
The Impact of MicroRNAs during Inflammatory Bowel Disease: Effects on the Mucus Layer and Intercellular Junctions for Gut Permeability


Abstract
:1. Introduction
2. MicroRNAs and Disease
2.1. Inflammatory Bowel Disease
2.2. Biomarkers and Treatments
2.3. Gut Immunity
3. Permeability of the Gut Epithelial Barrier
3.1. Protection by the Gut Mucosa
3.1.1. General Characteristics
3.1.2. The Mucus Layer
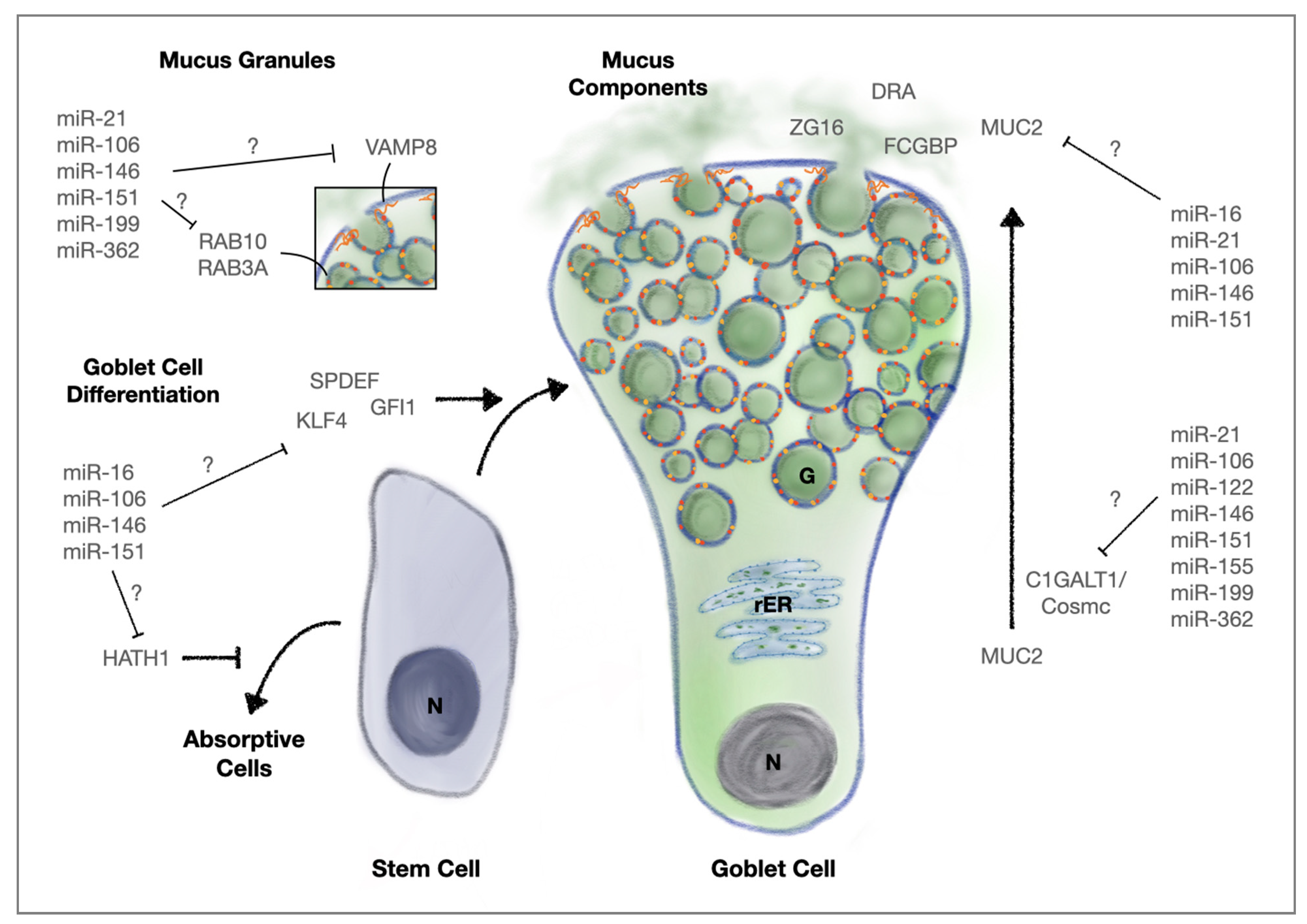
3.1.3. MicroRNAs, Goblet Cells and Mucus Secretion
3.2. Cell–Cell Interactions within the Gut Epithelium
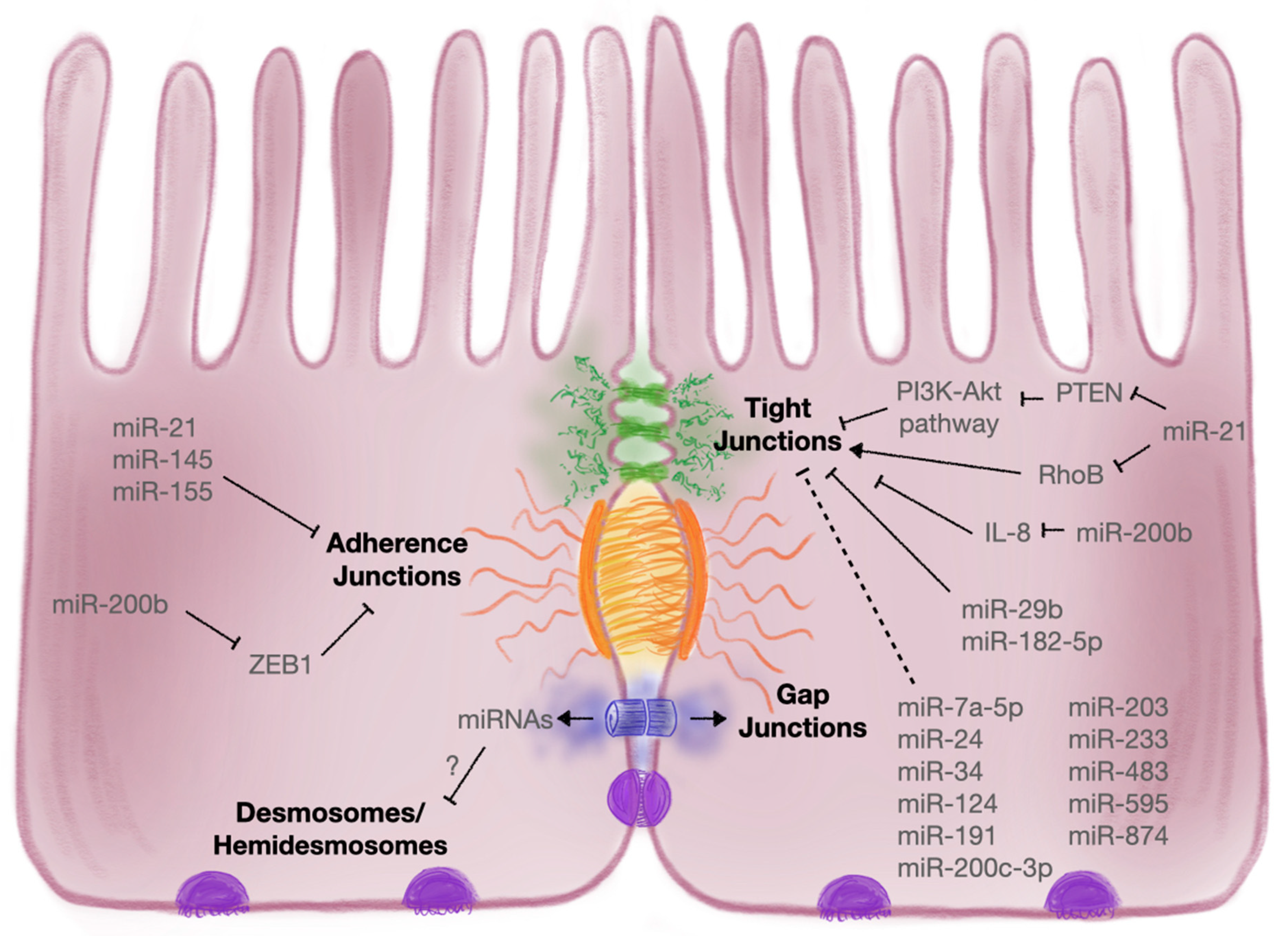
3.2.1. Intercellular Junctions
3.2.2. Tight Junctions
3.2.3. Adherens Junctions

{kind=link}
{kind=link}
| Intercellular Junction | Target | MicroRNA | Description | References |
|---|---|---|---|---|
| Tight Junction | Claudin-1 | miR-29 | Uc.173 and CircHIPK3 sequestered miR-29, which targets claudin-1 | [171] |
| miR-200b | MiR-200b supressed IL-8 secretion and thereby attenuated TJ dysfunction via claudin-1 | [180] | ||
| miR-874 | Suppression of claudin-1 has been attributed to miR-874 | [170] | ||
| Claudin-2 | miR-182-5p | Claudin-2 was shown to be a target of miR-182-5p | [169] | |
| Claudin-4 | miR-21-5p | Intestinal symbiotic flora upregulated miR-21-5p to target inhibitors of the PI3K–Akt, AP-1 and ERK pathways to upregulate ARF4 and reduce claudin-4 | [162] | |
| Claudin-8 | miR-233 | MiR-233 downregulated claudin-8; HMC-1 derived exosomes enriched in miR-223 inhibited claudin-8 in various intestinal epithelial cell lines | [173,174] | |
| Occludin | miR-21-5p | Intestinal symbiotic flora upregulated miR-21-5p to target inhibitors of the PI3K–Akt, AP-1 and ERK pathways to upregulate ARF4 and reduce occludin; also negatively affects occludin by targeting RhoB | [48,162] | |
| miR-29b | Uc.173 and CircHIPK3 sequestered miR-29, which targets occludin | [171] | ||
| miR-34 | PlncRNA1 and miR-34 cooperatively regulated expression of occludin in vitro during DSS-induced colitis | [168] | ||
| miR-200c-3p | IL-1β-induced upregulation of miR-200c-3p in UC patients decreased levels of occludin | [166] | ||
| miR-874 | Suppression of occludin has been attributed to miR-874 | [170] | ||
| ZO1 | miR-7a-5p | MiR-7a-5p antagomir increased ZO1 expression and promoted barrier recovery within TNBS-induced colitis models | [179] | |
| miR-21-5p | ZO1 shown to be indirectly regulated by miR-21 | [165] | ||
| miR-34 | PlncRNA1 and miR-34 cooperatively regulated expression of ZO1 in vitro during DSS-induced colitis | [168] | ||
| miR-191a | TNF-induced miR-191a expression led to decreased levels of ZO1 in IEC-6 cells | [177] | ||
| miR-200b | MiR-200b supressed IL-8 secretion and thereby attenuated TJ dysfunction via ZO1 | [180] | ||
| ZO2 | miR-203 | ZO2 levels were impacted when inhibitors for miR-203, miR-483-3p and miR-595 were used on T84 monolayers | [175] | |
| miR-483 | ||||
| miR-595 | ||||
| Cingulin | miR-24 | Overexpression of miR-24 led to decreased levels of cingulin, which negatively correlated with disease severity in UC patients | [176] | |
| JAM-A | miR-320a | Elevated miR-320a increased TER and JAM-A expression in T84 cells | [181] | |
| Other | miR-1 | Health benefits of salvianolic acid B were suggested to restore barrier function and TJ protein expression via induction of miR-1 | [183] | |
| miR-93 | MiR-93 targeted protein tyrosine kinase 6 and attenuated TNF/IFNγ-induced barrier dysfunction during IBD | [182] | ||
| miR-122 | TNF-induced miR-122 increased gut permeability | [167] | ||
| miR-124 | Suppression of AHR protein and TJ proteins by miR-124 was associated with intestinal barrier disruption | [178] | ||
| Adherens Junctions | E-Cadherin | miR-21 | A negative correlation between levels of E-cadherin and miR-21 was found in UC patients; exosome-derived miR-21a-5p from abnormally polarised macrophages decreased levels of E-cadherin in the epithelium, exacerbating DSS-induced colitis in mice | [195,196,197] |
| miR-130a-3p | Upregulated circRNA_102610 decreased miR-130a-3p, promoted TGF-β1-induced EMT in vitro and downregulated E-cadherin expression | [198] | ||
| miR-155 | E-cadherin was negatively affected by miR-155, leading to epithelial injury in early-life inflammatory stressor rat models | [199] | ||
| miR-200b | MiR-200b directly targets ZEB1, a negative regulator of AJ formation, and thus increased E-cadherin expression | [200] | ||
| Epithelial Membrane Protein 1 | miR-145 | Other AJ proteins such as epithelial membrane protein 1 were shown to be targeted by miR-145 | [59] |
3.2.4. Desmosomes and Hemidesmosomes
3.2.5. Gap Junctions
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lee, R.C.; Feinbaum, R.L.; Ambros, V. The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 1993, 75, 843–854. [Google Scholar] [CrossRef]
- Wightman, B.; Ha, I.; Ruvkun, G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell 1993, 75, 855–862. [Google Scholar] [CrossRef]
- Bhaskaran, M.; Mohan, M. MicroRNAs: History, biogenesis, and their evolving role in animal development and disease. Vet. Pathol. 2014, 51, 759–774. [Google Scholar] [CrossRef] [Green Version]
- Ha, M.; Kim, V.N. Regulation of microRNA biogenesis. Nat. Rev. Mol. Cell Biol. 2014, 15, 509–524. [Google Scholar] [CrossRef]
- Mohr, A.M.; Mott, J.L. Overview of microRNA biology. Semin. Liver Dis. 2015, 35, 3–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Brien, J.; Hayder, H.; Zayed, Y.; Peng, C. Overview of MicroRNA Biogenesis, mechanisms of actions, and circulation. Front. Endocrinol. (Lausanne) 2018, 9, 402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, P.W.; Huang, H.-D.; Hsu, S.-D.; Lin, L.-Z.; Tsou, A.-P.; Tseng, C.-P.; Stadler, P.F.; Washietl, S.; Hofacker, I.L. MiRNAMap: Genomic maps of microRNA genes and their target genes in mammalian genomes. Nucleic Acids Res. 2006, 34, D135–D139. [Google Scholar] [CrossRef] [Green Version]
- Zeidler, M.; Huttenhofer, A.; Kress, M.; Kummer, K.K. Intragenic microRNAs Autoregulate their host genes in both direct and indirect ways—A cross-species analysis. Cells 2020, 9, 232. [Google Scholar] [CrossRef] [Green Version]
- Grimson, A.; Farh, K.K.; Johnston, W.K.; Garrett-Engele, P.; Lim, L.P.; Bartel, D.P. MicroRNA targeting specificity in mammals: Determinants beyond seed pairing. Mol. Cell 2007, 27, 91–105. [Google Scholar] [CrossRef] [Green Version]
- Friedman, R.C.; Farh, K.K.; Burge, C.B.; Bartel, D.P. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2009, 19, 92–105. [Google Scholar] [CrossRef] [Green Version]
- Liu, G.; Zhang, R.; Xu, J.; Wu, C.I.; Lu, X. Functional conservation of both CDS- and 3’-UTR-located microRNA binding sites between species. Mol. Biol. Evol. 2015, 32, 623–628. [Google Scholar] [CrossRef] [Green Version]
- Gu, W.; Xu, Y.; Xie, X.; Wang, T.; Ko, J.H.; Zhou, T. The role of RNA structure at 5’ untranslated region in microRNA-mediated gene regulation. RNA 2014, 20, 1369–1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stavast, C.J.; Erkeland, S.J. The non-canonical aspects of microRNAs: Many roads to gene regulation. Cells 2019, 8, 1465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ali Syeda, Z.; Langden, S.S.S.; Munkhzul, C.; Lee, M.; Song, S.J. Regulatory mechanism of microRNA expression in cancer. Int. J. Mol. Sci. 2020, 21, 1723. [Google Scholar] [CrossRef] [Green Version]
- Cakmak, H.A.; Demir, M. MicroRNA and cardiovascular diseases. Balkan Med. J. 2020, 37, 60–71. [Google Scholar] [PubMed]
- Paraskevi, A.; Theodoropoulos, G.; Papaconstantinou, I.; Mantzaris, G.; Nikiteas, N.; Gazouli, M. Circulating microRNA in inflammatory bowel disease. J. Crohns Colitis 2012, 6, 900–904. [Google Scholar] [CrossRef]
- Wani, S.; Man Law, I.K.; Pothoulakis, C. Role and mechanisms of exosomal miRNAs in IBD pathophysiology. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 319, G646–G654. [Google Scholar] [CrossRef]
- Bi, K.; Zhang, X.; Chen, W.; Diao, H. MicroRNAs regulate intestinal immunity and gut microbiota for gastrointestinal health: A comprehensive review. Genes 2020, 11, 1075. [Google Scholar] [CrossRef]
- Baumgart, D.C.; Carding, S.R. Inflammatory bowel disease: Cause and immunobiology. Lancet 2007, 369, 1627–1640. [Google Scholar] [CrossRef]
- Podolsky, D.K. Inflammatory bowel disease. N. Engl. J. Med. 2002, 347, 417–429. [Google Scholar] [CrossRef]
- van Heel, D.A.; Fisher, S.A.; Kirby, A.; Daly, M.J.; Rioux, J.D.; Lewis, C.M.; Genome Scan Meta-Analysis Group of the, I.B.D.I.G.C. Inflammatory bowel disease susceptibility loci defined by genome scan meta-analysis of 1952 affected relative pairs. Hum. Mol. Genet. 2004, 13, 763–770. [Google Scholar] [CrossRef] [Green Version]
- Ramos, G.P.; Papadakis, K.A. Mechanisms of disease: Inflammatory bowel diseases. Mayo Clin. Proc. 2019, 94, 155–165. [Google Scholar] [CrossRef] [Green Version]
- Abraham, C.; Cho, J.H. Inflammatory bowel disease. N. Engl. J. Med. 2009, 361, 2066–2078. [Google Scholar] [CrossRef]
- Van Der Kraak, L.; Gros, P.; Beauchemin, N. Colitis-associated colon cancer: Is it in your genes? World J. Gastroenterol. 2015, 21, 11688–11699. [Google Scholar] [CrossRef]
- Choy, M.C.; Visvanathan, K.; De Cruz, P. An overview of the Innate and adaptive immune system in inflammatory bowel disease. Inflamm. Bowel Dis. 2017, 23, 2–13. [Google Scholar] [CrossRef] [PubMed]
- Di Sabatino, A.; Biancheri, P.; Rovedatti, L.; MacDonald, T.T.; Corazza, G.R. New pathogenic paradigms in inflammatory bowel disease. Inflamm. Bowel Dis. 2012, 18, 368–371. [Google Scholar] [CrossRef] [PubMed]
- Baumgart, D.C.; Sandborn, W.J. Crohn’s disease. Lancet 2012, 380, 1590–1605. [Google Scholar] [CrossRef] [Green Version]
- Peeters, M.; Joossens, S.; Vermeire, S.; Vlietinck, R.; Bossuyt, X.; Rutgeerts, P. Diagnostic value of anti-saccharomyces cerevisiae and antineutrophil cytoplasmic autoantibodies in inflammatory bowel disease. Am. J. Gastroenterol. 2001, 96, 730–734. [Google Scholar] [CrossRef]
- Lewis, J.D. The utility of biomarkers in the diagnosis and therapy of inflammatory bowel disease. Gastroenterology 2011, 140, 1817–1826. [Google Scholar] [CrossRef] [Green Version]
- Torres, J.; Petralia, F.; Sato, T.; Wang, P.; Telesco, S.E.; Choung, R.S.; Strauss, R.; Li, X.J.; Laird, R.M.; Gutierrez, R.L.; et al. Serum biomarkers identify patients who will develop inflammatory bowel diseases up to 5 years before diagnosis. Gastroenterology 2020, 159, 96–104. [Google Scholar] [CrossRef]
- Cichon, C.; Sabharwal, H.; Ruter, C.; Schmidt, M.A. MicroRNAs regulate tight junction proteins and modulate epithelial/endothelial barrier functions. Tissue Barriers 2014, 2, e944446. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weber, J.A.; Baxter, D.H.; Zhang, S.; Huang, D.Y.; Huang, K.H.; Lee, M.J.; Galas, D.J.; Wang, K. The microRNA spectrum in 12 body fluids. Clin. Chem. 2010, 56, 1733–1741. [Google Scholar] [CrossRef] [PubMed]
- Krissansen, G.W.; Yang, Y.; McQueen, F.M.; Leung, E.; Peek, D.; Chan, Y.C.; Print, C.; Dalbeth, N.; Williams, M.; Fraser, A.G. Overexpression of miR-595 and miR-1246 in the sera of patients with active forms of inflammatory bowel disease. Inflamm. Bowel Dis. 2015, 21, 520–530. [Google Scholar] [CrossRef] [PubMed]
- Jensen, M.D.; Andersen, R.F.; Christensen, H.; Nathan, T.; Kjeldsen, J.; Madsen, J.S. Circulating microRNAs as biomarkers of adult Crohn’s disease. Eur. J. Gastroenterol. Hepatol. 2015, 27, 1038–1044. [Google Scholar] [CrossRef]
- Kalla, R.; Adams, A.T.; Ventham, N.T.; Kennedy, N.A.; White, R.; Clarke, C.; Ivens, A.; Bergemalm, D.; Vatn, S.; Lopez-Jimena, B.; et al. Whole blood profiling of T-cell derived miRNA allows the development of prognostic models in inflammatory bowel disease. J. Crohns Colitis 2020, 14, 1724–1733. [Google Scholar] [CrossRef]
- Schaefer, J.S.; Attumi, T.; Opekun, A.R.; Abraham, B.; Hou, J.; Shelby, H.; Graham, D.Y.; Streckfus, C.; Klein, J.R. MicroRNA signatures differentiate Crohn’s disease from ulcerative colitis. BMC Immunol. 2015, 16, 5. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.; Welker, N.C.; Zhao, Z.; Li, Y.; Zhang, J.; Reuss, S.A.; Zhang, X.; Lee, H.; Liu, Y.; Bronner, M.P. Novel specific microRNA biomarkers in idiopathic inflammatory bowel disease unrelated to disease activity. Mod. Pathol. 2014, 27, 602–608. [Google Scholar] [CrossRef] [Green Version]
- Konstantinidis, A.; Pardali, D.; Adamama-Moraitou, K.K.; Gazouli, M.; Dovas, C.I.; Legaki, E.; Brellou, G.D.; Savvas, I.; Jergens, A.E.; Rallis, T.S.; et al. Colonic mucosal and serum expression of microRNAs in canine large intestinal inflammatory bowel disease. BMC Vet. Res. 2020, 16, 69. [Google Scholar] [CrossRef]
- Cordes, F.; Demmig, C.; Bokemeyer, A.; Bruckner, M.; Lenze, F.; Lenz, P.; Nowacki, T.; Tepasse, P.; Schmidt, H.H.; Schmidt, M.A.; et al. MicroRNA-320a monitors intestinal disease activity in patients with inflammatory bowel disease. Clin. Transl. Gastroenterol. 2020, 11, e00134. [Google Scholar] [CrossRef]
- James, J.P.; Riis, L.B.; Malham, M.; Hogdall, E.; Langholz, E.; Nielsen, B.S. MicroRNA biomarkers in IBD-differential diagnosis and prediction of Colitis-associated cancer. Int. J. Mol. Sci. 2020, 21, 7893. [Google Scholar] [CrossRef]
- Omidbakhsh, A.; Saeedi, M.; Khoshnia, M.; Marjani, A.; Hakimi, S. Micro-RNAs -106a and -362-3p in Peripheral blood of inflammatory bowel disease patients. Open. Biochem. J. 2018, 12, 78–86. [Google Scholar] [CrossRef] [PubMed]
- Zahm, A.M.; Thayu, M.; Hand, N.J.; Horner, A.; Leonard, M.B.; Friedman, J.R. Circulating microRNA is a biomarker of pediatric Crohn disease. J. Pediatr. Gastroenterol. Nutr. 2011, 53, 26–33. [Google Scholar] [CrossRef] [Green Version]
- Duttagupta, R.; DiRienzo, S.; Jiang, R.; Bowers, J.; Gollub, J.; Kao, J.; Kearney, K.; Rudolph, D.; Dawany, N.B.; Showe, M.K.; et al. Genome-wide maps of circulating miRNA biomarkers for ulcerative colitis. PLoS ONE 2012, 7, e31241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, F.; Guo, N.J.; Tian, H.; Marohn, M.; Gearhart, S.; Bayless, T.M.; Brant, S.R.; Kwon, J.H. Peripheral blood microRNAs distinguish active ulcerative colitis and Crohn’s disease. Inflamm. Bowel Dis. 2011, 17, 241–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zahm, A.M.; Hand, N.J.; Tsoucas, D.M.; Le Guen, C.L.; Baldassano, R.N.; Friedman, J.R. Rectal microRNAs are perturbed in pediatric inflammatory bowel disease of the colon. J. Crohns Colitis 2014, 8, 1108–1117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fasseu, M.; Treton, X.; Guichard, C.; Pedruzzi, E.; Cazals-Hatem, D.; Richard, C.; Aparicio, T.; Daniel, F.; Soule, J.C.; Moreau, R.; et al. Identification of restricted subsets of mature microRNA abnormally expressed in inactive colonic mucosa of patients with inflammatory bowel disease. PLoS ONE 2010, 5, e13160. [Google Scholar] [CrossRef]
- Coskun, M.; Bjerrum, J.T.; Seidelin, J.B.; Troelsen, J.T.; Olsen, J.; Nielsen, O.H. MiR-20b, miR-98, miR-125b-1*, and let-7e* as new potential diagnostic biomarkers in ulcerative colitis. World J. Gastroenterol. 2013, 19, 4289–4299. [Google Scholar] [CrossRef] [Green Version]
- Wu, F.; Zikusoka, M.; Trindade, A.; Dassopoulos, T.; Harris, M.L.; Bayless, T.M.; Brant, S.R.; Chakravarti, S.; Kwon, J.H. MicroRNAs are differentially expressed in ulcerative colitis and alter expression of macrophage inflammatory peptide-2 alpha. Gastroenterology 2008, 135, 1624–1635. [Google Scholar] [CrossRef]
- Wu, F.; Zhang, S.; Dassopoulos, T.; Harris, M.L.; Bayless, T.M.; Meltzer, S.J.; Brant, S.R.; Kwon, J.H. Identification of microRNAs associated with ileal and colonic Crohn’s disease. Inflamm. Bowel Dis. 2010, 16, 1729–1738. [Google Scholar] [CrossRef] [Green Version]
- Koukos, G.; Polytarchou, C.; Kaplan, J.L.; Oikonomopoulos, A.; Ziring, D.; Hommes, D.W.; Wahed, R.; Kokkotou, E.; Pothoulakis, C.; Winter, H.S.; et al. A microRNA signature in pediatric ulcerative colitis: Deregulation of the miR-4284/CXCL5 pathway in the intestinal epithelium. Inflamm. Bowel Dis. 2015, 21, 996–1005. [Google Scholar] [CrossRef] [Green Version]
- Peck, B.C.; Weiser, M.; Lee, S.E.; Gipson, G.R.; Iyer, V.B.; Sartor, R.B.; Herfarth, H.H.; Long, M.D.; Hansen, J.J.; Isaacs, K.L.; et al. MicroRNAs classify different disease behavior phenotypes of Crohn’s Disease and may have prognostic utility. Inflamm. Bowel Dis. 2015, 21, 2178–2187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Goten, J.; Vanhove, W.; Lemaire, K.; Van Lommel, L.; Machiels, K.; Wollants, W.J.; De Preter, V.; De Hertogh, G.; Ferrante, M.; Van Assche, G.; et al. Integrated miRNA and mRNA expression profiling in inflamed colon of patients with ulcerative colitis. PLoS ONE 2014, 9, e116117. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Wang, C.; Liu, Y.; Tang, L.; Zheng, M.; Xu, C.; Song, J.; Meng, X. MiR-122 targets NOD2 to decrease intestinal epithelial cell injury in Crohn’s disease. Biochem. Biophys. Res. Commun. 2013, 438, 133–139. [Google Scholar] [CrossRef]
- Pathak, S.; Grillo, A.R.; Scarpa, M.; Brun, P.; D’Inca, R.; Nai, L.; Banerjee, A.; Cavallo, D.; Barzon, L.; Palu, G.; et al. MiR-155 modulates the inflammatory phenotype of intestinal myofibroblasts by targeting SOCS1 in ulcerative colitis. Exp. Mol. Med. 2015, 47, e164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Zhu, F.; Li, H.; Fan, H.; Wu, H.; Dong, Y.; Chu, S.; Tan, C.; Wang, Q.; He, H.; et al. MiR-155 contributes to intestinal barrier dysfunction in DSS-induced mice colitis via targeting HIF-1alpha/TFF-3 axis. Aging 2020, 12, 14966–14977. [Google Scholar] [CrossRef]
- Chen, G.; Cao, S.; Liu, F.; Liu, Y. miR-195 plays a role in steroid resistance of ulcerative colitis by targeting Smad7. Biochem. J. 2015, 471, 357–367. [Google Scholar] [CrossRef] [PubMed]
- Fujioka, S.; Nakamichi, I.; Esaki, M.; Asano, K.; Matsumoto, T.; Kitazono, T. Serum microRNA levels in patients with Crohn’s disease during induction therapy by infliximab. J. Gastroenterol. Hepatol. 2014, 29, 1207–1214. [Google Scholar] [CrossRef]
- Cao, B.; Zhou, X.; Ma, J.; Zhou, W.; Yang, W.; Fan, D.; Hong, L. Role of MiRNAs in inflammatory bowel disease. Dig. Dis. Sci. 2017, 62, 1426–1438. [Google Scholar] [CrossRef]
- McKenna, L.B.; Schug, J.; Vourekas, A.; McKenna, J.B.; Bramswig, N.C.; Friedman, J.R.; Kaestner, K.H. MicroRNAs control intestinal epithelial differentiation, architecture, and barrier function. Gastroenterology 2010, 139, 1654–1664. [Google Scholar] [CrossRef] [Green Version]
- Mizoguchi, E.; Low, D.; Ezaki, Y.; Okada, T. Recent updates on the basic mechanisms and pathogenesis of inflammatory bowel diseases in experimental animal models. Intest. Res. 2020, 18, 151–167. [Google Scholar] [CrossRef]
- Jiminez, J.A.; Uwiera, T.C.; Douglas Inglis, G.; Uwiera, R.R. Animal models to study acute and chronic intestinal inflammation in mammals. Gut. Pathog. 2015, 7, 29. [Google Scholar] [CrossRef] [Green Version]
- Bazzoni, F.; Rossato, M.; Fabbri, M.; Gaudiosi, D.; Mirolo, M.; Mori, L.; Tamassia, N.; Mantovani, A.; Cassatella, M.A.; Locati, M. Induction and regulatory function of miR-9 in human monocytes and neutrophils exposed to proinflammatory signals. Proc. Natl. Acad. Sci. USA 2009, 106, 5282–5287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, J.; Qiao, Y.; Wang, P.; Li, S.; Zhao, W.; Gao, C. MicroRNA-210 negatively regulates LPS-induced production of proinflammatory cytokines by targeting NF-kappaB1 in murine macrophages. FEBS Lett. 2012, 586, 1201–1207. [Google Scholar] [CrossRef] [Green Version]
- Brain, O.; Owens, B.M.; Pichulik, T.; Allan, P.; Khatamzas, E.; Leslie, A.; Steevels, T.; Sharma, S.; Mayer, A.; Catuneanu, A.M.; et al. The intracellular sensor NOD2 induces microRNA-29 expression in human dendritic cells to limit IL-23 release. Immunity 2013, 39, 521–536. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ghorpade, D.S.; Sinha, A.Y.; Holla, S.; Singh, V.; Balaji, K.N. NOD2-nitric oxide-responsive microRNA-146a activates Sonic hedgehog signaling to orchestrate inflammatory responses in murine model of inflammatory bowel disease. J. Biol. Chem. 2013, 288, 33037–33048. [Google Scholar] [CrossRef] [Green Version]
- Chuang, A.Y.; Chuang, J.C.; Zhai, Z.; Wu, F.; Kwon, J.H. NOD2 expression is regulated by microRNAs in colonic epithelial HCT116 cells. Inflamm. Bowel Dis. 2014, 20, 126–135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujita, S.; Ito, T.; Mizutani, T.; Minoguchi, S.; Yamamichi, N.; Sakurai, K.; Iba, H. MiR-21 Gene expression triggered by AP-1 is sustained through a double-negative feedback mechanism. J. Mol. Biol. 2008, 378, 492–504. [Google Scholar] [CrossRef]
- Sheedy, F.J. Turning 21: Induction of miR-21 as a Key Switch in the inflammatory response. Front. Immunol. 2015, 6, 19. [Google Scholar] [CrossRef] [Green Version]
- Barrett, J.C.; Hansoul, S.; Nicolae, D.L.; Cho, J.H.; Duerr, R.H.; Rioux, J.D.; Brant, S.R.; Silverberg, M.S.; Taylor, K.D.; Barmada, M.M.; et al. Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat. Genet. 2008, 40, 955–962. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Ma, Y.; Shi, C.; Chen, H.; Zhang, H.; Chen, N.; Zhang, P.; Wang, F.; Yang, J.; Yang, J.; et al. Overexpression of miR-21 in patients with ulcerative colitis impairs intestinal epithelial barrier function through targeting the Rho GTPase RhoB. Biochem. Biophys. Res. Commun. 2013, 434, 746–752. [Google Scholar] [CrossRef]
- Adams, A.T.; Kennedy, N.A.; Hansen, R.; Ventham, N.T.; O’Leary, K.R.; Drummond, H.E.; Noble, C.L.; El-Omar, E.; Russell, R.K.; Wilson, D.C.; et al. Two-stage genome-wide methylation profiling in childhood-onset Crohn’s Disease implicates epigenetic alterations at the VMP1/MIR21 and HLA loci. Inflamm. Bowel Dis. 2014, 20, 1784–1793. [Google Scholar] [CrossRef] [Green Version]
- Shi, C.; Liang, Y.; Yang, J.; Xia, Y.; Chen, H.; Han, H.; Yang, Y.; Wu, W.; Gao, R.; Qin, H. MicroRNA-21 knockout improve the survival rate in DSS induced fatal colitis through protecting against inflammation and tissue injury. PLoS ONE 2013, 8, e66814. [Google Scholar]
- Johnston, D.G.W.; Williams, M.A.; Thaiss, C.A.; Cabrera-Rubio, R.; Raverdeau, M.; McEntee, C.; Cotter, P.D.; Elinav, E.; O’Neill, L.A.J.; Corr, S.C. Loss of microRNA-21 influences the gut microbiota, causing reduced susceptibility in a murine model of colitis. J. Crohns Colitis 2018, 12, 835–848. [Google Scholar] [CrossRef] [PubMed]
- Ando, Y.; Mazzurana, L.; Forkel, M.; Okazaki, K.; Aoi, M.; Schmidt, P.T.; Mjosberg, J.; Bresso, F. Downregulation of microRNA-21 in Colonic CD3+ T cells in UC remission. Inflamm. Bowel Dis. 2016, 22, 2788–2793. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singh, U.P.; Murphy, A.E.; Enos, R.T.; Shamran, H.A.; Singh, N.P.; Guan, H.; Hegde, V.L.; Fan, D.; Price, R.L.; Taub, D.D.; et al. MiR-155 deficiency protects mice from experimental colitis by reducing T helper type 1/type 17 responses. Immunology 2014, 143, 478–489. [Google Scholar] [CrossRef] [PubMed]
- He, C.; Yu, T.; Shi, Y.; Ma, C.; Yang, W.; Fang, L.; Sun, M.; Wu, W.; Xiao, F.; Guo, F.; et al. MicroRNA 301A promotes intestinal inflammation and colitis-associated cancer development by inhibiting BTG1. Gastroenterology 2017, 152, 1434–1448. [Google Scholar] [CrossRef]
- Sohn, J.J.; Schetter, A.J.; Yfantis, H.G.; Ridnour, L.A.; Horikawa, I.; Khan, M.A.; Robles, A.I.; Hussain, S.P.; Goto, A.; Bowman, E.D.; et al. Macrophages, nitric oxide and microRNAs are associated with DNA damage response pathway and senescence in inflammatory bowel disease. PLoS ONE 2012, 7, e44156. [Google Scholar] [CrossRef] [Green Version]
- Muenchau, S.; Deutsch, R.; de Castro, I.J.; Hielscher, T.; Heber, N.; Niesler, B.; Lusic, M.; Stanifer, M.L.; Boulant, S. Hypoxic environment promotes barrier formation in human intestinal epithelial cells through regulation of microRNA 320a expression. Mol. Cell Biol. 2019, 39, e00553. [Google Scholar] [CrossRef] [Green Version]
- Wu, F.; Dong, F.; Arendovich, N.; Zhang, J.; Huang, Y.; Kwon, J.H. Divergent influence of microRNA-21 deletion on murine colitis phenotypes. Inflamm. Bowel Dis. 2014, 20, 1972–1985. [Google Scholar] [CrossRef]
- Jeker, L.T.; Zhou, X.; Gershberg, K.; de Kouchkovsky, D.; Morar, M.M.; Stadthagen, G.; Lund, A.H.; Bluestone, J.A. MicroRNA 10a marks regulatory T cells. PLoS ONE 2012, 7, e36684. [Google Scholar] [CrossRef]
- de Kouchkovsky, D.; Esensten, J.H.; Rosenthal, W.L.; Morar, M.M.; Bluestone, J.A.; Jeker, L.T. MicroRNA-17-92 regulates IL-10 production by regulatory T cells and control of experimental autoimmune encephalomyelitis. J. Immunol. 2013, 191, 1594–1605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, L.F.; Boldin, M.P.; Chaudhry, A.; Lin, L.L.; Taganov, K.D.; Hanada, T.; Yoshimura, A.; Baltimore, D.; Rudensky, A.Y. Function of miR-146a in controlling Treg cell-mediated regulation of Th1 responses. Cell 2010, 142, 914–929. [Google Scholar] [CrossRef] [Green Version]
- Chinen, I.; Nakahama, T.; Kimura, A.; Nguyen, N.T.; Takemori, H.; Kumagai, A.; Kayama, H.; Takeda, K.; Lee, S.; Hanieh, H.; et al. The aryl hydrocarbon receptor/microRNA-212/132 axis in T cells regulates IL-10 production to maintain intestinal homeostasis. Int. Immunol. 2015, 27, 405–415. [Google Scholar] [CrossRef] [PubMed]
- Steiner, D.F.; Thomas, M.F.; Hu, J.K.; Yang, Z.; Babiarz, J.E.; Allen, C.D.; Matloubian, M.; Blelloch, R.; Ansel, K.M. MicroRNA-29 regulates T-box transcription factors and interferon-gamma production in helper T cells. Immunity 2011, 35, 169–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guerau-de-Arellano, M.; Smith, K.M.; Godlewski, J.; Liu, Y.; Winger, R.; Lawler, S.E.; Whitacre, C.C.; Racke, M.K.; Lovett-Racke, A.E. Micro-RNA dysregulation in multiple sclerosis favours pro-inflammatory T-cell-mediated autoimmunity. Brain 2011, 134, 3578–3589. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, A.; Schambach, F.; DeJong, C.S.; Hammond, S.M.; Reiner, S.L. Micro-RNA-155 inhibits IFN-gamma signaling in CD4+ T cells. Eur. J. Immunol. 2010, 40, 225–231. [Google Scholar] [CrossRef]
- Takahashi, H.; Kanno, T.; Nakayamada, S.; Hirahara, K.; Sciume, G.; Muljo, S.A.; Kuchen, S.; Casellas, R.; Wei, L.; Kanno, Y.; et al. TGF-beta and retinoic acid induce the microRNA miR-10a, which targets Bcl-6 and constrains the plasticity of helper T cells. Nat. Immunol. 2012, 13, 587–595. [Google Scholar] [CrossRef]
- O’Connell, R.M.; Kahn, D.; Gibson, W.S.; Round, J.L.; Scholz, R.L.; Chaudhuri, A.A.; Kahn, M.E.; Rao, D.S.; Baltimore, D. MicroRNA-155 promotes autoimmune inflammation by enhancing inflammatory T cell development. Immunity 2010, 33, 607–619. [Google Scholar] [CrossRef] [Green Version]
- Du, C.; Liu, C.; Kang, J.; Zhao, G.; Ye, Z.; Huang, S.; Li, Z.; Wu, Z.; Pei, G. MicroRNA miR-326 regulates TH-17 differentiation and is associated with the pathogenesis of multiple sclerosis. Nat. Immunol. 2009, 10, 1252–1259. [Google Scholar] [CrossRef]
- Mycko, M.P.; Cichalewska, M.; Machlanska, A.; Cwiklinska, H.; Mariasiewicz, M.; Selmaj, K.W. MicroRNA-301a regulation of a T-helper 17 immune response controls autoimmune demyelination. Proc. Natl. Acad. Sci. USA 2012, 109, E1248–E1257. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Flach, H.; Onizawa, M.; Wei, L.; McManus, M.T.; Weiss, A. Negative regulation of Hif1a expression and TH17 differentiation by the hypoxia-regulated microRNA miR-210. Nat. Immunol. 2014, 15, 393–401. [Google Scholar] [CrossRef] [PubMed]
- Koralov, S.B.; Muljo, S.A.; Galler, G.R.; Krek, A.; Chakraborty, T.; Kanellopoulou, C.; Jensen, K.; Cobb, B.S.; Merkenschlager, M.; Rajewsky, N.; et al. Dicer ablation affects antibody diversity and cell survival in the B lymphocyte lineage. Cell 2008, 132, 860–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.Z.; Li, L.; Lodish, H.F.; Bartel, D.P. MicroRNAs modulate hematopoietic lineage differentiation. Science 2004, 303, 83–86. [Google Scholar] [CrossRef] [Green Version]
- Suri, K.; Bubier, J.A.; Wiles, M.V.; Shultz, L.D.; Amiji, M.M.; Hosur, V. Role of microRNA in inflammatory bowel disease: Clinical evidence and the development of preclinical animal models. Cells 2021, 10, 2204. [Google Scholar] [CrossRef]
- Bischoff, S.C.; Barbara, G.; Buurman, W.; Ockhuizen, T.; Schulzke, J.D.; Serino, M.; Tilg, H.; Watson, A.; Wells, J.M. Intestinal permeability--a new target for disease prevention and therapy. BMC Gastroenterol. 2014, 14, 189. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.Y.; Wasinger, V.C.; Yau, Y.Y.; Chuang, E.; Yajnik, V.; Leong, R.W.L. Molecular pathophysiology of epithelial barrier dysfunction in inflammatory bowel diseases. Proteomes 2018, 6, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalla, R.; Ventham, N.T.; Kennedy, N.A.; Quintana, J.F.; Nimmo, E.R.; Buck, A.H.; Satsangi, J. MicroRNAs: New players in IBD. Gut 2015, 64, 504–517. [Google Scholar] [CrossRef] [Green Version]
- Puertolas-Balint, F.; Schroeder, B.O. Does an apple a day also keep the microbes away? The interplay between diet, microbiota, and host defense peptides at the intestinal mucosal barrier. Front. Immunol. 2020, 11, 1164. [Google Scholar] [CrossRef] [PubMed]
- van der Flier, L.G.; Clevers, H. Stem cells, self-renewal, and differentiation in the intestinal epithelium. Annu. Rev. Physiol. 2009, 71, 241–260. [Google Scholar] [CrossRef]
- Snoeck, V.; Goddeeris, B.; Cox, E. The role of enterocytes in the intestinal barrier function and antigen uptake. Microbes Infect. 2005, 7, 997–1004. [Google Scholar] [CrossRef]
- Hansson, G.C. Mucins and the microbiome. Annu. Rev. Biochem. 2020, 89, 769–793. [Google Scholar] [CrossRef] [Green Version]
- Simmonds, N.; Furman, M.; Karanika, E.; Phillips, A.; Bates, A.W. Paneth cell metaplasia in newly diagnosed inflammatory bowel disease in children. BMC Gastroenterol. 2014, 14, 93. [Google Scholar] [CrossRef] [PubMed]
- Nevo, S.; Kadouri, N.; Abramson, J. Tuft cells: From the mucosa to the thymus. Immunol. Lett. 2019, 210, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Birchenough, G.M.; Johansson, M.E.; Gustafsson, J.K.; Bergstrom, J.H.; Hansson, G.C. New developments in goblet cell mucus secretion and function. Mucosal Immunol. 2015, 8, 712–719. [Google Scholar] [CrossRef] [Green Version]
- Moszak, M.; Szulinska, M.; Bogdanski, P. You are what you eat—The relationship between diet, microbiota, and metabolic disorders—A review. Nutrients 2020, 12, 1096. [Google Scholar] [CrossRef] [Green Version]
- Conlon, M.A.; Bird, A.R. The impact of diet and lifestyle on gut microbiota and human health. Nutrients 2014, 7, 17–44. [Google Scholar] [CrossRef]
- Mentella, M.C.; Scaldaferri, F.; Pizzoferrato, M.; Gasbarrini, A.; Miggiano, G.A.D. Nutrition, IBD and gut microbiota: A review. Nutrients 2020, 12, 944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schutte, A.; Ermund, A.; Becker-Pauly, C.; Johansson, M.E.; Rodriguez-Pineiro, A.M.; Backhed, F.; Muller, S.; Lottaz, D.; Bond, J.S.; Hansson, G.C. Microbial-induced meprin beta cleavage in MUC2 mucin and a functional CFTR channel are required to release anchored small intestinal mucus. Proc. Natl. Acad. Sci. USA 2014, 111, 12396–12401. [Google Scholar] [CrossRef] [Green Version]
- Hansson, G.C. Mucus and mucins in diseases of the intestinal and respiratory tracts. J. Intern. Med. 2019, 285, 479–490. [Google Scholar] [CrossRef] [Green Version]
- Ermund, A.; Gustafsson, J.K.; Hansson, G.C.; Keita, A.V. Mucus properties and goblet cell quantification in mouse, rat and human ileal Peyer’s patches. PLoS ONE 2013, 8, e83688. [Google Scholar] [CrossRef] [Green Version]
- Johansson, M.E.; Phillipson, M.; Petersson, J.; Velcich, A.; Holm, L.; Hansson, G.C. The inner of the two Muc2 mucin-dependent mucus layers in colon is devoid of bacteria. Proc. Natl. Acad. Sci. USA 2008, 105, 15064–15069. [Google Scholar] [CrossRef] [Green Version]
- Van der Sluis, M.; De Koning, B.A.; De Bruijn, A.C.; Velcich, A.; Meijerink, J.P.; Van Goudoever, J.B.; Buller, H.A.; Dekker, J.; Van Seuningen, I.; Renes, I.B.; et al. Muc2-deficient mice spontaneously develop colitis, indicating that MUC2 is critical for colonic protection. Gastroenterology 2006, 131, 117–129. [Google Scholar] [CrossRef]
- Wu, M.; Wu, Y.; Li, J.; Bao, Y.; Guo, Y.; Yang, W. The dynamic changes of gut microbiota in Muc2 deficient mice. Int. J. Mol. Sci. 2018, 19, 2809. [Google Scholar] [CrossRef] [Green Version]
- van der Post, S.; Jabbar, K.S.; Birchenough, G.; Arike, L.; Akhtar, N.; Sjovall, H.; Johansson, M.E.V.; Hansson, G.C. Structural weakening of the colonic mucus barrier is an early event in ulcerative colitis pathogenesis. Gut 2019, 68, 2142–2151. [Google Scholar] [CrossRef] [Green Version]
- Johansson, M.E.; Gustafsson, J.K.; Holmen-Larsson, J.; Jabbar, K.S.; Xia, L.; Xu, H.; Ghishan, F.K.; Carvalho, F.A.; Gewirtz, A.T.; Sjovall, H.; et al. Bacteria penetrate the normally impenetrable inner colon mucus layer in both murine colitis models and patients with ulcerative colitis. Gut 2014, 63, 281–291. [Google Scholar] [CrossRef]
- Kim, Y.S.; Ho, S.B. Intestinal goblet cells and mucins in health and disease: Recent insights and progress. Curr. Gastroenterol. Rep. 2010, 12, 319–330. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Yu, M. Role of goblet cells in intestinal barrier and mucosal immunity. J. Inflamm. Res. 2021, 14, 3171–3183. [Google Scholar] [CrossRef] [PubMed]
- Gersemann, M.; Becker, S.; Kubler, I.; Koslowski, M.; Wang, G.; Herrlinger, K.R.; Griger, J.; Fritz, P.; Fellermann, K.; Schwab, M.; et al. Differences in goblet cell differentiation between Crohn’ s disease and ulcerative colitis. Differentiation 2009, 77, 84–94. [Google Scholar] [CrossRef] [PubMed]
- Sticht, C.; De La Torre, C.; Parveen, A.; Gretz, N. MiRWalk: An online resource for prediction of microRNA binding sites. PLoS ONE 2018, 13, e0206239. [Google Scholar] [CrossRef] [PubMed]
- Ehrencrona, E.; van der Post, S.; Gallego, P.; Recktenwald, C.V.; Rodriguez-Pineiro, A.M.; Garcia-Bonete, M.J.; Trillo-Muyo, S.; Backstrom, M.; Hansson, G.C.; Johansson, M.E.V. The IgGFc-binding protein FCGBP is secreted with all GDPH sequences cleaved but maintained by interfragment disulfide bonds. J. Biol. Chem. 2021, 297, 100871. [Google Scholar] [CrossRef] [PubMed]
- Bergstrom, J.H.; Birchenough, G.M.; Katona, G.; Schroeder, B.O.; Schutte, A.; Ermund, A.; Johansson, M.E.; Hansson, G.C. Gram-positive bacteria are held at a distance in the colon mucus by the lectin-like protein ZG16. Proc. Natl. Acad. Sci. USA 2016, 113, 13833–13838. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, A.; Priyamvada, S.; Ge, Y.; Jayawardena, D.; Singhal, M.; Anbazhagan, A.N.; Chatterjee, I.; Dayal, A.; Patel, M.; Zadeh, K.; et al. A novel role of SLC26A3 in the maintenance of intestinal epithelial barrier integrity. Gastroenterology 2021, 160, 1240–1255. [Google Scholar] [CrossRef] [PubMed]
- Ding, X.; Li, D.; Li, M.; Wang, H.; He, Q.; Wang, Y.; Yu, H.; Tian, D.; Yu, Q. SLC26A3 (DRA) prevents TNF-alpha-induced barrier dysfunction and dextran sulfate sodium-induced acute colitis. Lab. Investig. 2018, 98, 462–476. [Google Scholar] [CrossRef] [PubMed]
- Anbazhagan, A.N.; Priyamvada, S.; Kumar, A.; Maher, D.B.; Borthakur, A.; Alrefai, W.A.; Malakooti, J.; Kwon, J.H.; Dudeja, P.K. Translational repression of SLC26A3 by miR-494 in intestinal epithelial cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2014, 306, G123–G131. [Google Scholar] [CrossRef] [PubMed]
- Song, L.; Chang, R.; Sun, X.; Lu, L.; Gao, H.; Lu, H.; Lin, R.; Xu, X.; Liu, Z.; Zhan, L. Macrophage-derived EDA-A2 inhibits intestinal stem cells by targeting miR-494/EDA2R/beta-catenin signaling in mice. Commun. Biol. 2021, 4, 213. [Google Scholar] [CrossRef]
- Mo, J.S.; Alam, K.J.; Kim, H.S.; Lee, Y.M.; Yun, K.J.; Chae, S.C. MicroRNA 429 regulates mucin gene expression and secretion in murine model of colitis. J. Crohns Colitis 2016, 10, 837–849. [Google Scholar] [CrossRef] [Green Version]
- Li, T.; Li, D.; Sha, J.; Sun, P.; Huang, Y. MicroRNA-21 directly targets MARCKS and promotes apoptosis resistance and invasion in prostate cancer cells. Biochem. Biophys. Res. Commun. 2009, 383, 280–285. [Google Scholar] [CrossRef]
- Green, T.D.; Crews, A.L.; Park, J.; Fang, S.; Adler, K.B. Regulation of mucin secretion and inflammation in asthma: A role for MARCKS protein? Biochim. Biophys. Acta 2011, 1810, 1110–1113. [Google Scholar] [CrossRef] [Green Version]
- Cornick, S.; Kumar, M.; Moreau, F.; Gaisano, H.; Chadee, K. VAMP8-mediated MUC2 mucin exocytosis from colonic goblet cells maintains innate intestinal homeostasis. Nat. Commun. 2019, 10, 4306. [Google Scholar] [CrossRef]
- Birchenough, G.M.; Nystrom, E.E.; Johansson, M.E.; Hansson, G.C. A sentinel goblet cell guards the colonic crypt by triggering Nlrp6-dependent Muc2 secretion. Science 2016, 352, 1535–1542. [Google Scholar] [CrossRef] [Green Version]
- Wlodarska, M.; Thaiss, C.A.; Nowarski, R.; Henao-Mejia, J.; Zhang, J.P.; Brown, E.M.; Frankel, G.; Levy, M.; Katz, M.N.; Philbrick, W.M.; et al. NLRP6 inflammasome orchestrates the colonic host-microbial interface by regulating goblet cell mucus secretion. Cell 2014, 156, 1045–1059. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cornick, S.; Moreau, F.; Gaisano, H.Y.; Chadee, K. Entamoeba histolytica-induced mucin exocytosis is mediated by VAMP8 and is critical in mucosal innate host defense. mBio 2017, 8, e01323. [Google Scholar] [CrossRef] [Green Version]
- Jones, L.C.; Moussa, L.; Fulcher, M.L.; Zhu, Y.; Hudson, E.J.; O’Neal, W.K.; Randell, S.H.; Lazarowski, E.R.; Boucher, R.C.; Kreda, S.M. VAMP8 is a vesicle SNARE that regulates mucin secretion in airway goblet cells. J. Physiol. 2012, 590, 545–562. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.; Zhang, X.; Sun, Y.; Shi, J.; Jiang, D.; Wang, J.; Liu, Y.; Hu, C.; Pan, J.; Zheng, L.; et al. MicroRNA-362-3p inhibits migration and invasion via targeting BCAP31 in cervical cancer. Front. Mol. Biosci. 2020, 7, 107. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Pineiro, A.M.; van der Post, S.; Johansson, M.E.; Thomsson, K.A.; Nesvizhskii, A.I.; Hansson, G.C. Proteomic study of the mucin granulae in an intestinal goblet cell model. J. Proteome Res. 2012, 11, 1879–1890. [Google Scholar] [CrossRef]
- De Iudicibus, S.; Lucafo, M.; Vitulo, N.; Martelossi, S.; Zimbello, R.; De Pascale, F.; Forcato, C.; Naviglio, S.; Di Silvestre, A.; Gerdol, M.; et al. High-throughput sequencing of microRNAs in glucocorticoid sensitive paediatric inflammatory bowel disease patients. Int. J. Mol. Sci. 2018, 19, 1399. [Google Scholar] [CrossRef] [Green Version]
- Reily, C.; Stewart, T.J.; Renfrow, M.B.; Novak, J. Glycosylation in health and disease. Nat. Rev. Nephrol. 2019, 15, 346–366. [Google Scholar] [CrossRef]
- Arike, L.; Hansson, C.G. The densely O-Glycosylated MUC2 mucin protects the intestine and provides food for the commensal bacteria. J. Mol. Biol. 2016, 428, 3221–3229. [Google Scholar] [CrossRef] [Green Version]
- Kawashima, H. Roles of the gel-forming MUC2 mucin and its O-glycosylation in the protection against colitis and colorectal cancer. Biol. Pharm. Bull. 2012, 35, 1637–1641. [Google Scholar] [CrossRef] [Green Version]
- Huang, L.; Sun, T.Y.; Hu, L.J.; Hu, S.L.; Sun, H.M.; Zhao, F.Q.; Wu, B.; Yang, S.; Ji, F.Q.; Zhou, D.S. Elevated miR-124-3p in the aging colon disrupts mucus barrier and increases susceptibility to colitis by targeting T-synthase. Aging Cell. 2020, 19, e13252. [Google Scholar] [CrossRef]
- Sun, X.; Zhan, M.; Sun, X.; Liu, W.; Meng, X. C1GALT1 in health and disease. Oncol. Lett. 2021, 22, 589. [Google Scholar] [CrossRef]
- Bergstrom, K.; Fu, J.; Johansson, M.E.; Liu, X.; Gao, N.; Wu, Q.; Song, J.; McDaniel, J.M.; McGee, S.; Chen, W.; et al. Core 1- and 3-derived O-glycans collectively maintain the colonic mucus barrier and protect against spontaneous colitis in mice. Mucosal. Immunol. 2017, 10, 91–103. [Google Scholar] [CrossRef] [Green Version]
- Sun, Q.; Lan, J.; Zhang, H.; Zhou, N.; Liang, Y.; Liu, X. MicroRNA196b targets COSMC in pediatric IgA nephropathy. Mol. Med. Rep. 2020, 21, 2260–2266. [Google Scholar]
- Gao, Y.; Yu, Z. MicroRNA16 inhibits interleukin13induced inflammatory cytokine secretion and mucus production in nasal epithelial cells by suppressing the IkappaB kinase beta/nuclear factorkappaB pathway. Mol. Med. Rep. 2018, 18, 4042–4050. [Google Scholar] [PubMed] [Green Version]
- Zhu, Y.M.; Wu, F.; Zhou, J.Y. Analysis the effect of miR-141-3p/HMGB1 in LPS-induced mucus production and the apoptosis in nasal epithelial cells. Kaohsiung J. Med. Sci. 2020, 36, 622–629. [Google Scholar] [CrossRef] [Green Version]
- Teng, Y.; Zhang, R.; Liu, C.; Zhou, L.; Wang, H.; Zhuang, W.; Huang, Y.; Hong, Z. MiR-143 inhibits interleukin-13-induced inflammatory cytokine and mucus production in nasal epithelial cells from allergic rhinitis patients by targeting IL13Ralpha1. Biochem. Biophys. Res. Commun. 2015, 457, 58–64. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Z.; Dai, L.L.; Wang, X.; Jia, L.Q.; Jing, X.G.; Li, P.F.; Liu, M.; Wang, H.; An, L. MicroRNA-145 down-regulates mucin 5AC to alleviate airway remodeling and targets EGFR to inhibit cytokine expression. Oncotarget 2017, 8, 46312–46325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Wang, P.; Chen, D.; Xu, Z.; Yang, L. CircARRDC3 contributes to interleukin13induced inflammatory cytokine and mucus production in nasal epithelial cells via the miR375/KLF4 axis. Mol. Med. Rep. 2021, 23, 1. [Google Scholar]
- Liu, Z.; Chen, X.; Wu, Q.; Song, J.; Wang, L.; Li, G. MiR-125b inhibits goblet cell differentiation in allergic airway inflammation by targeting SPDEF. Eur. J. Pharmacol. 2016, 782, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Malmhall, C.; Alawieh, S.; Lu, Y.; Sjostrand, M.; Bossios, A.; Eldh, M.; Radinger, M. MicroRNA-155 is essential for T(H)2-mediated allergen-induced eosinophilic inflammation in the lung. J. Allergy Clin. Immunol. 2014, 133, 1429–1438. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, E.; Li, X.; Zhang, M.; Tang, Z.; He, L.; Lv, K. MiR-155 contributes to Df1-induced asthma by increasing the proliferative response of Th cells via CTLA-4 downregulation. Cell Immunol. 2017, 314, 1–9. [Google Scholar] [CrossRef]
- Odenwald, M.A.; Turner, J.R. The intestinal epithelial barrier: A therapeutic target? Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Tili, E.; Michaille, J.J.; Piurowski, V.; Rigot, B.; Croce, C.M. MicroRNAs in intestinal barrier function, inflammatory bowel disease and related cancers-their effects and therapeutic potentials. Curr. Opin. Pharmacol. 2017, 37, 142–150. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Zeng, H.; Lei, L.; Tong, X.; Yang, L.; Yang, Y.; Li, S.; Zhou, Y.; Luo, L.; Huang, J.; et al. Tight junctions and their regulation by non-coding RNAs. Int. J. Biol. Sci. 2021, 17, 712–727. [Google Scholar] [CrossRef]
- Gunzel, D.; Yu, A.S. Claudins and the modulation of tight junction permeability. Physiol. Rev. 2013, 93, 525–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, L.; Weber, C.R.; Raleigh, D.R.; Yu, D.; Turner, J.R. Tight junction pore and leak pathways: A dynamic duo. Annu. Rev. Physiol. 2011, 73, 283–309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gassler, N.; Rohr, C.; Schneider, A.; Kartenbeck, J.; Bach, A.; Obermuller, N.; Otto, H.F.; Autschbach, F. Inflammatory bowel disease is associated with changes of enterocytic junctions. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 281, G216–G228. [Google Scholar] [CrossRef]
- Zeissig, S.; Burgel, N.; Gunzel, D.; Richter, J.; Mankertz, J.; Wahnschaffe, U.; Kroesen, A.J.; Zeitz, M.; Fromm, M.; Schulzke, J.D. Changes in expression and distribution of claudin 2, 5 and 8 lead to discontinuous tight junctions and barrier dysfunction in active Crohn’s disease. Gut 2007, 56, 61–72. [Google Scholar] [CrossRef]
- Blair, S.A.; Kane, S.V.; Clayburgh, D.R.; Turner, J.R. Epithelial myosin light chain kinase expression and activity are upregulated in inflammatory bowel disease. Lab. Investig. 2006, 86, 191–201. [Google Scholar] [CrossRef] [Green Version]
- Weber, C.R.; Nalle, S.C.; Tretiakova, M.; Rubin, D.T.; Turner, J.R. Claudin-1 and claudin-2 expression is elevated in inflammatory bowel disease and may contribute to early neoplastic transformation. Lab. Investig. 2008, 88, 1110–1120. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Shen, J.; Cheng, J.; Fan, X. MicroRNA-21 regulates intestinal epithelial tight junction permeability. Cell Biochem. Funct. 2015, 33, 235–240. [Google Scholar] [CrossRef] [PubMed]
- Nakata, K.; Sugi, Y.; Narabayashi, H.; Kobayakawa, T.; Nakanishi, Y.; Tsuda, M.; Hosono, A.; Kaminogawa, S.; Hanazawa, S.; Takahashi, K. Commensal microbiota-induced microRNA modulates intestinal epithelial permeability through the small GTPase ARF4. J. Biol. Chem. 2017, 292, 15426–15433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vega, F.M.; Colomba, A.; Reymond, N.; Thomas, M.; Ridley, A.J. RhoB regulates cell migration through altered focal adhesion dynamics. Open. Biol. 2012, 2, 120076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Li, C.; Chen, S.; Lin, H.; Zhao, H.; Liu, M.; Weng, J.; Liu, T.; Li, X.; Lei, C.; et al. MicroRNA-21 increases the expression level of occludin through regulating ROCK1 in prevention of intestinal barrier dysfunction. J. Cell Biochem. 2019, 120, 4545–4554. [Google Scholar] [CrossRef]
- Zhuang, Y.; Peng, H.; Mastej, V.; Chen, W. MicroRNA regulation of endothelial junction proteins and clinical consequence. Mediat. Inflamm. 2016, 2016, 5078627. [Google Scholar] [CrossRef]
- Rawat, M.; Nighot, M.; Al-Sadi, R.; Gupta, Y.; Viszwapriya, D.; Yochum, G.; Koltun, W.; Ma, T.Y. IL1B increases intestinal tight junction permeability by up-regulation of MIR200C-3p, which degrades occludin mRNA. Gastroenterology 2020, 159, 1375–1389. [Google Scholar] [CrossRef]
- Ye, D.; Guo, S.; Al-Sadi, R.; Ma, T.Y. MicroRNA regulation of intestinal epithelial tight junction permeability. Gastroenterology 2011, 141, 1323–1333. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Xue, H.; Lin, R.; Huang, Z. MiR-34c and PlncRNA1 mediated the function of intestinal epithelial barrier by regulating tight junction proteins in inflammatory bowel disease. Biochem. Biophys. Res. Commun. 2017, 486, 6–13. [Google Scholar] [CrossRef]
- Tang, S.; Guo, W.; Kang, L.; Liang, J. MiRNA-182-5p aggravates experimental ulcerative colitis via sponging Claudin-2. J. Mol. Histol. 2021, 1–10. [Google Scholar] [CrossRef]
- Zhi, X.; Tao, J.; Li, Z.; Jiang, B.; Feng, J.; Yang, L.; Xu, H.; Xu, Z. MiR-874 promotes intestinal barrier dysfunction through targeting AQP3 following intestinal ischemic injury. FEBS Lett. 2014, 588, 757–763. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.Y.; Cui, Y.H.; Xiao, L.; Chung, H.K.; Zhang, Y.; Rao, J.N.; Gorospe, M.; Wang, J.Y. Regulation of intestinal epithelial barrier function by long noncoding RNA uc.173 through interaction with microRNA 29b. Mol. Cell. Biol. 2018, 38, e00010-18. [Google Scholar] [CrossRef] [Green Version]
- Xiao, L.; Ma, X.X.; Luo, J.; Chung, H.K.; Kwon, M.S.; Yu, T.X.; Rao, J.N.; Kozar, R.; Gorospe, M.; Wang, J.Y. Circular RNA circHIPK3 promotes homeostasis of the intestinal epithelium by reducing microRNA 29b function. Gastroenterology 2021, 161, 1303–1317. [Google Scholar] [CrossRef]
- Wang, H.; Chao, K.; Ng, S.C.; Bai, A.H.; Yu, Q.; Yu, J.; Li, M.; Cui, Y.; Chen, M.; Hu, J.F.; et al. Pro-inflammatory miR-223 mediates the cross-talk between the IL23 pathway and the intestinal barrier in inflammatory bowel disease. Genome Biol. 2016, 17, 58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, M.; Zhao, J.; Cao, M.; Liu, R.; Chen, G.; Li, S.; Xie, Y.; Xie, J.; Cheng, Y.; Huang, L.; et al. Mast cells-derived MiR-223 destroys intestinal barrier function by inhibition of CLDN8 expression in intestinal epithelial cells. Biol. Res. 2020, 53, 12. [Google Scholar] [CrossRef] [Green Version]
- Veltman, K.; Hummel, S.; Cichon, C.; Sonnenborn, U.; Schmidt, M.A. Identification of specific miRNAs targeting proteins of the apical junctional complex that simulate the probiotic effect of E. coli Nissle 1917 on T84 epithelial cells. Int. J. Biochem. Cell Biol. 2012, 44, 341–349. [Google Scholar] [CrossRef] [PubMed]
- Soroosh, A.; Rankin, C.R.; Polytarchou, C.; Lokhandwala, Z.A.; Patel, A.; Chang, L.; Pothoulakis, C.; Iliopoulos, D.; Padua, D.M. MiR-24 is elevated in ulcerative colitis patients and regulates intestinal epithelial barrier function. Am. J. Pathol. 2019, 189, 1763–1774. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, L.; Zhang, R.; Chen, J.; Wu, Q.; Kuang, Z. Baicalin protects against TNF-alpha-induced injury by down-regulating miR-191a that targets the tight junction protein ZO-1 in IEC-6 Cells. Biol. Pharm. Bull. 2017, 40, 435–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, X.; Li, J.; Ma, J.; Jiao, C.; Qiu, X.; Cui, X.; Wang, D.; Zhang, H. MiR-124a mediates the impairment of intestinal epithelial integrity by targeting aryl hydrocarbon receptor in Crohn’s disease. Inflammation 2020, 43, 1862–1875. [Google Scholar] [CrossRef]
- Yin, B.; Tian-Chu, H.; Ling-Fen, X. Protection by microRNA-7a-5p antagomir against intestinal mucosal injury related to the JNK pathway in TNBS-Induced experimental colitis. Turk J. Gastroenterol. 2021, 32, 431–436. [Google Scholar] [CrossRef]
- Shen, Y.; Zhou, M.; Yan, J.; Gong, Z.; Xiao, Y.; Zhang, C.; Du, P.; Chen, Y. miR-200b inhibits TNF-alpha-induced IL-8 secretion and tight junction disruption of intestinal epithelial cells in vitro. Am. J. Physiol. Gastrointest. Liver Physiol. 2017, 312, G123–G132. [Google Scholar] [CrossRef] [Green Version]
- Cordes, F.; Bruckner, M.; Lenz, P.; Veltman, K.; Glauben, R.; Siegmund, B.; Hengst, K.; Schmidt, M.A.; Cichon, C.; Bettenworth, D. MicroRNA-320a strengthens intestinal barrier function and follows the course of experimental colitis. Inflamm. Bowel Dis. 2016, 22, 2341–2355. [Google Scholar] [CrossRef] [Green Version]
- Haines, R.J.; Beard, R.S., Jr.; Eitner, R.A.; Chen, L.; Wu, M.H. TNFalpha/IFNgamma mediated intestinal epithelial barrier dysfunction is attenuated by microRNA-93 downregulation of PTK6 in mouse colonic epithelial cells. PLoS ONE 2016, 11, e0154351. [Google Scholar] [CrossRef] [Green Version]
- Xiong, Y.; Wang, J.; Chu, H.; Chen, D.; Guo, H. Salvianolic acid B restored impaired barrier function via downregulation of MLCK by microRNA-1 in rat colitis model. Front. Pharmacol. 2016, 7, 134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Sadi, R.; Engers, J.; Abdulqadir, R. Talk about micromanaging! Role of microRNAs in intestinal barrier function. Am. J. Physiol. Gastrointest. Liver Physiol. 2020, 319, G170–G174. [Google Scholar] [CrossRef] [PubMed]
- Hartsock, A.; Nelson, W.J. Adherens and tight junctions: Structure, function and connections to the actin cytoskeleton. Biochim. Biophys. Acta 2008, 1778, 660–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takeichi, M. Dynamic contacts: Rearranging adherens junctions to drive epithelial remodelling. Nat. Rev. Mol. Cell Biol. 2014, 15, 397–410. [Google Scholar] [CrossRef]
- Hermiston, M.L.; Gordon, J.I. Inflammatory bowel disease and adenomas in mice expressing a dominant negative N-cadherin. Science 1995, 270, 1203–1207. [Google Scholar] [CrossRef]
- Larue, L.; Ohsugi, M.; Hirchenhain, J.; Kemler, R. E-cadherin null mutant embryos fail to form a trophectoderm epithelium. Proc. Natl. Acad. Sci. USA 1994, 91, 8263–8267. [Google Scholar] [CrossRef] [Green Version]
- Bondow, B.J.; Faber, M.L.; Wojta, K.J.; Walker, E.M.; Battle, M.A. E-cadherin is required for intestinal morphogenesis in the mouse. Dev. Biol. 2012, 371, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Schneider, M.R.; Dahlhoff, M.; Horst, D.; Hirschi, B.; Trulzsch, K.; Muller-Hocker, J.; Vogelmann, R.; Allgauer, M.; Gerhard, M.; Steininger, S.; et al. A key role for E-cadherin in intestinal homeostasis and Paneth cell maturation. PLoS ONE 2010, 5, e14325. [Google Scholar] [CrossRef] [Green Version]
- Boros, E.; Kellermayer, Z.; Balogh, P.; Strifler, G.; Vӧrӧs, A.; Sarlós, P.; Vincze, Á.; Varga, C.; Nagy, I. Elevated expression of AXL may contribute to the epithelial-to-mesenchymal transition in inflammatory bowel disease patients. Mediat. Inflamm. 2018, 2018, 3241406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Consortium, U.I.G.; Barrett, J.C.; Lee, J.C.; Lees, C.W.; Prescott, N.J.; Anderson, C.A.; Phillips, A.; Wesley, E.; Parnell, K.; Zhang, H.; et al. Genome-wide association study of ulcerative colitis identifies three new susceptibility loci, including the HNF4A region. Nat. Genet. 2009, 41, 1330–1334. [Google Scholar] [CrossRef]
- Vlad-Fiegen, A.; Langerak, A.; Eberth, S.; Muller, O. The Wnt pathway destabilizes adherens junctions and promotes cell migration via beta-catenin and its target gene cyclin D1. FEBS Open. Biol. 2012, 2, 26–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serafino, A.; Moroni, N.; Zonfrillo, M.; Andreola, F.; Mercuri, L.; Nicotera, G.; Nunziata, J.; Ricci, R.; Antinori, A.; Rasi, G.; et al. WNT-pathway components as predictive markers useful for diagnosis, prevention and therapy in inflammatory bowel disease and sporadic colorectal cancer. Oncotarget 2014, 5, 978–992. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, L.; Cao, R.; Liu, Y.; Wang, L.; Pan, B.; Lv, X.; Jiao, H.; Zhuang, Q.; Sun, X.; Xiao, R. MiR-21-5p links epithelial-mesenchymal transition phenotype with stem-like cell signatures via AKT signaling in keloid keratinocytes. Sci. Rep. 2016, 6, 28281. [Google Scholar] [CrossRef] [Green Version]
- Guz, M.; Dworzanski, T.; Jeleniewicz, W.; Cybulski, M.; Kozicka, J.; Stepulak, A.; Celinski, K. Elevated miRNA inversely correlates with E-cadherin gene expression in tissue biopsies from crohn disease patients in contrast to ulcerative colitis patients. Biomed. Res. Int. 2020, 2020, 4250329. [Google Scholar] [CrossRef]
- Lu, J.; Liu, D.; Tan, Y.; Deng, F.; Li, R. M1 macrophage exosomes MiR-21a-5p aggravates inflammatory bowel disease through decreasing E-cadherin and subsequent ILC2 activation. J. Cell Mol. Med. 2021, 25, 3041–3050. [Google Scholar] [CrossRef]
- Yin, J.; Ye, Y.L.; Hu, T.; Xu, L.J.; Zhang, L.P.; Ji, R.N.; Li, P.; Chen, Q.; Zhu, J.Y.; Pang, Z. Hsa_circRNA_102610 upregulation in Crohn’s disease promotes transforming growth factor-beta1-induced epithelial-mesenchymal transition via sponging of hsa-miR-130a-3p. World J. Gastroenterol. 2020, 26, 3034–3055. [Google Scholar] [CrossRef]
- Kline, K.T.; Lian, H.; Zhong, X.S.; Luo, X.; Winston, J.H.; Cong, Y.; Savidge, T.C.; Dashwood, R.H.; Powell, D.W.; Li, Q. Neonatal injury increases gut permeability by epigenetically suppressing E-Cadherin in adulthood. J. Immunol. 2020, 204, 980–989. [Google Scholar] [CrossRef]
- Chen, Y.; Xiao, Y.; Ge, W.; Zhou, K.; Wen, J.; Yan, W.; Wang, Y.; Wang, B.; Qu, C.; Wu, J.; et al. MiR-200b inhibits TGF-beta1-induced epithelial-mesenchymal transition and promotes growth of intestinal epithelial cells. Cell Death Dis. 2013, 4, e541. [Google Scholar] [CrossRef] [Green Version]
- Menzel, K.; Hausmann, M.; Obermeier, F.; Schreiter, K.; Dunger, N.; Bataille, F.; Falk, W.; Scholmerich, J.; Herfarth, H.; Rogler, G. Cathepsins B, L and D in inflammatory bowel disease macrophages and potential therapeutic effects of cathepsin inhibition in vivo. Clin. Exp. Immunol. 2006, 146, 169–180. [Google Scholar] [CrossRef]
- Schlegel, N.; Boerner, K.; Waschke, J. Targeting desmosomal adhesion and signalling for intestinal barrier stabilization in inflammatory bowel diseases-Lessons from experimental models and patients. Acta Physiol. 2021, 231, e13492. [Google Scholar] [CrossRef]
- Garrod, D.; Chidgey, M. Desmosome structure, composition and function. Biochim. Biophys. Acta 2008, 1778, 572–587. [Google Scholar] [CrossRef] [PubMed]
- Borradori, L.; Sonnenberg, A. Structure and function of hemidesmosomes: More than simple adhesion complexes. J. Investig. Dermatol. 1999, 112, 411–418. [Google Scholar] [CrossRef] [Green Version]
- Stutzmann, J.; Bellissent-Waydelich, A.; Fontao, L.; Launay, J.F.; Simon-Assmann, P. Adhesion complexes implicated in intestinal epithelial cell-matrix interactions. Microsc. Res. Tech. 2000, 51, 179–190. [Google Scholar] [CrossRef]
- Gross, A.; Pack, L.A.P.; Schacht, G.M.; Kant, S.; Ungewiss, H.; Meir, M.; Schlegel, N.; Preisinger, C.; Boor, P.; Guldiken, N.; et al. Desmoglein 2, but not desmocollin 2, protects intestinal epithelia from injury. Mucosal. Immunol. 2018, 11, 1630–1639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flemming, S.; Luissint, A.C.; Kusters, D.H.M.; Raya-Sandino, A.; Fan, S.; Zhou, D.W.; Hasegawa, M.; Garcia-Hernandez, V.; Garcia, A.J.; Parkos, C.A.; et al. Desmocollin-2 promotes intestinal mucosal repair by controlling integrin-dependent cell adhesion and migration. Mol. Biol. Cell 2020, 31, 407–418. [Google Scholar] [CrossRef]
- Burkard, N.; Meir, M.; Kannapin, F.; Otto, C.; Petzke, M.; Germer, C.T.; Waschke, J.; Schlegel, N. Desmoglein2 regulates Claudin2 expression by sequestering PI-3-Kinase in intestinal epithelial cells. Front. Immunol. 2021, 12, 756321. [Google Scholar] [CrossRef] [PubMed]
- Meir, M.; Salm, J.; Fey, C.; Schweinlin, M.; Kollmann, C.; Kannapin, F.; Germer, C.T.; Waschke, J.; Beck, C.; Burkard, N.; et al. Enteroids Generated from Patients with Severe Inflammation in Crohn’ s Disease Maintain Alterations of Junctional Proteins. J. Crohns Colitis 2020, 14, 1473–1487. [Google Scholar] [CrossRef]
- De Arcangelis, A.; Hamade, H.; Alpy, F.; Normand, S.; Bruyere, E.; Lefebvre, O.; Mechine-Neuville, A.; Siebert, S.; Pfister, V.; Lepage, P.; et al. Hemidesmosome integrity protects the colon against colitis and colorectal cancer. Gut 2017, 66, 1748–1760. [Google Scholar] [CrossRef]
- Alexander, D.B.; Goldberg, G.S. Transfer of biologically important molecules between cells through gap junction channels. Curr. Med. Chem. 2003, 10, 2045–2058. [Google Scholar] [CrossRef]
- Goodenough, D.A.; Paul, D.L. Gap junctions. Cold Spring Harb. Perspect. Biol. 2009, 1, a002576. [Google Scholar] [CrossRef]
- Martin, C.A.; el-Sabban, M.E.; Zhao, L.; Burakoff, R.; Homaidan, F.R. Gap junctional communication between murine macrophages and intestinal epithelial cell lines. Cell Adhes. Commun. 1998, 5, 437–449. [Google Scholar] [CrossRef]
- Martin, C.A.; el-Sabban, M.E.; Zhao, L.; Burakoff, R.; Homaidan, F.R. Adhesion and cytosolic dye transfer between macrophages and intestinal epithelial cells. Cell Adhes. Commun. 1998, 5, 83–95. [Google Scholar] [CrossRef]
- Hyun, J.; Romero, L.; Riveron, R.; Flores, C.; Kanagavelu, S.; Chung, K.D.; Alonso, A.; Sotolongo, J.; Ruiz, J.; Manukyan, A.; et al. Human intestinal epithelial cells express interleukin-10 through Toll-like receptor 4-mediated epithelial-macrophage crosstalk. J. Innate Immun. 2015, 7, 87–101. [Google Scholar] [CrossRef] [PubMed]
- Al-Ghadban, S.; Kaissi, S.; Homaidan, F.R.; Naim, H.Y.; El-Sabban, M.E. Cross-talk between intestinal epithelial cells and immune cells in inflammatory bowel disease. Sci. Rep. 2016, 6, 29783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morita, H.; Katsuno, T.; Hoshimoto, A.; Hirano, N.; Saito, Y.; Suzuki, Y. Connexin 26-mediated gap junctional intercellular communication suppresses paracellular permeability of human intestinal epithelial cell monolayers. Exp. Cell Res. 2004, 298, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Ey, B.; Eyking, A.; Gerken, G.; Podolsky, D.K.; Cario, E. TLR2 mediates gap junctional intercellular communication through connexin-43 in intestinal epithelial barrier injury. J. Biol. Chem. 2009, 284, 22332–22343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zong, L.; Zhu, Y.; Liang, R.; Zhao, H.B. Gap junction mediated miRNA intercellular transfer and gene regulation: A novel mechanism for intercellular genetic communication. Sci. Rep. 2016, 6, 19884. [Google Scholar] [CrossRef] [Green Version]


| MicroRNA | Expression Level | Sample | Biomarker | References |
|---|---|---|---|---|
| let-7f | upregulated | colonic tissue | diagnosed UC patients | [48] |
| miR-16 | downregulated | colonic tissue; plasma | active UC; diagnosis of CD | [48,49] |
| upregulated | serum and colonic mucosa; blood; biopsy; colonic tissue | canine IBD model; diagnosed IBD patients; diagnosed UC patients | [16,38,40,48] | |
| miR-20b | differential pattern | colonic mucosa | active vs. quiescence UC | [47] |
| miR-21 | upregulated | colonic tissue; blood; serum; saliva | diagnosed UC patients; diagnosed IBD patients; canine IBD model | [16,36,38,40,48] |
| miR-23a | upregulated | colonic tissue | diagnosed UC patients | [48] |
| miR-24 | upregulated | colonic tissue | diagnosed UC patients | [48] |
| miR-26b | differential pattern | colonic mucosa | active vs. quiescence UC | [47] |
| miR-29a | upregulated | colonic tissue | diagnosed UC patients | [48] |
| miR-31 | upregulated | colonic mucosa, saliva | diagnosed IBD and UC patients | [36,37] |
| miR-31-5p | differential pattern | colonic tissue | diagnostic marker for CD | [51] |
| miR-98 | differential pattern | colonic mucosa | active vs. quiescence UC | [47] |
| miR-99a | differential pattern | colonic mucosa | active vs. quiescence UC | [47] |
| miR-101 | upregulated | saliva | CD | [36] |
| miR-106a | upregulated | blood/biopsy | diagnosed IBD patients | [16,40,41] |
| miR-122 | upregulated | blood/biopsy; serum and colonic mucosa | diagnosed IBD patients; canine IBD model | [16,38,40] |
| miR-126 | upregulated | colonic tissue | diagnosed UC patients | [48] |
| miR-142-3p | upregulated | saliva | UC | [36] |
| miR-142-5p | differential pattern | serum | active vs. quiescence CD | [33] |
| downregulated | saliva | UC | [36] | |
| miR-146a | upregulated | colonic mucosa; serum | diagnosed IBD patients; canine IBD model | [37,38] |
| miR-147 | upregulated | serum and colonic mucosa | canine IBD model | [38] |
| miR-150 | differential pattern | colonic tissue | non-inflamed UC vs. non-inflamed CD | [46] |
| miR-151-5p | upregulated | blood/biopsy | diagnosed IBD patients | [16,39,40] |
| miR-155 | upregulated | blood/biopsy | diagnosed IBD patients | [16,39,40] |
| miR-192 | upregulated | serum; colonic tissue | canine IBD model; active UC | [38,48,49] |
| miR-192 | downregulated | colonic tissue | diagnosed UC patients | [48] |
| miR-195 | upregulated | colonic tissue | diagnosed UC patients | [48] |
| miR-196b | differential pattern | colonic tissue | non-inflamed UC vs. non-inflamed CD | [46] |
| miR-199a-3p | differential pattern | colonic tissue | non-inflamed UC vs. non-inflamed CD | [46] |
| miR-199a-5p | upregulated | blood/biopsy | diagnosed IBD patients | [16,40] |
| miR-199b-5p | differential pattern | colonic tissue | non-inflamed UC vs. non-inflamed CD | [46] |
| miR-203 | differential pattern | colonic tissue | active vs. quiescence UC; diagnostic marker for CD | [47,51] |
| miR-206 | upregulated | colonic mucosa | diagnosed IBD patients | [37] |
| miR-223 | upregulated | serum | canine IBD model | [38] |
| differential pattern | colonic tissue | non-inflamed UC vs. non-inflamed CD | [46] | |
| miR-320 | upregulated | blood/biopsy | diagnosed IBD patients | [16,39,40] |
| miR-320a | differential pattern | colonic tissue | non-inflamed UC vs. non-inflamed CD | [46] |
| miR-362-3p | upregulated | blood/biopsy | diagnosed IBD patients | [16,40,41] |
| miR-375 | downregulated | colonic tissue | diagnosed UC patients | [48] |
| miR-422b | downregulated | colonic tissue | diagnosed UC patients | [48] |
| miR-424 | upregulated | colonic mucosa | diagnosed IBD patients | [37] |
| miR-595 | differential pattern | serum | active vs. quiescence CD | [33] |
| upregulated | serum | non-specific biomarker for IBD | [33] | |
| miR-1246 | upregulated | serum | non-specific biomarker for IBD | [33] |
| differential pattern | serum | active vs. quiescence CD and UC | [33] | |
| miR-1307-3p | upregulated | blood (CD4+ T-cells) | disease progression in IBD | [35] |
| miR-3615 | upregulated | blood (CD4+ T-cells) | disease progression in IBD | [35] |
| miR-4284 | downregulated | colonic tissue | active UC | [50] |
| miR-4792 | expression | blood (CD4+ T-cells) | disease progression in IBD | [35] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stiegeler, S.; Mercurio, K.; Iancu, M.A.; Corr, S.C. The Impact of MicroRNAs during Inflammatory Bowel Disease: Effects on the Mucus Layer and Intercellular Junctions for Gut Permeability. Cells 2021, 10, 3358. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10123358
Stiegeler S, Mercurio K, Iancu MA, Corr SC. The Impact of MicroRNAs during Inflammatory Bowel Disease: Effects on the Mucus Layer and Intercellular Junctions for Gut Permeability. Cells. 2021; 10(12):3358. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10123358
Chicago/Turabian StyleStiegeler, Sarah, Kevin Mercurio, Miruna Alexandra Iancu, and Sinéad C. Corr. 2021. "The Impact of MicroRNAs during Inflammatory Bowel Disease: Effects on the Mucus Layer and Intercellular Junctions for Gut Permeability" Cells 10, no. 12: 3358. https://0-doi-org.brum.beds.ac.uk/10.3390/cells10123358