

Effects of Metal–Support Interaction in the Electrocatalysis of the Hydrogen Evolution Reaction of the Metal-Decorated Titanium Dioxide Supported Carbon
 and
and
Abstract
:
1. Introduction
2. Results and Discussion
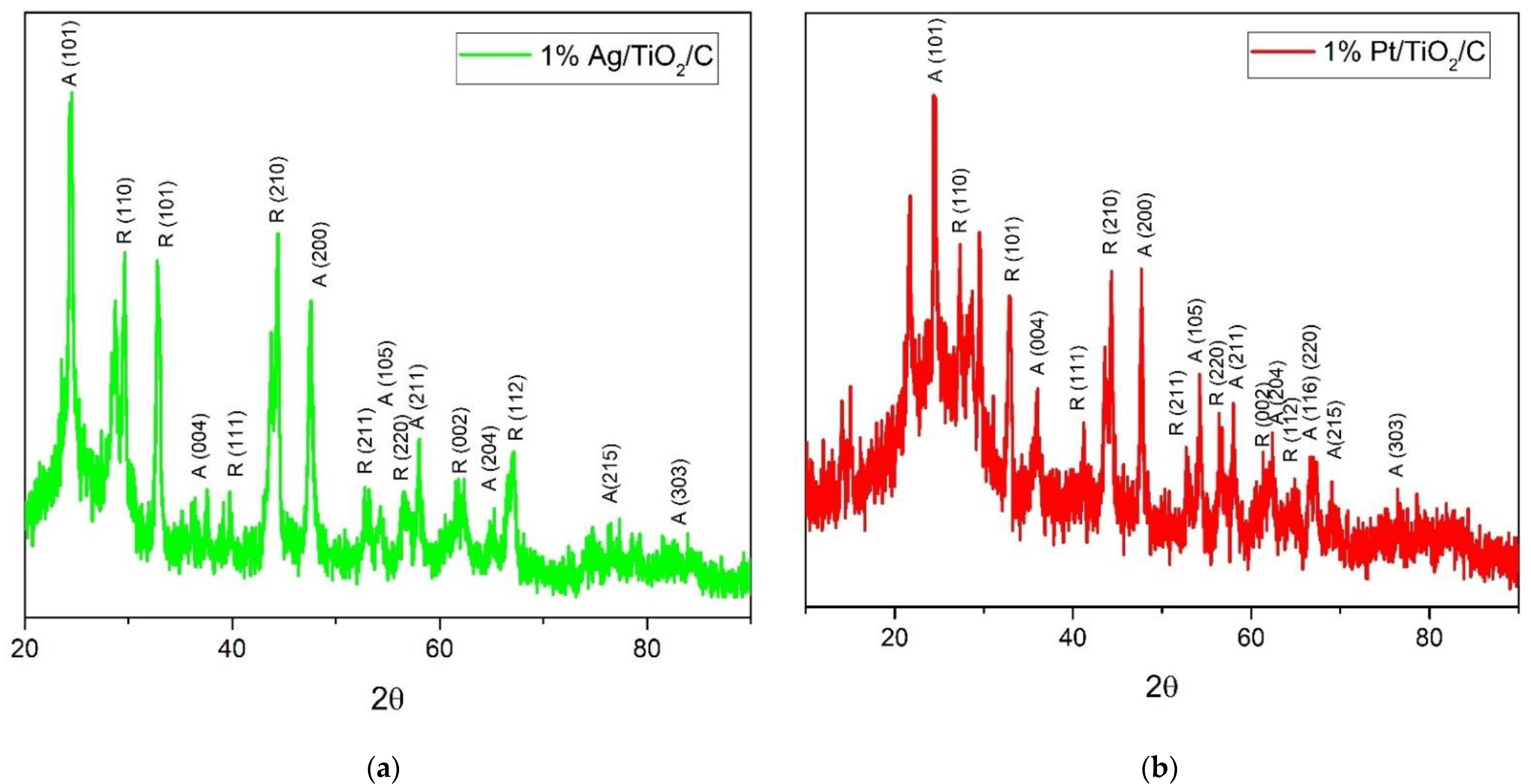
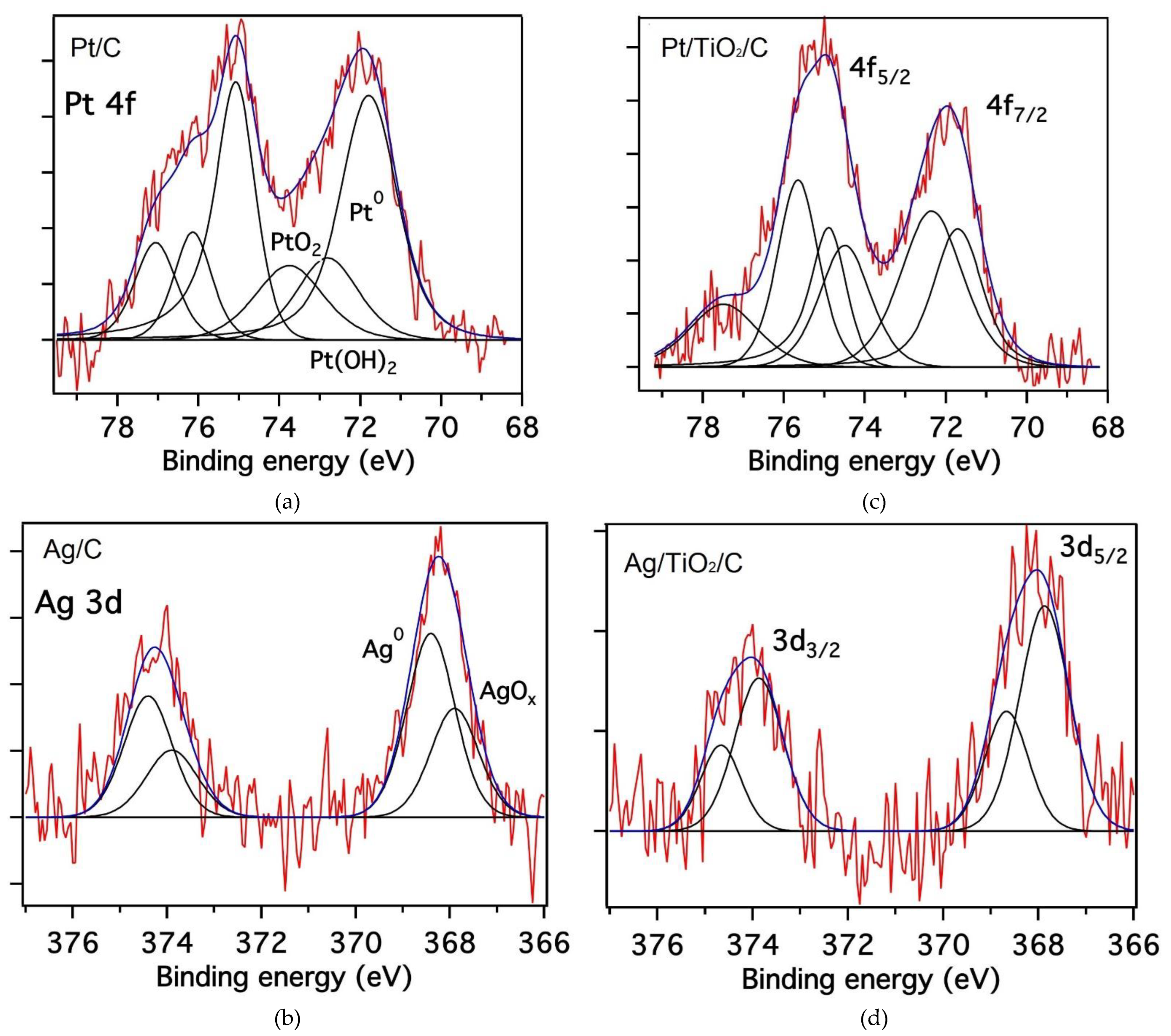
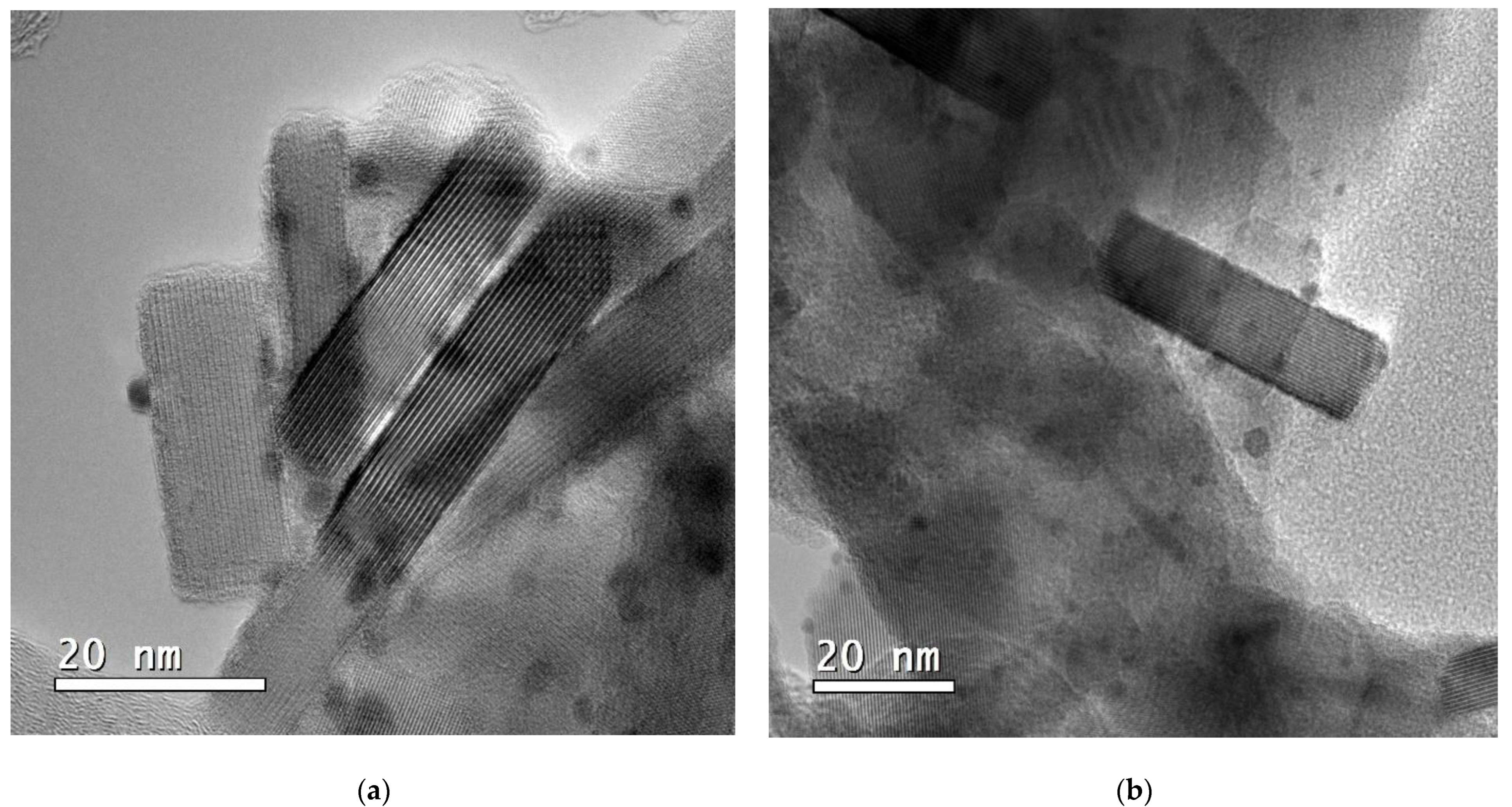
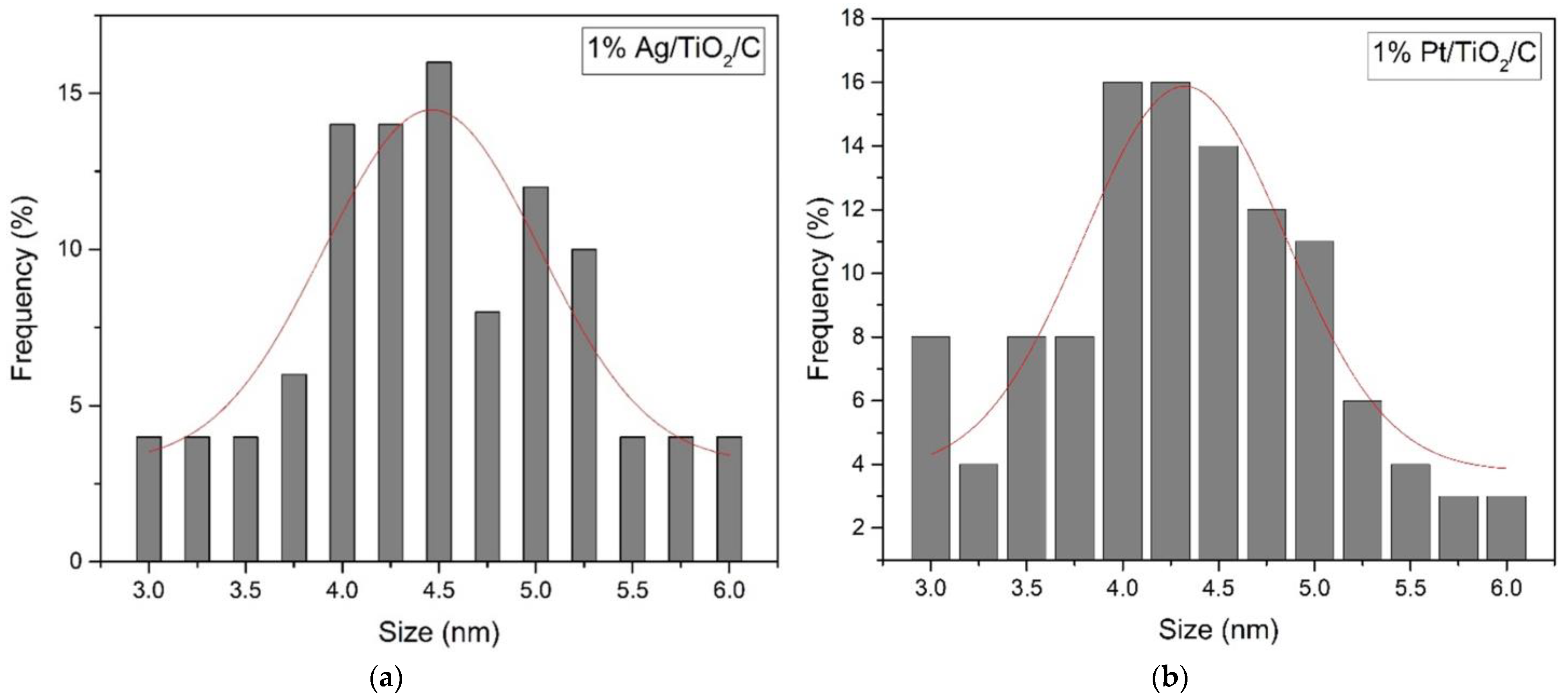
2.1. Catalyst Properties
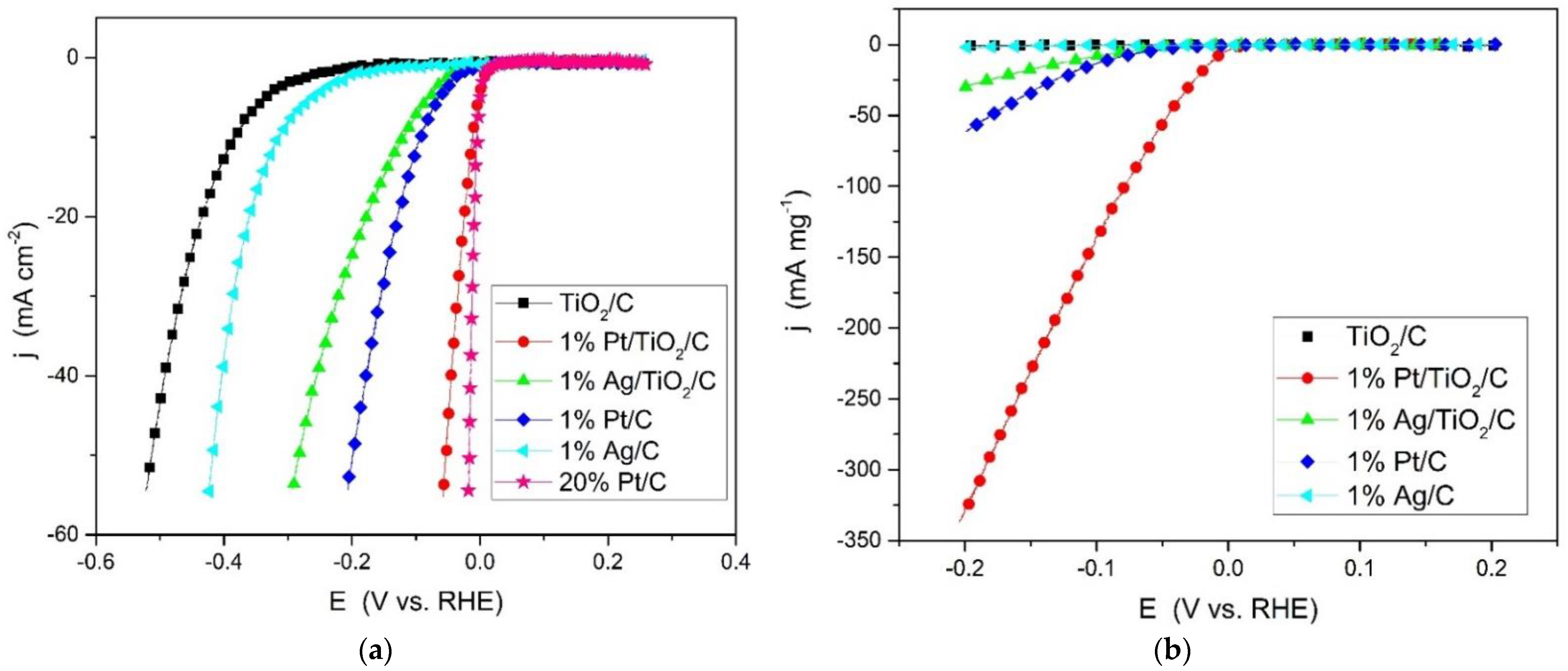
2.2. Hydrogen Evolution in Acid Medium
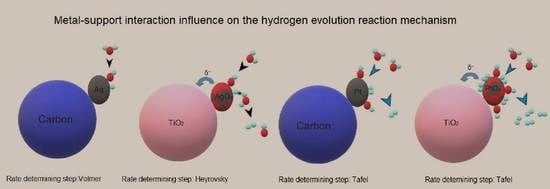
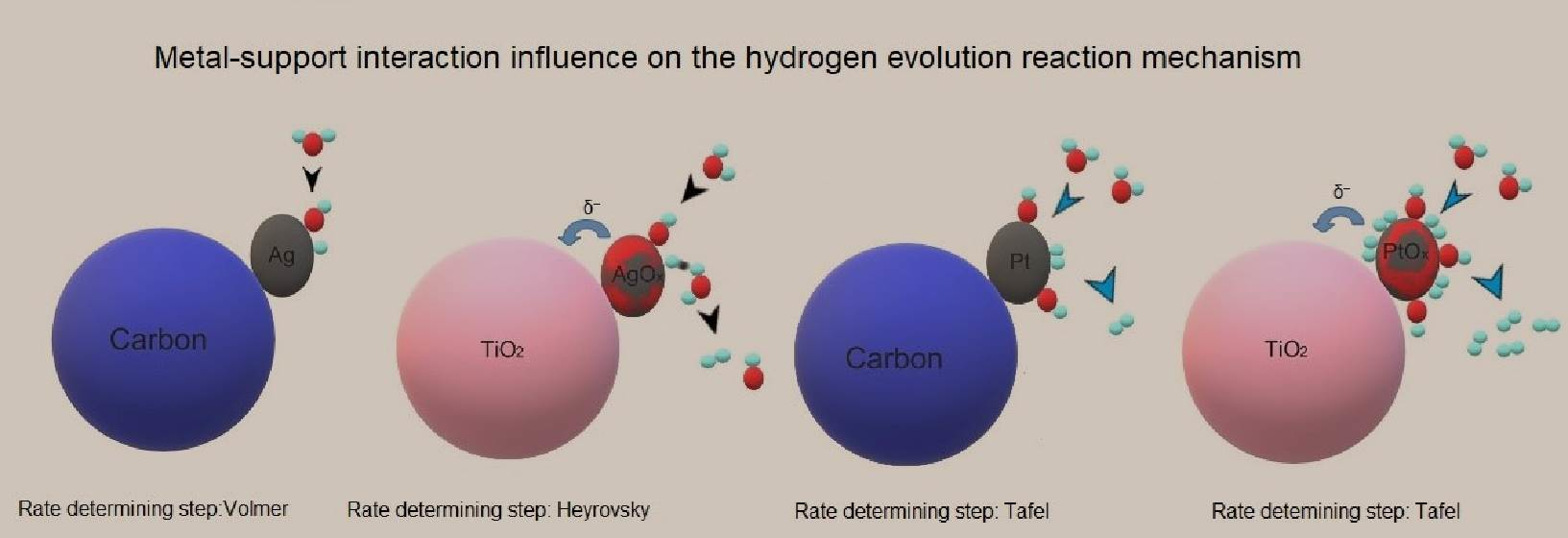
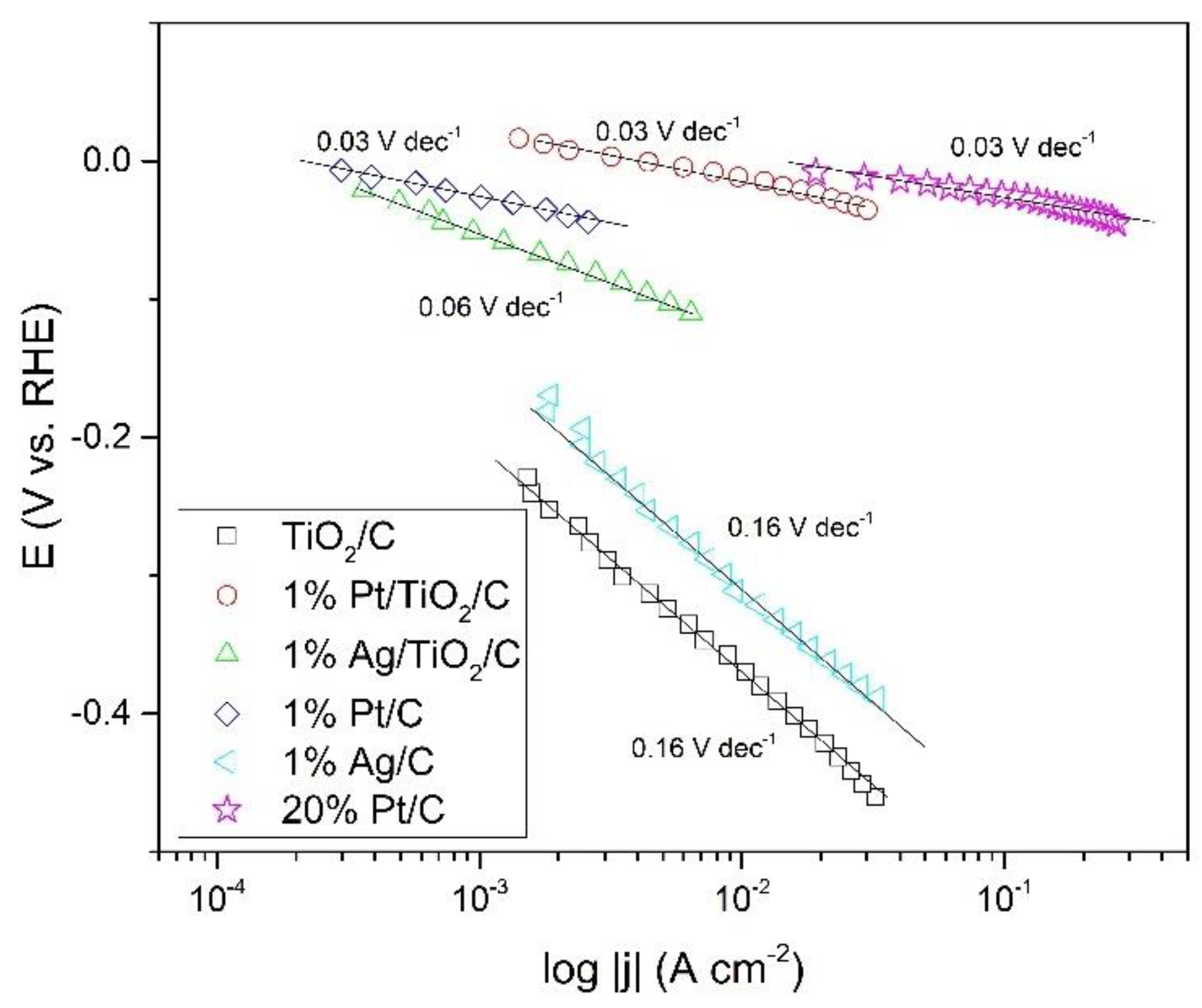
2.3. The Reaction Mechanism
2.4. Electronic Effects in HER
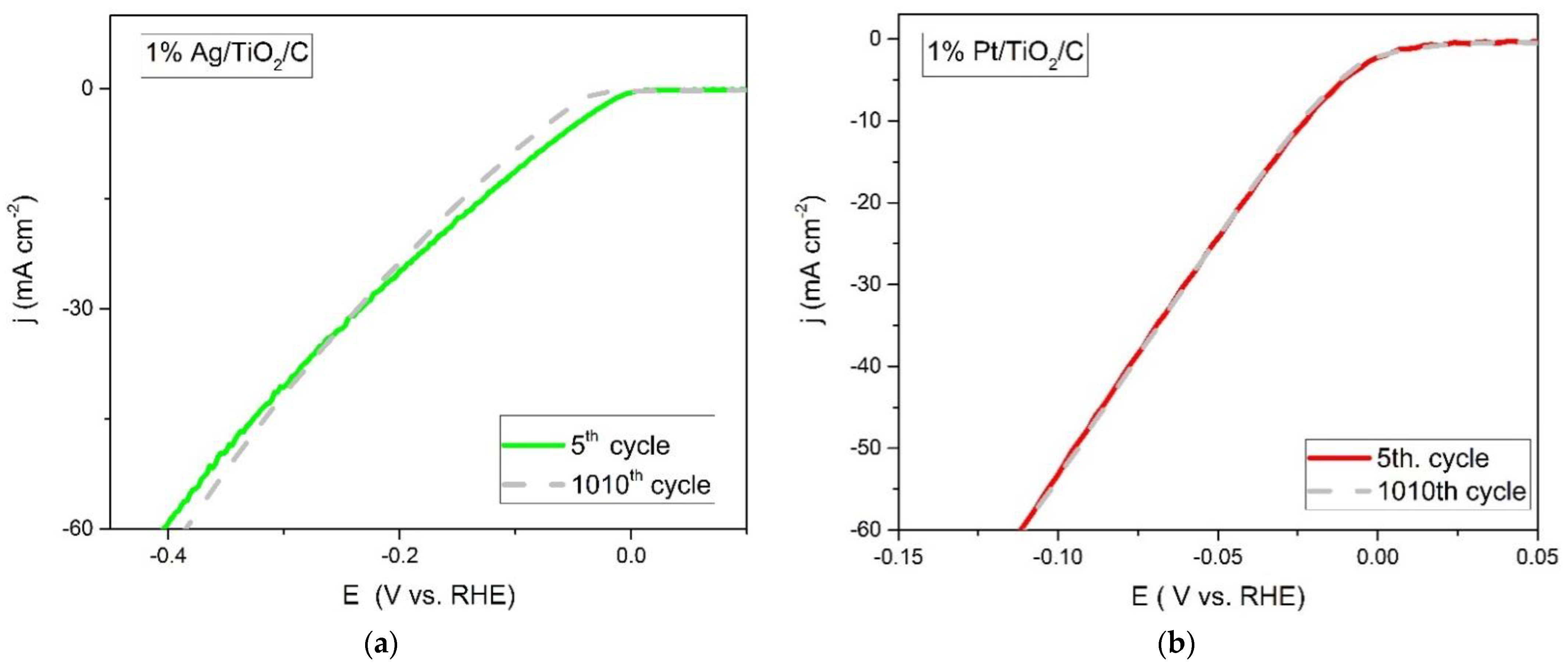
2.5. Catalyst Stability
3. Materials and Methods
3.1. Preparation of TiO2/C
3.2. Preparation of M/TiO2/C Catalysts
3.3. Preparation of M/C Catalysts
3.4. Catalyst Characterization
3.5. Electrochemical Measurements
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Strmcnik, D.; Lopes, P.P.; Genorio, B.; Stamenkovic, V.R.; Markovic, N.M. Design principles for hydrogen evolution reaction catalyst materials. Nano Energy 2016, 29, 29–36. [Google Scholar] [CrossRef] [Green Version]
- Zeng, M.; Li, Y. Recent advances in heterogeneous electrocatalysts for the hydrogen evolution reaction. J. Mater. Chem. A 2015, 3, 14942–14962. [Google Scholar] [CrossRef]
- Yan, Y.; Xia, B.Y.; Zhao, B.; Wang, X. A review on noble-metal-free bifunctional heterogeneous catalysts for overall electrochemical water splitting. J. Mater. Chem. A 2016, 4, 17587–17603. [Google Scholar] [CrossRef] [Green Version]
- Fang, S.; Zhu, X.; Liu, X.; Gu, J.; Liu, W.; Wang, D.; Yao, T. Uncovering near-free platinum single-atom dynamics during electrochemical hydrogen evolution reaction. Nat. Commun. 2020, 11, 1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, N.; Stambula, S.; Wang, D.; Banis, M.N.; Liu, J.; Riese, A.; Sun, X. Platinum single-atom and cluster catalysis of the hydrogen evolution reaction. Nat. Commun. 2016, 7, 13638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, X.P.; Wang, H.J.; Tang, S.F.; Lu, X.L.; Shu, M.; Si, R.; Lu, T.B. Engineering the coordination environment of single-atom platinum anchored on graphdiyne for optimizing electrocatalytic hydrogen evolution. Angew. Chem. Int. Ed. Engl. 2018, 57, 9382–9386. [Google Scholar] [CrossRef]
- Zeng, Z.; Su, Y.; Quan, X.; Choi, W.; Zhang, G.; Liu, N.; Zhang, S. Single-atom platinum confined by the interlayer nano space of carbon nitride for efficient photocatalytic hydrogen evolution. Nano Energy 2020, 69, 104409. [Google Scholar] [CrossRef]
- Wang, D.Y.; Gong, M.; Chou, H.L.; Pan, C.J.; Chen, H.A.; Wu, Y.; Wang, Y.L. Highly active and stable hybrid catalyst of cobalt-doped FeS2 nanosheets–carbon nanotubes for hydrogen evolution reaction. J. Am. Chem. Soc. 2015, 137, 1587–1592. [Google Scholar] [CrossRef]
- Danilov, F.I.; Tsurkan, A.V.; Vasil’eva, E.A.; Protsenko, V.S. Electrocatalytic activity of composite Fe/TiO2 electrodeposits for hydrogen evolution reaction in alkaline solutions. Int. J. Hydrogen Energy 2016, 41, 7363–7372. [Google Scholar] [CrossRef]
- Protsenko, V.S.; Bogdanov, D.A.; Korniy, S.A.; Kityk, A.A.; Baskevich, A.S.; Danilov, F.I. Application of a deep eutectic solvent to prepare nanocrystalline Ni and Ni/TiO2 coatings as electrocatalysts for the hydrogen evolution reaction. Int. J. Hydrogen Energy 2019, 44, 24604–24616. [Google Scholar] [CrossRef]
- Kullaiah, R.E.L.; Hegde, A.C. Effect of TiO2 nanoparticles on hydrogen evolution reaction activity of Ni coatings. Int. J. Miner. Metall. Mater. 2018, 25, 472–479. [Google Scholar] [CrossRef]
- Benea, L.; Pavlov, A.I. Ni-TiO2 nanocomposite coatings as cathode material for hydrogen evolution reaction. Optoelectron. Adv. Mater. Rapid Commun. 2013, 7, 895–899. [Google Scholar]
- Marino, T.; Figoli, A.; Molino, A.; Argurio, P.; Molinari, R. Hydrogen and oxygen evolution in a membrane photoreactor using suspended nanosized Au/TiO2 and Au/CeO2. ChemEngineering 2019, 3, 5. [Google Scholar] [CrossRef] [Green Version]
- Roy, N.; Leung, K.T.; Pradhan, D. Nitrogen doped reduced graphene oxide-based Pt–TiO2 nanocomposites for enhanced hydrogen evolution. J. Phys. Chem. C 2015, 119, 19117–19125. [Google Scholar] [CrossRef]
- Wang, Y.; Chen, L.; Yu, X.; Wang, Y.; Zheng, G. Superb Alkaline Hydrogen Evolution and Simultaneous Electricity Generation by Pt-Decorated Ni3N Nanosheets. Adv. Energy Mater. 2017, 7, 1601390. [Google Scholar] [CrossRef]
- Bukola, S.; Merzougui, B.; Akinpelu, A.; Zeama, M. Cobalt and nitrogen co-doped tungsten carbide catalyst for oxygen reduction and hydrogen evolution reactions. Electrochim. Acta 2016, 190, 1113–1123. [Google Scholar] [CrossRef]
- Sasaki, K.; Zhang, L.; Adzic, R.R. Niobium oxide-supported platinum ultra-low amount electrocatalysts for oxygen reduction. Phys. Chem. Chem. Phys. 2008, 10, 159–167. [Google Scholar] [CrossRef]
- Masuda, T.; Fukumitsu, H.; Fugane, K.; Togasaki, H.; Matsumura, D.; Tamura, K.; Uosaki, K. Role of Cerium Oxide in the Enhancement of Activity for the Oxygen Reduction Reaction at Pt–CeOx Nanocomposite Electrocatalyst -An in Situ Electrochemical X-ray Absorption Fine Structure Study. J. Phys. Chem. C 2012, 116, 10098–10102. [Google Scholar] [CrossRef] [Green Version]
- Lopes, T.; Kucernak, A.; Malko, D.; Ticianelli, E.A. Mechanistic insights into the oxygen reduction reaction on metal–N–C electrocatalysts under fuel cell conditions. ChemElectroChem 2016, 3, 1580–1590. [Google Scholar] [CrossRef] [Green Version]
- Do Rêgo, U.A.; Lopes, T.; Bott-Neto, J.L.; Tanaka, A.A.; Ticianelli, E.A. Oxygen reduction electrocatalysis on transition metal-nitrogen modified tungsten carbide nanomaterials. J. Electroanal. Chem. 2018, 810, 222–231. [Google Scholar] [CrossRef]
- Yu, B.; Jin, J.; Wu, H.; Wang, S.; Xia, Q.; LIU, H. Iron and nickel doped CoSe2 as efficient non precious metal catalysts for oxygen reduction. Int. J. Hydrogen Energy 2017, 42, 236–242. [Google Scholar] [CrossRef]
- Du, Y.; Cheng, G.; Luo, W. Colloidal synthesis of urchin-like Fe doped NiSe2 for efficient oxygen evolution. Nanoscale 2017, 9, 6821–6825. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Mahoney, E.G.; Zhao, S.; Yang, B.; Chen, J.G. Low loadings of platinum on transition metal carbides for hydrogen oxidation and evolution reactions in alkaline electrolytes. ChemComm 2016, 52, 3697–3700. [Google Scholar] [CrossRef] [PubMed]
- Hassan, A.; Paganin, V.A.; Ticianelli, E.A. Pt modified tungsten carbide as anode electrocatalyst for hydrogen oxidation in proton exchange membrane fuel cell: CO tolerance and stability. Appl. Catal. B 2015, 165, 611–619. [Google Scholar] [CrossRef]
- Gubán, D.; Borbáth, I.; Pászti, Z.; Sajó, I.; Drotár, E.; Hegedűs, M.; Tompos, A. Preparation and characterization of novel Ti0.7W0.3O2–C composite materials for Pt-based anode electrocatalysts with enhanced CO tolerance. Appl. Catal. B 2015, 174, 455–470. [Google Scholar] [CrossRef] [Green Version]
- Kim, G.J.; Kwon, D.W.; Hong, S.C. Effect of Pt particle size and valence state on the performance of Pt/TiO2 catalysts for CO oxidation at room temperature. J. Phys. Chem. C 2016, 120, 17996–18004. [Google Scholar] [CrossRef]
- Tauster, S.J.; Fung, S.C.; Baker, R.T.K.; Horsley, J.A. Strong interactions in supported-metal catalysts. Science 1981, 211, 1121–1125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ometto, F.B.; Carbonio, E.A.; Teixeira-Neto, É.; Villullas, H.M. Changes induced by transition metal oxides in Pt nanoparticles unveil the effects of electronic properties on oxygen reduction activity. J. Mater. Chem. A 2019, 7, 2075–2086. [Google Scholar] [CrossRef]
- Bruix, A.; Rodriguez, J.A.; Ramírez, P.J.; Senanayake, S.D.; Evans, J.; Park, J.B.; Illas, F. New Type of Strong Metal–Support Interaction and the Production of H2 through the Transformation of Water on Pt/CeO2 (111) and Pt/CeOx/TiO2 (110) Catalysts. J. Am. Chem. Soc. 2012, 134, 8968–8974. [Google Scholar] [CrossRef]
- Pan, Y.; Wen, M. Noble metals enhanced catalytic activity of anatase TiO2 for hydrogen evolution reaction. Int. J. Hydrogen Energy 2018, 43, 22055–22063. [Google Scholar] [CrossRef]
- Kim, J.H.; Chang, S.; Kim, Y.T. Compressive strain as the main origin of enhanced oxygen reduction reaction activity for Pt electrocatalysts on chromium-doped titania support. Appl. Catal. B 2014, 158, 112–118. [Google Scholar] [CrossRef]
- Timperman, L.; Lewera, A.; Vogel, W.; Alonso-Vante, N. Nanostructured platinum becomes alloyed at oxide-composite substrate. Electrochem. Commun. 2010, 12, 1772–1775. [Google Scholar] [CrossRef]
- Jiao, Y.; Zheng, Y.; Jaroniec, M.; Qiao, S.Z. Design of electrocatalysts for oxygen-and hydrogen-involving energy conversion reactions. Chem. Soc. Rev. 2015, 44, 2060–2086. [Google Scholar] [CrossRef] [PubMed]
- Ono, L.K.; Croy, J.R.; Heinrich, H.; Cuenya, B.R. Oxygen chemisorption, formation, and thermal stability of Pt oxides on Pt nanoparticles supported on SiO2/Si(001): Size effects. J. Phys. Chem. C 2011, 115, 16856–16866. [Google Scholar] [CrossRef]
- Agnihotri, S.; Mukherji, S.; Mukherji, S. Immobilized silver nanoparticles enhance contact killing and show highest efficacy: Elucidation of the mechanism of bactericidal action of silver. Nanoscale 2013, 5, 7328–7340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erraria, A.M.; Carapeto, A.P.; Rego, D.; Botelho, A.M. X-ray photoelectron spectroscopy: Silver saltsrevisited. Vacuum 2012, 86, 1988–1991. [Google Scholar] [CrossRef]
- Godoi, D.R.M.; Villullas, H.M.; Zhu, F.-C.; Jiang, Y.-X.; Sun, S.-G.; Guo, J.; Sun, L.; Chen, R. A comparative investigation of metal-support interactions on the catalytic activity of Pt nanoparticles for ethanol oxidation in alkaline medium. J. Power Sources 2016, 311, 81–90. [Google Scholar] [CrossRef] [Green Version]
- Lewera, A.; Timperman, L.; Roguska, A.; Alonso-Vante, N. Metal-support interactions between nanosized Pt and metal oxides (WO3 and TiO2) studied using X-ray photoelectron spectroscopy. J. Phys. Chem. C 2011, 115, 20153–20159. [Google Scholar] [CrossRef]
- Camacho, B.R.; Morais, C.; Valenzuela, M.A.; Alonso-Vante, N. Enhancing oxygen reduction reaction activity and stability of platinum via oxide-carbon composites. Catalysts 2013, 202, 36–43. [Google Scholar] [CrossRef]
- Kim, J.H.; Kwon, G.; Lim, H.; Zhu, C.; You, H.; Kim, Y.T. Effects of transition metal doping in Pt/M-TiO2 (M = V, Cr, and Nb) on oxygen reduction reaction activity. J. Power Sources 2016, 320, 188–195. [Google Scholar] [CrossRef]
- Bard, A.J.; Faulkner, L.R. Fundamentals and applications. Electrochem. Methods 2001, 2, 580–632. [Google Scholar]
- Bockris, J.O.; Reddy, A.K.N.; Gamboa-Aldeco, M.E. Modern Electrochemistry; Plenum Press: New York, NY, USA, 1998. [Google Scholar]
- Erdey-Grúz, T.; Volmer, M. Zur Theorie der Wasserstoff Überspannung Zeitschrift für Physikalische Chemie. Z. Für Phys. Chem. 1930, 150A, 203–213. [Google Scholar] [CrossRef]
- Heyrovský, J. A theory of overpotential. Recueil des Travaux Chimiques des Pays-Bas 1927, 46, 582–585. [Google Scholar] [CrossRef]
- Tafel, J. Über die Polarisation bei kathodischer Wasserstoffentwicklung. Z. Für Phys. Chem. 1905, 50U, 641–712. [Google Scholar] [CrossRef]
- Du, H.; Kong, R.M.; Guo, X.; Qu, F.; Li, J. Recent progress in transition metal phosphides with enhanced electrocatalysis for hydrogen evolution. Nanoscale 2018, 10, 21617–21624. [Google Scholar] [CrossRef] [PubMed]
- Masa, J.; Batchelor-Mcauley, C.; Schuhmann, W.; Compton, R.G. Koutecky-Levich analysis applied to nanoparticle modified rotating disk electrodes: Electrocatalysis or misinterpretation? Nano Res. 2014, 7, 71–78. [Google Scholar] [CrossRef]
- Armstrong, R.D.; Henderson, M. Impedance plane display of a reaction with an adsorbed intermediate. J. Electroanal. Chem. 1972, 39, 81–90. [Google Scholar] [CrossRef]
- Navarro-Flores, E.; Chong, Z.; Omanovic, S. Characterization of Ni, NiMo, NiW and NiFe electroactive coatings as electrocatalysts for hydrogen evolution in an acidic medium. J. Mol. Catal. A Chem. 2005, 226, 179–197. [Google Scholar] [CrossRef]
- Šimpraga, R.; Tremiliosi-Filho, G.; Qian, S.Y.; Conway, B.E. In situ determination of the ‘real are factor’in H2 evolution Electrocatalysis at porous Ni-Fe composite electrodes. J. Electroanal. Chem. 1997, 424, 141–151. [Google Scholar] [CrossRef]
- Castro, E.B.; de Giz, M.J.; Gonzalez, E.R.; Vilche, J.R. An electrochemical impedance study on the kinetics and mechanism of the hydrogen evolution reaction on nickel molybdenite electrodes. Electrochim. Acta 1997, 42, 951–959. [Google Scholar] [CrossRef]
- Xiong, L.; Manthiram, A. Nanostructured Pt-M/C (M = Fe and Co) catalysts prepared by microemulsion method for oxygen reduction in proton exchange membrane fuel cells. Electrochim. Acta 2005, 50, 2323–2329. [Google Scholar] [CrossRef]
- Santos, L.G.R.A.; Oliveira, C.H.F.; Moraes, I.R.; Ticianelli, E.A. Oxygen reduction reaction in acid medium on Pt-Ni/C prepared by microemulsion method. J. Electroanal. Chem. 2006, 596, 141–148. [Google Scholar] [CrossRef]
- Kumar, P.; Mittal, K.L. Handbook of Microemulsion Science and Technology; Marcel Dekker: New York, NY, USA, 1999. [Google Scholar]
- Capek, I. Preparation of metal nanoparticles in water-in-oil (w/o) microemulsions. Adv. Colloid Interface Sci. 2004, 110, 49–74. [Google Scholar] [CrossRef]
- Briggs, D.; Seah, M.P. Practical Surface Analysis, Auger and X-Ray Photoelectron Spectroscopy; John Wiley & Sons: Chichester, UK, 1990; Volume 1. [Google Scholar]
- Smith, G.C. Surface Analysis by Electron Spectroscopy; Plenum: New York, NY, USA, 1994. [Google Scholar]
- Scofield, J. Hrn. Hartree-Slater subshell photoionization cross-sections at 1254 and 1487 eV. J. Electron Spectrosc. Relat. Phenom. 1976, 8, 129–137. [Google Scholar] [CrossRef]
- Chastain, J.; King, J.R.; Roger, C. Handbook of X-ray photoelectron spectroscopy. Perkin-Elmer Corp. 1992, 40, 221. [Google Scholar]
- Naumkin, A.V.; Kraut-Vass, A.; Gaarenstroom, S.W.; Powell, C.J. NIST X-ray Photoelctron Spectroscopy Database. NIST Standard Reference Database 20, v. 4.1. Available online: htttp://srdata.nist.gov/XPS/ (accessed on 7 April 2021).









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Elements | Composition (wt.%) | ||||
|---|---|---|---|---|---|
| Pt/C | Ag/C | Pt/TiO2/C | Ag/TiO2/C | TiO2/C | |
| Oxygen (O 1s) | 7.9 | 6.2 | 15.4 | 16.8 | 19.8 |
| Carbon (C1s) | 91.3 | 93.6 | 80 | 75.8 | 73.7 |
| Platinum (Pt 4f) | 0.7 | - | 0.7 | - | - |
| Silver (Ag 3d) | - | 0.3 | - | 0.3 | - |
| Titanium (Ti 2p) | - | - | 3.9 | 7.2 | 6.5 |
| Catalyst | Pt 4f7/2—Binding Energy/eV | Species | % (atomic) |
| 1% Pt/C | 71.7 | Pt0 | 60 |
| 72.8 | Ptd+ | 40 | |
| 73.7 | |||
| 1% Pt/TiO2/C | 71.7 | Pt0 | 33 |
| 72.5 | Ptd+ | 67 | |
| 74.3 | |||
| Catalyst | Ag 3d5/2—Binding Energy/eV | Species | % (atomic) |
| 1% Ag /C | 368.4 | Ag0 | 63 |
| 367.9 | Agd+ | 37 | |
| 1% Ag/TiO2/C | 368.6 | Ag0 | 32 |
| 367.9 | Agd+ | 69 |
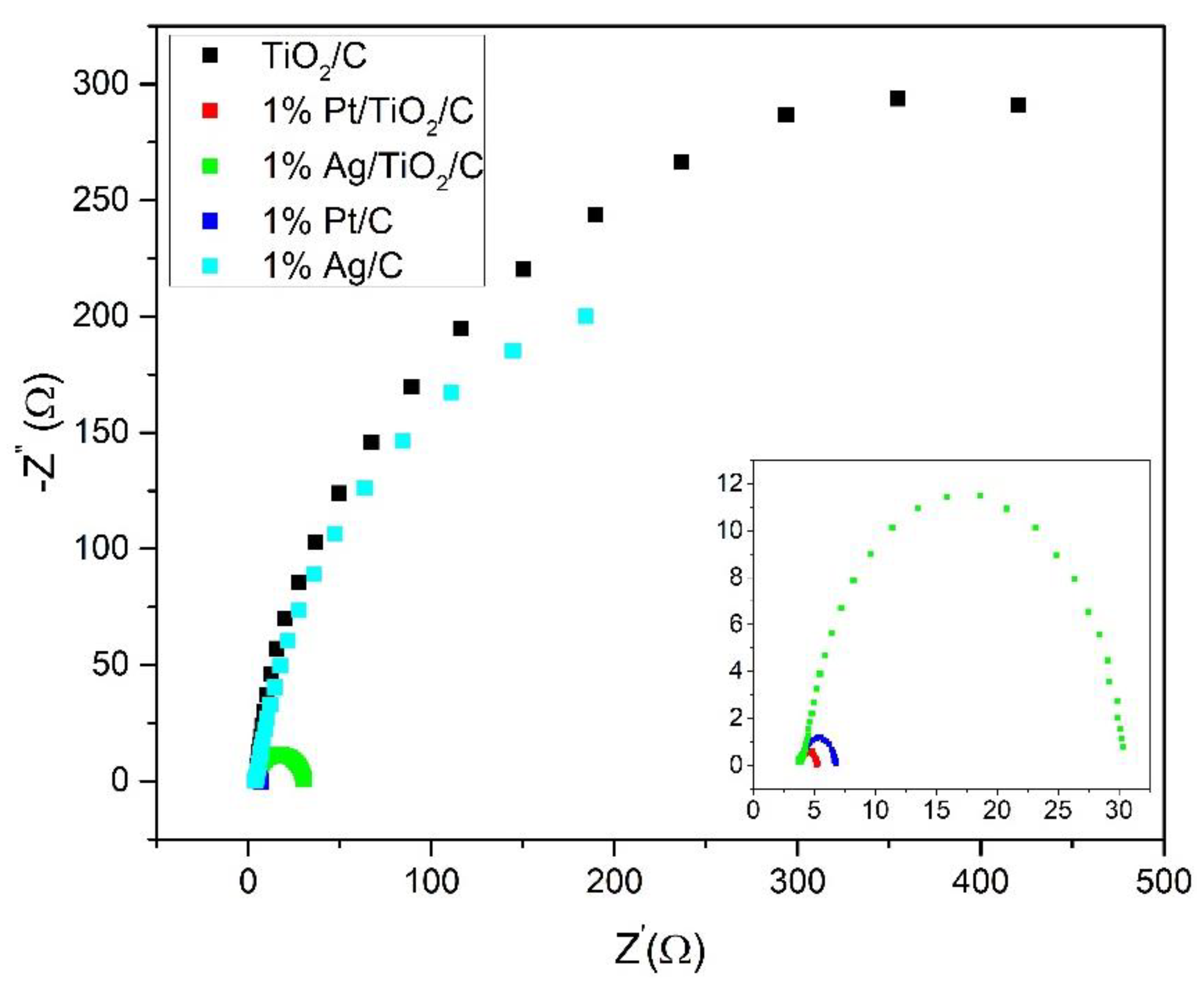
| Material | CPE-T | CPE-P | CP | R1 (Ω) | R2 (Ω) | RF (Ω) |
|---|---|---|---|---|---|---|
| TiO2/C | 0.0012 | 0.90 | 0.0005 | 3 | 4042 | 4045 |
| 1% Ag/TiO2/C | 0.0022 | 0.84 | 0.0013 | 1.6 | 25 | 26.6 |
| 1% Pt/TiO2/C | 0.0016 | 0.78 | 0.0004 | 0.6 | 0.9 | 1.5 |
| 1% Ag/C | 0.0034 | 0.85 | 0.0010 | 5.1 | 506 | 511 |
| 1% Pt/C | 0.0024 | 0.74 | 0.0006 | 0.5 | 2.6 | 3.1 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ometto, F.B.; Paganin, V.A.; Hammer, P.; Ticianelli, E.A. Effects of Metal–Support Interaction in the Electrocatalysis of the Hydrogen Evolution Reaction of the Metal-Decorated Titanium Dioxide Supported Carbon. Catalysts 2023, 13, 22. https://0-doi-org.brum.beds.ac.uk/10.3390/catal13010022
Ometto FB, Paganin VA, Hammer P, Ticianelli EA. Effects of Metal–Support Interaction in the Electrocatalysis of the Hydrogen Evolution Reaction of the Metal-Decorated Titanium Dioxide Supported Carbon. Catalysts. 2023; 13(1):22. https://0-doi-org.brum.beds.ac.uk/10.3390/catal13010022
Chicago/Turabian StyleOmetto, Felipe Berto, Valdecir Antonio Paganin, Peter Hammer, and Edson Antonio Ticianelli. 2023. "Effects of Metal–Support Interaction in the Electrocatalysis of the Hydrogen Evolution Reaction of the Metal-Decorated Titanium Dioxide Supported Carbon" Catalysts 13, no. 1: 22. https://0-doi-org.brum.beds.ac.uk/10.3390/catal13010022