
Snail Upregulates Transcription of FN, LEF, COX2, and COL1A1 in Hepatocellular Carcinoma: A General Model Established for Snail to Transactivate Mesenchymal Genes

 ,
,  , and
, and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture
2.2. Chemicals and Antibody
2.3. Constructions of Various Promoter Plasmids for Deletion Mapping
2.4. Site-Directed Mutagenesis on Promoters
2.5. Transwell and Wound Healing Migration Assays
2.6. Dual-Luciferase Promoter Assay
2.7. Chromatin Immunoprecipitation (ChIP) Assay
2.8. RT-PCR and Quantitative RT-PCR
2.9. Electrophoresis Mobility Shift Assay (EMSA)
2.10. Western Blot
2.11. shRNA Technology
2.12. Statistical Analysis
3. Results
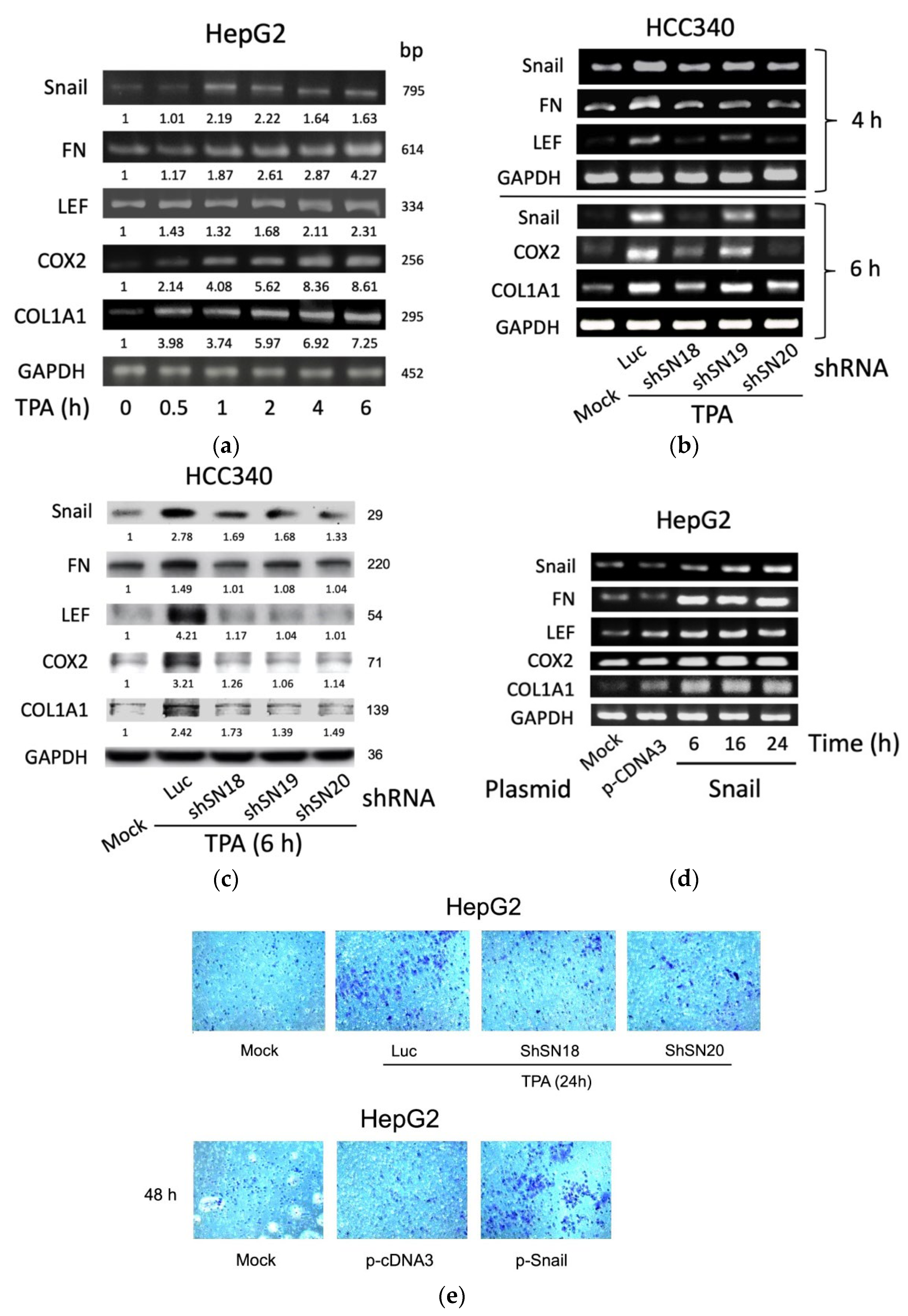
3.1. SNA Is Essential for Constitutive and TPA-Induced Gene Expression of FN, LEF, COX2, and COL1A1 in HepG2 and HCC340 Cells
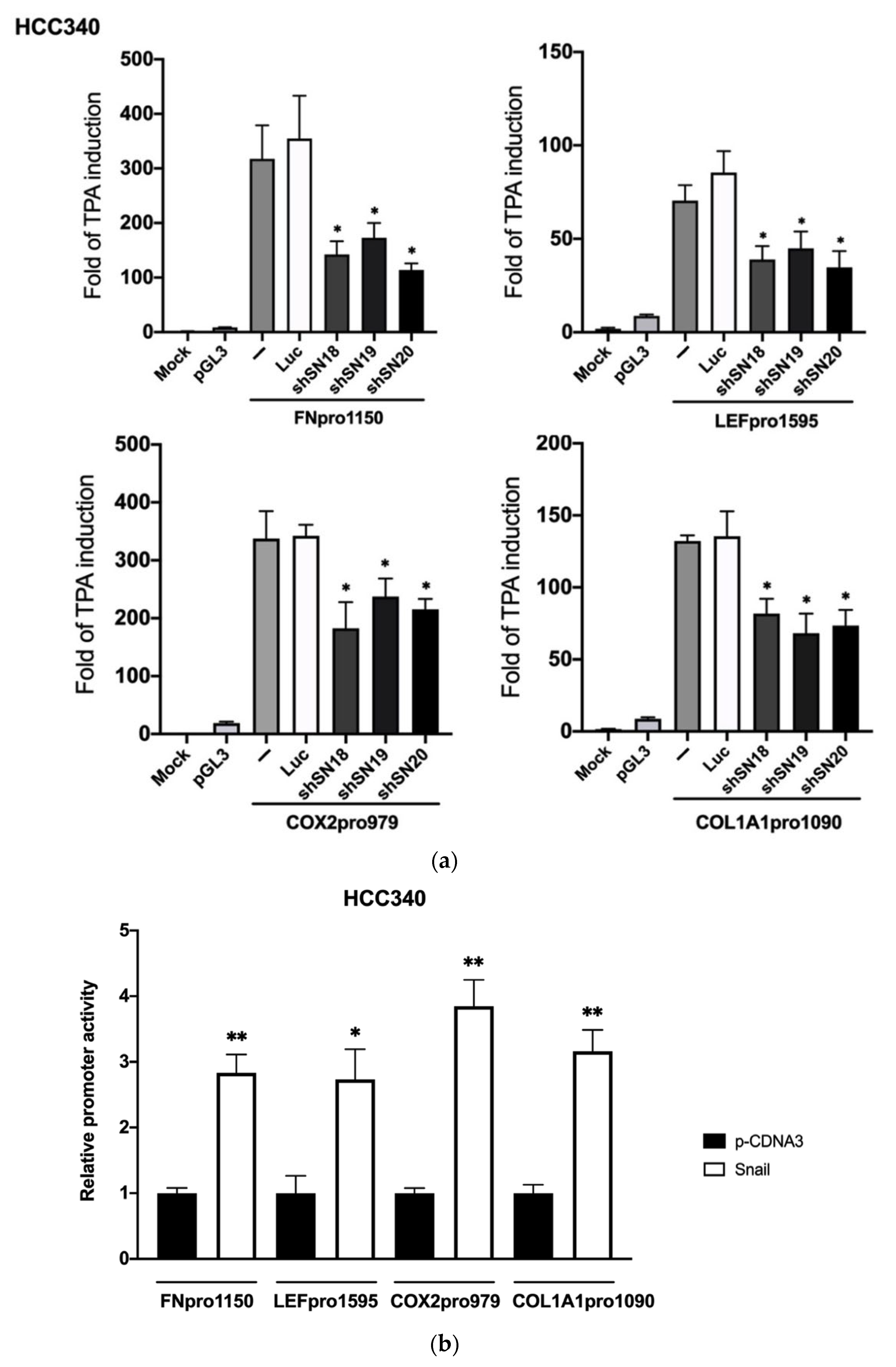
3.2. SNA Is Essential for Constitutive and TPA-Induced Transcriptional Activation of FN, LEF, COX2, and COL1A1 in HCC
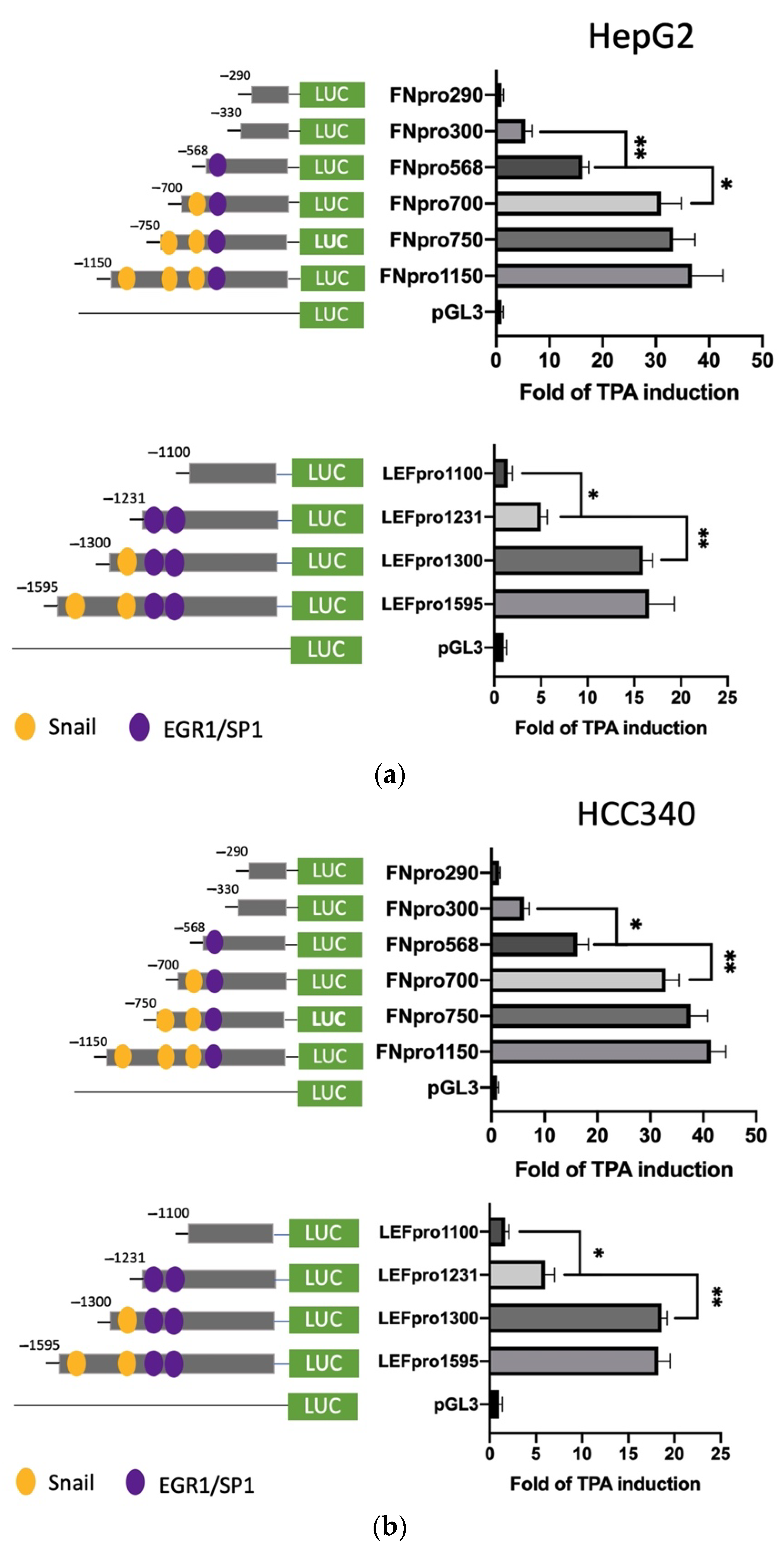
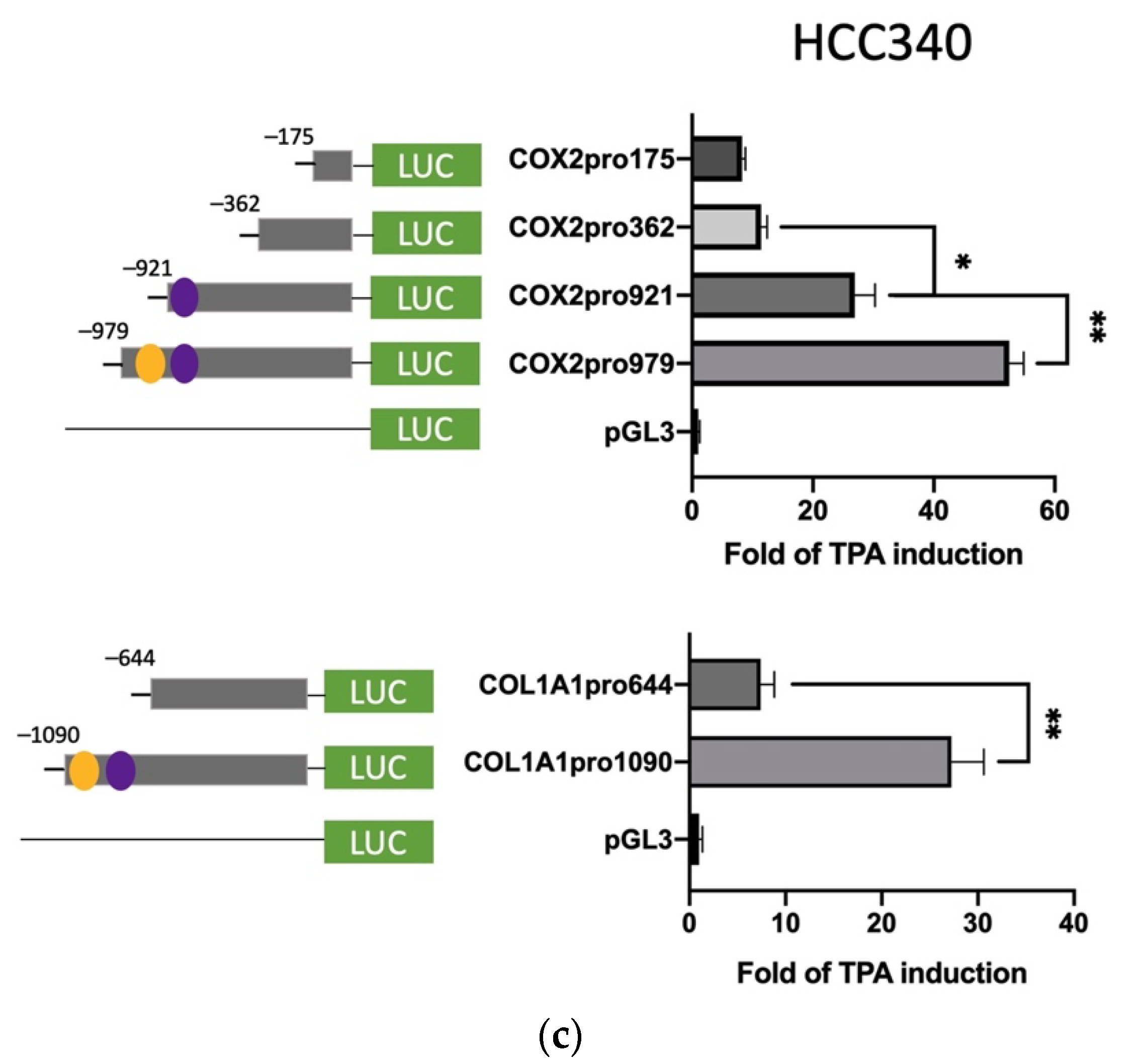
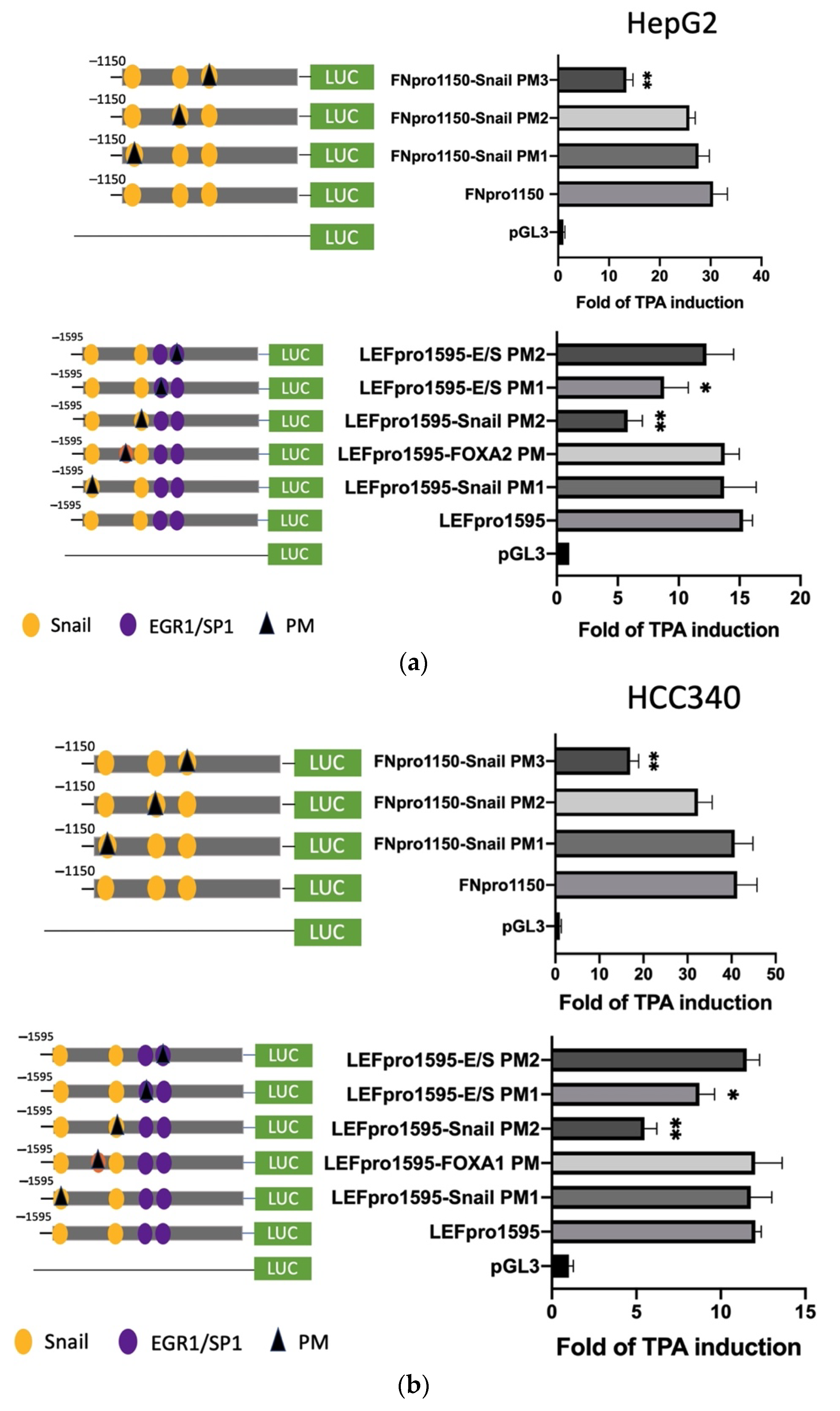
3.3. Deletion Mapping Identified SNA Motif and EGR1/SP1 Overlapping Regions within TPA-Response Element on FN, LEF, COX2, and COL1A1 Promoters
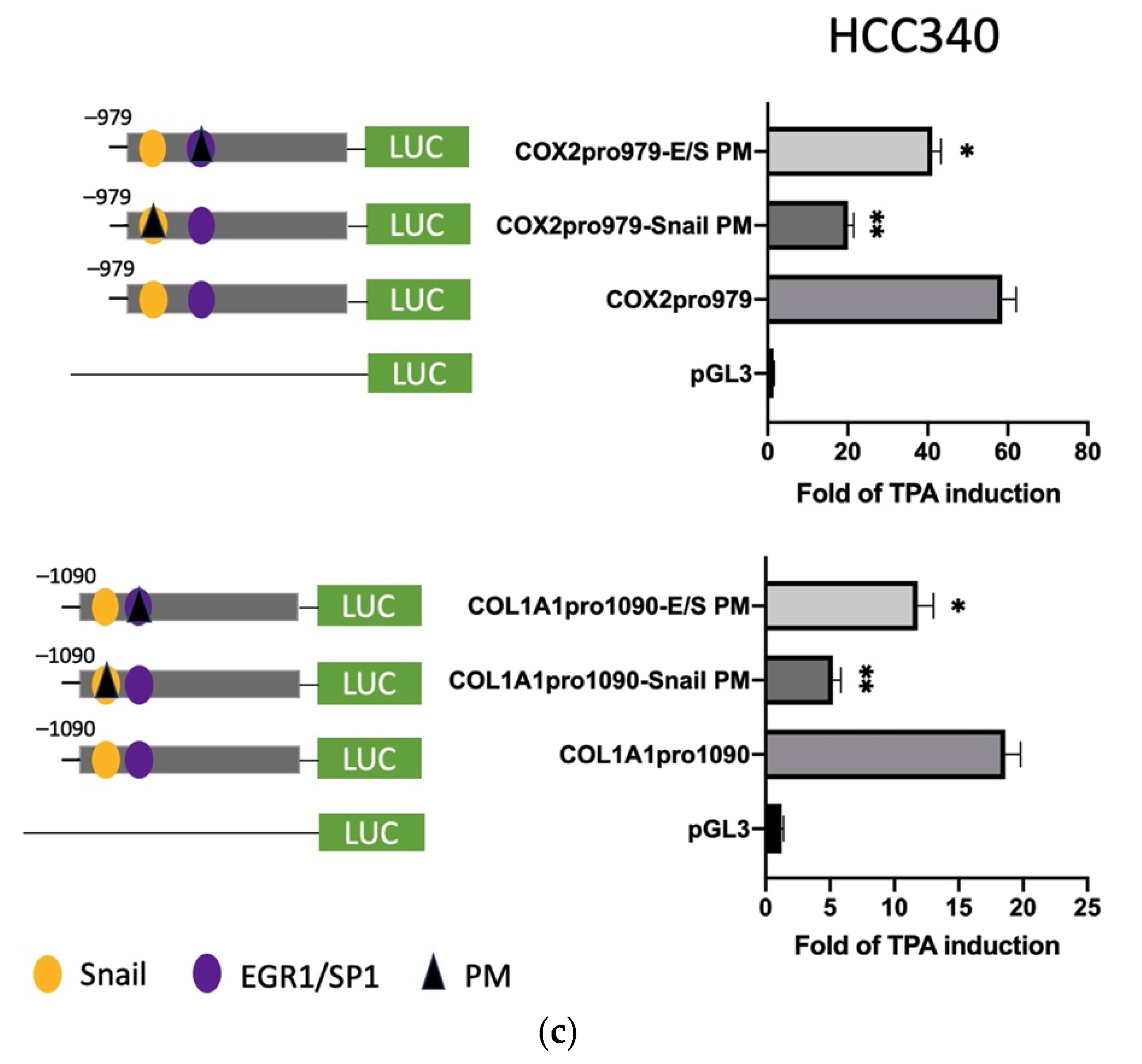
3.4. Mutagenesis on Promoter Validated SNA Binding Motif and E/S Overlapping Region for Transcription of FN, LEF, COX2, and COL1A1
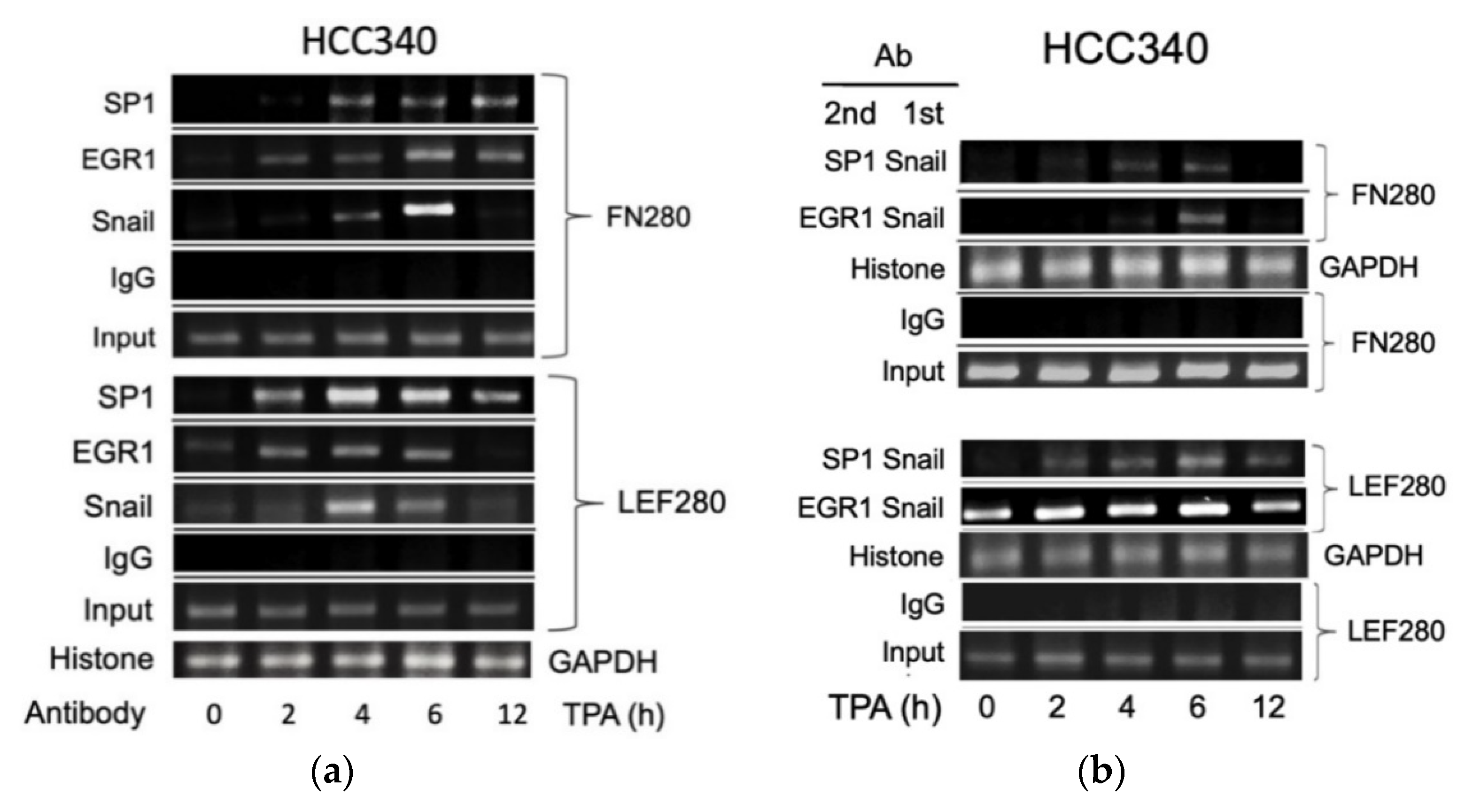
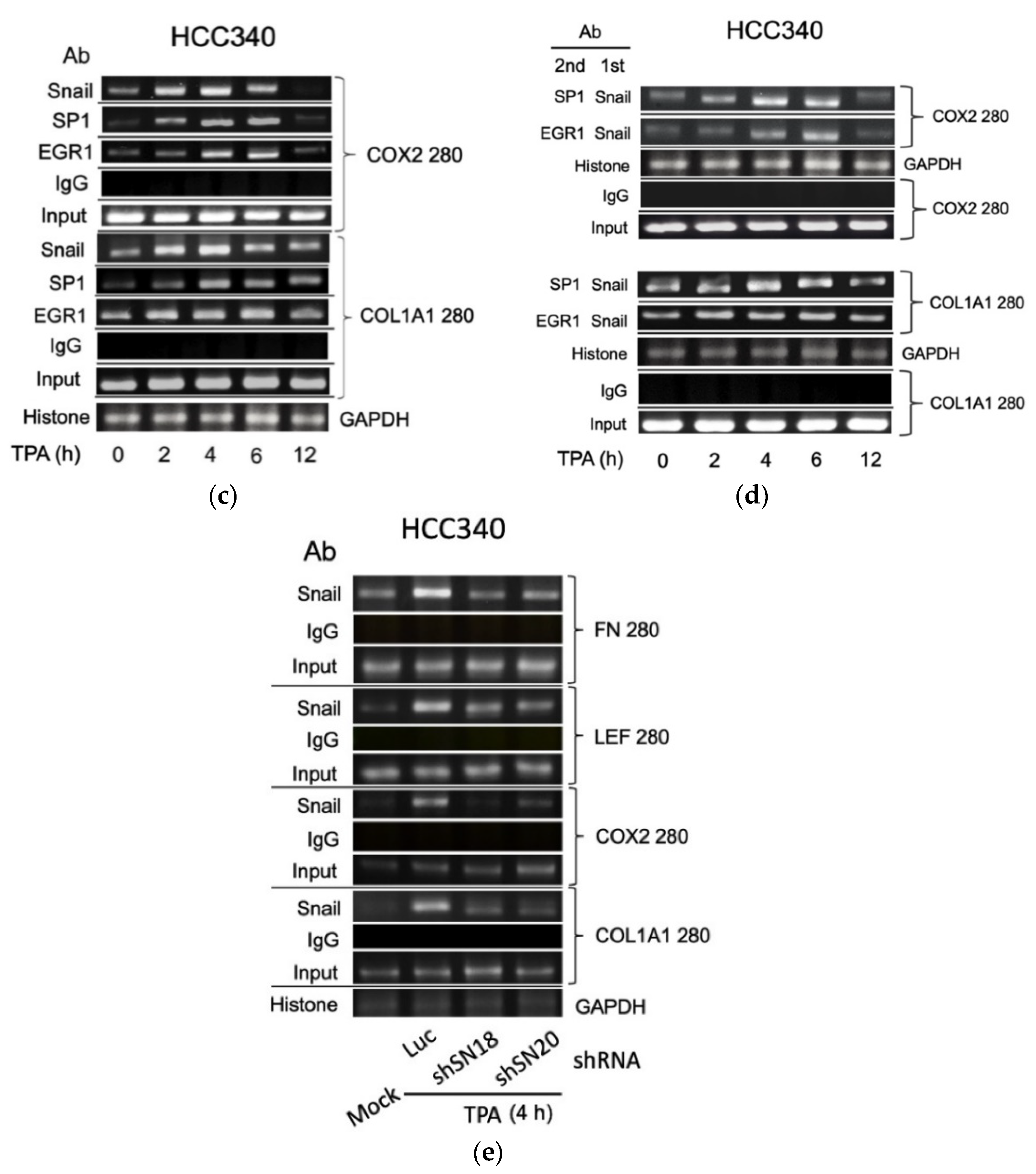
3.5. ChIP Validated the Binding of Key Transcription Factors on Putative Regions In Vivo
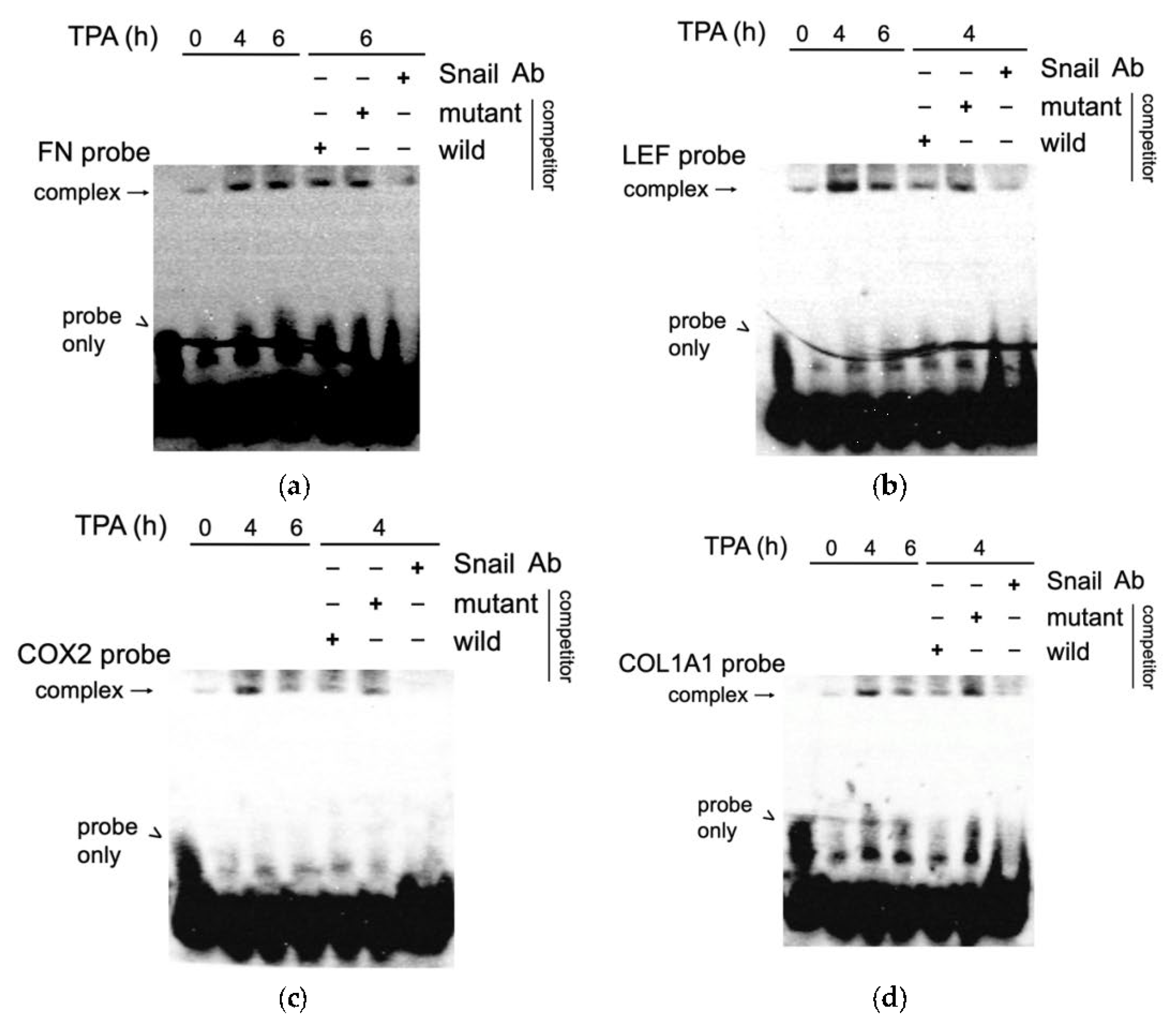
3.6. EMSA Assay Validated the Binding of SNA on Putative Regions of FN, LEF, COX2, and COL1A1 Promoter In Vitro
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
References
- Gupta, G.P.; Massagué, J. Cancer metastasis: Building a framework. Cell 2006, 127, 679–695. [Google Scholar] [CrossRef] [Green Version]
- Chen, D.; Zheng, X.; Jiao, X.; Gao, Y.; Zhang, K.; Liang, J. Transcriptional repressor snail and metastasis in hepatocellular carcinoma. Hepato Gastroenterol. 2012, 59, 1359–1365. [Google Scholar] [CrossRef]
- Jiao, W.; Miyazaki, K.; Kitajima, Y. Inverse correlation between E-cadherin and Snail expression in hepatocellular carcinoma cell lines in vitro and in vivo. Br. J. Cancer 2002, 86, 98–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, C.P.; Yu, S.; Shi, L.; Wang, S.; Li, Z.X.; Wang, Y.H.; Liang, J. FoxM1 promotes epithelial-mesenchymal transition of hepatocellular carcinoma by targeting Snai1. Mol. Med. Rep. 2017, 16, 5181–5188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, M.; Dong, X.; Zhang, D.; Chen, X.; Zhu, X. High expression of Snail and NF-κB predicts poor survival in Chinese hepatocellular carcinoma patients. Oncotarget 2017, 8, 4543. [Google Scholar] [CrossRef] [PubMed]
- Deng, G.; Zeng, S.; Ma, J.; Zhang, Y.; Qu, Y.; Han, Y.; Shen, H. The anti-tumor activities of Neferine on cell invasion and oxaliplatin sensitivity regulated by EMT via Snail signaling in hepatocellular carcinoma. Sci. Rep. 2017, 7, 41616. [Google Scholar] [CrossRef] [Green Version]
- Berghoff, A.S.; Ricken, G.; Widhalm, G.; Rajky, O.; Dieckmann, K.; Birner, P.; Preusser, M. Tumour-infiltrating lymphocytes and expression of programmed death ligand 1 (PD-L1) in melanoma brain metastases. Histopathology 2015, 66, 289–299. [Google Scholar] [CrossRef] [PubMed]
- Tang, Y.; Lv, P.; Sun, Z.; Han, L.; Zhou, W. 14-3-3β Promotes Migration and Invasion of Human Hepatocellular Carcinoma Cells by Modulating Expression of MMP2 and MMP9 through PI3K/Akt/NF-κB Pathway. PLoS ONE 2016, 11, e0146070. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Yang, P.; Yang, J.; Wen, Z.; Zhang, B.; Zheng, X. GIT1 is a novel prognostic biomarker and facilitates tumor progression via activating ERK/MMP9 signaling in hepatocellular carcinoma. Oncotargets Ther. 2015, 8, 3731. [Google Scholar] [CrossRef] [Green Version]
- Yan, X.L.; Jia, Y.L.; Chen, L.; Zeng, Q.; Zhou, J.N.; Fu, C.J.; Chen, H.X.; Yuan, H.F.; Li, Z.W.; Shi, L.; et al. Hepatocellular carcinoma-associated mesenchymal stem cells promote hepatocarcinoma progression: Role of the S100A4-miR155-SOCS1-MMP9 axis. Hepatology 2013, 57, 2274–2286. [Google Scholar] [CrossRef]
- Hashiguchi, M.; Ueno, S.; Sakoda, M.; Iino, S.; Hiwatashi, K.; Minami, K.; Ando, K.; Mataki, Y.; Maemura, K.; Shinchi, H.; et al. Clinical implication of ZEB-1 and E-cadherin expression in hepatocellular carcinoma (HCC). BMC Cancer 2013, 13, 572. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, T.A.; Jan, Y.J.; Ko, B.S.; Liang, S.M.; Chen, S.C.; Wang, J.; Hsu, C.; Wu, Y.M.; Liou, J.Y. 14-3-3ε overexpression contributes to epithelial-mesenchymal transition of hepatocellular carcinoma. PLoS ONE 2013, 8, e57968. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Herranz, N.; Pasini, D.; Díaz, V.M.; Francí, C.; Gutierrez, A.; Dave, N.; Escrivà, M.; Hernandez-Muñoz, I.; Di Croce, L.; Helin, K.; et al. Polycomb complex 2 is required for E-cadherin repression by the Snail1 transcription factor. Mol. Cell. Biol. 2008, 28, 4772–4781. [Google Scholar] [CrossRef] [Green Version]
- Tong, Z.T.; Cai, M.Y.; Wang, X.G.; Kong, L.L.; Mai, S.J.; Liu, Y.H.; Zhang, H.B.; Liao, Y.J.; Zheng, F.; Zhu, W.; et al. EZH2 supports nasopharyngeal carcinoma cell aggressiveness by forming a co-repressor complex with HDAC1/HDAC2 and Snail to inhibit E-cadherin. Oncogene 2012, 31, 583–594. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jorda, M.; Olmeda, D.; Vinyals, A.; Valero, E.; Cubillo, E.; Llorens, A.; Cano, A.; Fabra, A. Upregulation of MMP-9 in MDCK epithelial cell line in response to expression of the Snail transcription factor. J. Cell Sci. 2005, 118, 3371–3385. [Google Scholar] [CrossRef] [Green Version]
- Dave, N.; Guaita-Esteruelas, S.; Gutarra, S.; Frias, A.; Beltran, M.; Peiro, S.; de Herreros, A.G. Functional cooperation between Snail1 and twist in the regulation of ZEB1 expression during epithelial to mesenchymal transition. J. Biol. Chem. 2011, 286, 12024–12032. [Google Scholar] [CrossRef] [Green Version]
- Wu, W.S.; You, R.I.; Cheng, C.C.; Lee, M.C.; Lin, T.Y.; Hu, C.T. Snail collaborates with EGR-1 and SP-1 to directly activate transcription of MMP 9 and ZEB1. Sci. Rep. 2017, 7, 17753. [Google Scholar] [CrossRef]
- Hu, C.T.; Chang, T.Y.; Cheng, C.C.; Liu, C.S.; Wu, J.R.; Li, M.C.; Wu, W.S. Snail associates with EGR-1 and SP-1 to upregulate transcriptional activation of p15INK4b. FEBS J. 2010, 277, 1202–1218. [Google Scholar] [CrossRef]
- Kaufhold, S.; Bonavida, B. Central role of Snail1 in the regulation of EMT and resistance in cancer: A target for therapeutic intervention. J. Exp. Clin. Cancer Res. 2014, 33, 62. [Google Scholar] [CrossRef] [PubMed]
- Schmitt-Graeff, A.; Ertelt-Heitzmann, V.; Allgaier, H.P.; Olschewski, M.; Nitschke, R.; Haxelmans, S.; Koelble, K.; Behrens, J.; Blum, H.E. Coordinated expression of cyclin D1 and LEF-1/TCF transcription factor is restricted to a subset of hepatocellular carcinoma. Liver Int. 2005, 25, 839–847. [Google Scholar] [CrossRef]
- Chen, Y.; Chen, H.N.; Wang, K.; Zhang, L.; Huang, Z.; Liu, J.; Zhang, Z.; Luo, M.; Lei, Y.; Peng, Y.; et al. Ketoconazole exacerbates mitophagy to induce apoptosis by downregulating cyclooxygenase-2 in hepatocellular carcinoma. J. Hepatol. 2019, 70, 66–77. [Google Scholar] [CrossRef]
- Fishbein, A.; Wang, W.; Yang, H.; Yang, J.; Hallisey, V.M.; Deng, J.; Verheul, S.M.L.; Hwang, S.H.; Gartung, A.; Wang, Y.; et al. Resolution of eicosanoid/cytokine storm prevents carcinogen and inflammation-initiated hepatocellular cancer progression. Proc. Natl. Acad. Sci. USA 2020, 117, 21576–21587. [Google Scholar] [CrossRef]
- Yang, Y.; Zhu, J.; Gou, H.; Cao, D.; Jiang, M.; Hou, M. Clinical significance of Cox-2, Survivin and Bcl-2 expression in hepatocellular carcinoma (HCC). Med. Oncol. 2011, 28, 796–803. [Google Scholar] [CrossRef]
- Zhao, W.; Jiang, X.; Yang, S. lncRNA TUG1 Promotes Cell Proliferation, Migration, and Invasion in Hepatocellular Carcinoma via Regulating miR-29c-3p/COL1A1 Axis. Cancer Manag. Res. 2020, 12, 6837–6847. [Google Scholar] [CrossRef] [PubMed]
- Ma, H.P.; Chang, H.L.; Bamodu, O.A.; Yadav, V.K.; Huang, T.Y.; Wu, A.T.H.; Yeh, C.T.; Tsai, S.H.; Lee, W.H. Collagen 1A1 (COL1A1) Is a Reliable Biomarker and Putative Therapeutic Target for Hepatocellular Carcinogenesis and Metastasis. Cancers 2019, 11, 786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, Y.; Kim, S.H.; Kim, K.M.; Choi, E.K.; Kim, J.; Seo, H.R. Activated hepatic stellate cells play pivotal roles in hepatocellular carcinoma cell chemoresistance and migration in multicellular tumor spheroids. Sci. Rep. 2016, 6, 36750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, J.R.; Hu, C.T.; You, R.I.; Pan, S.M.; Cheng, C.C.; Lee, M.C.; Wu, C.C.; Chang, Y.J.; Lin, S.C.; Chen, C.S.; et al. Hydrogen peroxide inducible clone-5 mediates reactive oxygen species signaling for hepatocellular carcinoma progression. Oncotarget 2015, 6, 32526. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lyons, J.G.; Patel, V.; Roue, N.C.; Fok, S.Y.; Soon, L.L.; Halliday, G.M.; Gutkind, J.S. Snail up-regulates proinflammatory mediators and inhibits differentiation in oral keratinocytes. Cancer Res. 2008, 68, 4525–4530. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Skrzypek, K.; Majka, M. Interplay among SNAIL Transcription Factor, MicroRNAs, Long Non-Coding RNAs, and Circular RNAs in the Regulation of Tumor Growth and Metastasis. Cancers 2020, 12, 209. [Google Scholar] [CrossRef] [Green Version]
- Medici, D.; Hay, E.D.; Goodenough, D.A. Cooperation between snail and LEF-1 transcription factors is essential for TGF-beta1-induced epithelial-mesenchymal transition. Mol. Biol. Cell 2006, 17, 1871–1879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Li, M.; Xu, D.; Zhao, C.; Liu, G.; Wang, F. Overexpression of Snail in retinal pigment epithelial triggered epithelial-mesenchymal transition. Biochem. Biophys. Res. Commun. 2014, 446, 347–351. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.G.; Jung, E.; Heur, M. Fibroblast growth factor 2 induces proliferation and fibrosis via SNAI1-mediated activation of CDK2 and ZEB1 in corneal endothelium. J. Biol. Chem. 2018, 293, 3758–3769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Okumura, N.; Minamiyama, R.; Ho, L.T.; Kay, E.P.; Kawasaki, S.; Tourtas, T.; Schlötzer-Schrehardt, U.; Kruse, F.E.; Young, R.D.; Quantock, A.J.; et al. Involvement of ZEB1 and Snail1 in excessive production of extracellular matrix in Fuchs endothelial corneal dystrophy. Lab. Investig. 2015, 95, 1291–1304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shields, M.A.; Ebine, K.; Sahai, V.; Kumar, K.; Siddiqui, K.; Hwang, R.F.; Grippo, P.J.; Munshi, H.G. Snail cooperates with KrasG12D to promote pancreatic fibrosis. Mol. Cancer Res. 2013, 11, 1078–1087. [Google Scholar] [CrossRef] [Green Version]
- Stanisavljevic, J.; Porta-de-la-Riva, M.; Batlle, R.; de Herreros, A.G.; Baulida, J. The p65 subunit of NF-κB and PARP1 assist Snail1 in activating fibronectin transcription. J. Cell Sci. 2011, 124, 4161–4171. [Google Scholar] [CrossRef] [Green Version]
- Baritaki, S.; Chapman, A.; Yeung, K.; Spandidos, D.A.; Palladino, M.; Bonavida, B. Inhibition of epithelial to mesenchymal transition in metastatic prostate cancer cells by the novel proteasome inhibitor, NPI-0052: Pivotal roles of Snail repression and RKIP induction. Oncogene 2009, 28, 3573–3585. [Google Scholar] [CrossRef] [Green Version]
- Kwon, C.H.; Park, H.J.; Choi, J.H.; Lee, J.R.; Kim, H.K.; Jo, H.J.; Kim, H.S.; Oh, N.; Am Song, G.; Park, D.Y. Snail and serpinA1 promote tumor progression and predict prognosis in colorectal cancer. Oncotarget 2015, 6, 20312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, C.; Yao, J.; Mercola, D.; Adamson, E. The transcription factor EGR-1 directly transactivates the fibronectin gene and enhances attachment of human glioblastoma cell line U251. J. Biol. Chem. 2000, 275, 20315–30323. [Google Scholar] [CrossRef] [Green Version]
- Hu, J.; Shan, Z.; Hu, K.; Ren, F.; Zhang, W.; Han, M.; Li, Y.; Feng, K.; Lei, L.; Feng, Y. miRNA-223 inhibits epithelial-mesenchymal transition in gastric carcinoma cells via Sp1. Int. J. Oncol. 2016, 49, 325–335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Medici, D.; Hay, E.D.; Olsen, B.R. Snail and Slug promote epithelial-mesenchymal transition through beta-catenin-T-cell factor-4-dependent expression of transforming growth factor-beta3. Mol. Biol. Cell 2008, 19, 4875–4887. [Google Scholar] [CrossRef] [Green Version]
- Guaita, S.; Puig, I.; Francí, C.; Garrido, M.; Domínguez, D.; Batlle, E.; Sancho, E.; Dedhar, S.; de Herreros, A.G.; Baulida, J. Snail induction of epithelial to mesenchymal transition in tumor cells is accompanied by MUC1 repression and ZEB1 expression. J. Biol. Chem. 2002, 277, 39209–39216. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.H.; Liang, X.; Wang, T.S.; Liang, X.H.; Zuo, R.J.; Deng, W.B.; Zhang, Z.R.; Qin, F.N.; Zhao, Z.A.; Yang, Z.M. Heparin-binding epidermal growth factor-like growth factor (HB-EGF) induction on Snail expression during mouse decidualization. Mol. Cell. Endocrinol. 2013, 381, 272–279. [Google Scholar] [CrossRef] [PubMed]
- Osorio, L.A.; Farfán, N.M.; Castellón, E.A.; Contreras, H.R. SNAIL transcription factor increases the motility and invasive capacity of prostate cancer cells. Mol. Med. Rep. 2016, 13, 778–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Zhang, X.; Zehner, Z.E. c-Jun and the dominant-negative mutant, TAM67, induce vimentin gene expression by interacting with the activator Sp1. Oncogene 2003, 22, 8891–8901. [Google Scholar] [CrossRef] [Green Version]
- Zhao, J.; Geng, L.; Duan, G.; Xu, W.; Cheng, Y.; Huang, Z.; Zhou, Z.; Gong, S. REC8 inhibits EMT by downregulating EGR1 in gastric cancer cells. Oncol. Rep. 2018, 39, 1583–1590. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Assani, G.; Zhou, Y. Effect of modulation of epithelial-mesenchymal transition regulators Snail1 and Snail2 on cancer cell radiosensitivity by targeting of the cell cycle, cell apoptosis and cell migration/invasion. Oncol. Lett. 2019, 17, 23–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Škovierová, H.; Okajčeková, T.; Strnádel, J.; Vidomanová, E.; Halašová, E. Molecular regulation of epithelial-to-mesenchymal transition in tumorigenesis. Int. J. Mol. Med. 2018, 41, 1187–1200. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Liu, Y.; Lin, S.; Shang, J.; Liu, J.; Li, J.; Yuan, S.; Zhang, L. Early growth response gene-1 and hypoxia-inducible factor-1α affect tumor metastasis via regulation of tissue factor. Acta Oncol. 2013, 52, 842–851. [Google Scholar] [CrossRef]
- Peng, C.Y.; Liao, Y.W.; Lu, M.Y.; Yang, C.M.; Hsieh, P.L.; Yu, C.C. Positive Feedback Loop of SNAIL-IL-6 Mediates Myofibroblastic Differentiation Activity in Precancerous Oral Submucous Fibrosis. Cancers 2020, 12, 1611. [Google Scholar] [CrossRef]
- Pinto, M.T.; Melo, F.U.F.; Malta, T.M.; Rodrigues, E.S.; Placa, J.R.; Silva, W.A., Jr.; Panepucci, R.A.; Covas, D.T.; de Oliveira Rodrigues, C.; Kashima, S. Endothelial cells from different anatomical origin have distinct responses during SNAIL/TGF-β2-mediated endothelial-mesenchymal transition. Am. J. Transl. Res. 2018, 10, 4065. [Google Scholar]
- Gaut, L.; Robert, N.; Delalande, A.; Bonnin, M.A.; Pichon, C.; Duprez, D. EGR1 Regulates Transcription Downstream of Mechanical Signals during Tendon Formation and Healing. PLoS ONE 2016, 11, e0166237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duval, E.; Bouyoucef, M.; Leclercq, S.; Baugé, C.; Boumédiene, K. Hypoxia inducible factor 1 alpha down-regulates type i collagen through Sp3 transcription factor in human chondrocytes. IUBMB Life 2016, 68, 756–763. [Google Scholar] [CrossRef]
- Lee, S.W.; Won, J.Y.; Kim, W.J.; Lee, J.; Kim, K.H.; Youn, S.W.; Kim, J.Y.; Lee, E.J.; Kim, Y.J.; Kim, K.W.; et al. Snail as a potential target molecule in cardiac fibrosis: Paracrine action of endothelial cells on fibroblasts through snail and CTGF axis. Mol. Ther. 2013, 21, 1767–1777. [Google Scholar] [CrossRef] [Green Version]
- Li, H.Y.; Ju, D.; Zhang, D.W.; Li, H.; Kong, L.M.; Guo, Y.; Li, C.; Wang, X.L.; Chen, Z.N.; Bian, H. Activation of TGF-β1-CD147 positive feedback loop in hepatic stellate cells promotes liver fibrosis. Sci. Rep. 2015, 5, 16552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, X.; Yan, C.; Huang, Y.; Shi, D.; Fu, Z.; Qiu, J.; Yin, Y. Mouse double minute 2 (MDM2) upregulates Snail expression and induces epithelial-to-mesenchymal transition in breast cancer cells in vitro and in vivo. Oncotarget 2016, 7, 37177. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Liu, J.; Ying, X.; Lin, P.C.; Zhou, B.P. Twist-mediated Epithelial-mesenchymal Transition Promotes Breast Tumor Cell Invasion via Inhibition of Hippo Pathway. Sci. Rep. 2016, 6, 24606. [Google Scholar] [CrossRef] [Green Version]
- Peng, W.X.; Wan, Y.Y.; Gong, A.H.; Ge, L.; Jin, J.; Xu, M.; Wu, C.Y. Egr-1 regulates irradiation-induced autophagy through Atg4B to promote radioresistance in hepatocellular carcinoma cells. Oncogenesis 2017, 6, e292. [Google Scholar] [CrossRef] [Green Version]
- Lu, W.; Ma, Y.Y.; Shao, Q.Q.; Liang, J.; Qi, T.T.; Huang, Y.; Wang, Q.J. ROS/p53/miR-335-5p/Sp1 axis modulates the migration and epithelial to mesenchymal transition of JEG-3 cells. Mol. Med. Rep. 2020, 21, 1208–1216. [Google Scholar] [CrossRef]
- Zeng, Q.; Zhang, Y.; Zhang, W.; Guo, Q. Baicalein suppresses the proliferation and invasiveness of colorectal cancer cells by inhibiting Snail-induced epithelial-mesenchymal transition. Mol. Med. Rep. 2020, 21, 2544–2552. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer Sequence | Product Size |
|---|---|---|
| FN280 | F: 5′GGG AAG GGG GAG CGT CTT3′ | 280 bp |
| R: 5′CCC GCC CCA CCC CAC CCG3′ | ||
| LEF280 | F: 5′CTC GCC AAG TTG CCT GAT3′ | 280 bp |
| R: 5′CTC CCC ACT GCT TCT CCT3′ | ||
| COX2 280 | F: 5′GTC CAT CAG AAG GCA GGA AAC3′ | 280 bp |
| R: 5′CTA TAT GCA GCA CAT ACA TAC3′ | ||
| COL1A1 280 | F: 5′AGG GTC TCT AAG CAG CCC CTG3′ | 280 bp |
| R: 5′GAT GGA GTG GGG AGG CTG AGG3′ | ||
| GADPH | F: 5′TAC TAG CGG TTT TAC GGG CG3′ | 166 bp |
| R: 5′TCG AAC AGG AGG AGC AGA GAG CGA3′ |
| Gene | Primer Sequence | Product Size |
|---|---|---|
| Snail or | F: 5′AAGC TTCC ATGG CGCG CTCT TTCC TCGT CAGG AAGC CC3′ | 795 bp |
| R: 5′GGAT CCTC AGCG GGGA CATC CTGA GCAG CCGG ACTC TTG3′ | ||
| F: 5′TCCA GAGT TTAC CCTT CCAG CA3′ | 218 bp | |
| R: 5′CTTT CCCA CTGT CCTC ATCT G3′ | ||
| FN or | F: 5′GTGC CTGG GCAA CGCA3′ | 614 bp |
| R: 5′CCCG ACCC TGAC CGAA G3′ | ||
| F: 5′AAG GAG AAG ACC GGA CCA AT3′ | 314 bp | |
| R: 5′GGC TTG ATG GTT CTC TGG AT3′ | ||
| LEF | F: 5′TGG CAG CCC TAT TTC AGT TT3′ | 334 bp |
| R: 5′CAA AGG CTG TGC TTG CTT TT3′ | ||
| COX2 | F: 5′CGG TGA AAC TCT GGC TAG ACA G3′ | 256 bp |
| R: 5′GCA AAC CGT AGA TGC TCA GGG A3′ | ||
| COL1A1 | F: 5′TCT GCG ACA ACG GCA AGG TG3′ | 295 bp |
| R: 5′GAC GCC GGT GGT TTC TTG GT3′ | ||
| GADPH or | F: 5′ACC ACA GTC CAT GCC ATC AC3′ | 452 bp |
| R: 5′TCC ACC ACC CTG TTG CTG TA3′ | ||
| F: 5′ACGG ATTT GGTC GTAT TGGG3′ | 215 bp | |
| R: 5′TGAT TTTG GAGG GATC TCGC3′ |
| Biotin-Labeled Oligonucleotide Probe | ||
| FN | F: FN-585~-560 | 5′CCCCT TCGCT TCACA CAAGT CCAGC3′ |
| R: FN-585~-560 | 5′GCTGG ACTTG TGTGA AGCGA AGGGG3′ | |
| LEF | F: LEF-1297~-1272 | 5′CACAC CACAC TCACA CACCC CAAAA3′ |
| R: LEF-1297~-1272 | 5′TTTTG GGGTG TGTGA GTGTG GTGTG3′ | |
| COX2 | F: COX2-975~-950 | 5′CCCGT GGAGC TCACA TTAAC TATTT3′ |
| R: COX2-975~-950 | 5′AAATA GTTAA TGTGA GCTCC ACGGG3′ | |
| COL1A1 | F: COL1A1-1080~-1055 | 5′TCACC AATGA TCACA GGCCT CCCAC3′ |
| R: COL1A1-1080~-1055 | 5′GTGGG AGGCC TGTGA TCATT GGTGA3′ | |
| Unlabeled Oligonucleotide Competition Probe | ||
| FN | F: FN-Snail wild | 5′CCT TCGCT TCACA CAAGT CC3′ |
| R: FN-Snail wild | 5′GG ACTTG TGTGA AGCGA AGG3′ | |
| F: FN-Snail mutant | 5′CCT TCGCT TTGTA CAAGT CC3′ | |
| R: FN-Snail mutant | 5′GG ACTTG TACAA AGCGA AGG3′ | |
| LEF | F: LEF-Snail wild | 5′CAC CACAC TCACA CACCC CA3′ |
| R: LEF-Snail wild | 5′TG GGGTG TGTGA GTGTG GTG3′ | |
| F: LEF-Snail mutant | 5′CAC CACAC TTGTA CAAGT CC3′ | |
| R: LEF-Snail mutant | 5′TG GGGTG TACAA GTGTG GTG3′ | |
| COX2 | F: COX2-Snail wild | 5′GTG GAGC TCACA TTAAC TAT3′ |
| R: COX2-Snail wild | 5′ATA GTTAA TGTGA GCTC CAC3′ | |
| F: COX2-Snail mutant | 5′GTG GAGC TTGTA TTAAC TAT3′ | |
| R: COX2-Snail mutant | 5′ATA GTTAA TACAA GCTC CAC3′ | |
| COL1A1 | F: COL1A1-Snail wild | 5′CCA ATGA TCACA GGCCT CCC3′ |
| R: COL1A1-Snail wild | 5′GGG AGGCC TGTGA TCAT TGG3′ | |
| F: COL1A1-Snail mutant | 5′CCA ATGA TTGTA GGCCT CCC3′ | |
| R: COL1A1-Snail mutant | 5′GGG AGGCC TACAA TCAT TGG3′ | |
| Mesenchymal Genes Promoter | Proposed Transcription Factors | Proposed Binding Motif | References |
|---|---|---|---|
| FN | Snail | −576 TCACA −570 | [36,37] |
| E/S overlapping | −360 CGGCGGGCGGGCGGGC −345 | [38,39] | |
| MMP9 | Snail | −818 TCACA −812 | [15,17] |
| E/S overlapping | −797 GAGCCCCCCACCCCCC −781 | [17] | |
| LEF | Snail | −1287 TCACA −1281 | [40,41] |
| E/S overlapping | −1195 CCCTCACCCCCCGCCT −1179 | ||
| ZEB1 | Snail | −1065 TCACA −1059 | [17] |
| E/S overlapping | −933 AAGAGGGCGGGGAGCG −915 | [17] | |
| COX2 | Snail | −968 TCACA −962 | [42] |
| E/S overlapping | −911 CCTTTCCCGCCTCTC −896 | ||
| VIMENTIN | Snail | −2318 TCACA −2312 | [43] |
| E/S overlapping | −1976 CCCCCTGCCGCCACC−1960 | [44,45] | |
| VITRONECTIN | Snail | −1373 TCACA −1367 | [46,47] |
| E/S overlapping | −1238 GCACCCGCCCACCAC −1222 | [48] | |
| COL1A1 | Snail | −1071 TCACA −1065 | [49,50] |
| E/S overlapping | −993 CCCCAATCCCCACCTC −976 | [51,52] | |
| α-SMA | Snail | −689 TGTGA −683 | [31,53] |
| E/S overlapping | −507 GCTCTCTCCCCGCCCC −490 | [54] | |
| N-cadherin | Snail | −311 TCACA −305 | [55,56] |
| E/S overlapping | −289 CCCCCGCCCCCTCCCC −272 | [57,58] | |
| TWIST1 | Snail | −723 TCACA −717 | [59] |
| E/S overlapping | −606 CCCCGCGCCCGCCGGA −590 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ly, T.M.; Chen, Y.-C.; Lee, M.-C.; Hu, C.-T.; Cheng, C.-C.; Chang, H.-H.; You, R.-I.; Wu, W.-S. Snail Upregulates Transcription of FN, LEF, COX2, and COL1A1 in Hepatocellular Carcinoma: A General Model Established for Snail to Transactivate Mesenchymal Genes. Cells 2021, 10, 2202. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10092202
Ly TM, Chen Y-C, Lee M-C, Hu C-T, Cheng C-C, Chang H-H, You R-I, Wu W-S. Snail Upregulates Transcription of FN, LEF, COX2, and COL1A1 in Hepatocellular Carcinoma: A General Model Established for Snail to Transactivate Mesenchymal Genes. Cells. 2021; 10(9):2202. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10092202
Chicago/Turabian StyleLy, Tam Minh, Yen-Cheng Chen, Ming-Che Lee, Chi-Tan Hu, Chuan-Chu Cheng, Hsin-Hou Chang, Ren-In You, and Wen-Sheng Wu. 2021. "Snail Upregulates Transcription of FN, LEF, COX2, and COL1A1 in Hepatocellular Carcinoma: A General Model Established for Snail to Transactivate Mesenchymal Genes" Cells 10, no. 9: 2202. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10092202