Unravelling Heterogeneity of Amplified Human Amniotic Fluid Stem Cells Sub-Populations

 ,
,  ,
,  , , and
, , and 
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Amniotic Fluid Collection
2.2. Adult Human Tissue Isolation and Cell Culture
2.3. Cellular Proliferation
2.4. hAFSCs Sub-Populations Sorting
2.5. Immunofluorescence and Confocal Microscopy
2.6. Differentiation Protocol
2.7. Alizarin Red S Staining and Mineralization
2.8. CFU Analysis
2.9. Senescence Assay
2.10. RNA Isolation and Quantification
2.11. cDNA Retrotranscription and qRT-PCR
2.12. RNA and DNA Quality Check
2.13. Library Preparation and RNAseq
2.14. ELISA Assays
2.15. Mononuclear Cell Separation
2.16. Apoptosis Assay of PBMC Exposed to AFSC CM
2.17. SDS PAGE and Western Blot
2.18. Statistical Analysis
2.19. Ethics Approval and Consent to Participate
3. Results
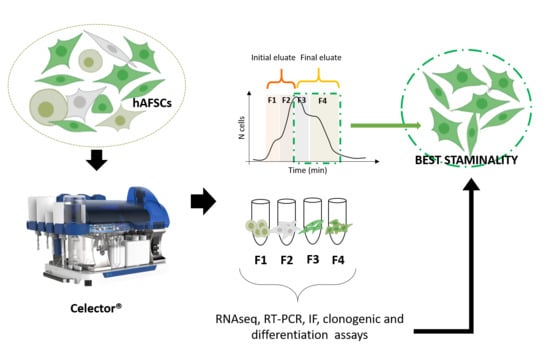
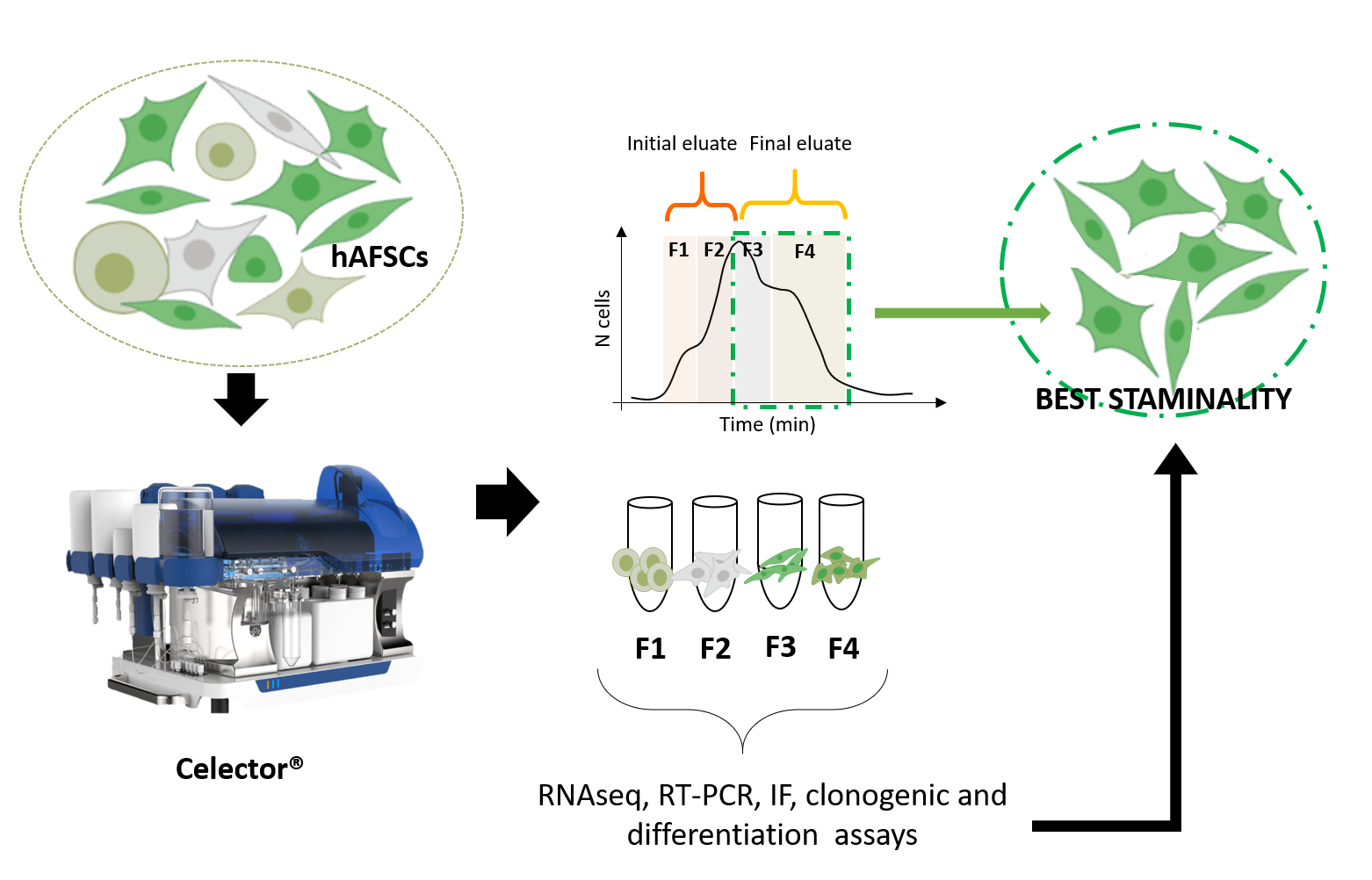
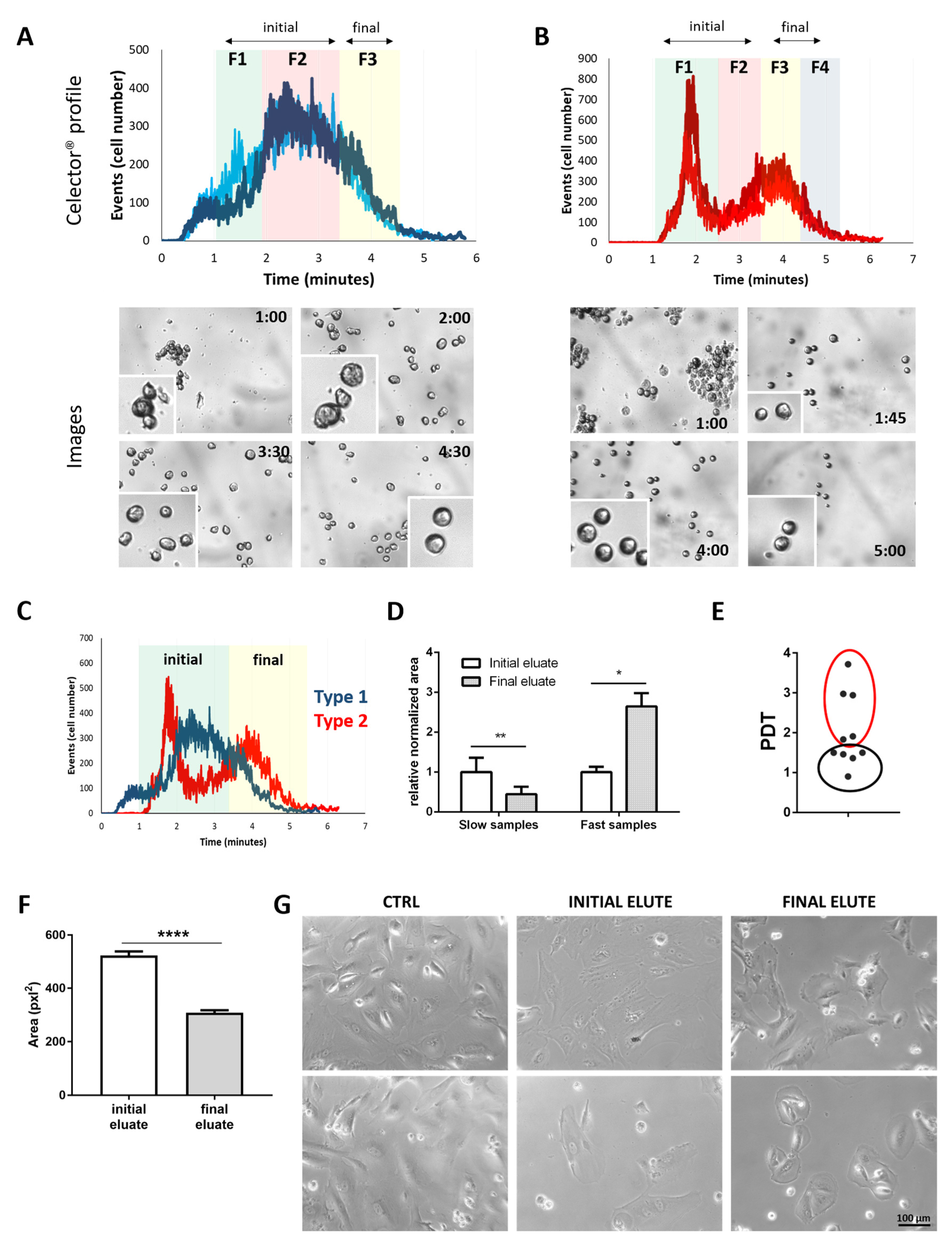
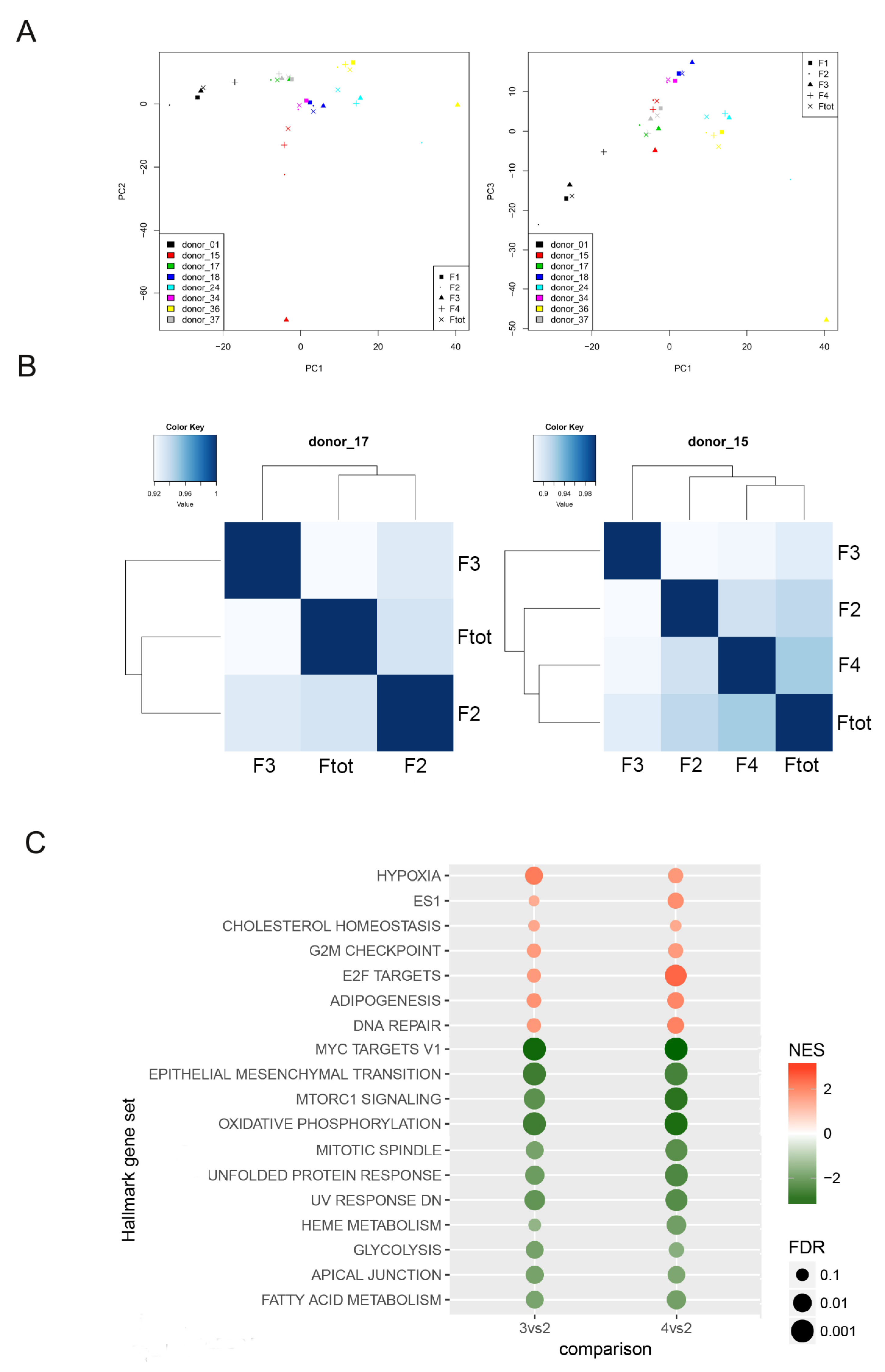
3.1. Sorting of hAFSCs by Celector® Technology
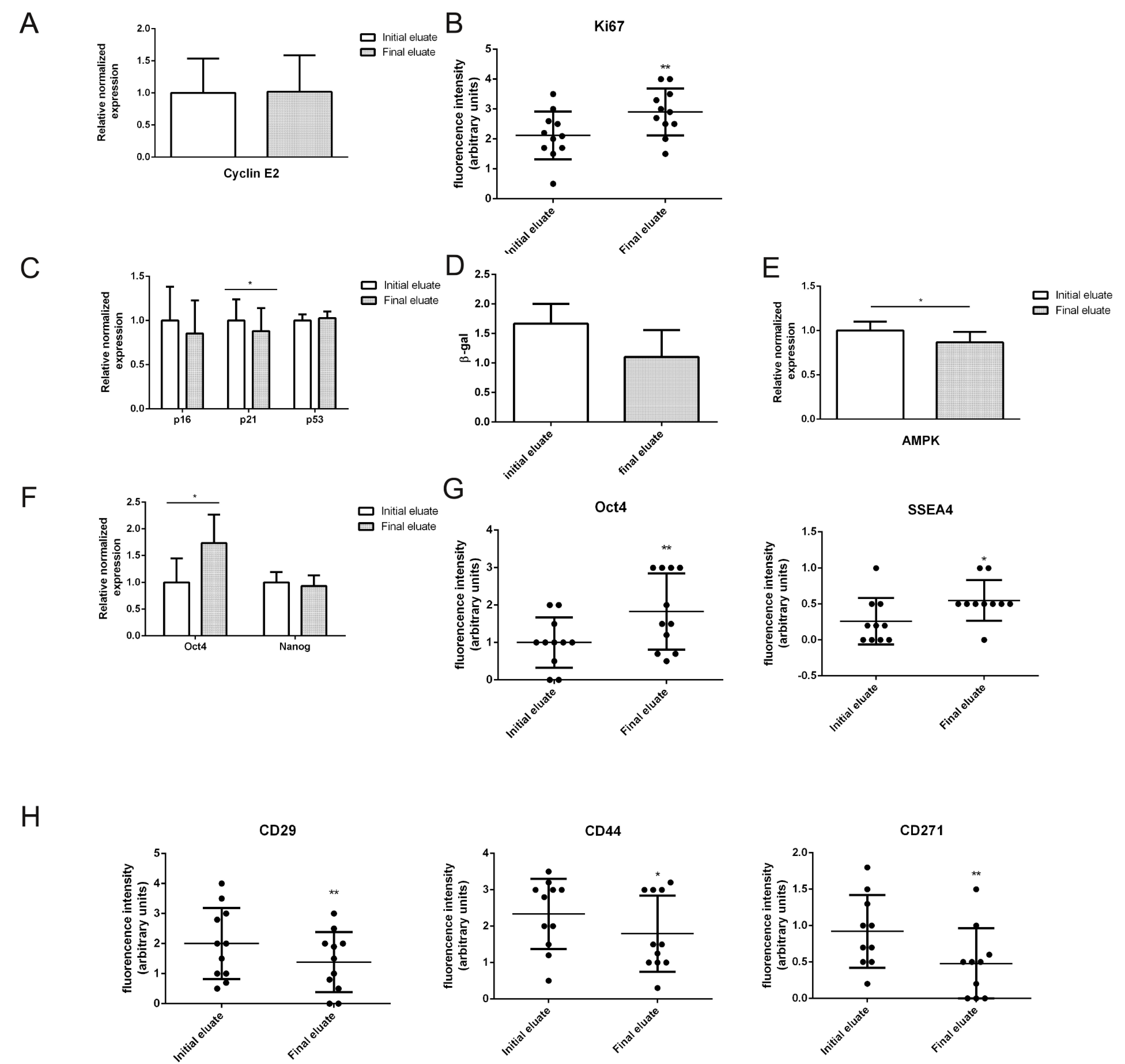
3.2. Specific Characteristics of hAFSCs Fractions
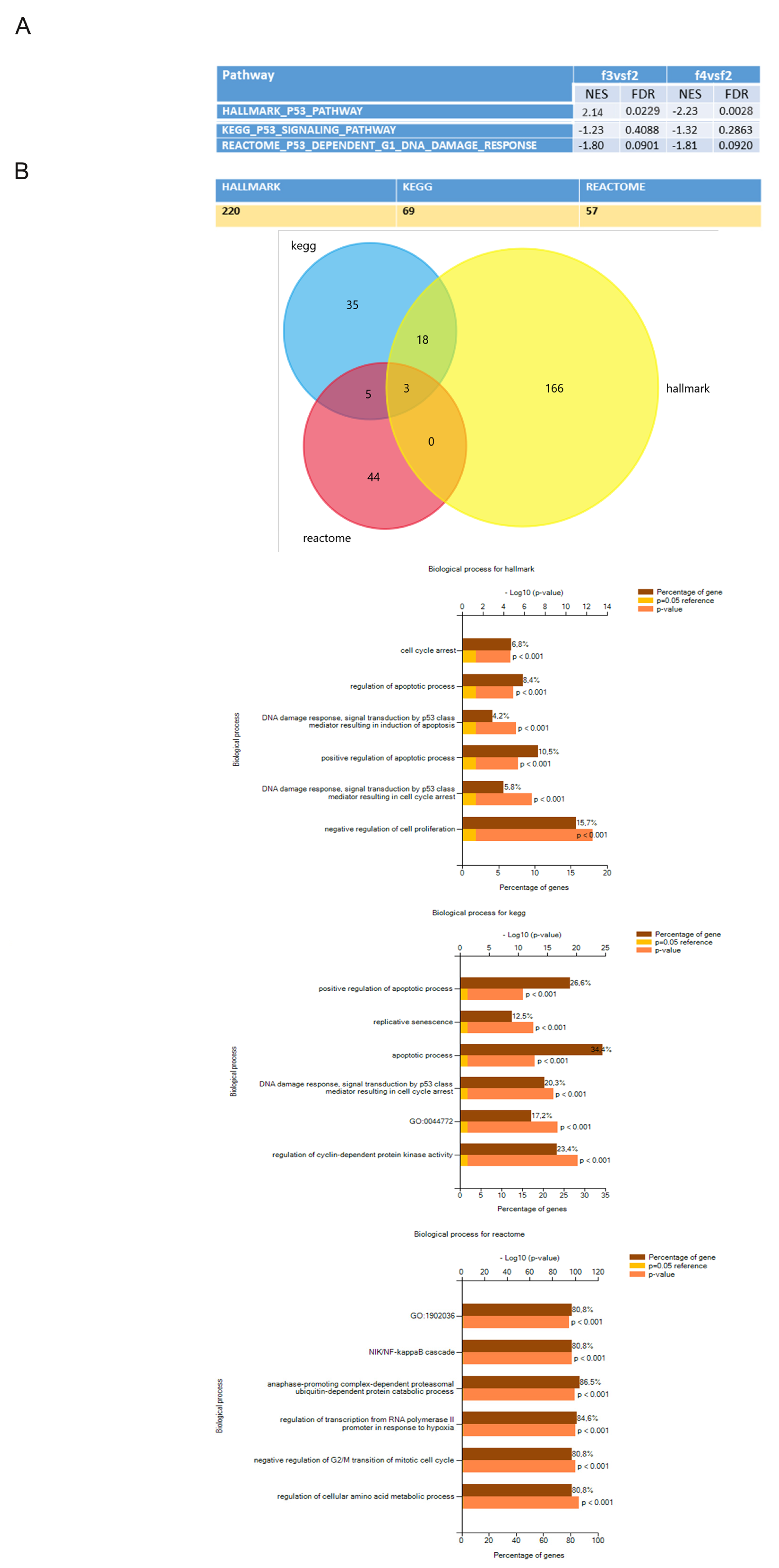
3.3. Transcriptomic Profiles of Different hAFSCs Fractions
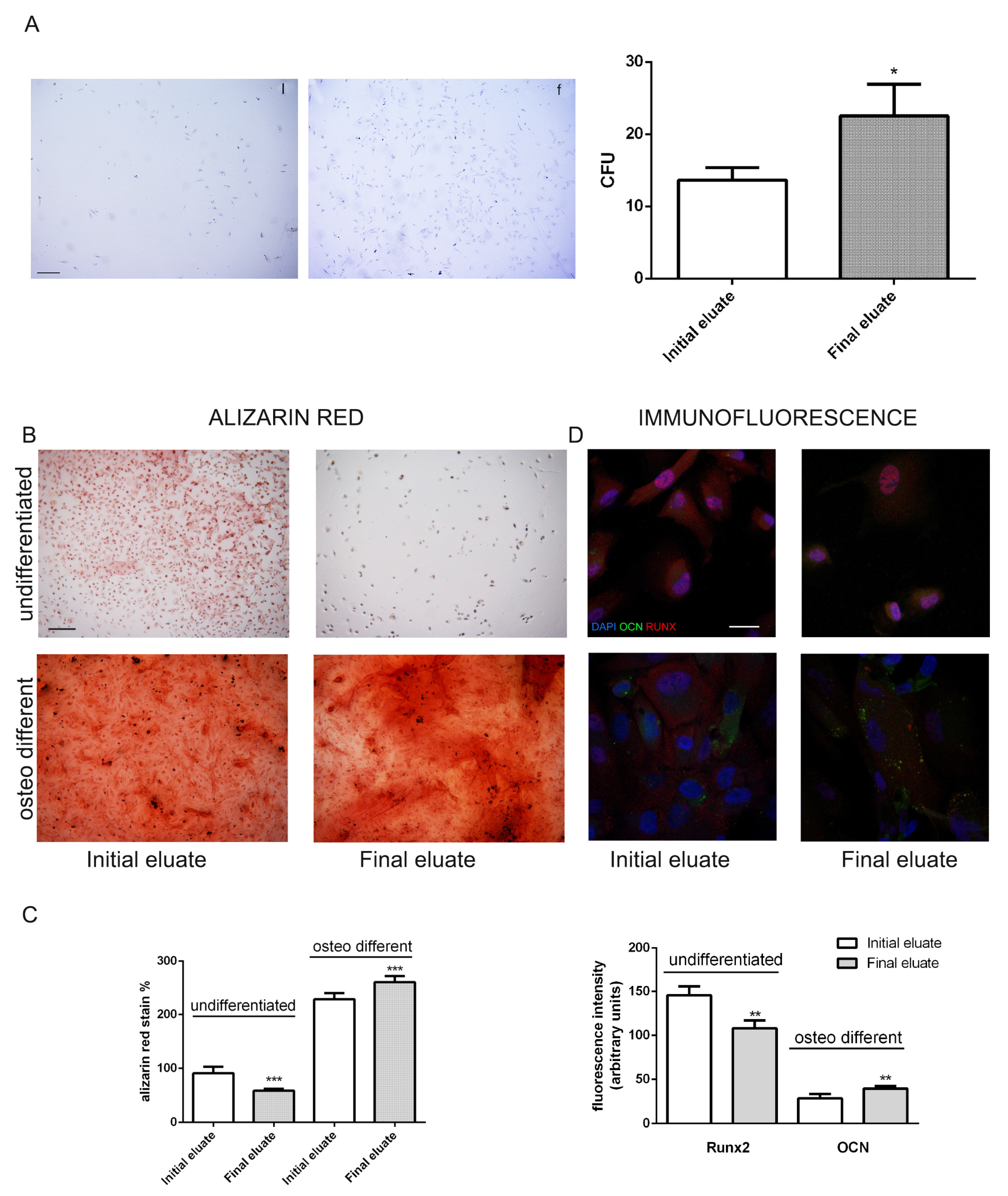
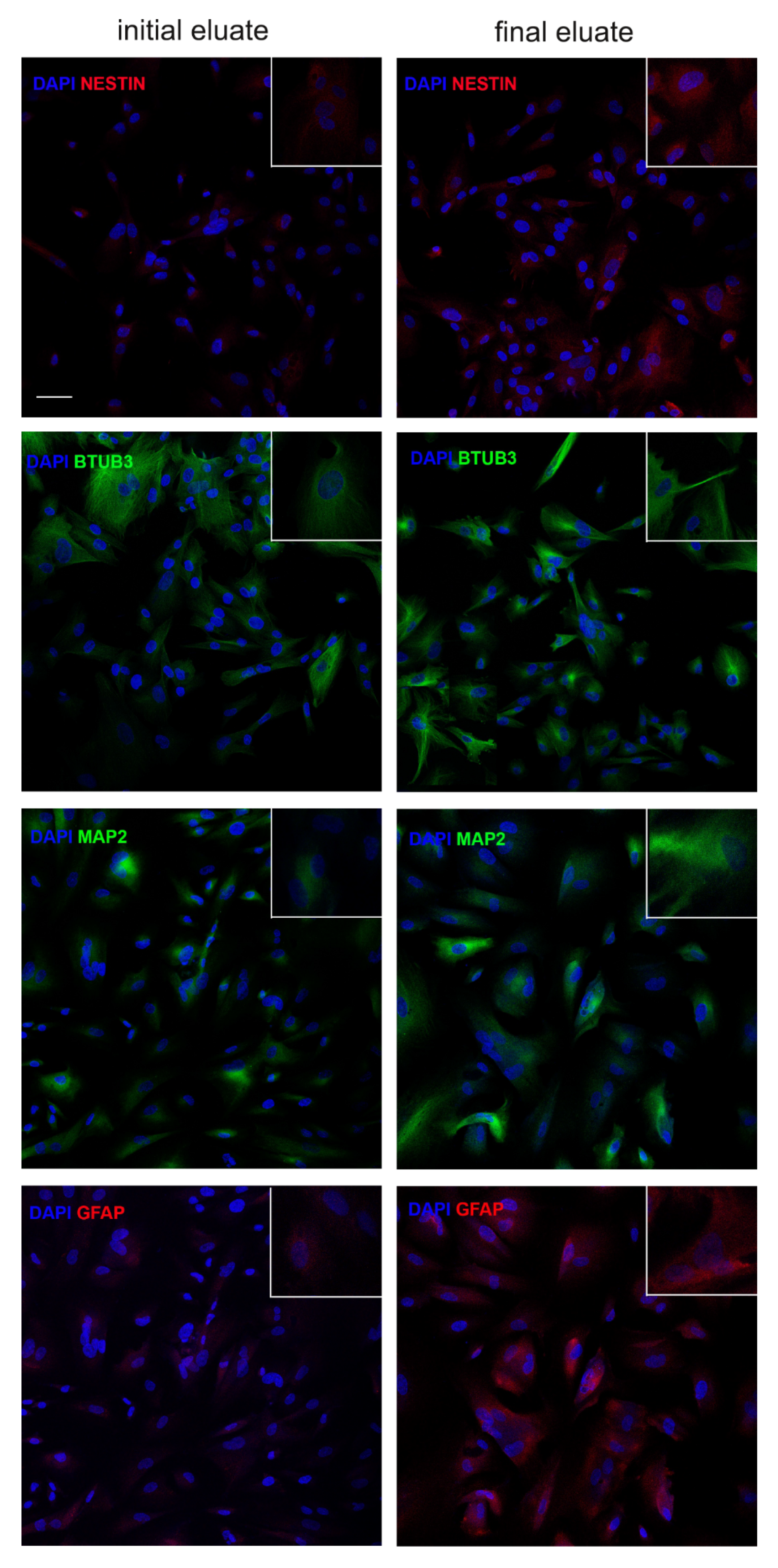
3.4. Clonogenic and Differentiation Ability of hAFSCs Fractions
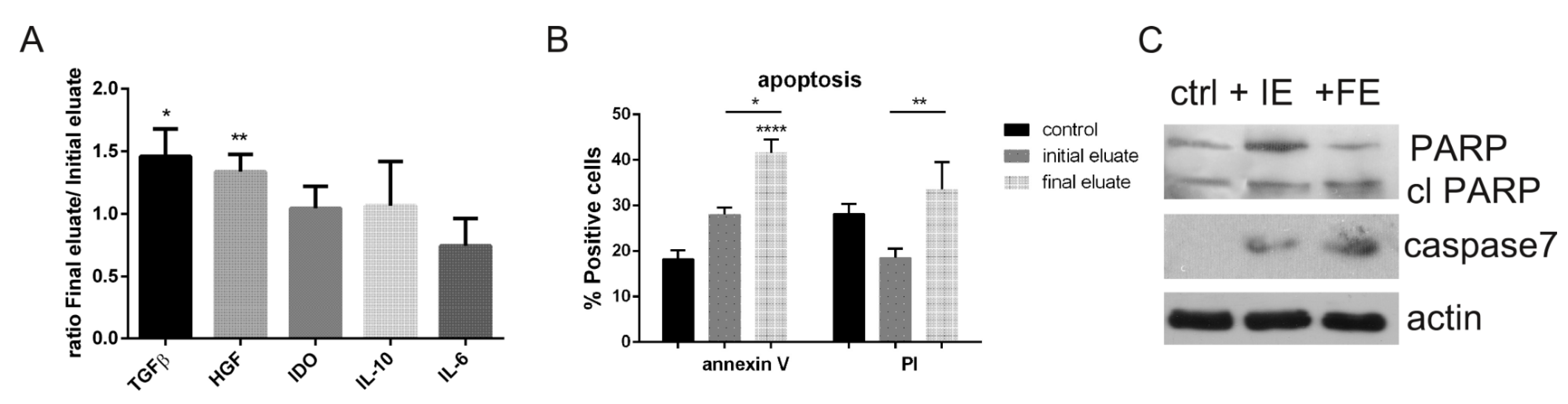
3.5. Analysis of the Secretome Produced by hAFSC Fractions
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| hAFSCs | Human amniotic fluid stem cells |
| CFU | Colony forming unit |
| CPM | Counts Per Million mapped reads |
| DAPI | 4′,6-diamidino-2-phenylindole |
| EDTA | Ethylenediaminetetraacetic acid |
| FBS | foetal bovine serum |
| MSCs | mesenchymal stem cells |
| OCN | osteocalcin |
| PBS | phosphate buffered saline |
| PCA | principal component analysis |
| RNAseq | RNA sequencing |
| RPKM | Reads Per Kilobase per Million mapped reads |
| SE | Standard Error |
| UPR | unfolded protein response |
References
- Lin, F.; Moran, A.; Igarashi, P. Intrarenal cells, not bone marrow–derived cells, are the major source for regeneration in postischemic kidney. J. Clin. Investig. 2005, 115, 1756–1764. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Xu, Y.; Zhao, J.; Zhang, Z.; Yang, R.; Xie, J.; Liu, X.; Qi, S. Conditioned medium from hypoxic bone marrow-derived mesenchymal stem cells enhances wound healing in mice. PLoS ONE 2014, 9, e96161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Blanc, K.; Tammik, L.; Sundberg, B.; Haynesworth, S.E.; Ringdén, O. Mesenchymal Stem Cells Inhibit and Stimulate Mixed Lymphocyte Cultures and Mitogenic Responses Independently of the Major Histocompatibility Complex. Scand. J. Immunol. 2003, 57, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Mo, M.; Wang, S.; Zhou, Y.; Li, H.; Wu, Y. Mesenchymal stem cell subpopulations: Phenotype, property and therapeutic potential. Cell. Mol. Life Sci. 2016, 73, 3311–3321. [Google Scholar] [CrossRef] [PubMed]
- Lukomska, B.; Stanaszek, L.; Zuba-Surma, E.; Legosz, P.; Sarzynska, S.; Drela, K. Challenges and Controversies in Human Mes-enchymal Stem Cell Therapy. Stem Cells Int. 2019, 2019, 9628536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dufrane, D. Impact of Age on Human Adipose Stem Cells for Bone Tissue Engineering. Cell Transplant. 2017, 26, 1496–1504. [Google Scholar] [CrossRef]
- Liu, M.; Lei, H.; Dong, P.; Fu, X.; Yang, Z.; Yang, Y.; Ma, J.; Liu, X.; Cao, Y.; Xiao, R. Adipose-derived mesenchymal stem cells from the elderly exhibit decreased migration and differentiation abilities with senescent properties. Cell Transplant. 2017, 26, 1505–1519. [Google Scholar] [CrossRef] [Green Version]
- Kokai, L.E.; Traktuev, D.O.; Zhang, L.; Merfeld-Clauss, S.; Dibernardo, G.; Lu, H.; Marra, K.G.; Donnenberg, A.; Donnenberg, V.; Meyer, E.M.; et al. Adipose Stem Cell Function Maintained with Age: An Intra-Subject Study of Long-Term Cryopreserved Cells. Aesthetic Surg. J. 2016, 37, 454–463. [Google Scholar] [CrossRef] [Green Version]
- Pachón-Peña, G.; Serena, C.; Ejarque, M.; Petriz, J.; Duran, X.; Oliva-Olivera, W.; Simó, R.; Tinahones, F.J.; Fernández-Veledo, S.; Vendrell, J. Obesity Determines the Immunophenotypic Profile and Functional Characteristics of Human Mesenchymal Stem Cells From Adipose Tissue. Stem Cells Transl. Med. 2016, 5, 464–475. [Google Scholar] [CrossRef]
- Wagner, W.; Ho, A.D.; Zenke, M. Different Facets of Aging in Human Mesenchymal Stem Cells. Tissue Eng. Part B Rev. 2010, 16, 445–453. [Google Scholar] [CrossRef]
- Antonucci, I.; Di Pietro, R.; Alfonsi, M.; Centurione, M.A.; Centurione, L.; Sancilio, S.; Pelagatti, F.; D’Amico, M.A.; Di Baldassarre, A.; Piattelli, A.; et al. Human Second Trimester Amniotic Fluid Cells are Able to Create Embryoid Body-Like Structures in Vitro and to Show Typical Expression Profiles of Embryonic and Primordial Germ Cells. Cell Transplant. 2014, 23, 1501–1515. [Google Scholar] [CrossRef] [PubMed]
- Roubelakis, M.G.; Pappa, K.I.; Bitsika, V.; Zagoura, D.; Vlahou, A.; Papadaki, H.A.; Antsaklis, A.; Anagnou, N.P. Molecular and Proteomic Characterization of Human Mesenchymal Stem Cells Derived from Amniotic Fluid: Comparison to Bone Marrow Mesenchymal Stem Cells. Stem Cells Dev. 2007, 16, 931–952. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramasamy, T.S.; Velaithan, V.; Yeow, Y.; Sarkar, F.H. Stem Cells Derived from Amniotic Fluid: A Potential Pluripotent-Like Cell Source for Cellular Therapy? Curr. Stem Cell Res. Ther. 2018, 13, 252–264. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lesage, F.; Zia, S.; Jiménez, J.A.; Deprest, J.; Toelen, J. The amniotic fluid as a source of mesenchymal stem cells with lung-specific characteristics. Prenat. Diagn. 2017, 37, 1093–1099. [Google Scholar] [CrossRef] [PubMed]
- Da Sacco, S.; Sedrakyan, S.; Boldrin, F.; Giuliani, S.; Parnigotto, P.; Habibian, R.; Warburton, D.; De Filippo, R.E.; Perinet, L. Human amniotic fluid as a potential new source of organ specific precursor cells for future regenerative medicine applications. J. Urol. 2010, 183, 1193–1200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Coppi, P.; Bartsch, G.; Siddiqui, M.M.; Xu, T.; Santos, C.C.; Perin, L.; Perin, L.; Mostoslavsky, G.; Serre, A.C.; Snyder, E.Y.; et al. Isolation of amniotic stem cell lines with potential for therapy. Nat. Biotechnol. 2007, 25, 100–106. [Google Scholar] [CrossRef]
- Sart, S.; Song, L.; Li, Y. Controlling Redox Status for Stem Cell Survival, Expansion, and Differentiation. Oxid. Med. Cell. Longev. 2015, 2015, 105135. [Google Scholar] [CrossRef] [Green Version]
- Roda, B.; Reschiglian, P.; Zattoni, A.; Alviano, F.; Lanzoni, G.; Costa, R.; Di Carlo, A.; Marchionni, C.; Franchina, M.; Bonsi, L.; et al. A tag-less method of sorting stem cells from clini-cal specimens and separating mesenchymal from epithelial progenitor cells. Cytom. Part B Clin. Cytom. 2009, 76, 285–290. [Google Scholar] [CrossRef]
- Lattuada, D.; Roda, B.; Pignatari, C.; Magni, R.; Colombo, F.; Cattaneo, A.; Zattoni, A.; Cetin, I.; Reschiglian, P.; Bolis, G. A tag-less method for direct isolation of human umbilical vein endothelial cells by gravitational field-flow fractionation. Anal. Bioanal. Chem. 2012, 405, 977–984. [Google Scholar] [CrossRef]
- Beretti, F.; Zavatti, M.; Casciaro, F.; Comitini, G.; Franchi, F.; Barbieri, V.; La Sala, G.B.; Maraldi, T. Amniotic fluid stem cell exosomes: Therapeutic perspective. BioFactors 2018, 44, 158–167. [Google Scholar] [CrossRef]
- Casciaro, F.; Beretti, F.; Zavatti, M.; McCubrey, J.A.; Ratti, S.; Marmiroli, S.; Follo, M.Y.; Maraldi, T. Nuclear Nox4 interaction with prelamin A is as-sociated with nuclear redox control of stem cell aging. Aging 2018, 10, 2911–2934. [Google Scholar] [CrossRef] [PubMed]
- Guida, M.; Maraldi, T.; Resca, E.; Beretti, F.; Zavatti, M.; Bertoni, L.; La Sala, G.B.; De Pol, A. Inhibition of Nuclear Nox4 Activity by Plumbagin: Effect on Proliferative Capacity in Human Amniotic Stem Cells. Oxid. Med. Cell. Longev. 2013, 2013, 680816. [Google Scholar] [CrossRef] [PubMed]
- Maraldi, T.; Riccio, M.; Pisciotta, A.; Zavatti, M.; Carnevale, G.; Beretti, F.; La Sala, G.B.; Motta, A.; De Pol, A. Human amniotic fluid-derived and dental pulp-derived stem cells seeded into collagen scaffold repair critical-size bone defects promoting vascularization. Stem Cell Res. Ther. 2013, 4, 53. [Google Scholar] [CrossRef] [Green Version]
- Maraldi, T.; Bertoni, L.; Riccio, M.; Zavatti, M.; Carnevale, G.; Resca, E.; Guida, M.; Beretti, F.; La Sala, G.B.; De Pol, A. Human amniotic fluid stem cells: Neural differentiation in vitro and in vivo. Cell Tissue Res. 2014, 357, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zavatti, M.; Resca, E.; Bertoni, L.; Maraldi, T.; Guida, M.; Carnevale, G.; Ferrari, A.; De Pol, A. Ferutinin promotes proliferation and osteoblastic differentiation in human amniotic fluid and dental pulp stem cells. Life Sci. 2013, 92, 993–1003. [Google Scholar] [CrossRef] [PubMed]
- Zavatti, M.; Beretti, F.; Casciaro, F.; Comitini, G.; Franchi, F.; Barbieri, V.; Bertoni, L.; De Pol, A.; La Sala, G.B.; Maraldi, T. Development of a novel method for amniotic fluid stem cell storage. Cytotherapy 2017, 19, 1002–1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben-Porath, I.; Thomson, M.W.; Carey, V.J.; Ge, R.; Bell, G.W.; Regev, A.; Weinberg, R.A. An embryonic stem cell-like gene expression signa-ture in poorly differentiated aggressive human tumors. Nat. Genet. 2008, 40, 499–507. [Google Scholar] [CrossRef] [PubMed]
- Maraldi, T.; Guida, M.; Beretti, F.; Resca, E.; Carpino, G.; Cardinale, V.; Gentile, R.; Ardizzoni, A.; Murgia, A.; Alvaro, D.; et al. Human biliary tree stem/progenitor cells immunomodulation: Role of hepatocyte growth factor. Hepatol. Res. 2017, 47, 465–479. [Google Scholar] [CrossRef]
- Sobecki, M.; Mrouj, K.; Colinge, J.; Gerbe, F.; Jay, P.; Krasinska, L.; Dulic, V.; Fisher, D. Cell-Cycle Regulation Accounts for Variability in Ki-67 Expression Levels. Cancer Res. 2017, 77, 2722–2734. [Google Scholar] [CrossRef] [Green Version]
- Hernandez-Segura, A.; Nehme, J.; DeMaria, M. Hallmarks of Cellular Senescence. Trends Cell Biol. 2018, 28, 436–453. [Google Scholar] [CrossRef]
- Xiao, B.; Sanders, M.J.; Underwood, E.; Heath, R.; Mayer, F.V.; Carmena, D.; Jing, C.; Walker, P.A.; Eccleston, J.F.; Haire, L.F.; et al. Structure of mammalian AMPK and its regula-tion by ADP—Supplementary Information. Nature 2011, 472, 1–22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barilani, M.; Banfi, F.; Sironi, S.; Ragni, E.; Guillaumin, S.; Polveraccio, F.; Rosso, L.; Moro, M.; Astori, G.; Pozzobon, M.; et al. Low-affinity Nerve Growth Factor Receptor (CD271) Heterogeneous Expression in Adult and Fetal Mesenchymal Stromal Cells. Sci. Rep. 2018, 8, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hetz, C. The unfolded protein response: Controlling cell fate decisions under ER stress and beyond. Nat. Rev. Mol. Cell Biol. 2012, 13, 89–102. [Google Scholar] [CrossRef] [PubMed]
- Childs, B.G.; Baker, D.J.; Kirkland, J.L.; Campisi, J.; Deursen, J.M. Senescence and apoptosis: Dueling or complementary cell fates? EMBO Rep. 2014, 15, 1139–1153. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmitz, M.L.; Shaban, M.S.; Albert, B.V.; Gökçen, A.; Kracht, M. The crosstalk of Endoplasmic Reticulum (ER) stress pathways with NF-κB: Complex mechanisms relevant for cancer, inflammation and infection. Biomedicines 2018, 6, 58. [Google Scholar] [CrossRef] [Green Version]
- Maraldi, T.; Beretti, F.; Guida, M.; Zavatti, M.; De Pol, A. Role of hepatocyte growth factor in the immunomodulation potential of amniotic fluid stem cells. Stem Cells Transl. Med. 2015, 4, 539–547. [Google Scholar] [CrossRef]
- Tulasne, D.; Foveau, B. The shadow of death on the MET tyrosine kinase receptor. Cell Death Differ. 2007, 15, 427–434. [Google Scholar] [CrossRef] [Green Version]
- Maraldi, T.; Guida, M.; Zavatti, M.; Resca, E.; Bertoni, L.; La Sala, G.B.; Pol, A.D. Nuclear Nox4 Role in Stemness Power of Human Am-niotic Fluid Stem Cells. Oxid. Med. Cell. Longev. 2015, 2015, 101304. [Google Scholar] [CrossRef] [Green Version]
- McLeod, C.; Mauck, R.L. On the origin and impact of mesenchymal stem cell heterogeneity: New insights and emerging tools for single cell analysis. Eur. Cells Mater. 2017, 34, 217–231. [Google Scholar] [CrossRef]
- Colter, D.C.; Sekiya, I.; Prockop, D.J. Identification of a subpopulation of rapidly self-renewing and multipotential adult stem cells in colonies of human marrow stromal cells. Proc. Natl. Acad. Sci. USA 2001, 98, 7841–7845. [Google Scholar] [CrossRef] [Green Version]
- Romani, R.; Fallarino, F.; Pirisinu, I.; Calvitti, M.; Caselli, A.; Fiaschi, T.; Gamberi, T.; Matino, D.; Talesa, V.N.; Donti, E.; et al. Comparative proteomic analysis of two distinct stem-cell populations from human amniotic fluid. Mol. BioSyst. 2015, 11, 1622–1632. [Google Scholar] [CrossRef] [PubMed]
- Altschuler, S.J.; Wu, L.F. Cellular Heterogeneity: Do Differences Make a Difference? Cell 2010, 141, 559–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.; Relan, N.K.; Przywara, D.A.; Schuger, L. Embryonic mesenchymal cells share the potential for smooth muscle differen-tiation: Myogenesis is controlled by the cell’s shape. Development 1999, 126, 3027–3033. [Google Scholar] [PubMed]
- Chen, C.; Liu, Y.; Zheng, P. mTOR Regulation and Therapeutic Rejuvenation of Aging Hematopoietic Stem Cells. Sci. Signal. 2009, 2, ra75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morita, M.; Gravel, S.P.; Chénard, V.; Sikström, K.; Zheng, L.; Alain, T.; Gandin, V.; Avizonis, D.; Arguello, M.; Zakaria, C.; et al. mTORC1 controls mitochondrial activity and bio-genesis through 4E-BP-dependent translational regulation. Cell Metab. 2013, 18, 698–711. [Google Scholar] [CrossRef] [Green Version]
- Balbi, C.; Bollini, S. Fetal and perinatal stem cells in cardiac regeneration: Moving forward to the paracrine era. Placenta 2017, 59, 96–106. [Google Scholar] [CrossRef]
- Hunter, C.A.; Jones, S.A. IL-6 as a keystone cytokine in health and disease. Nat. Immunol. 2015, 16, 448–457. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Sequence | RefSeq Accession n |
|---|---|---|
| hNANOG | Fw cagtctggacactggctgaa Rv cacgtggtttccaaacaaga Probe 55 | NM_024865 |
| hPOU5F1 (OCT4) | Fw tgagtagtcccttcgcaagc Rv gagaaggcgaaatccgaag Probe 60 | NM_002701.5 |
| hCDKN2A (p16) | Fw gagcagcatggagccttc Rv cgtaactattcggtgcgttg Probe 67 | NM_000077.4 |
| hCDKN1A (p21) | Fw tcactgtcttgtacccttgtgc Rv ccgttttcgaccctgagag Probe 32 | NM_000389.4 |
| hTP53 | Fw gctcaagactggcgctaaaa Rv gtcaccgtcgtggaaagc Probe 32 | NM_001276760.1 |
| AMPK | Fw tctcaggaggagagctatttgatt Rv gaacagacgccgactttcttt Probe 42 | NM_006251.5 |
| hCCNE2 (cyclin E2) | Fw ccccaagaagcccagataat Rv caggtggccaacaattcct Probe 35 | NM_057749.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Casciaro, F.; Zia, S.; Forcato, M.; Zavatti, M.; Beretti, F.; Bertucci, E.; Zattoni, A.; Reschiglian, P.; Alviano, F.; Bonsi, L.; et al. Unravelling Heterogeneity of Amplified Human Amniotic Fluid Stem Cells Sub-Populations. Cells 2021, 10, 158. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10010158
Casciaro F, Zia S, Forcato M, Zavatti M, Beretti F, Bertucci E, Zattoni A, Reschiglian P, Alviano F, Bonsi L, et al. Unravelling Heterogeneity of Amplified Human Amniotic Fluid Stem Cells Sub-Populations. Cells. 2021; 10(1):158. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10010158
Chicago/Turabian StyleCasciaro, Francesca, Silvia Zia, Mattia Forcato, Manuela Zavatti, Francesca Beretti, Emma Bertucci, Andrea Zattoni, Pierluigi Reschiglian, Francesco Alviano, Laura Bonsi, and et al. 2021. "Unravelling Heterogeneity of Amplified Human Amniotic Fluid Stem Cells Sub-Populations" Cells 10, no. 1: 158. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10010158