
The Phosphatidylserine Receptor TIM-1 Enhances Authentic Chikungunya Virus Cell Entry

 ,
,  , , , , and
, , , , and 
Abstract
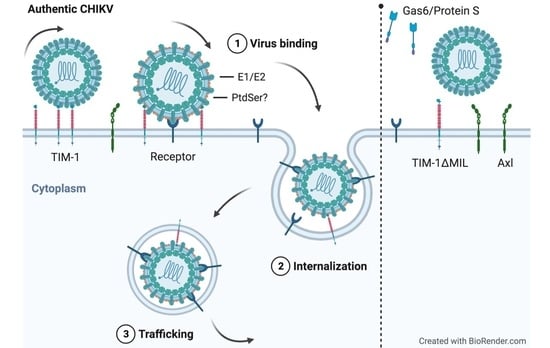
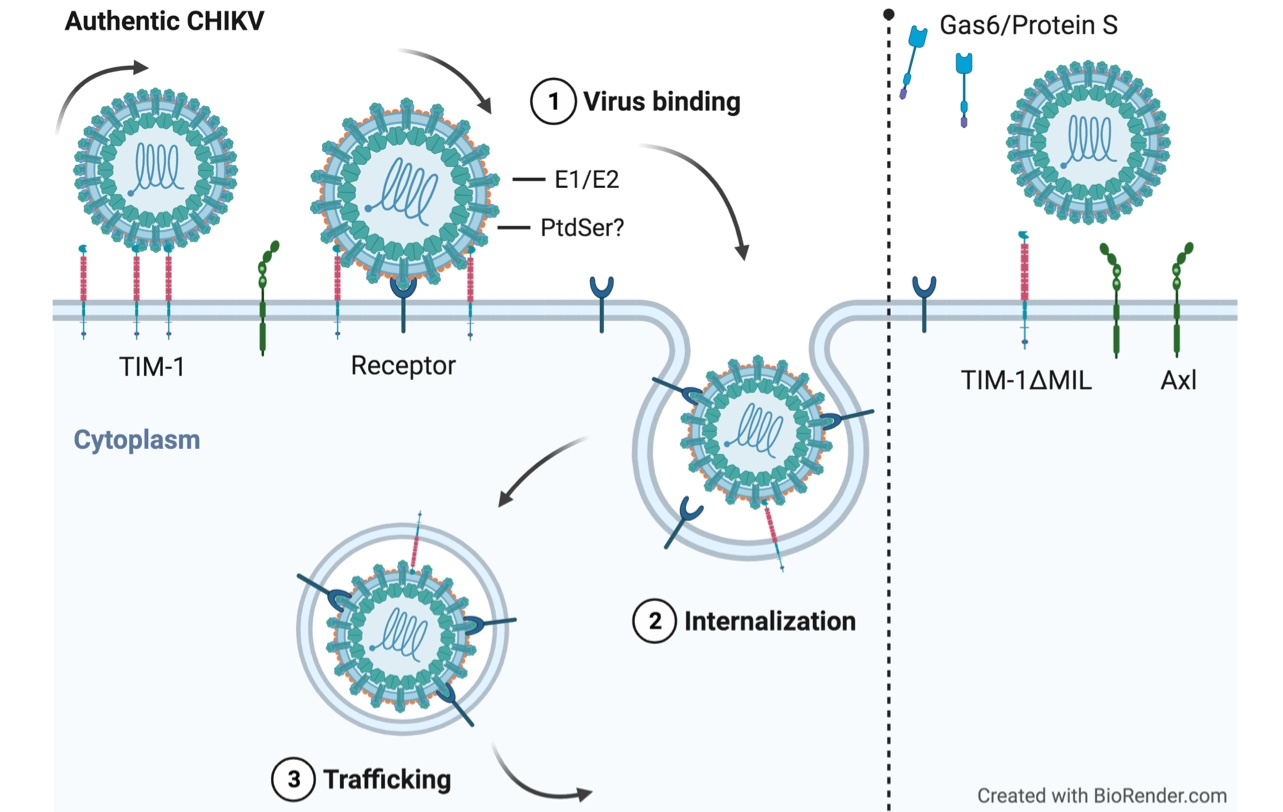
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Cells and Viruses
2.2. Plasmids and Antibodies
2.3. RNA Transfection by Electroporation
2.4. Generation of Lentiviral Vectors and Transduction of Cells
2.5. RNA Interference
2.6. Cell Viability and Proliferation Assay
2.7. Cell Culture Derived CHIKV Stock Production and Titration
2.8. Infection and Antibody Inhibition Assay
2.9. Virus Binding and Endosomal Escape Assays
2.10. Confocal Microscopy and Live Cell Imaging
2.11. Single Particle Tracking
2.12. Western Blot Analysis
2.13. Luciferase Assay
2.14. Surface Staining and Flow Cytometry
2.15. Statistical Analysis
3. Results
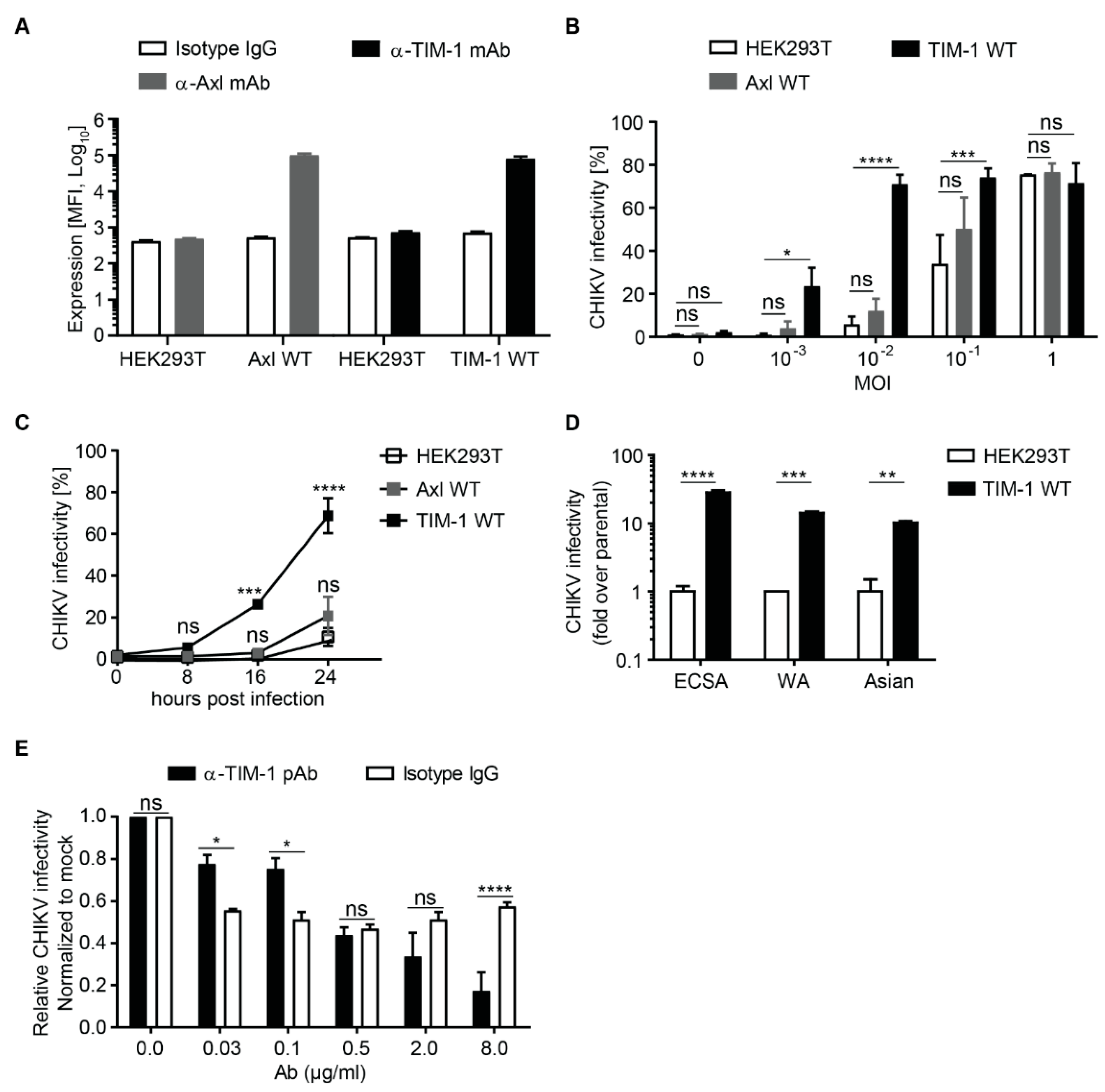
3.1. Ectopic TIM-1 Expression Enhances CHIKV Infection in HEK293T Cells
3.2. The TIM-1 Phosphatidylserine-Binding Domain Is Crucial for TIM-1-Dependent Infection
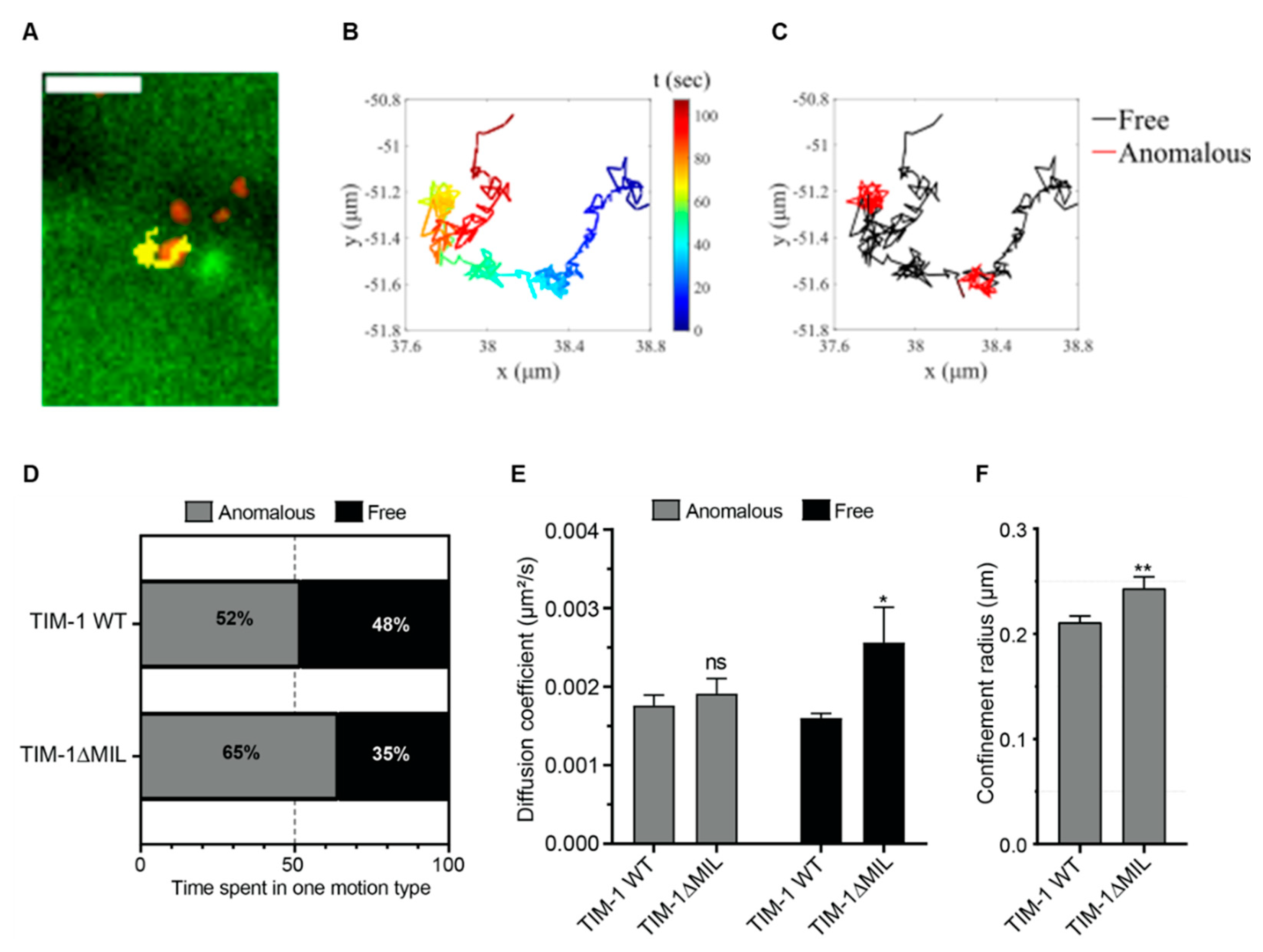
3.3. Single Particle Tracking of CHIKV Confirms PtdSer Domain Requirement
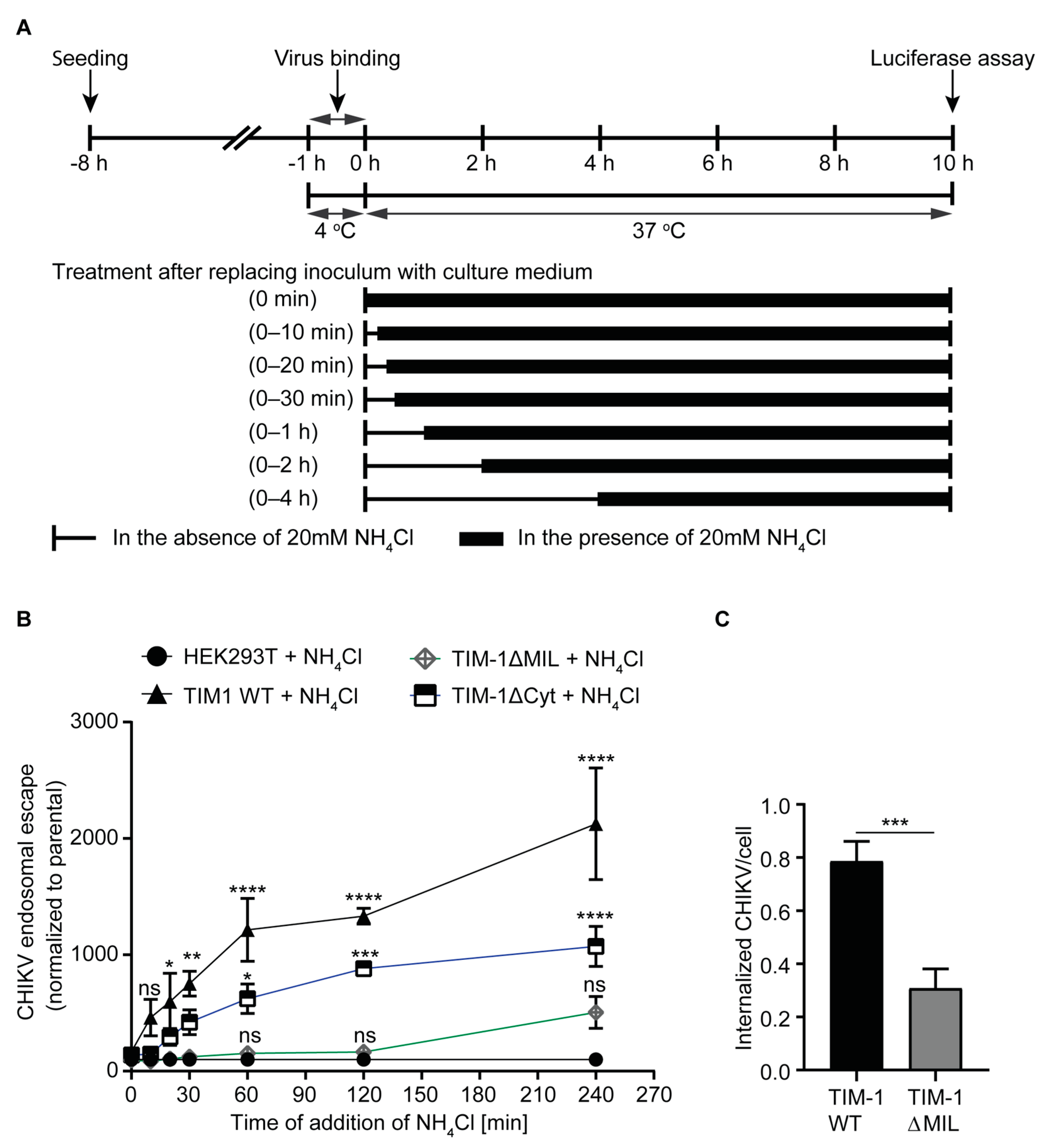
3.4. Entry Kinetics of CHIKV Are Altered by TIM-1
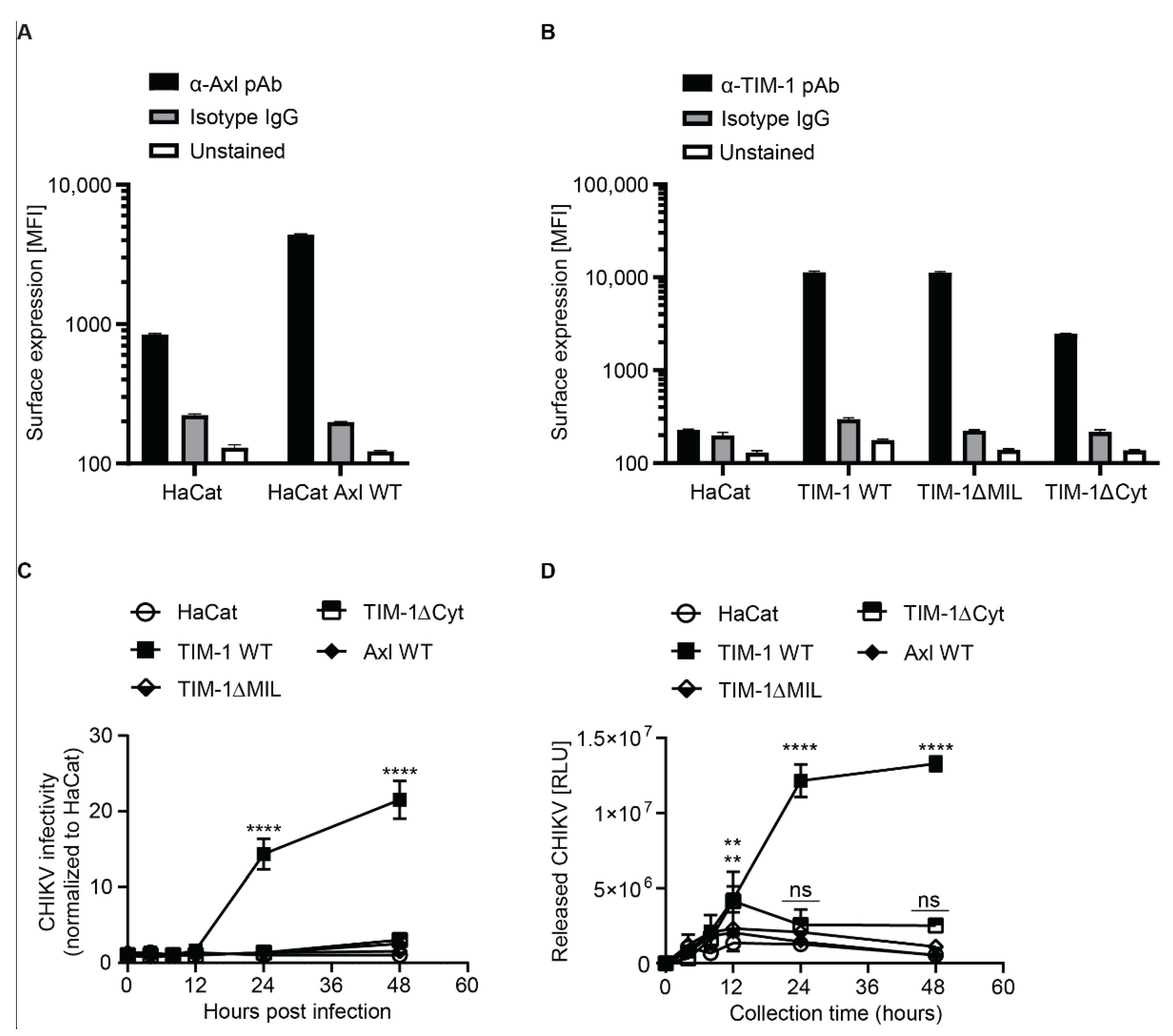
3.5. TIM-1 Expression Renders Keratinocyte Derived HaCat Cells Permissive to CHIKV
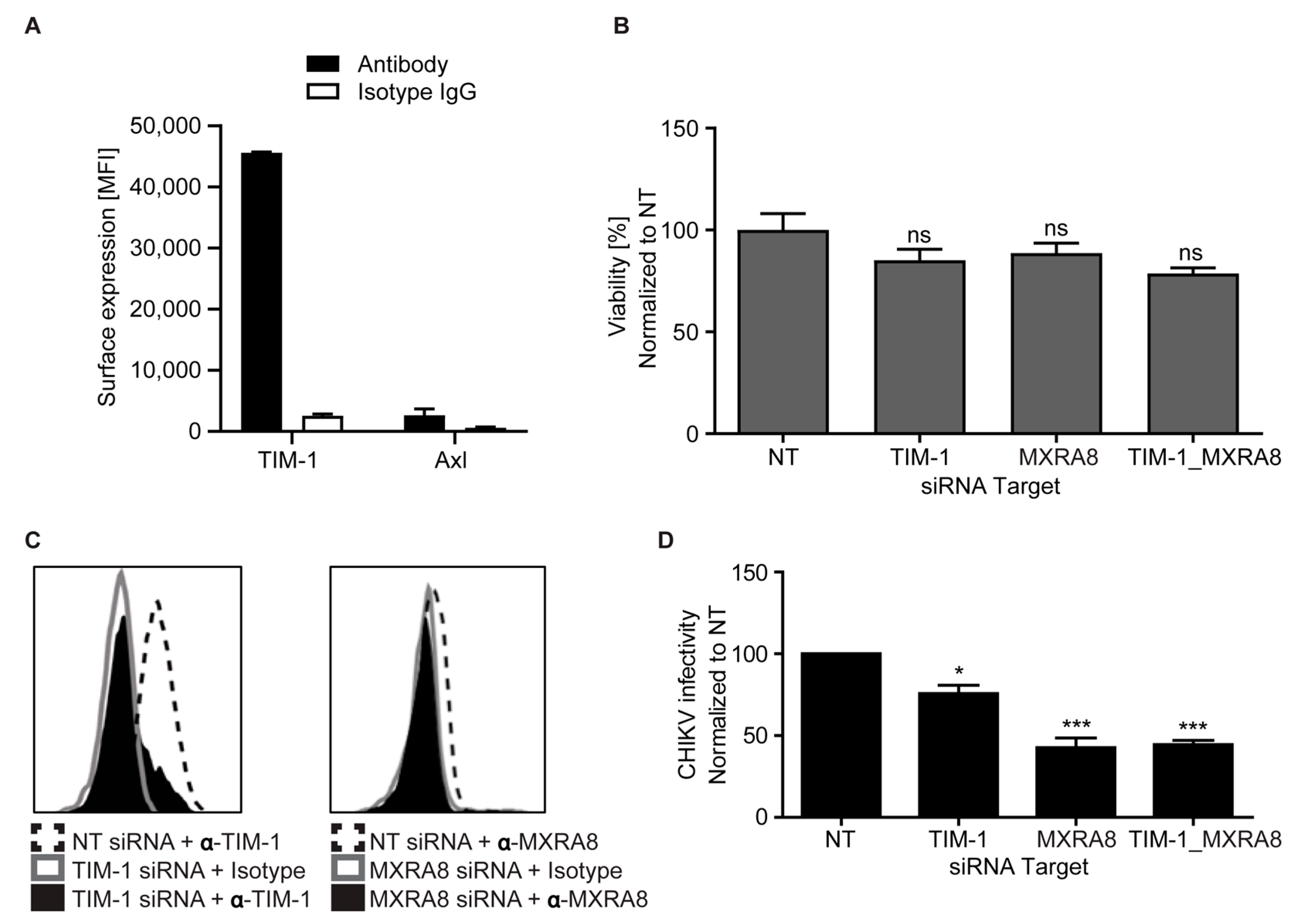
3.6. Endogenous TIM-1 Mediates CHIKV Infection of Hepatoma Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Morrison, T.E. Reemergence of chikungunya virus. J. Virol. 2014, 88, 11644–11647. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wahid, B.; Ali, A.; Rafique, S.; Idrees, M. Global expansion of chikungunya virus: Mapping the 64-year history. Int. J. Infect. Dis. 2017, 58, 69–76. [Google Scholar] [CrossRef] [Green Version]
- Tsetsarkin, K.A.; Vanlandingham, D.L.; McGee, C.E.; Higgs, S. A single mutation in chikungunya virus affects vector specificity and epidemic potential. PLoS Pathog. 2007, 3, e201. [Google Scholar] [CrossRef] [PubMed]
- Powers, A.M.; Brault, A.C.; Tesh, R.B.; Weaver, S.C. Re-emergence of Chikungunya and O’nyong-nyong viruses: Evidence for distinct geographical lineages and distant evolutionary relationships. J. Gen. Virol. 2000, 81, 471–479. [Google Scholar] [CrossRef]
- Thiberville, S.-D.; Moyen, N.; Dupuis-Maguiraga, L.; Nougairede, A.; Gould, E.A.; Roques, P.; de Lamballerie, X. Chikungunya fever: Epidemiology, clinical syndrome, pathogenesis and therapy. Antivir. Res. 2013, 99, 345–370. [Google Scholar] [CrossRef]
- Dupuis-Maguiraga, L.; Noret, M.; Brun, S.; Le Grand, R.; Gras, G.; Roques, P. Chikungunya disease: Infection-associated markers from the acute to the chronic phase of arbovirus-induced arthralgia. PLoS Negl. Trop. Dis. 2012, 6, e1446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- van Duijl-Richter, M.K.S.; Hoornweg, T.E.; Rodenhuis-Zybert, I.A.; Smit, J.M. Early Events in Chikungunya Virus Infection-From Virus Cell Binding to Membrane Fusion. Viruses 2015, 7, 3647–3674. [Google Scholar] [CrossRef]
- Sourisseau, M.; Schilte, C.; Casartelli, N.; Trouillet, C.; Guivel-Benhassine, F.; Rudnicka, D.; Sol-Foulon, N.; Le Roux, K.; Prevost, M.-C.; Fsihi, H.; et al. Characterization of reemerging chikungunya virus. PLoS Pathog. 2007, 3, e89. [Google Scholar] [CrossRef]
- Solignat, M.; Gay, B.; Higgs, S.; Briant, L.; Devaux, C. Replication cycle of chikungunya: A re-emerging arbovirus. Virology 2009, 393, 183–197. [Google Scholar] [CrossRef] [Green Version]
- Wikan, N.; Sakoonwatanyoo, P.; Ubol, S.; Yoksan, S.; Smith, D.R. Chikungunya virus infection of cell lines: Analysis of the East, Central and South African lineage. PLoS ONE 2012, 7, e31102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weaver, S.C.; Osorio, J.E.; Livengood, J.A.; Chen, R.; Stinchcomb, D.T. Chikungunya virus and prospects for a vaccine. Expert Rev. Vaccines 2012, 11, 1087–1101. [Google Scholar] [CrossRef]
- Schnierle, B.S. Cellular attachment and entry factors for chikungunya virus. Viruses 2019, 11, 1078. [Google Scholar] [CrossRef] [Green Version]
- Holmes, A.C.; Basore, K.; Fremont, D.H.; Diamond, M.S. A molecular understanding of alphavirus entry. PLoS Pathog. 2020, 16, e1008876. [Google Scholar] [CrossRef]
- Fongsaran, C.; Jirakanwisal, K.; Kuadkitkan, A.; Wikan, N.; Wintachai, P.; Thepparit, C.; Ubol, S.; Phaonakrop, N.; Roytrakul, S.; Smith, D.R. Involvement of ATP synthase β subunit in chikungunya virus entry into insect cells. Arch. Virol. 2014, 159, 3353–3364. [Google Scholar] [CrossRef] [PubMed]
- Wintachai, P.; Wikan, N.; Kuadkitkan, A.; Jaimipuk, T.; Ubol, S.; Pulmanausahakul, R.; Auewarakul, P.; Kasinrerk, W.; Weng, W.-Y.; Panyasrivanit, M.; et al. Identification of prohibitin as a Chikungunya virus receptor protein. J. Med. Virol. 2012, 84, 1757–1770. [Google Scholar] [CrossRef]
- Gardner, C.L.; Hritz, J.; Sun, C.; Vanlandingham, D.L.; Song, T.Y.; Ghedin, E.; Higgs, S.; Klimstra, W.B.; Ryman, K.D. Deliberate attenuation of chikungunya virus by adaptation to heparan sulfate-dependent infectivity: A model for rational arboviral vaccine design. PLoS Negl. Trop. Dis. 2014, 8, e2719. [Google Scholar] [CrossRef] [Green Version]
- Silva, L.A.; Khomandiak, S.; Ashbrook, A.W.; Weller, R.; Heise, M.T.; Morrison, T.E.; Dermody, T.S. A single-amino-acid polymorphism in Chikungunya virus E2 glycoprotein influences glycosaminoglycan utilization. J. Virol. 2014, 88, 2385–2397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moller-Tank, S.; Kondratowicz, A.S.; Davey, R.A.; Rennert, P.D.; Maury, W. Role of the phosphatidylserine receptor TIM-1 in enveloped-virus entry. J. Virol. 2013, 87, 8327–8341. [Google Scholar] [CrossRef] [Green Version]
- Moller-Tank, S.; Maury, W. Phosphatidylserine receptors: Enhancers of enveloped virus entry and infection. Virology 2014, 468, 565–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, R.; Kim, A.S.; Fox, J.M.; Nair, S.; Basore, K.; Klimstra, W.B.; Rimkunas, R.; Fong, R.H.; Lin, H.; Poddar, S.; et al. Mxra8 is a receptor for multiple arthritogenic alphaviruses. Nature 2018, 557, 570–574. [Google Scholar] [CrossRef]
- Weber, C.; Berberich, E.; von Rhein, C.; Henß, L.; Hildt, E.; Schnierle, B.S. Identification of functional determinants in the chikungunya virus E2 protein. PLoS Negl. Trop. Dis. 2017, 11, e0005318. [Google Scholar] [CrossRef] [PubMed]
- Basore, K.; Kim, A.S.; Nelson, C.A.; Zhang, R.; Smith, B.K.; Uranga, C.; Vang, L.; Cheng, M.; Gross, M.L.; Smith, J.; et al. Cryo-EM Structure of Chikungunya Virus in Complex with the Mxra8 Receptor. Cell 2019, 177, 1725–1737. [Google Scholar] [CrossRef]
- Song, H.; Zhao, Z.; Chai, Y.; Jin, X.; Li, C.; Yuan, F.; Liu, S.; Gao, Z.; Wang, H.; Song, J.; et al. Molecular basis of arthritogenic alphavirus receptor MXRA8 binding to chikungunya virus envelope protein. Cell 2019, 177, 1714–1724. [Google Scholar] [CrossRef] [PubMed]
- Ravichandran, K.S. Beginnings of a good apoptotic meal: The find-me and eat-me signaling pathways. Immunity 2011, 35, 445–455. [Google Scholar] [CrossRef] [Green Version]
- Wang, Q.; Imamura, R.; Motani, K.; Kushiyama, H.; Nagata, S.; Suda, T. Pyroptotic cells externalize eat-me and release find-me signals and are efficiently engulfed by macrophages. Int. Immunol. 2013, 25, 363–372. [Google Scholar] [CrossRef] [Green Version]
- Pietkiewicz, S.; Schmidt, J.H.; Lavrik, I.N. Quantification of apoptosis and necroptosis at the single cell level by a combination of Imaging Flow Cytometry with classical Annexin V/propidium iodide staining. J. Immunol. Methods 2015, 423, 99–103. [Google Scholar] [CrossRef]
- Erwig, L.-P.; Henson, P.M. Immunological consequences of apoptotic cell phagocytosis. Am. J. Pathol. 2007, 171, 2–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ichimura, T.; Asseldonk, E.J.P.V.; Humphreys, B.D.; Gunaratnam, L.; Duffield, J.S.; Bonventre, J.V. Kidney injury molecule-1 is a phosphatidylserine receptor that confers a phagocytic phenotype on epithelial cells. J. Clin. Investig. 2008, 118, 1657–1668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brooks, C.R.; Yeung, M.Y.; Brooks, Y.S.; Chen, H.; Ichimura, T.; Henderson, J.M.; Bonventre, J.V. KIM-1-/TIM-1-mediated phagocytosis links ATG5-/ULK1-dependent clearance of apoptotic cells to antigen presentation. EMBO J. 2015, 34, 2441–2464. [Google Scholar] [CrossRef] [Green Version]
- Mercer, J.; Helenius, A. Apoptotic mimicry: Phosphatidylserine-mediated macropinocytosis of vaccinia virus. Ann. N. Y. Acad. Sci. 2010, 1209, 49–55. [Google Scholar] [CrossRef]
- Amara, A.; Mercer, J. Viral apoptotic mimicry. Nat. Rev. Microbiol. 2015, 13, 461–469. [Google Scholar] [CrossRef]
- Niu, J.; Jiang, Y.; Xu, H.; Zhao, C.; Zhou, G.; Chen, P.; Cao, R. TIM-1 Promotes Japanese Encephalitis Virus Entry and Infection. Viruses 2018, 10, 630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jemielity, S.; Wang, J.J.; Chan, Y.K.; Ahmed, A.A.; Li, W.; Monahan, S.; Bu, X.; Farzan, M.; Freeman, G.J.; Umetsu, D.T.; et al. TIM-family proteins promote infection of multiple enveloped viruses through virion-associated phosphatidylserine. PLoS Pathog. 2013, 9, e1003232. [Google Scholar] [CrossRef] [Green Version]
- Meertens, L.; Carnec, X.; Lecoin, M.P.; Ramdasi, R.; Guivel-Benhassine, F.; Lew, E.; Lemke, G.; Schwartz, O.; Amara, A. The TIM and TAM families of phosphatidylserine receptors mediate dengue virus entry. Cell Host Microbe 2012, 12, 544–557. [Google Scholar] [CrossRef] [Green Version]
- Nagata, K.; Ohashi, K.; Nakano, T.; Arita, H.; Zong, C.; Hanafusa, H.; Mizuno, K. Identification of the product of growth arrest-specific gene 6 as a common ligand for Axl, Sky, and Mer receptor tyrosine kinases. J. Biol. Chem. 1996, 271, 30022–30027. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anderson, H.A.; Maylock, C.A.; Williams, J.A.; Paweletz, C.P.; Shu, H.; Shacter, E. Serum-derived protein S binds to phosphatidylserine and stimulates the phagocytosis of apoptotic cells. Nat. Immunol. 2003, 4, 87–91. [Google Scholar] [CrossRef] [PubMed]
- Dejarnac, O.; Hafirassou, M.L.; Chazal, M.; Versapuech, M.; Gaillard, J.; Perera-Lecoin, M.; Umana-Diaz, C.; Bonnet-Madin, L.; Carnec, X.; Tinevez, J.-Y.; et al. TIM-1 Ubiquitination Mediates Dengue Virus Entry. Cell Rep. 2018, 23, 1779–1793. [Google Scholar] [CrossRef] [PubMed]
- Bauer, T.; Zagórska, A.; Jurkin, J.; Yasmin, N.; Köffel, R.; Richter, S.; Gesslbauer, B.; Lemke, G.; Strobl, H. Identification of Axl as a downstream effector of TGF-β1 during Langerhans cell differentiation and epidermal homeostasis. J. Exp. Med. 2012, 209, 2033–2047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boukamp, P.; Petrussevska, R.T.; Breitkreutz, D.; Hornung, J.; Markham, A.; Fusenig, N.E. Normal keratinization in a spontaneously immortalized aneuploid human keratinocyte cell line. J. Cell Biol. 1988, 106, 761–771. [Google Scholar] [CrossRef] [Green Version]
- Hoornweg, T.E.; van Duijl-Richter, M.K.S.; Ayala Nuñez, N.V.; Albulescu, I.C.; van Hemert, M.J.; Smit, J.M. Dynamics of Chikungunya Virus Cell Entry Unraveled by Single-Virus Tracking in Living Cells. J. Virol. 2016, 90, 4745–4756. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ooi, Y.S.; Stiles, K.M.; Liu, C.Y.; Taylor, G.M.; Kielian, M. Genome-wide RNAi screen identifies novel host proteins required for alphavirus entry. PLoS Pathog. 2013, 9, e1003835. [Google Scholar] [CrossRef] [Green Version]
- Bernard, E.; Solignat, M.; Gay, B.; Chazal, N.; Higgs, S.; Devaux, C.; Briant, L. Endocytosis of chikungunya virus into mammalian cells: Role of clathrin and early endosomal compartments. PLoS ONE 2010, 5, e11479. [Google Scholar] [CrossRef] [Green Version]
- Lee, R.C.H.; Hapuarachchi, H.C.; Chen, K.C.; Hussain, K.M.; Chen, H.; Low, S.L.; Ng, L.C.; Lin, R.; Ng, M.M.-L.; Chu, J.J.H. Mosquito cellular factors and functions in mediating the infectious entry of chikungunya virus. PLoS Negl. Trop. Dis. 2013, 7, e2050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, S.-C.; Chen, Y.-J.; Wang, Y.-M.; Tsui, P.-Y.; Kuo, M.-D.; Wu, T.-Y.; Lo, S.J. Cell-based analysis of Chikungunya virus E1 protein in membrane fusion. J. Biomed. Sci. 2012, 19, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, S.-C.; Chen, Y.-J.; Wang, Y.-M.; Kuo, M.-D.; Jinn, T.-R.; Chen, W.-S.; Chang, Y.-C.; Tung, K.-L.; Wu, T.-Y.; Lo, S.J. Cell-based analysis of Chikungunya virus membrane fusion using baculovirus-expression vectors. J. Virol. Methods 2011, 175, 206–215. [Google Scholar] [CrossRef] [PubMed]
- van Duijl-Richter, M.K.S.; Blijleven, J.S.; van Oijen, A.M.; Smit, J.M. Chikungunya virus fusion properties elucidated by single-particle and bulk approaches. J. Gen. Virol. 2015, 96, 2122–2132. [Google Scholar] [CrossRef]
- Zeng, X.; Mukhopadhyay, S.; Brooks, C.L. Residue-level resolution of alphavirus envelope protein interactions in pH-dependent fusion. Proc. Natl. Acad. Sci. USA 2015, 112, 2034–2039. [Google Scholar] [CrossRef] [Green Version]
- DuBridge, R.B.; Tang, P.; Hsia, H.C.; Leong, P.M.; Miller, J.H.; Calos, M.P. Analysis of mutation in human cells by using an Epstein-Barr virus shuttle system. Mol. Cell. Biol. 1987, 7, 379–387. [Google Scholar] [CrossRef]
- Blight, K.J.; McKeating, J.A.; Rice, C.M. Highly permissive cell lines for subgenomic and genomic hepatitis C virus RNA replication. J. Virol. 2002, 76, 13001–13014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsetsarkin, K.; Higgs, S.; McGee, C.E.; De Lamballerie, X.; Charrel, R.N.; Vanlandingham, D.L. Infectious clones of Chikungunya virus (La Réunion isolate) for vector competence studies. Vector Borne Zoonotic Dis. 2006, 6, 325–337. [Google Scholar] [CrossRef]
- Vanlandingham, D.L.; Tsetsarkin, K.; Hong, C.; Klingler, K.; McElroy, K.L.; Lehane, M.J.; Higgs, S. Development and characterization of a double subgenomic chikungunya virus infectious clone to express heterologous genes in Aedes aegypti mosquitoes. Insect Biochem. Mol. Biol. 2005, 35, 1162–1170. [Google Scholar] [CrossRef]
- Levitt, N.H.; Ramsburg, H.H.; Hasty, S.E.; Repik, P.M.; Cole, F.E.; Lupton, H.W. Development of an attenuated strain of chikungunya virus for use in vaccine production. Vaccine 1986, 4, 157–162. [Google Scholar] [CrossRef]
- Jin, J.; Sherman, M.B.; Chafets, D.; Dinglasan, N.; Lu, K.; Lee, T.-H.; Carlson, L.-A.; Muench, M.O.; Simmons, G. An attenuated replication-competent chikungunya virus with a fluorescently tagged envelope. PLoS Negl. Trop. Dis. 2018, 12, e0006693. [Google Scholar] [CrossRef] [PubMed]
- Atasheva, S.; Krendelchtchikova, V.; Liopo, A.; Frolova, E.; Frolov, I. Interplay of acute and persistent infections caused by Venezuelan equine encephalitis virus encoding mutated capsid protein. J. Virol. 2010, 84, 10004–10015. [Google Scholar] [CrossRef] [Green Version]
- Bergqvist, J.; Forsman, O.; Larsson, P.; Näslund, J.; Lilja, T.; Engdahl, C.; Lindström, A.; Gylfe, Å.; Ahlm, C.; Evander, M.; et al. Detection and isolation of Sindbis virus from mosquitoes captured during an outbreak in Sweden, 2013. Vector Borne Zoonotic Dis. 2015, 15, 133–140. [Google Scholar] [CrossRef] [Green Version]
- Jaalouk, D.E.; Crosato, M.; Brodt, P.; Galipeau, J. Inhibition of histone deacetylation in 293GPG packaging cell line improves the production of self-inactivating MLV-derived retroviral vectors. Virol. J. 2006, 3, 27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palsson, B.; Andreadis, S. The physico-chemical factors that govern retrovirus-mediated gene transfer. Exp. Hematol. 1997, 25, 94–102. [Google Scholar]
- Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxicity assays. J. Immunol. Methods 1983, 65, 55–63. [Google Scholar] [CrossRef]
- Tinevez, J.-Y.; Perry, N.; Schindelin, J.; Hoopes, G.M.; Reynolds, G.D.; Laplantine, E.; Bednarek, S.Y.; Shorte, S.L.; Eliceiri, K.W. TrackMate: An open and extensible platform for single-particle tracking. Methods 2017, 115, 80–90. [Google Scholar] [CrossRef]
- Vega, A.R.; Freeman, S.A.; Grinstein, S.; Jaqaman, K. Multistep track segmentation and motion classification for transient mobility analysis. Biophys. J. 2018, 114, 1018–1025. [Google Scholar] [CrossRef]
- Alberione, M.P.; Moeller, R.; Kirui, J.; Ginkel, C.; Doepke, M.; Ströh, L.J.; Machtens, J.-P.; Pietschmann, T.; Gerold, G. Single-nucleotide variants in human CD81 influence hepatitis C virus infection of hepatoma cells. Med. Microbiol. Immunol. 2020, 209, 499–514. [Google Scholar] [CrossRef]
- Kuchroo, V.K.; Umetsu, D.T.; DeKruyff, R.H.; Freeman, G.J. The TIM gene family: Emerging roles in immunity and disease. Nat. Rev. Immunol. 2003, 3, 454–462. [Google Scholar] [CrossRef]
- Santiago, C.; Ballesteros, A.; Tami, C.; Martínez-Muñoz, L.; Kaplan, G.G.; Casasnovas, J.M. Structures of T Cell immunoglobulin mucin receptors 1 and 2 reveal mechanisms for regulation of immune responses by the TIM receptor family. Immunity 2007, 26, 299–310. [Google Scholar] [CrossRef] [Green Version]
- Kane, L.P. T cell Ig and mucin domain proteins and immunity. J. Immunol. 2010, 184, 2743–2749. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Jia, L.; Ma, Z.-H.; Ma, Q.-H.; Yang, X.-H.; Zhao, Y.-F. Axl glycosylation mediates tumor cell proliferation, invasion and lymphatic metastasis in murine hepatocellular carcinoma. World J. Gastroenterol. 2012, 18, 5369–5376. [Google Scholar] [CrossRef] [PubMed]
- Lauter, M.; Weber, A.; Torka, R. Targeting of the AXL receptor tyrosine kinase by small molecule inhibitor leads to AXL cell surface accumulation by impairing the ubiquitin-dependent receptor degradation. Cell Commun. Signal. 2019, 17, 59. [Google Scholar] [CrossRef] [Green Version]
- Brindley, M.A.; Hunt, C.L.; Kondratowicz, A.S.; Bowman, J.; Sinn, P.L.; McCray, P.B.; Quinn, K.; Weller, M.L.; Chiorini, J.A.; Maury, W. Tyrosine kinase receptor Axl enhances entry of Zaire ebolavirus without direct interactions with the viral glycoprotein. Virology 2011, 415, 83–94. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hunt, C.L.; Kolokoltsov, A.A.; Davey, R.A.; Maury, W. The Tyro3 receptor kinase Axl enhances macropinocytosis of Zaire ebolavirus. J. Virol. 2011, 85, 334–347. [Google Scholar] [CrossRef] [Green Version]
- Zapatero-Belinchón, F.J.; Dietzel, E.; Dolnik, O.; Döhner, K.; Costa, R.; Hertel, B.; Veselkova, B.; Kirui, J.; Klintworth, A.; Manns, M.P.; et al. Characterization of the Filovirus-Resistant Cell Line SH-SY5Y Reveals Redundant Role of Cell Surface Entry Factors. Viruses 2019, 11, 275. [Google Scholar] [CrossRef] [Green Version]
- Kao, F.T.; Puck, T.T. Genetics of somatic mammalian cells. IV. Properties of Chinese hamster cell mutants with respect to the requirement for proline. Genetics 1967, 55, 513–524. [Google Scholar] [CrossRef]
- Esko, J.D.; Stewart, T.E.; Taylor, W.H. Animal cell mutants defective in glycosaminoglycan biosynthesis. Proc. Natl. Acad. Sci. USA 1985, 82, 3197–3201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorchakov, R.; Wang, E.; Leal, G.; Forrester, N.L.; Plante, K.; Rossi, S.L.; Partidos, C.D.; Adams, A.P.; Seymour, R.L.; Weger, J.; et al. Attenuation of Chikungunya virus vaccine strain 181/clone 25 is determined by two amino acid substitutions in the E2 envelope glycoprotein. J. Virol. 2012, 86, 6084–6096. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohkuma, S.; Poole, B. Fluorescence probe measurement of the intralysosomal pH in living cells and the perturbation of pH by various agents. Proc. Natl. Acad. Sci. USA 1978, 75, 3327–3331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohkuma, S.; Poole, B. Cytoplasmic vacuolation of mouse peritoneal macrophages and the uptake into lysosomes of weakly basic substances. J. Cell Biol. 1981, 90, 656–664. [Google Scholar] [CrossRef] [PubMed]
- Fedeli, C.; Torriani, G.; Galan-Navarro, C.; Moraz, M.-L.; Moreno, H.; Gerold, G.; Kunz, S. Axl can serve as entry factor for lassa virus depending on the functional glycosylation of dystroglycan. J. Virol. 2018, 92, e01613–e01617. [Google Scholar] [CrossRef] [Green Version]
- Chua, H.H.; Abdul Rashid, K.; Law, W.C.; Hamizah, A.; Chem, Y.K.; Khairul, A.H.; Chua, K.B. A fatal case of chikungunya virus infection with liver involvement. Med. J. Malays. 2010, 65, 83–84. [Google Scholar]
- Matusali, G.; Colavita, F.; Bordi, L.; Lalle, E.; Ippolito, G.; Capobianchi, M.R.; Castilletti, C. Tropism of the chikungunya virus. Viruses 2019, 11, 175. [Google Scholar] [CrossRef] [Green Version]
- Bhattacharyya, S.; Zagórska, A.; Lew, E.D.; Shrestha, B.; Rothlin, C.V.; Naughton, J.; Diamond, M.S.; Lemke, G.; Young, J.A.T. Enveloped viruses disable innate immune responses in dendritic cells by direct activation of TAM receptors. Cell Host Microbe 2013, 14, 136–147. [Google Scholar] [CrossRef] [Green Version]
- Morizono, K.; Xie, Y.; Olafsen, T.; Lee, B.; Dasgupta, A.; Wu, A.M.; Chen, I.S.Y. The soluble serum protein Gas6 bridges virion envelope phosphatidylserine to the TAM receptor tyrosine kinase Axl to mediate viral entry. Cell Host Microbe 2011, 9, 286–298. [Google Scholar] [CrossRef] [Green Version]
- Morizono, K.; Chen, I.S.Y. Role of phosphatidylserine receptors in enveloped virus infection. J. Virol. 2014, 88, 4275–4290. [Google Scholar] [CrossRef] [Green Version]
- Freeman, G.J.; Casasnovas, J.M.; Umetsu, D.T.; DeKruyff, R.H. TIM genes: A family of cell surface phosphatidylserine receptors that regulate innate and adaptive immunity. Immunol. Rev. 2010, 235, 172–189. [Google Scholar] [CrossRef] [Green Version]
- Bishop, N.E.; Anderson, D.A. Early interactions of hepatitis A virus with cultured cells: Viral elution and the effect of pH and calcium ions. Arch. Virol. 1997, 142, 2161–2178. [Google Scholar] [CrossRef] [PubMed]
- Kondratowicz, A.S.; Lennemann, N.J.; Sinn, P.L.; Davey, R.A.; Hunt, C.L.; Moller-Tank, S.; Meyerholz, D.K.; Rennert, P.; Mullins, R.F.; Brindley, M.; et al. T-cell immunoglobulin and mucin domain 1 (TIM-1) is a receptor for Zaire Ebolavirus and Lake Victoria Marburgvirus. Proc. Natl. Acad. Sci. USA 2011, 108, 8426–8431. [Google Scholar] [CrossRef] [Green Version]
- Yuan, S.; Cao, L.; Ling, H.; Dang, M.; Sun, Y.; Zhang, X. TIM-1 acts a dual-attachment receptor for ebolavirus by interacting directly with viral GP and the PS on the viral envelope. Protein Cell 2015, 6, 814–824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, D.-H.; Garcia, G.; Situ, K.; Chua, B.A.; Hong, M.L.O.; Do, E.A.; Ramirez, C.M.; Harui, A.; Arumugaswami, V.; Morizono, K. Development of a blocker of the universal phosphatidylserine- and phosphatidylethanolamine-dependent viral entry pathways. Virology 2021, 560, 17–33. [Google Scholar] [CrossRef]
- Shih, S.C.; Sloper-Mould, K.E.; Hicke, L. Monoubiquitin carries a novel internalization signal that is appended to activated receptors. EMBO J. 2000, 19, 187–198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, C.H.R.; Mohamed Hussain, K.; Chu, J.J.H. Macropinocytosis dependent entry of Chikungunya virus into human muscle cells. PLoS Negl. Trop. Dis. 2019, 13, e0007610. [Google Scholar] [CrossRef]
- Marsh, M.; Kielian, M.C.; Helenius, A. Semliki forest virus entry and the endocytic pathway. Biochem. Soc. Trans. 1984, 12, 981–983. [Google Scholar] [CrossRef] [Green Version]
- Kielian, M.; Chanel-Vos, C.; Liao, M. Alphavirus Entry and Membrane Fusion. Viruses 2010, 2, 796–825. [Google Scholar] [CrossRef] [Green Version]
- Pirtle, E.C.; Beran, G.W. Virus survival in the environment. Rev. Sci. Tech. 1991, 10, 733–748. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Ablan, S.D.; Miao, C.; Zheng, Y.-M.; Fuller, M.S.; Rennert, P.D.; Maury, W.; Johnson, M.C.; Freed, E.O.; Liu, S.-L. TIM-family proteins inhibit HIV-1 release. Proc. Natl. Acad. Sci. USA 2014, 111, E3699–E3707. [Google Scholar] [CrossRef] [Green Version]
- Amitai, A.; Chakraborty, A.K.; Kardar, M. The low spike density of HIV may have evolved because of the effects of T helper cell depletion on affinity maturation. PLoS Comput. Biol. 2018, 14, e1006408. [Google Scholar] [CrossRef]
- Bernard, E.; Hamel, R.; Neyret, A.; Ekchariyawat, P.; Molès, J.-P.; Simmons, G.; Chazal, N.; Desprès, P.; Missé, D.; Briant, L. Human keratinocytes restrict chikungunya virus replication at a post-fusion step. Virology 2015, 476, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Qiao, L.; Hou, Z.; Luo, G. TIM-1 Promotes Hepatitis C Virus Cell Attachment and Infection. J. Virol. 2017, 91, e01583–e01616. [Google Scholar] [CrossRef] [Green Version]
- Bailly, V.; Zhang, Z.; Meier, W.; Cate, R.; Sanicola, M.; Bonventre, J.V. Shedding of kidney injury molecule-1, a putative adhesion protein involved in renal regeneration. J. Biol. Chem. 2002, 277, 39739–39748. [Google Scholar] [CrossRef] [Green Version]
- Aquino, R.S.; Park, P.W. Glycosaminoglycans and infection. Front. Biosci. (Landmark Ed.) 2016, 21, 1260–1277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.; Yang, Y.-F.; Yang, Y.; Zou, P.; Chen, J.; He, Y.; Shui, S.-L.; Cui, Y.-R.; Bai, R.; Liang, Y.-J.; et al. AXL promotes Zika virus infection in astrocytes by antagonizing type I interferon signalling. Nat. Microbiol. 2018, 3, 302–309. [Google Scholar] [CrossRef] [PubMed]
- Meertens, L.; Labeau, A.; Dejarnac, O.; Cipriani, S.; Sinigaglia, L.; Bonnet-Madin, L.; Le Charpentier, T.; Hafirassou, M.L.; Zamborlini, A.; Cao-Lormeau, V.-M.; et al. Axl mediates ZIKA virus entry in human glial cells and modulates innate immune responses. Cell Rep. 2017, 18, 324–333. [Google Scholar] [CrossRef]
- Sasaki, T.; Knyazev, P.G.; Clout, N.J.; Cheburkin, Y.; Göhring, W.; Ullrich, A.; Timpl, R.; Hohenester, E. Structural basis for Gas6-Axl signalling. EMBO J. 2006, 25, 80–87. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.-Y.; Wang, Z.; Zhen, Z.-D.; Feng, K.-H.; Guo, J.; Gao, N.; Fan, D.-Y.; Han, D.-S.; Wang, P.-G.; An, J. Axl is not an indispensable factor for Zika virus infection in mice. J. Gen. Virol. 2017, 98, 2061–2068. [Google Scholar] [CrossRef]
- Strange, D.P.; Jiyarom, B.; Pourhabibi Zarandi, N.; Xie, X.; Baker, C.; Sadri-Ardekani, H.; Shi, P.-Y.; Verma, S. Axl promotes zika virus entry and modulates the antiviral state of human sertoli cells. MBio 2019, 10, e01372–e01419. [Google Scholar] [CrossRef] [Green Version]
- Khan, M.; Dhanwani, R.; Patro, I.K.; Rao, P.V.L.; Parida, M.M. Cellular IMPDH enzyme activity is a potential target for the inhibition of Chikungunya virus replication and virus induced apoptosis in cultured mammalian cells. Antivir. Res. 2011, 89, 1–8. [Google Scholar] [CrossRef]
- Baer, A.; Lundberg, L.; Swales, D.; Waybright, N.; Pinkham, C.; Dinman, J.D.; Jacobs, J.L.; Kehn-Hall, K. Venezuelan Equine Encephalitis Virus Induces Apoptosis through the Unfolded Protein Response Activation of EGR1. J. Virol. 2016, 90, 3558–3572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhanwani, R.; Khan, M.; Bhaskar, A.S.B.; Singh, R.; Patro, I.K.; Rao, P.V.L.; Parida, M.M. Characterization of Chikungunya virus infection in human neuroblastoma SH-SY5Y cells: Role of apoptosis in neuronal cell death. Virus Res. 2012, 163, 563–572. [Google Scholar] [CrossRef]
- Nayak, T.K.; Mamidi, P.; Kumar, A.; Singh, L.P.K.; Sahoo, S.S.; Chattopadhyay, S.; Chattopadhyay, S. Regulation of Viral Replication, Apoptosis and Pro-Inflammatory Responses by 17-AAG during Chikungunya Virus Infection in Macrophages. Viruses 2017, 9, 3. [Google Scholar] [CrossRef] [PubMed]
- Krejbich-Trotot, P.; Denizot, M.; Hoarau, J.-J.; Jaffar-Bandjee, M.-C.; Das, T.; Gasque, P. Chikungunya virus mobilizes the apoptotic machinery to invade host cell defenses. FASEB J. 2011, 25, 314–325. [Google Scholar] [CrossRef]
- Pialoux, G.; Gaüzère, B.-A.; Jauréguiberry, S.; Strobel, M. Chikungunya, an epidemic arbovirosis. Lancet Infect. Dis. 2007, 7, 319–327. [Google Scholar] [CrossRef]
- Meyers, J.H.; Chakravarti, S.; Schlesinger, D.; Illes, Z.; Waldner, H.; Umetsu, S.E.; Kenny, J.; Zheng, X.X.; Umetsu, D.T.; DeKruyff, R.H.; et al. TIM-4 is the ligand for TIM-1, and the TIM-1-TIM-4 interaction regulates T cell proliferation. Nat. Immunol. 2005, 6, 455–464. [Google Scholar] [CrossRef]
- Umetsu, S.E.; Lee, W.-L.; McIntire, J.J.; Downey, L.; Sanjanwala, B.; Akbari, O.; Berry, G.J.; Nagumo, H.; Freeman, G.J.; Umetsu, D.T.; et al. TIM-1 induces T cell activation and inhibits the development of peripheral tolerance. Nat. Immunol. 2005, 6, 447–454. [Google Scholar] [CrossRef]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kirui, J.; Abidine, Y.; Lenman, A.; Islam, K.; Gwon, Y.-D.; Lasswitz, L.; Evander, M.; Bally, M.; Gerold, G. The Phosphatidylserine Receptor TIM-1 Enhances Authentic Chikungunya Virus Cell Entry. Cells 2021, 10, 1828. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10071828
Kirui J, Abidine Y, Lenman A, Islam K, Gwon Y-D, Lasswitz L, Evander M, Bally M, Gerold G. The Phosphatidylserine Receptor TIM-1 Enhances Authentic Chikungunya Virus Cell Entry. Cells. 2021; 10(7):1828. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10071828
Chicago/Turabian StyleKirui, Jared, Yara Abidine, Annasara Lenman, Koushikul Islam, Yong-Dae Gwon, Lisa Lasswitz, Magnus Evander, Marta Bally, and Gisa Gerold. 2021. "The Phosphatidylserine Receptor TIM-1 Enhances Authentic Chikungunya Virus Cell Entry" Cells 10, no. 7: 1828. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10071828