
Remdesivir and Ledipasvir among the FDA-Approved Antiviral Drugs Have Potential to Inhibit SARS-CoV-2 Replication


 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
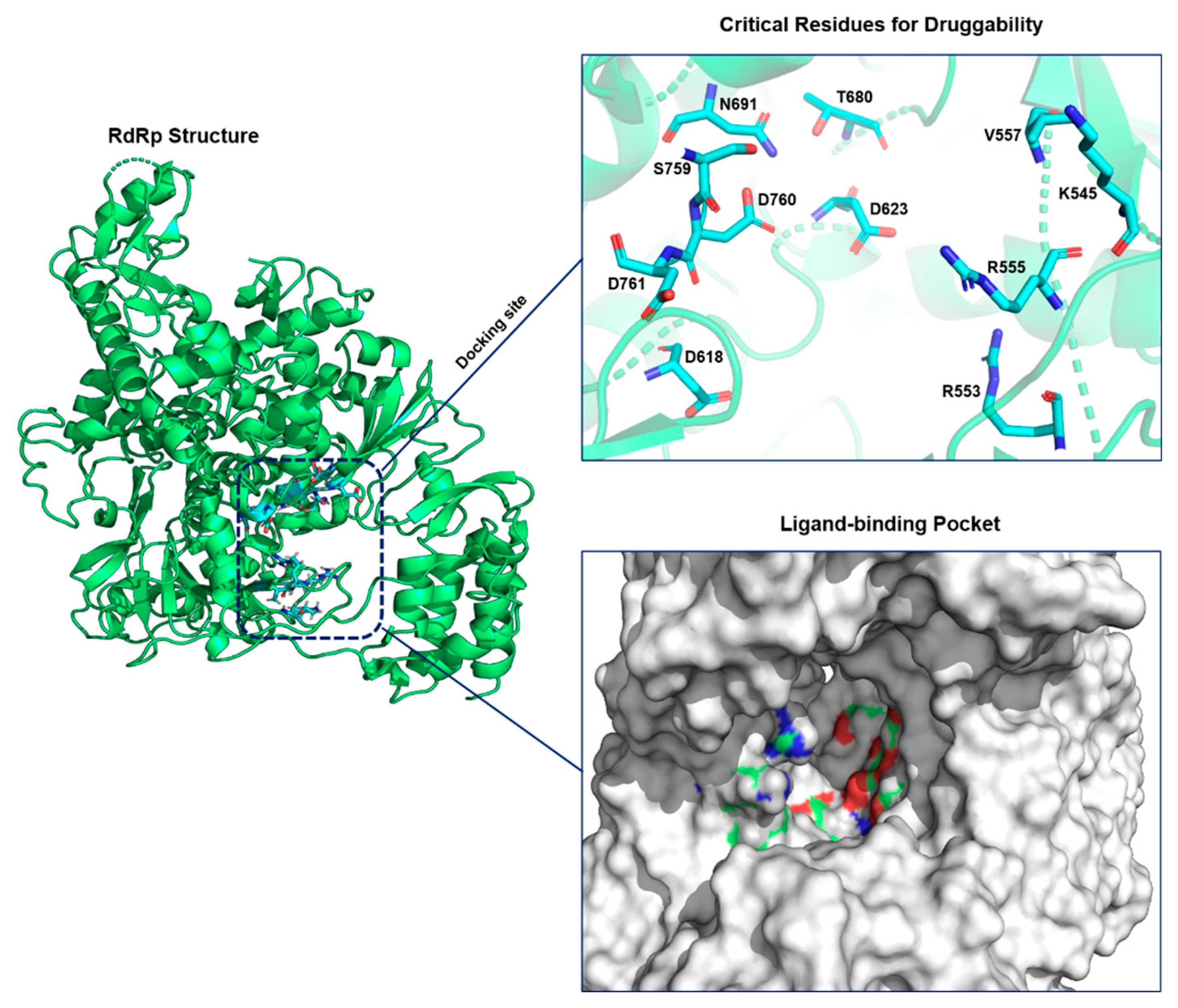
2.1. Protein Model Preparation and Active-Site Identification
2.2. Library Preparation
2.3. Virtual Screening and Molecular Docking
2.4. The MD Simulation Protocol
2.4.1. Ligand Topology Generation
2.4.2. MD Simulations
2.5. Post-MD Simulation Data Analysis and Visualization
2.6. Principal Component Analysis (PCA)
2.7. The Free Energy Landscape (FEL)
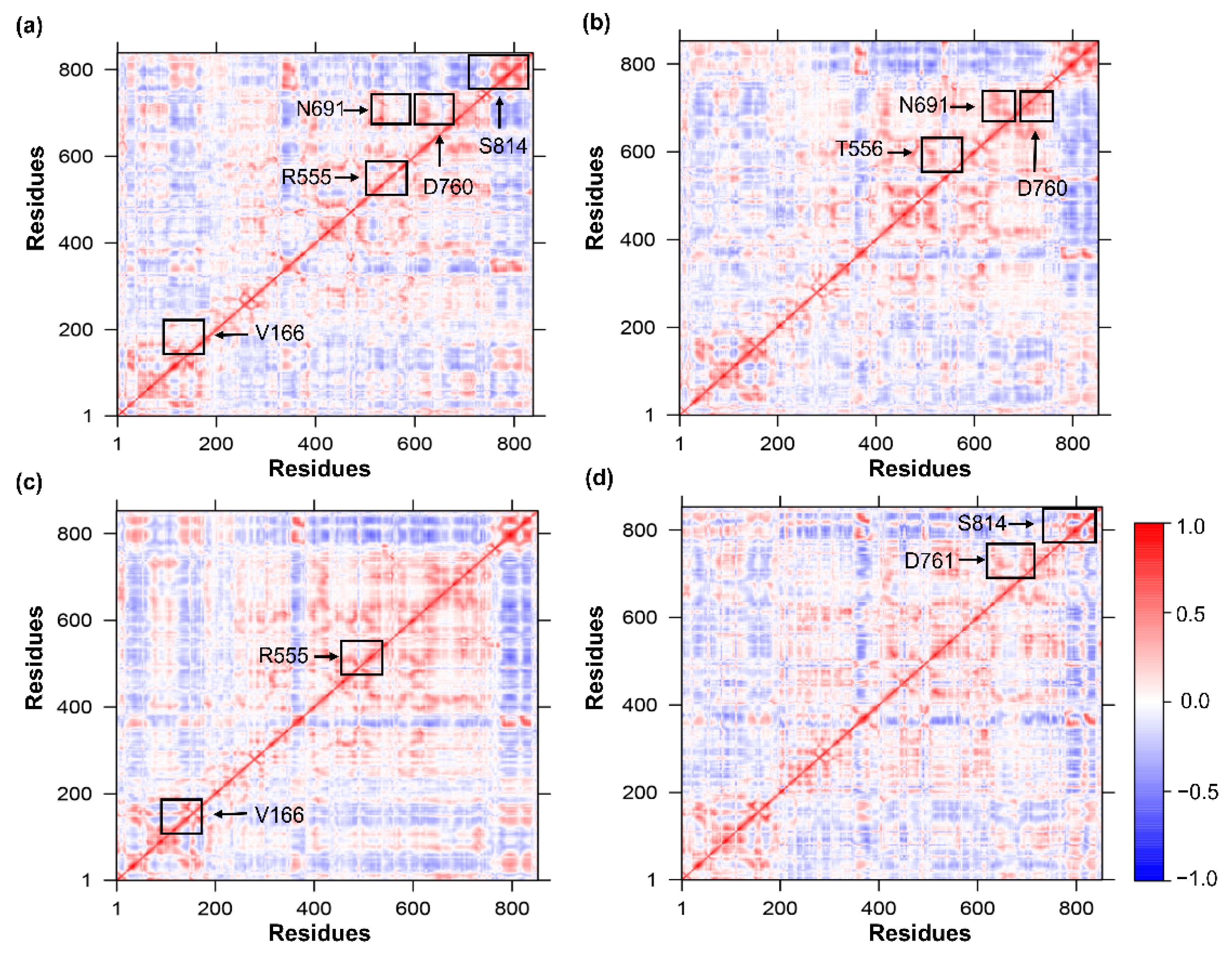
2.8. The Dynamic Cross-Correlation Matrix (DCCM)
2.9. MM-PBSA Calculation
2.10. In Vitro Activity of the Antiviral Drugs
3. Results and Discussion
3.1. Virtual Screening and Molecular Docking
3.2. The Analysis of the Interaction of Top Hits with RdRp
3.3. Measurement of Variations in the Cα Atoms of RdRp in the Presence and Absence of Drugs
3.4. Calculation of Equilibrium Conformation of Systems
3.5. Analysis of Essential Dynamics of Protein
3.6. Protein Folding Dynamics Exploration
3.7. Time-Correlated Protein Domain Motions and Molecular Flexibility
3.8. Binding Free Energy Calculations
3.9. In Vitro Antiviral Activity
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Fehr, A.R.; Perlman, S. Coronaviruses: An overview of their replication and pathogenesis. In Coronaviruses: Methods and Protocols; Springer: New York, NY, USA, 2015; Volume 1282, pp. 1–23. ISBN 9781493924387. [Google Scholar]
- Ye, Z.W.; Yuan, S.; Yuen, K.S.; Fung, S.Y.; Chan, C.P.; Jin, D.Y. Zoonotic origins of human coronaviruses. Int. J. Biol. Sci. 2020, 16, 1686–1697. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aftab, S.O.; Ghouri, M.Z.; Masood, M.U.; Haider, Z.; Khan, Z.; Ahmad, A.; Munawar, N. Analysis of SARS-CoV-2 RNA-dependent RNA polymerase as a potential therapeutic drug target using a computational approach. J. Transl. Med. 2020, 18. [Google Scholar] [CrossRef] [PubMed]
- Su, S.; Wong, G.; Shi, W.; Liu, J.; Lai, A.C.K.; Zhou, J.; Liu, W.; Bi, Y.; Gao, G.F. Epidemiology, Genetic Recombination, and Pathogenesis of Coronaviruses. Trends Microbiol. 2016, 24, 490–502. [Google Scholar] [CrossRef] [Green Version]
- Beniac, D.R.; Andonov, A.; Grudeski, E.; Booth, T.F. Architecture of the SARS coronavirus prefusion spike. Nat. Struct. Mol. Biol. 2006, 13, 751–752. [Google Scholar] [CrossRef] [Green Version]
- Delmas, B.; Laude, H. Assembly of coronavirus spike protein into trimers and its role in epitope expression. J. Virol. 1990, 64, 5367–5375. [Google Scholar] [CrossRef] [Green Version]
- Nal, B.; Chan, C.; Kien, F.; Siu, L.; Tse, J.; Chu, K.; Kam, J.; Staropoli, S.; Crescenzo-Chaigne, B.; Escriou, N.; et al. Differential maturation and subcellular localization of severe acute respiratory syndrome coronavirus surface proteins S, M and E. J. Gen. Virol. 2005, 86, 1423–1434. [Google Scholar] [CrossRef] [Green Version]
- Neuman, B.W.; Kiss, G.; Kunding, A.H.; Bhella, D.; Baksh, M.F.; Connelly, S.; Droese, B.; Klaus, J.P.; Makino, S.; Sawicki, S.G.; et al. A structural analysis of M protein in coronavirus assembly and morphology. J. Struct. Biol. 2011, 174, 11–22. [Google Scholar] [CrossRef]
- Wu, J.; Liu, W.; Gong, P.; Gong, P. A structural overview of RNA-dependent RNA polymerases from the Flaviviridae family. Int. J. Mol. Sci. 2015, 16, 12943–12957. [Google Scholar] [CrossRef] [Green Version]
- Smertina, E.; Urakova, N.; Strive, T.; Frese, M. Calicivirus RNA-dependent RNA polymerases: Evolution, structure, protein dynamics, and function. Front. Microbiol. 2019, 10, 1280. [Google Scholar] [CrossRef]
- Ago, H.; Adachi, T.; Yoshida, A.; Yamamoto, M.; Habuka, N.; Yatsunami, K.; Miyano, M. Crystal structure of the RNA-dependent RNA polymerase of hepatitis C virus. Structure 1999, 7, 1417–1426. [Google Scholar] [CrossRef] [Green Version]
- Elfiky, A.A. Anti-HCV, nucleotide inhibitors, repurposing against COVID-19. Life Sci. 2020, 248. [Google Scholar] [CrossRef]
- Nguyen, T.M.; Zhang, Y.; Pandolfi, P.P. Virus against virus: A potential treatment for 2019-nCov (SARS-CoV-2) and other RNA viruses. Cell Res. 2020, 30, 189–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kruse, R.L. Therapeutic strategies in an outbreak scenario to treat the novel coronavirus originating in Wuhan, China. F1000Research 2020, 9, 72. [Google Scholar] [CrossRef] [Green Version]
- Lai, M.M.; Baric, R.S.; Makino, S.; Keck, J.G.; Egbert, J.; Leibowitz, J.L.; Stohlman, S.A. Recombination between nonsegmented RNA genomes of murine coronaviruses. J. Virol. 1985, 56, 449–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Q.; Wu, J.; Nie, J.; Li, X.; Huang, W.; Correspondence, Y.W.; Zhang, L.; Hao, H.; Liu, S.; Zhao, C.; et al. The Impact of Mutations in SARS-CoV-2 Spike on Viral Infectivity and Antigenicity. Cell 2020, 182, 1284–1294. [Google Scholar] [CrossRef]
- Xu, X.; Liu, Y.; Weiss, S.; Arnold, E.; Sarafianos, S.G.; Ding, J. Molecular model of SARS coronavirus polymerase: Implications for biochemical functions and drug design. Nucleic Acids Res. 2003, 31, 7117–7130. [Google Scholar] [CrossRef] [Green Version]
- Hansen, J.L.; Long, A.M.; Schultz, S.C. Structure of the RNA-dependent RNA polymerase of poliovirus. Structure 1997, 5, 1109–1122. [Google Scholar] [CrossRef] [Green Version]
- Bressanelli, S.; Tomei, L.; Roussel, A.; Incitti, I.; Vitale, R.L.; Mathieu, M.; De Francesco, R.; Rey, F.A. Crystal structure of the RNA-dependent RNA polymerase of hepatitis C virus. Proc. Natl. Acad. Sci. USA 1999, 96, 13034–13039. [Google Scholar] [CrossRef] [Green Version]
- Kinsella, E.; Martin, S.G.; Grolla, A.; Czub, M.; Feldmann, H.; Flick, R. Sequence determination of the Crimean-Congo hemorrhagic fever virus L segment. Virology 2004, 321, 23–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, B.; Li, Z.; Jenni, S.; Rahmeh, A.A.; Morin, B.M.; Grant, T.; Grigorieff, N.; Harrison, S.C.; Whelan, S.P.J. Structure of the L Protein of Vesicular Stomatitis Virus from Electron Cryomicroscopy. Cell 2015, 162, 314–327. [Google Scholar] [CrossRef] [Green Version]
- Elfiky, A.A. Zika viral polymerase inhibition using anti-HCV drugs both in market and under clinical trials. J. Med. Virol. 2016, 88, 2044–2051. [Google Scholar] [CrossRef] [PubMed]
- Elfiky, A.A.; Ismail, A. Molecular dynamics and docking reveal the potency of novel GTP derivatives against RNA dependent RNA polymerase of genotype 4a HCV. Life Sci. 2019, 238. [Google Scholar] [CrossRef] [PubMed]
- Yang, P.L.; Gao, M.; Lin, K.; Liu, Q.; Villareal, V.A. Anti-HCV drugs in the pipeline. Curr. Opin. Virol. 2011, 1, 607–616. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, Y.; Yan, L.; Huang, Y.; Liu, F.; Zhao, Y.; Cao, L.; Wang, T.; Sun, Q.; Ming, Z.; Zhang, L.; et al. Structure of the RNA-dependent RNA polymerase from COVID-19 virus. Science 2020, 368, 779–782. [Google Scholar] [CrossRef] [Green Version]
- Molecular Operating Environment (MOE), version 2019.01; Chemical Computing Group ULC: Montreal, QC, Canada, 2021; H3A 2R7.
- De Clercq, E.; Li, G. Approved antiviral drugs over the past 50 years. Clin. Microbiol. Rev. 2016, 29, 695–747. [Google Scholar] [CrossRef] [Green Version]
- Chaudhuri, S.; Symons, J.A.; Deval, J. Innovation and trends in the development and approval of antiviral medicines: 1987–2017 and beyond. Antivir. Res. 2018, 155, 76–88. [Google Scholar] [CrossRef] [PubMed]
- Merck Molecular Force Field (MMFF94)—CHARMM v35b1 Documentation. Available online: http://www.charmm-gui.org/charmmdoc/mmff.html (accessed on 12 February 2021).
- Kaminski, G.; Jorgensen, W.L. Performance of the AMBER94, MMFF94, and OPLS-AA force fields for modeling organic liquids. J. Phys. Chem. 1996, 100, 18010–18013. [Google Scholar] [CrossRef]
- LLC The {PyMOL} Molecular Graphics System; Version 1.8; Schrödinger: Mannheim, Germany, 2015.
- Yu, W.; He, X.; Vanommeslaeghe, K.; MacKerell, A.D. Extension of the CHARMM general force field to sulfonyl-containing compounds and its utility in biomolecular simulations. J. Comput. Chem. 2012, 33, 2451–2468. [Google Scholar] [CrossRef] [Green Version]
- Huang, J.; Mackerell, A.D. CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 2013, 34, 2135–2145. [Google Scholar] [CrossRef] [Green Version]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical sampling through velocity rescaling. J. Chem. Phys. 2007, 126. [Google Scholar] [CrossRef] [Green Version]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A smooth particle mesh Ewald method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef] [Green Version]
- Kutzner, C.; Páll, S.; Fechner, M.; Esztermann, A.; De Groot, B.L.; Grubmüller, H. Best bang for your buck: GPU nodes for GROMACS biomolecular simulations. J. Comput. Chem. 2015, 36, 1990–2008. [Google Scholar] [CrossRef] [Green Version]
- Bahar, I.; Atilgan, A.R.; Demirel, M.C.; Erman, B. Vibrational dynamics of folded proteins: Significance of slow and fast motions in relation to function and stability. Phys. Rev. Lett. 1998, 80, 2733–2736. [Google Scholar] [CrossRef] [Green Version]
- Pronk, S.; Páll, S.; Schulz, R.; Larsson, P.; Bjelkmar, P.; Apostolov, R.; Shirts, M.R.; Smith, J.C.; Kasson, P.M.; Van Der Spoel, D.; et al. GROMACS 4.5: A high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 2013, 29, 845–854. [Google Scholar] [CrossRef]
- Seeber, M.; Cecchini, M.; Rao, F.; Settanni, G.; Caflisch, A. Wordom: A program for efficient analysis of molecular dynamics simulations. Bioinformatics 2007, 23, 2625–2627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dash, R.; Ali, M.C.; Dash, N.; Azad, M.A.K.; Hosen, S.M.Z.; Hannan, M.A.; Moon, I.S. Structural and Dynamic Characterizations Highlight the Deleterious Role of SULT1A1 R213H Polymorphism in Substrate Binding. Int. J. Mol. Sci. 2019, 20, 6256. [Google Scholar] [CrossRef] [Green Version]
- Maisuradze, G.G.; Liwo, A.; Scheraga, H.A. Relation between free energy landscapes of proteins and dynamics. J. Chem. Theory Comput. 2010, 6, 583–595. [Google Scholar] [CrossRef] [Green Version]
- Wan, H.; Hu, J.; Li, K.; Tian, X.; Chang, S. Molecular Dynamics Simulations of DNA-Free and DNA-Bound TAL Effectors. PLoS ONE 2013, 8, e76045. [Google Scholar] [CrossRef] [Green Version]
- Frauenfelder, H.; Sligar, S.G.; Wolynes, P.G. The energy landscapes and motions of proteins. Science 1991, 254, 1598–1603. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grant, B.J.; Rodrigues, A.P.C.; ElSawy, K.M.; McCammon, J.A.; Caves, L.S.D. Bio3d: An R package for the comparative analysis of protein structures. Bioinformatics 2006, 22, 2695–2696. [Google Scholar] [CrossRef] [Green Version]
- Upadhyay, S.K. Dynamics of Gal80p in the Gal80p-Gal3p complex differ significantly from the dynamics in the Gal80p-Gal1p complex: Implications for the higher specificity of Gal3p. Mol. Biosyst. 2014, 10, 3120–3129. [Google Scholar] [CrossRef] [PubMed]
- Chillemi, G.; D’Annessa, I.; Fiorani, P.; Losasso, C.; Benedetti, P.; Desideri, A. Thr729 in human topoisomerase I modulates anti-cancer drug resistance by altering protein domain communications as suggested by molecular dynamics simulations. Nucleic Acids Res. 2008, 36, 5645–5651. [Google Scholar] [CrossRef] [Green Version]
- Kumari, R.; Kumar, R.; Lynn, A. G-mmpbsa -A GROMACS tool for high-throughput MM-PBSA calculations. J. Chem. Inf. Model. 2014, 54, 1951–1962. [Google Scholar] [CrossRef]
- Hou, T.; Wang, J.; Li, Y.; Wang, W. Assessing the performance of the molecular mechanics/Poisson Boltzmann surface area and molecular mechanics/generalized Born surface area methods. II. the accuracy of ranking poses generated from docking. J. Comput. Chem. 2011, 32, 866–877. [Google Scholar] [CrossRef] [Green Version]
- Homeyer, N.; Gohlke, H. Free energy calculations by the Molecular Mechanics Poisson-Boltzmann Surface Area method. Mol. Inform. 2012, 31, 114–122. [Google Scholar] [CrossRef]
- Miller, B.R.; McGee, T.D.; Swails, J.M.; Homeyer, N.; Gohlke, H.; Roitberg, A.E. MMPBSA.py: An efficient program for end-state free energy calculations. J. Chem. Theory Comput. 2012, 8, 3314–3321. [Google Scholar] [CrossRef]
- Doublié, S.; Ellenberger, T. The mechanism of action of T7 DNA polymerase. Curr. Opin. Struct. Biol. 1998, 8, 704–712. [Google Scholar] [CrossRef] [Green Version]
- Koulgi, S.; Jani, V.; Uppuladinne, M.V.N.; Sonavane, U.; Joshi, R. Remdesivir-bound and ligand-free simulations reveal the probable mechanism of inhibiting the RNA dependent RNA polymerase of severe acute respiratory syndrome coronavirus 2. RSC Adv. 2020, 10, 26792–26803. [Google Scholar] [CrossRef]
- Boehr, D.D.; Nussinov, R.; Wright, P.E. The role of dynamic conformational ensembles in biomolecular recognition. Nat. Chem. Biol. 2009, 5, 789–796. [Google Scholar] [CrossRef] [Green Version]
- Karshikoff, A.; Nilsson, L.; Ladenstein, R. Rigidity versus flexibility: The dilemma of understanding protein thermal stability. FEBS J. 2015, 282, 3899–3917. [Google Scholar] [CrossRef]
- Hinsen, K. Structural flexibility in proteins: Impact of the crystal environment. Bioinformatics 2008, 24, 521–528. [Google Scholar] [CrossRef] [Green Version]
- Du, X.; Li, Y.; Xia, Y.L.; Ai, S.M.; Liang, J.; Sang, P.; Ji, X.L.; Liu, S.Q. Insights into protein–ligand interactions: Mechanisms, models, and methods. Int. J. Mol. Sci. 2016, 17, 144. [Google Scholar] [CrossRef]
- Rifai, E.A.; Van Dijk, M.; Vermeulen, N.P.E.; Yanuar, A.; Geerke, D.P. A Comparative Linear Interaction Energy and MM/PBSA Study on SIRT1-Ligand Binding Free Energy Calculation. J. Chem. Inf. Model. 2019, 59, 4018–4033. [Google Scholar] [CrossRef] [PubMed]
- Poli, G.; Granchi, C.; Rizzolio, F.; Tuccinardi, T. Application of MM-PBSA Methods in Virtual Screening. Molecules 2020, 25, 1971. [Google Scholar] [CrossRef] [Green Version]
- Mulangu, S.; Dodd, L.E.; Davey, R.T.; Tshiani Mbaya, O.; Proschan, M.; Mukadi, D.; Lusakibanza Manzo, M.; Nzolo, D.; Tshomba Oloma, A.; Ibanda, A.; et al. A Randomized, Controlled Trial of Ebola Virus Disease Therapeutics. N. Engl. J. Med. 2019, 381, 2293–2303. [Google Scholar] [CrossRef]
- Scott, L.J. Ledipasvir/Sofosbuvir: A Review in Chronic Hepatitis C. Drugs 2018, 78, 245–256. [Google Scholar] [CrossRef]
- Menon, R.M.; Polepally, A.R.; Khatri, A.; Awni, W.M.; Dutta, S. Clinical Pharmacokinetics of Paritaprevir. Clin. Pharmacokinet. 2017, 56, 1125–1137. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, D.; Du, G.; Du, R.; Zhao, J.; Jin, Y.; Fu, S.; Gao, L.; Cheng, Z.; Lu, Q.; et al. Remdesivir in adults with severe COVID-19: A randomised, double-blind, placebo-controlled, multicentre trial. Lancet 2020, 395, 1569–1578. [Google Scholar] [CrossRef]
- Warren, T.K.; Jordan, R.; Lo, M.K.; Ray, A.S.; Mackman, R.L.; Soloveva, V.; Siegel, D.; Perron, M.; Bannister, R.; Hui, H.C.; et al. Therapeutic efficacy of the small molecule GS-5734 against Ebola virus in rhesus monkeys. Nature 2016, 531, 381–385. [Google Scholar] [CrossRef]
- New drugs: Ledipasvir with sofosbuvir. Aust. Prescr. 2015, 38, 217–225. [CrossRef] [Green Version]
- Wang, M.; Cao, R.; Zhang, L.; Yang, X.; Liu, J.; Xu, M.; Shi, Z.; Hu, Z.; Zhong, W.; Xiao, G. Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 2020, 30, 269–271. [Google Scholar] [CrossRef] [PubMed]
- Sayad, B.; Sobhani, M.; Khodarahmi, R. Sofosbuvir as Repurposed Antiviral Drug Against COVID-19: Why Were We Convinced to Evaluate the Drug in a Registered/Approved Clinical Trial? Arch. Med. Res. 2020, 51, 577–581. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Drug Name | Hydrogen Bonds | Bond Length (Å) | Force Field | MOE Docking Score (kcal/mol) |
|---|---|---|---|---|
| Ledipasvir | R555 | 2.1 and 2.6 | CHARMM36 | −8.9 |
| V166 | 2.2 | |||
| Remdesivir | T556 | 2.1 and 2.4 | CHARMM36 | −8.3 |
| D760 | 2.3 and 2.5 | |||
| N691 | 2.5 | |||
| Paritaprevir | D761 | 2.1 | CHARMM36 | −9.4 |
| S814 | 2.0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pirzada, R.H.; Haseeb, M.; Batool, M.; Kim, M.; Choi, S. Remdesivir and Ledipasvir among the FDA-Approved Antiviral Drugs Have Potential to Inhibit SARS-CoV-2 Replication. Cells 2021, 10, 1052. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10051052
Pirzada RH, Haseeb M, Batool M, Kim M, Choi S. Remdesivir and Ledipasvir among the FDA-Approved Antiviral Drugs Have Potential to Inhibit SARS-CoV-2 Replication. Cells. 2021; 10(5):1052. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10051052
Chicago/Turabian StylePirzada, Rameez Hassan, Muhammad Haseeb, Maria Batool, MoonSuk Kim, and Sangdun Choi. 2021. "Remdesivir and Ledipasvir among the FDA-Approved Antiviral Drugs Have Potential to Inhibit SARS-CoV-2 Replication" Cells 10, no. 5: 1052. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10051052