
Effect of Binding Modules Fused to Cutinase on the Enzymatic Synthesis of Polyesters

 , , , , and
, , , , and
Abstract
:1. Introduction
2. Results
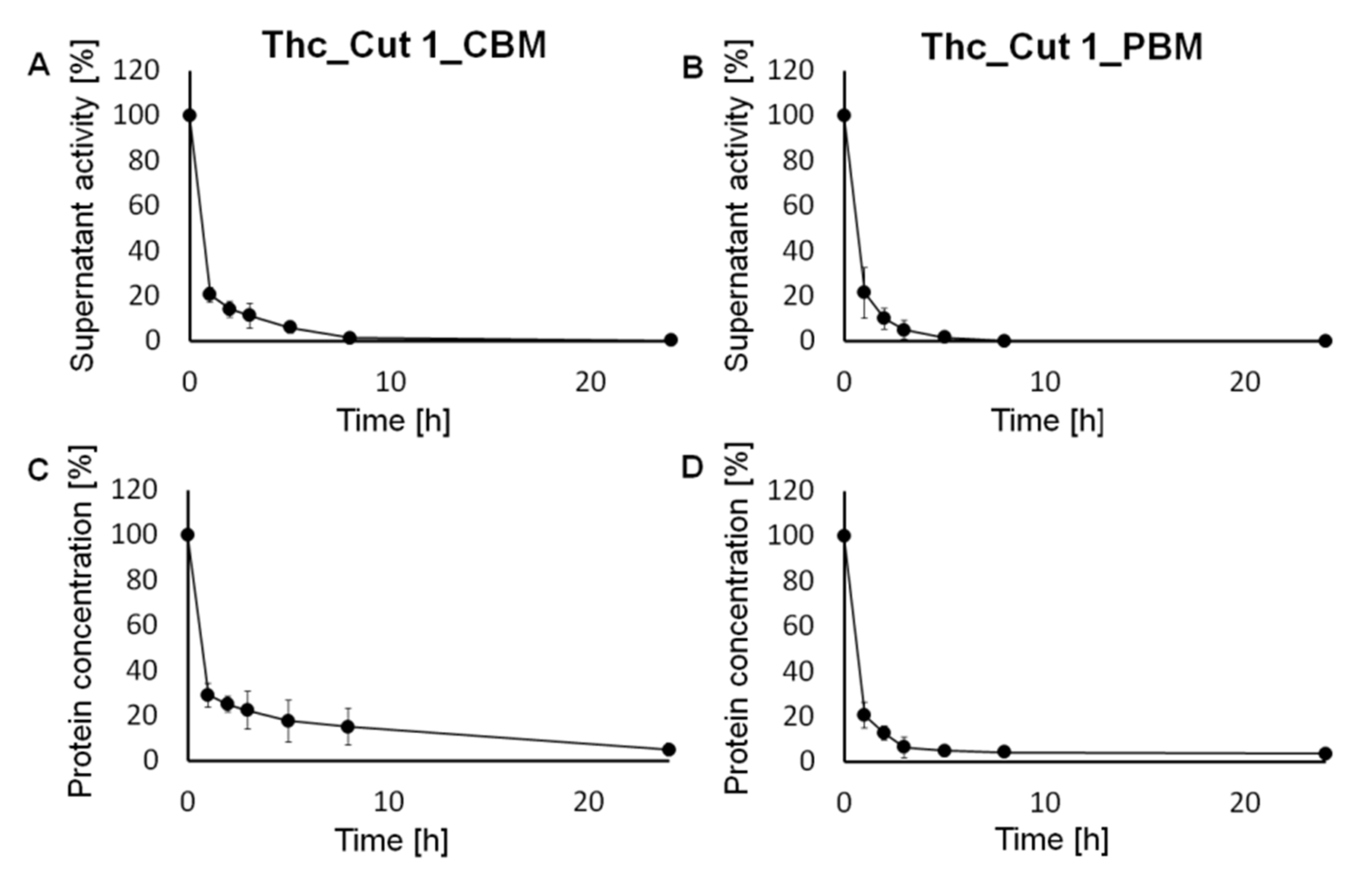
2.1. Covalent Immobilization of the Biocatalyst
2.2. Polycondensation Reactions in Bulk Systems
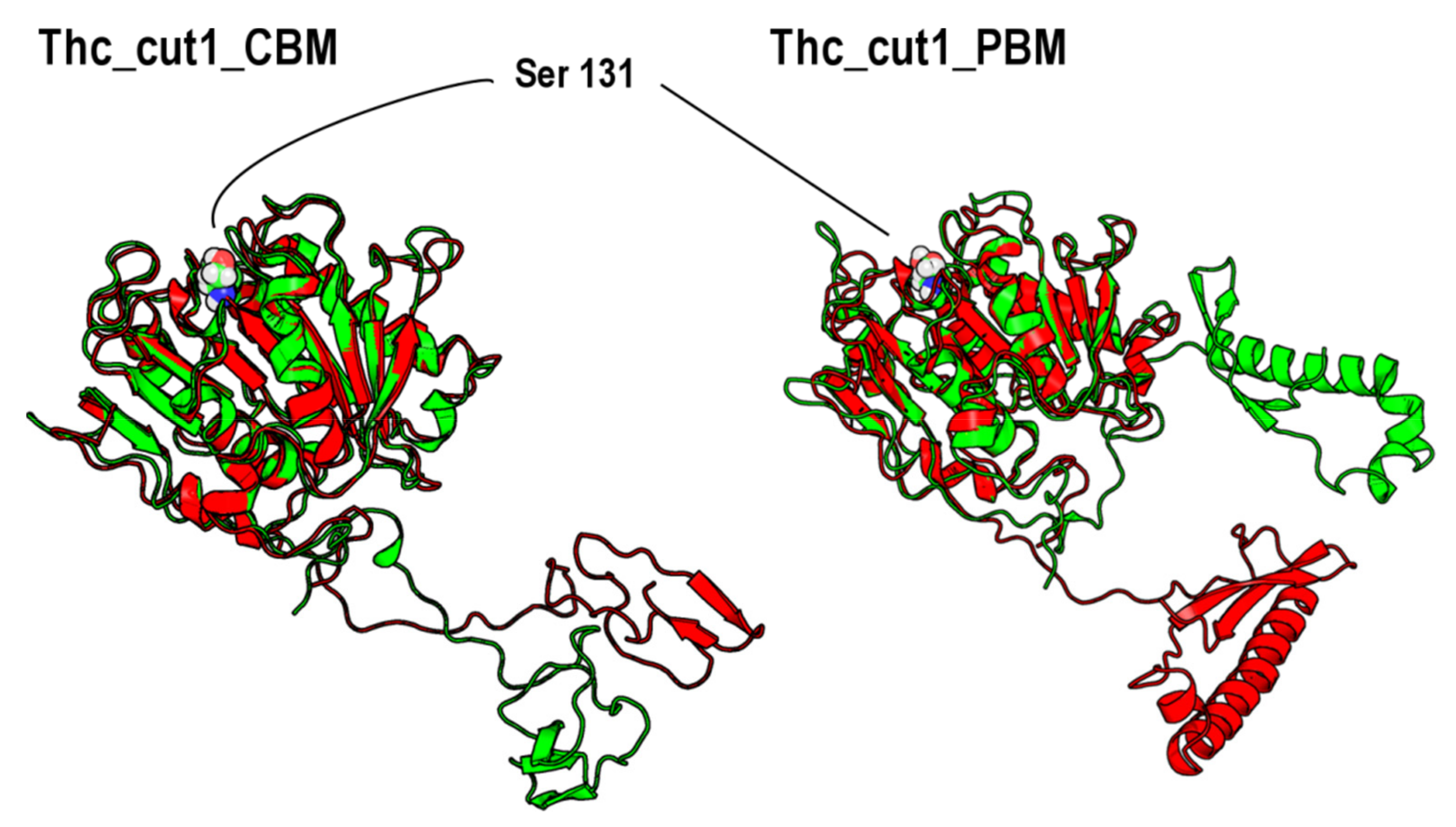
2.3. Enzyme Modelling & Surface Analysis
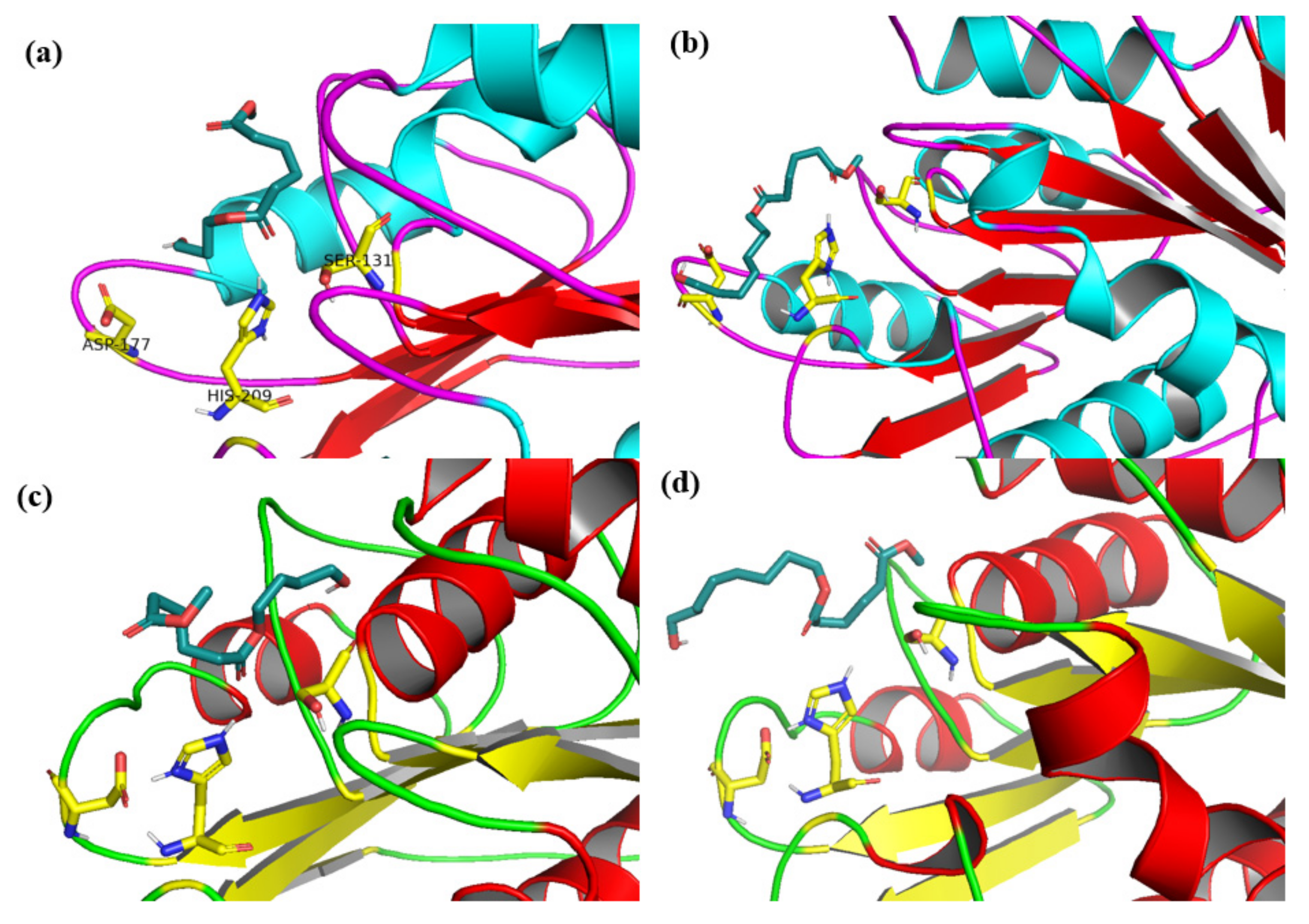
2.4. Docking Analysis
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Enzymes
3.3. Activity Assay for Free and Immobilized Cutinases
3.4. Protein Quantification
3.5. Immobilization of Thc_Cut1_CBM and Thc_Cut1_PBM on Epoxy-Activated Beads
3.6. Enzymatic Polycondensation of DMA and Various Diols
3.7. GPC
3.8. 1H-NMR
3.9. Enzyme Structures
3.10. Molecular Dynamic Simulations
3.11. Surface Analysis
3.12. Docking Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding

Data Availability Statement
Conflicts of Interest
References
- De María, P.D. Biocatalysis, sustainability, and industrial applications: Show me the metrics. Curr. Opin. Green Sustain. Chem. 2021, 31, 100514. [Google Scholar] [CrossRef]
- Steiner, K.; Schwab, H. Recent advances in rational approaches for enzyme engineering. Comput. Struct. Biotechnol. J. 2012, 2, e201209010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pellis, A.; Acero, E.H.; Ferrario, V.; Ribitsch, D.; Guebitz, G.M.; Gardossi, L. The Closure of the Cycle: Enzymatic Synthesis and Functionalization of Bio-Based Polyesters. Trends Biotechnol. 2016, 34, 316–328. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, R.A.; Brady, D. Green Chemistry, Biocatalysis, and the Chemical Industry of the Future. ChemSusChem 2022, 202102628. [Google Scholar] [CrossRef] [PubMed]
- Pellis, A.; Acero, E.H.; Weber, H.; Obersriebnig, M.; Breinbauer, R.; Srebotnik, E.; Guebitz, G.M. Biocatalyzed approach for the surface functionalization of poly(L-lactic acid) films using hydrolytic enzymes. Biotechnol. J. 2015, 10, 1739–1749. [Google Scholar] [CrossRef]
- Nyanhongo, G.S.; Rodríguez, R.D.; Prasetyo, E.N.; Caparrós, C.; Ribeiro, C.; Sencadas, V.; Lanceros-Mendez, S.; Acero, E.H.; Guebitz, G.M. Bioactive albumin functionalized polylactic acid membranes for improved biocompatibility. React. Funct. Polym. 2013, 73, 1399–1404. [Google Scholar] [CrossRef] [Green Version]
- Acero, E.H.; Ribitsch, D.; Steinkellner, G.; Gruber, K.; Greimel, K.; Eiteljoerg, I.; Trotscha, E.; Wei, R.; Zimmermann, W.; Zinn, M.; et al. Enzymatic Surface Hydrolysis of PET: Effect of Structural Diversity on Kinetic Properties of Cutinases from Thermobifida. Macromolecules 2011, 44, 4632–4640. [Google Scholar] [CrossRef] [Green Version]
- Barth, M.; Oeser, T.; Wei, R.; Then, J.; Schmidt, J.; Zimmermann, W. Effect of hydrolysis products on the enzymatic degradation of polyethylene terephthalate nanoparticles by a polyester hydrolase from Thermobifida fusca. Biochem. Eng. J. 2015, 93, 222–228. [Google Scholar] [CrossRef]
- Pellis, A.; Haernvall, K.; Pichler, C.M.; Ghazaryan, G.; Breinbauer, R.; Guebitz, G.M. Enzymatic hydrolysis of poly(ethylene furanoate). J. Biotechnol. 2016, 235, 47–53. [Google Scholar] [CrossRef]
- Feder, D.; Gross, R.A. Exploring Chain Length Selectivity in HIC-Catalyzed Polycondensation Reactions. Biomacromolecules 2010, 11, 690–697. [Google Scholar] [CrossRef]
- Hunsen, M.; Abul, A.; Xie, W.; Gross, R. Humicola insolens cutinase-catalyzed lactone ring-opening polymerizations: Kinetic and mechanistic studies. Biomacromolecules 2008, 9, 518–522. [Google Scholar] [CrossRef] [PubMed]
- Pellis, A.; Ferrario, V.; Zartl, B.; Brandauer, M.; Gamerith, C.; Acero, E.H.; Ebert, C.; Gardossi, L.; Guebitz, G.M. Enlarging the tools for efficient enzymatic polycondensation: Structural and catalytic features of cutinase 1 from: Thermobifida cellulosilytica. Catal. Sci. Technol. 2016, 6, 3430–3442. [Google Scholar] [CrossRef] [Green Version]
- Bloom, J.D.; Meyer, M.M.; Meinhold, P.; Otey, C.R.; MacMillan, D.; Arnold, F.H. Evolving strategies for enzyme engineering. Curr. Opin. Struct. Biol. 2005, 15, 447–452. [Google Scholar] [CrossRef] [PubMed]
- Then, J.; Wei, R.; Oeser, T.; Barth, M.; Belisário-Ferrari, M.R.; Schmidt, J.; Zimmermann, W. Ca2+ and Mg2+ binding site engineering increases the degradation of polyethylene terephthalate films by polyester hydrolases from Thermobifida fusca. Biotechnol. J. 2015, 10, 592–598. [Google Scholar] [CrossRef] [PubMed]
- Wei, R.; Oeser, T.; Schmidt, J.; Meier, R.; Barth, M.; Then, J.; Zimmermann, W. Engineered bacterial polyester hydrolases efficiently degrade polyethylene terephthalate due to relieved product inhibition. Biotechnol. Bioeng. 2016, 113, 1658–1665. [Google Scholar] [CrossRef]
- Roth, C.; Wei, R.; Oeser, T.; Then, J.; Föllner, C.; Zimmermann, W.; Sträter, N. Structural and functional studies on a thermostable polyethylene terephthalate degrading hydrolase from Thermobifida fusca. Appl. Microbiol. Biotechnol. 2014, 98, 7815–7823. [Google Scholar] [CrossRef]
- Herrero Acero, E.; Ribitsch, D.; Dellacher, A.; Zitzenbacher, S.; Marold, A.; Steinkellner, G.; Gruber, K.; Schwab, H.; Guebitz, G.M. Surface engineering of a cutinase from Thermobifida Cellulosilytica for improved polyester hydrolysis. Biotechnol. Bioeng. 2013, 110, 2581–2590. [Google Scholar] [CrossRef]
- Ribitsch, D.; Yebra, A.O.; Zitzenbacher, S.; Wu, J.; Nowitsch, S.; Steinkellner, G.; Greimel, K.; Doliska, A.; Oberdorfer, G.; Gruber, C.C.; et al. Fusion of Binding Domains to Thermobifida cellulosilytica Cutinase to Tune Sorption Characteristics and Enhancing PET Hydrolysis. Biomacromolecules 2013, 14, 1769–1776. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, S.; Xu, M.; Cavoco-Paulo, A.P.; Wu, J.; Chen, J. Characterization of thermobifida fusca cutinase-carbohydrate-binding module fusion proteins and their potential application in bioscouring. Appl. Environ. Microbiol. 2010, 76, 6870–6876. [Google Scholar] [CrossRef] [Green Version]
- Ribitsch, D.; Acero, E.H.; Przylucka, A.; Zitzenbacher, S.; Marold, A.; Gamerith, C.; Tscheließnig, R.; Jungbauer, A.; Rennhofer, H.; Lichtenegger, H.; et al. Enhanced cutinase-catalyzed hydrolysis of polyethylene terephthalate by covalent fusion to hydrophobins. Appl. Environ. Microbiol. 2015, 81, 3586–3592. [Google Scholar] [CrossRef] [Green Version]
- Pellis, A.; Guarneri, A.; Brandauer, M.; Acero, E.H.; Peerlings, H.; Gardossi, L.; Guebitz, G.M. Exploring mild enzymatic sustainable routes for the synthesis of bio-degradable aromatic-aliphatic oligoesters. Biotechnol. J. 2016, 11, 642–647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takwa, M.; Larsen, M.W.; Hult, K.; Martinelle, M. Rational redesign of Candida antarctica lipase B for the ring opening polymerization of d,d-lactide. Chem. Commun. 2011, 47, 7392–7394. [Google Scholar] [CrossRef] [PubMed]
- Cantone, S.; Ferrario, V.; Corici, L.; Ebert, C.; Fattor, D.; Spizzo, P.; Gardossi, L. Efficient immobilisation of industrial biocatalysts: Criteria and constraints for the selection of organic polymeric carriers and immobilisation methods. Chem. Soc. Rev. 2013, 42, 6262–6276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferrario, V.; Ebert, C.; Knapic, L.; Fattor, D.; Basso, A.; Spizzo, P.; Gardossi, L. Conformational Changes of Lipases in Aqueous Media: A Comparative Computational Study and Experimental Implications. Adv. Synth. Catal. 2011, 353, 2466–2480. [Google Scholar] [CrossRef]
- Pellis, A.; Corici, L.; Sinigoi, L.; D’Amelio, N.; Fattor, D.; Ferrario, V.; Ebert, C.; Gardossi, L. Towards feasible and scalable solvent-free enzymatic polycondensations: Integrating robust biocatalysts with thin film reactions. Green Chem. 2015, 17, 1756–1766. [Google Scholar] [CrossRef] [Green Version]
- Kaufmann, K.W.; Lemmon, G.H.; Deluca, S.L.; Sheehan, J.H.; Meiler, J. Practically Useful: What the Rosetta Protein Modeling Suite Can Do for You. Biochemistry 2010, 49, 2987–2998. [Google Scholar] [CrossRef]
- Kim, D.E.; Chivian, D.; Baker, D. Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 2004, 32, W526–W531. [Google Scholar] [CrossRef] [Green Version]
- Ferrario, V.; Pleiss, J. Simulation of protein diffusion: A sensitive probe of protein–solvent interactions. J. Biomol. Struct. Dyn. 2019, 37, 1534–1544. [Google Scholar] [CrossRef]
- Hess, B.; Kutzner, C.; van der Spoel, D.; Lindahl, E. GROMACS 4: Algorithms for Highly Efficient, Load-Balanced, and Scalable Molecular Simulation. J. Chem. Theory Comput. 2008, 4, 435–447. [Google Scholar] [CrossRef] [Green Version]
- Goodford, P.J. A Computational Procedure for Determining Energetically Favorable Binding Sites on Biologically Important Macromolecules. J. Med. Chem. 1985, 28, 849–857. [Google Scholar] [CrossRef]
- Morris, G.M.; Ruth, H.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolinsky, T.J.; Nielsen, J.E.; McCammon, J.A.; Baker, N.A. PDB2PQR: An automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004, 32, W665–W667. [Google Scholar] [CrossRef] [PubMed]
- Søndergaard, C.R.; Olsson, M.H.M.; Rostkowski, M.; Jensen, J.H. Improved Treatment of Ligands and Coupling Effects in Empirical Calculation and Rationalization of pKa Values. J. Chem. Theory Comput. 2011, 7, 2284–2295. [Google Scholar] [CrossRef] [PubMed]
- Kaminski, G.A.; Friesner, R.A.; Tirado-Rives, J.; Jorgensen, W.L. Evaluation and Reparametrization of the OPLS-AA Force Field for Proteins via Comparison with Accurate Quantum Chemical Calculations on Peptides. J. Phys. Chem. B 2001, 105, 6474–6487. [Google Scholar] [CrossRef]
- Jorgensen, W.L.; Chandrasekhar, J.; Madura, J.D.; Impey, R.W.; Klein, M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983, 79, 926. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577. [Google Scholar] [CrossRef] [Green Version]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef] [Green Version]
- Berendsen, H.J.C.; Postma, J.P.M.; van Gunsteren, W.F.; DiNola, A.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684. [Google Scholar] [CrossRef] [Green Version]
- Stewart, J.J.P. Optimization of parameters for semiempirical methods V: Modification of NDDO approximations and application to 70 elements. J. Mol. Model. 2007, 13, 1173–1213. [Google Scholar] [CrossRef] [Green Version]
- DeLano, W.L. The PyMOL Molecular Graphics System; Delano Scientific, S.C., Ed.; 2002; Available online: https://pymol.org/2/ (accessed on 3 February 2022).







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme | Bound Enzyme (%) | Ref. | |
|---|---|---|---|
| Thc_Cut1 | >99 * | >99 δ | [12] |
| Thc_Cut1_PBM | >99 * | 97 δ | This work |
| Thc_Cut1_CBM | >99 * | 95 δ | |
| Preparation | Diol | Conversion [%] ʎ | Mw * | Mn * | PD * |
|---|---|---|---|---|---|
| iCBM | BDO | 56 | 300 | 200 | 1.44 |
| ODO | 21 | 300 | 300 | 1.13 | |
| iPBM | BDO | 56 | 500 | 500 | 1.11 |
| ODO | 21 | 400 | 300 | 1.20 |
| Preparation | Diol | Conversion [%] ʎ | Mw * | Mn * | Ð * |
|---|---|---|---|---|---|
| Thc_Cut1_CBM | BDO | 67 | 700 | 600 | 1.29 |
| ODO | 60 | 1500 | 1000 | 1.59 | |
| Thc_Cut1_PBM | BDO | 68 | 900 | 700 | 1.37 |
| ODO | 74 | 1600 | 1000 | 1.53 |
| Mutant | Substrate | E*abs [kcal/mol] | Clustering |
|---|---|---|---|
| Thc_Cut1_PBM | BDO-DMA | −3.34 | 53 |
| ODO-DMA | −4.04 | 29 | |
| Thc_Cut1_CBM | BDO-DMA | −3.36 | 12 |
| ODO-DMA | −2.22 | 39 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferrario, V.; Todea, A.; Wolansky, L.; Piovesan, N.; Guarneri, A.; Ribitsch, D.; Guebitz, G.M.; Gardossi, L.; Pellis, A. Effect of Binding Modules Fused to Cutinase on the Enzymatic Synthesis of Polyesters. Catalysts 2022, 12, 303. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12030303
Ferrario V, Todea A, Wolansky L, Piovesan N, Guarneri A, Ribitsch D, Guebitz GM, Gardossi L, Pellis A. Effect of Binding Modules Fused to Cutinase on the Enzymatic Synthesis of Polyesters. Catalysts. 2022; 12(3):303. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12030303
Chicago/Turabian StyleFerrario, Valerio, Anamaria Todea, Lisa Wolansky, Nicola Piovesan, Alice Guarneri, Doris Ribitsch, Georg M. Guebitz, Lucia Gardossi, and Alessandro Pellis. 2022. "Effect of Binding Modules Fused to Cutinase on the Enzymatic Synthesis of Polyesters" Catalysts 12, no. 3: 303. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12030303