
Effect of Brønsted Acid on the Reactivity and Selectivity of the Oxoiron(V) Intermediates in C-H and C=C Oxidation Reactions
 , and
, and
Abstract
:
1. Introduction

2. Results
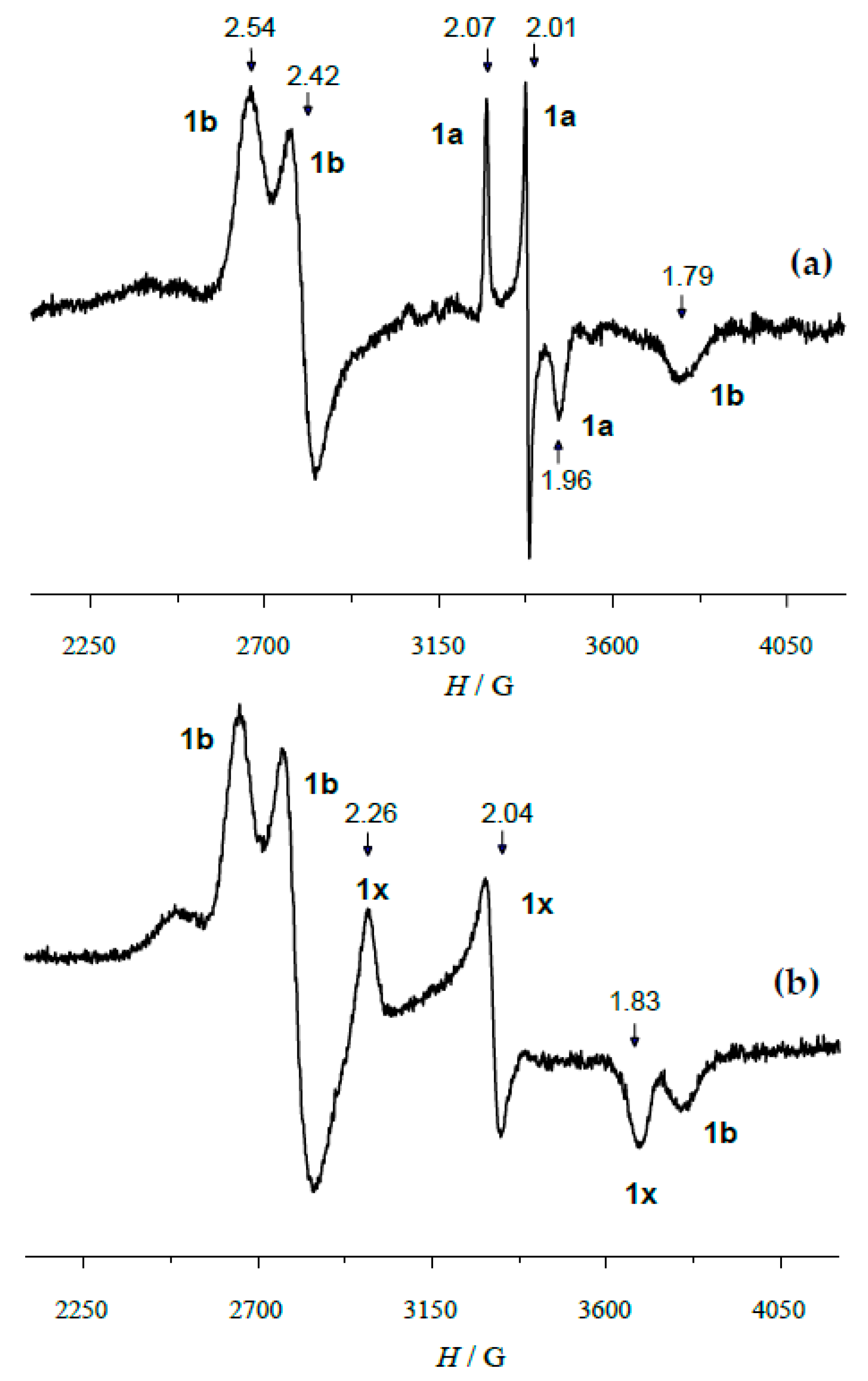
2.1. Effect of HClO4 on the Catalyst Systems 1/H2O2/AA and 2/H2O2/AA: EPR Data
2.2. Enantioselective Oxidation of Benzylideneacetone by the Catalyst Systems 1/H2O2/AA and 2/H2O2/AA
2.3. Regioselective Oxidation of Adamantane by the Catalyst Systems 1/H2O2/AA and 2/H2O2/AA
2.4. Chemo- and Regioselective Oxidation of (+)-Sclareolide by the Catalyst Systems 1/H2O2/AA and 2/H2O2/AA
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Instrumentation
4.3. Sample Preparation for EPR Measurements
4.4. General Catalytic Oxidation Procedure
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Guengerich, F.P. Mechanisms of Cytochrome P450-Catalyzed Oxidations. ACS Catal. 2018, 8, 10964–10976. [Google Scholar] [CrossRef]
- Huang, X.; Groves, J.T. Oxygen Activation and Radical Transformations in Heme Proteins and Metalloporphyrins. Chem. Rev. 2018, 118, 2491–2553. [Google Scholar] [CrossRef]
- Guo, M.; Corona, T.; Ray, K.; Nam, W. Heme and Nonheme High-Valent Iron and Manganese Oxo Cores in Biological and Abiological Oxidation Reactions. ACS Cent. Sci. 2019, 5, 13–28. [Google Scholar] [CrossRef]
- Larson, V.A.; Battistella, B.; Ray, K.; Lehnert, N.; Nam, W. Iron and Manganese Oxo Complexes, Oxo Wall and Beyond. Nat. Rev. Chem. 2020, 4, 404–419. [Google Scholar] [CrossRef]
- Baglia, R.A.; Zaragoza, J.P.T.; Goldberg, D.P. Biomimetic Reactivity of Oxygen-Derived Manganese and Iron Porphyrinoid Complexes. Chem. Rev. 2017, 117, 13320–13352. [Google Scholar] [CrossRef]
- Yokota, S.; Fujii, H. Critical Factors in Determining the Heterolytic versus Homolytic Bond Cleavage of Terminal Oxidants by Iron(III) Porphyrin Complexes. J. Am. Chem. Soc. 2018, 140, 5127–5137. [Google Scholar] [CrossRef]
- Rittle, J.; Green, M.T. Cytochrome P450 Compound I: Capture, Characterization, and C-H Bond Activation Kinetics. Science 2010, 330, 933–937. [Google Scholar] [CrossRef]
- Yosca, T.H.; Ledray, A.P.; Ngo, J.; Green, M.T. A New Look at the Role of Thiolate Ligation in Cytochrome P450. J. Biol. Inorg. Chem. 2017, 22, 209–220. [Google Scholar] [CrossRef]
- Mittra, K.; Green, M.T. Reduction Potentials of P450 Compounds I and II: Insight into the Thermodynamics of C-H Bond Activation. J. Am. Chem. Soc. 2019, 141, 5504–5510. [Google Scholar] [CrossRef]
- Sacramento, J.J.D.; Goldberg, D.P. Factors Affecting Hydrogen Atom Transfer Reactivity of Metal-Oxo Porphyrinoid Complexes. Acc. Chem. Res. 2018, 51, 2641–2652. [Google Scholar] [CrossRef]
- Nam, W.; Lee, Y.-M.; Fukuzumi, S. Hydrogen Atom Transfer Reactions of Mononuclear Nonheme Metal-Oxygen Intermediates. Acc. Chem. Res. 2018, 51, 2014–2022. [Google Scholar] [CrossRef]
- Kal, S.; Que, L. Dioxygen Activation by Nonheme Iron Enzymes with the 2-His-1-Carboxylate Facial Triad That Generate High-Valent Oxoiron Oxidants. J. Biol. Inorg. Chem. 2017, 22, 339–365. [Google Scholar] [CrossRef]
- Engelmann, X.; Monte-Pérez, I.; Ray, K. Oxidation Reactions with Bioinspired Mononuclear Non-Heme Metal-Oxo Complexes. Angew. Chem. Int. Ed. 2016, 55, 7632–7649. [Google Scholar] [CrossRef]
- Puri, M.; Que, L. Toward the Synthesis of More Reactive S = 2 Non-Heme Oxoiron(IV) Complexes. Acc. Chem. Res. 2015, 48, 2443–2452. [Google Scholar] [CrossRef]
- Nam, W.; Lee, Y.-M.; Fukuzumi, S. Tuning Reactivity and Mechanism in Oxidation Reactions by Mononuclear Nonheme Iron(IV)-Oxo Complexes. Acc. Chem. Res. 2014, 47, 1146–1154. [Google Scholar] [CrossRef]
- McDonald, A.R.; Que, L. High-Valent Nonheme Iron-Oxo Complexes: Synthesis, Structure, and Spectroscopy. Coord. Chem. Rev. 2013, 257, 414–428. [Google Scholar] [CrossRef]
- Ray, K.; Pfaff, F.F.; Wang, B.; Nam, W. Status of Reactive Non-Heme Metal-Oxygen Intermediates in Chemical and Enzymatic Reactions. J. Am. Chem. Soc. 2014, 136, 13942–13958. [Google Scholar] [CrossRef]
- Guo, M.; Lee, Y.-M.; Fukuzumi, S.; Nam, W. Biomimetic Metal-Oxidant Adducts as Active Oxidants in Oxidation Reactions. Coord. Chem. Rev. 2021, 435, 213807. [Google Scholar] [CrossRef]
- Lee, J.L.; Ross, D.L.; Barman, S.K.; Ziller, J.W.; Borovik, A.S. C-H Bond Cleavage by Bioinspired Nonheme Metal Complexes. Inorg. Chem. 2021, 60, 13759–13783. [Google Scholar] [CrossRef]
- Kal, S.; Xu, S.; Que, L. Bio-inspired Nonheme Iron Oxidation Catalysis: Involvement of Oxoiron(V) Oxidants in Cleaving Strong C-H Bonds. Angew. Chem. Int. Ed. 2020, 59, 7332–7349. [Google Scholar] [CrossRef]
- Dantignana, V.; Company, A.; Costas, M. Oxoiron(V) Complexes of Relevance in Oxidation Catalysis of Organic Substrates. Isr. J. Chem. 2020, 60, 1004–1018. [Google Scholar] [CrossRef]
- Devi, T.; Lee, Y.-M.; Nam, W.; Fukuzumi, S. Metal Ion-Coupled Electron-Transfer Reactions of Metal-Oxygen Complexes. Coord. Chem. Rev. 2020, 410, 213219. [Google Scholar] [CrossRef]
- Liu, Y.; Lau, T.-C. Activation of Metal Oxo and Nitrido Complexes by Lewis Acids. J. Am. Chem. Soc. 2019, 141, 3755–3766. [Google Scholar] [CrossRef]
- De Oliveira, F.T.; Chanda, A.; Banerjee, D.; Shan, X.; Mondal, S.; Que, L., Jr.; Bominaar, E.L.; Munck, E.; Collins, T.J. Chemical and Spectroscopic Evidence for an FeV-Oxo Complex. Science 2007, 315, 835–838. [Google Scholar] [CrossRef]
- Lyakin, O.Y.; Bryliakov, K.P.; Britovsek, G.J.P.; Talsi, E.P. EPR Spectroscopic Trapping of the Active Species of Nonheme Iron-Catalyzed Oxidation. J. Am. Chem. Soc. 2009, 131, 10798–10799. [Google Scholar] [CrossRef]
- Kundu, S.; Thompson, J.V.K.; Ryabov, A.D.; Collins, T.J. On the Reactivity of Mononuclear Iron(V)Oxo Complexes. J. Am. Chem. Soc. 2011, 133, 18546–18549. [Google Scholar] [CrossRef]
- Ghosh, M.; Singh, K.K.; Panda, C.; Weitz, A.; Hendrich, M.P.; Collins, T.J.; Dhar, B.B.; Sen Gupta, S. Formation of a Room Temperature Stable FeV(O) Complex: Reactivity Toward Unactivated C-H Bonds. J. Am. Chem. Soc. 2014, 136, 9524–9527. [Google Scholar] [CrossRef]
- Collins, T.J.; Ryabov, A.D. Targeting of High-Valent Iron-TAML Activators at Hydrocarbons and Beyond. Chem. Rev. 2017, 117, 9140–9162. [Google Scholar] [CrossRef]
- Ghosh, M.; Pattanayak, S.; Dhar, B.B.; Singh, K.K.; Panda, C.; Sen Gupta, S. Selective C-H Bond Oxidation Catalyzed by the Fe-BTAML Complex: Mechanistic Implications. Inorg. Chem. 2017, 56, 10852–10860. [Google Scholar] [CrossRef]
- Warner, G.R.; Somasundar, Y.; Jansen, K.C.; Kaaret, E.Z.; Weng, C.; Burton, A.E.; Mills, M.R.; Shen, L.Q.; Ryabov, A.D.; Pros, G.; et al. Bioinspired, Multidisciplinary, Iterative Catalyst Design Creates the Highest Performance Peroxidase Mimics and the Field of Sustainable Ultradilute Oxidation Catalysis (SUDOC). ACS Catal. 2019, 9, 7023–7037. [Google Scholar] [CrossRef]
- Pattanayak, S.; Cantú Reinhard, F.G.; Rana, A.; Gupta, S.S.; de Visser, S.P. The Equatorial Ligand Effect on the Properties and Reactivity of Iron(V) Oxo Intermediates. Chem. Eur. J. 2019, 25, 8092–8104. [Google Scholar] [CrossRef] [PubMed]
- Serrano-Plana, J.; Oloo, W.N.; Acosta-Rueda, L.; Meier, K.K.; Verdejo, B.; García-España, E.; Basallote, M.G.; Münck, E.; Que, L., Jr.; Company, A.; et al. Trapping a Highly Reactive Nonheme Iron Intermediate That Oxygenates Strong C-H Bonds with Stereoretention. J. Am. Chem. Soc. 2015, 137, 15833–15842. [Google Scholar] [CrossRef] [PubMed]
- Fan, R.; Serrano-Plana, J.; Oloo, W.N.; Draksharapu, A.; Delgado-Pinar, E.; Company, A.; Martin-Diaconescu, V.; Borrell, M.; Lloret-Fillol, J.; García-España, E.; et al. Spectroscopic and DFT Characterization of a Highly Reactive Nonheme FeV-Oxo Intermediate. J. Am. Chem. Soc. 2018, 140, 3916–3928. [Google Scholar] [CrossRef] [PubMed]
- Pattanayak, S.; Jasniewski, A.J.; Rana, A.; Draksharapu, A.; Singh, K.K.; Weitz, A.; Hendrich, M.; Que, L.; Dey, A.; Sen Gupta, S. Spectroscopic and Reactivity Comparisons of a Pair of BTAML Complexes with FeV=O and FeIV=O Units. Inorg. Chem. 2017, 56, 6352–6361. [Google Scholar] [CrossRef] [PubMed]
- Dantignana, V.; Serrano-Plana, J.; Draksharapu, A.; Magallón, C.; Banerjee, S.; Fan, R.; Gamba, I.; Guo, Y.; Que, L.; Costas, M.; et al. Spectroscopic and Reactivity Comparisons between Nonheme Oxoiron(IV) and Oxoiron(V) Species Bearing the Same Ancillary Ligand. J. Am. Chem. Soc. 2019, 141, 15078–15091. [Google Scholar] [CrossRef]
- Fukuzumi, S.; Morimoto, Y.; Kotani, H.; Naumov, P.; Lee, Y.-M.; Nam, W. Crystal Structure of a Metal Ion-Bound Oxoiron(IV) Complex and Implications for Biological Electron Transfer. Nat. Chem. 2010, 2, 756–759. [Google Scholar] [CrossRef]
- Morimoto, Y.; Kotani, H.; Park, J.; Lee, Y.-M.; Nam, W.; Fukuzumi, S. Metal Ion-Coupled Electron Transfer of a Nonheme Oxoiron(IV) Complex: Remarkable Enhancement of Electron-Transfer Rates by Sc3+. J. Am. Chem. Soc. 2011, 133, 403–405. [Google Scholar] [CrossRef]
- Prakash, J.; Rohde, G.T.; Meier, K.K.; Jasniewski, A.J.; Van Heuvelen, K.M.; Münck, E.; Que, L. Spectroscopic Identification of an FeIII Center, Not FeIV, in the Crystalline Sc–O–Fe Adduct Derived from [FeIV(O)(TMC)]2+. J. Am. Chem. Soc. 2015, 137, 3478–3481. [Google Scholar] [CrossRef]
- Fukuzumi, S.; Ohkubo, K.; Lee, Y.-M.; Nam, W. Lewis Acid Coupled Electron Transfer of Metal-Oxygen Intermediates. Chem-Eur. J. 2015, 21, 17548–17559. [Google Scholar] [CrossRef]
- Park, J.; Lee, Y.-M.; Nam, W.; Fukuzumi, S. Brønsted Acid-Promoted C-H Bond Cleavage via Electron Transfer from Toluene Derivatives to a Protonated Nonheme Iron(IV)-Oxo Complex with No Kinetic Isotope Effect. J. Am. Chem. Soc. 2013, 135, 5052–5061. [Google Scholar] [CrossRef]
- Park, J.; Lee, Y.-M.; Ohkubo, K.; Nam, W.; Fukuzumi, S. Efficient Epoxidation of Styrene Derivatives by a Nonheme Iron(IV)-Oxo Complex via Proton-Coupled Electron Transfer with Triflic Acid. Inorg. Chem. 2015, 54, 5806–5812. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Morimoto, Y.; Lee, Y.-M.; Nam, W.; Fukuzumi, S. Metal Ion Effect on the Switch of Mechanism from Direct Oxygen Transfer to Metal Ion-Coupled Electron Transfer in the Sulfoxidation of Thioanisoles by a Non-Heme Iron(IV)-Oxo Complex. J. Am. Chem. Soc. 2011, 133, 5236–5239. [Google Scholar] [CrossRef] [PubMed]
- Morimoto, Y.; Park, J.; Suenobu, T.; Lee, Y.-M.; Nam, W.; Fukuzumi, S. Mechanistic Borderline of One-Step Hydrogen Atom Transfer versus Stepwise Sc3+-Coupled Electron Transfer from Benzyl Alcohol Derivatives to a Non-Heme Iron(IV)-Oxo Complex. Inorg. Chem. 2012, 51, 10025–10036. [Google Scholar] [CrossRef] [PubMed]
- Park, J.; Morimoto, Y.; Lee, Y.-M.; Nam, W.; Fukuzumi, S. Unified View of Oxidative C-H Bond Cleavage and Sulfoxidation by a Nonheme Iron(IV)-Oxo Complex via Lewis Acid-Promoted Electron Transfer. Inorg. Chem. 2014, 53, 3618–3628. [Google Scholar] [CrossRef]
- Kal, S.; Draksharapu, A.; Que, L. Sc3+ (or HClO4) Activation of a Nonheme FeIII-OOH Intermediate for the Rapid Hydroxylation of Cyclohexane and Benzene. J. Am. Chem. Soc. 2018, 140, 5798–5804. [Google Scholar] [CrossRef]
- Serrano-Plana, J.; Acuña-Parés, F.; Dantignana, V.; Oloo, W.N.; Castillo, E.; Draksharapu, A.; Whiteoak, C.J.; Martin-Diaconescu, V.; Basallote, M.G.; Luis, J.M.; et al. Acid-Triggered O-O Bond Heterolysis of a Nonheme FeIII(OOH) Species for the Stereospecific Hydroxylation of Strong C-H Bonds. Chem-Eur. J. 2018, 24, 5331–5340. [Google Scholar] [CrossRef]
- Lyakin, O.Y.; Zima, A.M.; Samsonenko, D.G.; Bryliakov, K.P.; Talsi, E.P. EPR Spectroscopic Detection of the Elusive FeV=O Intermediates in Selective Catalytic Oxofunctionalizations of Hydrocarbons Mediated by Biomimetic Ferric Complexes. ACS Catal. 2015, 5, 2702–2707. [Google Scholar] [CrossRef]
- Zima, A.M.; Lyakin, O.Y.; Bryliakov, K.P.; Talsi, E.P. Direct Reactivity Studies of Non-Heme Iron-Oxo Intermediates toward Alkane Oxidation. Catal. Commun. 2018, 108, 77–81. [Google Scholar] [CrossRef]
- Burger, R.M.; Peisach, J.; Horwitz, S.B. Activated Bleomycin. A Transient Complex of Drug, Iron, and Oxygen That Degrades DNA. J. Biol. Chem. 1981, 256, 11636–11644. [Google Scholar] [CrossRef]
- Zima, A.M.; Lyakin, O.Y.; Bryliakova, A.A.; Babushkin, D.E.; Bryliakov, K.P.; Talsi, E.P. Reactivity vs. Selectivity of Biomimetic Catalyst Systems of the Fe(PDP) Family through the Nature and Spin State of the Active Iron-Oxygen Species. Chem. Rec. 2022, 22. [Google Scholar] [CrossRef]
- Zima, A.M.; Babushkin, D.E.; Lyakin, O.Y.; Bryliakov, K.P.; Talsi, E.P. High-Spin and Low-Spin State Perferryl Intermediates: Reactivity-Selectivity Correlation in Fe(PDP) Catalyzed Oxidation of (+)-Sclareolide. ChemCatChem 2022, 14, e202101430. [Google Scholar] [CrossRef]
- Zima, A.M.; Lyakin, O.Y.; Bryliakov, K.P.; Talsi, E.P. High-Spin and Low-Spin Perferryl Intermediates in Fe(PDP)-Catalyzed Epoxidations. ChemCatChem 2019, 11, 5345–5352. [Google Scholar] [CrossRef]
- Zima, A.M.; Lyakin, O.Y.; Bryliakov, K.P.; Talsi, E.P. Low-Spin and High-Spin Perferryl Intermediates in Non-Heme Iron Catalyzed Oxidations of Aliphatic C-H Groups. Chem. Eur. J. 2021, 27, 7781–7788. [Google Scholar] [CrossRef] [PubMed]
- Zima, A.M.; Lyakin, O.Y.; Bushmin, D.S.; Soshnikov, I.E.; Bryliakov, K.P.; Talsi, E.P. Non-Heme Perferryl Intermediates: Effect of Spin State on the Epoxidation Enantioselectivity. Mol. Catal. 2021, 502, 111403. [Google Scholar] [CrossRef]
- Zima, A.M.; Lyakin, O.Y.; Ottenbacher, R.V.; Bryliakov, K.P.; Talsi, E.P. Dramatic Effect of Carboxylic Acid on the Electronic Structure of the Active Species in Fe(PDP)-Catalyzed Asymmetric Epoxidation. ACS Catal. 2016, 6, 5399–5404. [Google Scholar] [CrossRef]
- Xue, S.-S.; Li, X.-X.; Lee, Y.-M.; Seo, M.S.; Kim, Y.; Yanagisawa, S.; Kubo, M.; Jeon, Y.-K.; Kim, W.-S.; Sarangi, R.; et al. Enhanced Redox Reactivity of a Nonheme Iron(V)-Oxo Complex Binding Proton. J. Am. Chem. Soc. 2020, 142, 15305–15319. [Google Scholar] [CrossRef]
- Ottenbacher, R.V.; Samsonenko, D.G.; Talsi, E.P.; Bryliakov, K.P. Highly Enantioselective Bioinspired Epoxidation of Electron-Deficient Olefins with H2O2 on Aminopyridine Mn Catalysts. ACS Catal. 2014, 4, 1599–1606. [Google Scholar] [CrossRef]
- Zima, A.M.; Lyakin, O.Y.; Bryliakov, K.P.; Talsi, E.P. On the Nature of the Active Intermediates in Iron-Catalyzed Oxidation of Cycloalkanes with Hydrogen Peroxide and Peracids. Mol. Catal. 2018, 455, 6–13. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
 | |||||
|---|---|---|---|---|---|
| Entry | Catalyst | Carboxylic Acid | Strong Acid | Epoxide Yield (%) | ee (%) |
| 1 | 1 | — | HClO4 | 52 | 34 |
| 2 | 1 | AA | — | 61 | 35 |
| 3 | 1 | AA | HClO4 | 89 | 36 |
| 4 | 1 | EHA | — | 44 | 63 |
| 5 | 1 | EHA | HClO4 | 13 | 66 |
| 6 | 2 | — | HClO4 | 12 | 46 |
| 7 b | 2 | AA | — | 11 | 59 |
| 8 | 2 | AA | HClO4 | 5 | 51 |
| 9 | 2 | EHA | — | 58 | 85 |
| 10 | 2 | EHA | HClO4 | 2 | — c |
 | ||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst | Carboxylic Acid | Strong Acid | Conversion (%) | 1-ol:2-ol:2-one | 3°/2° b |
| 1 | 1 | — | HClO4 | 17.5 | 15.0:1.8:0.7 | 18.0 |
| 2 c | 1 | AA | — | 59.2 | 55.1:1.7:2.4 | 40.0 |
| 3 | 1 | AA | HClO4 | 72.1 | 67.3:0.5:4.3 | 42.1 |
| 4 | 2 | — | HClO4 | 9.8 | 8.1:1.3:0.4 | 14.3 |
| 5 c | 2 | AA | — | 48.4 | 43.3:4.0:1.1 | 25.4 |
| 6 | 2 | AA | HClO4 | 57.9 | 53.1:1.5:2.6 | 38.8 |
 | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Cat. | Conv of S (%) | Products Distribution (%) | Regioseletivity (%) | ||||||||||||
| S3=O | S2=O | S1=O | S2(eq)-OH | S3(eq)-OH | S3(ax)-OH | S1(eq)-OH | S1(ax)-OH | S7(eq)-OH | S2(eq)-OH−3=O | C1 | C2 | C3 | C7 | |||
| 1 a | 1 | 9.6 | 24.0 | 18.3 | 15.9 | 27.7 | 3.0 | 3.2 | 3.8 | — | 3.8 | — | 19.7 | 46.0 | 30.5 | 3.8 |
| 2 b | 1 | 20.6 | 24.3 | 19.8 | 12.6 | 31.1 | 2.4 | 2.4 | 3.0 | 0.5 | 3.0 | 0.9 | 16.1 | 50.9 | 29.1 | 3.0 |
| 3 c | 1 | 50 | 28.9 | 37.6 | 12.9 | 18.9 | — | 0.9 | — | — | 0.8 | — | 12.9 | 56.5 | 29.8 | 0.8 |
| 4 d | 2 | 9.4 | 19.1 | 10.6 | 4.3 | 53.2 | 3.2 | 3.2 | 2.1 | 1.0 | 3.3 | — | 7.4 | 63.8 | 25.5 | 3.3 |
| 5 e | 2 | 29 | 30.3 | 24.8 | 7.7 | 34.8 | — | — | — | — | 2.4 | — | 7.7 | 59.6 | 30.3 | 2.4 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zima, A.M.; Lyakin, O.Y.; Bryliakova, A.A.; Babushkin, D.E.; Bryliakov, K.P.; Talsi, E.P. Effect of Brønsted Acid on the Reactivity and Selectivity of the Oxoiron(V) Intermediates in C-H and C=C Oxidation Reactions. Catalysts 2022, 12, 949. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12090949
Zima AM, Lyakin OY, Bryliakova AA, Babushkin DE, Bryliakov KP, Talsi EP. Effect of Brønsted Acid on the Reactivity and Selectivity of the Oxoiron(V) Intermediates in C-H and C=C Oxidation Reactions. Catalysts. 2022; 12(9):949. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12090949
Chicago/Turabian StyleZima, Alexandra M., Oleg Y. Lyakin, Anna A. Bryliakova, Dmitrii E. Babushkin, Konstantin P. Bryliakov, and Evgenii P. Talsi. 2022. "Effect of Brønsted Acid on the Reactivity and Selectivity of the Oxoiron(V) Intermediates in C-H and C=C Oxidation Reactions" Catalysts 12, no. 9: 949. https://0-doi-org.brum.beds.ac.uk/10.3390/catal12090949