
Light-Dependent Translation Change of Arabidopsis psbA Correlates with RNA Structure Alterations at the Translation Initiation Region

 , , , ,
, , , ,  , ,
, ,  , and
, and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Plant Material
2.2. Determining Photosynthetic Parameters
2.3. Polysome Analysis
2.4. Ribosome Profiling (Ribo-seq)
2.5. RNA-seq
2.6. Determination of 3′-Ends of Plastid Transcripts
2.7. Processing of Ribo-seq and RNA-seq Reads
2.8. Calculation of Translation Efficiency and Analysis of Differential Gene Expression
2.9. Gel-Blot Analysis of Small RNAs
2.10. RNA Structure Probing with DMS
2.11. DMS-MaPseq Analysis
2.12. Structure Analysis of 16S rRNA
2.13. RNA Secondary Structure Probing with NAI-N3
2.14. In Vitro RNA Secondary Structure Probing with NAI-N3
2.15. SHAPE Data Analysis
2.16. Receiver Operating Characteristic (ROC) Curve
2.17. Data Analyses
3. Results
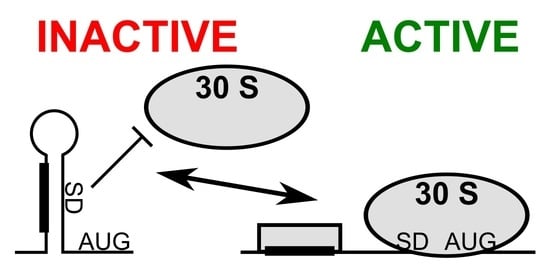
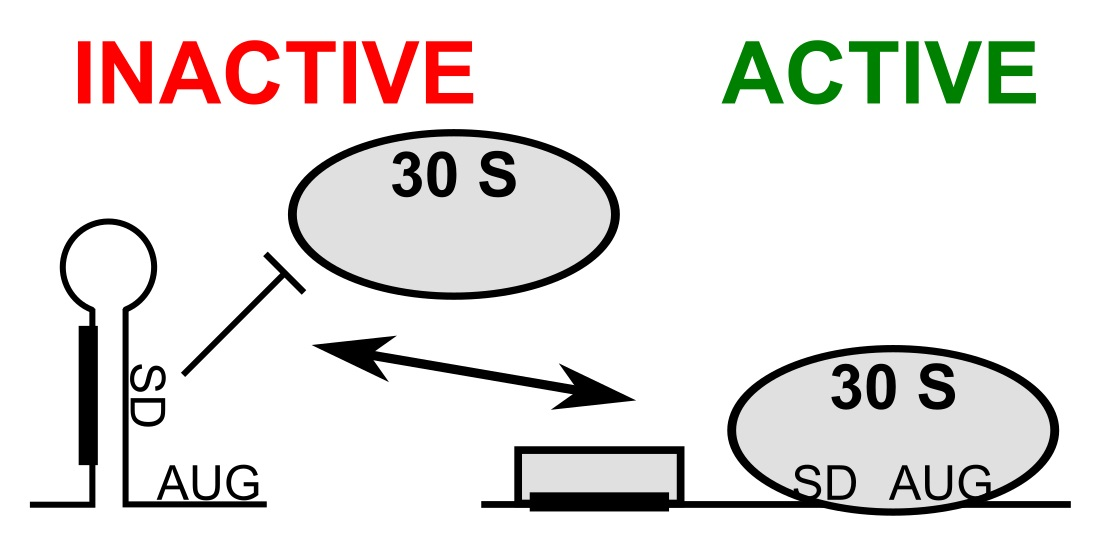
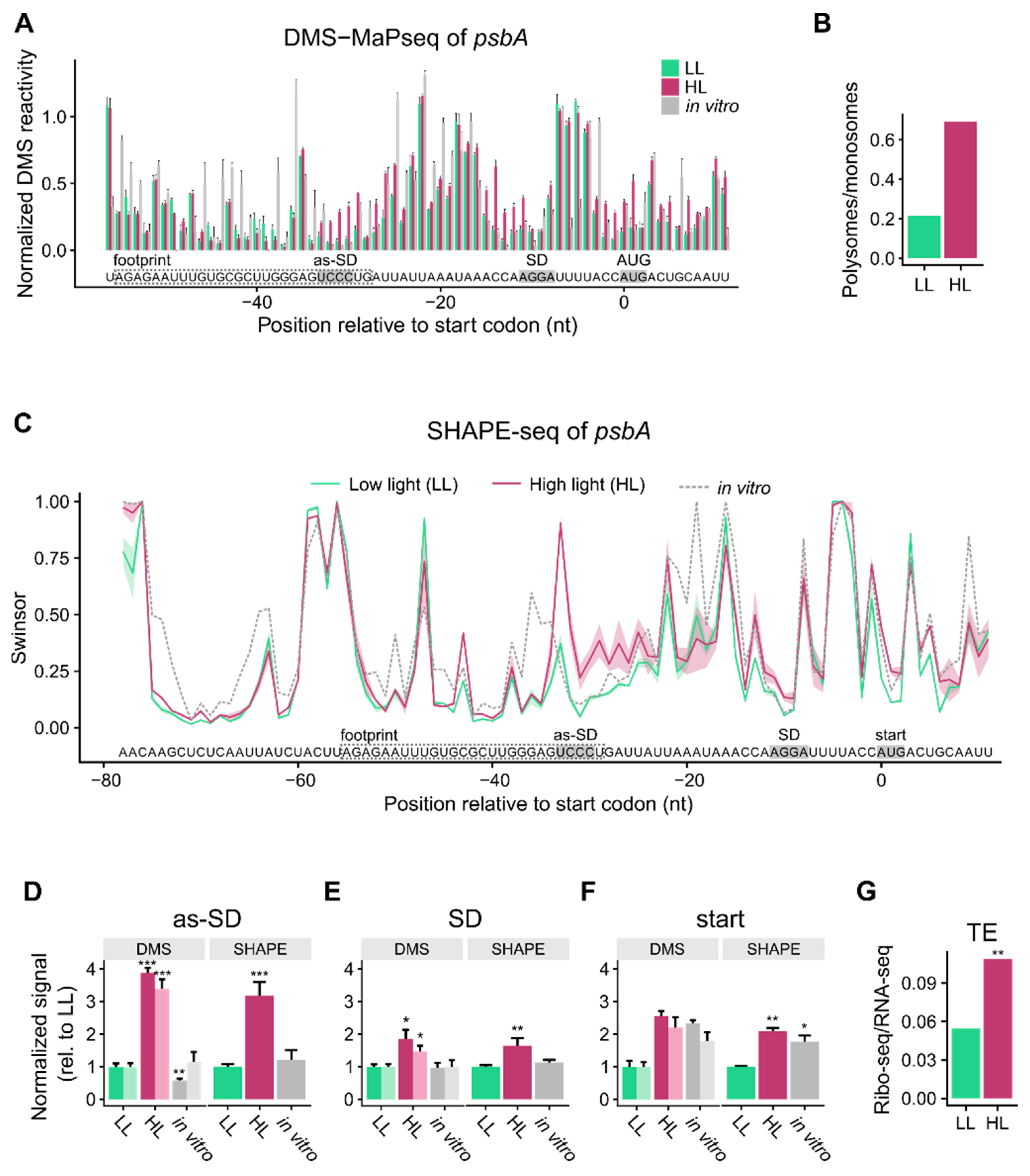
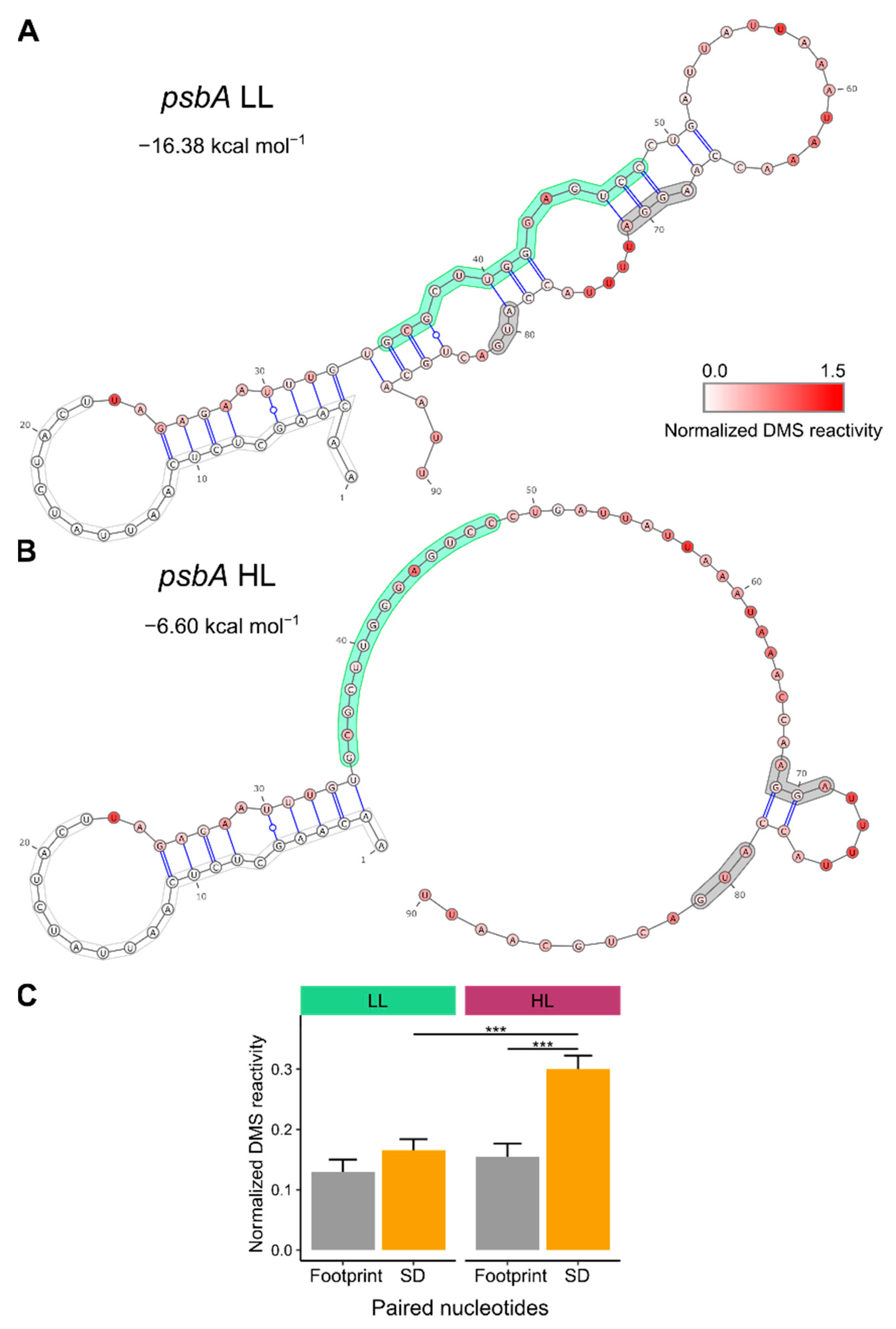
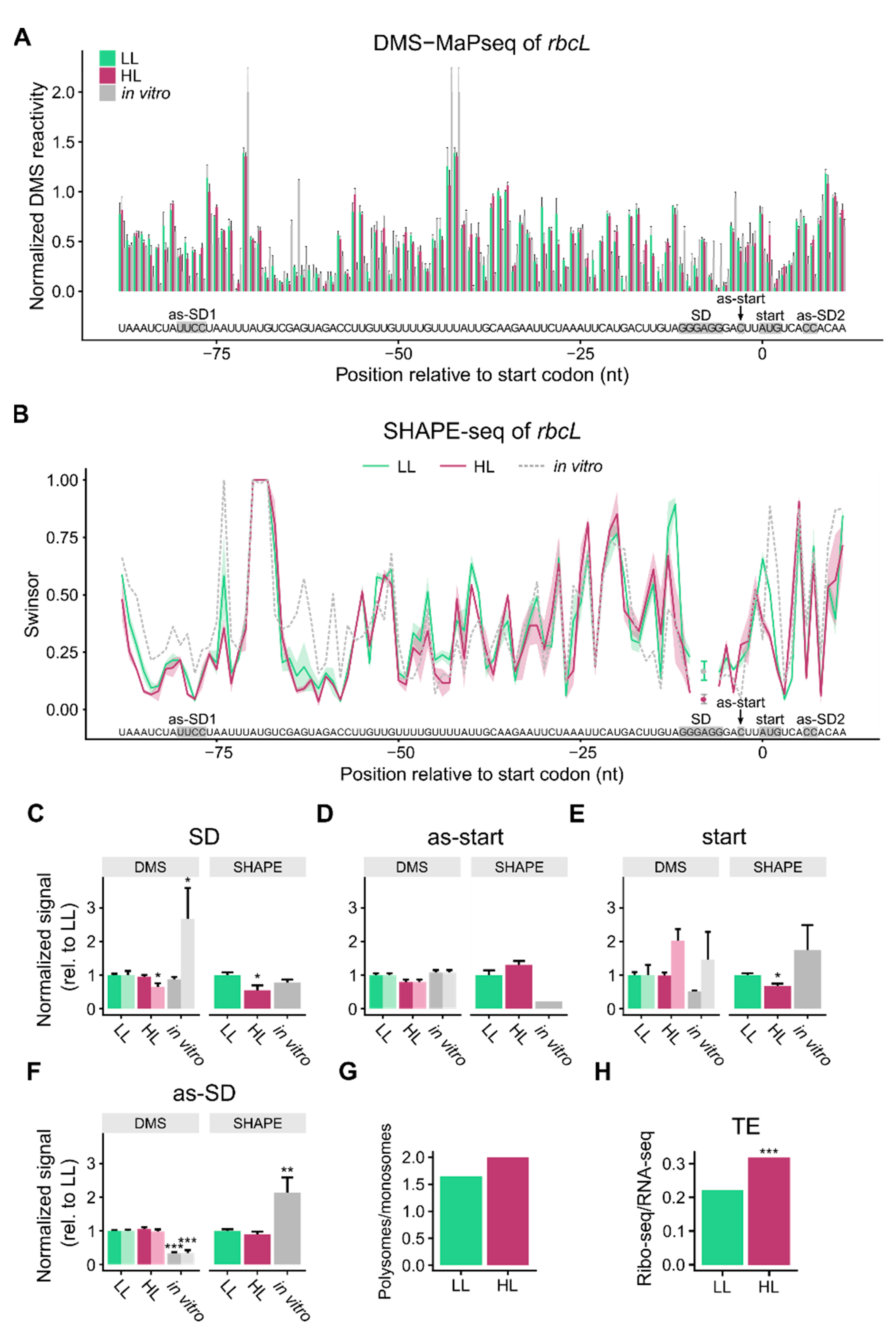
3.1. mRNA Secondary Structure Changes in the psbA Translation Initiation Region
3.2. Change of mRNA Secondary Structure of psbA Translation Initiation Region Likely Caused by Protein Binding
3.3. mRNA Secondary Structure of the Translation Initiation Regions of rbcL
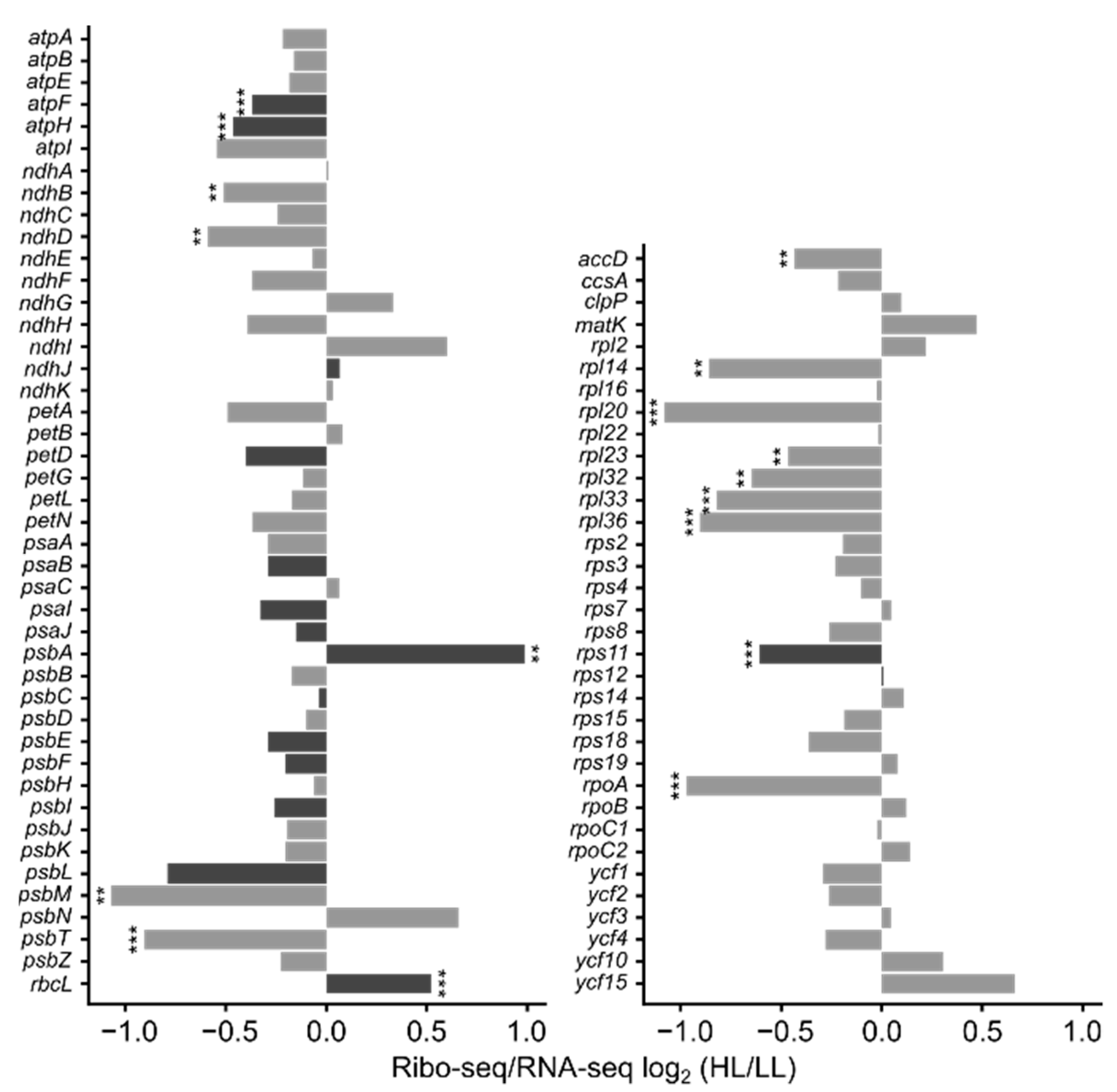
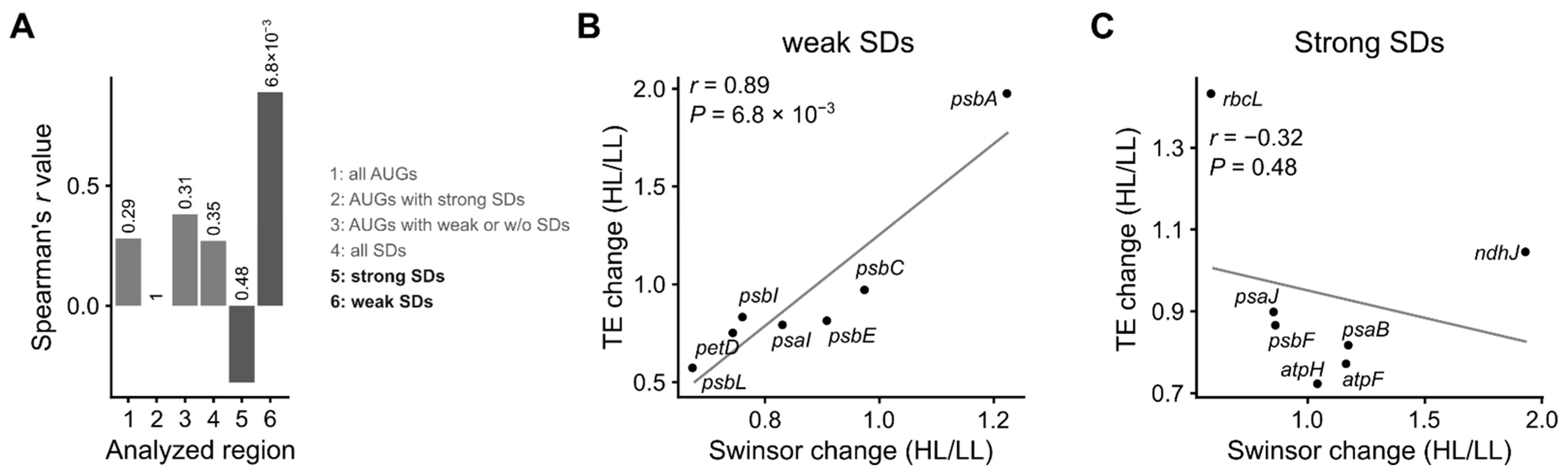
3.4. mRNA Secondary Structure and Translation Efficiency
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- De Smit, M.H.; van Duin, J. Secondary structure of the ribosome binding site determines translational efficiency: A quantitative analysis. Proc. Natl. Acad. Sci. USA 1990, 87, 7668–7672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kudla, G.; Murray, A.W.; Tollervey, D.; Plotkin, J.B. Coding-sequence determinants of gene expression in Escherichia coli. Science 2009, 324, 255–258. [Google Scholar] [CrossRef] [Green Version]
- Goodman, D.B.; Church, G.M.; Kosuri, S. Causes and effects of N-terminal codon bias in bacterial genes. Science 2013, 342, 475–479. [Google Scholar] [CrossRef] [Green Version]
- Mustoe, A.M.; Busan, S.; Rice, G.M.; Hajdin, C.E.; Peterson, B.K.; Ruda, V.M.; Kubica, N.; Nutiu, R.; Baryza, J.L.; Weeks, K.M. Pervasive Regulatory Functions of mRNA Structure Revealed by High-Resolution SHAPE Probing. Cell 2018, 173, 181–195.e18. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharyya, S.; Jacobs, W.M.; Adkar, B.V.; Yan, J.; Zhang, W.; Shakhnovich, E.I. Accessibility of the Shine-Dalgarno Sequence Dictates N-Terminal Codon Bias in E. coli. Mol. Cell 2018, 70, 894–905.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scharff, L.B.; Childs, L.; Walther, D.; Bock, R. Local absence of secondary structure permits translation of mRNAs that lack ribosome-binding sites. PLoS Genet. 2011, 7, e1002155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakagawa, S.; Niimura, Y.; Gojobori, T. Comparative genomic analysis of translation initiation mechanisms for genes lacking the Shine-Dalgarno sequence in prokaryotes. Nucleic Acids Res. 2017, 45, 3922–3931. [Google Scholar] [CrossRef] [Green Version]
- Scharff, L.B.; Ehrnthaler, M.; Janowski, M.; Childs, L.H.; Hasse, C.; Gremmels, J.; Ruf, S.; Zoschke, R.; Bock, R. Shine-Dalgarno Sequences Play an Essential Role in the Translation of Plastid mRNAs in Tobacco. Plant Cell 2017, 29, 3085–3101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, J.-D.; Lancaster, L.; Hodges, C.; Zeri, A.-C.; Yoshimura, S.H.; Noller, H.F.; Bustamante, C.; Tinoco, I. Following translation by single ribosomes one codon at a time. Nature 2008, 452, 598–603. [Google Scholar] [CrossRef] [Green Version]
- Tuller, T.; Veksler-Lublinsky, I.; Gazit, N.; Kupiec, M.; Ruppin, E.; Ziv-Ukelson, M. Composite effects of gene determinants on the translation speed and density of ribosomes. Genome Biol. 2011, 12, R110. [Google Scholar] [CrossRef] [Green Version]
- Gawroński, P.; Jensen, P.E.; Karpiński, S.; Leister, D.; Scharff, L.B. Pausing of Chloroplast Ribosomes Is Induced by Multiple Features and Is Linked to the Assembly of Photosynthetic Complexes. Plant Physiol. 2018, 176, 2557–2569. [Google Scholar] [CrossRef] [Green Version]
- Breaker, R.R. Riboswitches and Translation Control. Cold Spring Harb. Perspect. Biol. 2018, 10, a032797. [Google Scholar] [CrossRef]
- Neupert, J.; Karcher, D.; Bock, R. Design of simple synthetic RNA thermometers for temperature-controlled gene expression in Escherichia coli. Nucleic Acids Res. 2008, 36, e124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krajewski, S.S.; Narberhaus, F. Temperature-driven differential gene expression by RNA thermosensors. Biochim. Biophys. Acta 2014, 1839, 978–988. [Google Scholar] [CrossRef] [PubMed]
- Laursen, B.; Sørensen, H. Initiation of protein synthesis in bacteria. Microbiology 2005, 236, 747–771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duval, M.; Simonetti, A.; Caldelari, I.; Marzi, S. Multiple ways to regulate translation initiation in bacteria: Mechanisms, regulatory circuits, dynamics. Biochimie 2015, 114, 18–29. [Google Scholar] [CrossRef] [PubMed]
- Stampacchia, O.; Girard-Bascou, J.; Zanasco, J.L.; Zerges, W.; Bennoun, P.; Rochaix, J.-D. A nuclear-encoded function essential for translation of the chloroplast psaB mRNA in chlamydomonas. Plant Cell 1997, 9, 773–782. [Google Scholar]
- Klinkert, B.; Elles, I.; Nickelsen, J. Translation of chloroplast psbD mRNA in Chlamydomonas is controlled by a secondary RNA structure blocking the AUG start codon. Nucleic Acids Res. 2006, 34, 386–394. [Google Scholar] [CrossRef] [Green Version]
- Prikryl, J.; Rojas, M.; Schuster, G.; Barkan, A. Mechanism of RNA stabilization and translational activation by a pentatricopeptide repeat protein. Proc. Natl. Acad. Sci. USA 2011, 108, 415–420. [Google Scholar] [CrossRef] [Green Version]
- Hammani, K.; Cook, W.B.; Barkan, A. RNA binding and RNA remodeling activities of the half-a-tetratricopeptide (HAT) protein HCF107 underlie its effects on gene expression. Proc. Natl. Acad. Sci. USA 2012, 109, 5651–5656. [Google Scholar] [CrossRef] [Green Version]
- Barkan, A. Expression of Plastid Genes: Organelle-Specific Elaborations on a Prokaryotic Scaffold. Plant Physiol. 2011, 155, 1520–1532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugiura, M. Plastid mRNA translation. Methods Mol. Biol. 2014, 1132, 73–91. [Google Scholar] [PubMed]
- Sun, Y.; Zerges, W. Translational regulation in chloroplasts for development and homeostasis. Biochim. Biophys. Acta 2015, 1847, 809–820. [Google Scholar] [CrossRef] [Green Version]
- Zoschke, R.; Bock, R. Chloroplast Translation: Structural and Functional Organization, Operational Control, and Regulation. Plant Cell 2018, 30, 745–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirose, T.; Sugiura, M. Functional Shine-Dalgarno-Like Sequences for Translational Initiation of Chloroplast mRNAs. Plant Cell Physiol. 2004, 45, 114–117. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Ruf, S.; Hasse, C.; Childs, L.; Scharff, L.B.; Bock, R. Identification of cis-elements conferring high levels of gene expression in non-green plastids. Plant J. 2012, 72, 115–128. [Google Scholar] [CrossRef]
- Gawroński, P.; Pałac, A.; Scharff, L.B. Secondary Structure of Chloroplast mRNAs In Vivo and In Vitro. Plants 2020, 9, 323. [Google Scholar] [CrossRef] [Green Version]
- Fu, H.-Y.; Ghandour, R.; Ruf, S.; Zoschke, R.; Bock, R.; Schöttler, M.A. The availability of neither D2 nor CP43 limits the biogenesis of photosystem II in tobacco. Plant Physiol. 2020. [Google Scholar] [CrossRef]
- Chotewutmontri, P.; Barkan, A. Multilevel effects of light on ribosome dynamics in chloroplasts program genome-wide and psbA-specific changes in translation. PLoS Genet. 2018, 14, e1007555. [Google Scholar] [CrossRef]
- Schuster, M.; Gao, Y.; Schöttler, M.A.; Bock, R.; Zoschke, R. Limited Responsiveness of Chloroplast Gene Expression during Acclimation to High Light in Tobacco. Plant Physiol. 2020, 182, 424–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulo, P.; Sakurai, I.; Aro, E.-M. Strategies for psbA gene expression in cyanobacteria, green algae and higher plants: From transcription to PSII repair. Biochim. Biophys. Acta 2012, 1817, 247–257. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Aro, E.-M.; Millar, A.H. Mechanisms of Photodamage and Protein Turnover in Photoinhibition. Trends Plant Sci. 2018, 23, 667–676. [Google Scholar] [CrossRef]
- Danon, A.; Mayfield, S.P.Y. Light regulated translational activators: Identification of chloroplast gene specific mRNA binding proteins. EMBO J. 1991, 10, 3993–4001. [Google Scholar] [CrossRef]
- Schult, K.; Meierhoff, K.; Paradies, S.; Töller, T.; Wolff, P.; Westhoff, P. The nuclear-encoded factor HCF173 is involved in the initiation of translation of the psbA mRNA in Arabidopsis thaliana. Plant Cell 2007, 19, 1329–1346. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Link, S.; Meierhoff, K.; Westhoff, P. The Atypical Short-Chain Dehydrogenases HCF173 and HCF244 are Jointly Involved in Translational Initiation of the psbA mRNA of Arabidopsis thaliana. Plant Physiol. 2012, 160, 2202–2218. [Google Scholar] [CrossRef] [Green Version]
- McDermott, J.J.; Watkins, K.P.; Williams-Carrier, R.; Barkan, A. Ribonucleoprotein Capture by in Vivo Expression of a Designer Pentatricopeptide Repeat Protein in Arabidopsis. Plant Cell 2019, 31, 1723–1733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chotewutmontri, P.; Williams-Carrier, R.; Barkan, A. Exploring the Link between Photosystem II Assembly and Translation of the Chloroplast psbA mRNA. Plants 2020, 9, 152. [Google Scholar] [CrossRef] [Green Version]
- Chotewutmontri, P.; Barkan, A. Light-induced psbA translation in plants is triggered by photosystem II damage via an assembly-linked autoregulatory circuit. Proc. Natl. Acad. Sci. USA 2020, 117, 21775–21784. [Google Scholar] [CrossRef] [PubMed]
- Klughammer, C.; Schreiber, U. Complementary PS II quantum yields calculated from simple fluorescence parameters measured by PAM fluorometry and the Saturation Pulse method. PAM Appl. Notes 2008, 1, 27–35. [Google Scholar]
- Barkan, A. Nuclear Mutants of Maize with Defects in Chloroplast Polysome Assembly Have Altered Chloroplast RNA Metabolism. Plant Cell 1993, 5, 389–402. [Google Scholar] [CrossRef]
- Oh, E.; Becker, A.H.; Sandikci, A.; Huber, D.; Chaba, R.; Gloge, F.; Nichols, R.J.; Typas, A.; Gross, C.A.; Kramer, G.; et al. Selective ribosome profiling reveals the cotranslational chaperone action of trigger factor in vivo. Cell 2011, 147, 1295–1308. [Google Scholar] [CrossRef] [Green Version]
- Zoschke, R.; Watkins, K.P.; Barkan, A. A rapid ribosome profiling method elucidates chloroplast ribosome behavior in vivo. Plant Cell 2013, 25, 2265–2275. [Google Scholar] [CrossRef] [Green Version]
- Marquardt, S.; Escalante-Chong, R.; Pho, N.; Wang, J.; Churchman, L.S.; Springer, M.; Buratowski, S. A chromatin-based mechanism for limiting divergent noncoding transcription. Cell 2014, 157, 1712–1723. [Google Scholar] [CrossRef] [Green Version]
- Tillich, M.; Lehwark, P.; Pellizzer, T.; Ulbricht-Jones, E.S.; Fischer, A.; Bock, R.; Greiner, S. GeSeq—Versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 2017, 45, W6–W11. [Google Scholar] [CrossRef]
- Roy, S.; Ueda, M.; Kadowaki, K.; Tsutsumi, N. Different status of the gene for ribosomal protein S16 in the chloroplast genome during evolution of the genus Arabidopsis and closely related species. Genes Genet. Syst. 2010, 85, 319–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef] [PubMed]
- Zhong, Y.; Karaletsos, T.; Drewe, P.; Sreedharan, V.T.; Kuo, D.; Singh, K.; Wendel, H.-G.; Rätsch, G. RiboDiff: Detecting changes of mRNA translation efficiency from ribosome footprints. Bioinformatics 2017, 33, 139–141. [Google Scholar] [CrossRef]
- Patro, R.; Duggal, G.; Love, M.I.; Irizarry, R.A.; Kingsford, C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 2017, 14, 417–419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soneson, C.; Love, M.I.; Robinson, M.D. Differential analyses for RNA-seq: Transcript-level estimates improve gene-level inferences. F1000Research 2015, 4, 1521. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef] [Green Version]
- Loizeau, K.; Qu, Y.; Depp, S.; Fiechter, V.; Ruwe, H.; Lefebvre-Legendre, L.; Schmitz-Linneweber, C.; Goldschmidt-Clermont, M. Small RNAs reveal two target sites of the RNA-maturation factor Mbb1 in the chloroplast of Chlamydomonas. Nucleic Acids Res. 2014, 42, 3286–3297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, M.; Pages, H.; Obenchain, V.; Hayden, N. Rsamtools: Binary Alignment (BAM), FASTA, Variant Call (BCF), and Tabix File Import. 2017. Available online: https://bioconductor.org/packages/release/bioc/html/Rsamtools.html (accessed on 1 February 2021).
- Mathews, D.H.; Turner, D.H.; Watson, R.M. RNA Secondary Structure Prediction. Curr. Protoc. Nucleic Acid Chem. 2016, 67, 11.2.1–11.2.19. [Google Scholar] [CrossRef] [Green Version]
- Darty, K.; Denise, A.; Ponty, Y. VARNA: Interactive drawing and editing of the RNA secondary structure. Bioinformatics 2009, 25, 1974–1975. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, T.; Shi, J.; Bhushan, S. Unique localization of the plastid-specific ribosomal proteins in the chloroplast ribosome small subunit provides mechanistic insights into the chloroplastic translation. Nucleic Acids Res. 2017, 45, 8581–8595. [Google Scholar] [CrossRef] [PubMed]
- Flynn, R.A.; Zhang, Q.C.; Spitale, R.C.; Lee, B.; Mumbach, M.R.; Chang, H.Y. Transcriptome-wide interrogation of RNA secondary structure in living cells with icSHAPE. Nat. Protoc. 2016, 11, 273–290. [Google Scholar] [CrossRef] [Green Version]
- Poulsen, L.D.; Kielpinski, L.J.; Salama, S.R.; Krogh, A.; Vinther, J. SHAPE Selection (SHAPES) enrich for RNA structure signal in SHAPE sequencing-based probing data. RNA 2015, 21, 1042–1052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahashi, H.; Lassmann, T.; Murata, M.; Carninci, P. 5’ end-centered expression profiling using cap-analysis gene expression and next-generation sequencing. Nat. Protoc. 2012, 7, 542–561. [Google Scholar] [CrossRef] [Green Version]
- Martin, M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet. J. 2011, 17, 10. [Google Scholar] [CrossRef]
- Kielpinski, L.J.; Sidiropoulos, N.; Vinther, J. Reproducible Analysis of Sequencing-Based RNA Structure Probing Data with User-Friendly Tools. Methods Enzymol. 2015, 558, 153–180. [Google Scholar]
- Starmer, J.; Stomp, A.; Vouk, M.; Bitzer, D. Predicting Shine-Dalgarno sequence locations exposes genome annotation errors. PLoS Comput. Biol. 2006, 2, e57. [Google Scholar] [CrossRef] [Green Version]
- Cannone, J.J.; Subramanian, S.; Schnare, M.N.; Collett, J.R.; D’Souza, L.M.; Du, Y.; Feng, B.; Lin, N.; Madabusi, L.V.; Müller, K.M.; et al. The comparative RNA web (CRW) site: An online database of comparative sequence and structure information for ribosomal, intron, and other RNAs. BMC Bioinform. 2002, 3, 2. [Google Scholar]
- Robin, X.; Turck, N.; Hainard, A.; Tiberti, N.; Lisacek, F.; Sanchez, J.-C.; Müller, M. pROC: An open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinform. 2011, 12, 77. [Google Scholar] [CrossRef]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation Statistical Computing: Vienna, Austria, 2018. [Google Scholar]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis, 2nd ed.; Use, R., Ed.; Springer International Publishing: Cham, Germany, 2016; ISBN 978-3-319-24275-0. [Google Scholar]
- Mitchell, D.; Assmann, S.M.; Bevilacqua, P.C. Probing RNA structure in vivo. Curr. Opin. Struct. Biol. 2019, 59, 151–158. [Google Scholar] [CrossRef]
- Mustoe, A.M.; Lama, N.N.; Irving, P.S.; Olson, S.W.; Weeks, K.M. RNA base-pairing complexity in living cells visualized by correlated chemical probing. Proc. Natl. Acad. Sci. USA 2019, 116, 24574–24582. [Google Scholar] [CrossRef] [PubMed]
- Wells, S.E.; Hughes, J.M.; Igel, A.H.; Ares, M. Use of dimethyl sulfate to probe RNA structure in vivo. Methods Enzymol. 2000, 318, 479–493. [Google Scholar] [PubMed]
- Ding, Y.; Tang, Y.; Kwok, C.K.; Zhang, Y.; Bevilacqua, P.C.; Assmann, S.M. In vivo genome-wide profiling of RNA secondary structure reveals novel regulatory features. Nature 2014, 505, 696–700. [Google Scholar] [CrossRef] [PubMed]
- Zubradt, M.; Gupta, P.; Persad, S.; Lambowitz, A.M.; Weissman, J.S.; Rouskin, S. DMS-MaPseq for genome-wide or targeted RNA structure probing in vivo. Nat. Methods 2017, 14, 75–82. [Google Scholar] [CrossRef]
- Wang, Z.; Wang, M.; Wang, T.; Zhang, Y.; Zhang, X. Genome-wide probing RNA structure with the modified DMS-MaPseq in Arabidopsis. Methods 2019, 155, 30–40. [Google Scholar] [CrossRef]
- Spitale, R.C.; Crisalli, P.; Flynn, R.A.; Torre, E.A.; Kool, E.T.; Chang, H.Y. RNA SHAPE analysis in living cells. Nat. Chem. Biol. 2013, 9, 18–20. [Google Scholar] [CrossRef] [Green Version]
- Spitale, R.C.; Flynn, R.A.; Torre, E.A.; Kool, E.T.; Chang, H.Y. RNA structural analysis by evolving SHAPE chemistry. Wiley Interdiscip. Rev. RNA 2014, 5, 867–881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Merino, E.J.; Wilkinson, K.A.; Coughlan, J.L.; Weeks, K.M. RNA structure analysis at single nucleotide resolution by Selective 2′-Hydroxyl Acylation and Primer Extension (SHAPE). J. Am. Chem. Soc. 2005, 127, 4223–4231. [Google Scholar] [CrossRef] [PubMed]
- McGinnis, J.L.; Dunkle, J.A.; Cate, J.H.D.; Weeks, K.M. The mechanisms of RNA SHAPE chemistry. J. Am. Chem. Soc. 2012, 134, 6617–6624. [Google Scholar] [CrossRef] [Green Version]
- Ruwe, H.; Wang, G.; Gusewski, S.; Schmitz-Linneweber, C. Systematic analysis of plant mitochondrial and chloroplast small RNAs suggests organelle-specific mRNA stabilization mechanisms. Nucleic Acids Res. 2016, 44, 7406–7417. [Google Scholar] [CrossRef] [Green Version]
- Kwok, C.K.; Ding, Y.; Tang, Y.; Assmann, S.M.; Bevilacqua, P.C. Determination of in vivo RNA structure in low-abundance transcripts. Nat. Commun. 2013, 4, 2971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talkish, J.; May, G.; Lin, Y.; Woolford, J.L.; McManus, C.J. Mod-seq: High-throughput sequencing for chemical probing of RNA structure. RNA 2014, 20, 713–720. [Google Scholar] [CrossRef] [Green Version]
- Spitale, R.C.; Flynn, R.A.; Zhang, Q.C.; Crisalli, P.; Lee, B.; Jung, J.-W.; Kuchelmeister, H.Y.; Batista, P.J.; Torre, E.A.; Kool, E.T.; et al. Structural imprints in vivo decode RNA regulatory mechanisms. Nature 2015, 519, 486–490. [Google Scholar] [CrossRef] [Green Version]
- Kenyon, J.C.; Prestwood, L.J.; Lever, A.M.L. A novel combined RNA-protein interaction analysis distinguishes HIV-1 Gag protein binding sites from structural change in the viral RNA leader. Sci. Rep. 2015, 5, 14369. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ingolia, N.T.; Ghaemmaghami, S.; Newman, J.R.S.; Weissman, J.S. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 2009, 324, 218–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, H.; Fu, M.; Duan, Z.; Duan, S.; Li, M.; Dong, X.; Liu, B.; Feng, D.; Wang, J.; Peng, L.; et al. LOW PHOTOSYNTHETIC EFFICIENCY 1 is required for light-regulated photosystem II biogenesis in Arabidopsis. Proc. Natl. Acad. Sci. USA 2018, 115, E6075–E6084. [Google Scholar] [CrossRef] [Green Version]
- Wittenberg, G.; Levitan, A.; Klein, T.; Dangoor, I.; Keren, N.; Danon, A. Knockdown of the Arabidopsis thaliana chloroplast protein disulfide isomerase 6 results in reduced levels of photoinhibition and increased D1 synthesis in high light. Plant J. 2014, 78, 1003–1013. [Google Scholar] [CrossRef] [PubMed]
- Williams-Carrier, R.; Brewster, C.; Belcher, S.E.; Rojas, M.; Chotewutmontri, P.; Ljungdahl, S.; Barkan, A. The Arabidopsis pentatricopeptide repeat protein LPE1 and its maize ortholog are required for translation of the chloroplast psbJ RNA. Plant J. 2019, 99, 56–66. [Google Scholar] [CrossRef] [PubMed]
- Hey, D.; Grimm, B. ONE-HELIX PROTEIN2 (OHP2) Is Required for the Stability of OHP1 and Assembly Factor HCF244 and Is Functionally Linked to PSII Biogenesis. Plant Physiol. 2018, 177, 1453–1472. [Google Scholar] [CrossRef] [Green Version]
- Myouga, F.; Takahashi, K.; Tanaka, R.; Nagata, N.; Kiss, A.Z.; Funk, C.; Nomura, Y.; Nakagami, H.; Jansson, S.; Shinozaki, K. Stable Accumulation of Photosystem II Requires ONE-HELIX PROTEIN1 (OHP1) of the Light Harvesting-Like Family. Plant Physiol. 2018, 176, 2277–2291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verhounig, A.; Karcher, D.; Bock, R. Inducible gene expression from the plastid genome by a synthetic riboswitch. Proc. Natl. Acad. Sci. USA 2010, 107, 6204–6209. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gawroński, P.; Enroth, C.; Kindgren, P.; Marquardt, S.; Karpiński, S.; Leister, D.; Jensen, P.E.; Vinther, J.; Scharff, L.B. Light-Dependent Translation Change of Arabidopsis psbA Correlates with RNA Structure Alterations at the Translation Initiation Region. Cells 2021, 10, 322. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10020322
Gawroński P, Enroth C, Kindgren P, Marquardt S, Karpiński S, Leister D, Jensen PE, Vinther J, Scharff LB. Light-Dependent Translation Change of Arabidopsis psbA Correlates with RNA Structure Alterations at the Translation Initiation Region. Cells. 2021; 10(2):322. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10020322
Chicago/Turabian StyleGawroński, Piotr, Christel Enroth, Peter Kindgren, Sebastian Marquardt, Stanisław Karpiński, Dario Leister, Poul Erik Jensen, Jeppe Vinther, and Lars B. Scharff. 2021. "Light-Dependent Translation Change of Arabidopsis psbA Correlates with RNA Structure Alterations at the Translation Initiation Region" Cells 10, no. 2: 322. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10020322