
The Genomic Landscape of Thyroid Cancer Tumourigenesis and Implications for Immunotherapy
by
 and
and
Amandeep Singh
1,2,3,*, 
Jeehoon Ham
1,3,4,
Joseph William Po
4,5,
Navin Niles
1,4,6,
Tara Roberts
7 and
Cheok Soon Lee
1,2,3,4,8,9,10 1
Discipline of Pathology, School of Medicine, Western Sydney University, Campbelltown 2560, Australia
2
Department of Anatomical Pathology, Liverpool Hospital, Liverpool 2170, Australia
3
Cancer Pathology Laboratory, Ingham Institute for Applied Medical Research, Liverpool 2170, Australia
4
CONCERT Biobank, Ingham Institute for Applied Medical Research, Liverpool 2170, Australia
5
Surgical Innovation Unit, Department of Surgery, Westmead Hospital, Westmead 2145, Australia
6
Head and Neck Surgery, Liverpool Hospital, Liverpool 2170, Australia
7
School of Medicine, Western Sydney University and Ingham Institute for Applied Medical Research, Liverpool 2170, Australia
8
South Western Sydney Clinical School, University of New South Wales, Liverpool 2170, Australia
9
Department of Tissue Pathology and Diagnostic Oncology, Royal Prince Alfred Hospital, Camperdown 2050, Australia
10
Central Clinical School, University of Sydney, Camperdown 2050, Australia
*
Author to whom correspondence should be addressed.
Cells 2021, 10(5), 1082; https://0-doi-org.brum.beds.ac.uk/10.3390/cells10051082
Submission received: 23 March 2021
/
Revised: 23 April 2021
/
Accepted: 27 April 2021
/
Published: 1 May 2021
(This article belongs to the Special Issue 10th Anniversary of Cells—Advances in Cellular Pathology)
Abstract
:Thyroid cancer is the most prevalent endocrine malignancy that comprises mostly indolent differentiated cancers (DTCs) and less frequently aggressive poorly differentiated (PDTC) or anaplastic cancers (ATCs) with high mortality. Utilisation of next-generation sequencing (NGS) and advanced sequencing data analysis can aid in understanding the multi-step progression model in the development of thyroid cancers and their metastatic potential at a molecular level, promoting a targeted approach to further research and development of targeted treatment options including immunotherapy, especially for the aggressive variants. Tumour initiation and progression in thyroid cancer occurs through constitutional activation of the mitogen-activated protein kinase (MAPK) pathway through mutations in BRAF, RAS, mutations in the phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K) pathway and/or receptor tyrosine kinase fusions/translocations, and other genetic aberrations acquired in a stepwise manner. This review provides a summary of the recent genetic aberrations implicated in the development and progression of thyroid cancer and implications for immunotherapy.
1. Introduction
Thyroid cancer (TC) is the most common endocrine malignancy, constituting 2.1% of all newly diagnosed cancer cases worldwide [1]. TC incidence has increased 3-fold over the past three decades, with the incidence-based mortality rate increasing at 1.1% per year overall [2]. The incidence is higher in developed countries, and in females with a female to male ratio of 3:1 with a 5.1% incidence in females worldwide in 2018 [2,3]. It was the 7th commonest cancer in women in Australia in 2019 with higher prevalence in 15- to 24-year-olds [4]. Between 1982 and 2019, the age-standardised incidence rate of thyroid cancer increased by 392% (from 2.7 to 13 per 100,000 persons) [4]. This increase is in both genders, all age groups and ethnicities with papillary thyroid carcinomas accounting for the most cases [5,6].
Increased medical surveillance and advent of new radiological diagnostic techniques (low-cost ultrasonography for a more specific thyroid nodule screening), have contributed to increased detection of clinically occult cancers [7]. The increased incidence is attributed to overdetection of small indolent tumours and a concurrent increase in subsequent surgical intervention rates; therefore, patients are overtreated for low-risk tumours [5,7]. The American Thyroid Association treatment guidelines base treatment on risk stratification to prevent overtreatment, and includes assessment of clinical factors (age, gender, radiation exposure and family history), pathological parameters (type and size of tumour, lymphovascular invasion, extrathyroidal extension and lymph node metastases) and molecular markers (including BRAFV600E, RAS, TP53 and TERT) [8].
Importantly, overtreatment may subject patients to possible treatment-related complications without a meaningful improvement in clinical outcomes. Moreover, the cost of interventions in the indolent tumour setting can impose a significant undue financial burden to the healthcare system. Therefore, it is imperative to identify ‘high-risk’ patients to guide appropriate treatment and prevent overtreatment of ‘low-risk’ patients.
2. Classification
Thyroid tumours are classified histologically based on criterion implemented by the World Health Organisation (WHO) [9]. The primary tumours are epithelial in origin, developing from the follicular or parafollicular cells. The follicular-derived carcinomas are well differentiated carcinomas (DTC)–papillary carcinomas (PTC), follicular carcinomas (FTC) and Hürthle cell carcinoma (HCC), poorly-differentiated carcinomas (PDTC) and undifferentiated anaplastic carcinomas (ATC). PTCs are diagnosed primarily based on nuclear morphology, and are further subclassified morphologically and architecturally with certain subtypes correlating with an aggressive biology.
DTCs are believed to develop from pre-existing benign and/or borderline follicular neoplasms that display high-risk morphology, notably vascular and capsular invasion, in combination with specific genetic aberrations, whilst the clinically aggressive PDTCs and ATCs develop from dedifferentiation of the DTCs. The borderline/intermediate follicular neoplasms include “follicular tumour of uncertain malignant potential” in which conclusive malignant features are equivocal, and “non-invasive follicular thyroid neoplasm with papillary-like nuclear features (NIFTP)”, an encapsulated circumscribed tumour with stringent histopathologic diagnostic criteria. This criterion constitutes histologically imperative features including well circumscription or encapsulation, PTC nuclear features, a follicular growth pattern and an absence of capsular or vascular invasion. In addition, specific exclusion features including presence of true papillae (>1%) and/or psammomatous calcifications; solid, insular or trabecular growth pattern (>30%); increased mitoses (≥3 mitoses per 10 high-power fields) and tumours necrosis [9]. NIFTP was formerly termed non-invasive encapsulated follicular variant of PTC (EFVPTC); its reclassification initiated by the Endocrine Pathology Society working group through an international multi-institutional study that established preponderance of RAS genetic aberrations (similar to follicular-derived lesions), deficient common PTC BRAFV600E genetic aberrations and indolent nature of EFVPTC, preventing overdiagnosis and overtreatment of these tumours [10]. Its incidence is variable, lowest in Asian countries at around 1.6%, and highest in Western countries at around 13.3% [11]. Retrospective studies that re-evaluated previously diagnosed EFVPTC cases after reclassification, determined 1.3–40.7% of non-invasive EFVPTC conformed to the histologic criterion for NIFTP [10,12,13]; the variability in numbers attributed to differing prevalence in the Asian and non-Asians studies.
DTCs represent more than 85% of thyroid carcinomas of which PTCs are most prevalent, with a 5-year survival rate of 97.7% in younger age groups with reduction in survival with advancing age. PDTCs and ATCs constitute 5–10% of thyroid cancers with a poor 5-year survival rate of 5%, and develop from dedifferentiation of DTCs [9,14]. The overall improved survival rate in DTCs is attained from implementation of appropriate management plan including surgery, radioactive iodine (RAI) ablation and lifelong thyroid stimulating hormone (TSH) suppressive therapy, and a favourable tumour biology. Currently, administration of radioactive 131 I for ablation of residual thyroid tissue is selectively offered to low and intermediate-risk patients with increased serum thyroglobulin level at 6 months post-surgery and following TSH suppressive therapy or with unfavourable prognostic factors [15,16]. Despite this, 20% and 10% of DTC patients develop locoregional recurrences and distant metastases respectively within 10 years, and these are usually RAI-refractory and generally surgically unresectable with overall 3-year survival of less than 50%. These patients are treated with antiangiogenic multitarget tyrosine kinase inhibitors including lenvatinib and sorafenib, and specific inhibitors for BRAF-, MEK- or ALK-mutant tumours [16]. ATCs have an aggressive clinical course, with patient life expectancy of about 6 to 12 months despite the utilisation of systemic therapies such as adjuvant external beam radiation therapy (EBRT), intensity-modulated radiation therapy (IMRT) with radio-sensitising chemotherapy regimens [9,16].
3. Thyroid Cancer Pathogenesis
Thyroid cancers, benign and intermediate (NIFTP) entities develop from follicular epithelial cells, and it is theorised that DTCs develop from benign (example follicular adenomas in the case of follicular thyroid carcinomas [FTCs]) and intermediate lesions (NIFTP). PDTCs and ATCs develop from dedifferentiated of DTCs with approximately 10% of PTCs transforming into these forms through different genetic aberrations accumulated in a stepwise manner [17]. Conversely, medullary thyroid cancer (MTC) occurs in the neuroendocrine parafollicular cells (C cells), and is solely induced by RET proto-oncogene mutations [17].
4. Genetics of Thyroid Cancers
Multiple genes are implicated in the development of thyroid neoplasms, both benign and malignant as illustrated in Figure 1 and Table 1. Specific mutations or rearrangements occur in these genes which are responsible for cell proliferation, survival and differentiation through different pathways. Approximately 90% of mutations are mutually exclusive activating mutations in oncogenes RAS (~13%) and BRAF (~60%), and rearrangements involving RET, ALK and NTRK genes (~5%); whilst the remaining 10% are loss-of-function mutations affecting tumour suppressor genes such as PTEN, PPARγ and TP53 [17,18,19] Targeted therapies are in use for tumours with some of these mutations. The Cancer Genome Atlas (TCGA) reported comprehensive genetic aberrations in 97% of PTCs, including driver genes EIF1AX, CHEK2 and PPM1D, members of the phosphoinositide 3-kinase (PI3K) pathway and other gene fusions [19]. This leaves 3% of PTCs (termed “dark matter”) which remain genetically uncharacterised. Understanding the prevalence and the clinicopathologic relevance of these genetic aberrations have allowed for the pursuance of new targeted therapies.
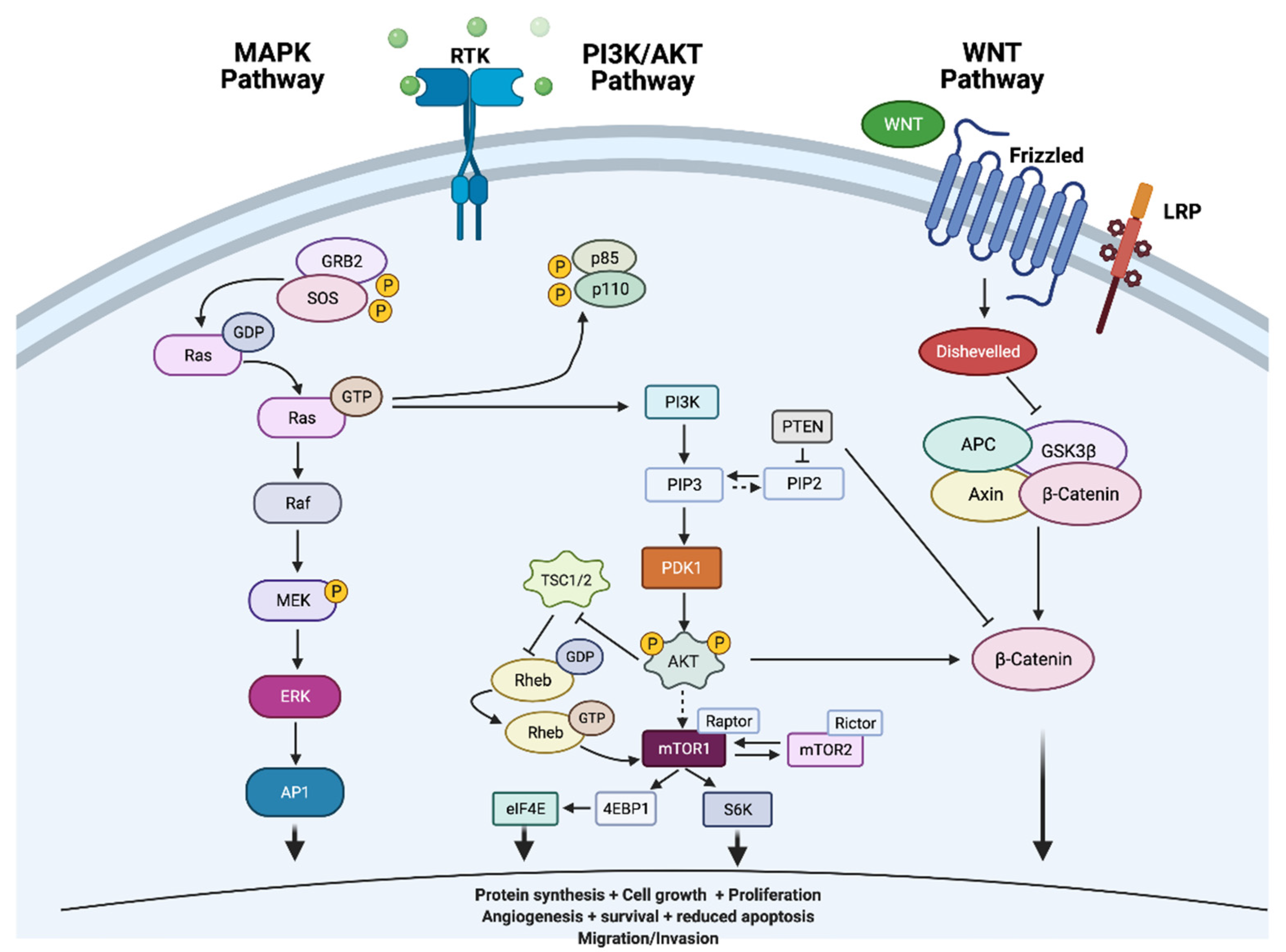
Tumourigenesis of thyroid tumours involves dysregulation of cell signalling pathways mitogen-activated protein kinase (MAPK) and phosphatidylinositol-3 kinase (PI3K)/Ak strain transforming (AKT)/mammalian target of rapamycin (mTOR) signalling pathways. The commonest oncogenic drivers of these pathways include BRAF and RAS point mutations (Figure 2 adapted from [20] [Created with BioRender.com]).
MAPK and PI3K/AKT pathways are dependent on activity of mutually exclusive RAS, BRAF and RET/PTC point mutations (BRAF/RAS/PIK3CA) and rearrangements (RET/PTC and TRK) which drive thyroid cancer oncogenesis. The prevalence of these genetic aberrations varies in the different TCs, with the highest prevalence of up to 62% (most frequently BRAFV600E) in PTCs [20]. BRAFV600E mutations are especially found in tall cell variant or infiltrative PTCs which express an aggressive biology, whilst less fatal RAS mutations are generally detected in encapsulated and follicular variants of PTCs and adenomas [21,22].
5. Tumour Initiation
5.1. The Mitogen-Activated Protein Kinase (MAPK) Pathway
The MAP kinase pathway is a signal transduction pathway, constitutive activation of which is essential in the development of TCs. MAPKs regulate vital cellular functions involved in cell proliferation, differentiation and development through key protein including receptor tyrosine kinases (RET, anaplastic lymphoma kinase [ALK], vascular endothelial growth factor receptor [VEGFR] and neurotrophic receptor tyrosine kinase [NTRK1/3]), RAS, rapidly accelerated fibrosarcoma (RAF), mitogen-activated protein kinase (MEK) and extracellular signal-regulated kinase (ERK) as illustrated in Figure 2 below [13]. The binding of a growth factor to a receptor tyrosine kinase receptor activates downstream pathways leading to modification in cell proliferation, differentiation and survival.
5.2. RAS Mutations
The rat sarcoma viral oncogenes homolog (RAS) gene is a commonly mutated protooncogene that codes for protein isoforms, NRAS, HRAS and KRAS, which are ubiquitously expressed at different levels in different tissue types. RAS proteins act as effector molecules in the MAPK and PI3K/AKT/mTOR signalling cascades and are oncogenically activated in numerous human cancers [25,26]. The RAS molecules transmit mitogen signals from the tyrosine kinase membrane receptors (RTKs) to transcription factors via downstream effectors. The RAS protooncogenes encode 21kDa G-proteins called p21RAS GTPases that are bound to guanosine diphosphate (GDP) in their inactive form and to guanosine triphosphate (GTP) when active. A group of proteins called ‘GAPs’ (guanine nucleotide exchange factors [GEFs] and GTPase activating proteins) promote conformational changes to the active form by allowing release of GDP, thereby enabling binding of GTP. This conformation change allows transduction of signals from growth factor receptors. Point mutations in the GTP-binding domain (codons 12 and 13) or the GTPase domain (codon 61) cause substitution of certain protein residues that affect GTPase activity, locking p21RAS in the activated form, initiating tumour development. Approximately 99% of RAS mutations involve codons 12, 13 or 61 [26,27].
RAS initiating mutations inhibit apoptosis promoting transient proliferation of neoplastic epithelial cells in the presence of TSH. This TSH-mediated dependent cell growth is inhibited via programmed cell death through the extracellular signal-regulated kinase (ERK) and c-Jun N-terminal kinases (JNK) signal transduction pathways [28]. Conversely, acute expression of RAS in association with absent/low TSH promotes TSH-independent cell growth, highlighting the dependence of thyrocytes expressing oncogenic RAS on TSH levels. RAS activation causes DNA damage and induces dedifferentiation in a dose-dependent manner with increased RAS expression inhibiting thyroid-specific genes that are vital in the maintenance of differentiation (examples transcription factors TTF-1 and PAX8) [28,29,30]. Transgenic mice with human NRAS oncogene expression in the thyrocytes under the control of Tg promoter (Tg-NRAS) developed thyroid adenomas (11%) and invasive follicular carcinomas (40%), with approximately 25% showing poor differentiation, lymphovascular invasion and increased metastatic risk [28,30].
Of its three isoforms NRAS, HRAS and KRAS, the most common mutation is the NRAS exon2 (codon 61) mutation, which is associated with higher risk of metastasis [31,32,33,34]. RAS mutations are documented in both benign and malignant thyroid follicular epithelium with characteristically follicular growth pattern, with higher frequencies in FTC of up to 57%, follicular adenomas 30%, hyperplastic nodules 5.6%, goitres 7–25%, Hürthle cell adenomas 0–4%, NIFTP 29.6–56.6%), and lesser in frequency in PTC of follicular variant with rates of 1.7–20% [10,13,22,32,33,35,36,37]. There is a preferential mutation in the RAS subtypes in different thyroid neoplasms with NRAS mutations characteristically identified in follicular variants of PTCs, FTCs and ATCs, while Hürthle cell carcinomas (HCC; 15–25%) and medullary thyroid carcinomas (MTCs) commonly harbour HRAS mutations [32,38,39]. About 28–55% and 24–52% of PDTCs and ATCs harbour NRAS, HRAS or KRAS mutations, which are mutually exclusive to BRAF mutations and RET/PTC gene fusions [17,40,41].
The development of these tumours is mediated through TSH-independent growth, TSH-dependent apoptosis, DNA damage and de-differentiation, mainly through the RAF/MEK/ERK pathway, stress-activated protein kinase (SAPK)/JNK pathway (apoptosis), and unknown pathway/s involved in RAS-induced dedifferentiation through inhibition of TTF-1 and/or PAX-8. Clinically, the mutational status of RAS proto-oncogene is valuable particularly in RAI-refractory thyroid tumours due to advent of MAPK kinase MEK1 and MEK2 inhibitor selumetinib [42].
5.3. BRAF Mutations
RAF is a serine-threonine kinase with three isoforms—ARAF, BRAF and CRAF (RAF1). These isoforms are differentially activated by RAS, initiating downstream activation of MAPK pathway effectors [43]. The BRAF isoform has the highest affinity for both MEK1 and MEK2 [44]. BRAF is an initiator mutation, with common valine-to-glutamate substitution at residue 600 (V600E) detected in about 60% of PTCs and is less prevalent in PDTCs (12–33% prevalence) and ATCs (25–29%) [20,45,46,47,48,49,50,51]. These BRAF-mutated PDTCs and ATCs arise from pre-existing PTCs. Infrequent BRAFK601E mutation, BRAF rearrangement and deletions or in-frame insertions represent 1–2% of the remaining cases. The rearrangement occurs through paracentric inversion of chromosome 7q that results in in-frame fusion between exons 1–8 of AKAP9 gene and exons 9–18 of BRAF. This AKAP9-BRAF rearrangement induces kinase activity, and exists in 11% of PTCs related to radiation exposure and only 1% of those unrelated to radiation exposure [52].
BRAF mutations are an early event in tumourigenesis with a presence in papillary microcarcinomas and genotype heterogeneity in TCs [24,53,54]. BRAFV600E mutated PTCs have classic papillary morphology or the more aggressive tall-cell morphology [53]. These mutated tumours exhibit reduced expression of genes involved in thyroid hormone biosynthesis, namely thyroglobulin, thyroid peroxidase and sodium iodide symporter, and are refractory to RAI therapy [54,55,56]. BRAFV600E-mutated tumours typically demonstrate extrathyroidal extension and are generally of advanced stage with TERT promoter mutation with evidence of lymph node and distant metastases [53,57,58,59].
BRAF activation induces apoptosis, dedifferentiation and promotes TSH-dependent cell growth, however, a resultant balance in apoptosis and synthesis results in no net growth. Through induction of genomic instability, there is acquisition of secondary genetic events that decrease the expression of TSH receptor (TSHR) [60]. Clinically, the higher affinity of BRAF for MEK suggests utilisation of selective MEK inhibitors, such as dabrafenib and trametinib, in addition to BRAF inhibitors to preferentially inhibit MAPK pathway-driven growth of BRAF-mutated thyrocytes [45,46,58,60,61].
5.4. RET/PTC Rearrangements
RET, a proto-oncogene located on chromosome 10q11.2 encodes a transmembrane tyrosine kinase receptor which is normally expressed in neural crest-derived cells, including thyroid parafollicular (‘C’) and follicular cells. The RET gene is activated by fusion with 5′ portion of heterogeneous genes, initiating the expression of the 3′ portion of the RET gene that codes for the tyrosine kinase domain of the receptor, producing constitutively active chimeric forms of the receptor. At least 10 rearrangements with different partner genes are known, and the commonest are RET/PTC1 (formed through paracentric inversion of chromosome 10 long arm which fuses with CCDC6/H4 gene) [62], RET/PTC2 (formed by a reciprocal translocation between chromosomes 10 and 17, leading to juxtaposition of c-RET tyrosine kinase domain with a regulatory subunit of R1acAMP-dependent protein kinase A) [63] and RET/PTC3 (results from fusion with NCOA4/RFG/ELE1 gene) [64]. RET/PTC1 and RET/PTC3 rearrangements account for >90% of all rearrangements [65] and occur in higher frequencies in younger patients and those exposed to radiation [66,67,68,69]. These rearrangements activate both the MAPK and PI3K/AKT pathways.
RET-PTC rearrangement is present in 6.8–32.9% of PTCs and 12.9% of PDTCs [69,70,71,72,73]. RET/PTC expressing PTCs can be divided into four groups—(1) lacking RET/PTC rearrangements (28%), (2) balanced RET expression with very low levels of RET/PTC1 (24%), (3) unbalanced RET exons 10–11 and 12–13 expression with high RET/PTC1 but no RET/PTC3 expression (28%), and (4) unbalanced RET expression with high RET/PTC1 and low RET/PTC3 expression (20%) [74]. These rearrangements are associated with distinct tumour biologic properties, with RET/PTC1 tumours displaying the typical papillary architecture, small size and better prognosis; by contrast, RET/PTC3 tumours are solid and aggressive [75]. RET rearrangements can occur sporadically in follicular adenomas with a prevalence of 17–63.2% by RT-PCR in Hashimoto’s thyroiditis especially in the metaplastic oxyphil cells [67]. In benign thyroid nodules bearing RET/PTC rearrangements, there is a 4.3-fold increase in size within a timeframe of about 36 months [76]. This cellular proliferation is through the MAPK pathway and/or through expression of chemokines CXCL1 and CXCL10 and their receptors that modulate cellular growth by an autocrine/paracrine mechanism [77,78]. RET/PTC activation-linked biological events include apoptosis and dedifferentiation and lack of TSH-independent growth.
The existence of RET/PTC rearrangements in thyroiditis potentially govern early tumourigenesis, highlighting the role of proinflammatory markers in the development of tumours. Furthermore, the existence of RET/PTC rearrangements in benign lesions in variable frequencies suggests this to be an initial event. Increased prevalence of RET/PTC rearrangements in PTCs, with a relatively low prevalence in PDTCs contradicts a potential role of these rearrangements in tumour progression. Therefore, it is implied that additional genetic aberrations occur for progression into aggressive carcinomas. Selpercatinib is a highly selective RET kinase inhibitor utilised in patients with RET fusion-positive PTC and RET-mutant MTC [79].
5.5. EIF1AX Mutations
Eukaryotic translation initiation factor 1A X-linked (EIF1AX) encodes a translation initiation factor, change-of-function or gain-of-function mutations (in exons 2, 5 and 6) of which were recognised originally in uveal melanomas. These mutations occur in a mutually exclusive with other driver mutations in both benign and malignant TCs including FTCs (17%), HCCs (11%) and PTCs (1–2%; commonly follicular variant). Conversely, 11% of PDTCs and 9–30% of ATCs also harbour these mutations, and are almost consistently associated with RAS and BRAF mutations [17,19,38,80,81,82]. EIF1AX and RAS mutations cooperate to drive thyroid tumourigenesis [82]. Co-expression with RAS mutations occurs in advanced tumours, which coharbour TERT promoter or TP53 mutations, and are present in approximately 50% of ATCs. EIF1AX/RAS-mutated tumours with either TERT promoter or TP53 mutations are larger and aggressive with early metastasis and confer worse survival in PDTCs and ATCs [82]. In thyroid carcinomas, there is a prevalence of hotspot splice-site EIF1AX-A113splice mutation which initiates eukaryotic initiation factor 2 alpha (EIF2α) suppression by dephosphorylation through induction of activating transcription factor 4 (ATF4; a cellular stress sensor), increasing protein synthesis. EIF1AX-A113splice also augments cellular myelocytomatosis oncogene (c-Myc) stabilisation by RAS. C-MYC in collaboration with ATF4 induce production of amino acids which sensitise mTOR signalling and protein synthesis [82]. Combinational treatment of mTOR (AZD8055) with either MEK (trametinib) or BRD4 (JQ1) inhibitors to EIF1AX-A113splice knock-in tumour cell lines result in reduced c-MYC and mTOR protein levels and consequent tumour size reduction [82]. In vivo studies are required to further elucidate the potential use of these inhibitors in humans.
6. Tumour Progression
6.1. The Phosphatidylinositol 3-Kinase (PI3K)/Ak Strain Transforming (AKT)/Mammalian Target of Rapamycin (mTOR) Pathway
The PI3K/AKT pathway constitutes PIK3CA, PIK3C2G, PIK3CGM, PIK3C3, PIK3R1, PRIK3R2, AKT1, AKT3, TSC1, TSC2, PTEN and the mTOR signalling complex proteins, alterations of which exist in diverse human malignancies. PI3K/AKT pathway is activated by binding of RAS to the p110 catalytic subunits of PI3K of which PIK3CA (α-type) and PIK3CB (β-type) are the most frequently expressed subunits in tissues. The other common mechanism of activation is through activation of receptor tyrosine kinases by numerous growth factors, leading to activation of the p110 catalytic subunits(s), formation of phosphatidylinositol-3, 4, 5-triphosphate (PIP3) which localises AKT to the cell membrane. Phosphorylation of AKT initiates a downstream activation of protein effectors including the mammalian target of rapamycin (mTOR). Phosphatase and tensin homolog deleted on chromosome ten (PTEN) is a key negative regulator of the pathway, a role achieved through dephosphorylation of PIP3. Activating mutations or amplification of one of the protein genes, generally PIK3CA gene, and/or inactivating mutations (PTEN) result in constitutive activation of the pathway, a feature distinguishable in less differentiated tumours [34,83,84,85].
PI3K/AKT pathway’s role in development of thyroid cancer is explained through PTEN (the pathway regulator and a major tumour suppressor) germline mutations which notably exist in Cowden disease, a disease characterised by hamartomatous growths, benign thyroid diseases and development of cancers in numerous organs including thyroid [34]. Loss of heterozygosity in PTEN occurs in 7% of follicular adenomas and 27% of follicular carcinomas [85,86]. The role of sporadic PTEN mutations in thyroid neoplasms is undetermined.
Activation of PI3K/AKT pathway through PTEN and/or PIK3CA mutation(s) lead to the development of carcinomas especially in the presence of BRAFV600E mutation [18,87,88]. PIK3CA mutations are prevalent in 5–25% of ATCs and 0–11% in PDTCs; AKT1 mutations in 0–8% of ATCs and 0–13% of PDTCs; and PTEN mutations in 10–15% of ATCs. The difference in mutations across thyroid subtypes is attributed to tumour heterogeneity or impurity due to high macrophage infiltration in ATCs [17,89]. These mutations are uncommon in DTCs, with an 11% prevalence of mutated PIK3CA in FTCs, and 3% PIK3CA and 2% PTEN mutations in PTCs [90]. They are mutually exclusive with BRAF or RAS mutations in DTCs, whilst occur in combination in PDTCs and ATCs [34]. This observation supports that accumulation of genetic alterations in the PI3K/AKT pathway that potentiates progression of DTCs into PDTCs and ATCs.
Inhibitors of certain PI3K/AKT pathway effectors have been developed, and could be utilised in combination with mainstream chemotherapeutic agents for treatment of thyroid cancers.
6.2. PAX8/PPARγ, ALK and NTRK Rearrangements
This rearrangement involves fusion of thyroid-specific transcription factor PAX8 gene (on chromosome 3p25) with PPARγ gene (on chromosome 2q13) [91]. PPARγ gene is a transcription factor ubiquitously expressed in adipocytes and functions in lipid metabolism and regulation of adipocyte differentiation [92]. In thyroid neoplasms, PAX8/PPARγ rearrangement is present in 4–33% of follicular adenomas, 30–58% of FTCs, 0.3% HCC and 37.5% of PTCs of follicular variant [19,32,91,92,93,94].
ALK rearrangement is common in PDTCs with a prevalence of 16%; striatin (STRN)-ALK rearrangement is the commonest and rarely EMAP like 4 (EML4)-ALK [95,96]. Neurotrophic tyrosine kinase receptor (NTRK) fusion oncogenes (NTRK3/ETV6, NTRK1-TPR and NTRK1-LMNA and NTRK1-TMP3) are recognised in up to 26% of PTCs. NTRK gene fusion positive advanced solid tumours with no conventional treatments are treated with entrectinib or larotrectinib [61,97].
6.3. Telomerase Reverse Transcriptase (TERT) Promoter Mutations
Telomerase is expressed in germline cells and its activation promotes cancer development. Activation of telomerase reverse transcriptase (TERT), the catalytic protein subunit of telomerase, is induced by mutations in its promoter region, namely C250T and C228T, the latter being more prevalent, which promotes tumourigenesis by enabling replicative immortality in tumour cells [18]. These mutations co-exist with BRAFV600E or RAS mutations and are associated with aggressive phenotype with higher invasive and metastatic capability, treatment resistance and poor survival [17,18,98,99,100,101,102]. TERT is overexpressed by binding of BRAFV600E-induced E26 transformation-specific/E-twenty-six (ETS) transcription factors to the ETS-binding site produced by the mutation. TERT-promoter mutations occur in 9–10.6% non-metastatic PTCs, but their incidence increase incredibly to 60% in metastatic PTCs [18]. These tumours have aggressive clinicopathological features including extrathyroidal extension and lymph node and/or distant metastases [19,103,104]. Genetic analysis studies found the prevalence of clonal TERT promoter mutations in 61–73% of ATCs and 40% of PDTCs, lending further credence to its importance in tumour progression [17]. Recently, TERT rearrangements, which are mutually exclusive with TERT promoter mutations, have been identified in some aggressive cancers such as glioblastoma, neuroblastoma and melanoma as late driver mutations [105,106,107,108]. This finding highlights the important role that TERT promoter mutations play as a late driver in tumour progression in ATCs. Implementation of molecular diagnostic assessment of these mutations could assist in the identification of high-risk patients.
6.4. TP53 Mutations
Inactivating point mutations of the tumour protein p53 (TP53) tumour suppressor gene that encode p53 protein are prevalent in human cancers including thyroid [109]. Of the thyroid cancers, p53 inactivation is particularly common in PDTCs and ATCs, with a prevalence of 8–35% in PDTCs and significantly higher prevalence of up to 73% in ATCs, especially in association with BRAFV600E mutation [17,19,20]. TP53 alterations are infrequent in metastatic PTCs (13%) or FTC (8%) [109]. This implies that p53 inactivation in association with other oncogenes, for example BRAFV600E, induces a malignant phenotype in thyrocytes with loss of differentiation and, therefore, is crucial in late-stage progression of thyroid cancer. This knowledge has been utilised in transgenic mouse studies, one of which demonstrated BRAFV600E and TP53-mutant mice harboured PTCs with accelerated growth and poorer prognostic features including progression into less differentiated PTDCs and ATCs [110]. The latter findings have also been replicated in humans [111,112]. TP53 is a late driver mutation.
6.5. CDKN2A
Cyclin dependent kinase inhibitor 2A (CDKN2A) encodes p16INK4a, a tumour suppressor gene that regulates the cell cycle. Inactivation of the CDKN2A gene due to copy number loss (CNL), likely secondary to epigenetic silencing, homozygous loss or truncating mutations is linked to cancer progression. CDKN2A mutations in thyroid cancer are mostly associated with advanced status with an incidence of 15–23% in ATCs [18]. CDKN2A loss is associated with advanced DTCs, has a higher prevalence in ATCs, and is associated with poor survival [18]. Cyclin D-cyclin-dependent kinase 4/6 (CDK4/6) inhibitor palbociclib could potentially be utilised in ATCs [61].
6.6. Mismatch Repair Gene Deficiency
Inactivation of DNA mismatch repair gene(s) encoding MutL-homolog DNA mismatch repair (MMR) enzymes MLH1, MLH3, PMS1 and PMS2 results in microsatellite instability. This inactivation is attributed to persistent oxidative stress which also results in genomic damage and inefficient DNA repair. The prevalence of microsatellite instability (MSI) in thyroid neoplasms is exceptionally rare, and is recognised in about 2.3–2.5% FTCs and is absent or exceedingly rare in other thyroid tumours [113,114,115]. However, Pozdeyev et al. noted presence of MMR DNA deficiency in up to 46% of thyroid cancers with high mutational burden especially ATCs, and these tumours lacked RAS, BRAF or RET oncogenes [18]. The mechanism of the MLH1 DNA mismatch repair (MMR) silencing in PTC is idiopathic, but is likely promoted by FOXO1 suppression, FTC and FTA retain MMR activity because of its separate tumorigenic pathways [114,115]. Numerous other studies have produced conflicting results. Santos et al. and Mitmaker et al. documented rates of 37–90% in benign lesions, 64–84% in PTCs and 62.5–87% in FTCs [116,117]. In contrast, other studies documented an almost complete absence of MSI [118,119]. The disparate results are likely attributed to false positives from the use of polyacrylamide gels, whist recent use of next generation sequencing support low prevalence of MSI. Further studies are required to understand the role of mismatch repair gene deficiency in thyroid neoplasms as they respond to anti-PD-L1 (programmed cell death 1 ligand 1) immunotherapy [120].
6.7. SWI/SNF Chromatin Remodelling Complex
SWI/SNF complexes consist of 12–15 subunits including ARID1A, ARID1B, ARID2, ARID5B, SMARCB1, SMARCA4, SMARCA2, PBRM1 and ATRX. These complexes interact with co-activators, co-repressors and transcription factors to mobilise nucleosomes, remodel chromatin and repair DNA. The transcriptional regulation role includes development of numerous cell lineages including T-cells and neural cells. An estimated 20% of all human tumours harbour mutations in these genes. Most of the mutations lead to loss of function of the affected tumour suppressor protein with studies highlighting that there is oncogenic activation of the residual complexes, promoting growth by possibly acquiring gain of function and subsequent gene expression facilitating transformation. Loss-of-function mutations in even one of the subunits are identified in 6% of PDTCs and 36% of ATCs [17,121,122].
6.8. Wnt Signalling Pathway
This pathway includes proteins encoded by CTNNB1(β-catenin), AXIN1 and APC genes that function in cell adhesion and transcription. β-catenin is an essential component of the Wnt signalling pathway, which is activated in numerous tumours [123,124]. APC binds β-catenin and recruits casein kinase I and glycogen synthase kinase-3 (GSK3) that phosphorylate β-catenin for subsequent ubiquitination and degradation by proteasome. This allows constitutive downregulation of β-catenin. β-catenin degradation is inhibited by the Wnt signalling pathway, a process achieved through inhibition of β-catenin phosphorylation, thus permitting translocation of β-catenin to the nucleus to function as a transcription factor. This process is enhanced through blockage of β-catenin binding to cadherin, overactivity of the Wnt pathway or defect in the GSK3β-axin-APC mediated degradation of β-catenin (secondary to mutations in APC or CTNNB1 genes) [125,126]. Additionally, TERT positively regulates this pathway, leading to its activation and resistance to antigrowth signals with subsequent cellular proliferation. Approximately 25% and 65% of PDTCs and ATCs contain these aberrations, with evident nuclear localisation of β-catenin, an infrequent process in DTCs [127]. Furthermore, E-cadherin loss is prominent in PDTCs and ATCs [128,129]. Therefore, β-catenin mutations and loss of E-cadherin expression initiate the tumour dedifferentiation process and further progression.
6.9. Epigenetic Modifications in Thyroid Neoplasms
Epigenetic modifications include DNA methylation and histone deacetylation, which regulate gene expression. DNA methylation is a covalent modification of cytosine residues that are present at the dinucleotide sequence CpG, which if unmethylated lead to increased gene transcription, and in contrast hypermethylation of vital gene promoter regions result in heritable inhibition of gene transcription [130].
In thyroid neoplasms, aberrant methylation of thyroid-specific tumour suppressor genes drives dedifferentiation and occurs in the initial phase of tumourigenesis. Reduced or absent TSH-promoted iodine uptake is linked to silencing of thyroid-stimulating hormone receptor (TSHR) expression. Silencing of the TSHR gene is secondary to hypermethylation of the TSHR promoter and is identified in 59–87% of PTCs and 47–50% of FTCs, and PTCs with metastasis [131,132,133]. Methylation is absent in normal human thyroid cells and adenomas. The silenced cell lines if treated with a demethylating agent can partially restore TSHR expression and subsequent TSH-promoted iodine uptake, permitting the use of and enhancing the effectiveness of RAI when used in conjunction with a demethylating agent. Kim et al. showed 90% reduction in thyroid hormone receptor β (THRB) mRNA expression in differentiated thyroid carcinomas, in particularly those with advanced histologic features suggesting an inverse correlation, which when treated with demethylating agents 5′-aza-2′ deoxynucleotide and/or zebularine induced 5.6 fold increase in re-expression of the THRB gene and concurrent inhibition of tumour growth by inhibition of cell proliferation and migration through the suppression of β-catenin signalling pathway [133]. Silencing of this gene (THRB or THRA) through promoter hypermethylation is recognised in lung, breast, colon and lymphoid tumours [134,135,136,137,138].
In addition to TSHR, numerous tumour suppressor genes silenced through aberrant methylation include genes encoding cyclin-dependent kinase inhibitors p15INKa and p16INK4b [139], RASSF1A [140], RARβ-2(retinoic acid receptor β-2), ECAD, NIS-I, ATM, DAPK (death-associated protein kinase), TIMP3 (tissue inhibitor of metalloproteinase-3), SLC26A and SLC5A8 (sodium monocarboxylate transporter) [141,142]; the latter four are associated with aggressive features.
6.10. Copy Number Variations
TCGA discovered presence of somatic copy number variations in 27.2% of TCs deficient of fusions or driver mutations [19]. These variations are variably common in invasive FVPTCs and tall cell variant PTCs. The number of variations correlate with tumour differentiation, with reduced prevalence in well-differentiated tumours (up to 27.1%). The common variations include loses in 22q (includes NF2 and CHEK2) and 10q, and gains in 1q and oncogenic drivers BRAF (in BRAF wild-type tumours) and TERT. Chromosome 1q and TERT amplifications are correlated with aggressive tumour biology [19,143]. ATCs harboured significantly higher genetic variations and tumour mutational burden, with no correlation with age. Conversely, PTCs gain genetic variations with increasing age [18].
6.11. Other Genetic Aberrations–Mitochondrial DNA and Genomic Haploidisation
Mitochondrial DNA (mtDNA) incorporate16,569 base pairs and encodes 13 constituents of the apparatus of cellular energy production or the electron transport chain, 2 RNA classes and 22 mitochondrial transfer RNAs. mtDNA has a 10–20 times more mutational rate than nuclear DNA, secondary to oxidative stress. One of these mutations characteristically seen in Hürthle cell tumours is a deletion of 4977 bp called “common deletion,” [144]. Complex 1 mtDNA mutations (loss of function and missense) affect 60% of HCCs [145].
Gopal et al. identified a near-haploid chromosmal content and chromosomal losses that lead to loss of heterozygosity across a large part of the genome in 54% HCCs. In regards, to ploidy, 41.46% were “near-haploid” (mean ploidy <1.6), 46.34% “quiet” (mean ploidy of >1.6 to <2.5) and 12.2% “complex” (mean ploidy >2.5) [145].
Both mtDNA mutations and near-haploid status are important driver events in tumourigenesis of HCCs. These aberrations or their metabolic consequences could be therapeutically targeted, a potential future direction for further research.

{kind=link}
{kind=link}
Table 1.
Genomic aberration landscape in benign thyroid lesions and thyroid cancer subtypes.
| Genetic Aberration | Benign/Borderline | Follicular Thyroid Carcinoma (FTC)/Hürthle Cell Carcinoma (HCC) | Papillary Thyroid Carcinoma (PTC) | Poorly Differentiated Thyroid Carcinoma (PDTC) | Anaplastic Thyroid Carcinoma (ATC) | Clinical Implication |
|---|---|---|---|---|---|---|
| RAS Point Mutations | 28.1–30% (Follicular adenoma) [33,35] 5.6% (Hyperplastic nodule [HN]) [33,35] 7–25% (Goitres [G]) [33,35] 0–4% (Hürthle cell adenoma [HCA])) [32] 29.6–55.6% (Non-invasive follicular thyroid neoplasm with papillary like nuclear features [NIFTP]) [10,13,22] | 20–57% (Follicular thyroid carcinoma [FTC]) [33] 15–25% (Hürthle cell carcinoma [HCC]) [32,38] | 1.7–52% (follicular variant of papillary thyroid carcinoma [FVPTC] [19,32,36] 13% (classic variant of papillary thyroid carcinoma [CVPTC]) [14] | 28–55% [22,40] | 23–52% [17,19,40] | Downstream Mitogen-activated protein kinase (MEK)1/2 inhibitor (selumetinib) [42] Higher metastasis risk with N-Rat sarcoma (RAS) codon 61 mutation147 Follicular morphology |
| V-raf murine sarcoma viral oncogene homolog B1(BRAF) activating mutations (most common is p.V600E; others are p.K601E and small deletions) and fusion (AKAP9-BRAF) | 3.7% NIFTP [10] | Up to 62% (mostly CVPTC) [20,45,46,47,48,49,50,51] | 12–33% [20,45,46,47,48,49,50,51] | 25–29% [20,51] | Selective MEK inhibitors (dabrafenib and trametinib) and BRAF inhibitors (vemurafenib and dabrafenib) Classic and tall cell morphology [20,53,54,55] Refractiveness to radioactive iodine (RAI) [52,53,55] Increased unfavourable prognostic Factors [47,52,53,55] | |
| Rearranged during transfection (RET)-PTC rearrangements | 17–63.2% (HT) | 6.8–32.9% [69,70,71,72,73] | 12.9% [69,70,71,72,73] | Selective RET kinase inhibitors (e.g., selpercatinib) [79] | ||
| Eukaryotic translation initiation factor 1A X-(E1F1AX) activating mutations | 5–10% (FA) [81,82] 0–5% (HN) [81,82] | 17% (FTC) [81,82] 11% (HCC) [38] | 1–2% (mostly FVPTC) [18,80,82] | 5–15% [17,80,81,82] | 9–30% [17,80,81,82]2 | Co-expression with RAS mutations to drive tumourigenesis [82] Co-expression with tumour portein (TP) 53/Telomerase reverse transcriptase (TERT) mutations in biologically aggressive tumours [82] |
| Paired box gene 8-peroxisome proliferator-activated receptor (PAX8-PPARγ) rearrangement | 4–33% (FA) [19,32,91,92,93,94] 22% NIFTP [10] | 30–58% (FTC) [19,32,91,92,93,94] 0–3% (HCC) [38] | 37.5% (FVPTC), <1% (CVPTC) [19,32,92,93,94] | Follicular phenotype | ||
| TERT promoter | 1–35% [19] | 9–15% [19] | 40% [17] | 73% [17] | Usually aggressive biology | |
| TP53 | 8% [109] | 13% [109] | 8–35% [17,19,20] | Up to 73% [17,19,20] | Usually aggressive biology | |
| Cyclin-dependent kinase inhibitor 2A/2B (CDKN2A/2B) | 15–23% [89] | Aggressive biology. Possible utilisation of Cyclin dependent kinases (CDK) 4/6 inhibitor (palbociclib) [61] | ||||
| Catenin beta 1 (CTNNB1) activating mutations | Up to 25% [127,128,129] | Up to 65% [127,128,129] | Usually aggressive biology | |||
| Anaplastic lymphoma kinase (ALK) fusions (STRN or EML4) or activating mutations | 0.8% [20] | Up to 16% [95,96] | 0–10% [95,96] | ALK inhibitors | ||
| Tyrosine kinase (NTRK)1/3 fusions | 0–5% [19] | 1.3–26% [19,90] | Targeted therapies (entrectinib or larotrectinib) [61] | |||
| Others | Phosphatase and tensin homolog (PTEN) loss of heterozygosity (7% FAs) [85,86] THADA (22% NIFTP) [11] | Phosphatidylinositol-4,5-bisphophate 3-kinase catalytic subunit alpha (PIK3C) (0–11% FTC) [19] PTEN (0–27%) [85,86] Thyroid stimulating hormone receptor (TSHR) BRAFK601E Copy number variations (CNVs) Mismatch repair (MMR) genes mtDNA and diploidies (HCC) | PIK3CA (3%) [19,90] PTEN (2%) [90] BRAFK601E Thyroid adenoma-associated protein (THADA) (5%) [19] TSHR MMR genes [115,116] Copy Number variations (CNVs) | PIK3CA (0–11%) [17,90] PTEN (5–20%) [17,89] Ak strain transforming (AKT)1 (0–13%) [17,89] Switch/sucrose non-fermentable (SWI/SNF) complex subunit mutations CNVs | PIK3CA (5–25%) [17,89] PTEN (10–15%) [17,89] AKT1 (0–8%) [17,89] Ataxia telangiectasia mutated (ATM), retinoblastoma 1 (RB1), Multiple endocrine neoplasia (MEN1) Neurofibromatosis (NF1), NF2, AT-rich interacting domain containing protein 2 (ARID2), MMR genes, V-kit Hardy-Zuckerman 4 feline sarcoma viral oncogene homolog (KIT), SWI-SNF complex subunit mutations CNVs | AKT1 mutation is present in metastatic or recurrent RAI-refractory tumours [89] |
7. Tumour Microenvironment and Programmed Cell Death 1 Ligand 1 (PD-L1) Expression
The tumour microenvironment (TME) in thyroid cancer plays a crucial role in tumour progression and metastasis. Its constituent immune cells include regulatory T-cells/Tregs, cytotoxic CD8+ T-cells, helper T-cells, tumour-associated macrophages, mast cells, natural killer cells, dendritic cells and cytokines [146].
CD4+Foxp3+CD25+ Tregs are exclusively enriched in the TME through their recruitment by cytokines/chemokines and certain growth factors, namely vascular endothelial growth factor and transforming growth factor-β, exerting an anti-tumour immune response and tumour evasion [146,147]. These specific T-regs are associated with aggressive clinicopathological features in TCs, and are notably less potent in benign conditions (e.g., multinodular goitre and Hashimoto’s thyroiditis) [148,149]. Tumour cells express ligands that are recognised by CD8+ T-cell receptors, which can be evaded to allow tumour death. Two subsets of tumours are recognised—(i) T-cell-inflamed phenotype consisting of tumour infiltrating T-cells, chemokines and interferon-1, and (ii) non-T-cell-inflamed phenotype [146]. The former is associated with BRAF and RET/PTC positive TCs. Macrophages and other myeloid cells produce cytokines that assist in tumour immune evasion. Tumour-promoting M2 macrophages are induced in tumours by specific cytokines including interleukin-4 (IL-4), IL-10, IL-13 and macrophage colony-stimulating factor (M-CSF). These M2 macrophages produce IL-10, IL-13 and tumour growth factor-β (TGF-β) which suppress immune response, promoting tumourigenesis; and also produce CCL22 that recruits CD4+Foxp3+CD25+ Treg cells. Tregs suppress immune system to maintain homeostasis and self-tolerance by inhibiting T-cell proliferation and cytokine production. Studies established high M2 macrophage infiltration in ATCs with expression of about 68 genes associated with M2 macrophages [17,146,150]. Furthermore, tumour infiltrating CD8+effector T-cells, including functionally exhausted ones significantly increase with tumour progression with higher quantities detected in PDTCs and ATCs, and are infrequent or absent in PTCs and normal thyroid tissue. Therefore, two immune signature types in thyroid cancer are introduced based on the immune cell infiltration levels: ATC-like and PDTC-like [151].
Upregulation of CD274 (encodes PD-L1), an immune checkpoint protein ligand for programmed cell death-1 (PD-1) has been reported in ATCs. PD-1 is a transmembrane protein which is expressed on immune cells [152]. Expression of PD-L1 on the surface of tumour cells allows for engagement with PD-1+ T cells, resulting in T cell dysfunction by exhaustion, anergy, apoptosis and IL-10 expression. There is consequential evasion of the host immune-mediated tumour destruction, resulting in tumour spread, relapse and metastasis [153]. PD-L1 expression in DTCs, PDTCs and ATCs is associated with poor survival, especially in co-existence with BRAFV600E mutation [154,155], and is noted in 6.1% PTCs, 7.6% FTCs and 22.2% ATCs [156,157,158]. Monoclonal antibodies targeting both PD-1 and PD-L1 have illustrated reduced tumour growth and increased survival [156,157,158,159]. Recently identified PD-L1 regulatory proteins CMTM4 and CMTM6 are documented to enhance a PD-L1 tumour’s ability to evade the immune system by inhibiting T-cells. Any interference with their expression leads to consequent altered PD-L1 protein expression on tumour cells, suggesting a potential for development of future immunotherapeutic agents. The role of these proteins on the PD-1/PD-L1 axis further needs elucidation [160]. PD-L1 expression is higher in ATCs (65%), and less frequent or absent in DTCs. Role of PD-L1 inhibitors such as pembrolizumab are currently under investigation [161].
The utilisation of PD-1/PD-L1 immunotherapies with multiple kinase inhibitors has proven effective in the treatment of ATC. Gunda et al. documented that Lenvatinib, a multi-targeted tyrosine kinase, in combination with PD-L1 inhibitors promoted tumour size reduction and increased overall survival in murine model. Lenvatinib monotherapy promoted production of CD8+ T-cells, Tregs, tumour infiltrating macrophages and polymorphonuclear myeloid derived suppressor cells (PMN-MDSCs), with increased numbers of the latter postulated in treatment resistance. Combination therapy, also potentially including anti-Gr-1 antibody significantly reduced PMN-MDSCs, enhancing the treatment effect [162]. Human studies are required to evaluate the role of these combined therapies.
Studies are underway to determine the benefits of longitudinal use of PD-1/PD-L1 inhibitors such as cemiplimab with dabrafenib and trametinib, pembrolizumab with lenvatinib, and atezolizumab or spartalizumab with chemotherapy or targeted therapy [159].
A trial underway showed a 20% improvement in tumour size with use of a new NTRK inhibitor entrectinib. Tipifarnib (HRAS inhibitor) and palbociclib (cyclin D-cyclin-dependent kinase 4/6 inhibitor), both also currently being investigated, have shown reduction in tumour size [163]. Further development in targeted therapy for ATCs is required.
8. Summary
Thyroid carcinomas arise and progress through some mutually exclusive genetic aberrations in MAPK and PI3K pathways. De-differentiation of these tumours occurs through additional genetic mutations in different pathways involved in cellular function. Improved knowledge has conclusively revealed the association of these genetic aberrations with specific tumour histology and biological behaviour. This has promoted research in establishing the possibility of utilisation of some of these specific molecular aberrations as putative prognostic markers and also the development of targeted therapies for biologically aggressive tumours.
Author Contributions
Author contributions are as follows: Conceptualization, A.S., C.S.L. and T.R.; writing—original draft preparation, A.S., J.H.; writing—review and editing, J.H., J.W.P.; supervision, C.S.L., T.R., N.N.; project administration, C.S.L. All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Kitahara, C.M.; Sosa, J.A. The changing incidence of thyroid cancer. Nat. Rev. Endocrinol. 2016, 12, 646–653. [Google Scholar] [CrossRef] [PubMed]
- Seib, C.D.; Sosa, J.A. Evolving understanding of the epidemiology of thyroid cancer. Endocrinol. Metab. Clin. N. Am. 2019, 48, 23–35. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Pineros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef] [Green Version]
- Australian Institute of Health and Welfare 2019 Australian Government. Canberra, AIHW. Available online: http://www.aihw.gov.au/reports/cancer/cancer-in-australia-2019/contents/summary (accessed on 28 August 2020).
- Lim, H.; Devesa, S.S.; Sosa, J.A.; Check, D.; Kitahara, C.M. Trends in thyroid cancer incidence and mortality in the United States, 1974–2013. JAMA 2017, 317, 1338. [Google Scholar] [CrossRef]
- La Vecchia, C.; Malvezzi, M.; Bosetti, C.; Garavello, W.; Bertuccio, P.; Levi, F.; Negri, E. Thyroid cancer mortality and incidence: A global overview. Int. J. Cancer 2015, 136, 2187–2195. [Google Scholar] [CrossRef]
- Vaccarella, S.; Franceschi, S.; Bray, F.; Wild, C.; Plummer, M.; Dal Maso, L. The increase in thyroid cancer may be due to an increase in medical surveillance and the introduction of new diagnostic techniques, such as neck ultrasonography. N. Engl. J. Med. 2016, 375, 614–617. [Google Scholar] [CrossRef]
- Haugen, B.R.; Alexander, E.K.; Bible, K.C.; Doherty, G.M.; Mandel, S.J.; Nikiforov, Y.E.; Pacini, F.; Randolph, G.W.; Sawka, A.M.; Schlumberger, M. American Thyroid Association management guidelines for adult patients with thyroid nodules and differentiated thyroid cancer: The American Thyroid Association guidelines task force on thyroid nodules and differentiated thyroid cancer. Thyroid 2016, 26, 1–133. [Google Scholar] [CrossRef] [Green Version]
- Lloyd, R.V.; Osamura, R.Y.; Klöppel, G.; Rosai, J. World Health Organization Classification of Tumours of Endocrine Organs; IARC Press: Lyon, France, 2017. [Google Scholar]
- Nikiforov, Y.E.; Seethala, R.R.; Tallini, G.; Baloch, Z.W.; Basolo, F.; Thompson, L.D.; Barletta, J.A.; Wenig, B.M.; Al Ghuzlan, A.; Kakudo, K.; et al. Nomenclature revision for encapsulated follicular variant of papillary thyroid carcinoma: A paradigm shift to reduce overtreatment of indolent tumors. JAMA Oncol. 2016, 2, 1023–1029. [Google Scholar] [CrossRef] [Green Version]
- Bychkov, A.; Jung, C.K.; Liu, Z.; Kakudo, K. Noninvasive follicular thyroid neoplasm with papillary-like nuclear features in Asian practice: Perspectives for surgical pathology and cytopathology. Endocr. Pathol. 2018, 29, 276–288. [Google Scholar] [CrossRef] [Green Version]
- Golding, A.; Shively, D.; Bimston, D.N.; Harrell, R.M. Noninvasive encapsulated follicular variant of papillary thyroid cancer: Clinical lessons from a community-based endocrine surgical practice. Int. J. Surg. Pract. 2017, 2017, 4689465. [Google Scholar] [CrossRef] [Green Version]
- Seo, J.Y.; Park, J.H.; Pyo, J.Y.; Cha, Y.J.; Jung, C.K.; Song, D.E.; Kwak, J.J.; Park, S.Y.; Na, Y.N.; Kim, J.-H.; et al. A multi-institutional study of prevalence and clinicopathologic features of non-invasive follicular thyroid neoplasm with papillary-like nuclear features (NIFTP) in Korea. J. Pathol. Transl. Med. 2019, 53, 378–385. [Google Scholar] [CrossRef] [PubMed]
- Schlumberger, M.J. Papillary and follicular thyroid carcinoma. N. Engl. J. Med. 1998, 338, 297–306. [Google Scholar] [CrossRef] [PubMed]
- Haymart, M.R.; Esfandiari, N.H.; Stang, M.T.; Sosa, J.A. Controversies in the management of low-risk differentiated thyroid cancer. Endocr. Rev. 2017, 38, 351–378. [Google Scholar] [CrossRef] [PubMed]
- Lamartina, L.; Grani, G.; Durante, C.; Filetti, S. Recent advances in managing differentiated thyroid cancer. F1000Research 2018, 7, 86. [Google Scholar] [CrossRef] [Green Version]
- Landa, I.; Ibrahimpasic, T.; Boucai, L.; Sinha, R.; Knauf, J.A.; Shah, R.H.; Dogan, S.; Ricarte-Filho, J.C.; Krishnamoorthy, G.P.; Xu, B.; et al. Genomic and transcriptomic hallmarks of poorly differentiated and anaplastic thyroid cancers. J. Clin. Investig. 2016, 126, 1052–1066. [Google Scholar] [CrossRef] [Green Version]
- Pozdeyev, N.; Gay, L.M.; Sokol, E.S.; Hartmaier, R.; Deaver, K.E.; Davis, S.; French, J.D.; Borre, P.V.; LaBarbera, D.V.; Tan, A.-C.; et al. Genetic analysis of 779 advanced differentiated and anaplastic thyroid cancers. Clin. Cancer Res. 2018, 24, 3059–3068. [Google Scholar] [CrossRef] [Green Version]
- Agrawal, N.; Akbani, R.; Aksoy, B.A.; Ally, A.; Arachchi, H.S.; Asa, S.L.; Auman, J.T.; Balasundaram, M.; Balu, S.; Baylin, S.B.; et al. Integrated genomic characterization of papillary thyroid carcinoma. Cell 2014, 159, 676–690. [Google Scholar] [CrossRef] [Green Version]
- Hsiao, S.J.; Nikiforov, Y. Molecular approaches to thyroid cancer diagnosis. Endocr. Relat. Cancer 2014. [Google Scholar] [CrossRef] [PubMed]
- Adeniran, A.J.; Zhu, Z.; Gandhi, M.; Steward, D.L.; Fidler, J.P.; Giordano, T.J.; Biddinger, P.W.; Nikiforov, Y.E. Correlation between genetic alterations and microscopic features, clinical manifestations, and prognostic characteristics of thyroid papillary carcinomas. Am. J. Surg. Pathol. 2006, 30, 216–222. [Google Scholar] [CrossRef]
- Rivera, M.; Ricarte-Filho, J.; Knauf, J.; Shaha, A.; Tuttle, M.; Fagin, J.A.; Ghossein, R.A. Molecular genotyping of papillary thyroid carcinoma follicular variant according to its histological subtypes (encapsulated vs infiltrative) reveals distinct BRAF and RAS mutation patterns. Mod. Pathol. 2010, 23, 1191–1200. [Google Scholar] [CrossRef] [Green Version]
- Guerra, A.; Sapio, M.R.; Marotta, V.; Campanile, E.; Rossi, S.; Forno, I.; Fugazzola, L.; Budillon, A.; Moccia, T.; Fenzi, G.; et al. The primary occurrence of BRAFV600E is a rare clonal event in papillary thyroid carcinoma. J. Clin. Endocrinol. Metab. 2012, 97, 517–524. [Google Scholar] [CrossRef] [Green Version]
- Guerra, A.; Zeppa, P.; Bifulco, M.; Vitale, M. Concomitant BRAFV600E mutation and RET/PTC rearrangement is a frequent occurrence in papillary thyroid carcinoma. Thyroid 2014, 24, 254–259. [Google Scholar] [CrossRef] [PubMed]
- Morrison, D.K. MAP kinase pathways. Cold Spring Harbor Perspect. Biol. 2012, 4, a011254. [Google Scholar] [CrossRef]
- Castellano, E.; Santos, E. Functional specificity of RAS isoforms: So similar but so different. Genes Cancer 2011, 2, 216–231. [Google Scholar] [CrossRef] [Green Version]
- Prior, I.A.; Lewis, P.D.; Mattos, C. A comprehensive survey of RAS mutations in cancer. Cancer Res. 2012, 72, 2457–2467. [Google Scholar] [CrossRef] [Green Version]
- Shirokawa, J.M.; Elisei, R.; Knauf, J.A.; Hara, T.; Wang, J.; Saavedra, H.I.; Fagin, J.A. Conditional apoptosis induced by oncogenic rats in thyroid cells. Mol. Endocrinol. 2000, 14, 1725–1738. [Google Scholar] [CrossRef]
- De Vita, G.; Bauer, L.; da Costa, V.M.; De Felice, M.; Baratta, M.G.; De Menna, M.; Di Lauro, R. Dose-dependent inhibition of thyroid differentiation by RAS oncogenes. Mol. Endocrinol. 2005, 19, 76–89. [Google Scholar] [CrossRef] [Green Version]
- Vitagliano, D.; Portella, G.; Troncone, G.; Francione, A.; Rossi, C.; Bruno, A.; Giorgini, A.; Coluzzi, S.; Nappi, T.C.; Rothstein, J.L.; et al. Thyroid targeting of the N-ras (Gln61Lys) oncogene in transgenic mice results in follicular tumors that progress to poorly differentiated carcinomas. Oncogene 2006, 25, 5467–5474. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vasko, V.; Ferrand, M.; Di Cristofaro, J.; Carayon, P.; Henry, J.F.; de Micco, C. Specific pattern of RAS oncogene mutations in follicular thyroid tumours. J. Clin. Endocrinol. Metab. 2003, 88, 2745–2752. [Google Scholar] [CrossRef] [Green Version]
- Nikiforova, M.N.; Lynch, R.A.; Biddinger, P.W.; Alexander, E.K.; Dorn, G.W.; Tallini, G.; Kroll, T.G.; Nikiforov, Y.E. RAS point mutations and PAX8-PPARγ rearrangement in thyroid tumors: Evidence for distinct molecular pathways in thyroid follicular carcinoma. J. Clin. Endocrinol. Metab. 2003, 88, 2318–2326. [Google Scholar] [CrossRef] [Green Version]
- Fukahori, M.; Yoshida, A.; Hayashi, H.; Yoshihara, M.; Matsukuma, S.; Sakuma, Y.; Koizume, S.; Okamoto, N.; Kondo, T.; Masuda, M.; et al. The associations between RAS mutations and clinical characteristics in follicular thyroid tumors: New insights from a single center and a large patient cohort. Thyroid 2012, 22, 683–689. [Google Scholar] [CrossRef]
- Ricarte-Filho, J.C.; Ryder, M.; Chitale, D.A.; Rivera, M.; Heguy, A.; Ladanyi, M.; Janakiraman, M.; Solit, D.; Knauf, J.A.; Tuttle, R.M.; et al. Mutational profile of advanced primary and metastatic radioactive iodine-refractory thyroid cancers reveals distinct pathogenetic roles for BRAF, PIK3CA, and AKT1. Cancer Res. 2009, 69, 4885–4893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hans-Juergen, S.; Salama, S.; Al-Ahmadi, A.; Al-Mansouri, Z.; Mirza, Z.; Al-Ghamdi, K.; Al-Hamour, O.A.; Huwait, E.; Gari, M.; Al-Qahtani, M.H.; et al. Comprehensive survey of HRAS, KRAS, and NRAS mutations in proliferative thyroid lesions from an ethinically diverse population. Anticancer Res. 2013, 33, 4779–4784. [Google Scholar]
- Horie, H.; Yokogoshi, Y.; Tsuyuguchi, M.; Saito, S. Point mutations of RAS and Gs alpha subunit genes in thyroid tumors. Jpn. J. Cancer Res. 1995, 86, 737–745. [Google Scholar] [CrossRef] [PubMed]
- Hobbs, G.A.; Der, C.J.; Rossman, K.L. RAS isoforms and mutations in cancer at a glance. J. Cell Sci. 2016, 129, 1287–1292. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganly, I.; Makarov, V.; Deraje, S.; Dong, Y.; Reznik, E.; Seshan, V.; Nanjangud, G.; Eng, S.; Bose, P.; Kuo, F.; et al. Integrated genomic analysis of Hürthle cell cancer reveals oncogenic drivers, recurrent mitochondrial mutations, and unique chromosomal landscapes. Cancer Cell 2018, 34, 256–270.e5. [Google Scholar] [CrossRef] [Green Version]
- Moura, M.M.; Cavaco, B.M.; Pinto, A.E.; Leite, V. High prevalence of RAS mutations in RET-negative sporadic medullary thyroid carcinomas. J. Clin. Endocrinol. Metab. 2011, 96, E863–E868. [Google Scholar] [CrossRef] [Green Version]
- Garcia-Rostan, G.; Zhao, H.; Camp, R.L.; Pollan, M.; Herrero, A.; Pardo, J.; Wu, R.; Carcangiu, M.L.; Costa, J.; Tallini, G. Ras mutations are associated with aggressive tumor phenotypes and poor prognosis in thyroid cancer. J. Clin. Oncol. 2003, 21, 3226–3235. [Google Scholar] [CrossRef]
- Costa, A.M.; Herrero, A.; Fresno, M.F.; Heymann, J.; Alvarez, J.A.; Cameselle-Teijeiro, J.; García-Rostán, G. BRAF mutation associated with other genetic events identifies a subset of aggressive papillary thyroid carcinoma. Clin. Endocrinol. 2008, 68, 618–634. [Google Scholar] [CrossRef] [PubMed]
- Ho, A.L.; Grewal, R.K.; Leboeuf, R.; Sherman, E.J.; Pfister, D.G.; Deandreis, D.; Pentlow, K.S.; Zanzonico, P.B.; Haque, S.; Gavane, S.; et al. Selumetinib-enhanced radioiodine uptake in advanced thyroid cancer. N. Engl. J. Med. 2013, 368, 623–632. [Google Scholar] [CrossRef] [Green Version]
- Nikiforov, Y.E.; Nikiforova, M.N. Molecular genetics and diagnosis of thyroid cancer. Nat. Rev. Endocrinol. 2011, 7, 569–580. [Google Scholar] [CrossRef] [PubMed]
- Mitsutake, N.; Miyagishi, M.; Mitsutake, S.; Akeno, N.; Mesa, C., Jr.; Knauf, J.A.; Zhang, L.; Taira, K.; Fagin, J.A. BRAF mediates RET/PTC-induced mitogen-activated protein kinase activation in thyroid cells: Functional support for requirement of the RET/PTC-RAS-BRAF pathway in papillary thyroid carcinogenesis. Endocrinology 2006, 147, 1014–1019. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kimura, E.T.; Nikiforova, M.N.; Zhu, Z.; Knauf, J.A.; Nikiforov, Y.E.; Fagin, J.A. High prevalence of BRAF mutations in thyroid cancer: Genetic evidence for constitutive activation of the RET/PTC-RAS-BRAF signaling pathway in papillary thyroid carcinoma. Cancer Res. 2003, 63, 1454–1457. [Google Scholar]
- Cohen, Y.; Xing, M.; Mambo, E.; Guo, Z.; Wu, G.; Trink, B.; Beller, U.; Westra, W.H.; Ladenson, P.W.; Sidransky, D. BRAF mutation in papillary thyroid carcinoma. JNCI J. Natl. Cancer Inst. 2003, 95, 625–627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukushima, T.; Suzuki, S.; Mashiko, M.; Ohtake, T.; Endo, Y.; Takebayashi, Y.; Sekikawa, K.; Hagiwara, K.; Takenoshita, S. BRAF mutations in papillary carcinomas of the thyroid. Oncogene 2003, 22, 6455–6457. [Google Scholar] [CrossRef] [Green Version]
- Namba, H.; Nakashima, M.; Hayashi, T.; Hayashida, N.; Maeda, S.; Rogounovitch, T.I.; Ohtsuru, A.; Saenko, V.A.; Kanematsu, T.; Yamashita, S. Clinical implication of hot spot BRAF mutation, V599E, in papillary thyroid cancers. J. Clin. Endocrinol. Metab. 2003, 88, 4393–4397. [Google Scholar] [CrossRef] [Green Version]
- Soares, P.; Trovisco., V.; Rocha, A.S.; Lima, J.; Castro, P.; Preto, A.; Maximo, V.; Botelho, T.; Seruca, R.; Sobrinho-Simoes, M. BRAF mutations and RET/PTC rearrangements are alternative events in the etiopathogenesis of PTC. Oncogene 2003, 22, 4578–4580. [Google Scholar] [CrossRef] [Green Version]
- Xu, X.; Quiros, R.M.; Gattuso, P.; Ain, K.B.; Prinz, R.A. High prevalence of BRAF gene mutation in papillary thyroid carcinomas and thyroid tumor cell lines. Cancer Res. 2003, 63, 4561–4567. [Google Scholar]
- Xing, M.; Westra, W.H.; Tufano, R.P.; Cohen, Y.; Rosenbaum, E.; Rhoden, K.J.; Carson, K.A.; Vasko, V.; Larin, A.; Tallini, G. BRAF mutation predicts a poorer clinical prognosis for papillary thyroid cancer. J. Clin. Endocrinol. Metab. 2005, 90, 6373–6379. [Google Scholar] [CrossRef] [Green Version]
- Ciampi, R.; Knauf, J.A.; Kerler, R.; Gandhi, M.; Zhu, Z.; Nikiforova, M.N.; Rabes, H.M.; Fagin, J.A.; Nikiforov, Y.E. Oncogenic AKAP9-BRAF fusion is a novel mechanism of MAPK pathway activation in thyroid cancer. J. Clin. Investig. 2005, 115, 94–101. [Google Scholar] [CrossRef] [Green Version]
- Nikiforova, M.N.; Kimura, E.T.; Gandhi, M.; Biddinger, P.W.; Knauf, J.A.; Basolo, F.; Zhu, Z.; Giannini, R.; Salvatore, G.; Fusco, A.; et al. BRAF mutations in thyroid tumors are restricted to papillary carcinomas and anaplastic or poorly differentiated carcinomas arising from papillary carcinomas. J. Clin. Endocrinol. Metab. 2003, 88, 5399–5404. [Google Scholar] [CrossRef] [PubMed]
- Zheng, X.; Wei, S.; Han, Y.; Li, Y.; Yu, Y.; Yun, X.; Ren, X.; Gao, M. Papillary microcarcinoma of the thyroid: Clinical characteristics and BRAFV600E mutational status of 977 cases. Ann. Surg. Oncol. 2013, 20, 2266–2273. [Google Scholar] [CrossRef]
- Walczyk, A.; Kowalska, A.; Kowalik, A.; Sygut, J.; Wypiórkiewicz, E.; Chodurska, R.; Pieciak, L.; Gozdz, S. The BRAFV600E mutation in papillary thyroid microcarcinoma: Does the mutation have an impact on clinical outcome? Clin. Endocrinol. 2014, 80, 899–904. [Google Scholar] [CrossRef]
- Romei, C.; Ciampi, R.; Faviana, P.; Agate, L.; Molinaro, E.; Bottici, V.; Basolo, F.; Miccoli, P.; Pacini, F.; Pinchera, A.; et al. BRAFV600E mutation, but not RET/PTC rearrangements, is correlated with a lower expression of both thyroperoxidase and sodium iodide symporter genes in papillary thyroid cancer. Endocr. Relat. Cancer 2008, 15, 511–520. [Google Scholar] [CrossRef]
- Xing, M. BRAF mutation in papillary thyroid cancer: Pathogenic role, molecular bases, and clinical implications. Endocr. Rev. 2007, 28, 742–762. [Google Scholar] [CrossRef]
- Kim, T.H.; Park, Y.J.; Lim, J.A.; Ahn, H.Y.; Lee, E.K.; Lee, Y.J.; Kim, K.W.; Hahn, S.K.; Youn, Y.K.; Kim, K.H.; et al. The association of the BRAFV600E mutation with prognostic factors and poor clinical outcome in papillary thyroid cancer. Cancer 2012, 118, 1764–1773. [Google Scholar] [CrossRef]
- Li, C.; Lee, K.C.; Schneider, E.B.; Zeiger, M.A. BRAFV600E mutation and its association with clinicopathological features of papillary thyroid cancer: A meta-analysis. J. Clin. Endocrinol. Metab. 2012, 97, 4559–4570. [Google Scholar] [CrossRef] [Green Version]
- Riesco-Eizaguirre, G.; Gutiérrez-Martínez, P.; García-Cabezas, M.A.; Nistal, M.; Santisteban, P. The oncogene BRAF V600E is associated with a high risk of recurrence and less differentiated papillary thyroid carcinoma due to the impairment of Na+/I- targeting to the membrane. Endocr. Relat. Cancer 2006, 13, 257–269. [Google Scholar] [CrossRef]
- Cabanillas, M.E.; Ryder, M.; Jimenez, C. Targeted therapy for advanced thyroid cancer: Kinase inhibitors and beyond. Endocr. Rev. 2019, 40, 1573–1604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grieco, M.; Santoro, M.; Berlingieri, M.T.; Melillo, R.M.; Donghi, R.; Bongarzone, I.; Pierotti, M.A.; Della Porta, G.; Fusco, A.; Vecchio, G. PTC is a novel rearranged form of the ret proto-oncogene and is frequently detected in vivo in human thyroid papillary carcinomas. Cell 1990, 60, 557–563. [Google Scholar] [CrossRef]
- Bongarzone, I.; Monzini, N.; Borrello, M.G.; Carcano, C.; Ferraresi, G.; Arighi, E.; Mondellini, P.; Della Porta, G.; Pierotti, M.A. Molecular characterization of a thyroid tumor-specific transforming sequence formed by the fusion of ret tyrosine kinase and the regulatory subuniy RI alpha of cyclic AMP-dependent protein kinase A. Mol. Cell. Biol. 1993, 13, 358–366. [Google Scholar] [CrossRef] [Green Version]
- Santoro, M.; Dathan, N.A.; Berlingieri, M.T.; Bongarzone, I.; Paulin, C.; Grieco, M.; Pierotti, M.A.; Vecchio, G.; Fusco, A. Molecular characterization of RET/PTC3; a novel rearranged version of the RETproto-oncogene in a human thyroid papillary carcinoma. Oncogene 1994, 9, 509–516. [Google Scholar] [PubMed]
- Santoro, M.; Melillo, R.M.; Fusco, A. RET/PTC activation in papillary thyroid carcinoma: European Journal of Endocrinology Prize Lecture. Eur. J. Endocrinol. 2006, 155, 645–653. [Google Scholar] [CrossRef] [Green Version]
- Gertz, R.J.; Nikiforov, Y.; Rehrauer, W.; McDaniel, L.; Lloyd, R.V. Mutation in BRAF and other members of the MAPK pathway in papillary thyroid carcinoma in the pediatric population. Arch. Pathol. Lab. Med. 2016, 140, 134–139. [Google Scholar] [CrossRef] [Green Version]
- Hamatani, K.; Eguchi, H.; Ito, R.; Mukai, M.; Takahashi, K.; Taga, M.; Imai, K.; Cologne, J.; Soda, M.; Arihiro, K.; et al. RET/PTC rearrangements preferentially occurred in papillary thyroid cancer among atomic bomb survivors exposed to high radiation dose. Cancer Res. 2008, 68, 7176–7182. [Google Scholar] [CrossRef] [Green Version]
- Rabes, H.M.; Demidchik, E.P.; Sidorow, J.D.; Lengfelder, E.; Beimfohr, C.; Hoelzel, D.; Klugbauer, S. Pattern of radiation-induced RET and NTRK1 rearrangements in 191 post-chernobyl papillary thyroid carcinomas: Biological, phenotypic, and clinical implications. Clin. Cancer Res. 2000, 6, 1093–1103. [Google Scholar]
- Rhoden, K.J.; Johnson, C.; Brandao, G.; Howe, J.G.; Smith, B.R.; Tallini, G. Real-time quantitative RT-PCR identifies disticnt c-RET, RET/PTC1 and RET/PTC3 expression patterns in papillary thyroid carcinoma. Lab. Investig. 2004, 84, 1557–1570. [Google Scholar] [CrossRef]
- Kang, D.Y.; Kim, K.H.; Kim, J.M.; Kim, S.H.; Kim, J.Y.; Bail, H.W.; Kim, Y.S. High prevalence of RET, RAS, and ERK expression in Hashimoto’s thyroiditis and in papillary thyroid carcinoma in the Korean population. Thyroid 2007, 17, 1031–1038. [Google Scholar] [CrossRef] [PubMed]
- Zou, M.; Baitei, E.Y.; Alzahrani, A.S.; Binhumaid, F.S.; Alkhafaji, D.; Al-Rijjal, R.A.; Meyer, B.F.; Shi, Y. Concomitant RAS, RET/PTC, or BRAF mutations in advanced stage of papillary thyroid carcinoma. Thyroid 2014, 24, 1256–1266. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, Z.; Ciampi, R.; Nikiforova, M.N.; Gandhi, M.; Nikiforov, Y.E. Prevalence of RET/PTC rearrangements in thyroid papillary carcinomas: Effects of the detection methods and genetic heterogeneity. J. Clin. Endocrinol. Metab. 2006, 91, 3603–3610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santoro, M.; Papotti, M.; Chiappetta, G.; Garcia-Rostan, G.; Volante, M.; Johnson, C.; Camp, R.L.; Pentimalli, F.; Monaco, C.; Herrero, A.; et al. RET activation and clinicopathologic features in poorly differentiated thyroid tumors. J. Clin. Endocrinol. Metab. 2002, 87, 370–379. [Google Scholar] [CrossRef] [PubMed]
- Rhoden, K.J.; Unger, K.; Salvatore, G.; Yilmaz, Y.; Vovk, V.; Chiappetta, G.; Qumsiyeh, M.B.; Rothstein, J.L.; Fusco, A.; Santoro, M.; et al. RET/papillary thyroid cancer rearrangement in nonneoplastic thyrocytes: Follicular cells of Hashimoto’s thyroiditis share low-level recombination events with a subset of papillary carcinoma. J. Clin. Endocrinol. Metab. 2006, 91, 2414–2423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nikiforov, Y.E. RET/PTC rearragment in thyroid tumours. Endocr. Pathol. 2002, 13, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Sapio, M.R.; Guerra, A.; Marotta, V.; Campanile, E.; Formisano, R.; Deandrea, M.; Motta, M.; Limone, P.P.; Fenzi, G.; Rossi, G.; et al. High growth rate of benign thyroid nodules BearingRET/PTCRearrangements. J. Clin. Endocrinol. Metab. 2011, 96, E916–E919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melillo, R.M.; Castellone, M.D.; Guarino, V.; De Falco, V.; Cirafici, A.M.; Salvatore, G.; Caiazzo, F.; Basolo, F.; Giannini, R.; Kruhoffer, M.; et al. The RET/PTC-RAS-BRAF linear signaling cascade mediates the motile and mitogenic phenotype of thyroid cancer cells. J. Clin. Investig. 2005, 115, 1068–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Castellone, M.D.; Guarino, V.; De Falco, V.; Carlomagno, F.; Basolo, F.; Faviana, P.; Kruhoffer, M.; Orntoft, T.; Russell, J.P.; Rothstein, J.L.; et al. Functional expression of the CXCR4 chemokine receptor is induced by RET/PTC oncogenes and is a common event in human papillary thyroid carcinomas. Oncogene 2004, 23, 5958–5967. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wirth, L.J.; Sherman, E.; Robinson, B.; Solomon, B.; Kang, H.; Lorch, J.; Worden, F.; Brose, M.; Patel, J.; Leboulleux, S.; et al. Efficacy of selpercatinib in RET-altered thyroid cancers. N. Engl. J. Med. 2020, 383, 825–835. [Google Scholar] [CrossRef]
- Karunamurthy, A.; Panebianco, F.; Hsiao, S.J.; Vorhauer, J.; Nikiforova, M.N.; Chiosea, S.; Nikiforov, Y.E. Prevalence and phenotypic correlations of EIF1AX mutations in thyroid nodules. Endocr. Relat. Cancer 2016, 23, 295–301. [Google Scholar] [CrossRef] [Green Version]
- Nicolson, N.G.; Murtha, T.D.; Dong, W.; Paulsson, J.O.; Choi, J.; Barbieri, A.L.; Brown, T.C.; Kunstman, J.W.; Larsson, C.; Prasad, M.L.; et al. Comprehensive genetic analysis of follicular thyroid carcinoma predicts prognosis independent of histology. J. Clin. Endocrinol. Metab. 2018, 103, 2640–2650. [Google Scholar] [CrossRef] [Green Version]
- Krishnamoorthy, G.P.; Davidson, N.R.; Leach, S.D.; Zhao, Z.; Lowe, S.W.; Lee, G.; Landa, I.; Nagarajah, J.; Saqcena, M.; Singh, K.; et al. EIF1AX and RAS mutations cooperate to drive thyroid tumorigenesis through ATF4 and c-MYC. Cancer Discov. 2019, 9, 264–281. [Google Scholar] [CrossRef] [Green Version]
- Hou, P.; Liu, D.; Shan, Y.; Hu, S.; Studeman, K.; Condouris, S.; Wang, Y.; Trink, A.; El-Naggar, A.K.; Tallini, G.; et al. Genetic alterations and their relationship in the phosphatidylinositol 3-kinase/akt pathway in thyroid cancer. Clin. Cancer Res. 2007, 13, 1161–1170. [Google Scholar] [CrossRef] [Green Version]
- Eng, C. Genetics of Cowden syndrome: Through the looking glass of oncology. Int. J. Oncol. 1998, 12, 701–710. [Google Scholar] [CrossRef] [PubMed]
- Halachmi, N.; Halachmi, S.; Evron, E.; Cairns, P.; Okami, K.; Saji, M.; Westra, W.H.; Zeiger, M.A.; Jen, J.; Sidransky, D. Somatic mutations of the PTEN tumor suppressor gene in sporadic follicular thyroid tumors. Genes Chromosomes Cancer 1998, 23, 239–243. [Google Scholar] [CrossRef]
- Yeager, N.; Klein-Szanto, A.; Kimura, S.; Di Cristofano, A. Pten loss in the mouse thyroid causes goitre and follicular adenomas: Insights into thyroid function and Cowden disease pathogenesis. Cancer Res. 2007, 67, 959–966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charles, R.P.; Silva, J.; Iezza, G.; Phillips, W.A.; McMahon, M. Activating BRAF and PIK3CA mutations cooperate to promote anaplastic thyroid carcinogenesis. Mol. Cancer Res. 2014, 12, 979–986. [Google Scholar] [CrossRef] [Green Version]
- Shimamura, M.; Shibusawa, N.; Kurashige, T.; Mussazhanova, Z.; Matsuzaki, H.; Nakashima, M.; Yamada, M.; Nagayama, Y. Mouse models of sporadic thyroid cancer derived from BRAFV600E alone or in combination with PTEN haploinsufficiency under physiologic TSH levels. PLoS ONE 2018, 13, e0201365. [Google Scholar] [CrossRef]
- Garcia-Rostan, G.; Costa, A.M.; Pereira-Castro, I.; Salvatore, G.; Hernandez, R.; Hermsem, M.J.; Herrero, A.; Fusco, A.; Cameselle-Teijeiro, J.; Santoro, M. Mutation of the PIK3CA gene in anaplastic thyroid cancer. Cancer Res. 2005, 65, 10199–10207. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osaki, M.; Oshimura, M.; Ito, H. PI3K-Akt pathway: Its functions and alterations in human cancer. Apoptosis 2004, 9, 667–676. [Google Scholar] [CrossRef] [PubMed]
- Kroll, T.G.; Sarraf, P.; Pecciarini, L.; Chen, C.J.; Mueller, E.; Spiegelman, B.M.; Fletcher, J.A. PAX8-PPPAR-gamma 1 fusion oncogene in human thyroid carcinoma. Science 2000, 289, 1357–1360. [Google Scholar] [CrossRef] [PubMed]
- Nikiforova, M.N.; Biddinger, P.W.; Caudill, C.M.; Kroll, T.G.; Nikiforov, Y.E. PAX8-PPAR gamma rearrangement in thyroid tumors: RT-PCR and immunohistochemical analyses. Am. J. Surg. Pathol. 2002, 26, 1016–1023. [Google Scholar] [CrossRef]
- Castro, P.; Rebocho, A.P.; Soares, R.J.; Mgalhaes, J.; Roque, L.; Trovisco, V.; Vieira de Castro, I.; Cardoso-de-Oliveira, M.; Fonseca, E.; Soares, P.; et al. PAX8-PPAR gamma rearrangement is frequently detected in the follicular variant of papillary thyroid carcioma. J. Clin. Endocrinol. Metab. 2006, 92, 213–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marques, A.R.; Espadinha, C.; Catarino, A.L.; Moniz, S.; Pereira, T.; Sobrinho, L.G.; Leite, V. Expression of PAX-PPAR gamma 1 rearrangements in both follicular thyroid carcinomas and adenomas. J. Clin. Endocrinol. Metab. 2002, 87, 3947–3952. [Google Scholar]
- Kelly, L.M.; Barila, G.; Liu, P.; Evdokimova, V.N.; Trivedi, S.; Panebianco, F.; Gandhi, M.; Carty, S.E.; Hodak, S.P.; Luo, J.; et al. Identification of the transforming STRNALK fusion as a potential therapeutic target in the aggressive forms of thyroid cancer. Proc. Natl. Acad. Sci. USA 2014, 111, 4233–4238. [Google Scholar] [CrossRef] [Green Version]
- Panebianco, F.; Nikitski, A.V.; Nikiforova, M.N.; Kaya, C.; Yip, L.; Condello, V.; Wald, A.I.; Nikiforov, Y.E.; Chiosea, S.I. Characterization of thyroid cancer driven by known and novel ALK fusions. Endocr. Relat. Cancer 2019, 26, 803–814. [Google Scholar] [CrossRef] [PubMed]
- Doebele, R.C.; Drilon, A.; Paz-Ares, L.; Siena, S.; Shaw, A.T.; Farago, A.F.; Blakely, C.M.; Seto, T.; Cho, B.C.; Tosi, D.; et al. Entrectinib in patients with advanced or metastatic NTRK fusion-positive solid tumours: Integrated analysis of three phase 1-2 trials. Lancet Oncol. 2020, 21, 271–282. [Google Scholar] [CrossRef]
- Zehir, A.; Benayed, R.; Shah, R.H.; Syed, A.; Middha, S.; Kim, H.R.; Srinivasan, P.; Gao, J.; Chakravarty, D.; Devlin, S.M.; et al. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nat. Med. 2017, 23, 703–713. [Google Scholar] [CrossRef]
- Song, Y.S.; Lim, J.A.; Choi, H.; Won, J.K.; Moon, J.H.; Cho, S.W.; Lee, K.E.; Park, Y.J.; Yi, K.H.; Park, D.J.; et al. Prognostic effects of TERT promoter mutations are enhanced by coexistence with BRAF or RAS mutations and strengthen the risk prediction by the ATA or TNM staging system in differentiated thyroid cancer patients. Cancer 2016, 122, 1370–1379. [Google Scholar] [CrossRef]
- Liu, J.; Liu, R.; Shen, X.; Zhu, G.; Li, B.; Xing, M. The genetic duet of BRAF V600E and TERT promoter mutations robustly predicts loss of radioiodine avidity in recurrent papillary thyroid cancer. J. Nucl. Med. 2020, 61, 177–182. [Google Scholar] [CrossRef] [Green Version]
- Melo, M.; Gaspar Da Rocha, A.; Batista, R.; Vinagre, J.; Martins, M.J.; Costa, G.; Ribeiro, C.; Carrilho, F.; Leite, V.; Lobo, C.; et al. TERT, BRAF, and NRAS in primary thyroid cancer and metastatic disease. J. Clin. Endocrinol. Metab. 2017, 102, 1898–1907. [Google Scholar] [CrossRef]
- Melo, M.; da Rocha, A.G.; Vinagre, J.; Batista, R.; Peixoto, J.; Tavares, C.; Celestino, R.; Almeida, A.; Salgado, S.; Eloy, C.; et al. TERT promoter mutations are a major indicator of poor outcome in differentiated thyroid carcinomas. J. Clin. Endocrinol. Metab. 2014, 99, E754–E765. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.S.; Yoo, S.K.; Kim, H.H.; Jung, G.; Oh, A.R.; Cha, J.Y.; Kim, S.-J.; Cho, S.W.; Lee, K.E.; Seo, J.S.; et al. Interaction of BRAF-induced ETS factors with mutant TERT promoter in papillary thyroid cancer. Endocr. Relat. Cancer 2019, 26, 629–641. [Google Scholar] [CrossRef] [PubMed]
- Moon, S.; Song, Y.S.; Kim, Y.A.; Lim, J.A.; Cho, S.W.; Moon, J.H.; Hahn, S.; Park, D.J.; Park, Y.J. Effects of coexistent BRAFV600E and TERT promoter mutations on poor clinical outcomes in papillary thyroid cancer: A meta-analysis. Thyroid 2017, 27, 651–660. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Gong, Y.; Yan, S.; Chen, H.; Qin, S.; Gong, R. Association between TERT promoter mutations and clinical behaviors in differentiated thyroid carcinoma: A systematic review and meta-analysis. Endocrine 2020, 67, 44–57. [Google Scholar] [CrossRef] [Green Version]
- Diplas, B.H.; He, X.; Brosnan-Cashman, J.A.; Liu, H.; Chen, L.H.; Wang, Z.; Moure, C.J.; Killela, P.J.; Loriaux, D.B.; Lipp, E.S.; et al. The genomic landscape of TERT promoter wildtype-IDH wildtype glioblastoma. Nat. Commun. 2018, 9, 2087. [Google Scholar] [CrossRef]
- Peifer, M.; Hertwig, F.; Roels, F.; Dreidax, D.; Gartlgruber, M.; Menon, R.; Kramer, A.; Roncaioli, J.L.; Sand, F.; Heuckmann, J.M.; et al. Telomerase activation by genomic rearrangements in high-risk neuroblastoma. Nature 2015, 526, 700–704. [Google Scholar] [CrossRef]
- Liang, W.S.; Hendricks, W.; Kiefer, J.; Schmidt, J.; Sekar, S.; Carpten, J.; Craig, D.W.; Adkins, J.; Cuyugan, L.; Manojlovic, Z.; et al. Integrated genomic analyses reveal frequent TERT aberrations in acral melanoma. Genome Res. 2017, 27, 524–532. [Google Scholar] [CrossRef] [Green Version]
- Suzuki, K.; Matsubara, H. Recent advances in p53 research and cancer treatment. J. Biomed. Biotechnol. 2011, 3, 978312. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.K.; Song, Y.S.; Lee, E.K.; Hwang, J.; Kim, H.H.; Jung, G.; Kim, Y.A.; Kim, S.-J.; Cho, S.W.; Won, J.-K.; et al. Integrative analysis of genomic and transcriptomic characteristics associated with progression of aggressive thyroid. cancer. Nat. Commun. 2019, 10, 2764. [Google Scholar] [CrossRef] [Green Version]
- McFadden, D.G.; Vernon, A.; Santiago, P.M.; Martinez-McFaline, R.; Bhutkar, A.; Crowley, D.M.; McMahon, M.; Sadow, P.M.; Jacks, T. p53 constrains progression to anaplastic thyroid carcinoma in a Braf-mutant mouse model of papillary thyroid cancer. Proc. Natl. Acad. Sci. USA 2014, 111, E1600–E1609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zou, M.; Baitei, E.; Al-Rijjal, R.; Parhar, R.; Al-Mohanna, F.; Kimura, S.; Pritchard, C.; Binessa, H.A.; Alzahrani, A.S.; Al-Khalaf, H.H.; et al. TSH overcomes Braf V600E-induced senescence to promote tumor progression via downregulation of p53 expression in papillary thyroid cancer. Oncogene 2016, 35, 1909–1918. [Google Scholar] [CrossRef] [PubMed]
- Ravi, N.; Yang, M.; Gretarsson, S.; Jansson, C.; Mylona, N.; Sydow, S.R.; Woodward, E.L.; Ekblad, L.; Wennerberg, J.; Paulsson, K. Identification of targetable lesions in anaplastic thyroid cancer by genome profiling. Cancers 2019, 11, 402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paulsson, J.O.; Backman, S.; Wang, N.; Stenman, A.; Crona, J.; Thutkawkorapin, J.; Ghaderi, M.; Tham, E.; Stalberg, P.; Zedenius, J.; et al. Whole-genome sequencing of synchronous thyroid carcinomas identifies aberrant DNA repair in thyroid cancer dedifferentiation. J. Pathol. 2020, 250, 183–194. [Google Scholar] [CrossRef]
- Genutis, L.K.; Tomsic, J.; Bundschuh, R.A.; Brock, P.L.; Williams, M.D.; Roychowdhury, S.; Reeser, J.W.; Frankel, W.L.; Alsomali, M.; Routbort, M.J.; et al. Microsatellite instability occurs in a subset of follicular thyroid cancers. Thyroid 2019, 29, 523–529. [Google Scholar] [CrossRef]
- Santos, J.C.; Bastos, A.U.; Cerutti, J.M.; Ribeiro, M.L. Correlation of MLH1 and MGMT expression and promoter methylation with genomic instability in patients with thyroid carcinoma. BMC Cancer 2013, 13, 79. [Google Scholar] [CrossRef] [Green Version]
- Mitmaker, E.; Alvarado, C.; Bégin, L.R.; Trifiro, M. Microsatellite instability in benign and malignant thyroid neoplasms. J. Surg. Res. 2008, 150, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Nikiforov, Y.E.; Nikiforova, M.; Fagin, J.A. Prevalence of minisatellite and microsatellite instability in radiation-induced post-Chernobyl pediatric thyroid carcinomas. Oncogene 1998, 17, 1983–1988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lengfelder, E.; Bauchinger, M. Microsatellite instability and loss of heterozygosity in radiation-associated thyroid carcinomas of Belarussian children and adults. Carcinogenesis 1999, 20, 2247–2252. [Google Scholar] [CrossRef] [Green Version]
- <named-content content-type="background:white">Le, D.T.; Durham, J.N.; Smith, K.N.; Wang, H.; Bartlett, B.R.; Aulakh, L.K.; Lu, S.; Kemberling, H.; Wilt, C.; Luber, B.S.; et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017, 357, 409–413. [Google Scholar] [CrossRef] [Green Version]
- Helming, K.C.; Wang, X.; Roberts, C.W.M. Vulnerabilities of mutant SWI/SNF complexes in cancer. Cancer Cell 2014, 26, 309–317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- <named-content content-type="background:white">Wang, X.; Haswell, J.R.; Roberts, C.W.M. Molecular pathways: SWI/SNF (BAF) complexes are frequently mutated in cancer-mechanisms and potential therapeutic insights. Clin. Cancer Res. 2014, 20, 21–27. [Google Scholar]
- Fagin, J.A.; Mitsiades, N. Molecular pathology of thyroid cancer: Diagnostic and clinical implications. Best Pract. Res. Clin. Endocrinol. Metab. 2008, 22, 955–969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gavert, N.; Ben-Ze’ev, A. Beta-catenin signaling in biological control and cancer. J. Cell Biochem. 2007, 102, 820–828. [Google Scholar] [CrossRef]
- Polakis, P. The many ways of Wnt in cancer. Curr. Opin. Genet. Dev. 2007, 17, 45–51. [Google Scholar] [CrossRef] [PubMed]
- Cetta, F.; Montalto, G.; Gori, M.; Curia, M.C.; Cama, A.; Olschwang, S. Germline mutations of the APC gene in patients with familial adenomatous polyposis-associated thyroid carcinoma: Results from a European cooperative study. J. Clin. Endocrinol Metab. 2000, 85, 286–292. [Google Scholar] [PubMed] [Green Version]
- Von, W.R.; Rhein, A.; Werner, M.; Scheumann, G.F.; Dralle, H.; Potter, E.; Brabant, G.; Georgii, A. Immunohistochemical detection of E-cadherin in differentiated thyroid carcinomas correlates with clinical outcome. Cancer Res. 1997, 57, 2501–2507. [Google Scholar]
- Garcia-Rostan, G.; Camp, R.L.; Herrero, A.; Carcangiu, M.L.; Rimm, D.L.; Tallini, G. Beta-catenin dysregulation in thyroid neoplasms: Down-regulation, aberrant nuclear expression, and CTNNB1 exon 3 mutations are markers for aggressive tumor phenotypes and poor prognosis. Am. J. Pathol. 2001, 158, 987–996. [Google Scholar] [CrossRef]
- Garcia-Rostan, G.; Tallini, G.; Herrero, A.; D’Aquila, T.G.; Carcangiu, M.L.; Rimm, D.L. Frequent mutation and nuclear localization of beta-catenin in anaplastic thyroid carcinoma. Cancer Res. 1999, 59, 1811–1815. [Google Scholar] [PubMed]
- Baylin, S.B. Tying it all together: Epigenetics, genetics, cell cycle, and cancer. Science 1997, 277, 1948–1949. [Google Scholar] [CrossRef] [PubMed]
- Xing, M.; Usadel, H.; Cohen, Y.; Tokumaru, Y.; Guo, Z.; Westra, W.B.; Tong, B.C.; Tallinni, G.; Udelsman, R.; Califano, J.A.; et al. Methylation of the thyroid-stimulating hormone receptor gene in epithelial thyroid tumors: A marker of malignancy and a cause of gene silencing. Cancer Res. 2003, 63, 2316–2321. [Google Scholar]
- Kartal, K.O.S. Methylation status of TSHr in well-differentiated thyroid cancer by using cytologic material. BMC Cancer 2015, 15, 824–830. [Google Scholar] [CrossRef] [Green Version]
- Kim, G.W.; Zhu, X.; Kim, W.D.; Zhang, L.; Kebew, E.; Cheng, S. Reactivation of the silenced thyroid hormone receptor β gene expression delays thyroid tumor progression. Endocrinology 2013, 154, 25–35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Joseph, B.; Ji, M.; Liu, D.; Hou, P.; Xing, M. Lack of mutations in the thyroid hormone receptor (TR) α and β genes but frequent hypermethylation of theTRβ gene in differentiated thyroid tumors. J. Clin. Endocrinol. Metab. 2007, 92, 4766–4770. [Google Scholar] [CrossRef] [Green Version]
- Iwasaki, Y.; Sunaga, N.; Tomizawa, Y.; Imai, H.; Iijima, H.; Yanagitani, N.; Horiguchi, K.; Yamada, M.; Mori, M. Epigenetic inactivation of the thyroid hormone receptor β1 gene at 3p24.2 in lung cancer. Ann. Surg. Oncol. 2010, 17, 2222–2228. [Google Scholar] [CrossRef]
- Li, Z.; Meng, Z.H.; Chandrasekaran, R.; Kuo, W.L.; Collins, C.C.; Gray, J.W.; Dairkee, S.H. Biallelic inactivation of the thyroid hormone receptor β 1 gene in early stage breast cancer. Cancer Res. 2002, 62, 1939–1943. [Google Scholar] [PubMed]
- Hörkkö, T.T.; Tuppurainen, K.; George, S.M.; Jernvall, P.; Karttunen, T.J.; Mäkinen, M.J. Thyroid hormone receptor β 1 in normal colon and colorectal cancer-association with differentiation, polypoid growth type and K-ras mutations. Int. J. Cancer 2006, 118, 1653–1659. [Google Scholar] [CrossRef] [PubMed]
- Dunwell, T.L.; Hesson, L.B.; Pavlova., T.; Zabarovska., V.; Kashuba, V.; Catchpoole, D.; Chiaramonte, R.; Brini, A.T.; Griffiths, M.; Maher, E.R.; et al. Epigenetic analysis of childhood acute lymphoblastic leukemia. Epigenetics 2009, 4, 185–193. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boltze, C.; Zack, S.; Quednow, C.; Bettge, S.; Roessner, A.; Schneider-Stock, R. Hypermethylation of the CDKN2/p16INK4A promoter in thyroid carcinogenesis. Pathol. Res. Pract. 2003, 199, 399–404. [Google Scholar] [CrossRef]
- Shou, F.; Xu, F.; Li, G.; Zhao, Z.; Mao, Y.; Yang, F.; Wang, H.; Guo, H. RASSF1A promoter methylation is associated with incresaed risk of thyroid cancer: A meta-analysis. Onco Targets Ther. 2017, 10, 247–257. [Google Scholar] [CrossRef] [Green Version]
- Hu, S.; Liu, D.; Tufano, R.P.; Carson, K.A.; Rosenbaum, E.; Cohen, Y.; Holy, E.H.; Kiseljak-Vassiliades, K.; Rhoden, K.J.; Tolaney, S.; et al. Association of aberrant methyrlation of tumor suppressor genes with tumor aggressiveness and BRAF mutation in papillary thyroid cancer. Int J. Cancer 2006, 119, 2322–2329. [Google Scholar] [CrossRef]
- Xing, M.; Tokumaru, Y.; Wu, G.; Westra, W.B.; Ladenson, P.W.; Sidransky, D. Hypermethylation of the Pendred syndrome gene SLC26A is an early event in thyroid tumorigenesis. Cancer Res. 2003, 63, 2312–2315. [Google Scholar]
- McKelvey, B.A.; Zeiger, M.A.; Umbricht, C.B. Characterization of TERT and BRAF copy number variation in papillary thyroid carcinoma: An analysis of the cancer genome atlas study. Genes Chromosomes Cancer 2021, 60, 403–409. [Google Scholar] [CrossRef]
- Máximo, V.; Soares, P.; Lima, J.; Cameselle-Teijeiro, J.; Sobrinho-Simões, M. Mitochondrial DNA somatic mutations (point mutations and large deletions) and mitochondrial DNA variants in human thyroid pathology: A study with emphasis on Hürthle cell tumors. Am. J. Pathol. 2002, 160, 1857–1865. [Google Scholar] [CrossRef]
- Gopal, R.K.; Kübler, K.; Calvo, S.E.; Polak, P.; Livitz, D.; Rosebrock, D.; Sadow, P.M.; Campbell, B.; Donovan, S.E.; Amin, S.; et al. Widespread chromosomal losses and mitochondrial DNA alterations as genetic drivers in Hürthle cell carcinoma. Cancer Cell 2018, 34, 242–255.e5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gajewski, T.F.; Corrales, L.; Williams, J.; Horton, B.; Sivan, A.; Spranger, S. Cancer immunotherapy targets based on understanding the T-cell-inflamed versus non-T cell-inflamed tumor microenvironment. Adv. Exp. Med. Biol. 2017, 1036, 19–31. [Google Scholar] [PubMed]
- Yu, H.; Huang, X.; Liu, X.; Jin, H.; Zhang, G.; Zhang, Q.; Yu, J. Regulatory T cells and plasmacytoid dendritic cells contribute to the immune escape of papillary thyroid cancer coexisting with multinodular non-toxic goiter. Endocrine 2013, 44, 172–181. [Google Scholar] [CrossRef] [PubMed]
- Cunha, L.L.; Morari, E.C.; Nonogaki, S.; Soarees, F.A.; Vassallo, J.; Ward, L.S. Foxp3 expression is associated with aggressiveness in differentiated thyroid carcinomas. Clinics 2012, 67, 483–488. [Google Scholar] [CrossRef]
- Liu, Y.; Yun, X.; Gao, M.; Yu, Y.; Li, X. Analysis of regulatory T cells frequency in peripheral blood and tumor tissues in papillary thyroid carcinoma with and without Hashimoto’s thyroiditis. Clin. Transl. Oncol. 2015, 17, 274–280. [Google Scholar] [CrossRef]
- Coates, P.J.; Rundle, J.K.; Lorimore, S.A.; Wright, E.G. Indirect macrophage responses to ionizing radiation: Implications for genotype-dependent bystander signaling. Cancer Res. 2008, 68, 450–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giannini, R.; Moretti, S.; Ugolini, C.; Macerola, E.; Menicali, E.; Nucci, N.; Morelli, S.; Colella, R.; Mandarano, M.; Sidoni, A.; et al. Immune profiling of thyroid carcinomas suggests the existence of two major phenotypes: An ATC-like and a PDTC-like. J. Clin. Endocrinol. Metab. 2019, 104, 3557–3575. [Google Scholar] [CrossRef]
- Riella, L.V.; Paterson, A.M.; Sharpe, A.H.; Chandraker, A. Role of the PD-1 pathway in the immune response. Am. J. Transpl. 2012, 12, 2575–2587. [Google Scholar] [CrossRef] [PubMed]
- Zou, W.; Wolchok, J.D.; Chen, L. PD-L1 (N7-H1) and PD-1 pathway blockade for cancer therapy: Mechanisms, response biomarkers, and combinations. Sci. Transl. Med. 2016, 8, 328rv4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ahn, S.; Kim, T.H.; Kim, S.W.; Ki, C.S.; Jang, H.W.; Kim, J.S.; Kim, J.H.; Choe, J.-H.; Shin, J.H.; Hahn, S.Y.; et al. Comprehensive screening for PD-L1 expression in thyroid cancer. Endocr. Relat. Cancer 2017, 24, 97–106. [Google Scholar] [CrossRef] [Green Version]
- Angell, T.E.; Lechner, M.G.; Jang, J.K.; Correa, A.J.; Lopresti, J.S.; Epstein, A.L. BRAFV600E in papillary thyroid carcinoma is associated with increased programmed death Ligand 1 expression and suppressive immune cell infiltration. Thyroid 2014, 24, 1385–1393. [Google Scholar] [CrossRef] [Green Version]
- Chowdhury, S.; Veyhl, J.; Jessa, F.; Polyakova, O.; Alenzi, A.; MacMillan, C.; Ralhan, R.; Walfish, P.G. Programmed death-ligand 1 overexpression is a prognostic marker for aggressive papillary thyroid cancer and its variants. Oncotarget 2016, 7, 32318–32328. [Google Scholar] [CrossRef] [PubMed]
- Rosenbaum, M.W.; Gigliotti, B.J.; Pai, S.I.; Parangi, S.; Wachtel, H.; Mino-Kenudson, M.; Gunda, V.; Faquin, W.C. PD-L1 and IDO1 are expressed in poorly differentiated thyroid carcinoma. Endocr. Pathol. 2018, 29, 59–67. [Google Scholar] [CrossRef]
- Shi, R.; Qu, N.; Luo, T.; Xiang, J.; Liao, T.; Sun, G.; Wang, Y.; Wang, Y.-L.; Huang, C.-P.; Ji, Q.-H. Programmed death-ligand 1 expression in papillary thyroid cancer and its correlation with clinicopathologic factors and recurrence. Thyroid 2017, 27, 537–545. [Google Scholar] [CrossRef]
- Zwaenepoel, K.; Jacobs, J.; De Meulenaere, A.; Silence, K.; Smits, E.; Siozopoulou, V.; Hauben, E.; Rolfo, C.; Rottey, S.; Pauwels, P. CD 70 and PD-L1 in anaplastic thyroid cancer–promising targets for immunotherapy. Histopathology 2017, 71, 357–365. [Google Scholar] [CrossRef]
- Mezzadra, R.; Sun, C.; Jae, L.T.; Gomez-Eerland, R.; De Vries, E.; Wu, W.; Logtenberg, M.E.W.; Slagter, M.; Rozeman, E.A.; Hofland, I.; et al. Identification of CMTM6 and CMTM4 as PD-L1 protein regulators. Nature 2017, 549, 106–110. [Google Scholar] [CrossRef]
- Cantara, S.; Bertelli, E.; Occhini, R.; Regoli, M.; Brilli, L.; Pacini, F.; Castanga, M.G.; Toti, P. Blockade of the programmed death ligand 1 (PD-L1) as potential therapy for anaplastic thyroid cancer. Endocrine 2019, 64, 122–129. [Google Scholar] [CrossRef]
- Gunda, V.; Gigliotti, B.; Ashry, T.; Ndishabandi, D.; McCarthy, M.; Zhou, Z.; Amin, S.; Lee, K.E.; Stork, T.; Wirth, L.; et al. Anti-PD-1/PD-L1 therapy augments lenvatinib’s efficacy by favorably altering the immune microenvironment of murine anaplastic thyroid cancer. Int. J. Cancer 2019, 144, 2266–2278. [Google Scholar] [CrossRef] [PubMed]
- Lopes-Ventura, S.; Pojo, M.; Matias, A.T.; Moura, M.M.; Marques, I.J.; Leite, V.; Cavaco, B.M. The efficacy of HRAS and CDK4/6 inhibitors in anaplastic thyroid cancer cell lines. J. Endocrinol. Investig. 2018, 42, 527–540. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Genetic alterations involved in the evolution of thyroid cancers. Figure adapted from Pozdeyez et al. [18]. Rat sarcoma (RAS). V-raf murine sarcoma viral oncogene homolog B1(BRAF). Rearranged during transfection (RET). Anaplastic lymphoma kinase (ALK). Paired box gene 8-peroxisome proliferator-activated receptor (PAX8-PPARγ). Telomerase reverse transcriptase (TERT). Tumour protein 53 (TP53). Cyclin-dependent kinase inhibitor 2A/2B (CDKN2A/2B). (PIK3CA). Phosphatase and tensin homolog (PTEN). Ak strain transforming (AKT). Guanine nucleotide binding protein, alpha stimulating activity polypeptide (GNAS). Retinoblastoma1 (RB1). AT-rich interactive domain-containing protein 2 (ARID2). Neurofibromatosis1 (NF1). V-kit Hardy-Zuckerman 4 feline sarcoma viral oncogene homolog (KIT). Mismatch repair (MMR). Papillary thyroid carcinoma (PTC). Follicular thyroid carcinoma (FTC). Medullary thyroid carcinoma (MTC). Anaplastic thyroid carcinoma (ATC).
Figure 1.
Genetic alterations involved in the evolution of thyroid cancers. Figure adapted from Pozdeyez et al. [18]. Rat sarcoma (RAS). V-raf murine sarcoma viral oncogene homolog B1(BRAF). Rearranged during transfection (RET). Anaplastic lymphoma kinase (ALK). Paired box gene 8-peroxisome proliferator-activated receptor (PAX8-PPARγ). Telomerase reverse transcriptase (TERT). Tumour protein 53 (TP53). Cyclin-dependent kinase inhibitor 2A/2B (CDKN2A/2B). (PIK3CA). Phosphatase and tensin homolog (PTEN). Ak strain transforming (AKT). Guanine nucleotide binding protein, alpha stimulating activity polypeptide (GNAS). Retinoblastoma1 (RB1). AT-rich interactive domain-containing protein 2 (ARID2). Neurofibromatosis1 (NF1). V-kit Hardy-Zuckerman 4 feline sarcoma viral oncogene homolog (KIT). Mismatch repair (MMR). Papillary thyroid carcinoma (PTC). Follicular thyroid carcinoma (FTC). Medullary thyroid carcinoma (MTC). Anaplastic thyroid carcinoma (ATC).

Figure 2.
Commonly dysregulated cell signalling mitogen-activated protein kinase (MAPK), phosphatidylinositol-3 kinase (P13K)/Ak strain transforming (AKT) and wingless-related integration site (WNT) pathways in thyroid cancers.
Figure 2.
Commonly dysregulated cell signalling mitogen-activated protein kinase (MAPK), phosphatidylinositol-3 kinase (P13K)/Ak strain transforming (AKT) and wingless-related integration site (WNT) pathways in thyroid cancers.

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Singh, A.; Ham, J.; Po, J.W.; Niles, N.; Roberts, T.; Lee, C.S. The Genomic Landscape of Thyroid Cancer Tumourigenesis and Implications for Immunotherapy. Cells 2021, 10, 1082. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10051082
AMA Style
Singh A, Ham J, Po JW, Niles N, Roberts T, Lee CS. The Genomic Landscape of Thyroid Cancer Tumourigenesis and Implications for Immunotherapy. Cells. 2021; 10(5):1082. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10051082
Chicago/Turabian StyleSingh, Amandeep, Jeehoon Ham, Joseph William Po, Navin Niles, Tara Roberts, and Cheok Soon Lee. 2021. "The Genomic Landscape of Thyroid Cancer Tumourigenesis and Implications for Immunotherapy" Cells 10, no. 5: 1082. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10051082
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.