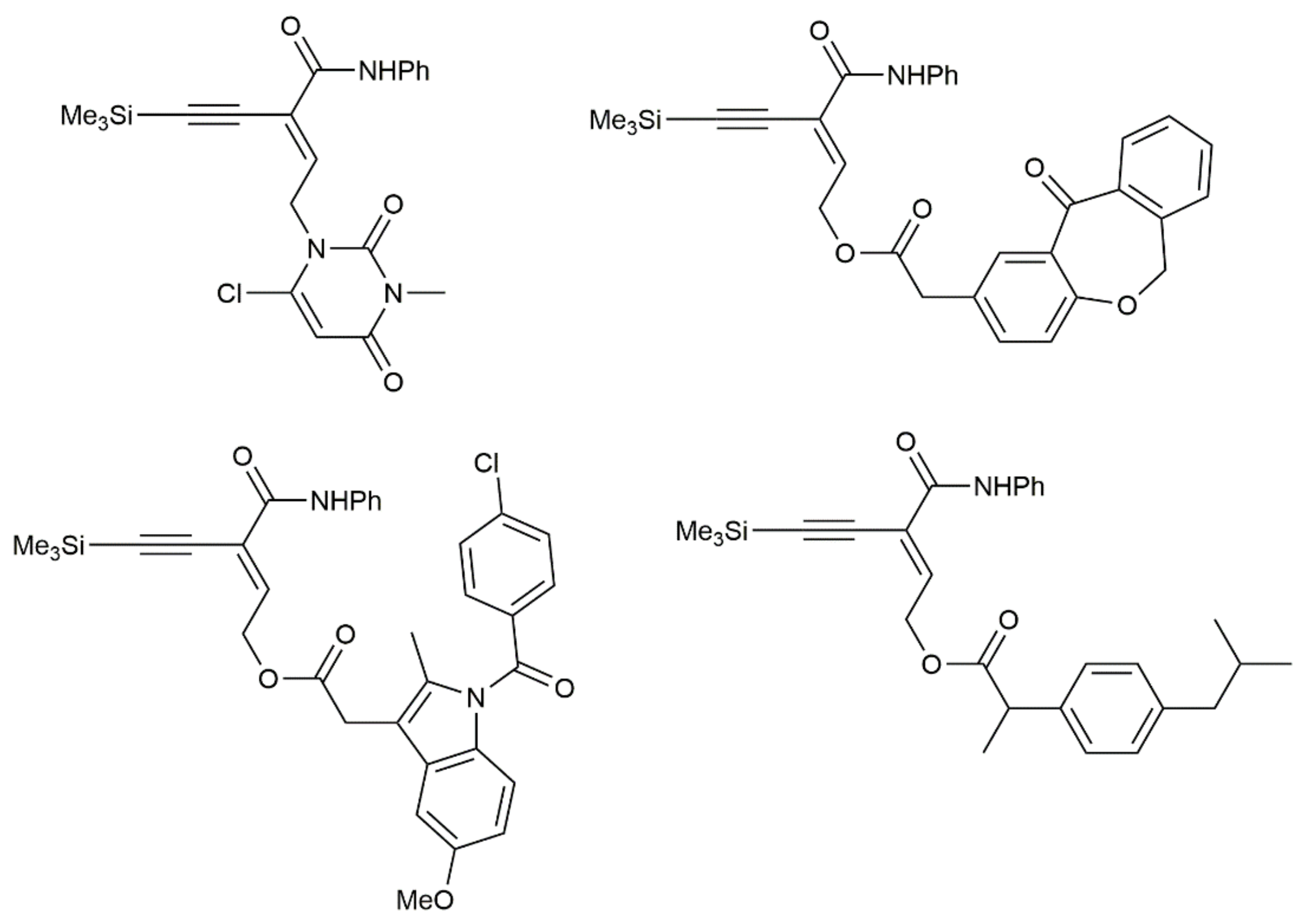
As illustrated in Figure 5, in which different amides containing biologically or pharmaceutically relevant skeletons are shown, the molecular complexity achieved by applying the Beller’s hydroaminocarbonylation reaction was very high [47].
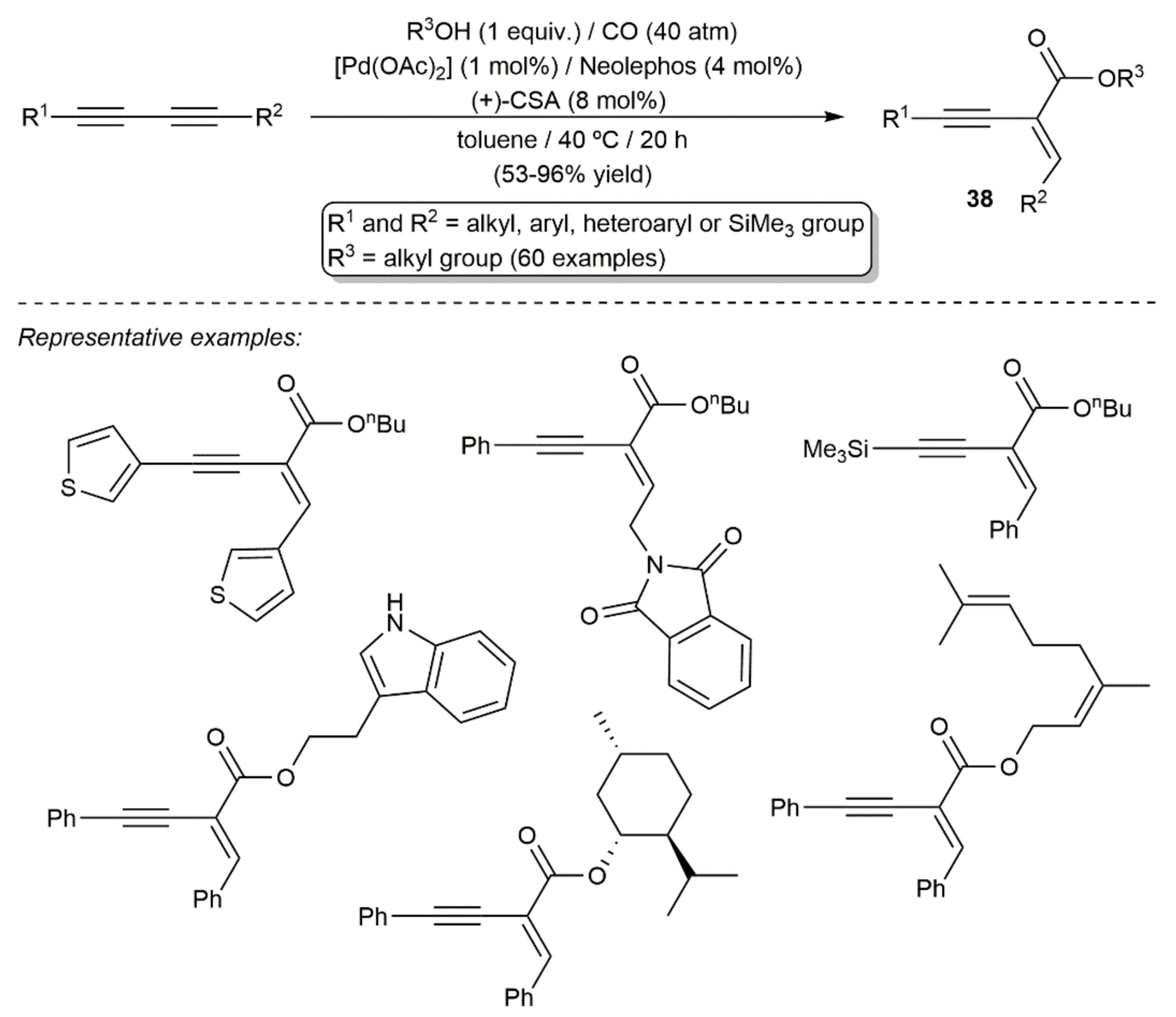
It should also be noted at this point that, by combining Neolephos with Pd(OAc)2 and (+)-10-camphorsulfonic acid (CSA), the group of Beller was also able to develop a wide-scope procedure for the monoalkoxycarbonylation of 1,3-diynes, allowing the access to functionalized 1,3-enynes of type 38 in good yields and with excellent chemoselectivity (Scheme 23) [48].
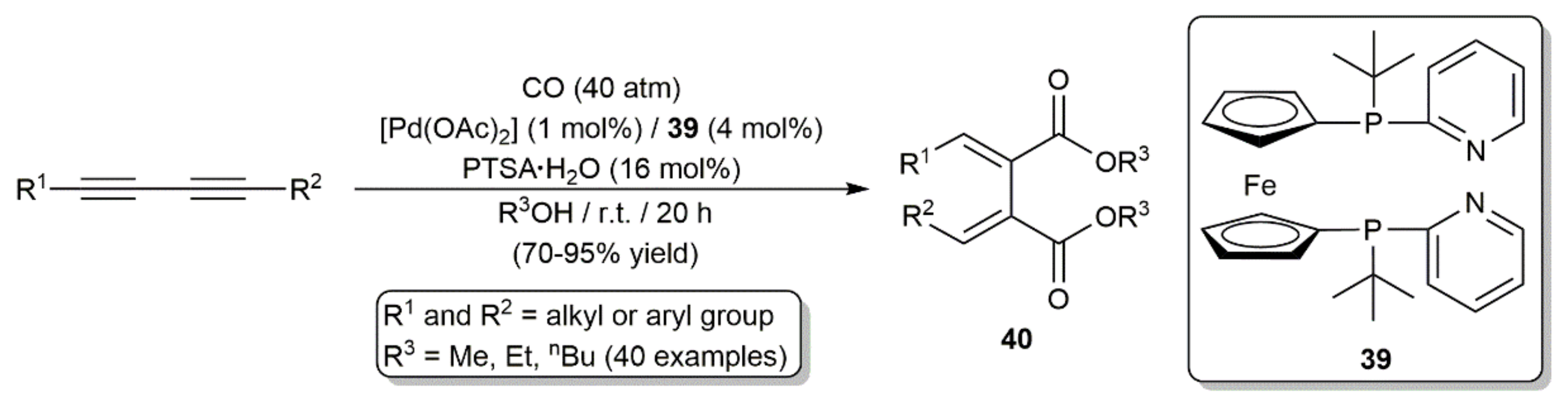
Palladium-catalyzed double alkoxycarbonylation of 1,3-diynes was additionally described by Beller and colleagues, the reactions leading to the regio- and stereoselective formation of 1,2,3,4-tetrasubstituted conjugated dienes 40 (Scheme 24) [49]. The ferrocene-based diphosphine 39, instead of Neolephos, was, in this case, employed as the auxiliary ligand.
6. Hydrosilylation Processes
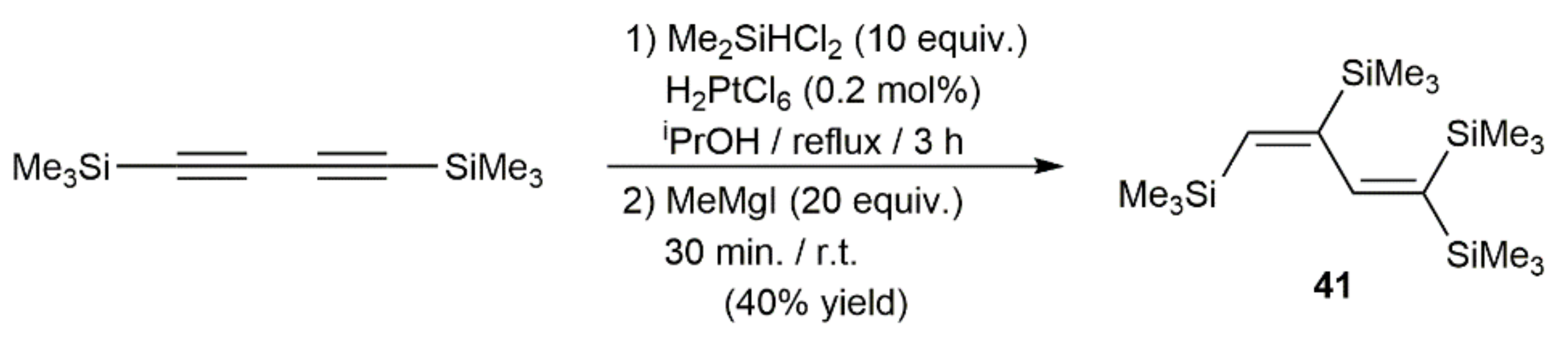
Alkyne hydrosilylation is one of the most efficient methods for preparing alkenyl-silicon derivatives and it has been for a long time a hot topic of research. The versatility of the catalytic hydrosilylation of carbon–carbon triple bonds has been exploited, not only for the preparation of useful synthetic intermediates, but also for the production of complex functional materials [50]. Noble metal catalysts based on Ru, Rh, Ir, Pd, and Pt have been extensively applied in these hydrofunctionalization reactions, and their mechanism of action perfectly established [51,52]. The first example of a 1,3-diyne hydrosilylation was described in 1968 by Bock and Seidl, who reported the preparation of 1,1,3,4-tetrakis(trimethylsilyl)butadiene 41 by reacting 1,4-bis(trimethylsilyl)-1,3-butadiyne with dichloromethylsilane in the presence of a catalytic amount of hexachloroplatinic(IV) acid (H2PtCl6; Speier´s catalyst), followed by methylation with MeMgI (Scheme 25) [53].
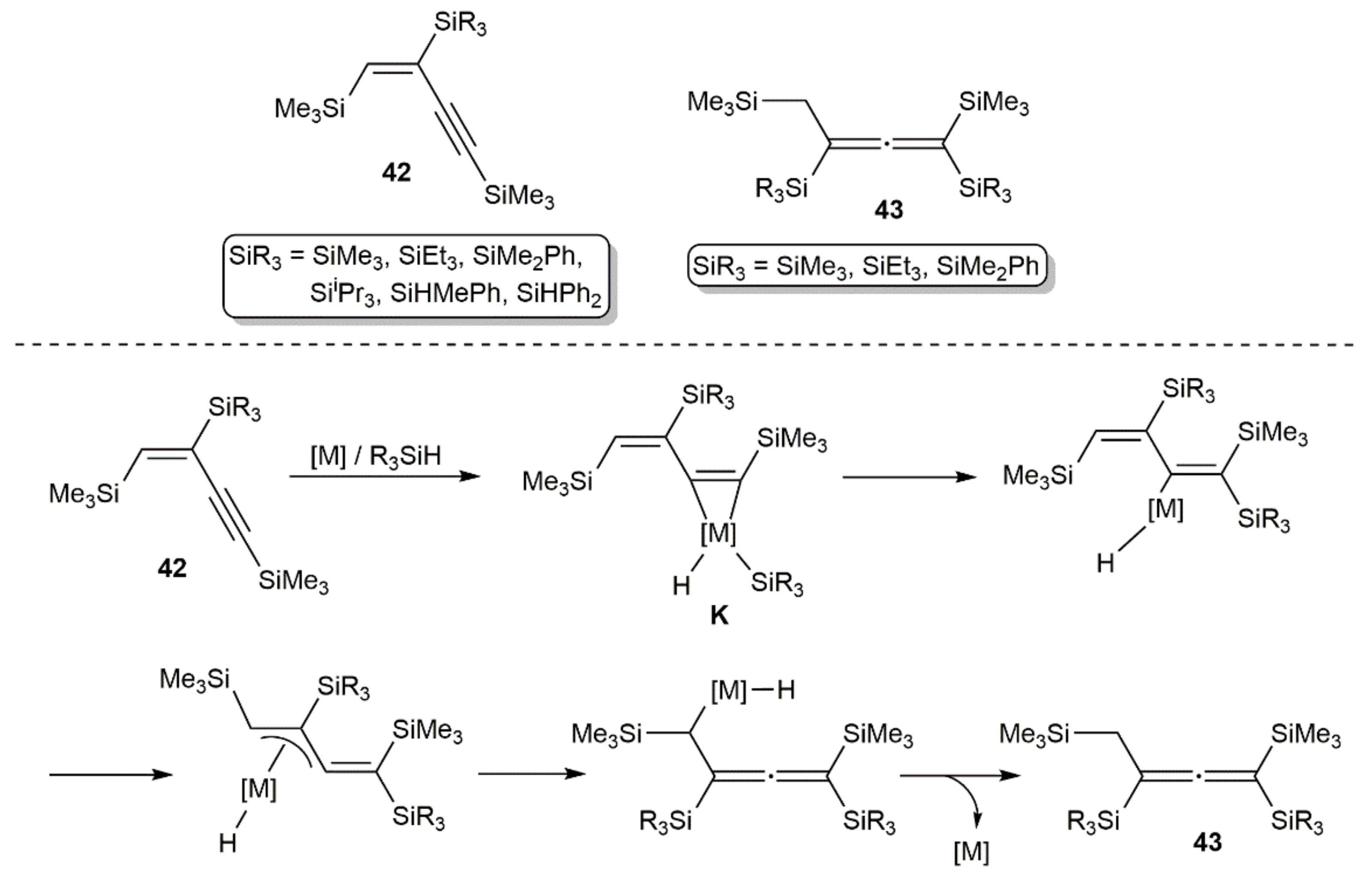
Some years later, Hiyama and colleagues studied the hydrosilylation of 1,4-bis(trimethylsilyl)-1,3-butadiyne with various hydrosilanes using H2PtCl6, [Pt(PPh3)4], [Pd(PPh3)4], [PdCl2(PPh3)2], or [RhCl(PPh3)3] as catalysts [54,55]. The reactions led to three different types of products, i.e., the diene 41, the allenes 42, or the butenynes 43 (see Figure 6), depending on the catalyst and hydrosilane employed. In accord with the results of Bock and Seidl, the diene 41 was selectively generated using H2PtCl6 and Me2SiHCl after treatment of the reaction mixture with MeMgBr. In the rest of the cases, compounds 42 or 43 were formed in different ratios, with allenes 43 resulting from the hydrosilylation of the primary products 42 via a metallacyclopropene intermediate K, whose generation is favored with the low-valent metal catalysts [Pt(PPh3)4], [Pd(PPh3)4], and [RhCl(PPh3)3]. Related mixtures of allene and butenyne products were obtained by Tillack and colleagues when studying the hydrosilylation of RC≡CC≡CR (R = SiMe3, Ph, tBu) with Ph2SiH2, PhMe2SiH, and Et3SiH in the presence of different low-valent Ni(0)-butadiyne catalysts [56]. In this point, the attempts made by Tillack’s group to generate the allenes in an enantioselective manner must also be noted, employing the dimeric rhodium(I) catalyst [{Rh(µ-Cl)(COD)}2] in combination with chiral mono- and diphosphine ligands [57,58]. Unfortunately, an enantiomeric excess up to 27% was achieved at best.
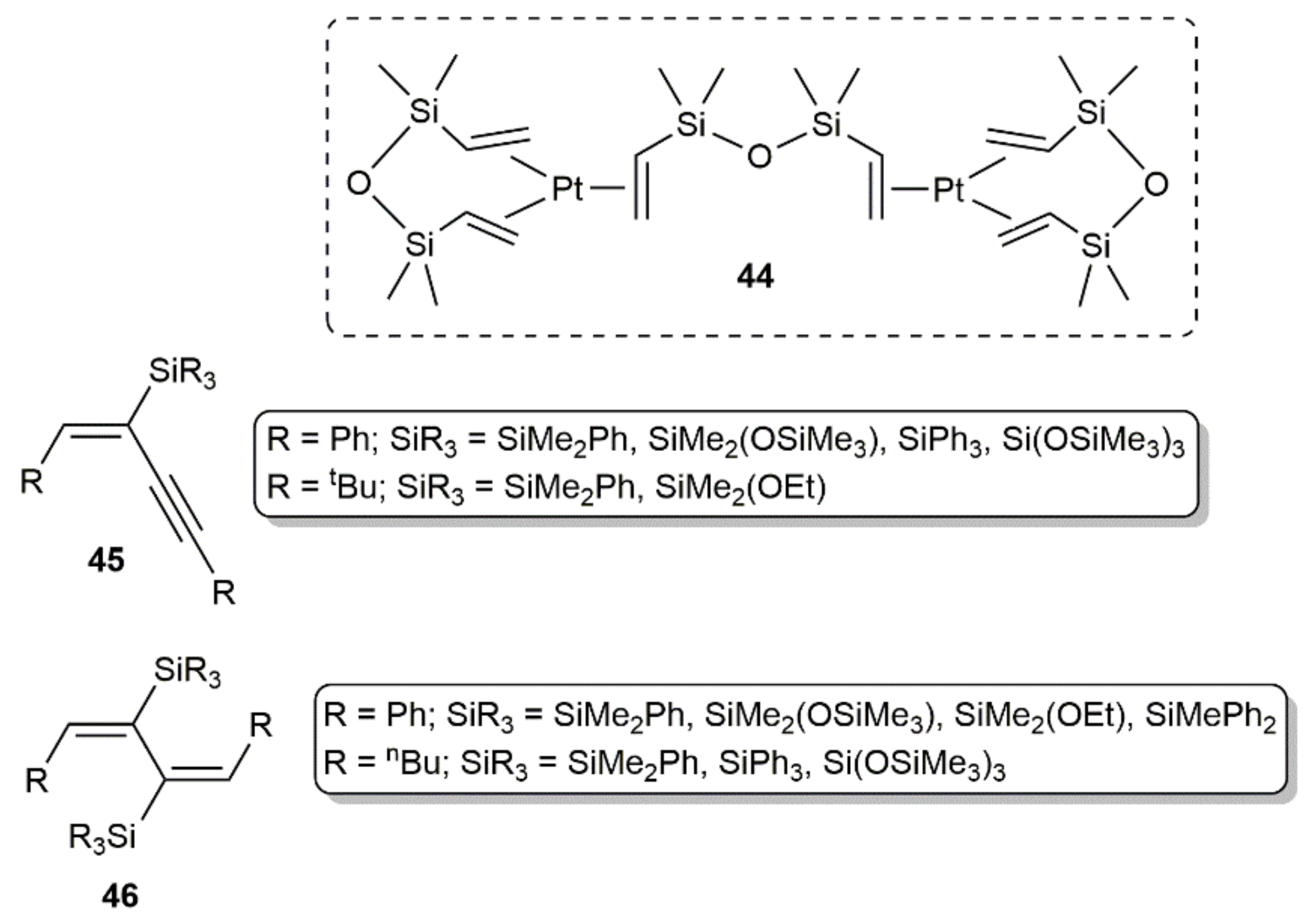
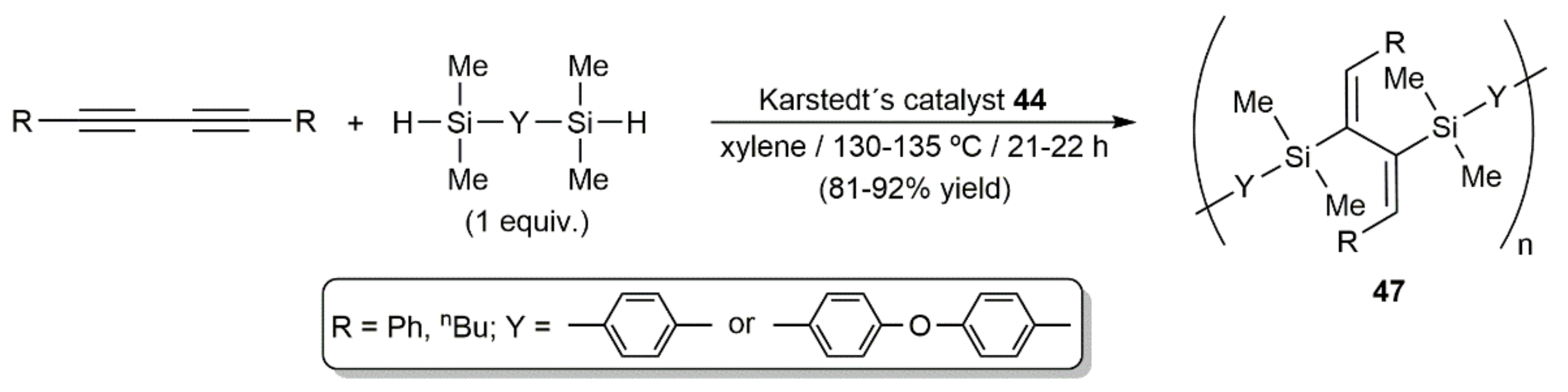
Employing the well-known hydrosilylation catalyst 44 (Karstedt´s catalyst; Figure 7), Perry and colleagues studied the hydrosilylation of diynes RC≡CC≡CR (R = Ph, nBu, tBu) with PhMe2SiH, (Me3SiO)Me2SiH, (EtO)Me2SiH, Ph2MeSiH, Ph3SiH, and (Me3SiO)3SiH [59]. Performing the reactions in toluene or xylene in the temperature range 80–140 °C with a diyne/silane ratio of 1:2, they obtained selectively the mono- and bis-silylated products 45 and 46 (see Figure 7), the formation of the latter being, in general, favored at high temperature. Nonetheless, the most important factor in guiding the reaction towards the formation of the mono- or dihydrosilylated products was found to be the steric hindrance between the diyne and silane substituents. Thus, the results obtained indicated that compounds 46 are only accessible when the steric constraints are not too high [59]. Regardless of the product obtained, addition of the Si–H bonds to the C≡C units proceeded in all the cases in a syn manner.
In the same work, the silicon-containing polymers 47 were also synthesized in high yield by bis-hydrosilylation of diynes RC≡CC≡CR (R = Ph, nBu) with 1,4-bis(dimethylsilyl)benzene and 4,4′-bis(dimethylsilyl)diphenyl ether (Scheme 26) [59].
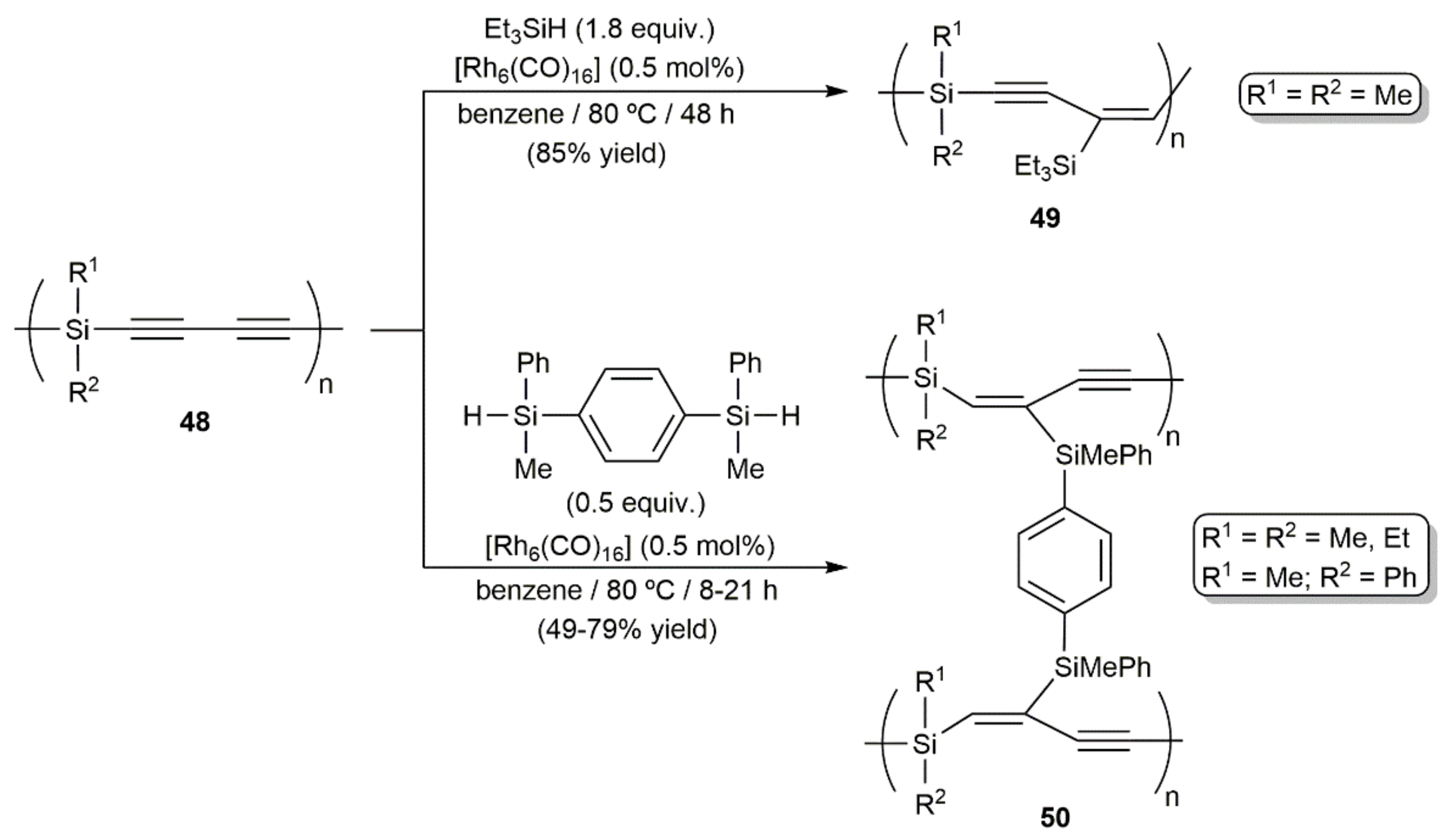
At this point, it must also be mentioned that access to other organosilicon polymers, i.e., compounds 49 and 50, through the stereoselective and regiospecific hydrosilylation of the poly[(silylene)but-1,3-diynes] derivatives 48 with triethylsilane and 1,4-bis(methylphenylsilyl)benzene, respectively, was described by Ishikawa and colleagues, employing, in this case, [Rh6(CO)16] as the catalyst (Scheme 27) [60,61].
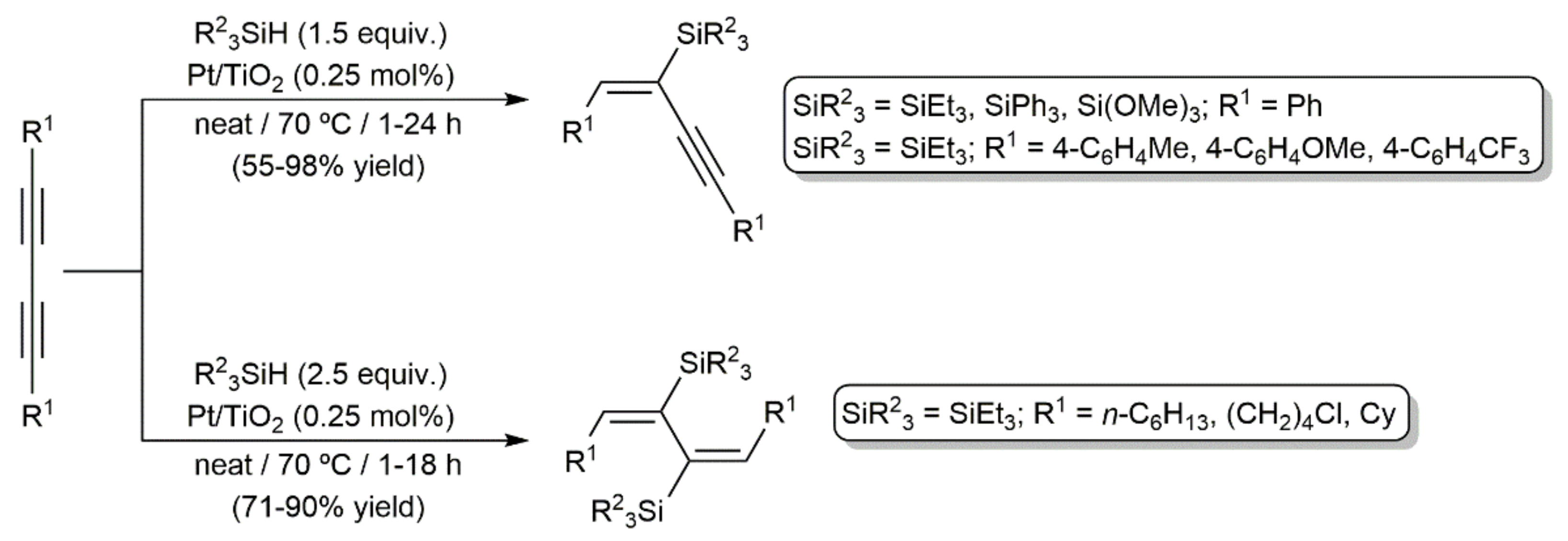
In an independent study, Walkowiak and colleagues explored the addition of triethyl- and triphenylsilane to diynes RC≡CC≡CR (R = Me, Ph, tBu, (CH2)8CO2Me, CMe2OSiMe3) catalyzed by the Karstedt´s catalyst 44, as well as [Pt(PPh3)4] and PtO2 [62]. By selecting appropriate reaction conditions (temperature, stoichiometry, and catalyst), and depending on the steric constraints of the reagents, they were able to synthesize mono- and bis-silylated products related to those obtained by Perry and colleagues (45 and 46 in Figure 7) with high yields and selectivities. In a later work, the same group also demonstrated the possibility of using a silsesquioxane-functionalized silane, i.e., (HSiMe2O)(iBu)7Si8O12, as a substrate in these Pt-catalyzed hydrosilylation processes [63]. On the other hand, as shown in Scheme 28, mono- and bis-addition products were also selectively obtained by using titania-supported platinum as the catalyst [64]. In addition to the stoichiometry employed, the aromatic or aliphatic nature of the starting 1,3-diyne was, in this case, key to obtaining one product or the other. As in the previous examples, the reactions were, again, highly stereo- and regioselective, leading to the monosilylated enynes and disilylated dienes with exclusive E-stereochemistry (syn addition) and with the silicon atoms bonded to the internal carbons of the diynic units.
In addition to PtO2 and Pt/TiO2, other heterogeneous catalysts capable of promoting the hydrosilylation of 1,3-diynes quoted in the literature are: impregnated platinum on magnetite, i.e., PtO/PtO2-Fe3O4 [65], Pd(0) nanoparticles supported on polymeric resins [66] or stabilized by imidazolium salts [67], Rh(0) nanoparticles stabilized by a nitrogen-rich polyethylene glycol (PEG) derivative [68] and a nickel(II)-containing porous organic polymer [69]. All of them showed an exquisite syn selectivity and, depending on the reaction conditions employed, led to the corresponding mono- or bis-addition products.
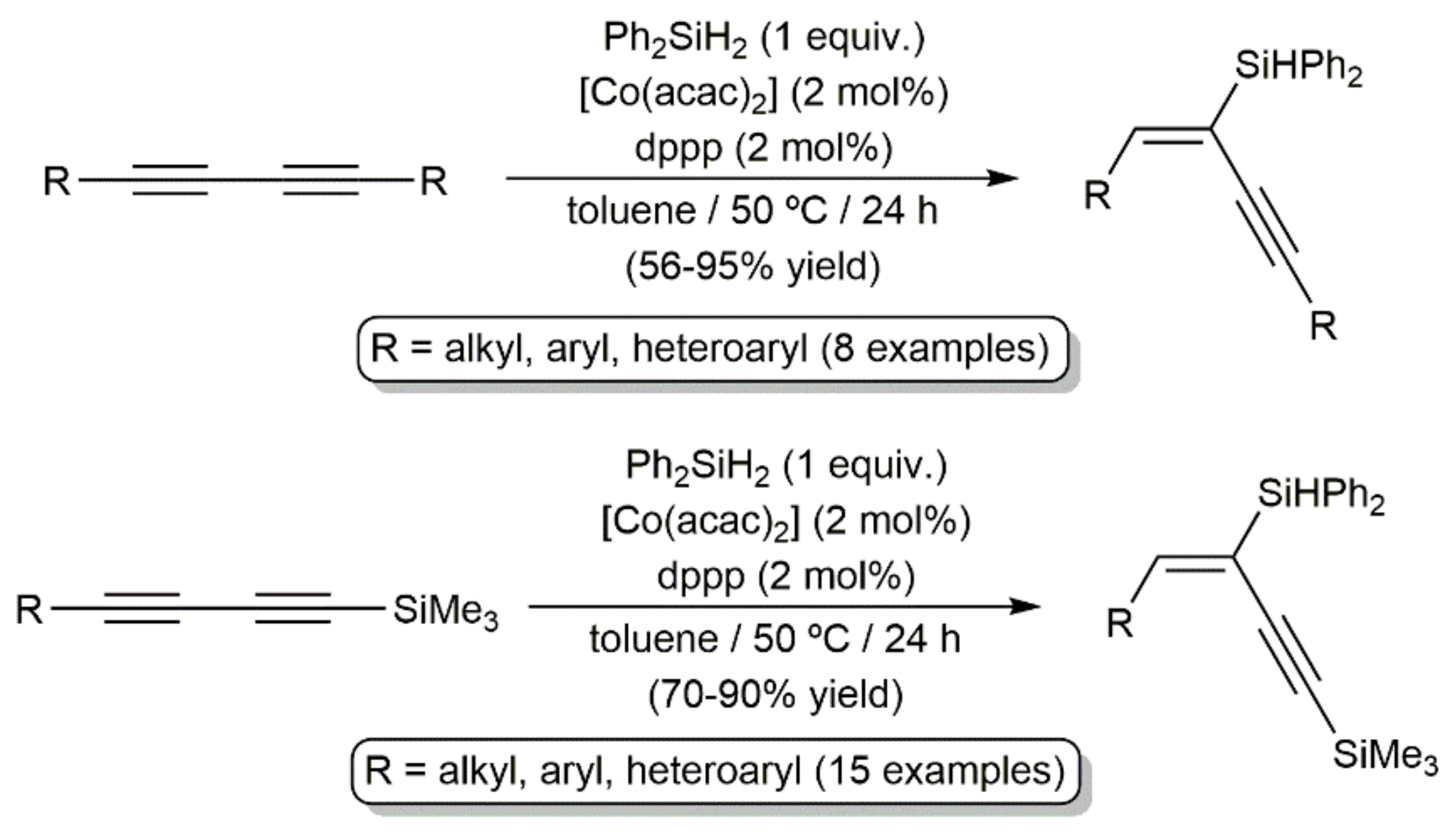
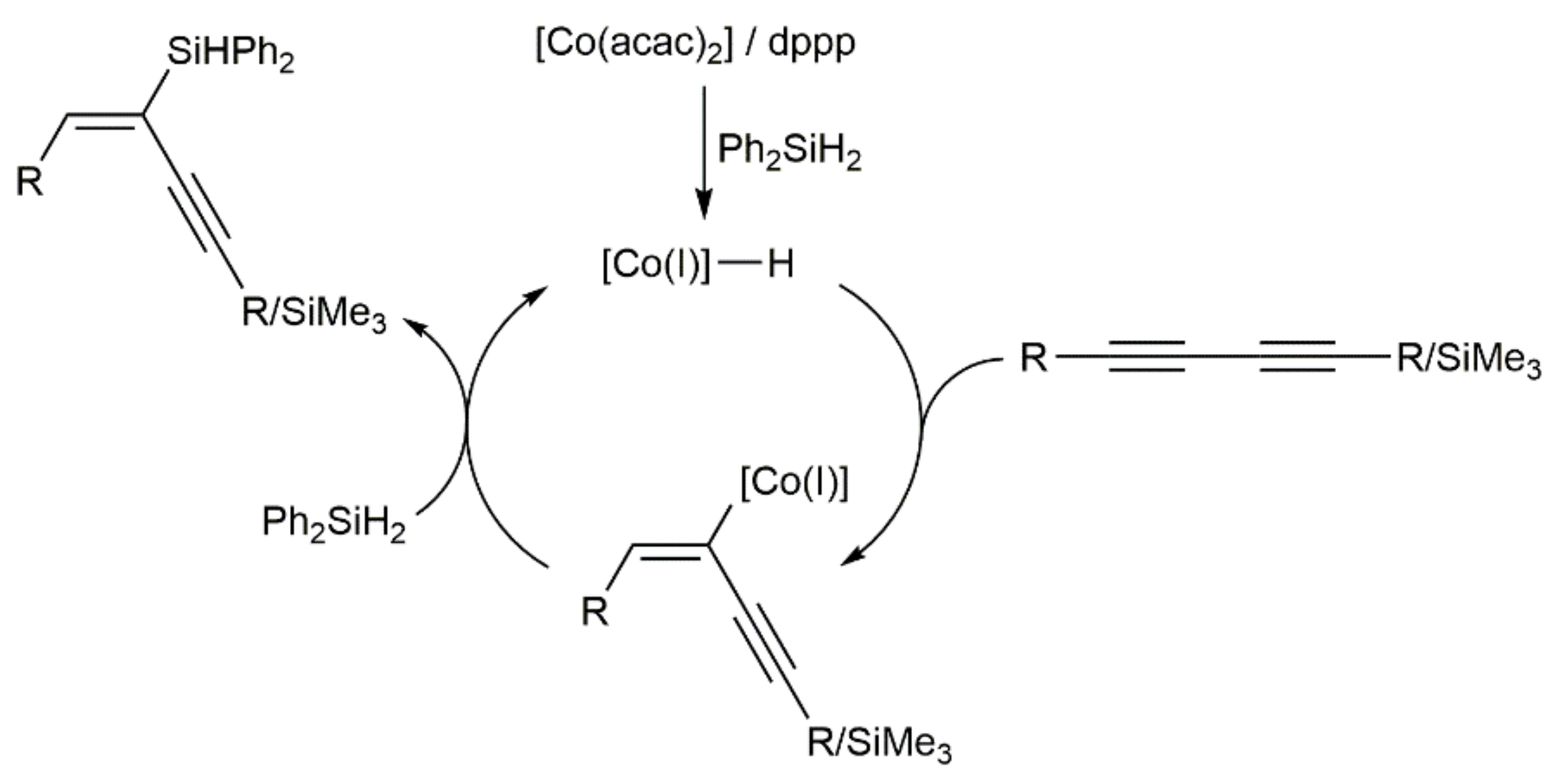
In another vein, the current demand for economical and environmentally benign catalysts has spurred the search for efficient cobalt-based systems able to promote the hydrosilylation of both terminal and internal alkynes [70,71]. In this context, Ge and colleagues reported in 2019 the regio- and stereoselective access to a broad range of silyl-functionalized 1,3-enynes by hydrosilylation of 1,3-diynes (syn addition), employing a catalyst generated from bench-stable Co(acac)2 and 1,3-bis(diphenylphosphino)propane (dppp) (Scheme 29) [72].
A classical hydrometallation pathway via a cobalt(I)-hydride intermediate was proposed by the authors for this co-catalyzed hydrosilylation of 1,3-diynes (see Scheme 30) [72].
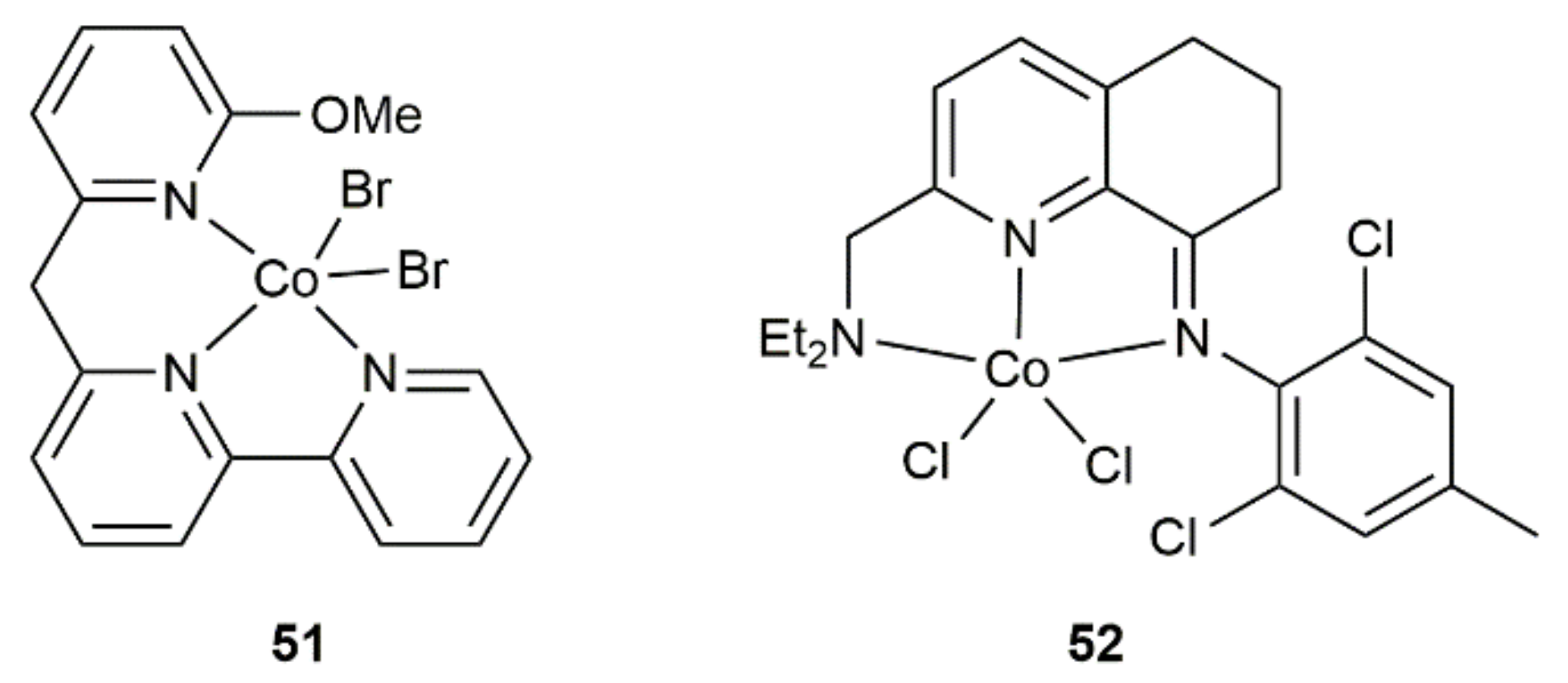
Related hydrosilylation reactions, featuring the same regio- and stereocontrol, were subsequently described with other cobalt-based catalytic systems, such as complexes 51 [73] and 52 [74] (see Figure 8) or the CoCl2/dppp combination [75]. In these cases, the reducing agent NaHBEt3 was systematically employed as a co-catalyst, allowing the reactions to proceed under milder conditions (r.t. or 30 °C) and shorter times (from 5 min to 1 h).
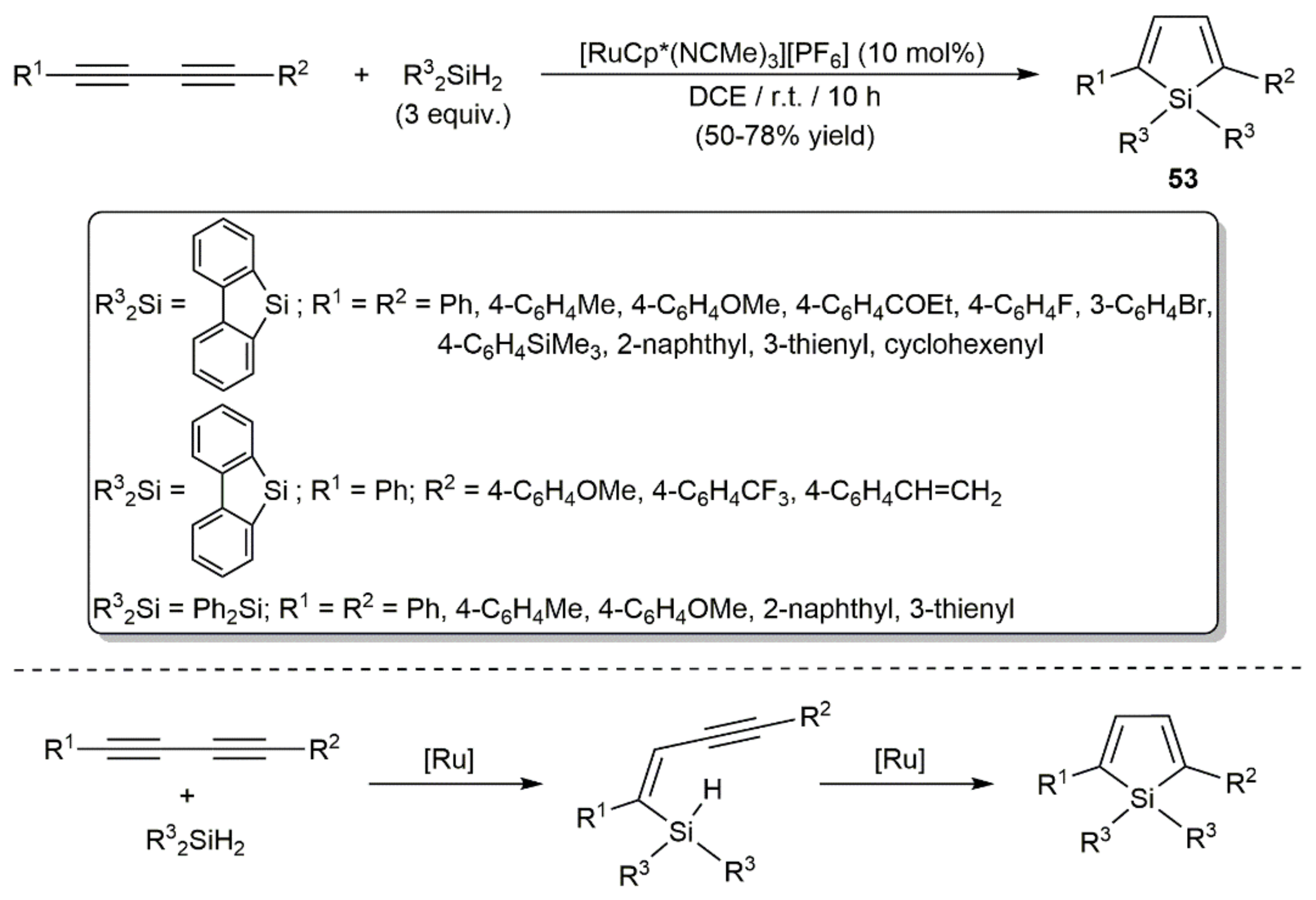
Access to 2,5-disubstituted heteroles (heteroatom-substituted cyclopentadienes) by [4+1]-type annulation reactions of 1,3-diynes with appropriate group 14–16 element compounds is well documented in the literature [76]. In line with this, Murakami and colleagues reported the preparation of several 2,5-disubstituted siloles 53 facing different symmetrically and unsymmetrically substituted 1,3-diynes to diphenylsilane or 9-silafluorene in the presence of catalytic amounts of the cationic ruthenium(II) complex [RuCp*(NCMe)3][PF6] (Cp* = pentamethylcyclopentadienyl) (Scheme 31) [77]. The reactions, which were conducted in 1,2-dichloroethane at room temperature, proceed through the sequential double anti-hydrosilylation of the diynes. By employing diphenylgermane Ph2GeH2 instead of Ph2SiH2 or 9-silafluorene and applying identical reaction conditions, 2,5-substituted germoles could be equally generated in moderate to high yields [78].
As shown in Scheme 32, the Ru(II) complex [RuCp*(MeCN)3][PF6] was also capable of promoting the anti-addition of the tertiary silane BnMe2SiH to the chiral diynol 54 [79]. The resulting enynyl-silane product 55 was isolated in excellent yield, with other possible isomers not being observed in the reaction crude.
7. Hydroamination and Hydroamidation Processes
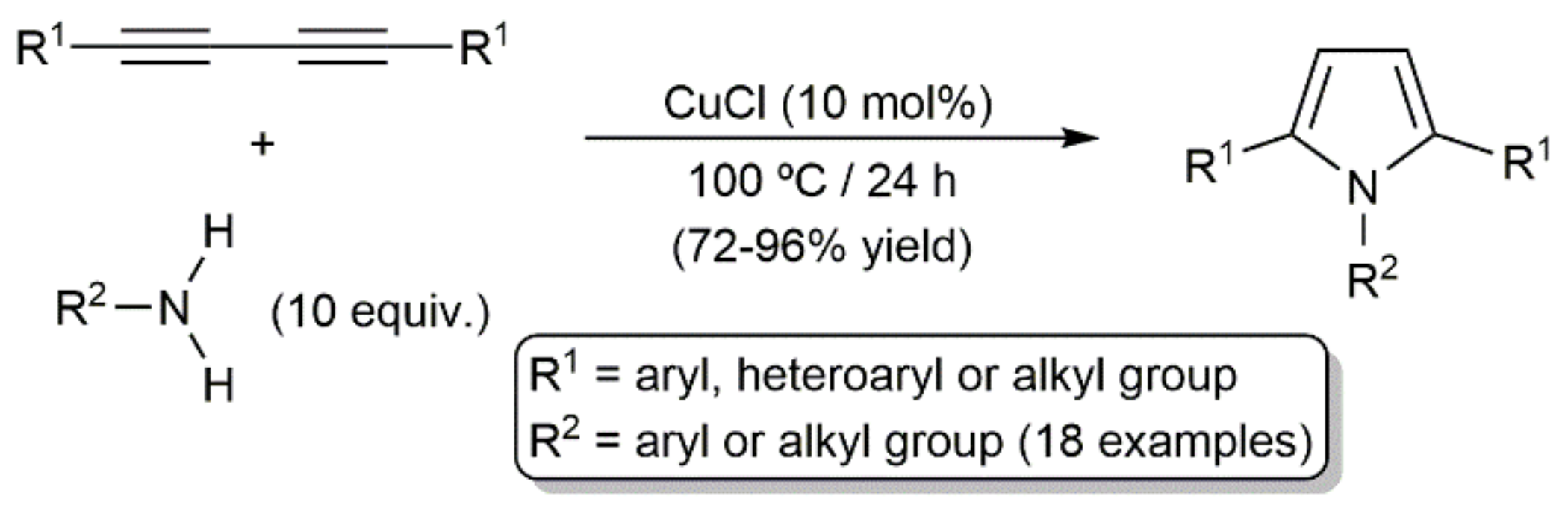
In line with the silole-ring formation reactions discussed above (see Scheme 31), access to 1,2,5-trisubstituted pyrroles by double addition of the N–H bonds of primary amines across 1,3-diynes is well documented. Such transformations were first reported in 1961, employing 0.1 mol% of CuCl as a catalyst at 150–180 °C [80,81], but, as shown in Scheme 33, they also proceed in high yields at a lower temperature (100 °C), increasing the amount of CuCl to 10 mol% [82].
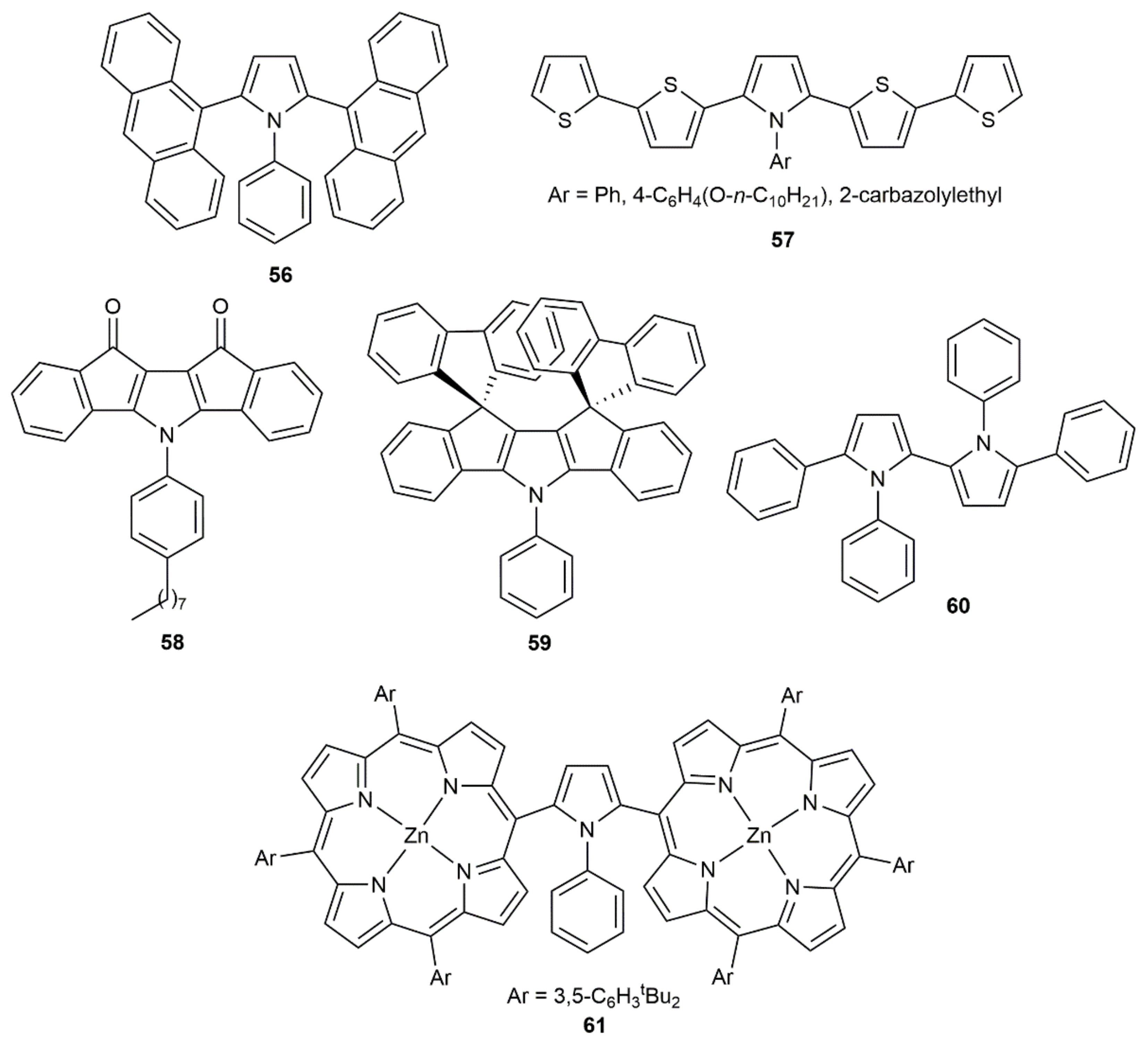
Although these pyrrole-ring formation reactions are also known to proceed in the presence of KOH [83], the synthetic utility of the process, exemplified with the preparation of compounds 56–61 featuring interesting photophysical properties (see Figure 9), has only been demonstrated using catalytic CuCl [84,85,86,87,88,89]. In all the cases, the central pyrrolic unit was generated through the initial copper-catalyzed annulation of a primary amine with a 1,3-diynic system.
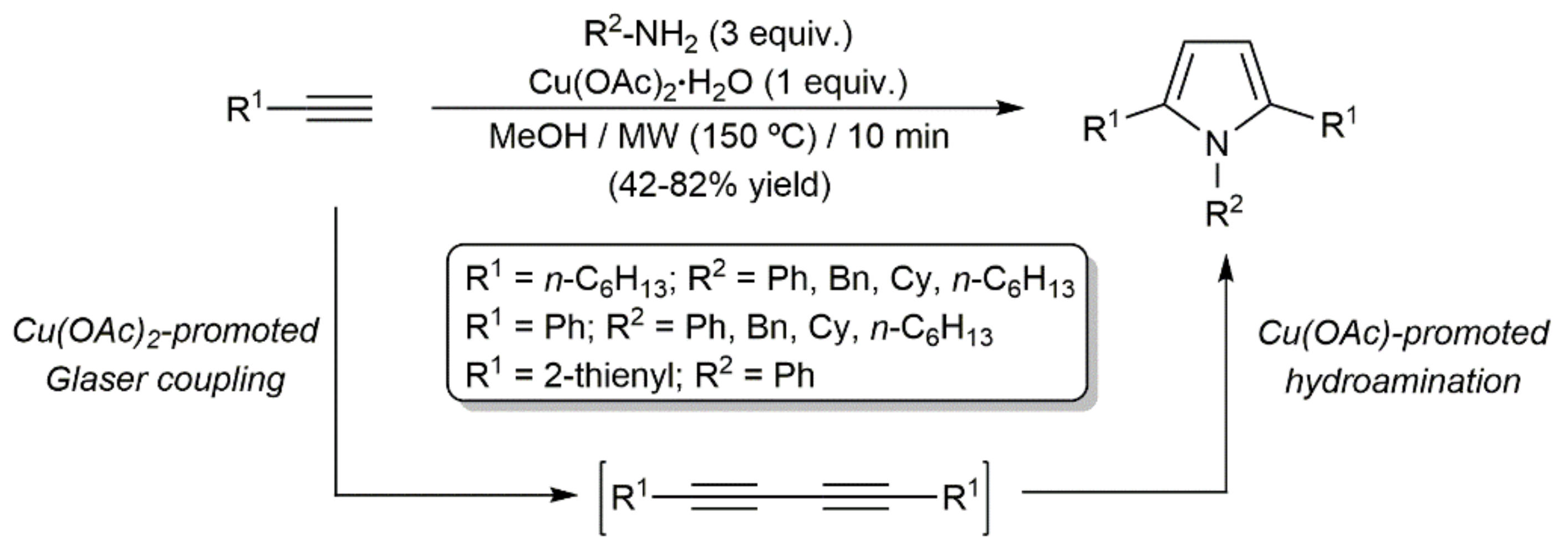
In this context, we must note that several 1,2,5-trisubstituted pyrroles were synthesized by Jun and colleagues by reacting terminal alkynes with primary amines in the presence of Cu(OAc)2·H2O (Scheme 34) [90]. The process was performed under microwave (MW) irradiation and involved the initial Cu(OAc)2-promoted Glaser homocoupling of the alkynes, followed by the double hydroamination of the resulting diynes. In the cyclization step, the Cu(I) salt Cu(OAc) generated during the homocoupling reaction was proposed by the authors to serve as the catalytically active species.
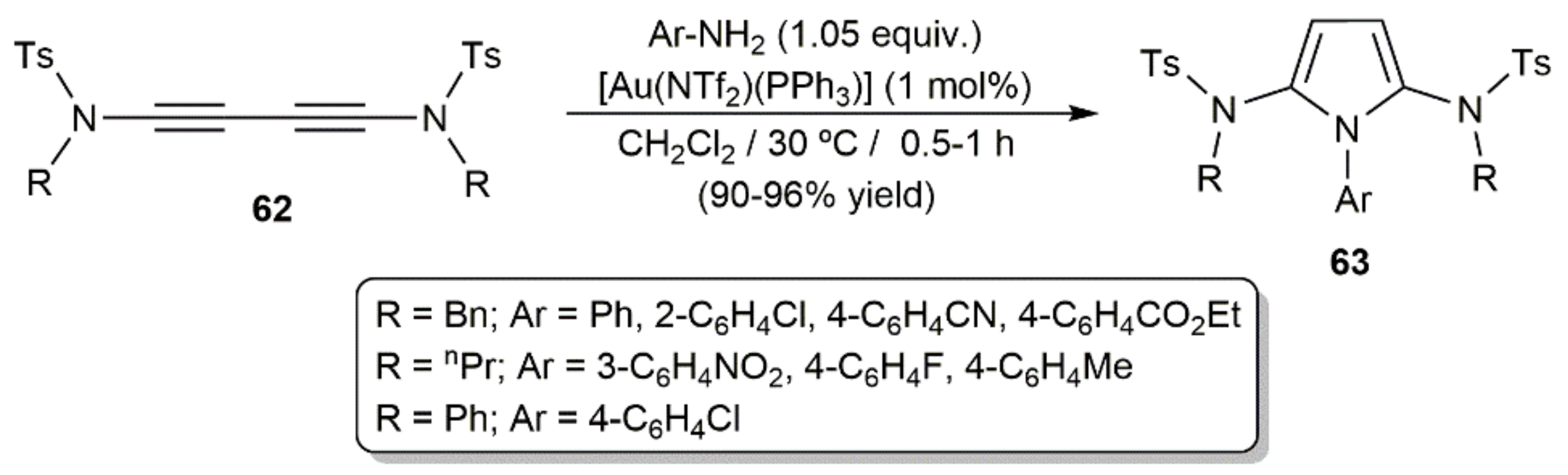
On the other hand, taking advantage of the well-known ability of gold complexes to promote the addition of N–H bonds across C–C multiple bonds [91], the access to pyrrole derivatives from diynes and primary amines has also been achieved under gold catalysis. Thus, Skrydstrup and colleagues synthesized the 2,5-diamidopyrroles 63 in excellent yields, short times, and under mild conditions when 1,3-diynamides R(Ts)NC≡CC≡CN(Ts)R (62; Ts = p-toluenesulfonyl) and different anilines were subjected to the catalytic action of the Au(I)-phosphine complex [Au(NTf2)(PPh3)] (Scheme 35) [92].
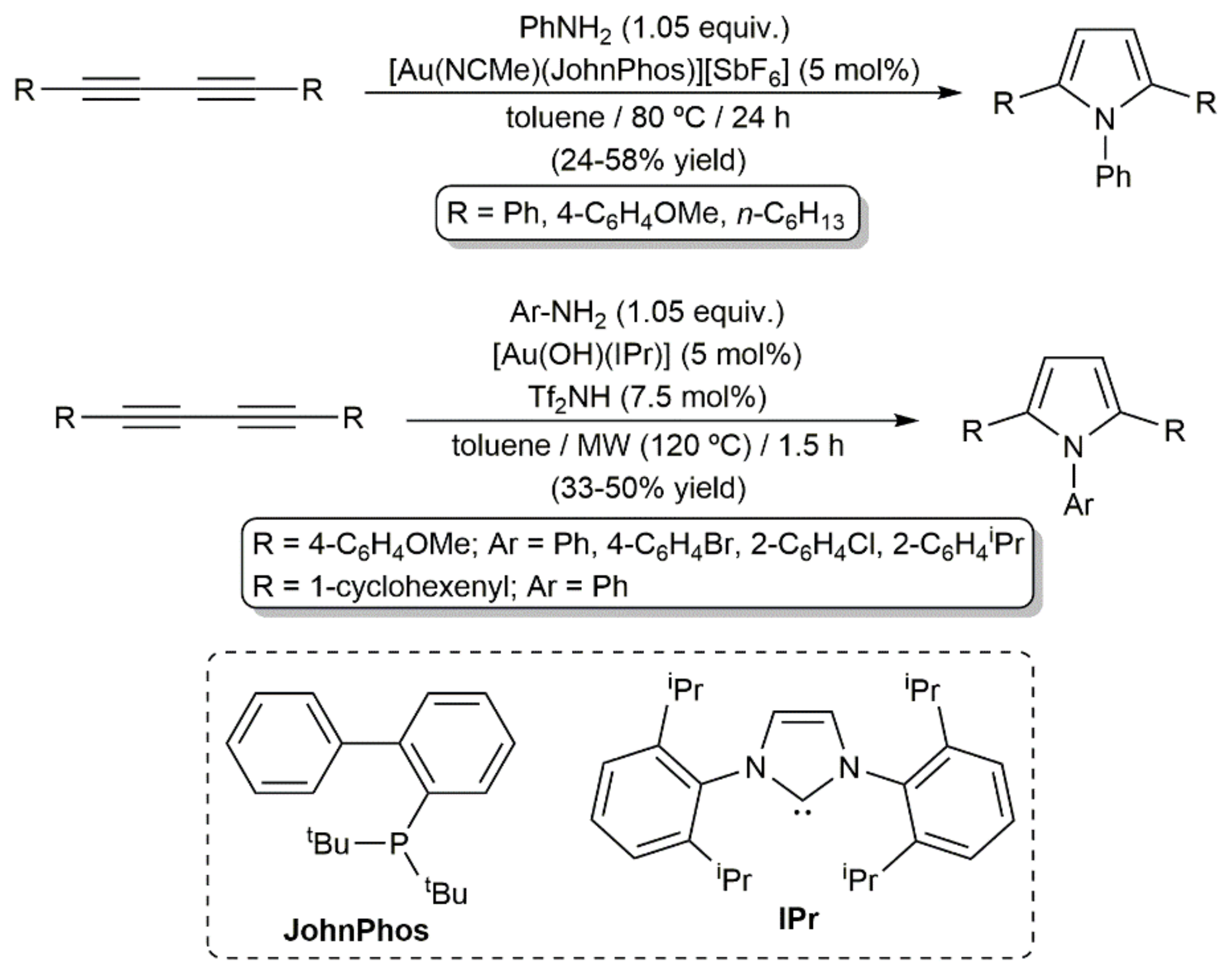
In the same work, the hydroamination of more classical alkyl- and aryl-substituted diynes with aniline was additionally explored, a process that proved to be more difficult due to the lower electronic activation of the C≡C bonds in comparison to those of compounds 62. In fact, only when the reactions were performed in toluene at 80 °C with 5 mol% of the cationic complex [Au(NCMe)(JohnPhos)][SbF6] could the desired pyrroles be accessed, albeit in modest yield and after remarkably longer times (Scheme 36) [92]. Comparable yields were reached by Nolan and colleagues when studying related hydroamination processes with complex [Au(OH)(IPr)] (IPr = 1,3-bis(2,6-diisopropylphenyl)imidazole-2-ylidene) under MW irradiation (see Scheme 37) [93].
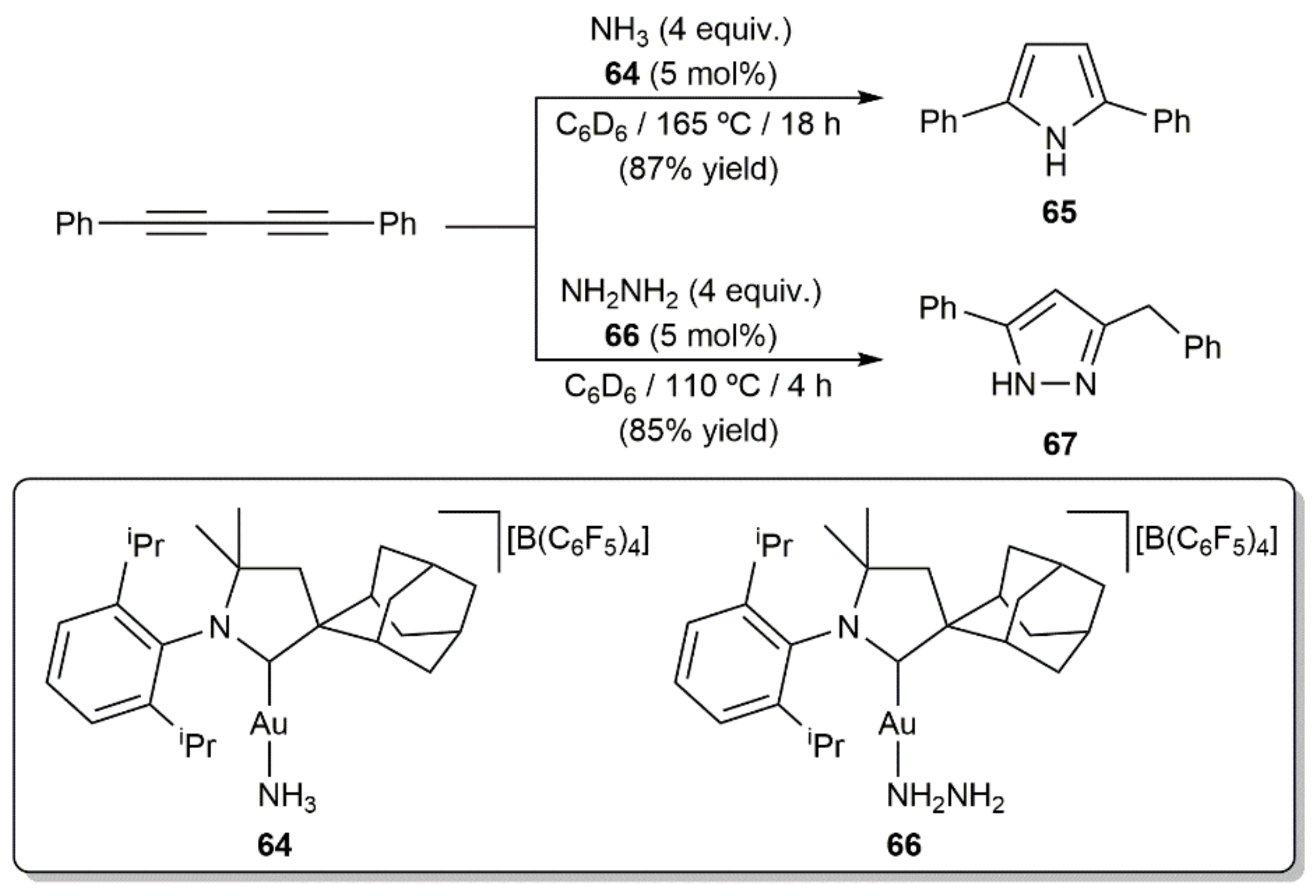
In addition to the examples just commented upon, Bertrand and colleagues successfully achieved the catalytic hydroamination of 1,4-diphenylbuta-1,3-diyne with parent ammonia and hydrazine to afford the N-unsubstituted pyrrole 65 [94] and the pyrazole derivative 67 [95], respectively, employing the cationic gold(I) complexes 64 and 66 having a bulky cyclic (alkyl)(amino)carbene ligand (Scheme 37). Within this context, we must mention that access to substituted pyrazoles through metal-free Cope-type hydroamination of 1,3-diynes with hydrazine derivative H2NNHR (R = H, alkyl or aryl group) under thermal or base-promoted conditions has been amply reported [96,97,98,99,100,101,102]. Similarly, catalyst-free protocols for the construction of isoxazole [96,100,101,103,104] and pyrimidine [105,106,107] scaffolds by Cope-type hydroamination of 1,3-diynes with hydroxylamine and guanidines, respectively, can also be found in the literature.
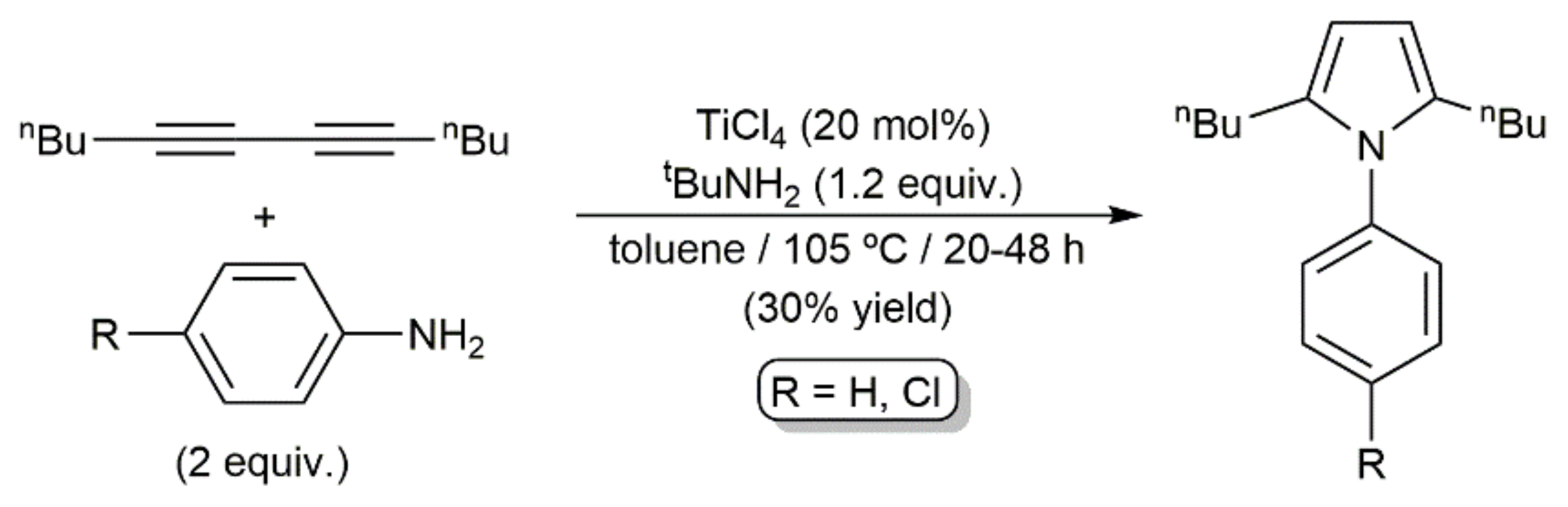
As shown in Scheme 38, a catalytic system composed of TiCl4 and tert-butylamine was also able to promote the coupling of an aliphatic 1,3-diyne with aromatic primary amines to afford pyrroles, albeit featuring a low efficiency [108].
On the other hand, intermolecular copper-catalyzed hydroamination reactions of aromatic 1,3-diynes with indole, imidazole, and pyrazole derivatives were described by Kundu and colleagues (Scheme 39) [109]. The reactions, conducted in DMSO at 100 °C with a CuI/Phen/Cs2CO3 (Phen = 1,10-phenantroline) combination, afforded the linear N-alkenynes 68 in high yields as mixtures of the corresponding E and Z isomers, with the latter being predominant in all the cases. Employing the same reaction conditions, addition of the N–H bond of both cyclic and acyclic amides to the diynes also took place, leading to the corresponding N-alkenyne products 69 with generally lower yields (Scheme 40). Interestingly, while a similar Z-stereoselectivity was observed when employing cyclic amides, the hydroamidation reactions with the acyclic ones afforded compounds 69 as the E-stereoisomers exclusively.
In 2011, Hua and colleagues developed an efficient and atom-economic approach to diamino-substituted naphthalene derivatives 70 through a CuCl-catalyzed coupling reaction of aromatic diynes and cyclic primary amines (Scheme 40) [110]. The process involves the initial hydroamination of one the C≡C bonds of the diyne to generate an N-alkenyne intermediate L, in which the remaining alkyne group and the ortho-C–H of the aryl substituent are activated by the copper catalyst, evolving into naphthalenes 70 through the intramolecular hydroarylation of the C≡C bond and subsequent reaction with a second equivalent of the amine.
Access to related diamino-substituted naphthalenes was later described by Lee and Jung starting from both cyclic and acyclic amines and aryl alkynyl carboxylic acids ArC≡CCO2H, which, under the action of CuCl/Cu(OTf)2, are transformed in situ into the corresponding 1,3-diynes ArC≡CC≡CAr [111]. 1,4-Bis(trimethylsilyl)-1,3-butadiyne was additionally employed as a precursor of compound ArC≡CC≡CAr in this type of transformation. A co-operative Pd/Cu catalytic system, enabling the initial double Sonogashira coupling of Me3SiC≡CC≡CSiMe3 with aryl halides, was, in this case, used to generate the naphthalene products in a one-pot manner [112].
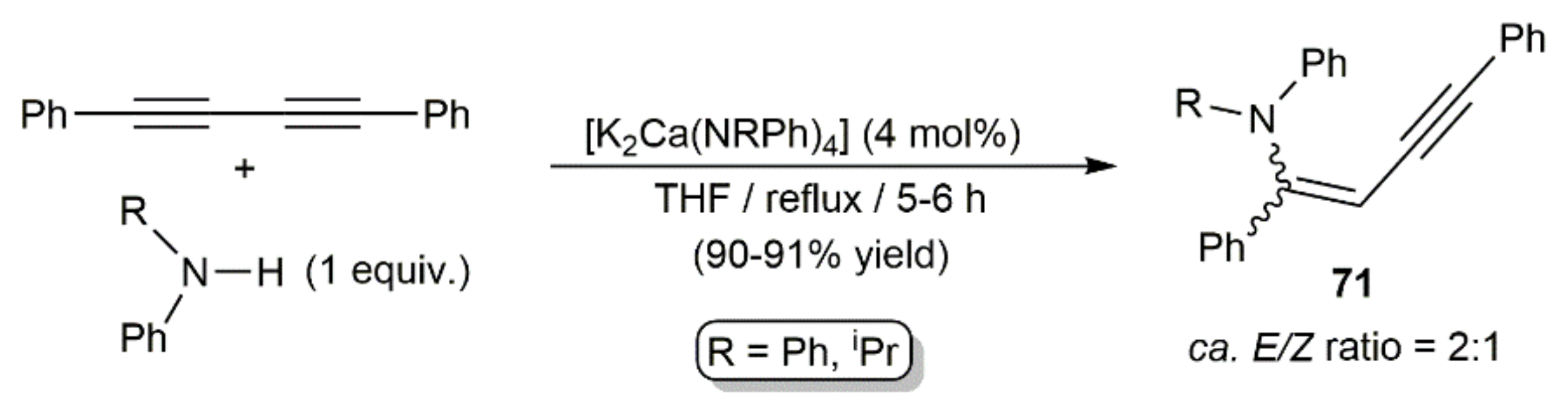
Hydroamination reactions of 1,4-diphenyl-1,3-butadiyne with s-block metal catalysts were studied by the group of Westerhausen. Thus, in an initial report, they found that monoaddition of the secondary amine HNRPh (R = Ph, iPr) to the diyne can be promoted by the respective heterobimetallic amides [K2Ca(NRPh)4] in refluxing THF, the reactions affording E/Z mixtures of the 1-anilino-1,4-diphenylbut-1-ene-3-ynes 71 in excellent yields (Scheme 41) [113].
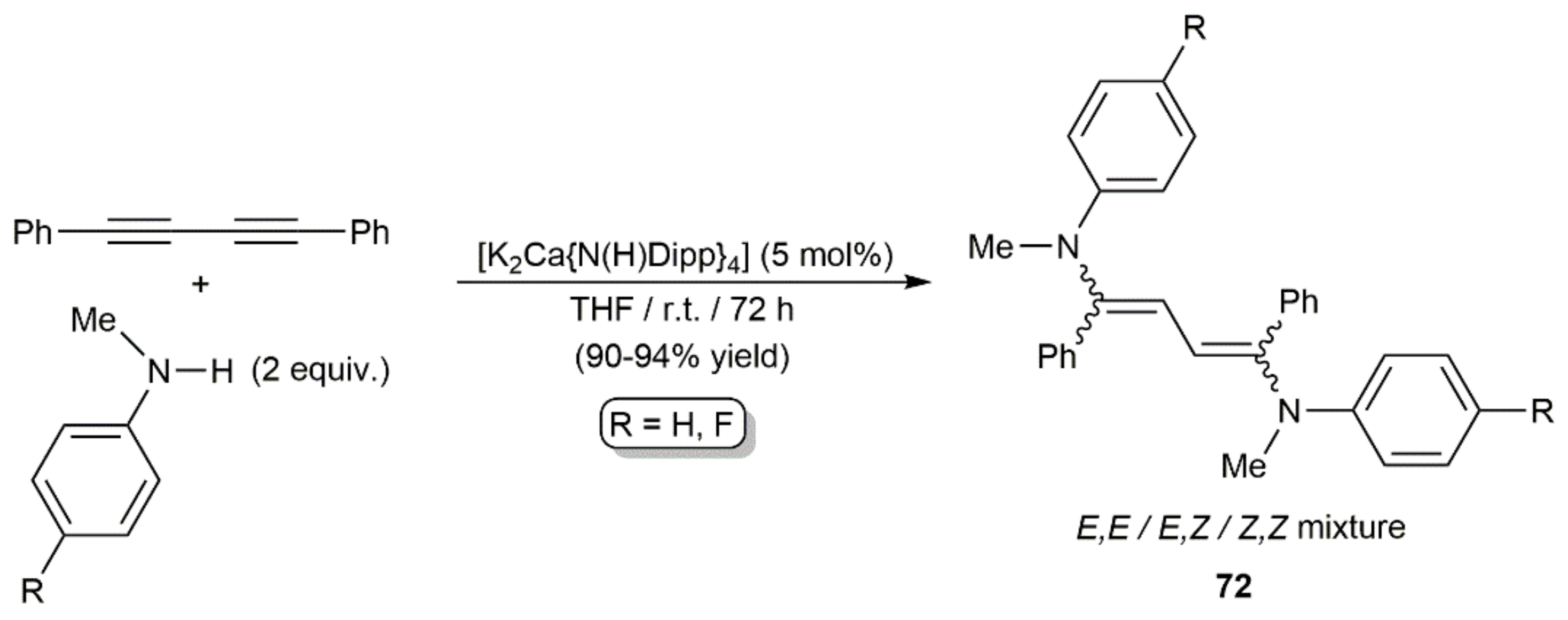
Later, they described related hydroamination reactions of PhC≡CC≡CPh with N-methylanilines HNMeAr (Ar = Ph, 4-C6H4F) under milder conditions (r.t.) using catalytic amounts of [K2Ca{N(H)Dipp}4] (Dipp = 2,6-diisopropylphenyl) [114]. This calciate complex allowed not only access to the monoaddition products (E/Z)-PhC≡CCH = CPh(NMeAr), but also to the diaddition ones 72 when the reactions were carried out with a twofold excess of the amines (Scheme 42). Dienes 72 were, in all the cases, generated as mixtures of the three possible stereoisomers.
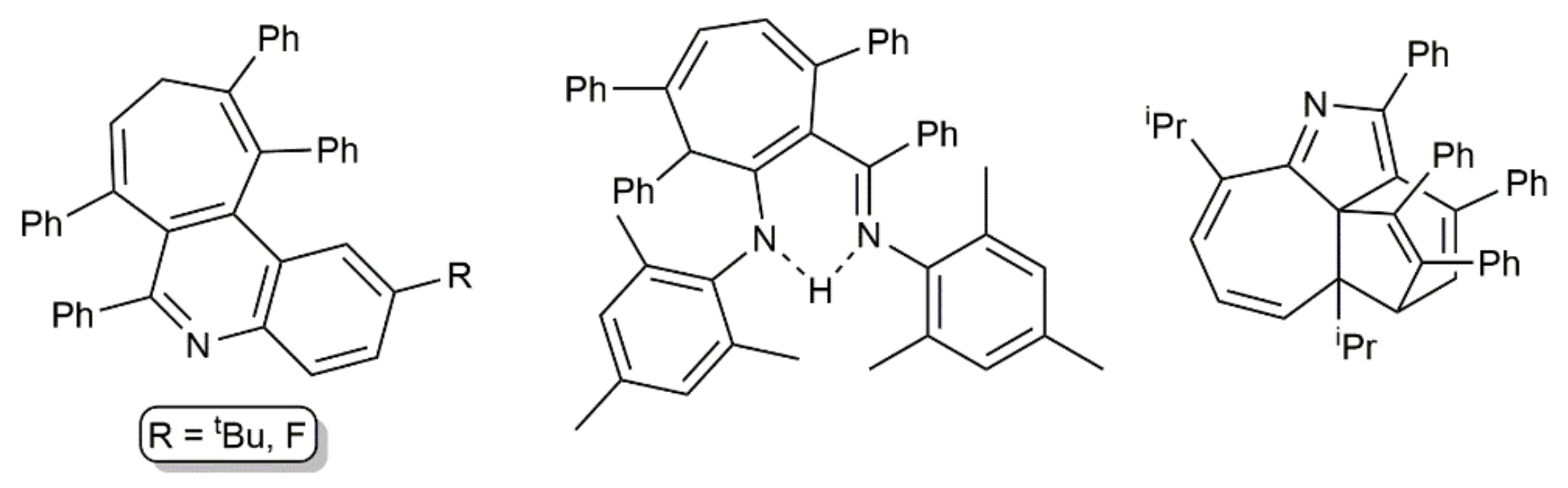
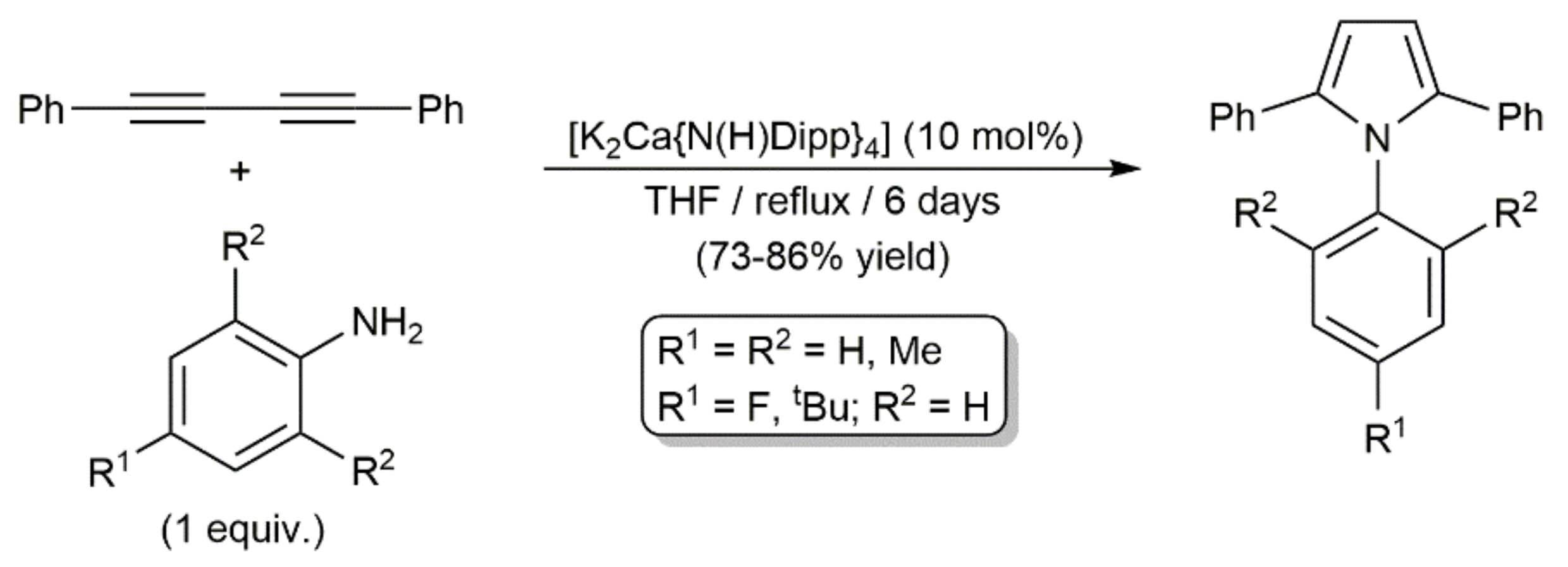
Complex [K2Ca{N(H)Dipp}4] also catalyzed the addition of N-unsubstituted anilines to PhC≡CC≡CPh. As shown in Scheme 43, the reactions conducted in refluxing THF for six days afforded the corresponding pyrrole derivatives in 73–86%, regardless of the substitution pattern of the aromatic ring [115]. Harsh conditions were needed for the intramolecular hydroamination of the second alkyne unit in the intermediate species (E/Z)-PhC≡CCH = CPh(NHAr) to proceed. Conversely, when these hydroamination processes were performed at room temperature, other reaction pathways took place, leading to, depending on the stoichiometry employed and the substituents in ortho-position of the anilines, different compounds, such as those depicted in Figure 10, whose formation was explained through radical mechanisms [115,116].
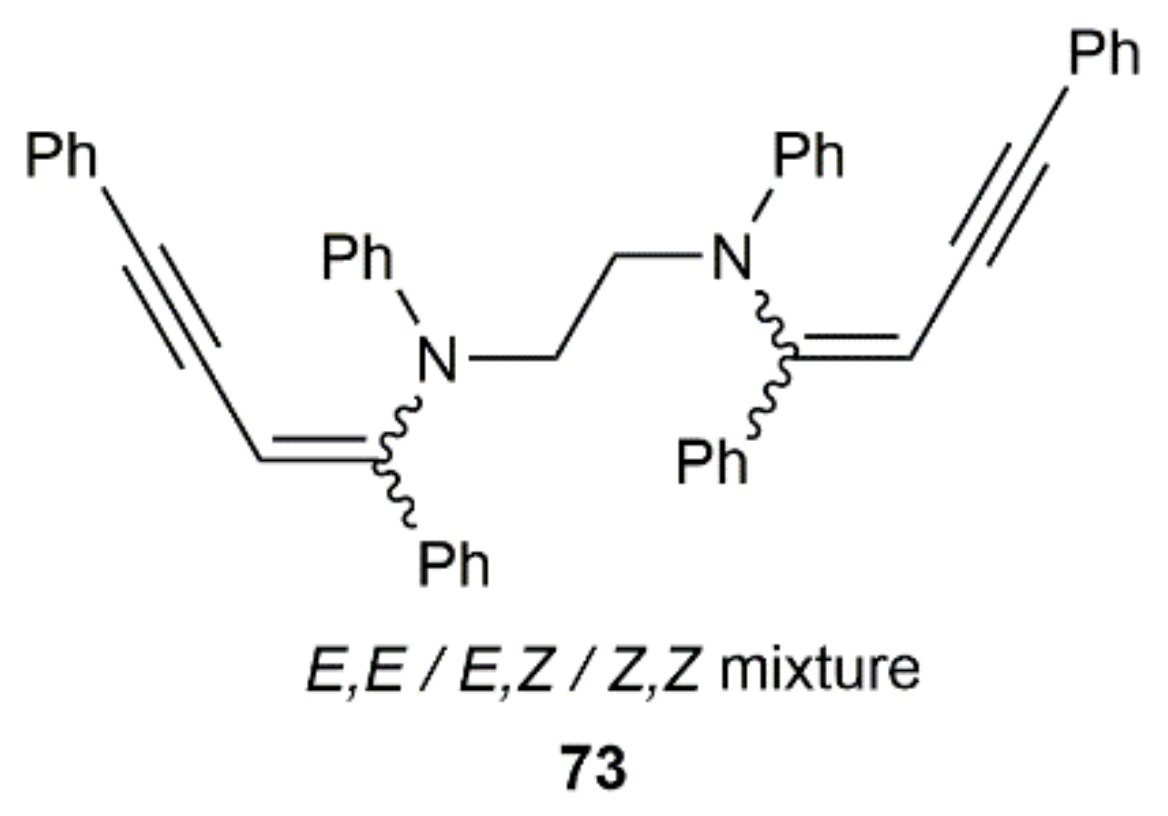
In an additional work, Westerhausen and colleagues attempted the generation of cyclic butadiene derivatives by double hydroamination of PhC≡CC≡CPh with a linear diamine, i.e., N,N′-diphenylethylenediamine. The expected cyclic product was not formed, the reaction yielding compound 73, resulting from the addition of the diamine to two molecules of the diyne (Figure 11) [117]. For this particular transformation, the resulting complex [K2Ca{N(H)Dipp}4] was completely inoperative, and a mixture of the potassium and calcium complexes [(THF)3K2{1,2-(PhN)2C2H4}] and [(THF)5Ca2{1,2-(PhN)2C2H4}2] in 2:1 ratio were, in this case, employed to promote the process.
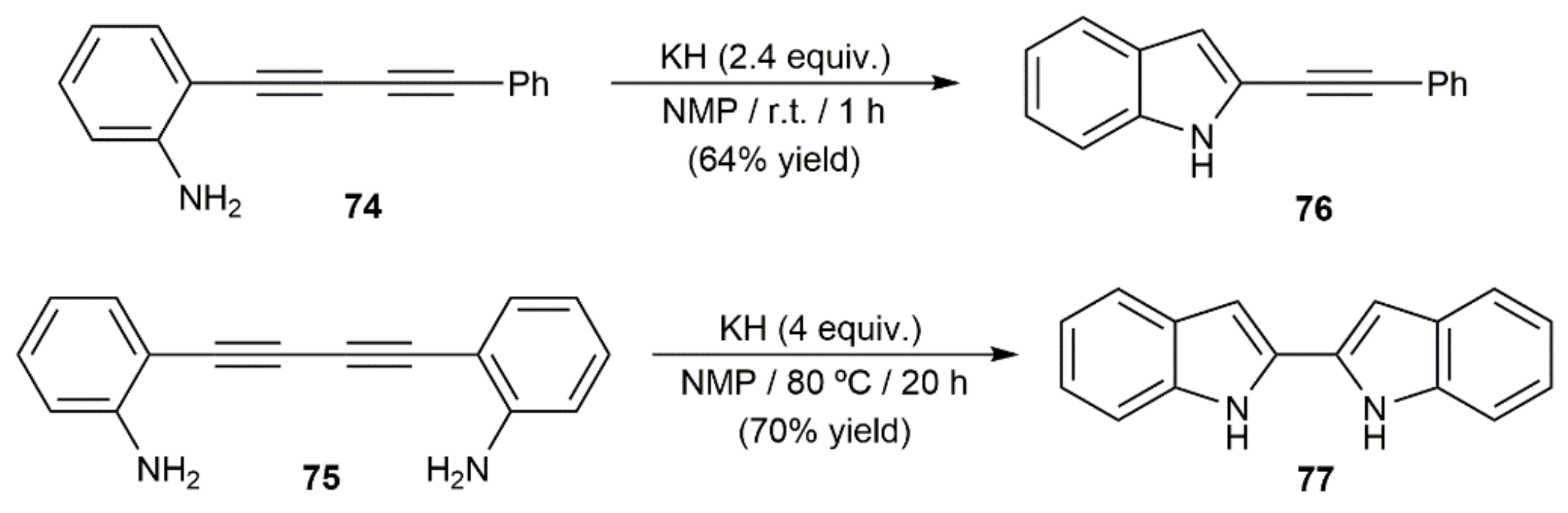
Intramolecular hydroamination reactions of 1,3-diynes functionalized with pendant amino groups have also been described. As exemplified in Scheme 44 with the 5-endo-dig-cyclization of the o-aniline-substituted diynes 74 and 75 into the indole derivatives 76 and 77, respectively, such reactions do not require metal catalysts to proceed, being conveniently promoted by a base [118,119]. However, we must note that conversion of 74 into 76 was successfully achieved under neutral conditions employing catalytic amounts of PdCl2 (5 mol%). Thus, after 12 h of heating at 80 °C in 1,2-dichloroethane, the 2-alkynylindole 76 could be isolated in 54% yield [120].
Shi and colleagues described the synthesis of a broad family of naphthol-indole derivatives 81 by coupling the related 1,3-diynes 78 with the sulfoxonium ylide 79, employing a catalytic system composed of the Rh(III) dimer [{RhCl(µ-Cl)Cp*}2] and the silver(I) salt AgSbF6 in combination with pivalic acid (PivOH) and zinc acetate (Scheme 45) [121]. Control experiments indicated that the process involves the initial AgSbF6-catalyzed cyclization of the diynes to generate the 2-alkynylindole intermediates 80. Subsequent reaction of ylide 79 with compounds 80 leads to the final naphthol-indole products 81 through a Rh-catalyzed Zn-assisted ortho C–H activation/annulation cascade.
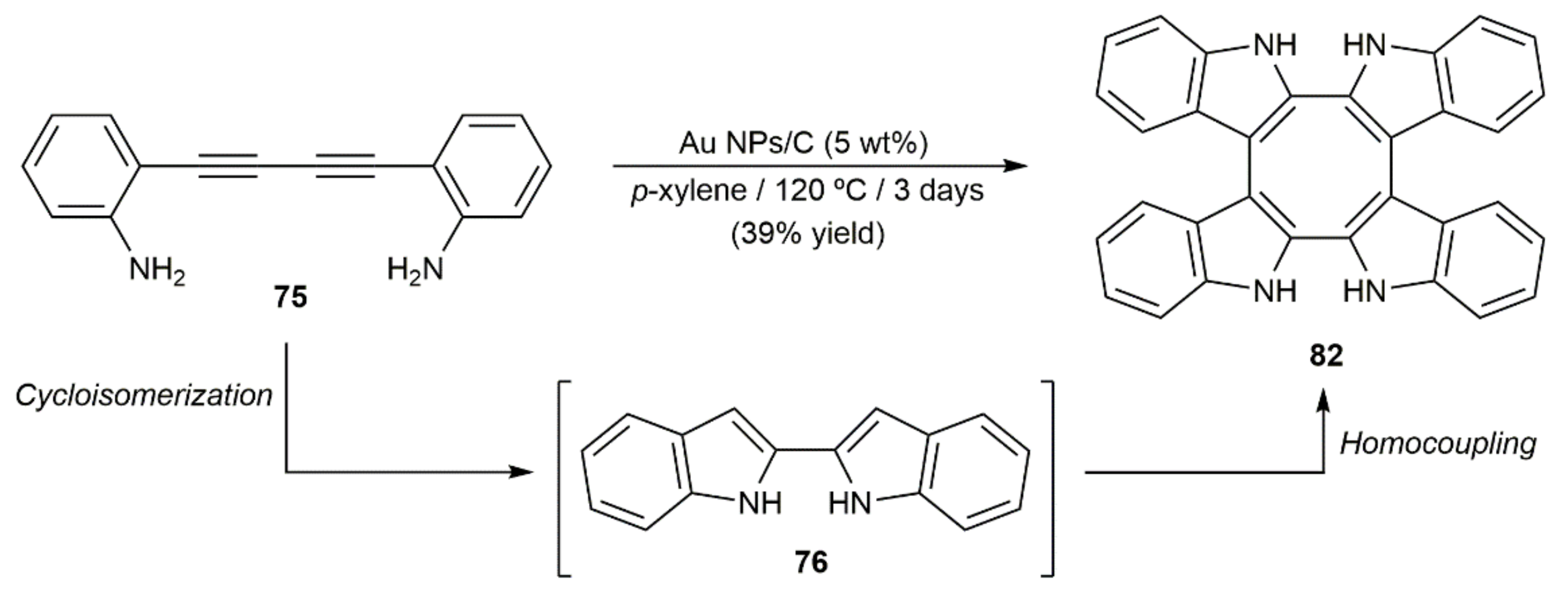
In the context of a broad study on the cycloisomerization of 2-alkynylanilines catalyzed by carbon-supported gold nanoparticles (NPs), the groups of Lopez-Sanchez and Helaja developed an effective cascade process to obtain 3,3′-biindoles by combining the cycloisomerization of the substrates with a subsequent indole homocoupling reaction [122]. As shown in Scheme 46, when the diyne 75 was subjected to the catalytic action of the supported Au NPs in p-xylene at 120 °C the 3,3,3′,3′-tetraindole 82 containing a cyclooctatetraene core was selectively formed, albeit in only 39% yield.
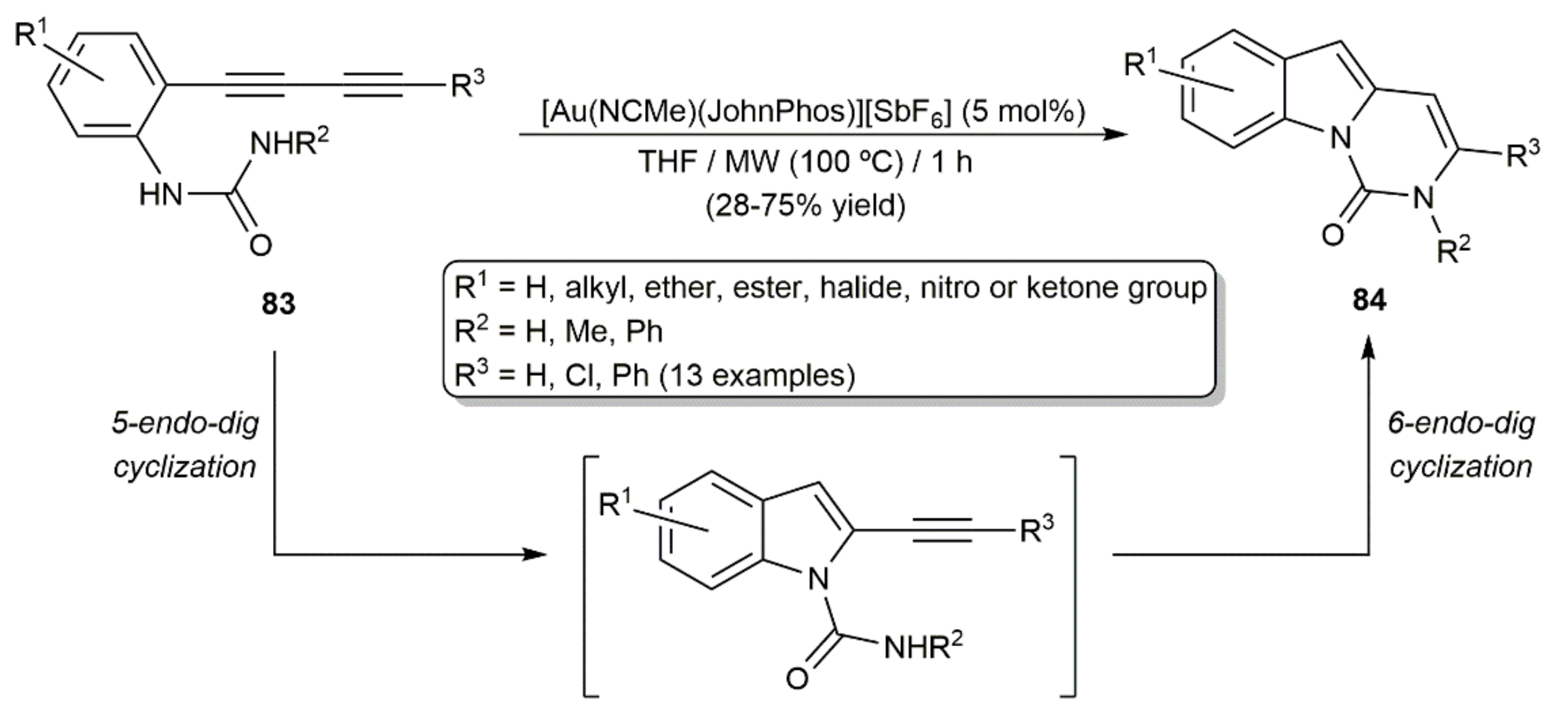
On the other hand, Banwell´s group elegantly combined two consecutive intramolecular hydroamidation processes with the o-(1,3-diynyl)-substituted N-aryl ureas 83 to produce the polycyclic pyrimido[1,6-a]indol-1(2H)-one derivatives 84 (Scheme 47) [123]. In these reactions, the initial 5-endo-dig cyclization of the substrates is followed by a 6-endo-dig one, both ring-closure processes being catalyzed by the cationic complex [Au(NCMe)(JohnPhos)][SbF6] under MW irradiation.
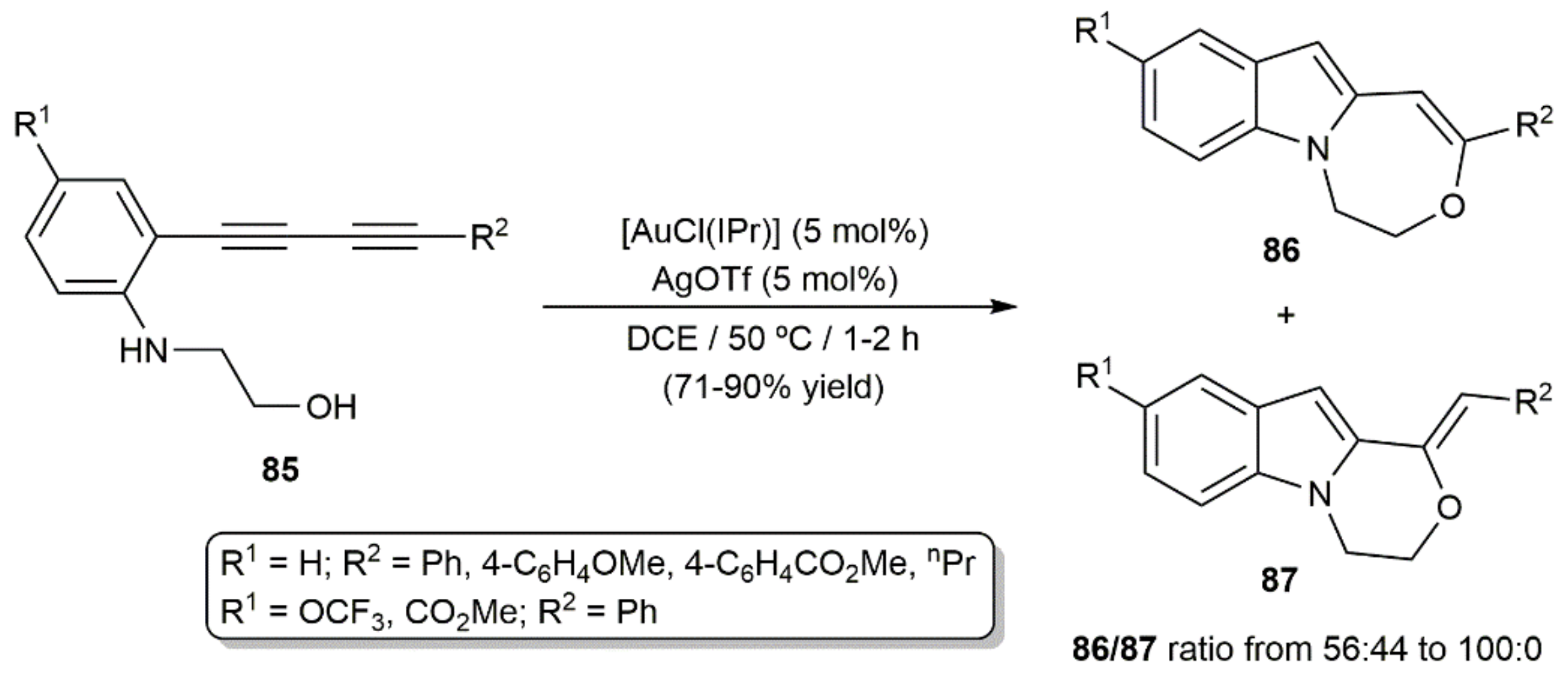
Uchiyama, Ohno, and colleagues developed a related cascade process with 1,3-diynes 85, in which the initial gold(I)-catalyzed 5-endo-dig indole formation reaction was followed by the hydroalkoxylation of the remaining C≡C bond (Scheme 48) [124]. Although in the last step a marked preference for the 7-endo-dig cyclization was observed, the oxepino[1,7-a]indole products 86 were not selectively formed and minor amounts of the isomeric species 87, resulting from a 6-exo-dig cyclization, were also generated in most of the cases.
8. Hydrophosphination and Hydrophosphorylation Processes
An early work by Märkl and Potthast in 1967 showed that 2,5-disubstituted phospholes can be easily obtained by double hydrophosphination of symmetrically substituted 1,3-diynes with phenylphosphine in the presence of catalytic amounts of phenyllithium (Scheme 49) [125]. In the process, in situ generated lithium phosphide PhPHLi adds to the diyne generating the lithiated phosphole intermediates M, which rapidly react with a second equivalent of the phosphine. In this way, the final reaction products 88 are formed and the nucleophile PhPHLi regenerated.
Applying the same protocol, but using LinBu instead of LiPh, the synthesis of 2,5-di(1-naphthyl)phosphole 89 [126], the chiral derivatives 90 [127] and 91 [128], and the ferrocenyl-substituted phospholes 92 [129,130,131] was later described by reacting the respective 1,3-diyne with a primary aryl phosphine (see Figure 12). Noncatalytic transformations allowing the access to phosphole derivatives from 1,3-diynes have additionally been described employing stoichiometric amounts of lithium-phosphide reagents [132,133,134]. Moreover, of note is the fact that, by using phenylarsane PhAsH2 instead a primary aryl phosphine, several arsoles were synthesized by Hauptmann and colleagues by LinBu-catalyzed addition to 1,3-diynes [135,136].
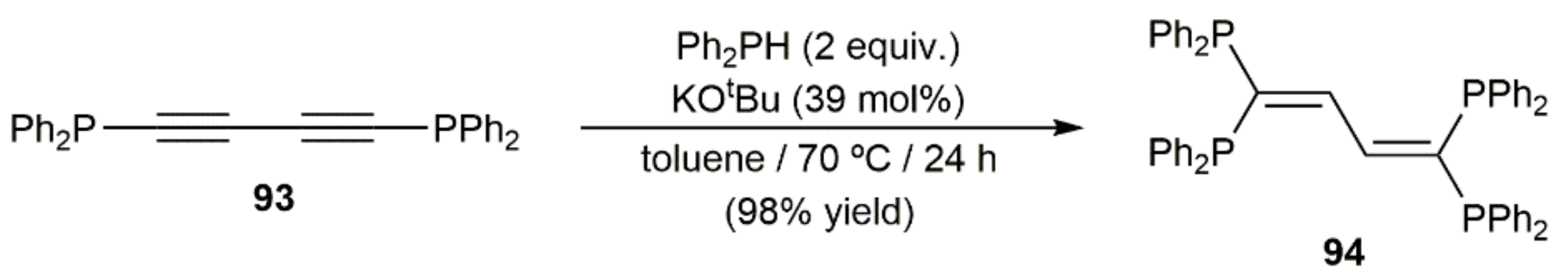
Base-catalyzed additions of secondary phosphines to 1,3-diynes can also be found in the literature. For example, Schmidbauer and colleagues reported in 1995 the high-yield preparation of 1,1,4,4-tetrakis(diphenylphosphino)-1,3-butadiene 94 by double hydrophosphination of 1,4-bis(diphenylphosphino)-1,3-butadiyne 93 with Ph2PH, using KOtBu as a promoter (Scheme 50) [137].
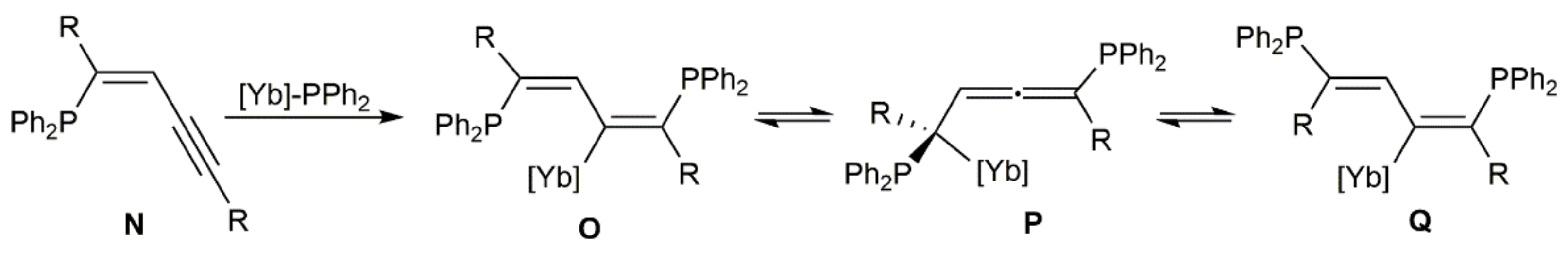
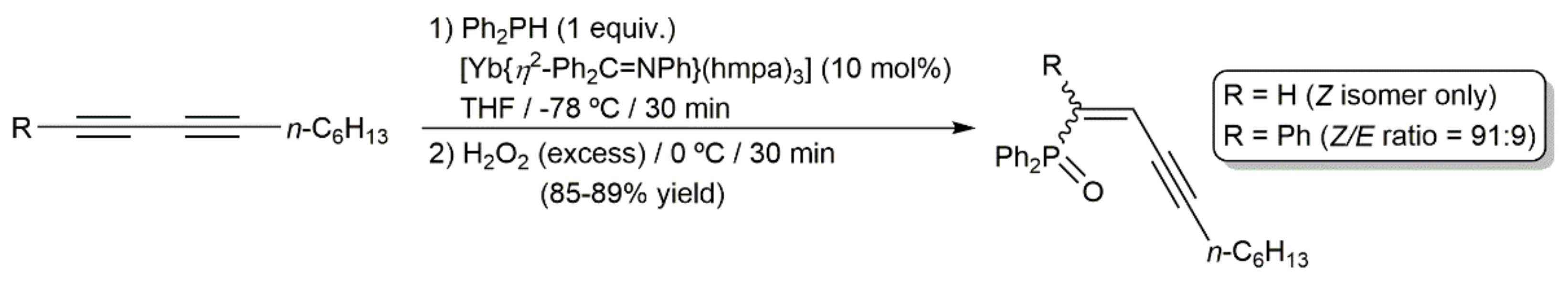
On the other hand, taking advantage of the ability of organolanthanides to promote intermolecular hydrophosphination reactions of multiple C–C bonds [138], Takaki and colleagues described the stereoselective synthesis of the (Z,Z)-bis(diphenylphosphinyl)diene 96 under mild conditions by double hydrophosphination of 7,9-hexadecadiyne with Ph2PH, catalyzed by the ytterbium–imine complex [Yb{η2-Ph2C = NPh}(hmpa)3] (hmpa = hexamethylphosphoramide), and subsequent oxidation with H2O2 of the initially formed bis(diphenylphosphino)diene 95 (Scheme 51) [139,140]. Starting from 2,2,7,7-tetramethyl-3,5-octadiyne and applying the same protocol, a mixture of the corresponding diene 97 and the allene 98 was formed, with the latter being largely major.
The hydrophosphination reactions depicted in Scheme 51 are believed to proceed through the sequential anti addition of in situ generated ytterbium-phosphide species [Yb]-PPh2 to the C≡C bonds of the diyne substrate [141]. In this way, enynes N are initially generated and rapidly transformed into the metallated dienes O, which evolve into compounds 96 and 97 by protodemetallation and oxidation (Scheme 52). Formation of allene 98 most probably results from the metallated species P, generated by isomerization of the corresponding O one.
These initial studies were subsequently extended to other 1,3-diynes using [Yb{η2-Ph2C = NPh}(hmpa)3] or [Yb{N(SiMe3)2}3(hmpa)2] as the catalyst [141]. As a general trend, the bis(diphenylphosphinyl)diene products were mostly generated as the corresponding (Z,Z)-isomers when disubstituted diynes were employed as substrates, while (E,Z)-butadienes were the major products starting from terminal 1,3-diynes HC≡CC≡CR (R = aryl or alkyl group). The different stereochemistry found for the latter seems to indicate that conversion of the dienylytterbium intermediates O into the isomeric Q ones via the metallated allenes P rapidly occurs in solution for these particular substrates (see Scheme 52). Moreover, of note is the fact that, as illustrated in Scheme 53, single hydrophosphination of unsymmetrical diynes was successfully achieved, with high regio- and stereocontrol, by performing the reactions at −78 °C with an equimolar amount of diphenylphosphine.
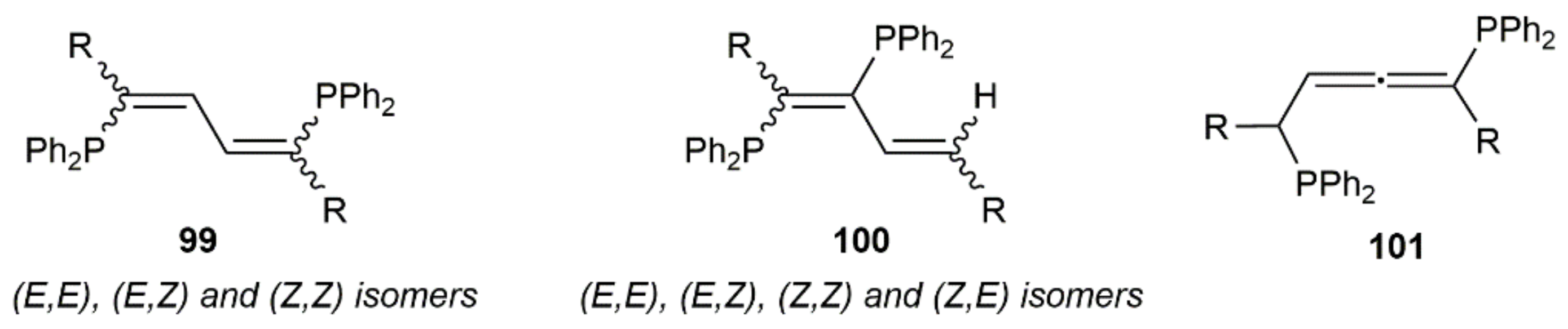
Double hydrophosphination reactions of 1,3-diynes RC≡CC≡CR (R = Me, tBu, SiMe3, Ph, 2,4,6-C6H2Me3) with diphenylphosphine catalyzed by the phosphido–calcium complex [Ca(THF)4(PPh2)2] were additionally described by Westerhausen and colleagues [142,143]. A low selectivity was, in general, observed, the reactions leading to complex mixtures containing the corresponding 1,4-bis(diphenylphosphino)-1,3-dienes 99 (major products), along with the 1,2-bis(diphenylphosphino)-1,3-dienes 100 (the four possible stereoisomers) and, in some cases, the allenic derivative 101 (see Figure 13).
As shown in Scheme 54, complete regio- and stereoselective transformations were reached when [Ca(THF)4(PPh2)2] was employed to promote the hydrophosphorylation of RC≡CC≡CR (R = Ph, tBu) with diphenylphosphine oxide, reactions from which the diene 102 and the alkyne 103 were isolated in high yields [144]. The structures of compounds 102 and 103 differ markedly from those generated in the hydrophosphination processes (see Figure 13), and the differences observed were suspected to be related with the higher stability of the intermediate species [Ca]-P( = O)Ph2 vs. the [Ca]-PPh2 ones involved in these P–H bond addition reactions. Thus, intermediates of type [Ca]-PPh2 can undergo dissociation and reorganization processes in solution, which would explain the low regio- and stereocontrol in the hydrophosphination processes. Moreover, the use of a phosphine oxide offers the possibility to maintain close contact between the calcium center and the corresponding phosphorylated intermediates, via Ca–O Lewis acid–base interactions, a key factor for the effective regiocontrol in the reactions depicted in Scheme 54.
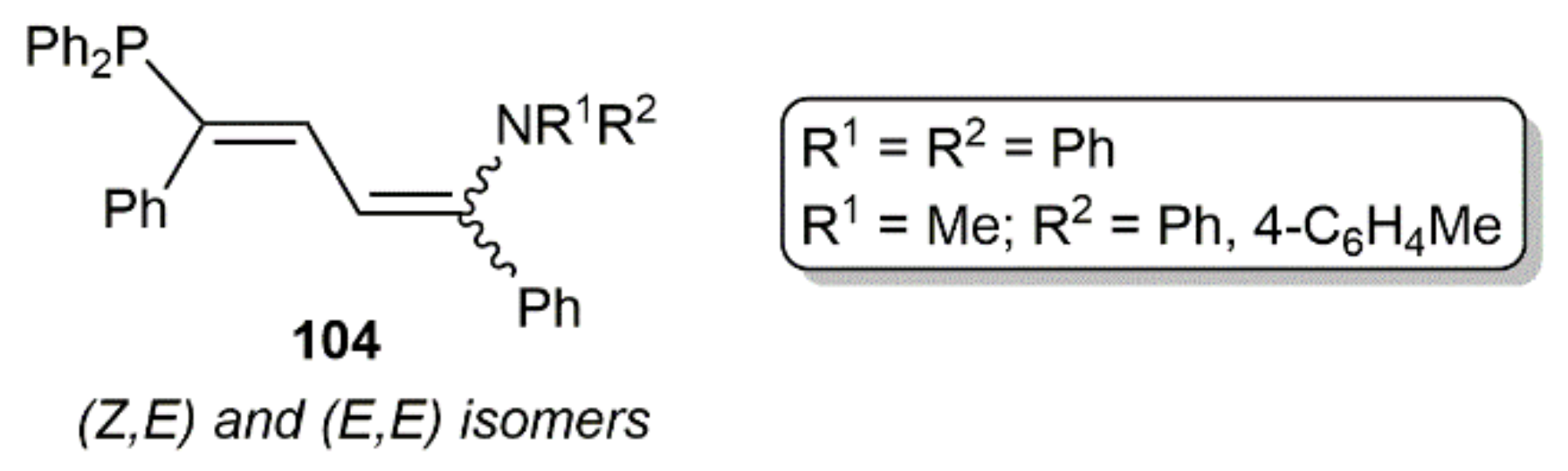
Within this section, we must finally mention the synthesis of the 1,3-butadiene derivatives 104 (see Figure 14) by Westerhausen´s group through the sequential hydroamination and hydrophosphination of 1,4-diphenyl-1,3-butadiyne catalyzed by [K2Ca{N(H)Dipp}4] [145]. Whereas the initial hydroamination step is regio- (amino group in 1-position) but not stereoselective (formation of E and Z isomers), the subsequent hydrophosphination one proceeded with complete regio- and sterocontrol.
9. Hydration Processes
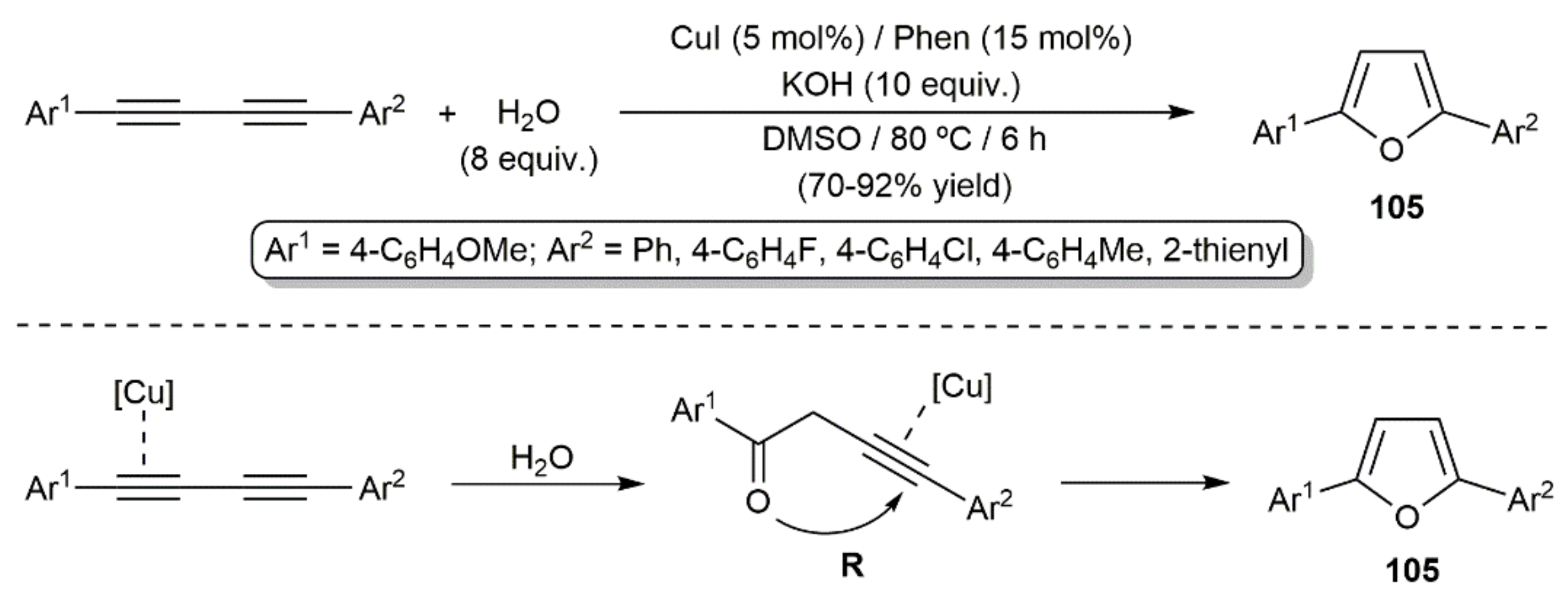
As in the case of simple alkynes [146], the Bronsted-acid-promoted hydration of 1,3-diynes leading to carbonyl compounds is well documented, particularly under photochemical conditions [147,148,149,150,151,152,153]. Although similar reaction products would also be expected when using metal-based catalysts [146], the works carried out by different authors have shown the preferential formation of furan derivatives as the result of the formal double addition of the water O–H bonds to the diyne. This was evidenced for the first time by Jiang and colleagues in 2012, with the synthesis of the 2,5-diaryl-substituted furans 105 (Scheme 55) [154]. The hydration process is catalyzed by a CuI, in conjunction with 1,10-phenanthroline and KOH, in dimethyl sulfoxide (DMSO) at 70 °C. A reaction pathway involving the initial formation of the ketone intermediate R by single hydration of the diyne, followed by a Cu(I)-catalyzed cyclization, was proposed by the authors (see Scheme 55). Moreover, of note is the fact that related symmetrically substituted 2,5-diaryl furans were accessed, under the same reaction conditions, starting from haloalkynes ArC≡CX, which, by means of the Cu(I) catalyst, are in situ converted into the respective 1,3-diynes ArC≡CC≡CAr by homocoupling.
A slight modification of this catalytic system was later shown to be capable of promoting the one-pot generation of 2,5-diaryl-substituted furans from aryl-substituted propiolic acids through the initial copper-catalyzed decarboxylative coupling of the substrates to generate the corresponding diynes and subsequent reaction with water (Scheme 56) [155]. Conversion of preformed 1,4-diaryl-1,3-diynes into the corresponding 2,5-disubstituted furans catalyzed by [Pd(PPh3)4] was also described by Hua and colleagues, the reactions requiring, again, the assistance of a base (KOH) [156]. It should be mentioned at this point that some studies showing the ability of KOH to promote, by itself, the hydration of 1,3-diynes into furans can be found in the literature [83,157,158].
On the other hand, hydration of 1,3-diynes under base-free conditions has been reported to occur in the presence of gold(I) complexes. This was evidenced for the first time by Skrydstrup and colleagues in 2010 with the synthesis of the 2,5-diamidofurans 106 from the corresponding 1,3-diynamides 62 upon the catalytic action of the Au(I)–phosphine complex [Au(NTf2)(PPh3)] (Scheme 57) [92].
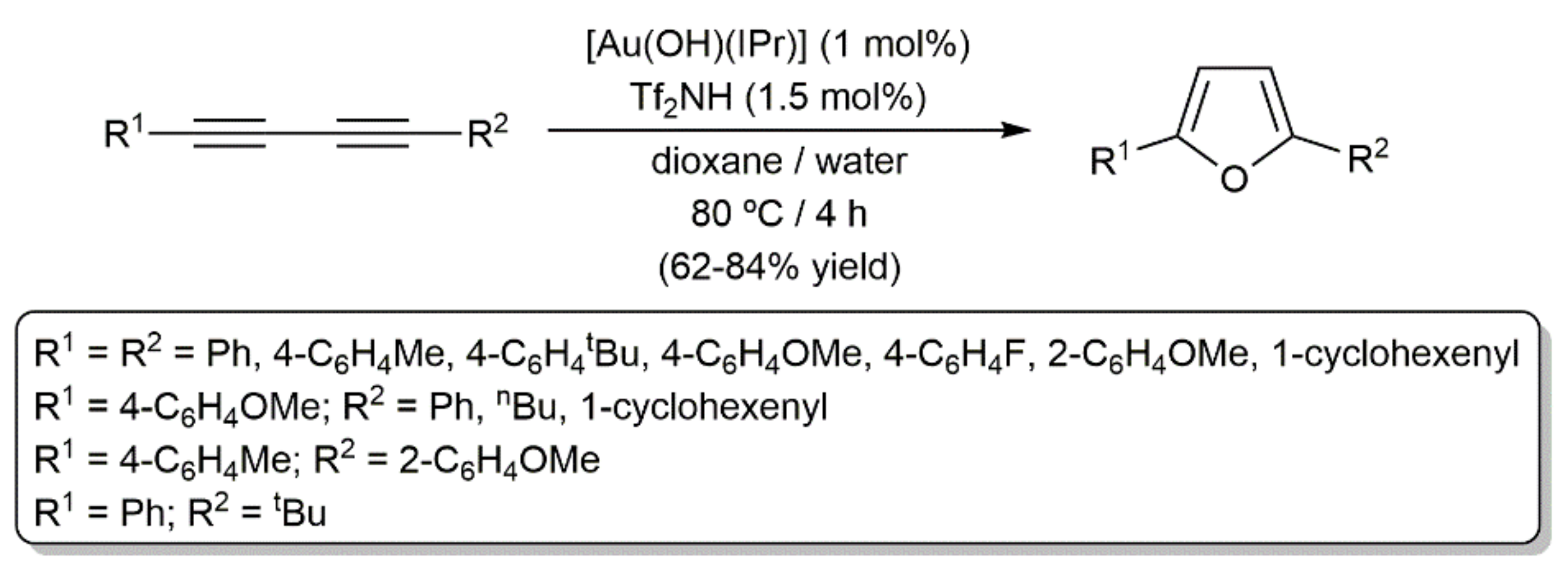
In a broader substrate-scope study, Nolan and colleagues successfully employed the NHC-gold(I) complex [Au(NTf2)(IPr)], generated in situ from the hydroxo-derivative [Au(OH)(IPr)] and trifluoromethanesulfonimide (Tf2NH), to promote the process [93]. As shown in Scheme 58, the reactions were conveniently conducted in a dioxane/water mixture and both symmetric and unsymmetric diynes, substituted with aromatic, aliphatic, or alkenyl groups, were tolerated. Moreover, of note is the fact that DFT calculations confirmed that a reaction pathway analogous to that commented on above for copper (see Scheme 56) also operates in the present case.
The utility of these gold(I)-catalyzed furan-ring formation reactions was fully demonstrated by Verniest and colleagues with the synthesis of the furan-bridged peptidic macrocycles 108 by hydration of diynes 107, reactions in which [Au(NTf2)(SPhos)], generated in situ from [AuCl(SPhos)] and AgNTf2, was used as the catalyst (Scheme 59) [101]. [Au(NTf2)(SPhos)] was additionally applied by Furstner´s group, in the context of the total synthesis of Ivorenolide B, to synthesize the 15-membered macrocyclic dilactone 109 (see Scheme 59) from the corresponding 1,3-diyne (20 in Scheme 11) [31].
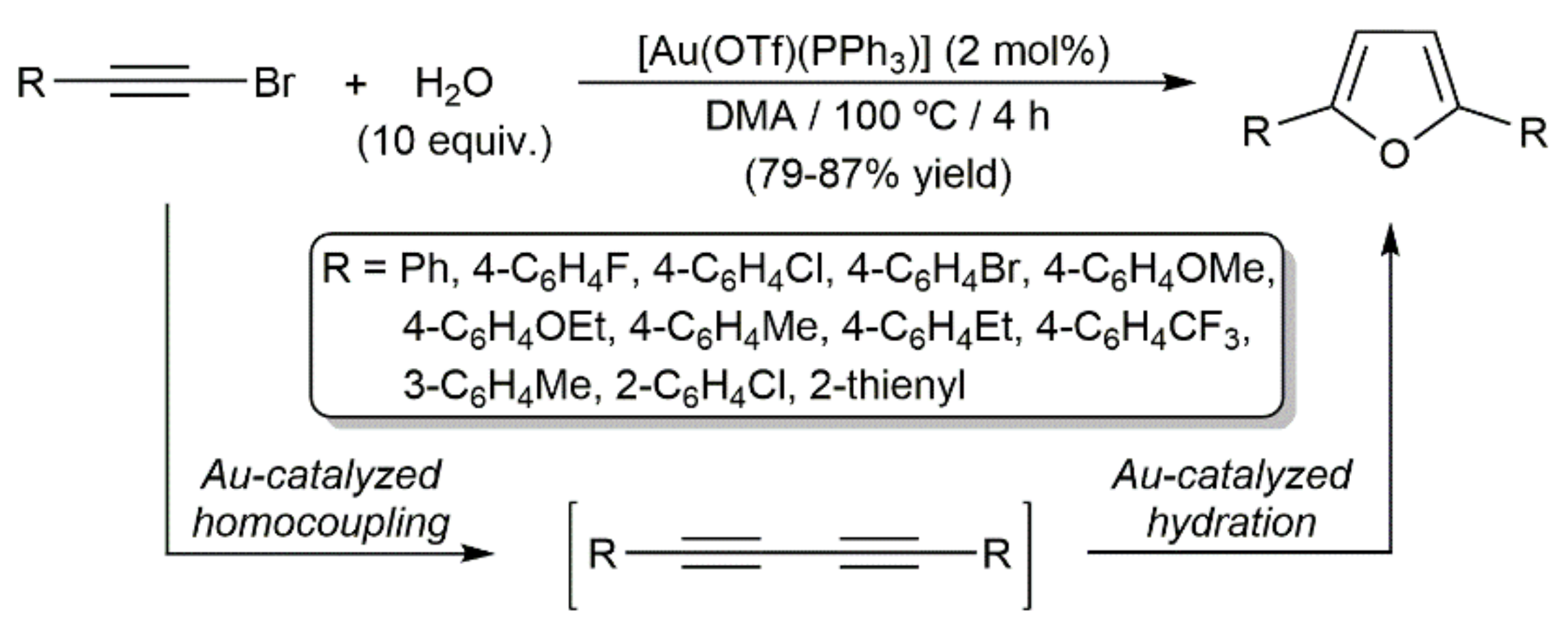
In addition, Guo demonstrated the possibility of preparing 2,5-substituted furans directly from bromoalkynes, employing the triflate-gold(I)–phosphine complex [Ag(OTf)(PPh3)] as a catalyst in dimethylacetamide (DMA) at 100 °C (Scheme 60) [159]. The domino process was operative with aromatic and heteroaromatic bromoalkynes, but completely ineffective with aliphatic ones. As demonstrated for PhC≡CI, which led to the corresponding furan in 87% yield under identical experimental conditions, this gold-catalyzed transformation can be extended to iodoalkynes.
10. Hydroalkoxylation Processes
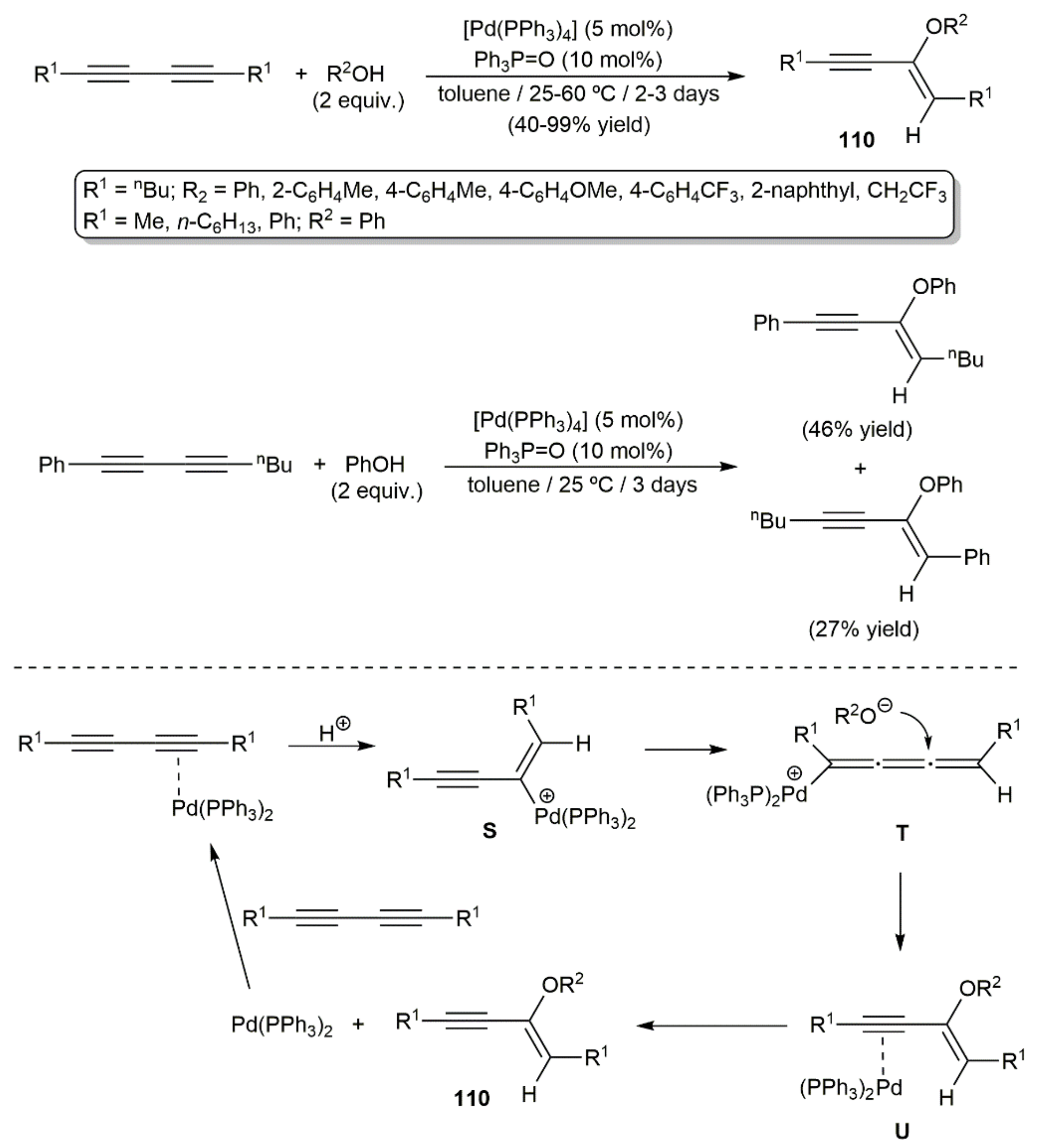
Only one example describing the intermolecular addition of alcohols to 1,3-diynes promoted by a transition metal can be currently found in the literature. It was reported by Yamamoto and colleagues in 2002, who found that the palladium(0) complex [Pd(PPh3)4] was able to catalyze the regio- and stereoselective trans addition of acidic alcohols (phenols or CF3CH2OH) to different symmetrically substituted diynes to afford the alkoxylated enynes 110 (Scheme 61) [160]. Only the monoaddition products were formed even over longer reaction periods or at higher temperatures. In the study, the addition of phenol to the nonsymmetrical substituted substrate octa-1,3-dinyl-1-ylbenzene was also explored, the reaction leading to mixtures of regioisomers, since the catalyst was not capable of discriminating between the two C≡C bonds (Scheme 61). A reaction pathway involving the initial activation of one of the C≡C bonds by co-ordination to the Pd(0) center, followed by protonation to generate the metallated intermediate S, was proposed by the authors. Rearrangement of S into the σ-cumulenyl palladium complex T then allows the nucleophilic attack of the alkoxyde. This attack occurs on the opposite side of the bulky PPh3 ligands, thus explaining the trans selectivity of the process. Intermediate U is formed in this way, from which the product is liberated after de-coordination of the catalytically active Pd(0) fragment. In favor of this mechanistic proposal is the fact that no reaction was observed when simple alkynes were employed as substrates. Moreover, of note is the fact that the exquisite trans selectivity observed in this Pd(0)-catalyzed hydroalkoxylation process contrasts with that of related metal-free addition reactions, where mixtures of Z/E isomers are usually obtained [161,162].
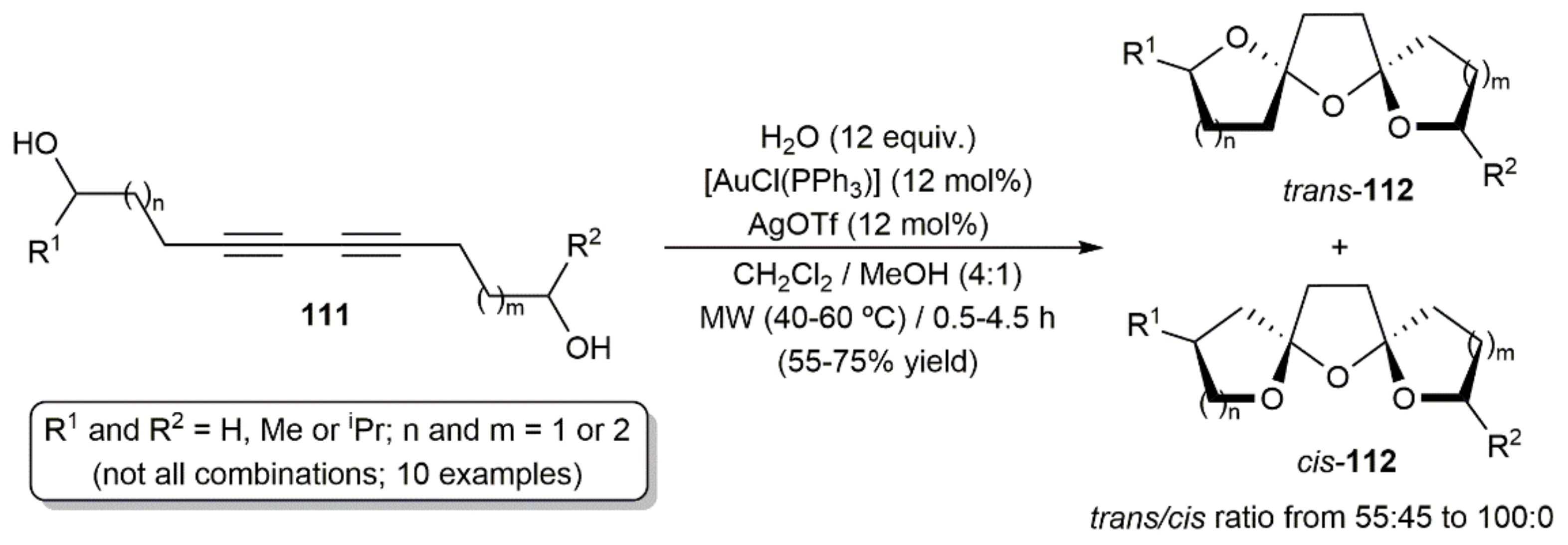
Regarding intramolecular processes, Lee and colleagues described the synthesis of a variety of bis-spiroketal derivatives 112 from the diynediols 111 through a gold(I)-catalyzed tandem addition of the tethered hydroxyl groups and one equivalent of water (Scheme 62) [163]. The hydroalkoxylation/hydration reactions, conducted under MW irradiation with the in situ generated complex [Au(OTf)(PPh3)], led, in most of the cases, to the bis-spiroketals 112 as separable mixtures of the corresponding trans (major) and cis (minor) isomers, which, once isolated, were found to interconvert in solution to reach an equilibrium. Preference for the C2 symmetrical trans isomers vs. the unsymmetrical cis ones was reasoned in terms of the most favorable anomeric effect in the former.
The reaction pathway leading to compounds 112 was elucidated by DFT calculations using 5,7-diyne-1,12-diol as a model [164]. As shown in Scheme 63, the pyranyl enol ether intermediate V is initially generated by a gold-mediated 6-exo-dig cyclization, evolving into the bis-enol ether W by a second 6-exo-dig addition. A subsequent dihydration of W leads to X, which converts into the final bis-spiroketal products by dehydrative ring closure. In complete agreement with the trans/cis ratio experimentally observed (69:31), the computations indicated a slightly lower energy for the trans isomer (1.08 Kcal/mol).
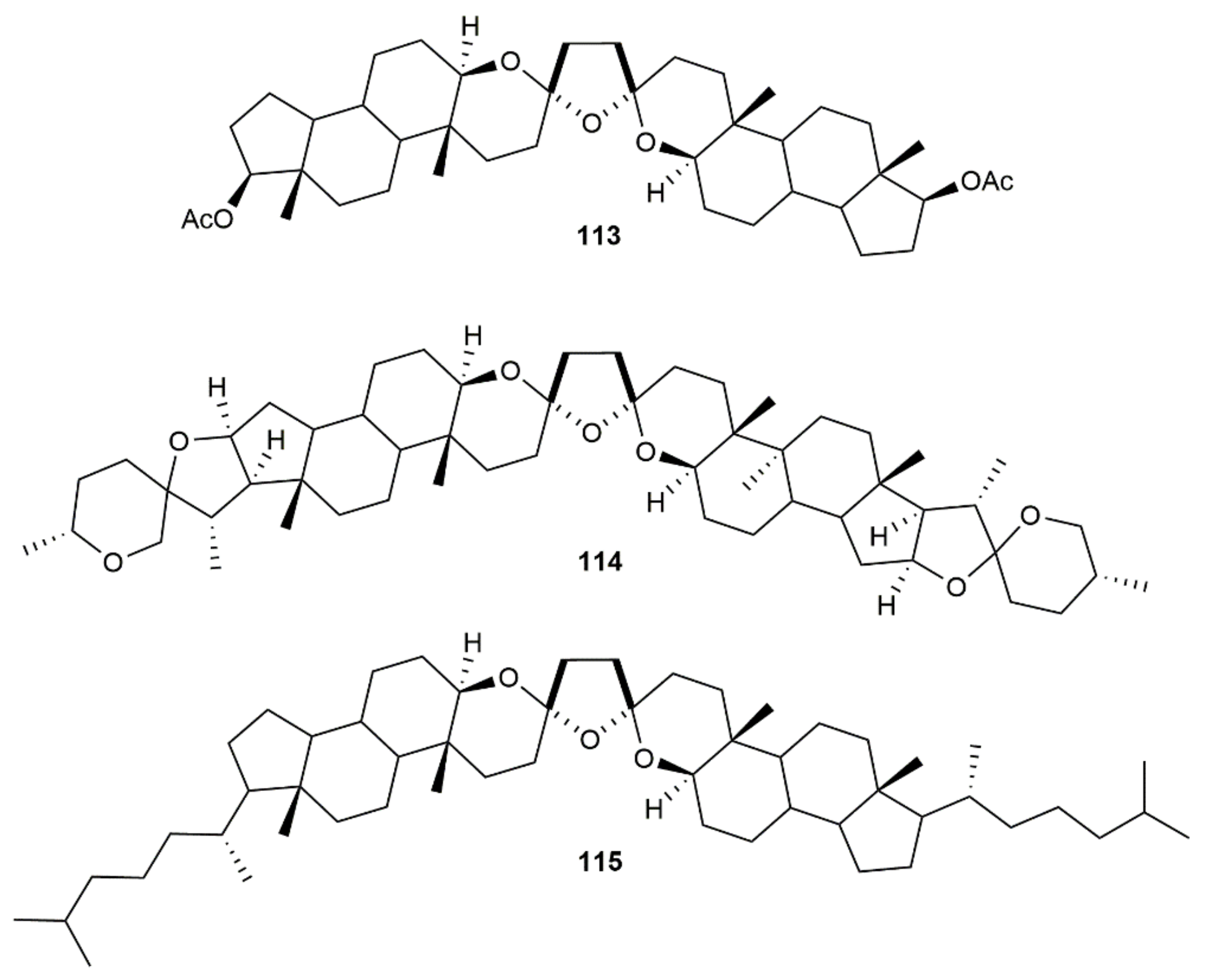
Employing the cationic gold(I) complex [Au(NCMe)(JohnPhos)][SbF6] as a catalyst, the related bis-spiroketals 113–115 were synthesized by Iglesias-Arteaga and colleagues through the hydroalkoxylation/hydration of the respective steroid-derived diynediols (Figure 15) [165]. The double spiroacetalization process proceeded in high yields (≥68%) and produced almost exclusively the C2 symmetrical trans distereoisomers (only in the case of 115 was a small amount of the unsymmetrical cis isomer formed). DFT calculations indicated a higher stability of the trans vs. cis diastereoisomers for these particular compounds (ca. 4 Kcal/mol), with the fusion of the side tetrahydropyrane rings to rigid scaffolds suppressing the conformational equilibrium between both isomers.
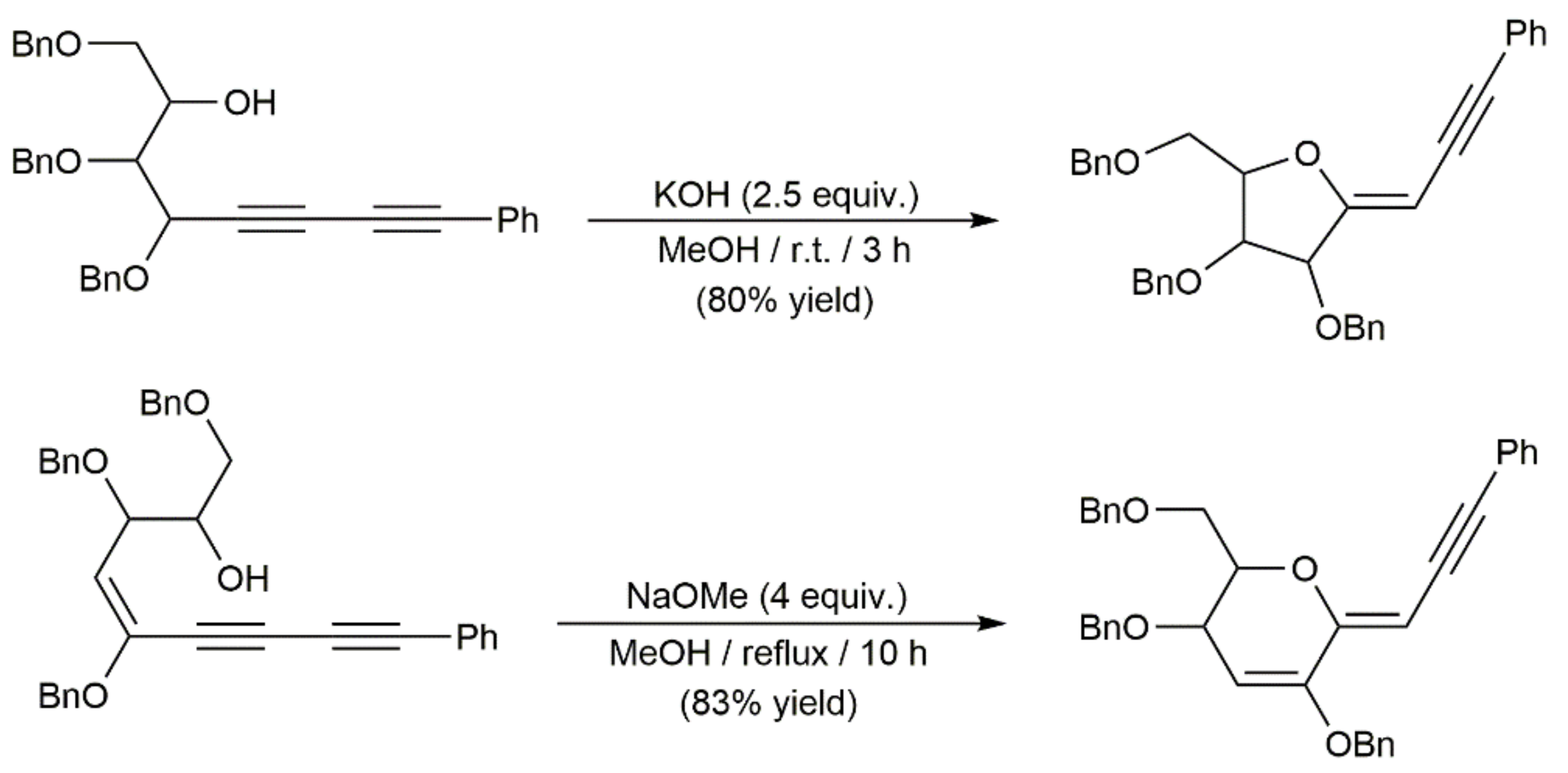
In addition to the gold-catalyzed reactions just commented on, several works describing the base-promoted cyclization of 1,3-diynic substrates bearing tethered hydroxyl groups can be found in the literature [166,167,168,169,170,171,172]. A couple of illustrative examples are given in Scheme 64 [172].
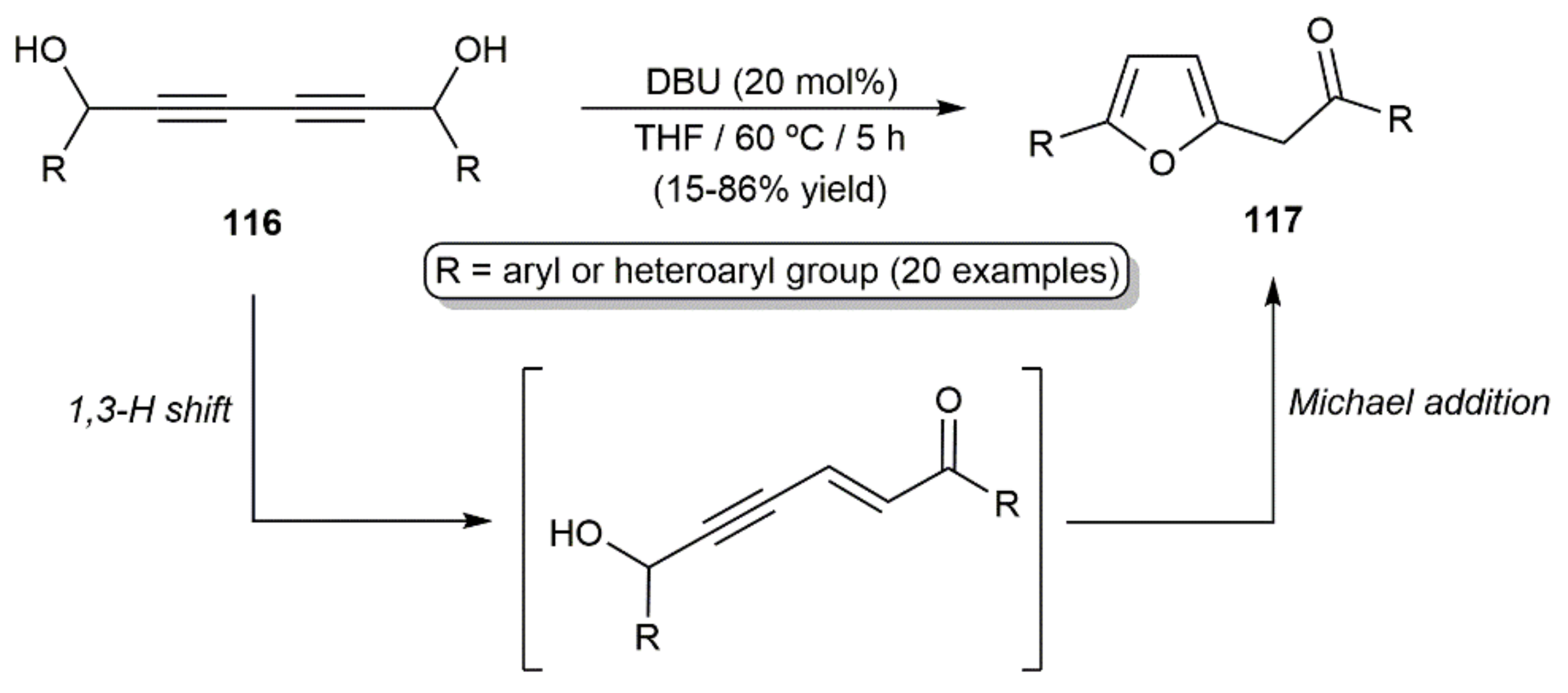
These cycloisomerization processes have been studied with substrates in which one of the diynic C≡C bonds and the hydroxyl group are connected through three to five carbon chain linkers, thus giving access to five- to seven-membered ring cyclic enol-ethers. A particular case is that of the diynyl-1,6-diols 116, which, in the presence of the DBU base (1,8-diazabicycloundec-7-ene), evolved into furans 117 through a cascade 1,3-H shift/intramolecular Michael addition reaction (Scheme 65) [173].
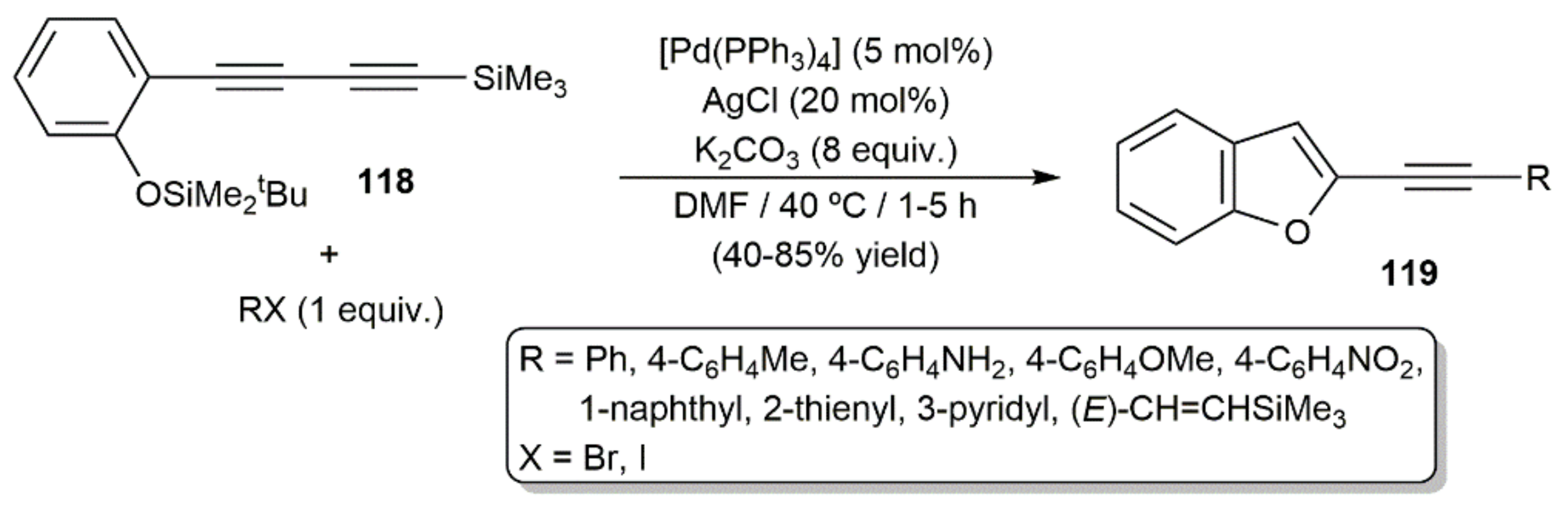
On the other hand, Fiandanese and colleagues developed a protocol for the expeditious synthesis of 2-alkynylbenzofurans 119 by reacting 2(4-trimethylsilyl)-1,3-butadiyn-1-yl)-tert-butyldimethylsilylphenol 118 with organic halides through a tandem Sonogashira coupling/cyclization reaction (Scheme 66) [119]. Given that, in the presence of disilylating agents, the cyclization of compound 118 into the parent 2-ethynyl-1-benzofuran was previously found to occur without the assistance of a metal [174], participation of the palladium catalyst in the cyclization step can probably be discarded.
11. Hydro-Oxycarbonylation Processes
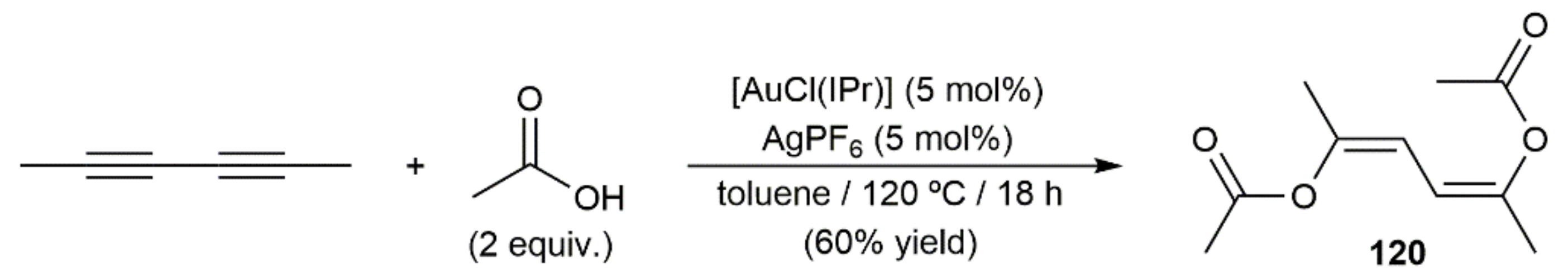
Addition of carboxylic acids to alkynes catalyzed by transition metals represents one of the most efficient ways to prepare synthetically useful enol esters [175,176,177,178]. Among the different catalytic systems reported to date, gold complexes have emerged in recent years as the most effective, as they can be successfully applied in the hydro-oxycarbonylation, not only of terminal alkynes, but also of the more challenging internal ones [179]. Indeed, the only example involving a 1,3-diyne as substrate that can be found in the literature made use of the in situ generated cationic NHC-gold(I) catalyst [Au(IPr)][SbF6] [180]. As shown in Scheme 67, it was able to promote the double addition of acetic acid to 2,4-hexadiyne to generate in a regio- and stereospecific manner the diene-diester 120. The Z configuration found for the C=C bonds of 120 is in complete accord with the marked preference of gold complexes to promote anti-type addition processes [179].
12. Hydrothiolation, Hydroselenation, and Hydrotelluration Processes
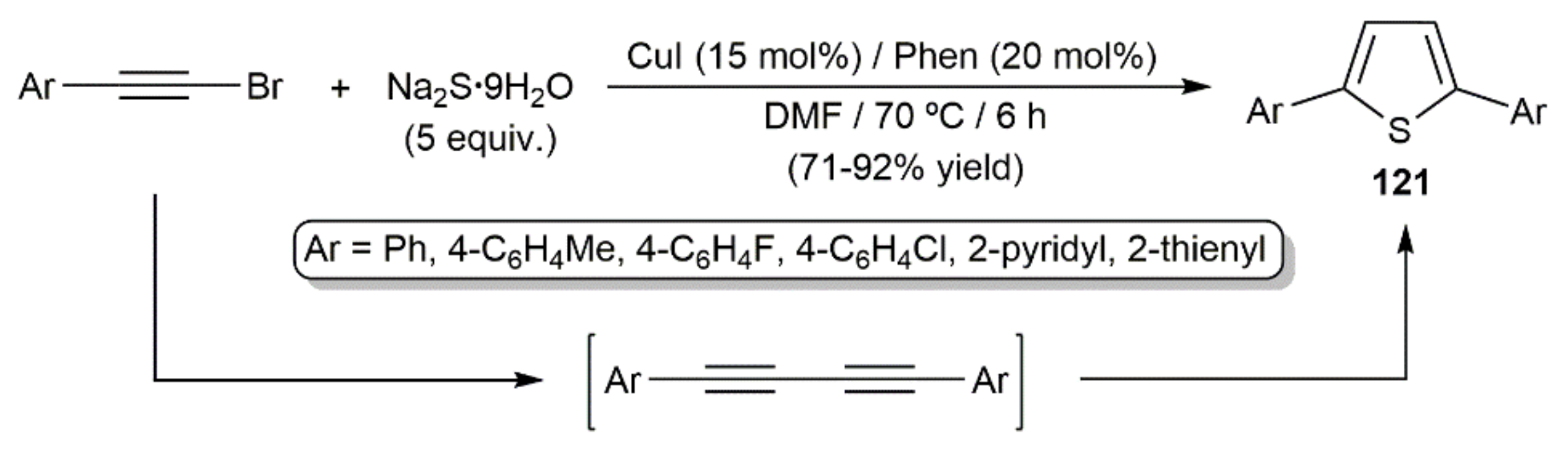
In 2012, Jiang and colleagues described the synthesis of different 2,5-diaryl-substituted thiophenes 121 by reacting aromatic bromoalkynes with sodium sufide hydrate, i.e., Na2S·9H2O, in the presence of CuI and the 1,10-phenanthroline ligand (Scheme 68) [154]. Compounds 121 are the result of the formal double hydrothiolation of the corresponding 1,3-diynes, generated in situ by copper-catalyzed homocoupling of the starting bromoalkynes.
Related thiophene-ring formation reactions using 1,3-diynes as starting materials have been described in the absence of any metal catalyst, the cyclization process taking place properly in the presence of a simple base (KOH, KOtBu or NH4OAc) and employing not only Na2S·9H2O, but also NaHS or S8 as the sulfur source [83,87,101,155,181,182,183,184,185,186]. Similarly, metal-free protocols to access selenophenes [187,188] and tellurophenes [189,190,191] from 1,3-diynes are known.
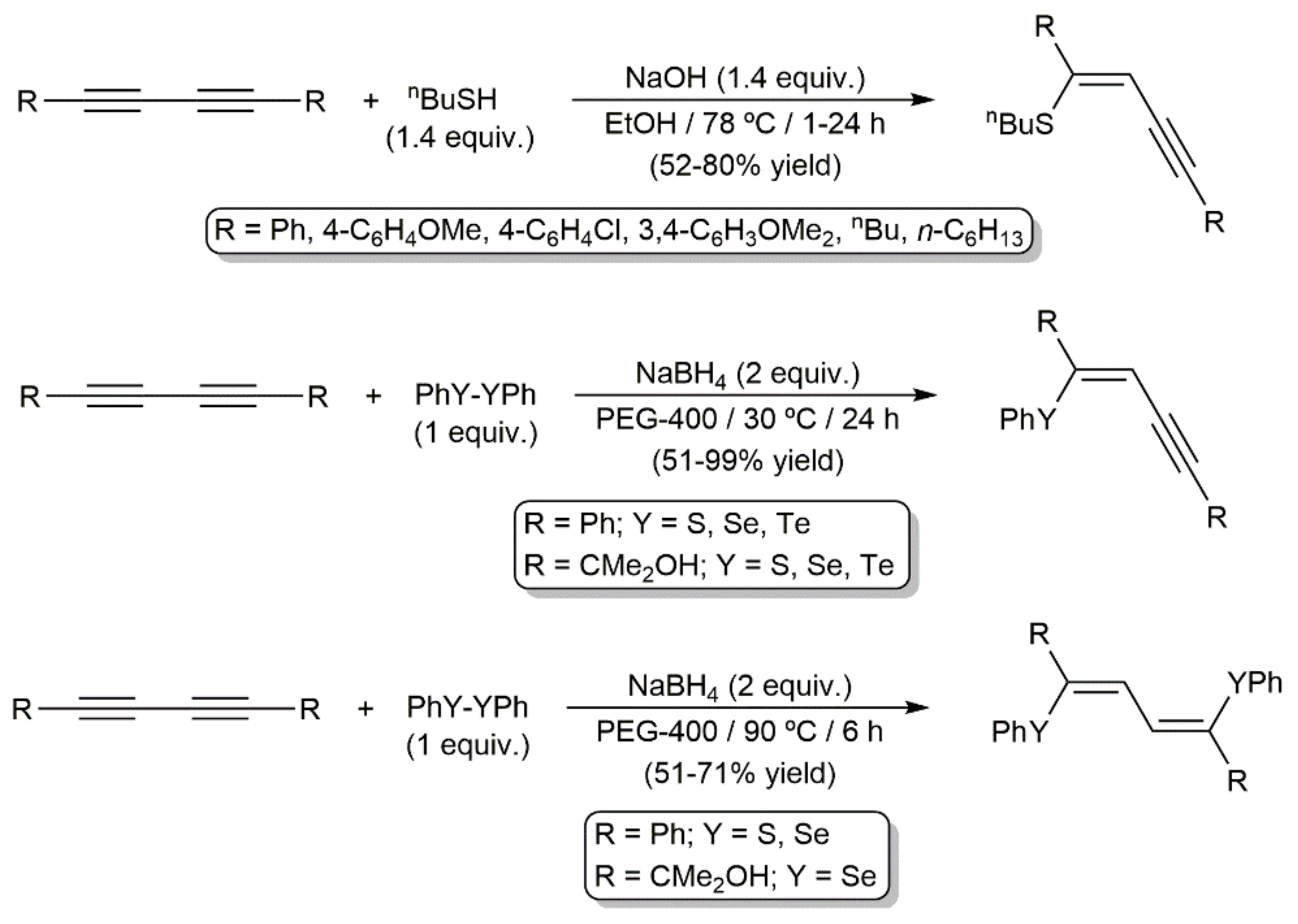
On the other hand, base-promoted mono- and dihydrochalcogenation reactions of 1,3-diynes with organic thiols RSH [192,193,194] and dichalcogenides (RSSR, RSeSeR, or RTeTeR) [195,196,197,198,199,200,201] to afford functionalized enynes or butadienes, respectively, can also be found in the literature. In general, the reaction products were obtained with high or complete Z-selectivity (anti-type addition). Illustrative examples are shown in Scheme 69.
13. Hydrohalogenation Processes
Very few works dealing with the addition of halogen–hydrogen bonds to 1,3-diynes can be currently found in the literature. Hydrohalogenations using aqueous or gaseous HX under CuCl catalysis were described in early reports, but the yields reached were, in general, very low and the reactions led, in most cases, to isomeric mixtures of products (a representative example is given in Scheme 70) [202,203,204].
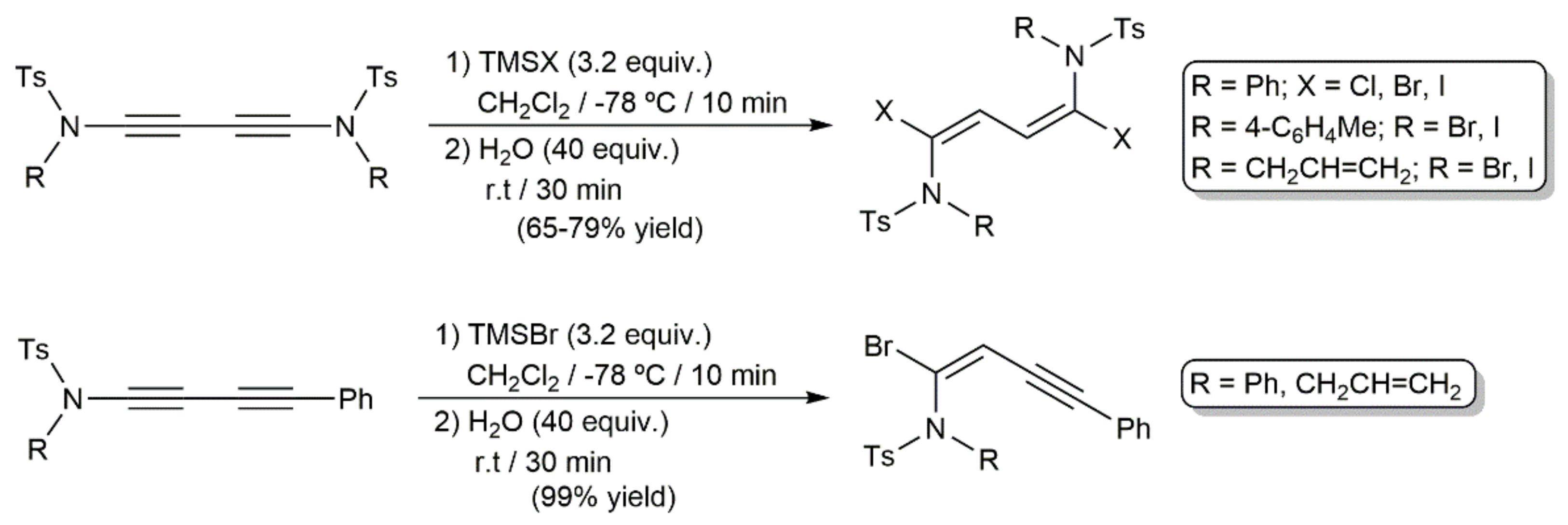
In a more recent study, regio- and stereoselective hydrohalogenation reactions of 1,3-diynes containing ynamide moieties were described by Iwasawa and colleagues under catalyst-free conditions (Scheme 71) [205]. The corresponding HX reagent was generated in situ by hydrolysis of the respective halotrimethylsilane Me3SiX (TMSX; X = Cl, Br, I). In complete accord with the lack of reactivity observed when the diyne PhC≡CC≡CPh was employed as a substrate, in the case of compound R(Ts)NC≡CC≡CPh, only the ynamide C≡C bond participated in the process. In all the cases, the addition proceeded in syn-mode, with the halogen attacking the electrophilic ynamide α carbon.
14. Summary
In this contribution, metal-catalyzed hydrofunctionalization reactions of conjugated 1,3-diynes have been comprehensively reviewed for this first time, thus complementing previous review articles and accounts covering the synthesis and applications of this particular class of alkynes [10,11,12,13,14,15,76]. The challenges associated with the presence of two reactive C≡C units, as the number of potential regio- and stereoisomeric products that can be formed increases, have made these substrates much less employed in hydrofunctionalization processes compared to classical monoynes. However, many examples collected in this review highlight the possibility of obtaining unique products through the selection of suitable catalytic systems, making 1,3-diynes interesting building blocks for obtaining functionalized enynes, dienes, as well as a wide variety of heterocyclic and carbocyclic compounds, in a selective manner. The enormous potential offered by 1,3-diynes in hydrofunctionalization processes will deserve undoubtedly more attention in the coming years and will continue to greatly contribute to modern synthetic chemistry.
Funding
Financial support from the Spanish Ministry of Economy, Industry and Competitiveness MINECO, grant number CTQ2016-75986-P, is gratefully acknowledged.
Conflicts of Interest
The author declares no conflict of interest.
References
- Houk, K.; Hopf, H.; Stang, P.; Nicholas, K.M.; Schore, N.; Regitz, M.; Nicolaou, K.C.; Gleiter, R.; Scott, L.R.; Grubbs, H.; et al. Modern Acetylene Chemistry; Stang, P.J., Diederich, F., Eds.; Wiley-VCH: Weinheim, Germany, 1995. [Google Scholar]
- Diederich, F.; Stang, P.J.; Tykwinski, R.R. (Eds.) Acetylene Chemistry: Chemistry, Biology and Material Science; Wiley-VCH: Weinheim, Germany, 2005. [Google Scholar]
- Trost, B.M.; Li, C.-J. (Eds.) Modern Alkyne Chemistry: Catalytic and Atom-Economic Transformations; Wiley-VCH: Weinheim, Germany, 2015. [Google Scholar]
- Alonso, F.; Beletskaya, I.P.; Yus, M. Transition-metal-catalyzed addition of heteroatom-hydrogen bonds to alkynes. Chem. Rev. 2004, 104, 3079–3160. [Google Scholar] [CrossRef]
- Beller, M.; Seayad, J.; Tillack, A.; Jiao, H. Catalytic Markovnikov and anti-Markovnikov functionalization of alkenes and alkynes: Recent developments and trends. Angew. Chem. Int. Ed. 2004, 43, 3368–3398. [Google Scholar] [CrossRef] [PubMed]
- Patil, N.T.; Kavthe, R.D.; Shinde, V.S. Transition metal-catalyzed addition of C-, N- and O-nucleophiles to unactivated C-C multiple bonds. Tetrahedron 2012, 68, 8079–8146. [Google Scholar] [CrossRef]
- Zeng, X. Recent advances in catalytic sequential reactions involving heteroelement addition to carbon-carbon multiple bonds. Chem. Rev. 2013, 113, 6864–6900. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Zi, W. Transition-metal catalyzed enantioselective functionalization of alkynes. Tetrahedron Lett. 2018, 59, 2205–2213. [Google Scholar] [CrossRef]
- Cadierno, V. Metal-catalyzed hydrofunctionalization reactions of haloalkynes. Eur. J. Inorg. Chem. 2020, 2020, 886–898. [Google Scholar] [CrossRef]
- Siemsen, P.; Livingston, R.C.; Diederich, F. Acetylenic coupling: A powerful tool in molecular construction. Angew. Chem. Int. Ed. 2000, 39, 2632–2657. [Google Scholar] [CrossRef]
- Maretina, I.A.; Trofimov, B.A. Diacetylene and its derivatives in heterocyclization reactions. Adv. Heterocycl. Chem. 2002, 82, 157–259. [Google Scholar]
- Shi, W.; Lei, A. 1,3-Diyne Chemistry: Synthesis and derivatizations. Tetrahedron Lett. 2014, 55, 2763–2772. [Google Scholar] [CrossRef] [Green Version]
- Nizami, T.A.; Hua, R. Cycloaddition of 1,3-butadiynes: Efficient synthesis of carbo- and heterocycles. Molecules 2014, 19, 13788–13802. [Google Scholar] [CrossRef] [PubMed]
- Shi, W. Conjugated diyne chemistry: Synthesis, natural existence and applications. Curr. Organocatalysis 2015, 2, 2–13. [Google Scholar] [CrossRef]
- Jiang, Y.; Liu, Y.; Zhang, W.; Cheng, H.; Kuang, C.; Chen, Z. Synthesis of 1,3-diynes. Prog. Chem. 2016, 28, 507–527. [Google Scholar]
- Rej, S.; Das, A.; Panda, T.K. Overview of regioselective and steroselective catalytic hydroboration of alkynes. Adv. Synth. Catal. 2021, 363, 4818–4840. [Google Scholar] [CrossRef]
- Zweifel, G.; Polston, N.L. Selective hydroboration of conjugated diynes with dialkylboranes. A convenient route to conjugated cis-enynes, α,β-acetylenic ketones, and cis,cis-dienes. J. Am. Chem. Soc. 1970, 92, 4068–4071. [Google Scholar] [CrossRef]
- Stracker, E.C.; Leong, W.; Miller, J.A.; Shoup, T.M.; Zweifel, G. Directive effects in hydroborations of 1-(trimethylsilyl)-1,3-diynes. Syntheses of (Z)-enynes and α-ketocetylenes. Tetrahedron Lett. 1989, 30, 6487–6490. [Google Scholar] [CrossRef]
- Shimoi, M.; Watanabe, T.; Maeda, K.; Curran, D.P.; Taniguchi, T. Radical trans-hydroboration of alkynes with N-heterocyclic carbene boranes. Angew. Chem. Int. Ed. 2018, 57, 9485–9490. [Google Scholar] [CrossRef]
- Takahashi, K.; Geib, S.J.; Maeda, K.; Curran, D.P.; Taniguchi, T. Radical trans-hydroboration of substituted 1,3-diynes with an N-heterocyclic carbene borane. Org. Lett. 2021, 23, 1071–1075. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Kim, Y.E.; Yun, J. Highly regio- and stereoselective synthesis of boron-substituted enynes via copper-catalyzed borylation of conjugated diynes. Org. Lett. 2015, 17, 860–863. [Google Scholar] [CrossRef]
- Sokolnicki, T.; Szyling, J.; Franczyk, A.; Walkowiak, J. Regio- and stereoselective synthesis of enynyl boronates via ruthenium-catalyzed hydroboration of 1,4-diaryl-substituted 1,3-diynes. Adv. Synth. Catal. 2020, 362, 177–183. [Google Scholar] [CrossRef] [Green Version]
- Sang, H.L.; Wu, C.; Phua, G.G.D.; Ge, S. Cobalt-catalyzed regiodivergent setereoselective hydroboration of 1,3-diynes to access boryl-functionalized enynes. ACS Catal. 2019, 9, 10109–10114. [Google Scholar] [CrossRef]
- Nevado, C.; Echavarren, A.M. Transition metal-catalyzed hydroarylation of alkynes. Synthesis 2005, 2005, 167–182. [Google Scholar] [CrossRef]
- Kitamura, T. Transition-metal-catalyzed hydroarylation reactions of alkynes through direct functionalization of C-H bonds: A convenient tool for organic synthesis. Eur. J. Org. Chem. 2009, 2009, 1111–1125. [Google Scholar] [CrossRef]
- Yamamoto, Y. Synthesis of heterocycles via transition-metal-catalyzed hydroarylation of alkynes. Chem. Soc. Rev. 2014, 43, 1575–1600. [Google Scholar] [CrossRef]
- Gorin, D.J.; Dubé, P.; Toste, F.D. Synthesis of benzonorcaradienes by gold(I)-catalyzed [4+3] annulation. J. Am. Chem. Soc. 2006, 128, 14480–14481. [Google Scholar] [CrossRef]
- Matsuda, Y.; Naoe, S.; Oishi, S.; Fujii, N.; Ohno, H. Formal [4+2] reaction of 1,3-diynes and pyrroles: Gold(I)-catalyzed indole synthesis by double hydroarylation. Chem. Eur. J. 2015, 21, 1463–1467. [Google Scholar] [CrossRef] [Green Version]
- Stylianakis, I.; Faza, O.N.; López, C.S.; Kolocouris, A. The key role of protodeauration in the gold-catalyzed reaction of 1,3-diynes with pyrrole and indole to form complex heterocycles. Org. Chem. Front. 2020, 7, 997–1005. [Google Scholar] [CrossRef]
- Matsuoka, J.; Matsuda, Y.; Kawada, Y.; Oishi, S.; Ohno, H. Total synthesis of dictyodendrins by the gold-catalyzed cascade cyclization of conjugated diynes and pyrroles. Angew. Chem. Int. Ed. 2017, 56, 7444–7448. [Google Scholar] [CrossRef] [PubMed]
- Ungeheuer, F.; Fürstner, A. Concise total synthesis of Ivorenolide B. Chem. Eur. J. 2015, 21, 11387–11392. [Google Scholar] [CrossRef] [Green Version]
- Mo, J.; Eom, D.; Lee, E.; Lee, P.H. Hybrid system of metal/Brønsted acid relay catalysis for the intramolecular double hydroarylation and cationic cyclization of diyne diethers and diamines. Org. Lett. 2012, 14, 3684–3687. [Google Scholar] [CrossRef]
- Zeng, H.; Hua, R. Palladium-catalyzed hydrophenylation of alkynes with sodium tetraphenylborate under mild conditions. J. Org. Chem. 2008, 73, 558–562. [Google Scholar] [CrossRef] [PubMed]
- Ha, H.; Shin, C.; Bae, S.; Joo, J.M. Divergent palladium-catalyzed cross-coupling of nitropyrazoles with terminal alkynes. Eur. J. Org. Chem. 2018, 2018, 2645–2650. [Google Scholar] [CrossRef]
- Yan, Z.; Yuan, X.-A.; Zhao, Y.; Zhu, C.; Xie, J. Selective hydroarylation of 1,3-diynes using dimeric manganese catalyst: Modular synthesis of Z-enynes. Angew. Chem. Int. Ed. 2018, 57, 12906–12910. [Google Scholar] [CrossRef] [PubMed]
- Yan, Z.; Zhu, C.; Xie, J. Manganese(I)-catalyzed selective functionalization of alkynes. Synlett 2018, 29, 124–128. [Google Scholar]
- Cembellin, S.; Dalton, T.; Pinkert, T.; Schäfers, F.; Glorius, F. Highly selective synthesis of 1,3-enynes, pyrroles, and furans by manganese(I)-catalyzed C-H activation. ACS Catal. 2020, 10, 197–202. [Google Scholar] [CrossRef]
- Gao, Y.; Zhang, M.; Wang, C.; Yang, Z.; Huang, X.; Feng, Q.C. Cobalt(II)-catalyzed hydroarylation of 1,3-diynes and internal alkynes with picolinamides promoted by alcohol. Chem. Commun. 2020, 56, 14231–14234. [Google Scholar] [CrossRef]
- Nakamura, M.; Endo, K.; Nakamura, E. Indium-catalyzed addition of active methylene compounds to 1-alkynes. J. Am. Chem. Soc. 2003, 125, 13002–13003. [Google Scholar] [CrossRef]
- Nakamura, M.; Endo, K.; Nakamura, E. A modular approach to α-arylated carbonyl compounds via indium tris(bistriflylamide)-catalyzed regioselective addition of β-keto esters to 1,3-diynes. Adv. Synth. Catal. 2005, 347, 1681–1686. [Google Scholar] [CrossRef]
- Rajanbabu, T.V. Hydrocyanation of alkenes and alkynes. Org. React. 2011, 75, 1–73. [Google Scholar]
- Peng, L.; Hu, Z.; Wang, H.; Wu, L.; Jiao, Y.; Tang, Z.; Xu, X. Direct cyanation, hydrocyanation, dicyanation and cyanofunctionalization of alkynes. RSC Adv. 2020, 10, 10232–10244. [Google Scholar] [CrossRef]
- Zhang, H.; Su, X.; Dong, K. Recent progress in transition-metal-catalyzed hydrocyanation of nonpolar alkenes and alkynes. Org. Biomol. Chem. 2020, 18, 391–399. [Google Scholar] [CrossRef] [PubMed]
- Sun, F.; Yang, C.; Ni, J.; Cheng, G.-J.; Fang, X. Ligand-controlled regiodivergent nickel-catalyzed hydrocyanation of silyl-substituted 1,3-diynes. Org. Lett. 2021, 23, 4045–4050. [Google Scholar] [CrossRef]
- Cai, S.; Zhang, H.; Huang, H. Transition-metal-catalyzed hydroaminocarbonylations of alkenes and alkynes. Trends Chem. 2021, 3, 218–230. [Google Scholar] [CrossRef]
- Ji, X.; Gao, B.; Zhou, X.; Liu, Z.; Huang, H. Palladium-catalyzed regioselective hydroaminocarbonylation of alkynes to α,β-unsaturated primary amides with ammonium chloride. J. Org. Chem. 2018, 83, 10134–10141. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Schneider, C.; Yang, J.; Wei, Z.; Jiao, H.; Franke, R.; Jackstell, R.; Beller, M. A general and highly selective palladium-catalyzed hydroamidation of 1,3-diynes. Angew. Chem. Int. Ed. 2021, 60, 371–379. [Google Scholar] [CrossRef]
- Liu, J.; Yang, J.; Schneider, C.; Franke, R.; Jackstell, R.; Beller, M. Tailored palladium catalyst for the selective synthesis of conjugated enynes by monocarbonylation of 1,3-diynes. Angew. Chem. Int. Ed. 2020, 59, 9032–9040. [Google Scholar] [CrossRef]
- Liu, J.; Yang, J.; Baumann, W.; Jackstell, R.; Beller, M. Stereoselective synthesis of highly substituted conjugated dienes via Pd-catalyzed carbonylation of 1,3-diynes. Angew. Chem. Int. Ed. 2019, 58, 10683–10687. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, J.C.; Trogler, W.C. Hydrosilylation of diynes as a route to functional polymers delocalized though silicon. Macromol. Chem. Phys. 2008, 209, 1527–1540. [Google Scholar] [CrossRef]
- Trost, B.M.; Ball, Z.T. Addition of metalloid hydrides to alkynes: Hydrometallation with boron, silicon, and tin. Synthesis 2005, 2005, 853–887. [Google Scholar] [CrossRef]
- He, P.; Hu, M.-Y.; Zhang, X.-Y.; Zhu, S.-F. Transition-metal-catalyzed stereo- and regioselective hydrosilylation of unsymmetrical alkynes. Synthesis 2022, 54, 49–66. [Google Scholar]
- Bock, H.; Seidl, H. “d-Orbital effects” in silicon-substituted π-electron systems. XII. Synthesis and properties of the isomeric bis(trimethylsilyl)-1,3-butadienes. J. Am. Chem. Soc. 1968, 90, 5694–5700. [Google Scholar] [CrossRef]
- Kusumoto, T.; Hiyama, T. Hydrosilylation of 1,4-bis(trimethylsilyl)-1,3-butadiyne. Chem. Lett. 1985, 14, 1405–1408. [Google Scholar] [CrossRef]
- Kusumoto, T.; Ando, K.; Hiyama, T. Hydrosilylation of 1,4-bis(trimethylsilyl)butadiyne and silyl-substituted butenynes. Bull. Chem. Soc. Jpn. 1992, 65, 1280–1290. [Google Scholar] [CrossRef]
- Tillack, A.; Pulst, S.; Baumann, W.; Baudisch, H.; Kortus, K.; Rosenthal, U. Hydrosilylierung von symmetrisch disubstituierten alkinen und butadiinen mit L2Ni(0)-butadiin-komplexen [Ph3P, (o-Tol-O)3P] als katalysatoren. J. Organomet. Chem. 1997, 532, 117–123. [Google Scholar] [CrossRef]
- Tillack, A.; Michalik, D.; Koy, C.; Michalik, M. Catalytic asymmetric hydrosilylation of butadiynes: A new synthesis of optically active allenes. Tetrahedron Lett. 1999, 40, 6567–6568. [Google Scholar] [CrossRef]
- Tillack, A.; Koy, C.; Michalik, D.; Fischer, C. A new asymmetric synthesis of optically active allenes via metal catalyzed hydrosilylation. J. Organomet. Chem. 2000, 603, 116–121. [Google Scholar] [CrossRef]
- Perry, R.J.; Karageorgis, M.; Hensler, J. Hydrosilylation reactions of 1,3-diynes and bis(silyl hydrides): Model studies and polymerizations. Macromolecules 2007, 40, 3929–3938. [Google Scholar] [CrossRef]
- Ishikawa, M.; Toyoda, E.; Horio, T.; Kunai, A. Polymeric organosilicon systems. 19. Preparation of branched polymers by selective hydrosilylation of poly[(silylene)but-1,3-diynes]. Organometallics 1994, 13, 26–27. [Google Scholar] [CrossRef]
- Kunai, A.; Toyoda, E.; Nagamoto, I.; Horio, T.; Ishikawa, M. Polymeric organosilicon systems. 25. Preparation of branched polymers by regiospecific hydrosilylation of poly[(silylene)diethynylenes] and their properties. Organometallics 1996, 15, 75–83. [Google Scholar] [CrossRef]
- Walkowiak, J.; Salamon, K.; Franczyk, A.; Stefanowska, K.; Szyling, J.; Kownacki, I. Pt-catalyzed hydrosilylation of 1,3-diynes with triorganosilanes: Regio- and stereoselective synthesis of mono- or bis-silylated adducts. J. Org. Chem. 2019, 84, 2358–2365. [Google Scholar] [CrossRef] [PubMed]
- Stefanowska, K.; Franczyk, A.; Szyling, J.; Walkowiak, J. Synthesis of functional 3-buten-1-ynes and 1,3-butadienes with silsesquioxane moiety via hydrosilylation of 1,3-diynes. ChemCatChem 2019, 11, 4848–4853. [Google Scholar] [CrossRef]
- Alonso, F.; Buitrago, R.; Moglie, Y.; Sepúlveda-Escribano, A.; Yus, M. Selective hydrosilylation of 1,3-diynes catalyzed by titania-supported platinum. Organometallics 2012, 31, 2336–2342. [Google Scholar] [CrossRef]
- Cano, R.; Yus, M.; Ramón, D.J. Impregnated platinum on magnetite as an efficient, fast, and reciclable catalyst for the hydrosilylation of alkynes. ACS Catal. 2012, 2, 1070–1078. [Google Scholar] [CrossRef]
- Reddy, C.B.; Shil, A.K.; Guha, N.R.; Sharma, D.; Das, P. Solid supported palladium(0) nanoparticles: An efficient heterogeneous catalyst for regioselective hydrosilylation of alkynes and Suzuki coupling of β-arylvinyl iodides. Catal. Lett. 2014, 144, 1530–1536. [Google Scholar] [CrossRef]
- Planellas, M.; Guo, W.; Alonso, F.; Yus, M.; Shafir, A.; Pleixats, R.; Parella, T. Hydrosilylation of internal alkynes catalyzed by tris-imidazolium salt-stabilized palladium nanoparticles. Adv. Synth. Catal. 2014, 356, 179–188. [Google Scholar] [CrossRef]
- Guo, W.; Pleixats, R.; Shafir, A.; Parella, T. Rhodium nanoflowers stabilized by a nitrogen-rich PEG-tagged substrate as recyclable catalyst for the stereoselective hydrosilylation of internal alkynes. Adv. Synth. Catal. 2015, 357, 89–99. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Jiang, Y.-N.; Lin, Z.-Y.; Zeng, J.-H.; Liu, Z.-K.; Zhan, Z.-P. Highly regio- and stereo-selective heterogeneous 1,3-diyne hydrosilylation controlled by a nickel-metalated porous organic polymer. Org. Chem. Front. 2021, 8, 4826–4832. [Google Scholar] [CrossRef]
- Sun, J.; Deng, L. Cobalt complex-catalyzed hydrosilylation of alkenes and alkynes. ACS Catal. 2016, 6, 290–300. [Google Scholar] [CrossRef]
- Tamang, S.R.; Findlater, M. Emergence and applications of base metals (Fe, Co and Ni) in hydroboration and hydrosilylation. Molecules 2019, 24, 3194. [Google Scholar] [CrossRef] [Green Version]
- Sang, H.L.; Hu, Y.; Ge, S. Cobalt-catalyzed regio- and stereoselective hydrosilylation of 1,3-diynes to access silyl-functionalized 1,3-enynes. Org. Lett. 2019, 21, 5234–5237. [Google Scholar] [CrossRef] [PubMed]
- Kong, D.; Hu, B.; Yang, M.; Chen, D.; Xia, H. Highly regio- and stereoselective tridentate NCNN cobalt-catalyzed 1,3-diyne hydrosilylation. Organometallics 2019, 38, 4341–4350. [Google Scholar] [CrossRef]
- Shen, S.; Zong, Z.; Sun, N.; Hu, B.; Shen, Z.; Hu, X.; Jin, L. Regio- and stereoselective cobalt-catalyzed hydroilylation of 1,3-diynes with primary and secondary silanes. Org. Chem. Front. 2021, 8, 6317–6322. [Google Scholar] [CrossRef]
- Kong, D.; Hu, B.; Yang, M.; Gong, D.; Xia, H.; Chen, D. Bis(phosphine)cobalt-catalyzed highly regio- and stereoselective hydrosilylation of 1,3-diynes. Organometallics 2020, 39, 4437–4443. [Google Scholar] [CrossRef]
- Matsuda, T. Synthesis of heterocycles via X-H bond addition to diynes. In Transition-Metal-Mediated Aromatic Ring Construction, 1st ed.; Tanaka, K., Ed.; John Wiley & Sons: Hoboken, NJ, USA, 2013; pp. 537–547. [Google Scholar]
- Matsuda, T.; Kadowaki, S.; Murakami, M. Ruthenium-catalyzed trans-hydrosilylation of 1,4-diaryl-1,3-diynes leading to 2,5-diarylsiloles. Chem. Commun. 2007, 2627–2629. [Google Scholar] [CrossRef]
- Matsuda, T.; Kadowaki, S.; Yamaguchi, Y.; Murakami, M. Ruthenium-catalyzed trans-hydrogermylation of alkynes: Formation of 2,5-disubstituted germoles through double trans-hydrogermylation of 1,3-diynes. Org. Lett. 2010, 12, 1056–1058. [Google Scholar] [CrossRef]
- Trost, B.M.; Chan, V.S.; Yamamoto, D. Enantioselective ProPhenol-catalyzed addition of 1,3-diynes to aldehydes to generate synthetically versatile building blocks and diyne natural products. J. Am. Chem. Soc. 2010, 132, 5186–5192. [Google Scholar] [CrossRef] [Green Version]
- Reisch, J.; Schulte, K.E. Pyrrol-derivate aus diacetylenen. Angew. Chem. 1961, 73, 241. [Google Scholar] [CrossRef]
- Schulte, K.E.; Reisch, J.; Walker, H. Eine neue pyrrolsynthese aus butadiin-derivaten. Chem. Ber. 1965, 98, 98–103. [Google Scholar] [CrossRef]
- Zheng, Q.; Hua, R. CuCl-catalyzed cycloaddition of 1,3-butadiynes with primary amines: An atom-economic process for the synthesis of 1,2,5-trisubstituted pyrroles. Tetrahedron Lett. 2010, 51, 4512–4514. [Google Scholar] [CrossRef]
- Zheng, Q.; Hua, R.; Jiang, J.; Zhang, L. A general approach to arylated furans, pyrroles, and thiophenes. Tetrahedron 2014, 70, 8252–8256. [Google Scholar] [CrossRef]
- Feng, X.; Tong, B.; Shen, J.; Shi, J.; Han, T.; Chen, L.; Zhi, J.; Lu, P.; Ma, Y.; Dong, Y. Aggregation-induced emission enhancement of aryl-substituted pyrrole derivatives. J. Phys. Chem. B 2010, 114, 16731–16736. [Google Scholar] [CrossRef]
- Krompiec, S.; Filapek, M.; Grudzka-Flak, I.; Slodek, A.; Kula, S.; Malecki, J.G.; Malarz, J.; Szafraniec-Gorol, G.; Penkala, M.; Schab-Balcerzak, E.; et al. Multifaceted strategy for the synthesis of diverse 2,2´-bithiophene derivatives. Molecules 2015, 20, 4565–4593. [Google Scholar] [CrossRef] [Green Version]
- Yang, C. Pyrrole-cored push-pull single chromophore. Tetrahedron Lett. 2010, 51, 2007–2009. [Google Scholar] [CrossRef]
- Kowada, T.; Kuwabara, T.; Ohe, K. Synthesis, structure, and optical properties of heteroarene-fused dispiro compounds. J. Org. Chem. 2010, 75, 906–913. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, S.; Kobayashi, T.; Ogura, K. Novel 1,1′,5,5′-tetraaryl-2,2′-bipyrroles: Their synthesis and physical properties. Heterocycles 2005, 66, 319–332. [Google Scholar] [CrossRef]
- Maeda, C.; Shinokubo, H.; Osuka, A. Synthesis of meso,meso’-pyrrole-bridged diporphyrins by Cu(I)-mediated annulation. Org. Lett. 2010, 12, 1820–1823. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.; Yi, L.; Jun, C.-H. Copper(II)-promoted, one-pot conversion of 1-alkynes with anhydrides or primary amines to the respective 2,5-disubstituted furans or pyrroles under microwave irradiation conditions. Adv. Synth. Catal. 2015, 357, 3485–3490. [Google Scholar] [CrossRef]
- Widenhoefer, R.A.; Han, X. Gold-catalyzed hydroamination of C-C multiple bonds. Eur. J. Org. Chem. 2006, 2006, 4555–4563. [Google Scholar] [CrossRef]
- Kramer, S.; Madsen, J.L.H.; Rottländer, M.; Skrydstrup, T. Access to 2,5-diamidopyrroles and 2,5-diamidofurans by Au(I)-catalyzed double hydroamination or hydration of 1,3-diynes. Org. Lett. 2010, 12, 2758–2761. [Google Scholar] [CrossRef] [PubMed]
- Num, P.; Dupuy, S.; Gaillard, S.; Poater, A.; Cavallo, L.; Nolan, S.P. Gold(I)-catalyzed synthesis of furans and pyrroles via alkyne hydration. Catal. Sci. Technol. 2011, 1, 58–61. [Google Scholar]
- Lavallo, V.; Frey, G.D.; Donnadieu, B.; Soleilhavoup, M.; Bertrand, G. Homogeneous catalytic hydroamination of alkynes and allenes with ammonia. Angew. Chem. Int. Ed. 2008, 47, 5224–5228. [Google Scholar] [CrossRef] [Green Version]
- Kinjo, R.; Donnadieu, B.; Bertrand, G. Gold-catalyzed hydroamination of alkynes and allenes with parent hydrazine. Angew. Chem. Int. Ed. 2011, 50, 5560–5563. [Google Scholar] [CrossRef] [PubMed]
- Paudler, W.W.; Zeiler, A.G. 1,3-butadiynes in the synthesis of heterocyclic compounds. I. 2,3-Dihydro-1,4-diazepine, pyrazole, and isoxazole derivatives. J. Org. Chem. 1969, 34, 999–1001. [Google Scholar] [CrossRef]
- Wang, L.; Yu, X.; Feng, X.; Bao, M. Synthesis of 3,5-disubstituted pyrazoles via Cope-type hydroamination of 1,3-dialkynes. J. Org. Chem. 2013, 78, 1693–1698. [Google Scholar] [CrossRef]
- Yu, X.; Huang, N.; Feng, X.; Yamamoto, Y.; Bao, M. Synthesis of 1,3,5-trisubstituted pyrazoles by the Cope-type hydroamination of 1,3-dialkynes with alkylhydrazines. Synthesis 2014, 46, 2422–2429. [Google Scholar] [CrossRef]
- Bassaco, M.M.; Fortes, M.P.; Kaufman, T.S.; Silveira, C.C. Metal-free synthesis of 3,5-disubstituted 1H- and 1-aryl-1H-pyrazoles from 1,3-diyne-indole derivatives employing two successive hydroaminations. RSC Adv. 2015, 5, 21112–21124. [Google Scholar] [CrossRef]
- Shen, R.; Yang, J.; Luo, B.; Zhang, L.; Han, L.-B. Copper-catalyzed allenylation-isomerization sequence of 1,4-diyn-3-yl acetates with P(O)H compounds: Facile synthesis of 1-phosphonyl 2,4-diynes. Adv. Synth. Catal. 2016, 358, 3897–3906. [Google Scholar] [CrossRef]
- Verlinden, S.; Ballet, S.; Verniest, G. Synthesis of heterocycle-bridged peptidic macrocycles through 1,3-diyne transformations. Eur. J. Org. Chem. 2016, 2016, 5807–5812. [Google Scholar] [CrossRef]
- Ötvös, S.B.; Georgiádes, Á.; Ozsvár, D.; Fülöp, F. Continuous-flow synthesis of 3,5-disubstituted pyrazoles via sequential alkyne homocoupling and Cope-type hydroamination. RSC Adv. 2019, 9, 8197–8203. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Yu, X.; Feng, X.; Bao, M. Synthesis of 3,5-disubstituted isoxazoles via Cope-type hydroamination of 1,3-dialkynes. Org. Lett. 2012, 14, 2418–2421. [Google Scholar] [CrossRef]
- Bassaco, M.M.; Fortes, M.P.; Back, D.F.; Kaufman, T.S.; Silveira, C.C. An eco-friendly synthesis of novel 3,5-disubstituted-1,2-isoxazoles in PEG-400, employing the Et3N-promoted hydroamination of symmetric and unsymmetric 1,3-diyne-indole derivatives. RSC Adv. 2014, 4, 60785–60797. [Google Scholar] [CrossRef]
- Kirillova, M.A.; Maretina, I.A.; Petrov, A.A. Reactions of alkadiynes with guanidine and its derivatives. Russ. J. Org. Chem. 1971, 7, 14–16. [Google Scholar]
- Singha, R.; Ray, J.K. Transition metal free synthesis of 2,4,6-trisubstitutd pyrimidines via Cope-type hydroamination of 1,4-diarylbuta-1,3-diynes. RSC Adv. 2014, 4, 44052–44056. [Google Scholar] [CrossRef]
- Zhang, L.; Zhao, M.; Zhao, X. The synthesis of carbonyl 2-amino-pyrimidines via tandem regioselective heterocyclization of 1,3-diynes with guanidine and selective oxidation. Chem. Commun. 2015, 51, 9370–9373. [Google Scholar] [CrossRef]
- Ackermann, L.; Born, R. TiCl4/t-BuNH2 as the sole catalyst for a hydroamination-based Fischer indole synthesis. Tetrahedron Lett. 2004, 45, 9541–9544. [Google Scholar] [CrossRef]
- Gupta, S.; Agarwal, P.K.; Saifuddin, M.; Kundu, B. Hydro-amination/-amidation of 1,3-diynes with indoles/azoles/amides under modified Ullmann conditions: Stereo- and regio-selective synthesis of N-alkenynes via N-H bond activation. Tetrahedron Lett. 2011, 52, 5752–5757. [Google Scholar] [CrossRef]
- Sun, H.; Wu, X.; Hua, R. Copper(I)-Catalyzed reaction of diaryl buta-1,3-diynes with cyclic amines: An atom-economic approach to amino-substituted naphthalene derivatives. Tetrahedron Lett. 2011, 52, 4408–4411. [Google Scholar] [CrossRef]
- Choi, J.; Park, K.; Lim, J.; Jung, H.M.; Lee, S. Copper-catalyzed synthesis of amino-substituted polycyclic aromatic hydrocarbons by the sequential reaction between aryl alkynyl carboxylic acids and amines. Asian J. Org. Chem. 2015, 4, 969–974. [Google Scholar] [CrossRef]
- Li, Y.; Qiu, S.; Fan, L.; Yin, G. Cooperative palladium and copper catalysis: One-pot synthesis of diamino-substituted naphthalenes from aryl halides, 1,4-bis(trimethylsilyl)butadiene and amines. ChemCatChem 2020, 12, 1230–1235. [Google Scholar] [CrossRef]
- Glock, C.; Görls, H.; Westerhausen, M. Calcite-mediated intermolecular hydroamination of diphenylbutadiyne with secondary anilines. Chem. Commun. 2012, 48, 7094–7096. [Google Scholar] [CrossRef] [Green Version]
- Younis, F.M.; Krieck, S.; Görls, H.; Westerhausen, M. Hydroamination of diphenylbutadiyne with secondary N-methyl-anilines using the dipotassium tetrakis(2,6-diisopropylanilino)calciate precatalyst. Dalton Trans. 2016, 45, 6241–6250. [Google Scholar] [CrossRef]
- Younis, F.M.; Krieck, S.; Görls, H.; Westerhausen, M. s-Block-metal-mediated hydroamination of diphenylbutadiyne with primary arylamines using dipotassium tetrakis(amino)calciate precatalyst. Organometallics 2015, 34, 3577–3585. [Google Scholar] [CrossRef]
- Glock, C.; Younis, F.M.; Ziemann, S.; Görls, H.; Imhof, W.; Kriek, S.; Westerhausen, M. 2,6-Diisopropylphenylamides of potassium and calcium: A primary amido ligand in s-block metal chemistry with an unprecedented catalytic reactivity. Organometallics 2013, 32, 2649–2660. [Google Scholar] [CrossRef]
- Ziemann, S.; Krieck, S.; Görls, H.; Westerhausen, M. 1,2-Bis(anilido)ethane complexes of calcium and potassium: Synthesis, structures, and catalytic activity. Organometallics 2018, 37, 924–933. [Google Scholar] [CrossRef]
- Koradin, C.; Dohle, W.; Rodriguez, A.L.; Schmid, B.; Knochel, P. Synthesis of polyfunctional indoles and related heterocycles mediated by cesium and potassium bases. Tetrahedron 2003, 59, 1571–1587. [Google Scholar] [CrossRef]
- Fiandanese, V.; Bottalico, D.; Marchese, G.; Punzi, A. A rapid synthesis of 2-alkynylindoles and 2-alkynylbenzofurans. Tetrahedron 2008, 64, 7301–7306. [Google Scholar] [CrossRef]
- Inamdar, S.M.; Gonnade, R.G.; Patil, N.T. Synthesis of annulated bis-indoles through Au(I)/Brønsted acid-catalyzed reactions of (1H-indol-3-yl)(aryl)methanols with 2-(arylethynyl)-1H-indoles. Org. Biomol. Chem. 2017, 15, 863–869. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Wei, Y.; Shi, M. Rhodium(III)-catalyzed cross coupling of sulfoxonium ylides and 1,3-diynes to produce naphthol-indole derivatives: An arene ortho C-H activation/annulation cascade. ChemCatChem 2020, 12, 5903–5906. [Google Scholar] [CrossRef]
- Perea-Buceta, J.E.; Wirtanen, T.; Laukkanen, O.-V.; Mäkelä, M.K.; Nieger, M.; Melchionna, M.; Huittinen, N.; Lopez-Sanchez, J.A.; Heleja, J. Cycloisomerization of 2-alkynylanilines to indoles by carbon-supported gold nanoparticles and subsequent homocoupling to 3,3′-biindoles. Angew. Chem. Int. Ed. 2013, 52, 11835–11839. [Google Scholar] [CrossRef]
- Sharp, P.P.; Banwell, M.G.; Renner, J.; Lohmann, K.; Willis, A.C. Consecutive gold(I)-catalyzed cyclization reactions of o-(buta-1,3-diyn-1-yl-)-substituted N-aryl ureas: A one-pot synthesis of pyrimido[1,6-a]indol-1(2H)-ones and related systems. Org. Lett. 2013, 15, 2616–2619. [Google Scholar] [CrossRef]
- Naoe, S.; Saito, T.; Uchiyama, M.; Oishi, S.; Fujii, N.; Ohno, H. Direct construction of fused indoles by gold-catalyzed cascade cyclization of conjugated diynes. Org. Lett. 2015, 17, 1774–1777. [Google Scholar] [CrossRef]
- Märkl, G.; Potthast, R. A simple synthesis of phospholes. Angew. Chem. Int. Ed. 1967, 6, 86. [Google Scholar] [CrossRef]
- Arkhypchuk, A.I.; Orthaber, A.; Ott, S. Tuning the optical properties of 1,1´-biphospholes by chemical alterations of the P-P bridge. Eur. J. Inorg. Chem. 2014, 2014, 1760–1766. [Google Scholar] [CrossRef]
- Ogasawara, M.; Yoshida, K.; Hayashi, T. Synthesis and characterization of a novel chiral phosphole and its derivatives. Organometallics 2001, 20, 1014–1019. [Google Scholar] [CrossRef]
- Ogasawara, M.; Ito, A.; Yoshida, K.; Hayashi, T. Synthesis of 2,5-bis(binaphthyl)phospholes and phosphametallocene derivatives and their application in palladium-catalyzed asymmetric hydrosilylation. Organometallics 2006, 25, 2715–2718. [Google Scholar] [CrossRef]
- Miesel, D.; Hildebrandt, A.; Korb, M.; Low, P.J.; Lang, H. Synthesis and (spectro)electrochemical properties of 2,5-diferrocenyl-1-phenyl-1H-phosphole. Organometallics 2013, 32, 2993–3002. [Google Scholar] [CrossRef] [Green Version]
- Miesel, D.; Hildebrandt, A.; Korb, M.; Wild, D.A.; Low, P.J.; Lang, H. Influence of P-bonded bulky substituents on electronic interactions in ferrocenyl-substituted phospholes. Chem. Eur. J. 2015, 21, 11545–11559. [Google Scholar] [CrossRef]
- Miesel, D.; Korb, M.; Hildebrandt, A.; Lang, H. Synthesis and crystal structure of an acetylenic ferrocenyl substituted phosphaalkene. Inorg. Chim. Acta 2018, 471, 741–745. [Google Scholar] [CrossRef]
- Klintuch, D.; Krekić, K.; Bruhn, C.; Benkő, Z.; Pietschnig, R. A rational synthetic approach to 2,5-diphenyl-β-silyl phospholes. Eur. J. Inorg. Chem. 2016, 2016, 718–725. [Google Scholar] [CrossRef]
- Roesler, F.; Kaban, B.; Klintuch, D.; Ha, U.-M.; Bruhn, C.; Hillmer, H.; Pietschnig, R. Tailoring phospholes for imprint of fluorescent 3D structures. Eur. J. Inorg. Chem. 2019, 2019, 4820–4825. [Google Scholar] [CrossRef] [Green Version]
- Klintuch, D.; Kirchmeier, A.; Bruhn, C.; Pietschnig, R. Synthetic access and luminescence tuning in a seies of β-H and β-silyl substituted phospholes. Dye. Pigm. 2020, 180, 108443. [Google Scholar] [CrossRef]
- Märkl, G.; Hauptmann, H. 1.2.5-Trisubstituierte arsole. Tetrahedron Lett. 1968, 9, 3257–3260. [Google Scholar] [CrossRef]
- Märkl, G.; Hauptmann, H.; Merz, A. Synthese von 1-phenyl-2,5-diaryl(dialkyl)-arsolen; umsetzung der arsole mit alkalimetallen und lithiumorganylen. J. Organomet. Chem. 1983, 249, 335–363. [Google Scholar] [CrossRef]
- Manhart, S.; Schier, A.; Paul, M.; Riede, J.; Schmidbaur, H. New organophosphorus ligands: Cyclopropanation of cumulenes bearing diphenylphosphanyl substituents. Chem. Ber. 1995, 128, 365–371. [Google Scholar] [CrossRef]
- Trifonov, A.A.; Basalov, I.V.; Kisel, A.A. Organolanthanides in catalytic intermolecular hydrophosphination and hydroamination of multiple C-C bonds. Dalton Trans. 2016, 45, 19172–19193. [Google Scholar] [CrossRef]
- Takaki, K.; Takeda, M.; Koshoji, G.; Shishido, T.; Takehira, K. Intermolecular hydrophosphination of alkynes and related carbon-carbon multiple bonds catalyzed by ytterbium-imine complexes. Tetrahedron Lett. 2001, 42, 6357–6360. [Google Scholar] [CrossRef] [Green Version]
- Takaki, K.; Koshoji, G.; Komeyama, K.; Takeda, M.; Shishido, T.; Kitani, A.; Takehira, K. Intermolecular hydrophosphination of alkynes and related carbon-carbon multiple bonds catalyzed by organoytterbiums. J. Org. Chem. 2003, 68, 6554–6565. [Google Scholar] [CrossRef]
- Komeyama, K.; Kobayashi, D.; Yamamoto, Y.; Takehira, K.; Takaki, K. Ytterbium-catalyzed dual intermolecular hydrophosphination: Synthesis of bis(phosphinyl)dienes and bis(alkenyl)phosphine oxides. Tetrahedron 2006, 62, 2511–2519. [Google Scholar] [CrossRef] [Green Version]
- Al-Shboul, T.M.A.; Görls, H.; Westerhausen, M. Calcium-mediated hydrophosphination of diphenylethyne and diphenylbutadiyne as well as crystal structure of 1,4-diphenyl-1,4-bis(diphenylphosphanyl)buta-1,3-diene. Inorg. Chem. Commun. 2008, 11, 1419–1421. [Google Scholar] [CrossRef]
- Al-Shboul, T.M.A.; Pálfi, V.K.; Yu, L.; Kretschmer, R.; Wimmer, K.; Fischer, R.; Görls, H.; Reiher, M.; Westerhausen, M. Catalytic synthesis of vinylphosphanes via calcium-mediated intermolecular hydrophosphanylation of alkynes and butadiynes. J. Organomet. Chem. 2011, 696, 216–227. [Google Scholar] [CrossRef]
- Al-Shboul, T.M.A.; Görls, H.; Krieck, S.; Westerhausen, M. Regiospecific calcium-mediated intermolecular hydrophosphanylation of butadiynes with diphenylphosphane oxide. Eur. J. Inorg. Chem. 2012, 2012, 5451–5455. [Google Scholar] [CrossRef]
- Younis, F.M.; Krieck, S.; Al-Shboul, T.M.A.; Görls, H.; Westerhausen, M. Calcium-mediated synthesis of 1-(diorganylamino)-1,4-diphenyl-4-(diphenylphosphanyl)buta-1,3-dienes. Inorg. Chem. 2016, 55, 4676–4682. [Google Scholar] [CrossRef] [PubMed]
- Hintermann, L.; Labonne, A. Catalytic hydration of alkynes and its application in synthesis. Synthesis 2007, 2007, 1121–1150. [Google Scholar] [CrossRef] [Green Version]
- Turbanova, E.S.; Porfir´eva, Y.I.; Petrov, A.A. Rules in the addition reactions of diacetylenes. VIII. Orientation of hydration of disubstituted diacetylenes. Russ. J. Gen. Chem. 1966, 2, 772–777. [Google Scholar]
- Sokolov, L.B.; Petrov, A.A. Rules in the addition reactions of diacetylenes. X. Orientation of hydration of conjugated monoalkyldiacetylenes. Russ. J. Gen. Chem. 1966, 2, 1003–1005. [Google Scholar]
- Constantino, M.G.; Donate, P.M.; Petragnani, N. Hydration of diacetylene compounds. Synthesis of a merine natural product: (±)-1-(2,6,6-trimethyl-4-hydroxycyclohexenyl)-1,3-butanedione. J. Org. Chem. 1986, 51, 387–390. [Google Scholar] [CrossRef]
- Shim, S.C.; Lee, T.S. Photohydration reaction of 1-(1-naphthyl)buta-1,3-diynes. J. Chem. Soc. Perkin Trans. 1990, 2, 1739–1743. [Google Scholar] [CrossRef]
- Baek, E.K.; Shim, S.C. Photohydration reaction of 1-(p-nitrophenyl)-5,5-dimethyl-1,3-hexadiyne. J. Phys. Org. Chem. 1995, 8, 699–705. [Google Scholar] [CrossRef]
- Baek, E.K.; Lee, S.T.; Chae, Y.S.; Shim, S.C. The photohydration of 1-(nitrophenyl)-5,5-dimethyl-1,3-hexadiynes: The nitro substituent effect on the excited states of diacetylenes. J. Photosci. 1995, 2, 73–76. [Google Scholar]
- Shim, S.C.; Chae, Y.S.; Baek, E.K.; Park, S.K. Substituent effects on the photohydration of 1-aryl-5,5-diemthyl-1,3-hexadiynes. J. Photochem. Photobiol. A 1997, 106, 155–160. [Google Scholar] [CrossRef]
- Jiang, H.; Zeng, W.; Li, Y.; Wu, W.; Huang, L.; Fu, W. Copper(I)-catalyzed synthesis of 2,5-disubstituted furans and thiophenes from haloalkynes or 1,3-diynes. J. Org. Chem. 2012, 77, 5179–5183. [Google Scholar] [CrossRef]
- Irudayanathan, F.M.; Raja, G.C.E.; Lee, S. Copper-catalyzed direct synthesis of furans and thiophenes via decarboxylative coupling of alkynyl carboxylic acids with H2O or Na2S. Tetrahedron 2015, 71, 4418–4425. [Google Scholar] [CrossRef]
- Zheng, Q.; Hua, R.; Yin, T. Palladium-catalyzed cycloaddition of 1,3-butadiynes with water: An alternative efficient catalytic system for atom-economic synthesis of 2,5-disubstituted furans. Curr. Org. Synth. 2013, 10, 161–164. [Google Scholar]
- Pérez, J.M.; Cano, R.; Yus, M.; Ramón, D.J. Copper-impregnated magnetite as a heterogeneous catalyst for the homocoupling of terminal alkynes. Synthesis 2013, 45, 1373–1379. [Google Scholar]
- Klukas, F.; Grunwald, A.; Menschel, F.; Müller, T.J.J. Rapid pseudo five-component synthesis of intensively blue luminescent 2,5-di(hetero)arylfurans via a Sonogashira-Glaser cyclization sequence. Beilstein J. Org. Chem. 2014, 10, 672–679. [Google Scholar] [CrossRef] [Green Version]
- Guo, P. Gold-catalyzed formation of C-O and C-C bonds: An efficient domino reaction synthesis of functionalized furans. Catal. Commun. 2015, 68, 58–60. [Google Scholar] [CrossRef]
- Camacho, D.H.; Saito, S.; Yamamoto, Y. “Anti-Wacker”-type hydroalkoxylation of diynes catalyzed by palladium(0). Tetrahedron Lett. 2002, 43, 1085–1088. [Google Scholar] [CrossRef]
- Lee, T.S.; Shim, S.C.; Kim, S.S. Photoreaction of 1,4-disubstituted-1,3-butadiyne with alcohols. Bull. Korean Chem. Soc. 1986, 7, 116–120. [Google Scholar]
- Schermann, G.; Vostrowsky, O.; Hirsch, A. Addition chemistry of rod-shaped 1,6-dicyanohexatriyne: Regioselectivity control by the remote cyano function. Eur. J. Org. Chem. 1999, 1999, 2491–2500. [Google Scholar] [CrossRef]
- Volchkov, I.; Sharma, K.; Cho, E.J.; Lee, D. Efficient one-step synthesis of bis-spiroketals from diynediols by π-Lewis acid-catalyzed hydroalkoxylation/hydration. Chem. Asian J. 2011, 6, 1961–1966. [Google Scholar] [CrossRef] [PubMed]
- Sharma, K. Unraveling the mechanism of tricyclic bis-spiroketal formation from diyne diol by DFT study. Lett. Org. Chem. 2019, 16, 392–395. [Google Scholar] [CrossRef]
- Valdez-García, R.M.; Alarcón-Manjarrez, C.; Galano, A.; Rodríguez-Molina, B.; Flores-Álamo, M.; Iglesias-Arteaga, M.A. Synthesis of dimeric steroid trioxabispiroacetals scaffolds by gold(I)-catalyzed hydroalkoxylation-hydration of diynediols. Eur. J. Org. Chem. 2019, 2019, 4916–4927. [Google Scholar] [CrossRef]
- Jones, E.R.H.; Stephenson, J.S. Chemistry of the higher fungi. Part IX. Polyacetylenic metabolites from Coprinus quadrifidus. J. Chem. Soc. 1959, 2197–2203. [Google Scholar] [CrossRef]
- Bohlmann, F.; Herbst, P.; Gleinig, H. Synthese von natürlich vorkommenden polyacetylenverbindungen mit endständigen dreifachbindungen. Chem. Ber. 1961, 94, 948–957. [Google Scholar] [CrossRef]
- Jones, E.R.H.; Stephenson, J.S.; Turner, W.B.; Whiting, M.C. Chemistry of the higher fungi. Part XIII. Synthesis of (a) a C9 triacetylenic epoxy-alcohol, a Coprinus quadrifidus metabolite and (b) a C9 triacetylenic 1,2-diol. The structure of biformyne 1. J. Chem. Soc. 1963, 2048–2055. [Google Scholar] [CrossRef]
- Cambie, R.C.; Hirschberg, A.; Jones, E.R.H.; Lowe, G. Chemistry of the higher fungi. Part XVI. Polyacetylenic metabolites from Aleurodiscus roseus. J. Chem. Soc. 1963, 4120–4130. [Google Scholar] [CrossRef]
- Jones, E.R.H.; Lowe, G.; Shannon, P.V.R. Natural acetylenes. Part XX. Tetra-acetylenic and other metabolites from Fistulina hepatica (Huds) Fr. J. Chem. Soc. C 1966, 139–144. [Google Scholar] [CrossRef]
- Miao, Z.; Xu, M.; Hoffmann, B.; Bernet, B.; Vasella, A. Functionalised bicyclic exo-glycals by alkynol cycloisomerisation of hydroxy 1,3-diynes and hydroxy haloalkynes. Helv. Chim. Acta 2005, 88, 1885–1912. [Google Scholar] [CrossRef]
- Xu, M.; Miao, Z.; Bernet, B.; Vasella, A. Functionalised monocyclic five- to seven-membered exo-glycals by alkynol cycloisomerisation of hydroxyl buta-1,3-diynes and 1-haloalkynes. Helv. Chim. Acta 2005, 88, 2918–2937. [Google Scholar] [CrossRef]
- Wang, Y.-H.; Liu, H.; Zhu, L.-L.; Li, X.X.; Chen, Z. Base-catalyzed cascade 1,3-H shift/cyclization reaction to construct polyaromatic furans. Adv. Synth. Catal. 2011, 353, 707–712. [Google Scholar] [CrossRef]
- Fiandanese, V.; Bottalico, D.; Marchese, G.; Punzi, A. A straightforward synthesis of indole and benzofuran derivatives. Tetrahedron 2008, 64, 53–60. [Google Scholar] [CrossRef]
- Gooβen, L.J.; Rodríguez, N.; Gooβen, K. Carboxylic acids as substrates in homogeneous catalysis. Angew. Chem. Int. Ed. 2008, 47, 3100–3120. [Google Scholar]
- Francos, J.; Cadierno, V. Metal-catalyzed intra- and intermolecular addition of carboxylic acids to alkynes in aqueous media: A review. Catalysts 2017, 7, 328. [Google Scholar] [CrossRef] [Green Version]
- González-Liste, P.J.; Francos, J.; García-Garrido, S.E.; Cadierno, V. The intermolecular hydro-oxycarbonylation of internal alkynes: Current state of the art. Arkivoc 2018, 2, 17–39. [Google Scholar] [CrossRef] [Green Version]
- Cadierno, V. Recent advances in the transition metal-catalyzed addition of carboxylic acids to alkynes. Curr. Org. Chem. 2021, 25, 2260–2303. [Google Scholar] [CrossRef]
- Cadierno, V. Gold-catalyzed addition of carboxylic acids to alkynes and allenes: Valuable tools for organic synthesis. Catalysts 2020, 10, 1206. [Google Scholar] [CrossRef]
- León, F.; Francos, J.; López-Serrano, J.; García-Garrido, S.E.; Cadierno, V.; Pizzano, A. Double asymmetric hydrogenation of conjugated dienes: A self-breeding chirality route for C2 symmetric 1,4-diols. Chem. Commun. 2019, 55, 786–789. [Google Scholar] [CrossRef] [PubMed]
- Schulte, K.E.; Reish, J.; Hörner, L. Thiophen-derivate aus polyacetylenen. Angew. Chem. 1960, 72, 920. [Google Scholar] [CrossRef]
- Tang, J.; Zhao, X. Synthesis of 2,5-disubstituted thiophenes via metal-free sulfur heterocyclization of 1,3-diynes with sodium hydrosulfide. RSC Adv. 2012, 2, 5488–5490. [Google Scholar] [CrossRef]
- Zhang, G.; Yi, H.; Chen, H.; Bian, C.; Liu, C.; Lei, A. Trisulfur radical anion as the key intermediate for the synthesis of thiophene via the interaction between elemental sulfur and NaOtBu. Org. Lett. 2014, 16, 6156–6159. [Google Scholar] [CrossRef]
- Rao, M.L.N.; Islam, S.K.; Dasgupta, P. Rapid access to unsymmetrical 1,3-diynes and 2,5-disubstituted thiophenes under ligand and Pd/Ni-free Cu-catalysis. RSC Adv. 2015, 5, 78090–78098. [Google Scholar] [CrossRef]
- Talbi, I.; Alayrac, C.; Lohier, J.-F.; Touil, S.; Witulski, B. Application of ynamides in the synthesis of 2-(tosylamido)- and 2,5-bis(tosylamido)thiophenes. Org. Lett. 2016, 18, 2656–2659. [Google Scholar] [CrossRef] [PubMed]
- Andrade, C.B.; Carvalho, D.B.; Trfzger, O.S.; Kassab, N.M.; Guerrero, P.G., Jr.; Barbosa, S.L.; Shiguemoto, C.Y.K.; Baroni, A.C.M. One-pot synthesis of unsymmetrical 1,3-butadiyne derivatives and their application in the synthesis of unsymmetrical 2,5-diarylthiophenes. Eur. J. Org. Chem. 2019, 2019, 696–704. [Google Scholar] [CrossRef]
- Curtis, R.F.; Hasnain, S.N.; Taylor, J.A. Selenophens from diacetylenes. Chem. Commun. 1968, 365a. [Google Scholar] [CrossRef]
- Barancelli, D.A.; Acker, C.I.; Menezes, P.H.; Zeni, G. Selective base-promoted synthesis of substituted selenophenes by carbocyclization of (Z)-benzylselenoenynes. Org. Biomol. Chem. 2011, 9, 1529–1537. [Google Scholar] [CrossRef] [PubMed]
- Mack, W. Synthesis of tellurophene and its 2,5-disubstituted derivatives. Angew. Chem. Int. Ed. 1966, 5, 896. [Google Scholar] [CrossRef]
- Ulman, A.; Manassen, J.; Frolow, F.; Rabinovich, D. Synthesis of new tetraphenylporphyrin molecules containing heteroatoms. III. Tetraphenyl-21-tellura-23-thiaprophyrin: An internally-bridged porphyrin. Tetrahedron Lett. 1978, 19, 1885–1886. [Google Scholar] [CrossRef]
- McCormick, T.M.; Jahnke, A.A.; Lough, A.J.; Seferos, D.S. Tellurophenes with delocalized π-systems and their extended valence adducts. J. Am. Chem. Soc. 2012, 134, 3542–3548. [Google Scholar] [CrossRef] [PubMed]
- Alves, D.; Sachini, M.; Jacob, R.G.; Lenardão, E.J.; Contreira, M.E.; Savegnano, L.; Perin, G. Synthesis of (Z)-organylthioenynes using KF/Al2O3/solvent as recyclable system. Tetrahedron Lett. 2011, 52, 133–135. [Google Scholar] [CrossRef] [Green Version]
- Santana, A.S.; Carvalho, D.B.; Casemiro, N.S.; Hurtado, G.R.; Viana, L.H.; Kassab, N.M.; Barbosa, S.L.; Marques, F.A.; Guerrero, P.G., Jr.; Baroni, A.C.M. Improvement in the synthesis of (Z)-organylthioenynes via hydrothiolation of buta-1,3-diynes: A comparative study using NaOH or TBAOH as base. Tetrahedron Lett. 2012, 53, 5733–5738. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Wu, J.; Li, H.; Sun, Q.; Xiong, L.; Yin, G. Highly regio- and stereoselective synthesis of bis-sulfanyl substituted conjugated dienes by copper-palladium cooperative catalysis. Org. Chem. Front. 2021, 8, 628–634. [Google Scholar] [CrossRef]
- Dabdoub, M.J.; Baroni, A.C.M.; Lenardão, E.J.; Gianeti, T.R.; Hurtado, G.R. Synthesis of (Z)-1-phenylseleno-1,4-diorganyl-1-buten-3-ynes: Hydroselenation of symmetrical and unsymmetrical 1,4-diorganyl-1,3-butadiynes. Tetrahedron 2001, 57, 4271–4276. [Google Scholar] [CrossRef]
- Alves, D.; Luchese, C.; Nogueira, C.W.; Zeni, G. Electrophilic cyclization of (Z)-selenoenynes: Synthesis and reactivity of 3-iodoselenophenes. J. Org. Chem. 2007, 72, 6726–6734. [Google Scholar] [CrossRef]
- Dabdoub, M.J.; Dabdoub, V.B.; Lenardão, E.J.; Hurtado, G.R.; Barbosa, S.L.; Guerrero, P.G., Jr.; Nazário, C.E.D.; Viana, L.H.; Santana, A.S.; Baroni, A.C.M. Synthesis of (Z)-1-organylthiobut-1-en-3-ynes: Hydrothiolation of symmetrical and unsymmetrical buta-1,3-diynes. Synlett 2009, 986–990. [Google Scholar] [CrossRef]
- Ishii, A.; Annaka, T.; Nakata, N. Convenient synthesis and photophysical properties of 1-thio- and 1-seleno-1,3-butadiene fluorophores in rigid dibenzobarrelene and benzobarrelene skeletons. Chem. Eur. J. 2012, 18, 6428–6432. [Google Scholar] [CrossRef] [PubMed]
- Venkateswarlu, C.; Chandrasekaran, S. Hydrochalcogenation of symmetrical and unsymmetrical buta-1,3-diynes with diaryl dichalcogenides: Facile entry to (Z)-1-(organylchalcogeno)but-1-en-3-yne derivatives. Synthesis 2015, 47, 395–410. [Google Scholar] [CrossRef]
- Lopes, E.F.; Gonçalves, L.C.; Vinueza, J.C.G.; Jacob, R.G.; Perin, G.; Santi, C.; Lenardão, E.J. DES as a green solvent to prepare 1,2-bis-organylseleno alkenes. Scope and limitations. Tetrahedron Lett. 2015, 56, 6890–6895. [Google Scholar] [CrossRef]
- Lara, R.G.; Soares, L.K.; Jacob, R.G.; Schumacher, R.F.; Perin, G. Selective synthesis of (Z)-chalcogenoenynes and (Z,Z)-1,4-bis-chalcogenbuta-1,3-dienes using PEG-400. J. Braz. Chem. Soc. 2016, 27, 2046–2054. [Google Scholar] [CrossRef]
- Profir´eva, Y.I.; Vasil´eva, L.A.; Turbanova, E.S.; Petrov, A.A. Electrophilic and radical addition of hydrogen halides to diacetylene and its homologs. Russ. J. Org. Chem. 1969, 5, 591–600. [Google Scholar]
- Radchenko, S.I. Reaction of alkylthioalkyldiacetylenes with hydrogen halides and alkyllithiums. Russ. J. Org. Chem. 1976, 12, 229–230. [Google Scholar]
- Radchenko, S.I.; Komarov, V.Y.; Ionin, B.I. Regioselectivity of addition of electrophilic reagents to conjugated acetylenic sulfides and selenides. Russ. J. Org. Chem. 1985, 21, 244–249. [Google Scholar]
- Ide, M.; Ohashi, K.; Mihara, S.; Iwasawa, T. Regio- and stereoselective hydrohalogenation of ynamide components in 1,3-butadiynes with in situ generated HX. Tetrahedron Lett. 2014, 55, 2130–2133. [Google Scholar] [CrossRef]
Scheme 1.
Metal-catalyzed inter- and intramolecular hydrofunctionalization of alkynes.

Figure 1.
The four reactive positions of conjugated 1,3-diynes.

Scheme 2.
Mono- and dihydroboration reactions of symmetrically alkyl-substituted 1,3-diynes.

Scheme 3.
Radical monohydroboration of 1,3-diynes with an N-heterocyclic carbene borane.

Scheme 4.
Proposed mechanism for the formation of the enynylboranates 7–8.

Scheme 5.
Copper-catalyzed hydroboration of 1,3-diynes.

Scheme 6.
Ruthenium-catalyzed hydroboration of 1,4-diaryl-substituted 1,3-diynes.

Scheme 7.
Cobalt-catalyzed ligand-controlled regio-divergent syn-hydroboration of a 1,3-diyne.

Scheme 8.
Proposed mechanisms for the cobalt-catalyzed hydroboration of PhC≡CC≡CSiMe3.

Scheme 9.
Gold(I)-catalyzed [4+3] annulation of the 1,3-diyne 14 with styrene derivatives.

Scheme 10.
Gold(I)-catalyzed synthesis of indoles and carbazoles from 1,3-diynes.

Figure 2.
Structures of dictyodendrins B, C, E, and F.

Scheme 11.
Gold(I)-catalyzed synthesis of the para-indolophane derivative 21.

Scheme 12.
Catalytic synthesis of 4,4′-bi(2H-chromenes), bi(2H-quinolines), and dioxafluoranthenes from 1,3-diynes.
Scheme 12.
Catalytic synthesis of 4,4′-bi(2H-chromenes), bi(2H-quinolines), and dioxafluoranthenes from 1,3-diynes.

Scheme 13.
Palladium-catalyzed hydrophenylation 1,4-bis(trimethylsilyl)buta-1,3-diyne.

Scheme 14.
Palladium-catalyzed homocoupling/hydroarylation of terminal alkynes with pyrazoles.

Scheme 15.
Proposed mechanism for the hydroarylation step involved in the tandem process leading to enynes 27.
Scheme 15.
Proposed mechanism for the hydroarylation step involved in the tandem process leading to enynes 27.

Scheme 16.
Mn(I)-catalyzed hydroarylation of 1,3-diynes 28 with arylboronic acids.

Figure 3.
Selected products generated by [MnBr(CO)5]-catalyzed hydrorarylation of 1,3-diynes with arenes and heteroarenes.
Figure 3.
Selected products generated by [MnBr(CO)5]-catalyzed hydrorarylation of 1,3-diynes with arenes and heteroarenes.

Scheme 17.
Mn(I)-catalyzed synthesis of furans and pyrroles by hydroarylation of 1,3-diynes.

Scheme 18.
Proposed mechanism for the hydroarylation of diynes with (hetero)arenes catalyzed by [MnBr(CO)5].
Scheme 18.
Proposed mechanism for the hydroarylation of diynes with (hetero)arenes catalyzed by [MnBr(CO)5].

Scheme 19.
Co-catalyzed hydroarylation of 1,3-diynes with picolinamides.

Figure 4.
Products resulting from the In-catalyzed addition of β-keto esters to 1,3-diynes.

Scheme 20.
Nickel-catalyzed regio-divergent hydrocyanation of silyl-substituted 1,3-butadiynes.

Scheme 21.
Pd-catalyzed hydroaminocarbonylation of 1,3-diynes with NH4Cl.

Scheme 22.
Pd-catalyzed hydroaminocarbonylation of 1,3-diynes with amines.

Figure 5.
The structure of some elaborated amides containing bio-relevant skeletons.

Scheme 23.
Pd-catalyzed monoalkoxycarbonylation of 1,3-diynes.

Scheme 24.
Pd-catalyzed access to tetrasubstituted dienes by alkoxycarbonylation of 1,3-diynes.

Scheme 25.
H2PtCl6-catalyzed hydrosilylation of 1,4-bis(trimethylsilyl)-1,3-butadiyne with dichloromethylsilane.
Scheme 25.
H2PtCl6-catalyzed hydrosilylation of 1,4-bis(trimethylsilyl)-1,3-butadiyne with dichloromethylsilane.

Figure 6.
Structure of compounds 42 and 43, and how 43 are formed from 42.

Figure 7.
Structure of the Karstedt´s catalyst 44 and the silylated compounds 45 and 46.

Scheme 26.
Synthesis of silicon-containing polymers by Pt-catalyzed bis-hydrosilylation of 1,3-diynes.
Scheme 26.
Synthesis of silicon-containing polymers by Pt-catalyzed bis-hydrosilylation of 1,3-diynes.

Scheme 27.
Rhodium-catalyzed hydrosilylation reactions of poly[(silylene)but-1,3-diynes] 48.

Scheme 28.
Hydrosilylation of 1,3-diyne catalyzed by the heterogeneous system Pt/TiO2.

Scheme 29.
Cobalt-catalyzed hydrosilylation of 1,3-diynes.

Scheme 30.
Proposed mechanism for the cobalt-catalyzed hydrosilylation of 1,3-diynes.

Figure 8.
Structure of the cobalt(II) complexes 51 and 52.

Scheme 31.
Ru-catalyzed synthesis of 2,5-disubstituted siloles by hydrosilylation of 1,3-diynes.

Scheme 32.
Ru-catalyzed hydrosilylation of the chiral diynol 14 with BnMe2SiH.

Scheme 33.
CuCl-catalyzed annulation reactions of 1,3-diynes with primary amines.

Figure 9.
Structure of the elaborated pyrrole derivatives 56–61.

Scheme 34.
Direct access to pyrroles from terminal alkynes and primary amines.

Scheme 35.
Gold-catalyzed synthesis of 2,5-amidopyrroles.

Scheme 36.
Gold-catalyzed synthesis of pyrroles from alkyl- and aryl-substituted 1,3-diynes.

Scheme 37.
Gold-catalyzed hydroamination reactions of 1,4-diphenylbuta-1,3-diyne with ammonia and hydrazine.
Scheme 37.
Gold-catalyzed hydroamination reactions of 1,4-diphenylbuta-1,3-diyne with ammonia and hydrazine.

Scheme 38.
TiCl4/tBuNH2-catalyzed pyrrole synthesis from dodeca-5,7-diyne and aromatic primary amines.
Scheme 38.
TiCl4/tBuNH2-catalyzed pyrrole synthesis from dodeca-5,7-diyne and aromatic primary amines.

Scheme 39.
Cu-catalyzed hydroamination and hydroamidation reactions of 1,3-diynes with indoles, azoles, and amides.
Scheme 39.
Cu-catalyzed hydroamination and hydroamidation reactions of 1,3-diynes with indoles, azoles, and amides.

Scheme 40.
Access to diamino-substituted naphthalenes from 1,3-diynes and cyclic amines.

Scheme 41.
Hydroamination 1,4-diphenyl-1,3-butadiyne with secondary amines catalyzed by heterobimetallic amides.
Scheme 41.
Hydroamination 1,4-diphenyl-1,3-butadiyne with secondary amines catalyzed by heterobimetallic amides.

Scheme 42.
Double hydroamination of 1,4-diphenyl-1,3-butadiyne with N-methylanilines.

Scheme 43.
Access to pyrrole derivatives using the calciate catalyst [K2Ca{N(H)Dipp}4].

Figure 10.
Other products generated from 1,4-diphenyl-1,3-butadiyne and anilines using the calciate catalyst [K2Ca{N(H)Dipp}4].
Figure 10.
Other products generated from 1,4-diphenyl-1,3-butadiyne and anilines using the calciate catalyst [K2Ca{N(H)Dipp}4].

Figure 11.
Structure of compound 73 generated from N,N′-diphenylethylenediamine and 1,4-diphenyl-1,3-butadiyne.
Figure 11.
Structure of compound 73 generated from N,N′-diphenylethylenediamine and 1,4-diphenyl-1,3-butadiyne.

Scheme 44.
Base-mediated intramolecular hydroamination of 1,3-diynes.

Scheme 45.
Access to naphthol-indoles 81 from 1,3-diynes 78 and the sulfoxonium ylide 79.

Scheme 46.
Au-catalyzed conversion of diyne 75 into tetraindole 82.

Scheme 47.
Gold-catalyzed synthesis of pyrimido[1,6-a]indol-1(2H)-one derivatives.

Scheme 48.
Gold-catalyzed hydroamination/hydroalkoxylation cyclization cascade of diynes 85.

Scheme 49.
LiPh-catalyzed synthesis of 2,5-disubstituted phospholes from 1,3-diynes and PhPH2.

Figure 12.
Structure of the 2,5-disubstituted phospholes 89–92.

Scheme 50.
Base-catalyzed double hydrophosphination of diyne 93 with Ph2PH.

Scheme 51.
Ytterbium-catalyzed double hydrophosphination reactions of diynes with Ph2PH.

Scheme 52.
Intermediate species involved in the Yb-catalyzed double hydrophosphination reactions of diynes.
Scheme 52.
Intermediate species involved in the Yb-catalyzed double hydrophosphination reactions of diynes.

Scheme 53.
Ytterbium-catalyzed single hydrophosphination reactions of diynes with Ph2PH.

Figure 13.
Products formed in the [Ca(THF)4(PPh2)2]-catalyzed hydrophosphination of 1,3-diynes.

Scheme 54.
[Ca(THF)4(PPh2)2]-catalyzed hydrophosphorylation of 1,3-diynes with Ph2P(= O)H.

Figure 14.
Structure of the 1,3-butadiene derivatives 104.

Scheme 55.
Copper-catalyzed hydration of 1,3-diynes.

Scheme 56.
Copper-catalyzed synthesis of 2,5-disubstituted furans from propiolic acids.

Scheme 57.
Gold-catalyzed synthesis of the 2,5-diamidofurans 106.

Scheme 58.
Hydration of 1,3-diynes catalyzed by an NHC-gold(I) complex.

Scheme 59.
Gold-catalyzed synthesis of the 2,5-diamidofurans 106.

Scheme 60.
Gold-catalyzed synthesis of 2,5-disubstituted furans from bromoalkynes.

Scheme 61.
Pd(0)-catalyzed intermolecular hydroalkoxylation of 1,3-diynes.

Scheme 62.
Gold-catalyzed synthesis of bis-spiroketals from diynediol derivatives.

Scheme 63.
Mechanism for the conversion of 5,7-diyne-1,12-diol into the bis-spiroketal isomers.

Figure 15.
Structure of the steroid-derived bis-spiroketals 113–115.

Scheme 64.
Base-promoted cycloisomerization of hydroxyl-containing 1,3-diynes.

Scheme 65.
Synthesis of 2,5-disubstituted furan derivatives from diynyl-1,6-diols.

Scheme 66.
Synthesis of 2-alkynylbenzofuran derivatives from 1,3-diyne 118.

Scheme 67.
Gold-catalyzed addition of acetic acid to 2,4-hexadiyne.

Scheme 68.
Copper-catalyzed synthesis of 2,5-disubstituted thiophenes from bromoalkynes.

Scheme 69.
Base-promoted hydrochalcogenation reactions of 1,3-diynes.

Scheme 70.
CuCl-catalyzed hydrochlorination of MeSC≡CC≡CnPr.

Scheme 71.
Hydrohalogenation of ynamide-containing 1,3-diynes with in situ generated HX.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Cadierno, V. Catalytic Hydrofunctionalization Reactions of 1,3-Diynes. Catalysts 2022, 12, 89. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12010089
AMA Style
Cadierno V. Catalytic Hydrofunctionalization Reactions of 1,3-Diynes. Catalysts. 2022; 12(1):89. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12010089
Chicago/Turabian StyleCadierno, Victorio. 2022. "Catalytic Hydrofunctionalization Reactions of 1,3-Diynes" Catalysts 12, no. 1: 89. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12010089
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.