
Paired Electrolysis of Acrylonitrile and 5-Hydroxymethylfurfural for Simultaneous Generation of Adiponitrile and 2,5-Furandicarboxylic Acid


Abstract
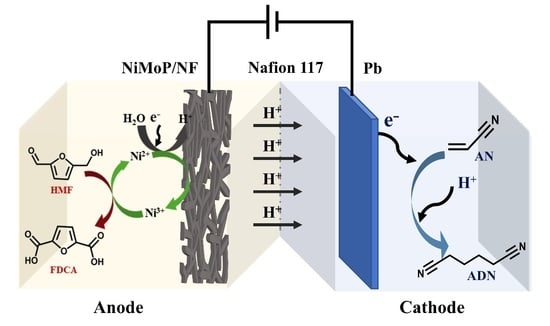
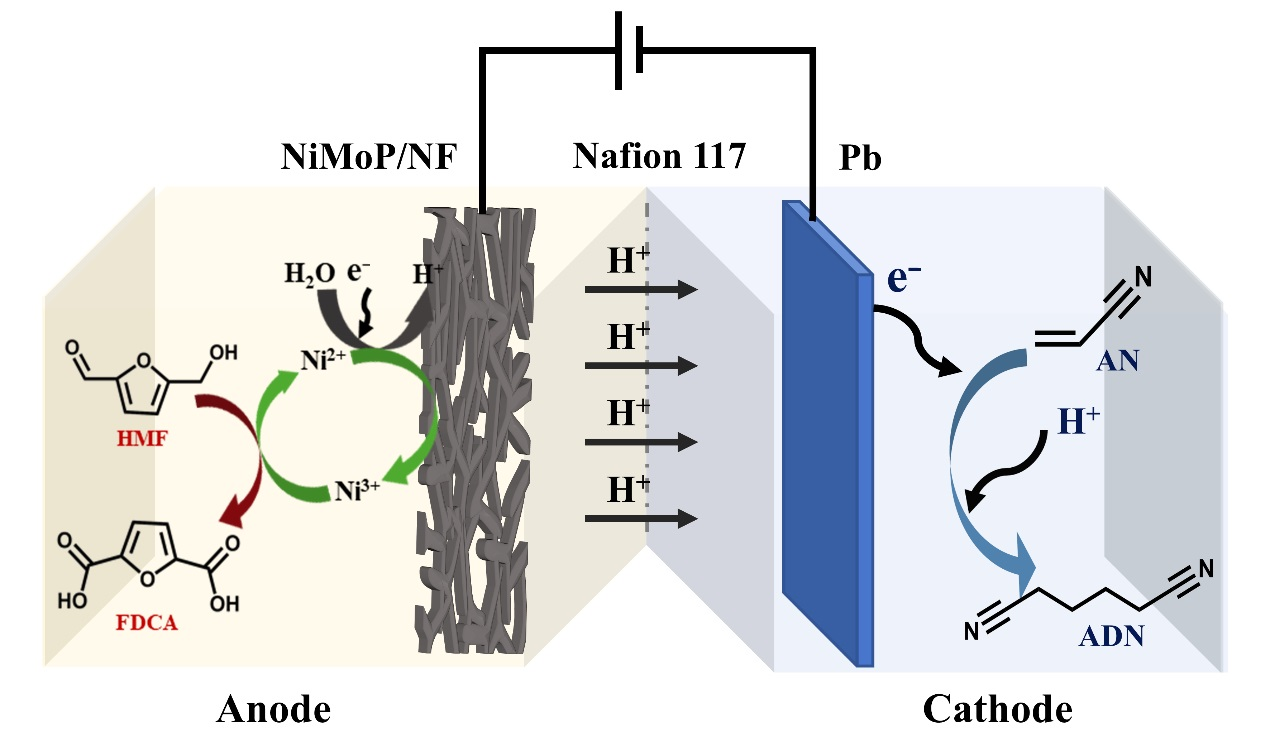
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
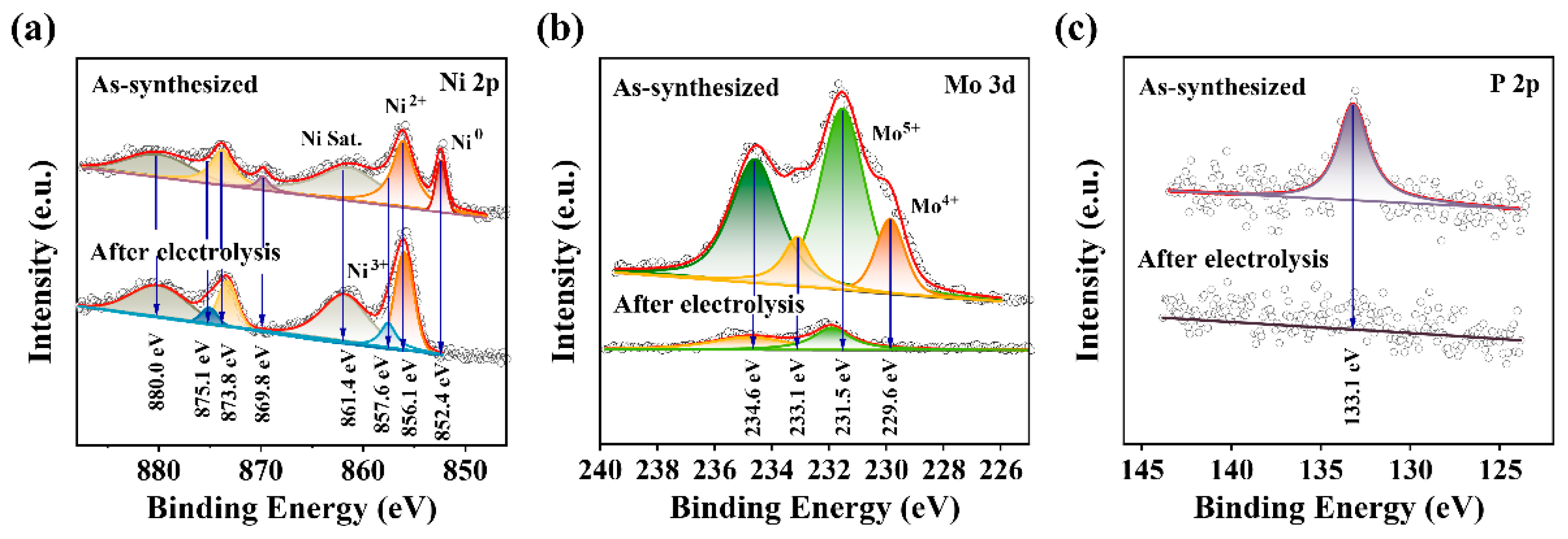
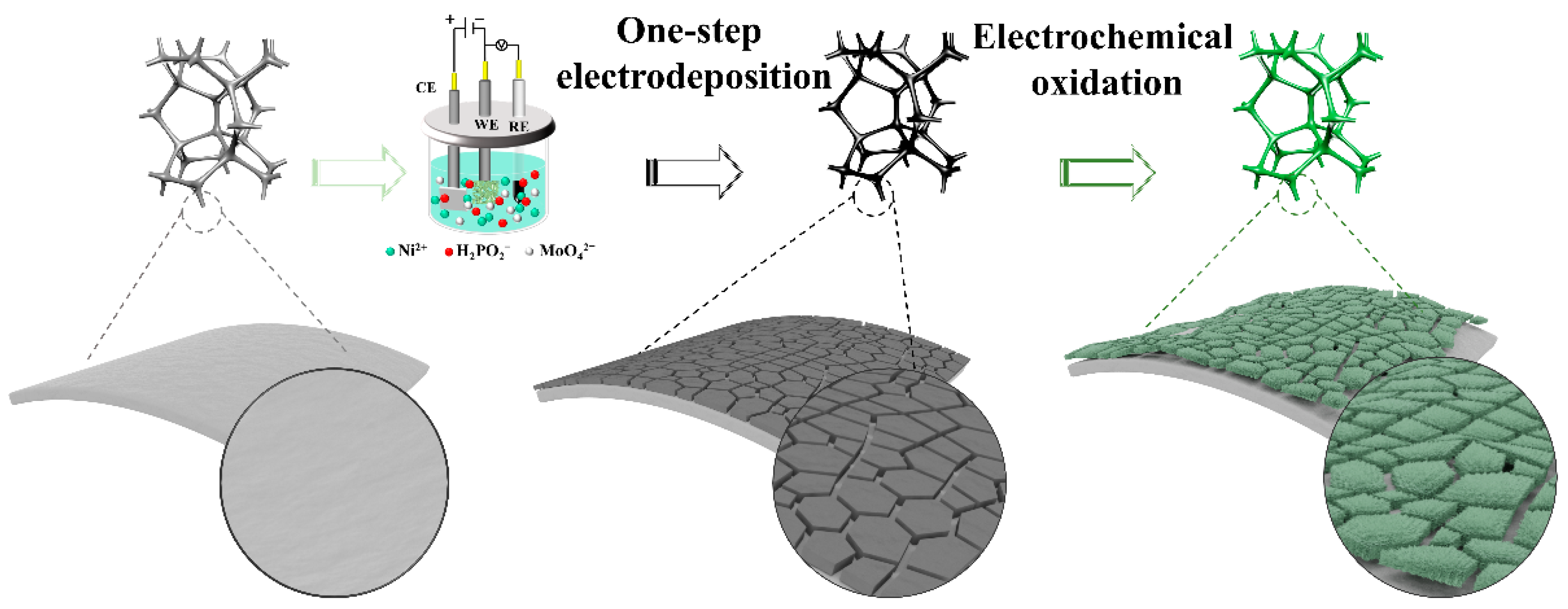
2.1. Preparation and Characterization of NiMoP/NF
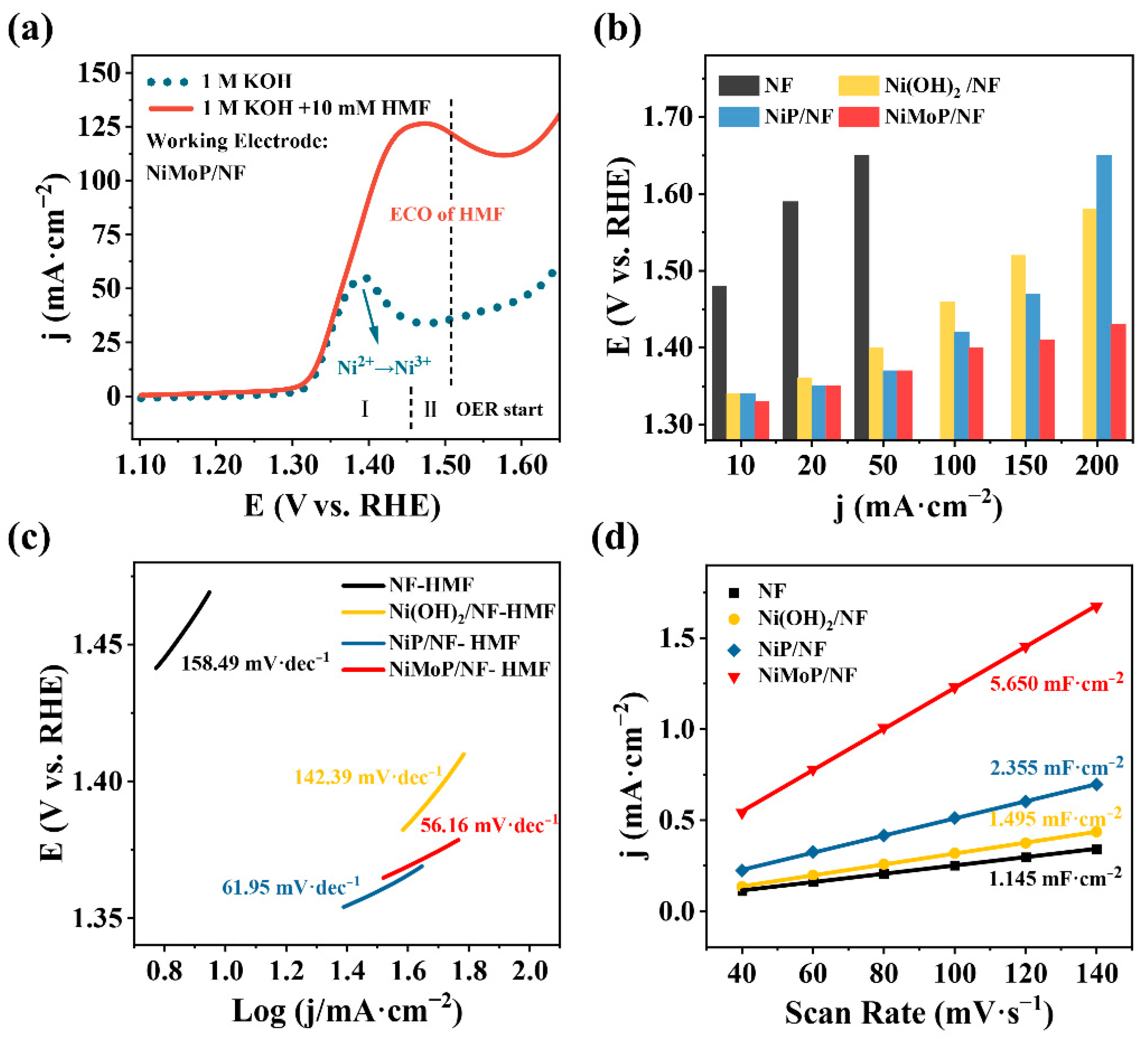
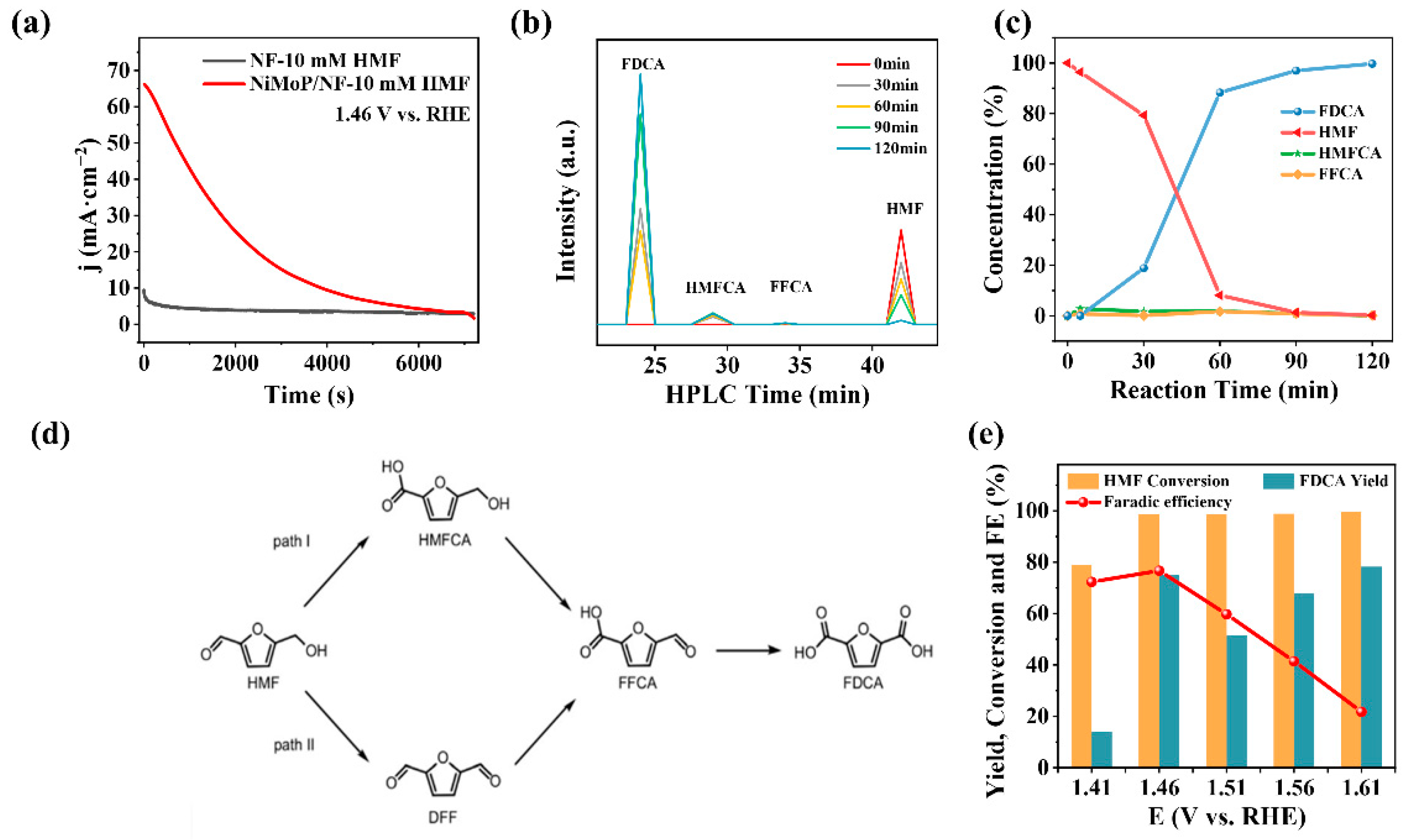
2.2. Electrocatalytic HMF Oxidation Performance of NiMoP/NF
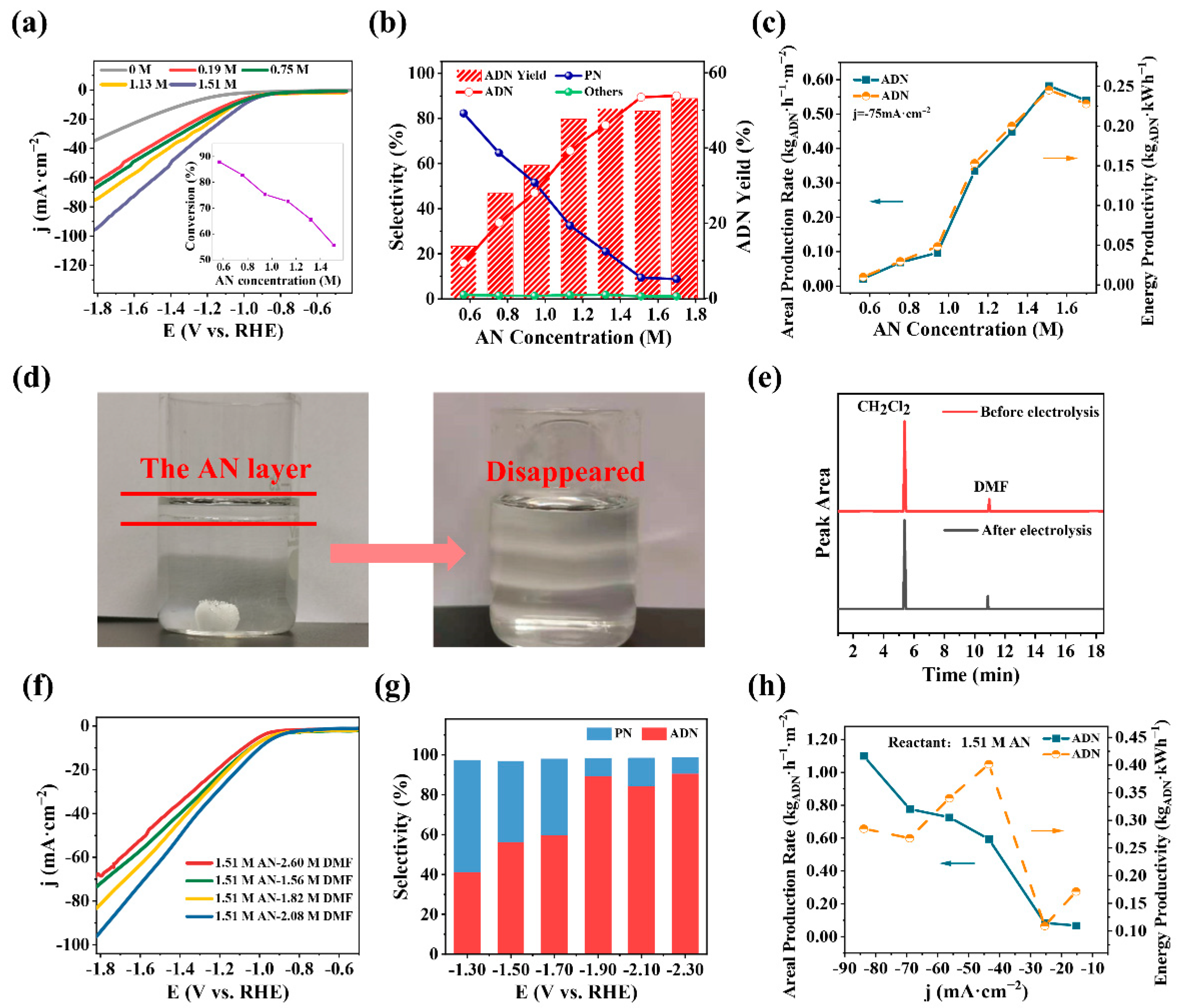
2.3. Electrohydrodimerization (EHD) of AN
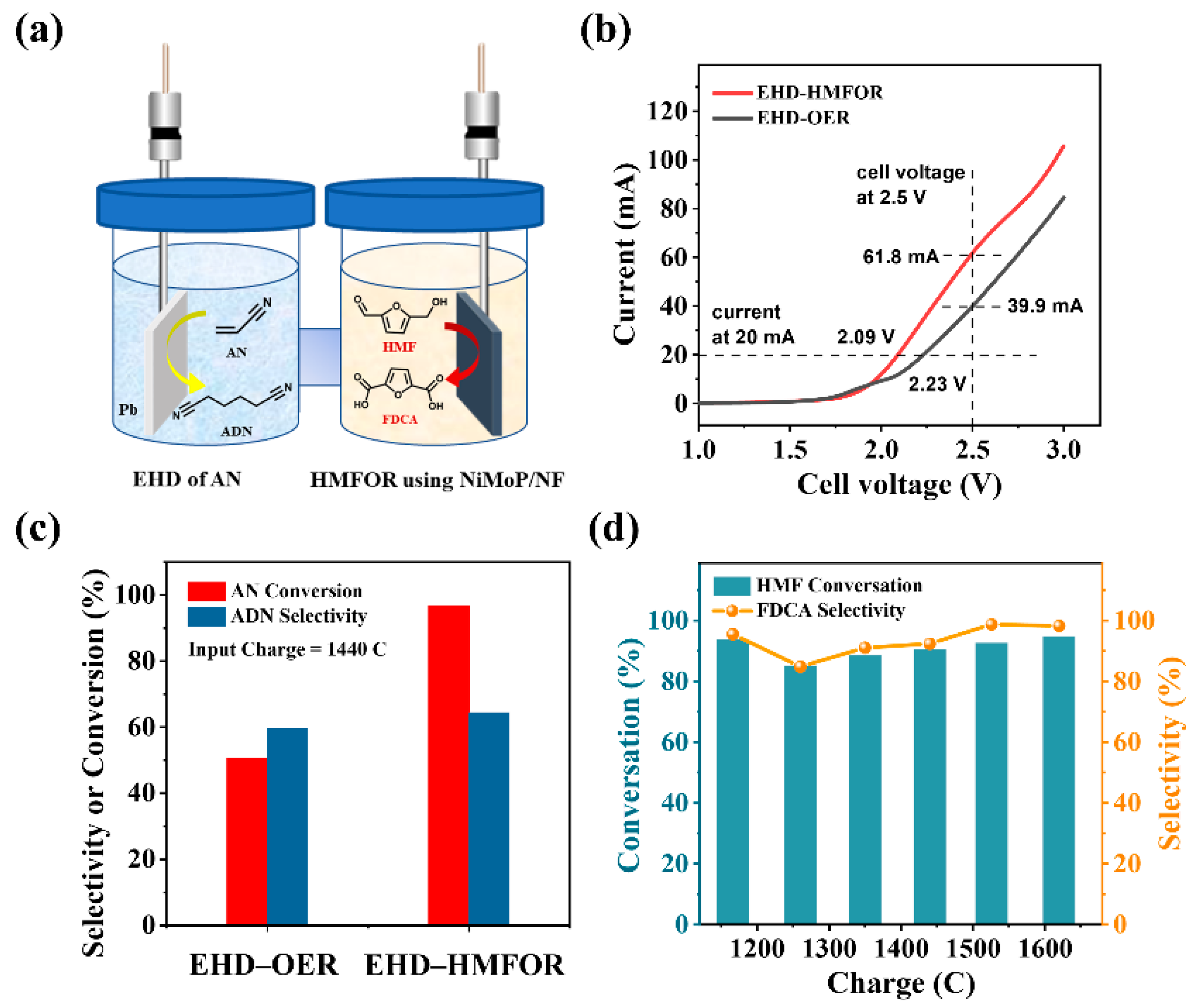
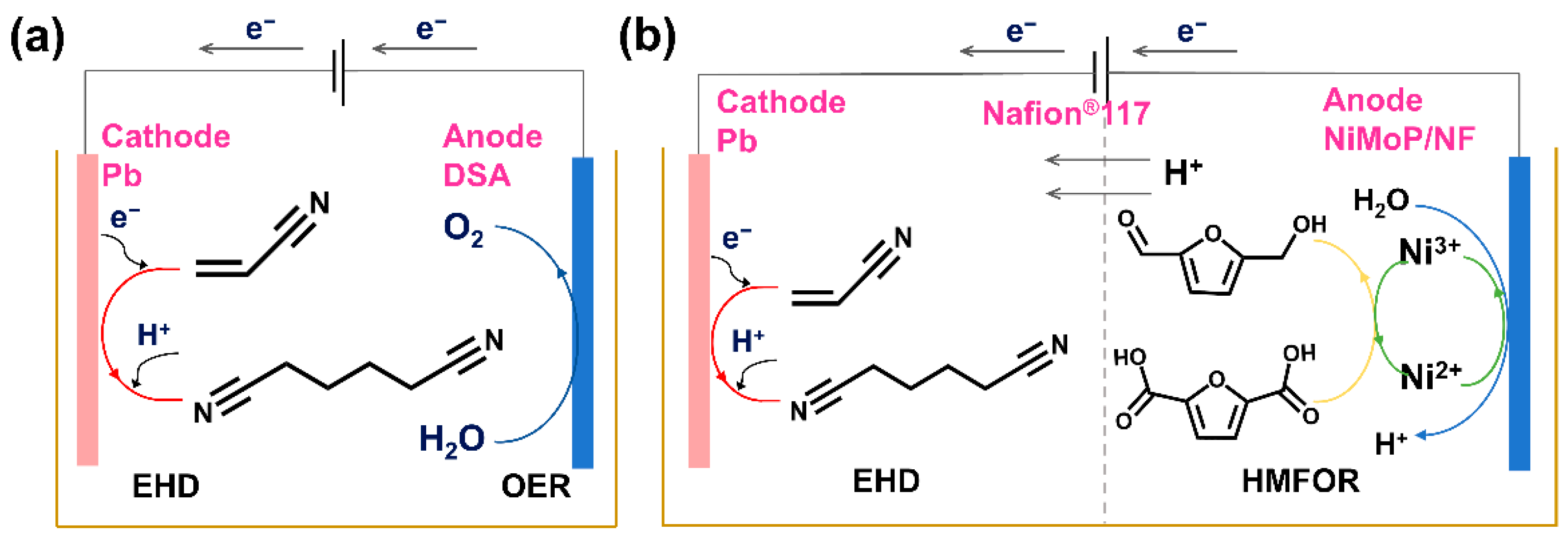
2.4. Paired Electrolysis of AN and HMF
3. Materials and Methods
3.1. Chemicals and Materials
3.2. Catalyst Synthesis Method
3.2.1. Synthesis of NiMoP/NF and NiP/NF
3.2.2. Synthesis of Ni(OH)2/NF Hydrothermal Method
3.3. Physical Characterization
3.4. Evaluation of Electrochemical Reactions
3.4.1. Anode Reaction—Electrocatalytic HMF Oxidation Reaction
- The electrochemical characterization of anode materials. The CV and LSV tests were performed in a three-electrode cell with a 25 mL electrolyte at room temperature. The as-prepared catalytic materials (1 cm × 1 cm), Pt sheet (1 cm × 1 cm), and Hg/HgO electrode (filled with 1.0 M KOH) acted as WE, CE and RE, respectively. The LSV experiments regarding electrocatalytic HMFOR and OER were conducted in a KOH electrolyte with and without HMF at a scan rate of 10 mV·s−1 within a 1.09–1.65 V (vs. RHE) anode potential window. To evaluate the ECSA of these monolithic catalysts, CV tests were conducted in the non-Faradaic region from 0.97 to 1.07 (vs. RHE) at 40, 60, 80, 100, 120, and 140 mV·s−1. CV tests were also employed to study HMFOR and OER by choosing a 1.05–1.60 V scan range at a 10 mV·s−1 scan rate for 10 cycles.
- The product distribution analysis of electrocatalytic HMFOR catalyzed by NiMoP/NF anode. In the three-electrode system, the product distribution analysis was carried out with CA tests in a 25 mL 1 M KOH electrolyte containing 10 mM HMF at around 30 °C. During the electrolysis, the electrolyte was magnetically stirred (1000 rpm) and deaerated with N2 to remove oxygen and hydrogen.
3.4.2. Cathode Reaction—Electrohydrodimerization of AN
3.4.3. The ANEHD–HMFOR Paired Electrolysis System in H-Cell
3.5. Quantification Methods
3.5.1. Cathode Samples Analyzed by Gas Chromatography (GC)
- The organic substances in the cathode solution (500 μL) were separated by liquid–liquid extraction using dichloromethane (1000 μL) as the extracting solvent. The upper oil phase was taken out, and 1 mL lower liquid was taken into the chromatographic bottle.
- Then the extracted samples were analyzed with Agilent 7890 GC equipped with a DB-FFAP (30 m × 0.320 mm × 0.50 μm) column and a flame ionization detector (FID). The detailed GC analyzing parameters are as follows: injection-port temperature = 280 °C, split ratio = 10:1, column temperature = 50 (5 min hold time before heating)–250 °C (20 °C/min heating rate), column N2 carrier gas flow rate = 0.896 mL/min, FID temperature = 300 °C, air flow rate = 400 mL/min, H2 flow rate = 30 mL/min.
3.5.2. Anode Samples Analyzed by HPLC
- Preparation of an anode sample: a 100 μL reaction solution and 900 μL mobile phase were mixed, filtered, and placed in an autosampler vial.
- The anode samples were analyzed by Waters HPLC equipped with a 2998 photodiode array detector. The mobile phase was 0.01 M H2SO4 with a 0.6 mL/min flow rate. The wavelength of the detector was set to 265 nm to quantify anode products.
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wu, S.; Zhang, H.; Huang, X.; Liao, Q.; Wei, Z. Acrylonitrile Conversion on Metal Cathodes: How Surface Adsorption Determines the Reduction Pathways. Ind. Eng. Chem. Res. 2021, 60, 8324–8330. [Google Scholar] [CrossRef]
- Luo, H.; Barrio, J.; Sunny, N.; Li, A.; Steier, L.; Shah, N.I.; Stephens, I.E.L.; Titirici, M.M. Progress and Perspectives in Photo- and Electrochemical-Oxidation of Biomass for Sustainable Chemicals and Hydrogen Production. Adv. Energy Mater. 2021, 11, 2101180. [Google Scholar] [CrossRef]
- Qi, Y.F.; Wang, K.Y.; Sun, Y.; Wang, J.; Wang, C. Engineering the Electronic Structure of NiFe Layered Double Hydroxide Nanosheet Array by Implanting Cationic Vacancies for Efficient Electrochemical Conversion of 5-Hydroxymethylfurfural to 2,5-Furandicarboxylic Acid. Acs Sustain. Chem. Eng. 2022, 10, 645–654. [Google Scholar] [CrossRef]
- Kunnikuruvan, S.; Nair, N.N. Mechanistic Insights into the Bronsted Acid-Catalyzed Dehydration of beta-D-Glucose to 5-Hydroxymethylfurfural under Ambient and Subcritical Conditions. ACS Catal. 2019, 9, 7250–7263. [Google Scholar] [CrossRef]
- Liu, W.J.; Dang, L.N.; Xu, Z.R.; Yu, H.Q.; Jin, S.; Huber, G.W. Electrochemical Oxidation of 5-Hydroxymethylfurfural with NiFe Layered Double Hydroxide (LDH) Nanosheet Catalysts. ACS Catal. 2018, 8, 5533–5541. [Google Scholar] [CrossRef]
- Yang, Y.C.; Mu, T.C. Electrochemical oxidation of biomass derived 5-hydroxymethylfurfural (HMF): Pathway, mechanism, catalysts and coupling reactions. Green Chem. 2021, 23, 4228–4254. [Google Scholar] [CrossRef]
- Chadderdon, D.J.; Xin, L.; Qi, J.; Qiu, Y.; Krishna, P.; More, K.L.; Li, W.Z. Electrocatalytic oxidation of 5-hydroxymethylfurfural to 2,5-furandicarboxylic acid on supported Au and Pd bimetallic nanoparticles. Green Chem. 2014, 16, 3778–3786. [Google Scholar] [CrossRef]
- You, B.; Jiang, N.; Liu, X.; Sun, Y. Simultaneous H2 Generation and Biomass Upgrading in Water by an Efficient Noble-Metal-Free Bifunctional Electrocatalyst. Angew. Chem. Int. Ed. 2016, 55, 9913–9917. [Google Scholar] [CrossRef]
- Jiang, N.; You, B.; Boonstra, R.; Terrero Rodriguez, I.M.; Sun, Y. Integrating electrocatalytic 5-hydroxymethylfurfural oxidation and hydrogen production via Co–P-derived electrocatalysts. ACS Energy Lett. 2016, 1, 386–390. [Google Scholar] [CrossRef]
- Zhang, N.N.; Zou, Y.Q.; Tao, L.; Chen, W.; Zhou, L.; Liu, Z.J.; Zhou, B.; Huang, G.; Lin, H.Z.; Wang, S.Y. Electrochemical Oxidation of 5-Hydroxymethylfurfural on Nickel Nitride/Carbon Nanosheets: Reaction Pathway Determined by In Situ Sum Frequency Generation Vibrational Spectroscopy. Angew. Chem. Int. Ed. 2019, 58, 15895–15903. [Google Scholar] [CrossRef]
- Barwe, S.; Weidner, J.; Cychy, S.; Morales, D.M.; Dieckhofer, S.; Hiltrop, D.; Masa, J.; Muhler, M.; Schuhmann, W. Electrocatalytic Oxidation of 5-(Hydroxymethyl)furfural Using High-Surface-Area Nickel Boride. Angew. Chem. Int. Ed. 2018, 57, 11460–11464. [Google Scholar] [CrossRef] [PubMed]
- Heidary, N.; Kornienko, N. Electrochemical biomass valorization on gold-metal oxide nanoscale heterojunctions enables investigation of both catalyst and reaction dynamics with operando surface-enhanced Raman spectroscopy. Chem. Sci. 2020, 11, 1798–1806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Zhou, Z.; Liu, J.; Zhu, B.; Hu, H.; Yang, Y.; Chen, G.; Gao, M.; Zhang, J. Sustainable biomass upgrading coupled with H2 generation over in-situ oxidized Co3O4 electrocatalysts. Appl. Catal. B 2022, 307, 121209. [Google Scholar] [CrossRef]
- Liu, H.; Lee, T.-H.; Chen, Y.; Cochran, E.W.; Li, W. Paired electrolysis of 5-(hydroxymethyl)furfural in flow cells with a high-performance oxide-derived silver cathode. Green Chem. 2021, 23, 5056–5063. [Google Scholar] [CrossRef]
- Abdel Hamid, Z.; Hassan, H.B. Influence of electrodeposition parameters on the characteristics of NiMoP film. Surf. Coat. Technol. 2012, 212, 37–45. [Google Scholar] [CrossRef]
- Toghraei, A.; Shahrabi, T.; Barati Darband, G. Electrodeposition of self-supported Ni-Mo-P film on Ni foam as an affordable and high-performance electrocatalyst toward hydrogen evolution reaction. Electrochim. Acta 2020, 335, 135643. [Google Scholar] [CrossRef]
- Xiang, Q.; Li, F.; Chen, W.; Ma, Y.; Wu, Y.; Gu, X.; Qin, Y.; Tao, P.; Song, C.; Shang, W. In situ vertical growth of Fe–Ni layered double-hydroxide arrays on Fe–Ni alloy foil: Interfacial layer enhanced electrocatalyst with small overpotential for oxygen evolution reaction. ACS Energy Lett. 2018, 3, 2357–2365. [Google Scholar] [CrossRef]
- Du, Y.; Pan, G.; Wang, L.; Song, Y. CoxNiyP embedded in nitrogen-doped porous carbon on Ni foam for efficient hydrogen evolution. Appl. Surf. Sci. 2019, 469, 61–67. [Google Scholar] [CrossRef]
- Wu, G.; Zheng, X.; Cui, P.; Jiang, H.; Wang, X.; Qu, Y.; Chen, W.; Lin, Y.; Li, H.; Han, X. A general synthesis approach for amorphous noble metal nanosheets. Nature Commun. 2019, 10, 4855. [Google Scholar] [CrossRef] [Green Version]
- Duan, Y.X.; Meng, F.L.; Liu, K.H.; Yi, S.S.; Li, S.J.; Yan, J.M.; Jiang, Q. Amorphizing of Cu nanoparticles toward highly efficient and robust electrocatalyst for CO2 reduction to liquid fuels with high faradaic efficiencies. Adv. Mater. 2018, 30, 1706194. [Google Scholar] [CrossRef]
- Che, Q.; Li, Q.; Chen, X.; Tan, Y.; Xu, X. Assembling amorphous (Fe-Ni)Cox-OH/Ni3S2 nanohybrids with S-vacancy and interfacial effects as an ultra-highly efficient electrocatalyst: Inner investigation of mechanism for alkaline water-to-hydrogen/oxygen conversion. Appl. Catal. B 2020, 263, 118338. [Google Scholar] [CrossRef]
- Fu, S.; Song, J.; Zhu, C.; Xu, G.-L.; Amine, K.; Sun, C.; Li, X.; Engelhard, M.H.; Du, D.; Lin, Y. Ultrafine and highly disordered Ni2Fe1 nanofoams enabled highly efficient oxygen evolution reaction in alkaline electrolyte. Nano Energy 2018, 44, 319–326. [Google Scholar] [CrossRef]
- Wang, M.; Wang, Y.; Mao, S.S.; Shen, S. Transition-metal alloy electrocatalysts with active sites modulated by metal-carbide heterophases for efficient oxygen evolution. Nano Energy 2021, 88, 106216. [Google Scholar] [CrossRef]
- Su, H.; Song, S.; Gao, Y.; Li, N.; Fu, Y.; Ge, L.; Song, W.; Liu, J.; Ma, T. In Situ Electronic Redistribution Tuning of NiCo2S4 Nanosheets for Enhanced Electrocatalysis. Adv. Funct. Mater. 2022, 32, 2109731. [Google Scholar] [CrossRef]
- Ito, Y.; Ohto, T.; Hojo, D.; Wakisaka, M.; Nagata, Y.; Chen, L.; Hu, K.; Izumi, M.; Fujita, J.-i.; Adschiri, T. Cooperation between holey graphene and NiMo alloy for hydrogen evolution in an acidic electrolyte. ACS Catal. 2018, 8, 3579–3586. [Google Scholar] [CrossRef]
- Liu, H.; He, Q.; Jiang, H.; Lin, Y.; Zhang, Y.; Habib, M.; Chen, S.; Song, L. Electronic Structure Reconfiguration toward Pyrite NiS2 via Engineered Heteroatom Defect Boosting Overall Water Splitting. ACS Nano 2017, 11, 11574–11583. [Google Scholar] [CrossRef]
- Luo, W.; Wang, Y.; Luo, L.; Gong, S.; Wei, M.; Li, Y.; Gan, X.; Zhao, Y.; Zhu, Z.; Li, Z. Single-Atom and Bimetallic Nanoalloy Supported on Nanotubes as a Bifunctional Electrocatalyst for Ultrahigh-Current-Density Overall Water Splitting. ACS Catal. 2022, 12, 1167–1179. [Google Scholar] [CrossRef]
- Wang, Z.; Xu, L.; Huang, F.; Qu, L.; Li, J.; Owusu, K.A.; Liu, Z.; Lin, Z.; Xiang, B.; Liu, X.; et al. Copper–Nickel Nitride Nanosheets as Efficient Bifunctional Catalysts for Hydrazine-Assisted Electrolytic Hydrogen Production. Adv. Energy Mater. 2019, 9, 1900390. [Google Scholar] [CrossRef]
- Zhang, X.; Zhao, H.; Li, C.; Li, S.; Liu, K.; Wang, L. Facile coordination driven synthesis of metal-organic gels toward efficiently electrocatalytic overall water splitting. Appl. Catal. B 2021, 299, 120641. [Google Scholar] [CrossRef]
- Razmjooei, F.; Singh, K.P.; Yang, D.-S.; Cui, W.; Jang, Y.H.; Yu, J.-S. Fe-Treated Heteroatom (S/N/B/P)-Doped Graphene Electrocatalysts for Water Oxidation. ACS Catal. 2017, 7, 2381–2391. [Google Scholar] [CrossRef]
- Senthil Raja, D.; Chuah, X.-F.; Lu, S.-Y. In Situ Grown Bimetallic MOF-Based Composite as Highly Efficient Bifunctional Electrocatalyst for Overall Water Splitting with Ultrastability at High Current Densities. Adv. Energy Mater. 2018, 8, 1801065. [Google Scholar] [CrossRef]
- Yu, X.; Zhang, M.; Tong, Y.; Li, C.; Shi, G. A Large-Scale Graphene–Bimetal Film Electrode with an Ultrahigh Mass Catalytic Activity for Durable Water Splitting. Adv. Energy Mater. 2018, 8, 1800403. [Google Scholar] [CrossRef]
- Taitt, B.J.; Nam, D.-H.; Choi, K.-S. A Comparative Study of Nickel, Cobalt, and Iron Oxyhydroxide Anodes for the Electrochemical Oxidation of 5-Hydroxymethylfurfural to 2,5-Furandicarboxylic Acid. ACS Catal. 2019, 9, 660–670. [Google Scholar] [CrossRef]
- Li, R.-Q.; Wang, B.-L.; Gao, T.; Zhang, R.; Xu, C.; Jiang, X.; Zeng, J.; Bando, Y.; Hu, P.; Li, Y.; et al. Monolithic electrode integrated of ultrathin NiFeP on 3D strutted graphene for bifunctionally efficient overall water splitting. Nano Energy 2019, 58, 870–876. [Google Scholar] [CrossRef]
- Zhou, J.; Dou, Y.; Wu, X.-Q.; Zhou, A.; Shu, L.; Li, J.-R. Alkali-Etched Ni(II)-Based Metal–Organic Framework Nanosheet Arrays for Electrocatalytic Overall Water Splitting. Small 2020, 16, 1906564. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Chen, S.; Yao, N.; Wang, Q.; Li, J.; Wei, Z. Integrating H2 generation with sewage disposal by an efficient anti-poisoning bifunctional electrocatalyst. Appl. Catal. B 2020, 277, 119175. [Google Scholar] [CrossRef]
- Wei, H.; Wang, J.; Lin, Q.; Zou, Y.; Chen, X.a.; Zhao, H.; Li, J.; Jin, H.; Lei, Y.; Wang, S. Incorporating ultra-small N-doped Mo2C nanoparticles onto 3D N-doped flower-like carbon nanospheres for robust electrocatalytic hydrogen evolution. Nano Energy 2021, 86, 106047. [Google Scholar] [CrossRef]
- Quan, L.; Li, S.; Zhao, Z.; Liu, J.; Ran, Y.; Cui, J.; Lin, W.; Yu, X.; Wang, L.; Zhang, Y.; et al. Hierarchically Assembling CoFe Prussian Blue Analogue Nanocubes on CoP Nanosheets as Highly Efficient Electrocatalysts for Overall Water Splitting. Small Methods 2021, 5, 2100125. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Xu, G.-Y.; Zhang, Y.-J.; Wang, L.; Zhang, J.; Li, J.-F.; Fu, X.-Z.; Luo, J.-L. Understanding the Roles of Electrogenerated Co3+ and Co4+ in Selectivity-Tuned 5-Hydroxymethylfurfural Oxidation. Angew. Chem. Int. Ed. 2021, 60, 20535–20542. [Google Scholar] [CrossRef]
- Fleischmann, M.; Korinek, K.; Pletcher, D. The oxidation of organic compounds at a nickel anode in alkaline solution. J. Electroanal. Chem. Interfacial Electrochem. 1971, 31, 39–49. [Google Scholar] [CrossRef]
- Zhou, Y.; Li, T.; Xi, S.; He, C.; Yang, X.; Wu, H. One-step Synthesis of Self-standing Ni3S2/Ni2P Heteronanorods on Nickel Foam for Efficient Electrocatalytic Hydrogen Evolution over a Wide pH Range. ChemCatChem 2018, 10, 5487–5495. [Google Scholar] [CrossRef]
- Zhang, W.; Zheng, J.; Gu, X.; Tang, B.; Li, J.; Wang, X. Facile synthesis, characterization and DFT studies of a nanostructured nickel–molybdenum–phosphorous planar electrode as an active electrocatalyst for the hydrogen evolution reaction. Nanoscale 2019, 11, 9353–9361. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Kang, X.; Li, M.; Xiang, K.; Wang, C.; Guo, Z.; Zhang, J.; Fu, X.-Z.; Luo, J.-L. Coupling efficient biomass upgrading with H2 production via bifunctional CuxS@NiCo-LDH core–shell nanoarray electrocatalysts. J. Mater. Chem. A 2020, 8, 1138–1146. [Google Scholar] [CrossRef]
- Gao, L.; Bao, Y.; Gan, S.; Sun, Z.; Song, Z.; Han, D.; Li, F.; Niu, L. Hierarchical Nickel-Cobalt-Based Transition Metal Oxide Catalysts for the Electrochemical Conversion of Biomass into Valuable Chemicals. ChemSusChem 2018, 11, 2547–2553. [Google Scholar] [CrossRef]
- Huang, X.; Tan, L.; Zhang, L.; Li, C.; Wei, Z. Coverage-dependent acrylonitrile adsorption and electrochemical reduction kinetics on Pb electrode. Chem. Eng. J. 2020, 382, 123006. [Google Scholar] [CrossRef]
- Wu, T.; Nguyen, B.H.; Daugherty, M.C.; Moeller, K.D. Paired Electrochemical Reactions and the On-Site Generation of a Chemical Reagent. Angew. Chem. 2019, 131, 3600–3603. [Google Scholar] [CrossRef]
- Anantharaj, S.; Ede, S.R.; Karthick, K.; Sam Sankar, S.; Sangeetha, K.; Karthik, P.E.; Kundu, S. Precision and correctness in the evaluation of electrocatalytic water splitting: Revisiting activity parameters with a critical assessment. Energy Environ. Sci. 2018, 11, 744–771. [Google Scholar] [CrossRef]









Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Qi, J.; An, Z.; Chen, X.; Li, C.; Du, Y.; Zhang, X.; Liang, C. Paired Electrolysis of Acrylonitrile and 5-Hydroxymethylfurfural for Simultaneous Generation of Adiponitrile and 2,5-Furandicarboxylic Acid. Catalysts 2022, 12, 694. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12070694
Qi J, An Z, Chen X, Li C, Du Y, Zhang X, Liang C. Paired Electrolysis of Acrylonitrile and 5-Hydroxymethylfurfural for Simultaneous Generation of Adiponitrile and 2,5-Furandicarboxylic Acid. Catalysts. 2022; 12(7):694. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12070694
Chicago/Turabian StyleQi, Ji, Ziying An, Xiao Chen, Chuang Li, Yan Du, Xiuhong Zhang, and Changhai Liang. 2022. "Paired Electrolysis of Acrylonitrile and 5-Hydroxymethylfurfural for Simultaneous Generation of Adiponitrile and 2,5-Furandicarboxylic Acid" Catalysts 12, no. 7: 694. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12070694