
The Distribution and Strength of Brönsted Acid Sites on the Multi-Aluminum Model of FER Zeolite: A Theoretical Study

Abstract
:
1. Introduction
2. Results and Discussions
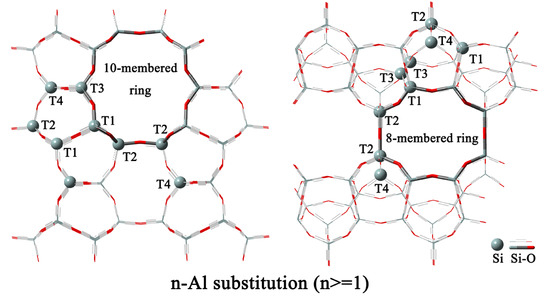
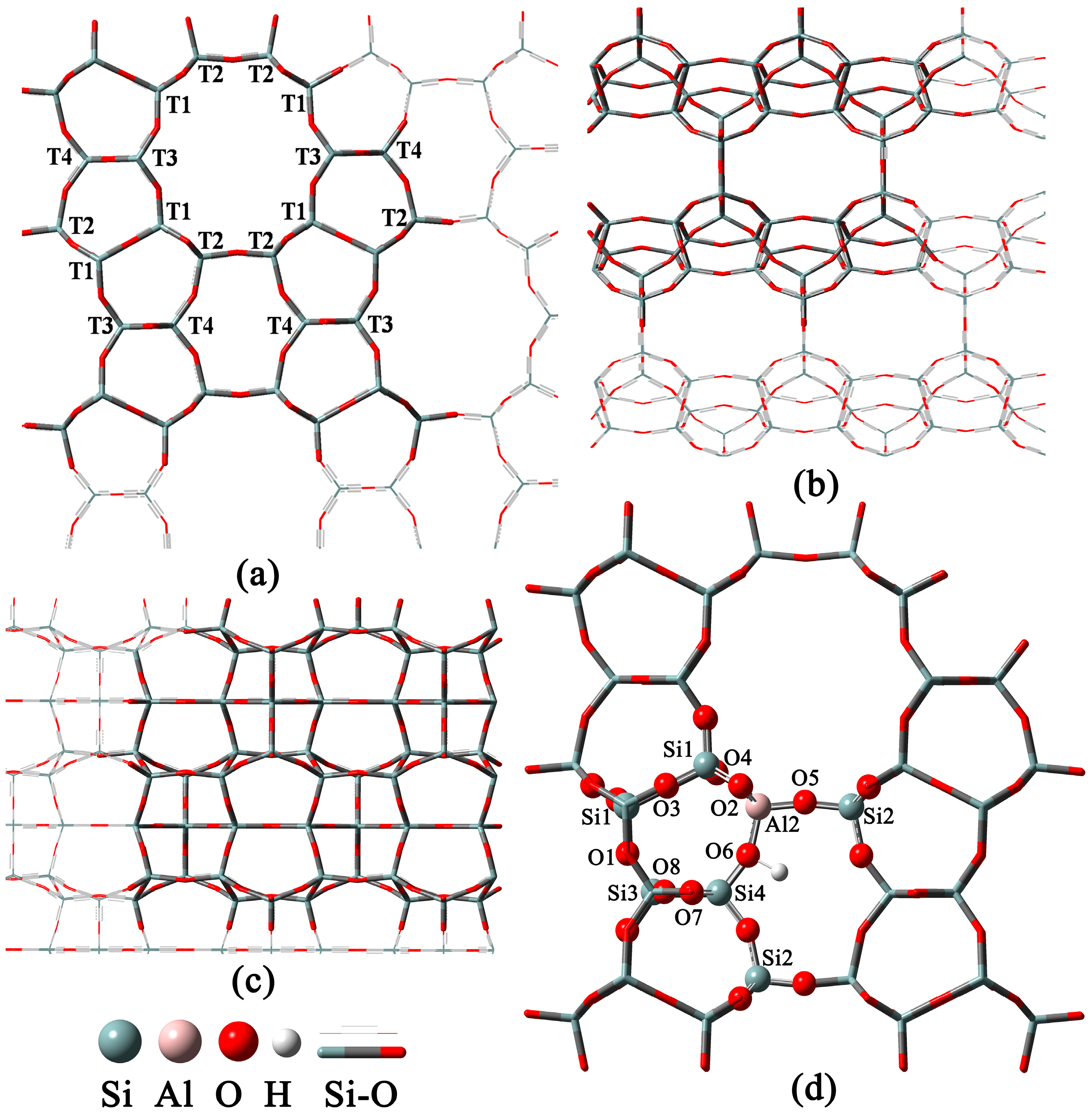
2.1. 1-Al Substitution
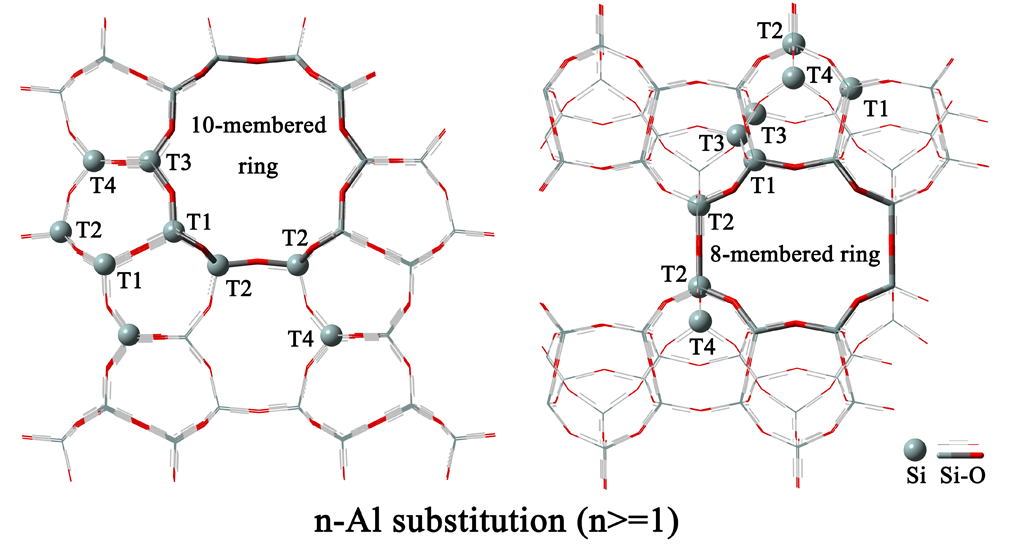
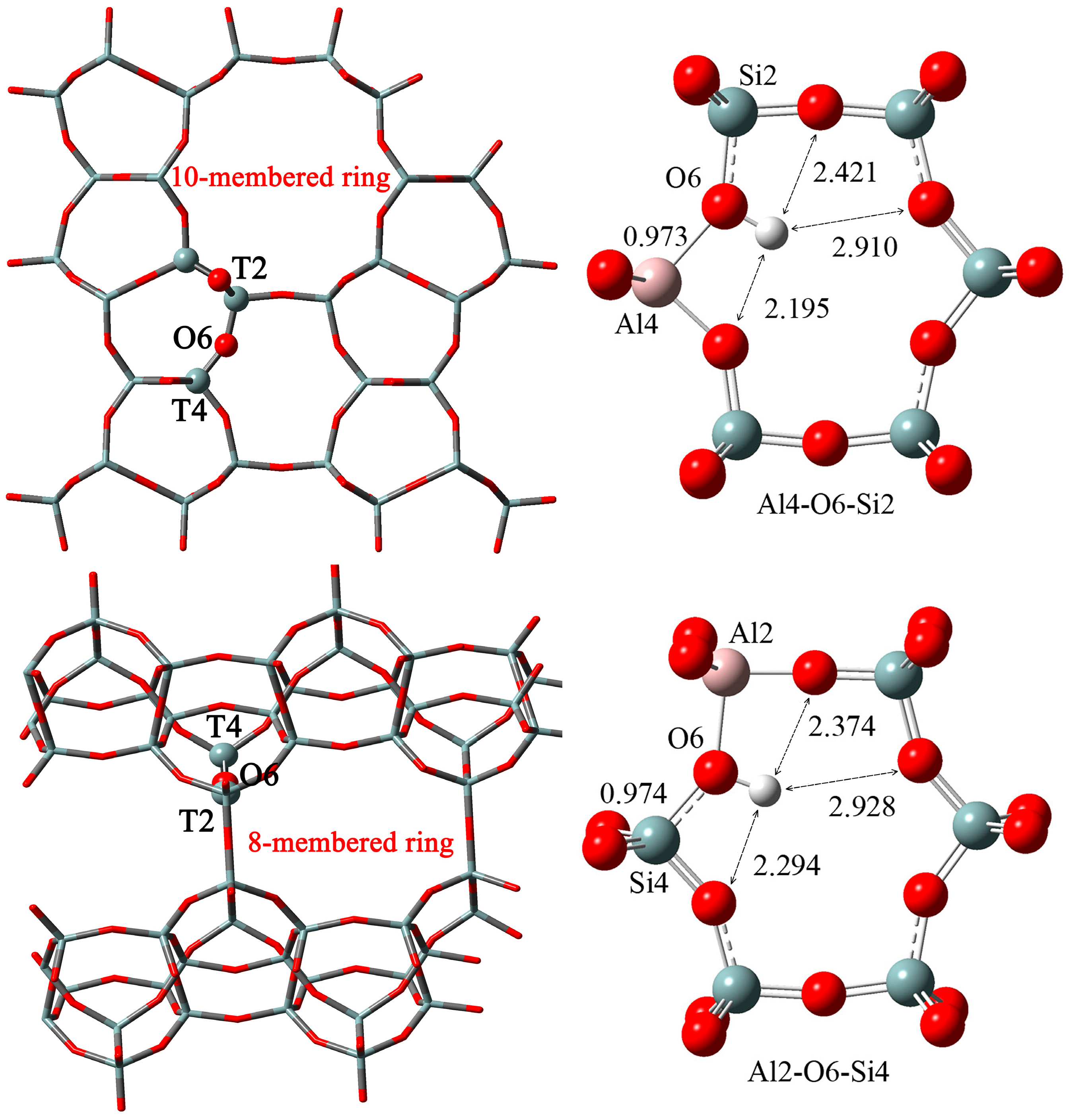
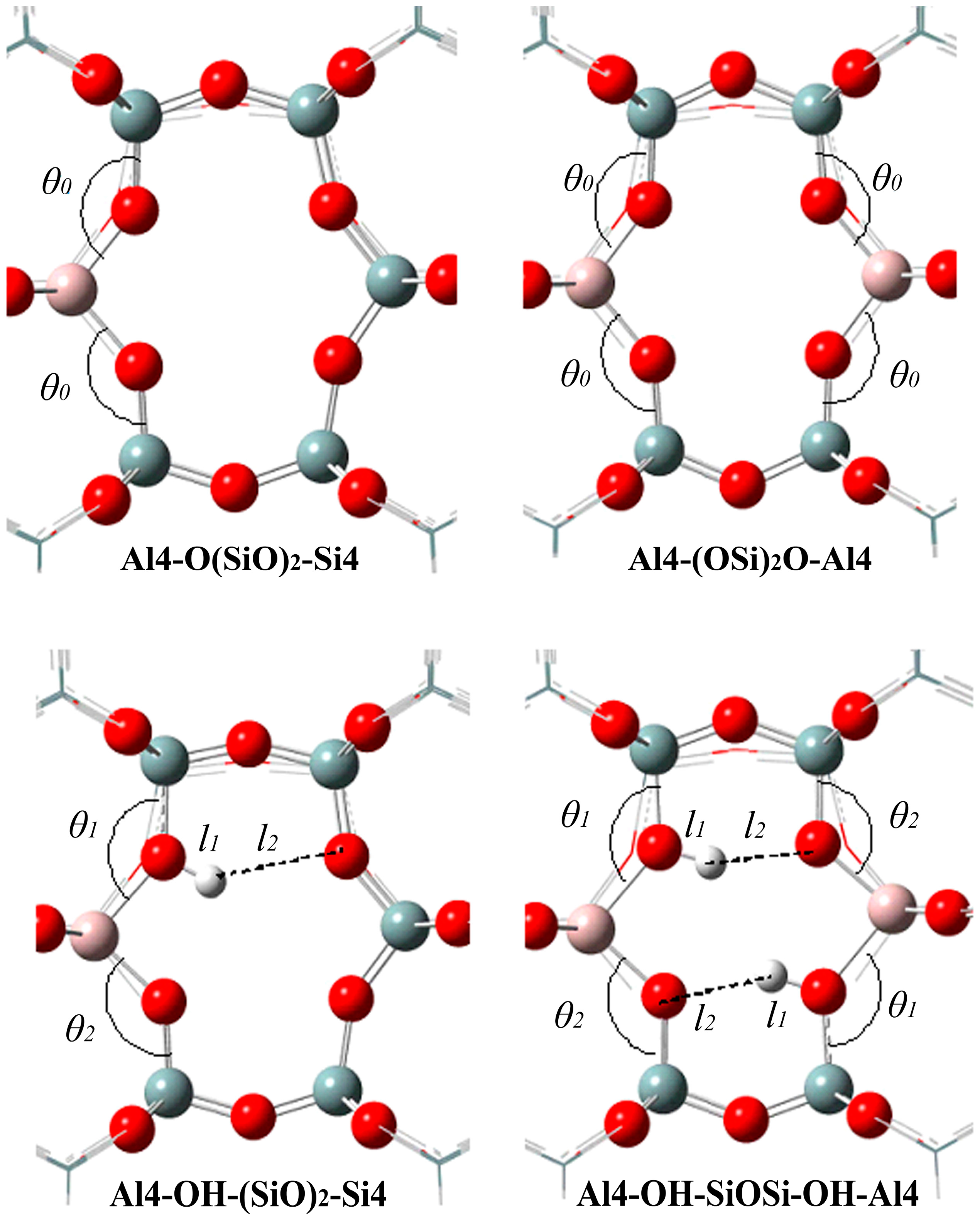
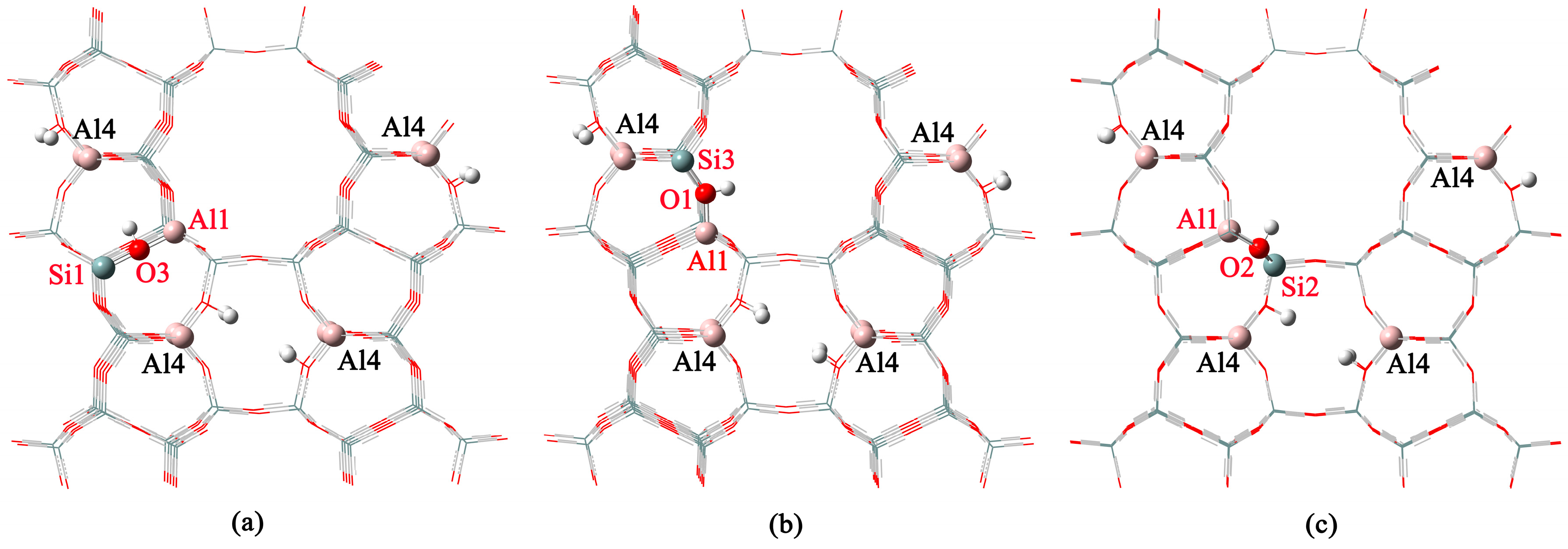
2.2. n-Al Substitution (n ≥ 2)
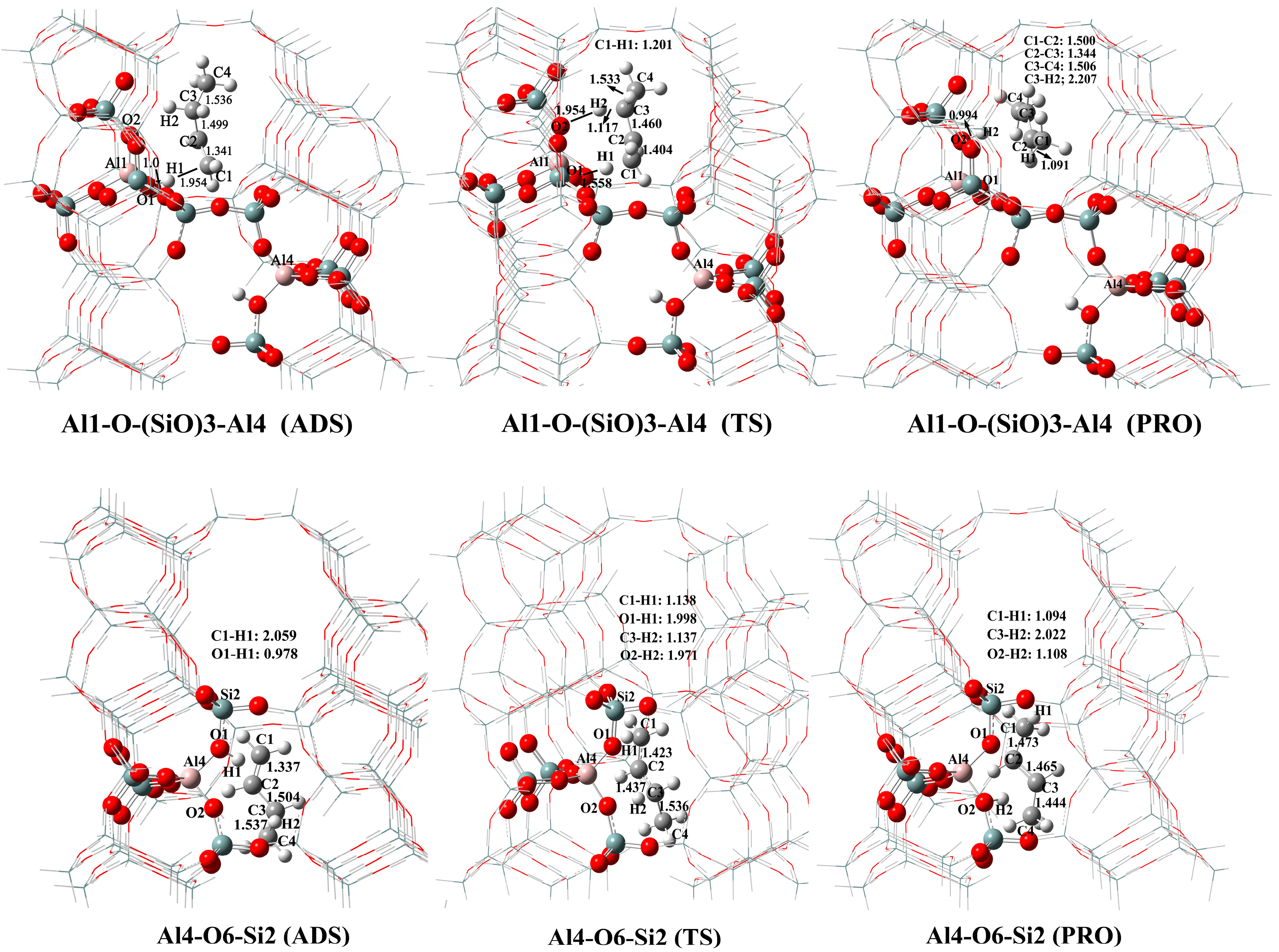
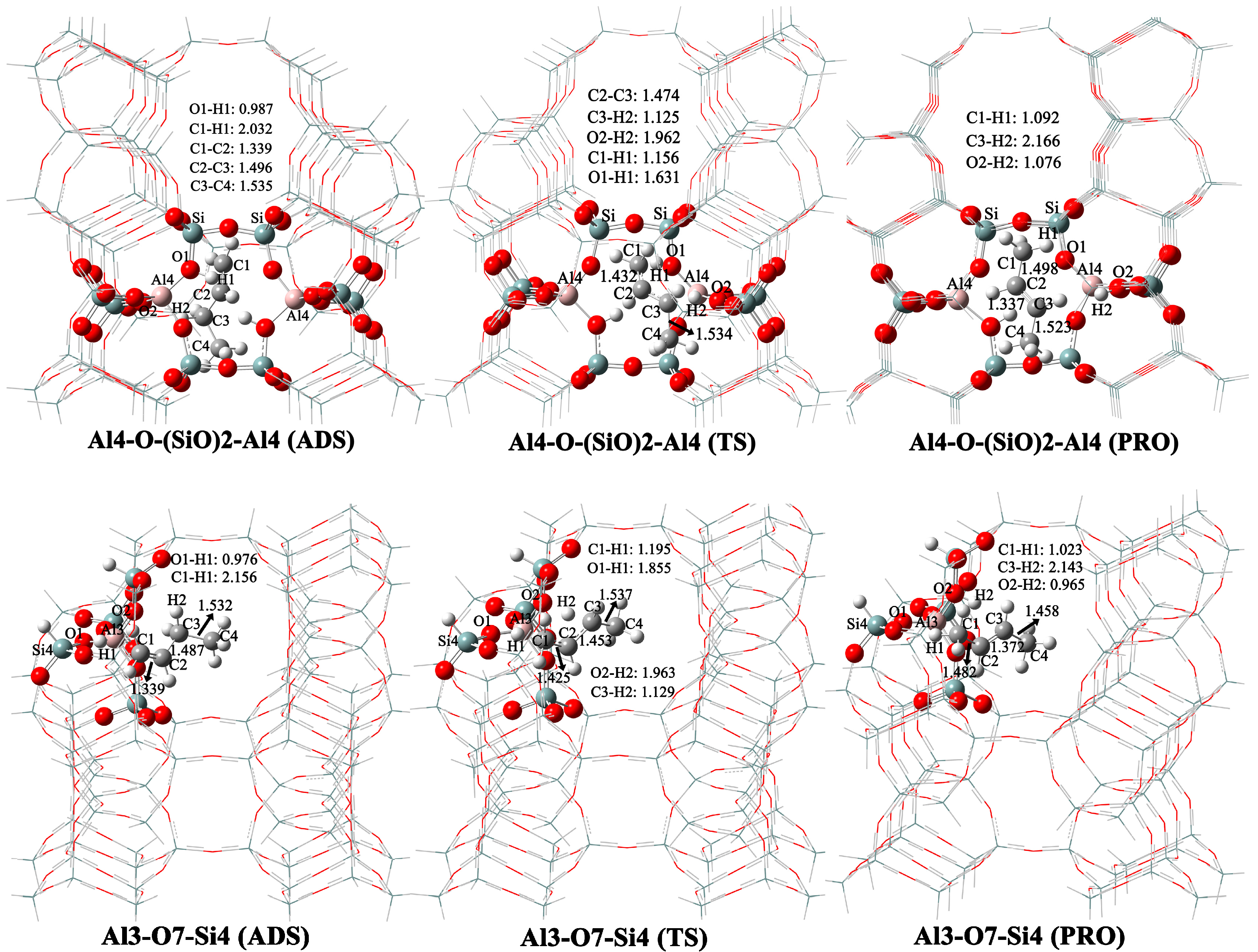
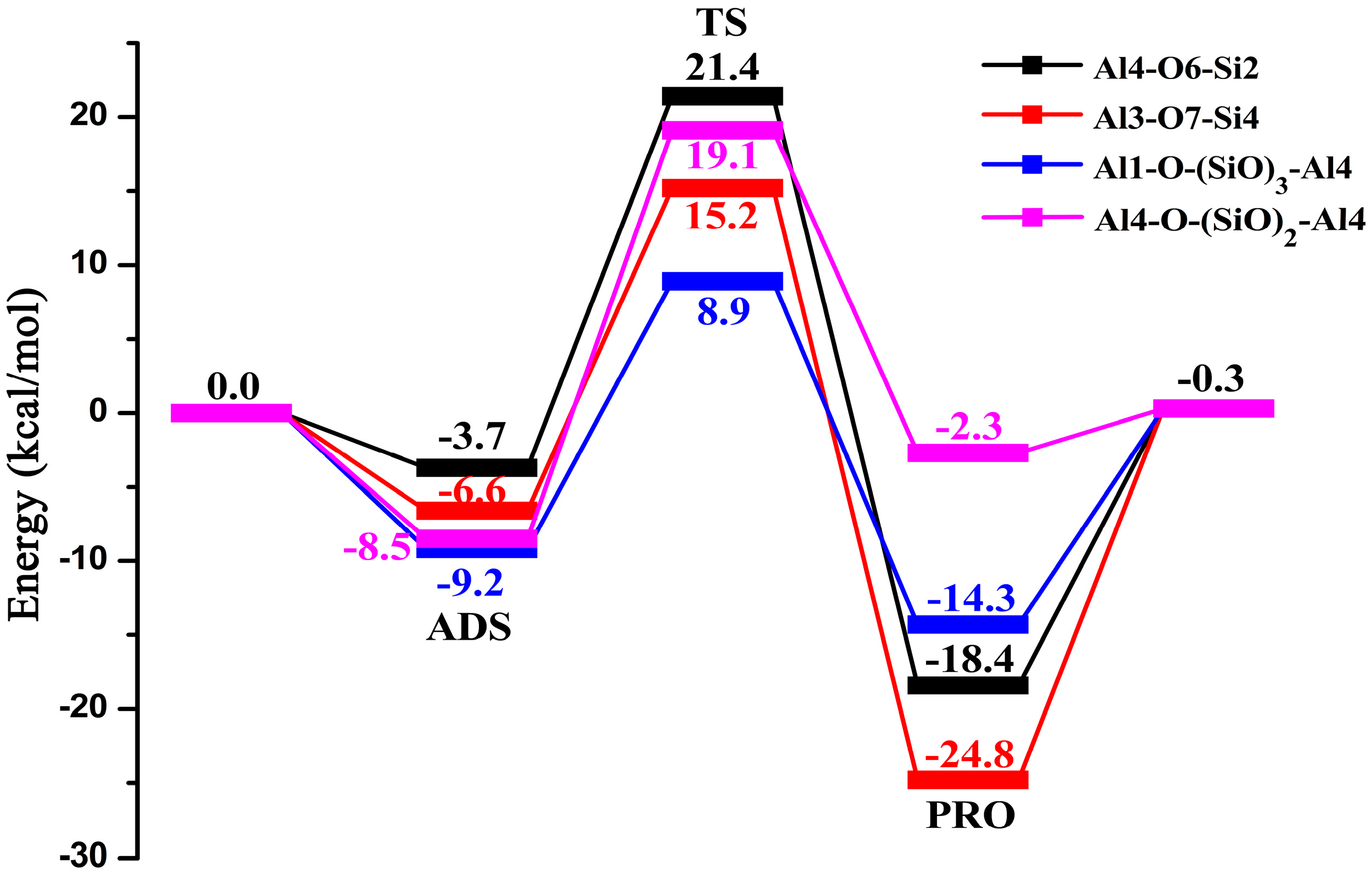
2.3. Butene Activation
3. Computational Details
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Pavlov, M.L.; Shavaleev, D.A.; Kutepov, B.I.; Travkina, O.S.; Pavlova, I.N.; Basimova, R.A.; Ershtein, A.S.; Gerzeliev, I.M. Synthesis and investigation of ZSM-5 zeolite-based catalysts for benzene alkylation with ethylene. Petrol. Chem. 2016, 56, 151–157. [Google Scholar] [CrossRef]
- Jin, W.K.; Kim, D.J.; Han, J.U.; Min, K.; Ji, M.K.; Yie, J.E. Preparation and characterization of zeolite catalysts for etherification reaction. Catal. Today 2003, 87, 195–203. [Google Scholar]
- Khitev, Y.P.; Ivanova, I.I.; Kolyagin, Y.G.; Ponomareva, O.A. Skeletal isomerization of 1-butene over micro/mesoporous materials based on FER zeolite. Appl. Catal. A 2012, 441–442, 124–135. [Google Scholar] [CrossRef]
- Sklenak, S.; Li, C.; Wichterlová, B.; Gábová, V.; Sierka, M.; Sauer, J. Aluminum siting in silicon-rich zeolite frameworks: A combined high-resolution 27Al NMR spectroscopy and quantum mechanics / molecular mechanics study of ZSM-5. Angew. Chem. Int. Ed. 2007, 46, 7286–7289. [Google Scholar] [CrossRef] [PubMed]
- Bokhoven, J.A.V.; Lee, T.L.; Drakopoulos, M.; Lamberti, C.; Thiess, S.; Zegenhagen, J. Determining the aluminium occupancy on the active T sites in zeolites using X-ray standing waves. Nat. Mater. 2008, 7, 551–555. [Google Scholar] [CrossRef] [PubMed]
- Houžvička, J.; Ponec, V. Skeletal isomerisation of N-butene on phosphorus containing catalysts. Appl. Catal. A 1996, 145, 95–109. [Google Scholar] [CrossRef]
- Maciver, D.S.; Wilmot, W.H.; Bridges, J.M. Catalytic aluminas: II. Catalytic properties of eta and gamma alumina. J. Catal. 1964, 3, 502–511. [Google Scholar] [CrossRef]
- Donk, S.V.; Bus, E.; Broersma, A.; Bitter, J.H.; Jong, K.P.D. Probing the accessible sites for n-butene skeletal isomerization over aged and selective H-ferrierite with d3-acetonitrile. J. Catal. 2002, 212, 86–93. [Google Scholar] [CrossRef]
- Hu, Y.; Liu, L.; Zhang, H.; Hu, L.; Zhang, C.; Zhang, H. Effect of crystal size on the skeletal isomerization of n-butene over H-FER zeolite. React. Kinet. Mech. Cat. 2014, 112, 241–248. [Google Scholar] [CrossRef]
- Gleeson, D. Skeletal isomerization of butene in ferrierite: Assessing the energetic and structural differences between carbenium and alkoxide based pathways. J. Phys. Chem. A 2011, 115, 14629–14636. [Google Scholar] [CrossRef] [PubMed]
- Oyoung, C.L.; Pellet, R.J.; Casey, D.G.; Ugolini, J.R.; Sawicki, R.A. Skeletal isomerization of 1-butene on 10-member ring zeolite catalysts. J. Catal. 1995, 151, 467–469. [Google Scholar] [CrossRef]
- Dědeček, J.; Kaucký, D.; Wichterlová, B.; Gonsiorová, O. Co2+ ions as probes of Al distribution in the framework of zeolites ZSM-5 study. Phys. Chem. Chem. Phys. 2002, 4, 5406–5413. [Google Scholar] [CrossRef]
- Dědeček, J.; Kaucký, D.; Wichterlová, B. Al distribution in ZSM-5 zeolites: An experimental study. Chem. Commun. 2001, 11, 970–971. [Google Scholar] [CrossRef]
- Sklenak, S.; Dedecek, J.; Li, C.; Wichterlová, B.; Gábová, V.; Sierka, M.; Sauer, J. Aluminium siting in the ZSM-5 framework by combination of high resolution 27Al NMR and DFT/MM calculations. Phys. Chem. Chem. Phys. 2009, 11, 1237–1247. [Google Scholar] [CrossRef] [PubMed]
- Sklenak, S.; Andrikopoulos, P.C.; Whittleton, S.R.; Jirglova, H.; Sazama, P.; Benco, L.; Bucko, T.; Hafner, J.; Sobalik, Z. Effect of the Al siting on the structure of Co(II) and Cu(II) cationic sites in ferrierite. A periodic DFT molecular dynamics and FTIR Study. J. Phys. Chem. C 2013, 117, 3958–3968. [Google Scholar] [CrossRef]
- Sastre, G.; Katada, N.; Suzuki, K.; Niwa, M. Computational study of Brønsted acidity of Faujasite: Effect of the Al content on the infrared OH stretching frequencies. J. Phys. Chem. C 2008, 63, 19293–19301. [Google Scholar] [CrossRef]
- Zhou, D.; Ying, B.; Yang, M.; Ning, H.; Gang, Y. DFT studies on the location and acid strength of Brönsted acid sites in MCM-22 zeolite. J. Mol. Catal. A 2006, 244, 11–19. [Google Scholar] [CrossRef]
- Lo, C.; Trout, B.L. Density functional theory characterization of acid sites in chabazite. J. Catal. 2004, 227, 77–89. [Google Scholar] [CrossRef]
- Dědeček, J.; Sklenak, S.; Li, C.; Wichterlová, B.; Gábová, V.; Brus, J.; Sierka, M.; Sauer, J. Effect of Al−Si−Al and Al−Si−Si−Al pairs in the ZSM-5 zeolite framework on the 27Al NMRspectra. A combined high-resolution 27Al NMR and DFT/MM study. J. Phys. Chem. C 2009, 113, 1447–1458. [Google Scholar] [CrossRef]
- Sazama, P.; Tabor, E.; Klein, P.; Wichterlova, B.; Sklenak, S.; Mokrzycki, L.; Pashkkova, V.; Ogura, M.; Dedecek, J. Al-rich beta zeolites distribution of Al atoms in the framework and related protonic and metal-ion species. J. Catal. 2016, 333, 102–114. [Google Scholar] [CrossRef]
- Mooiweer, H.H.; Jong, K.P.D.; Kraushaar-Czametzki, B.; Stork, W.H.J.; Krutzen, B.C.H. Skeletal isomerization of olefins with the zeolite ferrierite as catalyst. Stud. Surf. Sci. Catal. 1994, 84, 2327–2334. [Google Scholar]
- Houžvička, J.; Hansildaar, S.; Ponec, V. The shape selectivity in the skeletal isomerization of n-butene to isobutene. J. Catal. 1997, 167, 273–278. [Google Scholar] [CrossRef]
- Dedecek, J.; Lucero, M.J.; Li, C.; Gao, F.; Klein, P.; Urbanova, M.; Tvaruzkova, Z.; Sazama, P.; Sklenak, S. Complex analysis of the aluminum siting in the framework of silicon-rich zeolites. A case study on ferrierites. J. Phys. Chem. C 2011, 115, 11056–11064. [Google Scholar] [CrossRef]
- Simperler, A.; Bell, R.; Anderson, M. Probing the acid strength of Brönsted acidic zeolites with acetonitrile: Quantum chemical calculation of H1, N15 and C13 NMR shift parameters. J. Phys. Chem. B 2004, 108, 7142–7151. [Google Scholar] [CrossRef]
- Liu, R.; Zhang, J.; Sun, X.; Huang, C.; Chen, B. An oniom study on the distribution, local structure and strength of Brönsted acid sites in FER zeolite. Comput. Theor. Chem. 2014, 1027, 5–10. [Google Scholar] [CrossRef]
- Benco, L.; Bucko, T.; Grybos, R.; Hafner, J.; Sobalik, Z.; Dedecek, J.; Hrusak, J. Adsorption of NO in Fe2+-exchanged ferrierite. A density functional theory study. J. Phys. Chem. C 2007, 111, 586–595. [Google Scholar] [CrossRef]
- Kikhtyanin, O.; Kubička, D.; Čejkab, J. Toward understanding of the role of Lewis acidity in aldol condensation of acetone and furfural using MOF and zeolite catalysts. Catal. Today 2015, 243, 158–162. [Google Scholar] [CrossRef]
- Grajciar, L.; Arean, C.O.; Pulido, A.; Nachtigall, P. Periodic DFT investigation of the effect of aluminium content on the properties of the acid zeolite H-FER. Phys. Chem. Chem. Phys. 2010, 12, 1497–1506. [Google Scholar] [CrossRef] [PubMed]
- Feng, P.; Chen, X.F.; Li, X.J.; Zhao, D.; Xie, S.J.; Xu, L.Y.; He, G.Z. The distribution analysis on the proton siting and the acid strength of the zeolite ferrierite: A computational study. Microporous Mesoporous Mater. 2017, 239, 354–362. [Google Scholar] [CrossRef]
- Nachtiqall, P.; Bludsky, O.; Grajciar, L.; Nachtiqallova, D.; Delgado, M.R.; Arean, C.O. Computational and FTIR spectroscopic studies on carbon monoxide and dinitrogen adsorption on a high-silica H-FER zeolite. Phys. Chem. Chem. Phys. 2009, 11, 791–802. [Google Scholar] [CrossRef] [PubMed]
- Simperler, A.; Bell, R.G.; Foster, M.D.; Gray, A.E.; Lewis, D.W.; Anderson, M.W. Probing the acid strength of Brönsted acidic zeolites with acetonitrile: An atomistic and quantum chemical study. J. Phys. Chem. B 2004, 108, 7152–7161. [Google Scholar] [CrossRef]
- Takaishi, T.; Kato, M.; Itabashi, K. Stability of the Al-O-Si-O-Al linkage in a zeolitic framework. J. Phys. Chem. 2002, 98, 5742–5743. [Google Scholar] [CrossRef]
- Benco, L.; Bucko, T.; Hafner, J. Activity and reactivity of Fe2+ cations in the zeolite: Ab initio free-energy MD calculation of the N2O dissociation over iron-exchanged ferrierite. J. Phys. Chem. C 2009, 113, 18807–18816. [Google Scholar] [CrossRef]
- Zhao, G.; Teng, J.; Zhang, Y.; Xie, Z.; Yue, Y.; Chen, Q.; Yi, T. Synthesis of ZSM-48 zeolites and their catalytic performance in C4-olefin cracking reactions. Appl. Catal. A 2006, 299, 167–174. [Google Scholar] [CrossRef]
- Boronat, M.; Viruela, P.; Corma, A. Theoretical study of the mechanism of zeolite-catalyzed isomerization reactions of linear butenes. J. Phys. Chem. A 1998, 102, 982–989. [Google Scholar] [CrossRef]
- Li, H.Y.; Pu, M.; Liu, K.H.; Zhang, B.F.; Chen, B.H. A density functional theory study on double-bond isomerization of 1-butene to cis-2-butene catalyzed by zeolites. Chem. Phys. Lett. 2005, 404, 384–388. [Google Scholar] [CrossRef]
- Kondo, J.N.; Domen, K. IR observation of adsorption and reactions of olefins on H-form zeolites. J. Mol. Catal. A 2003, 199, 27–38. [Google Scholar] [CrossRef]
- Vaughan, P.A. The crystal structure of the zeolite ferrierite. Acta Crystallogr. 1966, 21, 983–990. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Revision C.01; Gaussian, Inc.: Wallingford, CT, USA, 2010.
- Wattanakit, C.; Nokbin, S.; Boekfa, B.; Pantu, P.; Limtrakul, J. Skeletal isomerization of 1-butene over ferrierite zeolite: A quantum chemical analysis of structures and reaction mechanisms. J. Phys. Chem. C 2012, 116, 5654–5663. [Google Scholar] [CrossRef]
- Ferrante, F.; Rubino, T.; Duca, D. Butene isomerization and double-bond migration on the H-ZSM-5 outer surface: A density functional theory study. J. Phys. Chem. C 2011, 115, 14862–14868. [Google Scholar] [CrossRef]
- Fellah, M.F.; Santen, R.A.V.; Onal, I. Oxidation of benzene to phenol by N2O on an Fe2+-ZSM-5 cluster: A density functional theory study. J. Phys. Chem. C 2009, 113, 15307–15313. [Google Scholar] [CrossRef]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number | Brönsted Acid Sites | SE(Al/Si) (kcal/mol) | ED (kcal/mol) |
|---|---|---|---|
| 0 | Al1-O4-Si1 | 22.2 | - |
| 1 | Al1-O1-Si3 | 8.3 | 5.8 |
| 2 | Al1-O2-Si2 | 5.3 | 8.7 |
| 3 | Al1-O3-Si1 | 7.0 | 7.1 |
| 4 | Al2-O2-Si1 | 7.1 | 4.5 |
| 5 | Al2-O5-Si2 | 8.7 | 1.9 |
| 6 | Al2-O6-Si4 | 8.2 | 2.9 |
| 7 | Al3-O1-Si1 | 7.2 | 3.7 |
| 8 | Al3-O7-Si4 | 10.9 | 0.0 |
| 9 | Al3-O8-Si3 | 7.2 | 3.7 |
| 10 | Al4-O6-Si2 | 0.0 | 12.0 |
| 11 | Al4-O7-Si3 | 11.4 | - |
| Substitution Site | SE(Al/Si) (kcal/mol) | ED (kcal/mol) | |
|---|---|---|---|
| T1-O(SiO)-T4 | Al1-OH-()-OH-Al4 | 17.1 | 3.8 |
| Al1-OH-()-Al4-OH | 16.8 | 3.1 | |
| T1-O(SiO)2-T4 | Al1-OH-()-OH-Al4 | 16.6 | 3.4 |
| Al1-OH-()-Al4-OH | 14.9 | 5.1 | |
| T1-O(SiO)3-T4 | Al1-OH-()-OH-Al4 | 15.1 | 0 |
| Al1-OH-()-O-Al4-OH | 15.3 | 5.0 | |
| T4-O(SiO)2-T4 | Al4-OH-()-Al4-OH | 0 | 7.7 |
| Al4-OH-()-OH-Al4 | 7.8 | 6.5 | |
| T1-T4-T4 (Al > 2) | Al1-O1-Si3 | 1.9 | 33.6 |
| Al1-O2-Si2 | 6.6 | 28.9 | |
| Al1-O3-Si1 | 3.6 | 31.9 | |
| Substitution Site | θ0/(°) | θ1/(°) | θ2/(°) | l1/Å | l2/Å |
|---|---|---|---|---|---|
| Al4-O(SiO)2-Si4 | 141.4 | - | - | - | - |
| Al4-(OSi)2O-Al4 | 143.9 | - | - | - | - |
| Al4-OH-(SiO)2-Si4 | - | 139.1 | 135.5 | 0.97 | 2.52 |
| Al4-OH-SiOSi-OH-Al4 | - | 136.3 | 130.3 | 0.98 | 1.99 |
© 2017 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
He, M.; Zhang, J.; Liu, R.; Sun, X.; Chen, B. The Distribution and Strength of Brönsted Acid Sites on the Multi-Aluminum Model of FER Zeolite: A Theoretical Study. Catalysts 2017, 7, 11. https://0-doi-org.brum.beds.ac.uk/10.3390/catal7010011
He M, Zhang J, Liu R, Sun X, Chen B. The Distribution and Strength of Brönsted Acid Sites on the Multi-Aluminum Model of FER Zeolite: A Theoretical Study. Catalysts. 2017; 7(1):11. https://0-doi-org.brum.beds.ac.uk/10.3390/catal7010011
Chicago/Turabian StyleHe, Miao, Jie Zhang, Rui Liu, Xiuliang Sun, and Biaohua Chen. 2017. "The Distribution and Strength of Brönsted Acid Sites on the Multi-Aluminum Model of FER Zeolite: A Theoretical Study" Catalysts 7, no. 1: 11. https://0-doi-org.brum.beds.ac.uk/10.3390/catal7010011