
Morphological Characteristics of Idiopathic Inflammatory Myopathies in Juvenile Patients

 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Subjects and Samples
2.2. Muscle Pathology Analysis
2.2.1. Histochemical, Enzymatic and Immunohistochemical Microscopy
2.2.2. Transmission Electron Microscopy (TEM)
2.3. Statistical Analysis
3. Results
3.1. Clinical Data
3.2. Muscle Pathology
3.2.1. Muscle Pathology Score and Inflammatory Cell Invasion Are Highly Variable in All IIM Subtypes
3.2.2. COX Deficient Fibers Are a Striking Pathology in DM Biopsies
3.2.3. Perimysial Alkaline Phosphatase (ALP) Positivity Is Specific for Anti-Jo-1–ASyS
3.2.4. Sarcolemmal Upregulation of MHC Class I, MHC Class II and Sarcolemmal Complement Deposits
3.2.5. Vascular Pathology and Upregulation of Proangiogenic Factor VEGF
3.2.6. Ultrastructural Pathology
3.2.7. IFN 1 Surrogate Marker MxA Upregulation Correlates with Endothelial Inclusions
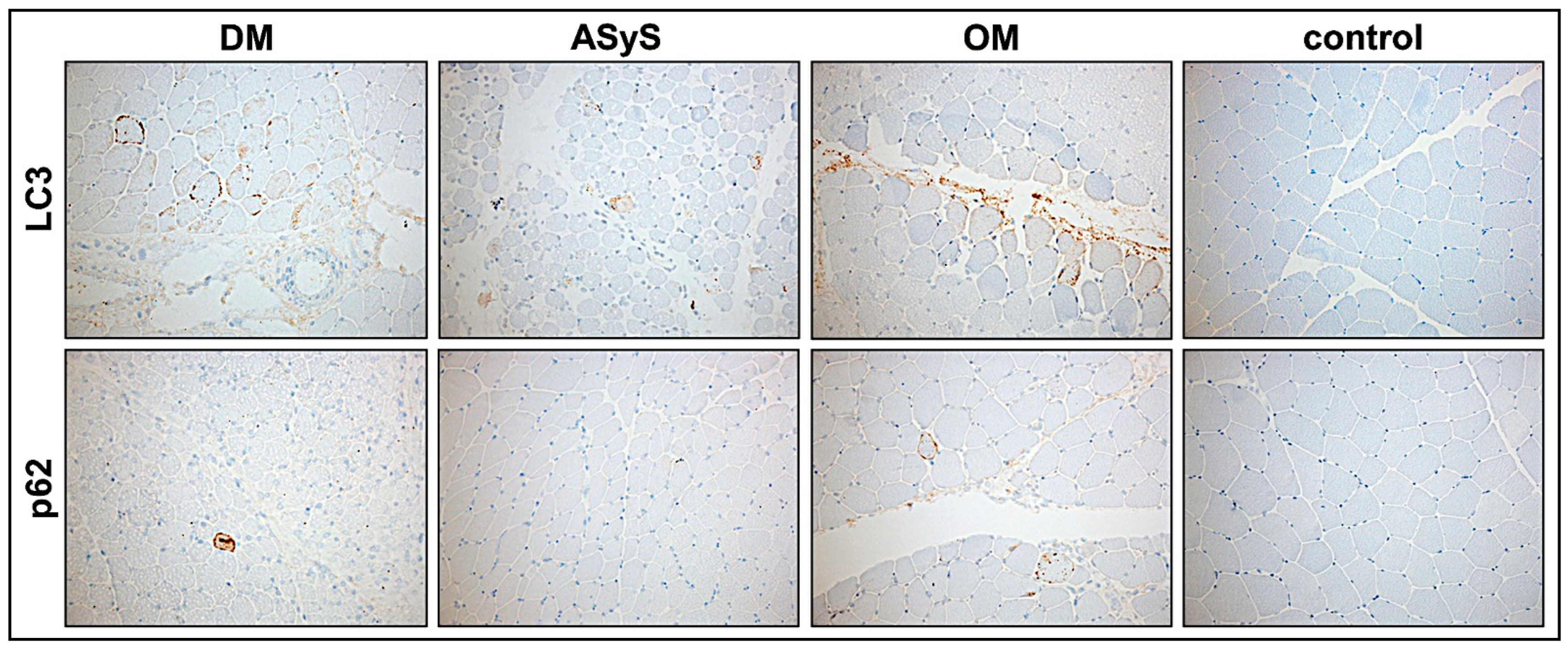
3.2.8. Moderate Activation of Autophagy in jIIM
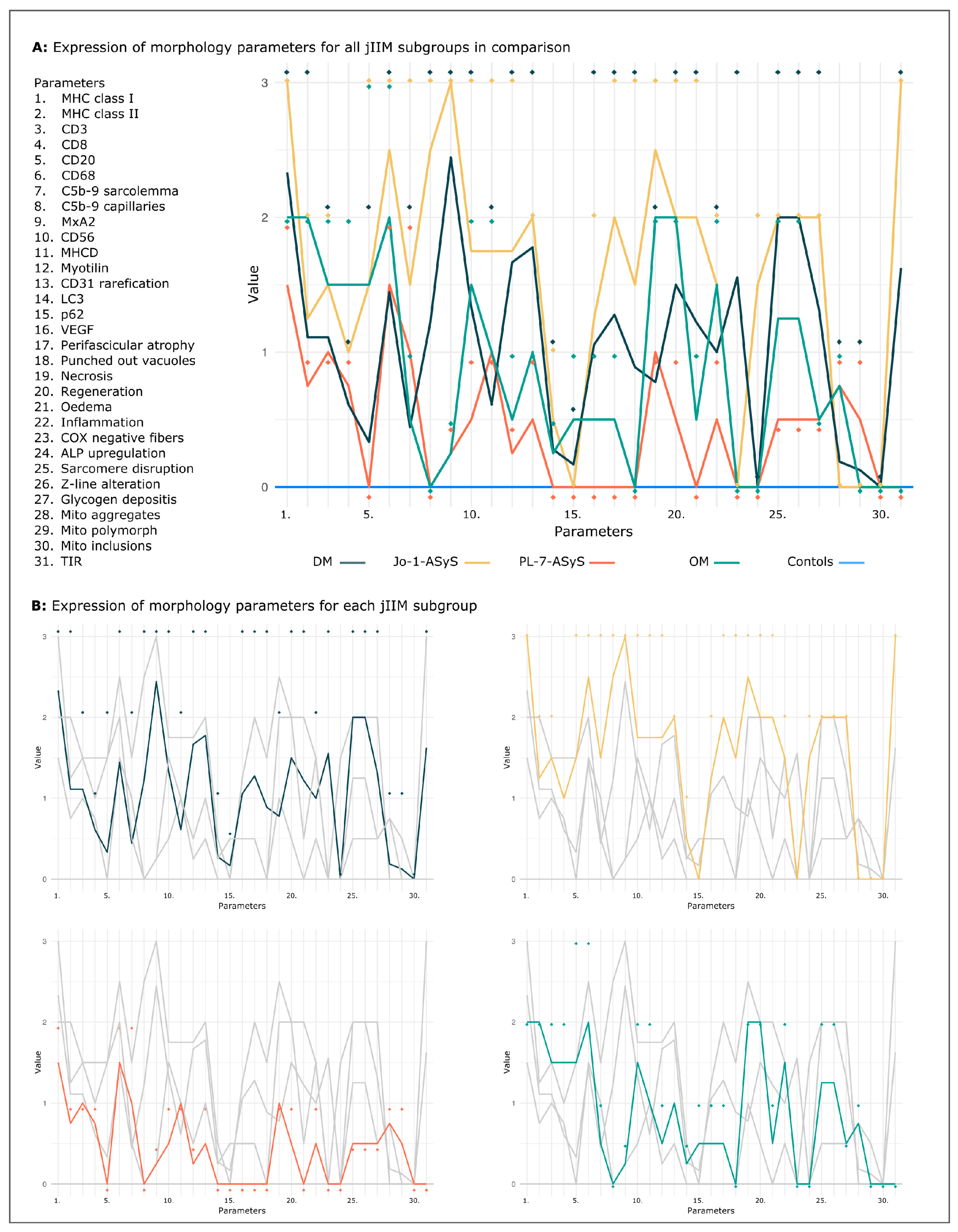
3.2.9. Common Staining Pattern and Discriminative Key Findings in jIIM Subtypes
4. Discussion
5. Limitations
6. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
Appendix A

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Diagnosis | VAS-Score | HE | COX-SDH | ALP | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Perifascicular Atrophy (PA) | Punched-Out-Vacuoles (POV) | Necrosis | Regeneration | Oedema | Inflammation | COX-Negative Fibers | Upregulation | |||
| P1 | DM | 4 | 1 | 0 | 1 | 2 | 1 | 1 | 2 | 0 |
| P2 | DM | 6 | 3 | 1 | 0 | 3 | 1 | 1 | 3 | 0 |
| P5 | DM | 6 | 1 | 3 | 1 | 1 | 3 | 2 | 3 | 0 |
| P10 | DM | 4 | 0.5 | 0 | 1 | 1 | 1 | 0.5 | 0 | 0 |
| P11 | DM | 1 | 0 | 0 | 0 | 0 | 0 | 0.5 | 0 | 0 |
| P12 | DM | 8 | 3 | 1 | 2 | 3 | 2 | 2 | 3 | 0 |
| P15 | DM | 2 | 0 | 0 | 1 | 0.5 | 1 | 0.5 | 0 | 0 |
| P4 | DM (NXP-2) | 9 | 3 | 3 | 1 | 3 | 2 | 1 | 3 | 0 |
| P13 | DM (NXP-2) | 1 | 0 | 0 | 0 | 0 | 0 | 0.5 | 0 | 0 |
| P8 | ASyS (Jo-1) | 4 | 1 | 0 | 2 | 1 | 1 | 1 | 0 | 1 |
| P14 | ASyS (Jo-1) | 9 | 3 | 3 | 3 | 3 | 3 | 2 | 0 | 2 |
| P6 | ASyS (PL-7) | 2 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 |
| P9 | ASyS (PL-7) | 2 | 0 | 0 | 1 | 1 | 0 | 0 | 0 | 0 |
| P3 | OM (SLE) | 4 | 1 | 0 | 2 | 2 | 0 | 1 | 0 | 0 |
| P7 | OM (SLE) | 6 | 0 | 0 | 2 | 2 | 1 | 2 | 0 | 0 |
| Diagnosis | Inflammation and Immunoactivation | Complement Deposits (c5b-9) | MxA | Regen- Eration | Sarcomere Disruption | Angiogenesis | Autophagy | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| MHC1 | MHC2 | CD3 | CD8 | CD20 | CD68 | Sarco-Lemma | Capil-laries | CD56 | MHCd | Myo-Tilin | CD31 Loss | VEGF | LC 3 | p62 | |||
| P1 | DM | 2 | 1 | 1 | 1 | 0 | 1 | 2 | 2 | 3 | 2 | 0 | 1 | 1 | 1 | 0 | 0 |
| P2 | DM | 2 | 1 | 1 | 1 | 0 | 1 | 1 | 0 | 3 | 1 | 1 | 2 | 2 | 2 | 0 | 0 |
| P5 | DM | 3 | 0.5 | 2 | 1 | 2 | 3 | 0 | 1 | 3 | 3 | 2 | 3 | 2 | 3 | 0.5 | 0.5 |
| P10 | DM | 3 | 3 | 2 | 1 | 0 | 1 | 0 | 3 | 2 | 0 | 0 | 2 | 2 | 1 | 0 | 0 |
| P11 | DM | 2 | 0 | 0.5 | 0 | 0 | 1 | 0 | 0.5 | 2 | 0 | 0 | 0 | 1 | 0 | 0 | 0 |
| P12 | DM | 3 | 2 | 1 | 0 | 0 | 2 | 0 | 0.5 | 3 | 2 | 2 | 3 | 3 | 2 | 1 | 0.5 |
| P15 | DM | 3 | 0.5 | 0.5 | 0.5 | 0 | 1 | 0 | 2 | 3 | 1 | 0 | 2 | 1 | 0 | 0 | 0 |
| P4 | DM (NXP-2) | 3 | 2 | 2 | 1 | 1 | 2 | 1 | 2 | 3 | 3 | 0.5 | 2 | 3 | 0.5 | 1 | 0.5 |
| P13 | DM (NXP-2) | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | 0 |
| P8 | ASyS (Jo-1) | 3 | 0.5 | 1 | 1 | 0 | 2 | 0 | 3 | 3 | 0.5 | 0.5 | 0.5 | 2 | 0.5 | 0 | 0 |
| P14 | ASyS (Jo-1) | 3 | 2 | 2 | 1 | 3 | 3 | 3 | 2 | 3 | 3 | 3 | 3 | 2 | 2 | 1 | 0 |
| P6 | ASyS (PL-7) | 2 | 0.5 | 1 | 1 | 0 | 2 | 2 | 0 | 0.5 | 0 | 1 | 0 | 1 | 0 | 0 | 0 |
| P9 | ASyS (PL-7) | 1 | 1 | 1 | 0.5 | 0 | 1 | 0 | 0 | 0 | 1 | 1 | 0.5 | 0 | 0 | 0 | 0 |
| P3 | OM (SLE) | 2 | 2 | 2 | 2 | 3 | 3 | 1 | 0 | 0.5 | 2 | 2 | 1 | 1 | 1 | 0.5 | 1 |
| P7 | OM (SLE) | 2 | 2 | 1 | 1 | 0 | 1 | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | 0 |
| Diagnosis | Myofibrillar Structure | Mitochondria | Endomysial Capillaries | Nuclear Inclusions | |||||
|---|---|---|---|---|---|---|---|---|---|
| Sarcomere Structure Disruption | Z-Line-Alteration | Glycogen Deposits | Aggregation | Polymorphism | Inclusions | Tubuloreticular Structures (TIR) | |||
| P1 | DM | 1 | 1 | 2 | 1 | 1 | 0 | 2 (3/10) | n.a. |
| P2 | DM | 3 | 3 | 0 | 0.5 | 0 | 0 | 2 (4/10) | n.a. |
| P5 | DM | n.a. | |||||||
| P10 | DM | 2 | 2 | 2 | 0 | 0 | 0 | 1 (2/10) | n.a. |
| P11 | DM | 1 | 1 | 3 | 0 | 0 | 0 | 3 (6/10) | n.a. |
| P12 | DM | 3 | 3 | 0.5 | 0 | 0 | 0 | 1 (2/10) | n.a. |
| P15 | DM | 3 | 3 | 2 | 0 | 0 | 0 | 1 (2/10) | n.a. |
| P4 | DM (NXP-2) | 3 | 3 | 1 | 0 | 0 | 0 | 3 (8/10) | n.a. |
| P13 | DM (NXP-2) | 0 | 0 | 0 | 0 | 0 | 0 | 0 (0/10) | n.a. |
| P8 | ASyS (Jo-1) | n.a. | |||||||
| P14 | ASyS (Jo-1) | 2 | 2 | 2 | 0 | 0 | 0 | 3 (3/5) | 3/410 |
| P6 | ASyS (PL-7) | 0.5 | 0.5 | 0.5 | 0.5 | 0 | 0 | 0 (0/10) | 0/205 |
| P9 | ASyS (PL-7) | 0.5 | 0.5 | 0.5 | 1 | 1 | 0 | 0 (0/10) | 0/210 |
| P3 | OM (SLE) | 0.5 | 0.5 | 0.5 | 0.5 | 0 | 0 | 0 (0/10) | n.a. |
| P7 | OM (SLE) | 2 | 2 | 0.5 | 1 | 0 | 0 | 0 (0/10) | n.a. |
References
- Ladislau, L.; Suarez-Calvet, X.; Toquet, S.; Landon-Cardinal, O.; Amelin, D.; Depp, M.; Rodero, M.P.; Hathazi, D.; Duffy, D.; Bondet, V.; et al. JAK inhibitor improves type I interferon induced damage: Proof of concept in dermatomyositis. Brain 2018, 141, 1609–1621. [Google Scholar] [CrossRef]
- Melki, I.; Devilliers, H.; Gitiaux, C.; Bondet, V.; Belot, A.; Bodemer, C.; Quartier, P.; Crow, Y.J.; Duffy, D.; Rodero, M.P.; et al. Circulating Interferon-α Measured With a Highly Sensitive Assay as a Biomarker for Juvenile Inflammatory Myositis Activity: Comment on the Article by Mathian et al. Arthritis Rheumatol. 2020, 72, 195–197. [Google Scholar] [CrossRef] [PubMed]
- Allenbach, Y.; Benveniste, O.; Goebel, H.H.; Stenzel, W. Integrated classification of inflammatory myopathies. Neuropathol. Appl. Neurobiol. 2017, 43, 62–81. [Google Scholar] [CrossRef]
- De Bleecker, J.L.; De Paepe, B.; Aronica, E.; de Visser, M.; Amato, A.; Benveniste, O.; De Bleecker, J.; de Boer, O.; Dimachkie, M.; Gherardi, R.; et al. 205th ENMC International Workshop: Pathology diagnosis of idiopathic inflammatory myopathies part II, 28–30 March 2014, Naarden, The Netherlands. Neuromuscul. Disord. 2015, 25, 268–272. [Google Scholar] [CrossRef] [PubMed]
- Mariampillai, K.; Granger, B.; Amelin, D.; Guiguet, M.; Hachulla, E.; Maurier, F.; Meyer, A.; Tohme, A.; Charuel, J.L.; Musset, L.; et al. Development of a New Classification System for Idiopathic Inflammatory Myopathies Based on Clinical Manifestations and Myositis-Specific Autoantibodies. JAMA Neurol. 2018, 75, 1528–1537. [Google Scholar] [CrossRef] [Green Version]
- Tanboon, J.; Uruha, A.; Stenzel, W.; Nishino, I. Where are we moving in the classification of idiopathic inflammatory myopathies? Curr. Opin. Neurol. 2020, 33, 590–603. [Google Scholar] [CrossRef] [PubMed]
- Siegert, E.; Uruha, A.; Goebel, H.H.; Preuße, C.; Casteleyn, V.; Kleefeld, F.; Alten, R.; Burmester, G.R.; Schneider, U.; Höppner, J.; et al. Systemic sclerosis-associated myositis features minimal inflammation and characteristic capillary pathology. Acta Neuropathol. 2021, 141, 917–927. [Google Scholar] [CrossRef]
- Fredi, M.; Cavazzana, I.; Franceschini, F. The clinico-serological spectrum of overlap myositis. Curr. Opin. Rheumatol. 2018, 30, 637–643. [Google Scholar] [CrossRef]
- Patwardhan, A.; Spencer, C.H. Biologics in refractory idiopathic inflammatory myositis (IIM): What experience in juvenile vs adult myositis tells us about the use of biologics in pediatric IIM. Mod. Rheumatol. 2021, 31, 933–948. [Google Scholar] [CrossRef]
- Rider, L.G.; Nistala, K. The juvenile idiopathic inflammatory myopathies: Pathogenesis, clinical and autoantibody phenotypes, and outcomes. J. Intern. Med. 2016, 280, 24–38. [Google Scholar] [CrossRef] [Green Version]
- Shah, M.; Mamyrova, G.; Targoff, I.N.; Huber, A.M.; Malley, J.D.; Rice, M.M.; Miller, F.W.; Rider, L.G. The clinical phenotypes of the juvenile idiopathic inflammatory myopathies. Medicine 2013, 92, 25–41. [Google Scholar] [CrossRef]
- Bottai, M.; Tjärnlund, A.; Santoni, G.; Werth, V.P.; Pilkington, C.; de Visser, M.; Alfredsson, L.; Amato, A.A.; Barohn, R.J.; Liang, M.H.; et al. EULAR/ACR classification criteria for adult and juvenile idiopathic inflammatory myopathies and their major subgroups: A methodology report. RMD Open 2017, 3, e000507. [Google Scholar] [CrossRef] [Green Version]
- Rider, L.G.; Shah, M.; Mamyrova, G.; Huber, A.M.; Rice, M.M.; Targoff, I.N.; Miller, F.W. The myositis autoantibody phenotypes of the juvenile idiopathic inflammatory myopathies. Medicine 2013, 92, 223–243. [Google Scholar] [CrossRef] [PubMed]
- Ueki, M.; Kobayashi, I.; Takezaki, S.; Tozawa, Y.; Okura, Y.; Yamada, M.; Kuwana, M.; Ariga, T. Myositis-specific autoantibodies in Japanese patients with juvenile idiopathic inflammatory myopathies. Mod. Rheumatol. 2019, 29, 351–356. [Google Scholar] [CrossRef] [PubMed]
- Yamasaki, Y.; Kobayashi, N.; Akioka, S.; Yamazaki, K.; Takezaki, S.; Nakaseko, H.; Ohara, A.; Nishimura, K.; Nishida, Y.; Sato, S.; et al. Clinical impact of myositis-specific autoantibodies on long-term prognosis of juvenile idiopathic inflammatory myopathies: Multicenter study. Rheumatology 2021, 60, 4821–4831. [Google Scholar] [CrossRef]
- Betteridge, Z.E.; McHugh, N.J. Myositis-specific autoantibodies: An important tool to support diagnosis of myositis. J. Intern. Med. 2016, 280, 8–23. [Google Scholar] [CrossRef]
- Bitencourt, N.; Solow, E.B.; Wright, T.; Bermas, B.L. Inflammatory myositis in systemic lupus erythematosus. Lupus 2020, 29, 776–781. [Google Scholar] [CrossRef]
- Turnier, J.L.; Pachman, L.M.; Lowe, L.; Tsoi, L.C.; Elhaj, S.; Menon, R.; Amoruso, M.C.; Morgan, G.A.; Gudjonsson, J.E.; Berthier, C.C.; et al. Comparison of Lesional Juvenile Myositis and Lupus Skin Reveals Overlapping Yet Unique Disease Pathophysiology. Arthritis Rheumatol. 2020, 73, 1062–1072. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Gunter-Rahman, F.; McGrath, J.A.; Lee, E.; de Jesus, A.A.; Targoff, I.N.; Huang, Y.; O’Hanlon, T.P.; Tsay, W.L.; Gadina, M.; et al. Expression of interferon-regulated genes in juvenile dermatomyositis versus Mendelian autoinflammatory interferonopathies. Arthritis Res. Ther. 2020, 6, 69. [Google Scholar] [CrossRef] [Green Version]
- Kim, H.; Sanchez, G.A.; Goldbach-Mansky, R. Insights from Mendelian Interferonopathies: Comparison of CANDLE, SAVI with AGS, Monogenic Lupus. J. Mol. Med. 2016, 94, 1111–1127. [Google Scholar] [CrossRef] [Green Version]
- Meyer, A.; Meyer, N.; Schaeffer, M.; Gottenberg, J.E.; Geny, B.; Sibilia, J. Incidence and prevalence of inflammatory myopathies: A systematic review. Rheumatology 2015, 54, 50–63. [Google Scholar] [CrossRef] [Green Version]
- Tansley, S.L.; McHugh, N.J.; Wedderburn, L.R. Adult and juvenile dermatomyositis: Are the distinct clinical features explained by our current understanding of serological subgroups and pathogenic mechanisms? Arthritis Res. Ther. 2013, 15, 211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wienke, J.; Deakin, C.T.; Wedderburn, L.R.; van Wijk, F.; van Royen-Kerkhof, A. Systemic and Tissue Inflammation in Juvenile Dermatomyositis: From Pathogenesis to the Quest for Monitoring Tools. Front. Immunol. 2018, 9, 2951. [Google Scholar] [CrossRef] [Green Version]
- Marhaug, G.; Shah, V.; Shroff, R.; Varsani, H.; Wedderburn, L.R.; Pilkington, C.A.; Brogan, P.A. Age-dependent inhibition of ectopic calcification: A possible role for fetuin-A and osteopontin in patients with juvenile dermatomyositis with calcinosis. Rheumatology 2008, 47, 1031–1037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sag, E.; Demir, S.; Bilginer, Y.; Talim, B.; Haliloglu, G.; Topaloglu, H.; Ozen, S. Clinical features, muscle biopsy scores, myositis specific antibody profiles and outcome in juvenile dermatomyositis. Semin. Arthritis Rheum. 2021, 51, 95–100. [Google Scholar] [CrossRef]
- Nguyen, M.; Do, V.; Yell, P.C.; Jo, C.; Liu, J.; Burns, D.K.; Wright, T.; Cai, C. Distinct tissue injury patterns in juvenile dermatomyositis auto-antibody subgroups. Acta Neuropathol. Commun. 2020, 8, 125. [Google Scholar] [CrossRef]
- Li, L.; Liu, C.; Cheng, L.; Yan, S.; Chen, H.; Li, Y. Assessment of diagnostic utility, clinical phenotypic associations, and prognostic significance of anti-NXP2 autoantibody in patients with idiopathic inflammatory myopathies: A systematic review and meta-analysis. Clin. Rheumatol. 2021, 40, 819–832. [Google Scholar] [CrossRef]
- Sanner, H.; Sjaastad, I.; Flato, B. Disease activity and prognostic factors in juvenile dermatomyositis: A long-term follow-up study applying the Paediatric Rheumatology International Trials Organization criteria for inactive disease and the myositis disease activity assessment tool. Rheumatology 2014, 53, 1578–1585. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolko, L.; Jiang, W.; Tawara, N.; Landon-Cardinal, O.; Anquetil, C.; Benveniste, O.; Allenbach, Y. The role of interferons type I, II and III in myositis: A review. Brain Pathol. 2021, 31, e12955. [Google Scholar] [CrossRef]
- Hou, C.; Durrleman, C.; Periou, B.; Barnerias, C.; Bodemer, C.; Desguerre, I.; Quartier, P.; Melki, I.; Rice, G.I.; Rodero, M.P.; et al. From Diagnosis to Prognosis: Revisiting the Meaning of Muscle ISG15 Overexpression in Juvenile Inflammatory Myopathies. Arthritis Rheumatol. 2021, 73, 1044–1052. [Google Scholar] [CrossRef] [PubMed]
- Allenbach, Y.; Leroux, G.; Suarez-Calvet, X.; Preusse, C.; Gallardo, E.; Hervier, B.; Rigolet, A.; Hie, M.; Pehl, D.; Limal, N.; et al. Dermatomyositis With or Without Anti-Melanoma Differentiation-Associated Gene 5 Antibodies: Common Interferon Signature but Distinct NOS2 Expression. Am. J. Pathol. 2016, 186, 691–700. [Google Scholar] [CrossRef]
- Hida, A.; Yamashita, T.; Hosono, Y.; Inoue, M.; Kaida, K.; Kadoya, M.; Miwa, Y.; Yajima, N.; Maezawa, R.; Arai, S.; et al. Anti-TIF1-gamma antibody and cancer-associated myositis: A clinicohistopathologic study. Neurology 2016, 87, 299–308. [Google Scholar] [CrossRef]
- Yasin, S.A.; Schutz, P.W.; Deakin, C.T.; Sag, E.; Varsani, H.; Simou, S.; Marshall, L.R.; Tansley, S.L.; McHugh, N.J.; Holton, J.L.; et al. Histological heterogeneity in a large clinical cohort of juvenile idiopathic inflammatory myopathy: Analysis by myositis autoantibody and pathological features. Neuropathol. Applied. Neurobiol. 2019, 45, 495–512. [Google Scholar] [CrossRef] [Green Version]
- Noguchi, E.; Uruha, A.; Suzuki, S.; Hamanaka, K.; Ohnuki, Y.; Tsugawa, J.; Watanabe, Y.; Nakahara, J.; Shiina, T.; Suzuki, N.; et al. Skeletal Muscle Involvement in Antisynthetase Syndrome. JAMA Neurol. 2017, 74, 992–999. [Google Scholar] [CrossRef] [PubMed]
- Hervier, B.; Benveniste, O. Clinical heterogeneity and outcomes of antisynthetase syndrome. Curr. Rheumatol. Rep. 2013, 15, 349. [Google Scholar] [CrossRef]
- Gallay, L.; Gayed, C.; Hervier, B. Antisynthetase syndrome pathogenesis: Knowledge and uncertainties. Curr. Opin. Rheumatol. 2018, 30, 664–673. [Google Scholar] [CrossRef]
- Gunawardena, H.; Betteridge, Z.E.; McHugh, N.J. Myositis-specific autoantibodies: Their clinical and pathogenic significance in disease expression. Rheumatology 2009, 48, 607–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rider, L.G.; Miller, F.W.; Targoff, I.N.; Sherry, D.D.; Samayoa, E.; Lindahl, M.; Wener, M.H.; Pachman, L.M.; Plotz, P.H. A broadened spectrum of juvenile myositis. Myositis-specific autoantibodies in children. Arthritis Rheum. 1994, 37, 1534–1538. [Google Scholar] [CrossRef]
- Mescam-Mancini, L.; Allenbach, Y.; Hervier, B.; Devilliers, H.; Mariampillay, K.; Dubourg, O.; Maisonobe, T.; Gherardi, R.; Mezin, P.; Preusse, C.; et al. Anti-Jo-1 antibody-positive patients show a characteristic necrotizing perifascicular myositis. Brain 2015, 138, 2485–2492. [Google Scholar] [CrossRef] [Green Version]
- Uruha, A.; Suzuki, S.; Suzuki, N.; Nishino, I. Perifascicular necrosis in anti-synthetase syndrome beyond anti-Jo-1. Brain 2016, 139, e50. [Google Scholar] [CrossRef] [Green Version]
- Stenzel, W.; Preuße, C.; Allenbach, Y.; Pehl, D.; Junckerstorff, R.; Heppner, F.L.; Nolte, K.; Aronica, E.; Kana, V.; Rushing, E.; et al. Nuclear actin aggregation is a hallmark of anti-synthetase syndrome-induced dysimmune myopathy. Neurology 2015, 84, 1346–1354. [Google Scholar] [CrossRef] [Green Version]
- Allenbach, Y.; Benveniste, O.; Stenzel, W.; Boyer, O. Immune-mediated necrotizing myopathy: Clinical features and pathogenesis. Nat. Rev. Rheumatol. 2020, 16, 689–701. [Google Scholar] [CrossRef]
- Mohassel, P.; Foley, A.R.; Donkervoort, S.; Fequiere, P.R.; Pak, K.; Bönnemann, C.G.; Mammen, A.L. Anti-3-hydroxy-3-methylglutaryl-coenzyme a reductase necrotizing myopathy masquerading as a muscular dystrophy in a child. Muscle Nerve 2017, 56, 1177–1181. [Google Scholar] [CrossRef]
- Tansley, S.L.; Betteridge, Z.E.; Simou, S.; Jacques, T.S.; Pilkington, C.; Wood, M.; Warrier, K.; Wedderburn, L.R.; McHugh, N.J. Anti-HMGCR Autoantibodies in Juvenile Idiopathic Inflammatory Myopathies Identify a Rare but Clinically Important Subset of Patients. J. Rheumatol. 2017, 44, 488–492. [Google Scholar] [CrossRef] [Green Version]
- Kishi, T.; Rider, L.G.; Pak, K.; Barillas-Arias, L.; Henrickson, M.; McCarthy, P.L.; Shaham, B.; Weiss, P.F.; Horkayne-Szakaly, I.; Targoff, I.N.; et al. Association of Anti-3-Hydroxy-3-Methylglutaryl-Coenzyme A Reductase Autoantibodies With DRB1*07:01 and Severe Myositis in Juvenile Myositis Patients. Arthritis Care Res. 2017, 69, 1088–1094. [Google Scholar] [CrossRef] [Green Version]
- Dubowitz, V.; Sewry, C.A.; Oldfords, A. Muscle Biopsy: A Practical Approach, 5th ed.; Elsevier Ltd.: Amsterdam, The Netherlands, 2021. [Google Scholar]
- Wedderburn, L.R.; Varsani, H.; Li, C.K.; Newton, K.R.; Amato, A.A.; Banwell, B.; Bove, K.E.; Corse, A.M.; Emslie-Smith, A.; Harding, B.; et al. International consensus on a proposed score system for muscle biopsy evaluation in patients with juvenile dermatomyositis: A tool for potential use in clinical trials. Arthritis Rheum. 2007, 57, 1192–1201. [Google Scholar] [CrossRef]
- Varsani, H.; Charman, S.C.; Li, C.K.; Marie, S.K.; Amato, A.A.; Banwell, B.; Bove, K.E.; Corse, A.M.; Emslie-Smith, A.M.; Jacques, T.S.; et al. Validation of a score tool for measurement of histological severity in juvenile dermatomyositis and association with clinical severity of disease. Ann. Rheum. Dis. 2015, 74, 204–210. [Google Scholar] [CrossRef] [Green Version]
- Kölbel, H.; Preuße, C.; Brand, L.; von Moers, A.; Della Marina, A.; Schuelke, M.; Roos, A.; Goebel, H.H.; Schara-Schmidt, U.; Stenzel, W. Inflammation, fibrosis and skeletal muscle regeneration in LGMDR9 are orchestrated by macrophages. Neuropathol. Appl. Neurobiol. 2021, 47, 856–866. [Google Scholar] [CrossRef]
- Aschman, T.; Schneider, J.; Greuel, S.; Meinhardt, J.; Streit, S.; Goebel, H.H.; Büttnerova, I.; Elezkurtaj, S.; Scheibe, F.; Radke, J.; et al. Association Between SARS-CoV-2 Infection and Immune-Mediated Myopathy in Patients Who Have Died. JAMA Neurol. 2021, 78, 948–960. [Google Scholar] [CrossRef]
- Preusse, C.; Allenbach, Y.; Hoffmann, O.; Goebel, H.H.; Pehl, D.; Radke, J.; Doeser, A.; Schneider, U.; Alten, R.H.; Kallinich, T.; et al. Differential roles of hypoxia and innate immunity in juvenile and adult dermatomyositis. Acta Neuropathol. Commun. 2016, 4, 45. [Google Scholar] [CrossRef] [Green Version]
- Mozaffar, T.; Pestronk, A. Myopathy with anti-Jo-1 antibodies: Pathology in perimysium and neighbouring muscle fibres. J. Neurol. Neurosurg. Psychiatry 2000, 68, 472–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sallum, A.M.; Kiss, M.H.; Silva, C.A.; Wakamatsu, A.; Sachetti, S.; Lotufo, S.; Matsumura, N.; Marie, S.K. MHC class I and II expression in juvenile dermatomyositis skeletal muscle. Clin. Exp. Rheumatol. 2009, 27, 519–526. [Google Scholar]
- Soponkanaporn, S.; Deakin, C.T.; Schutz, P.W.; Marshall, L.R.; Yasin, S.A.; Johnson, C.M.; Sag, E.; Tansley, S.L.; McHugh, N.J.; Wedderburn, L.R.; et al. Expression of myxovirus-resistance protein A: A possible marker of muscle disease activity and autoantibody specificities in juvenile dermatomyositis. Neuropathol. Appl. Neurobiol. 2019, 45, 410–420. [Google Scholar] [CrossRef] [PubMed]
- Udd, B.; Stenzel, W.; Oldfors, A.; Olivé, M.; Romero, N.; Lammens, M.; Kusters, B.; Sewry, C.; Goebel, H.H.; Evangelista, T. 1st ENMC European meeting: The EURO-NMD pathology working group Recommended Standards for Muscle Pathology Amsterdam, The Netherlands, 7 December 2018. Neuromuscul. Disord. 2019, 29, 483–485. [Google Scholar] [CrossRef] [Green Version]
- Benveniste, O.; Goebel, H.-H.; Stenzel, W. Biomarkers in Inflammatory Myopathies—An Expanded Definition. Front. Neurol. 2019, 10, 554. [Google Scholar] [CrossRef] [Green Version]
- Sag, E.; Kale, G.; Haliloglu, G.; Bilginer, Y.; Akcoren, Z.; Orhan, D.; Gucer, S.; Topaloglu, H.; Ozen, S.; Talim, B. Inflammatory milieu of muscle biopsies in juvenile dermatomyositis. Rheumatol. Int. 2021, 41, 77–85. [Google Scholar] [CrossRef]
- Baumann, M.; Gumpold, C.; Mueller-Felber, W.; Schoser, B.; Haberler, C.; Loescher, W.N.; Rostasy, K.; Fischer, M.B.; Wanschitz, J.V. Pattern of myogenesis and vascular repair in early and advanced lesions of juvenile dermatomyositis. Neuromuscul. Disord. 2018, 28, 973–985. [Google Scholar] [CrossRef]
- Gitiaux, C.; De Antonio, M.; Aouizerate, J.; Gherardi, R.K.; Guilbert, T.; Barnerias, C.; Bodemer, C.; Brochard-Payet, K.; Quartier, P.; Musset, L.; et al. Vasculopathy-related clinical and pathological features are associated with severe juvenile dermatomyositis. Rheumatology 2016, 55, 470–479. [Google Scholar] [CrossRef] [Green Version]
- Papadopoulou, C.; Hong, Y.; Krol, P.; Al Obaidi, M.; Pilkington, C.; Wedderburn, L.R.; Brogan, P.A.; Eleftheriou, D. The Vasculopathy of Juvenile Dermatomyositis: Endothelial injury, hypercoagulability, and increased arterial stiffness. Arthritis Rheumatol. 2021, 73, 1253–1266. [Google Scholar] [CrossRef]
- Lahoria, R.; Selcen, D.; Engel, A.G. Microvascular alterations and the role of complement in dermatomyositis. Brain 2016, 139, 1891–1903. [Google Scholar] [CrossRef] [Green Version]
- Meyer, A.; Laverny, G.; Allenbach, Y.; Grelet, E.; Ueberschlag, V.; Echaniz-Laguna, A.; Lannes, B.; Alsaleh, G.; Charles, A.L.; Singh, F.; et al. IFN-beta-induced reactive oxygen species and mitochondrial damage contribute to muscle impairment and inflammation maintenance in dermatomyositis. Acta Neuropathol. 2017, 134, 655–666. [Google Scholar] [CrossRef]
- Uruha, A.; Allenbach, Y.; Charuel, J.L.; Musset, L.; Aussy, A.; Boyer, O.; Mariampillai, K.; Landon-Cardinal, O.; Rasmussen, C.; Bolko, L.; et al. Diagnostic potential of sarcoplasmic MxA expression in subsets of dermatomyositis. Neuropathol. Appl. Neurobiol. 2019, 45, 513–522. [Google Scholar] [CrossRef]
- Inoue, M.; Tanboon, J.; Okubo, M.; Theerawat, K.; Saito, Y.; Ogasawara, M.; Indrawati, L.A.; Uruha, A.; Okiyama, N.; Fujimoto, M.; et al. Absence of sarcoplasmic myxovirus resistance protein A (MxA) expression in antisynthetase syndrome in a cohort of 194 cases. Neuropathol. Appl. Neurobiol. 2019, 45, 523–524. [Google Scholar] [CrossRef]
- Bronner, I.M.; Hoogendijk, J.E.; Veldman, H.; Ramkema, M.; van den Bergh Weerman, M.A.; Rozemuller, A.J.; de Visser, M. Tubuloreticular structures in different types of myositis: Implications for pathogenesis. Ultrastruct. Pathol. 2008, 32, 123–126. [Google Scholar] [CrossRef]
- Carpenter, S.; Karpati, G.; Rothman, S.; Watters, G. The childhood type of dermatomyositis. Neurology 1976, 26, 952–962. [Google Scholar] [CrossRef]
- Wong, D.; Kea, B.; Pesich, R.; Higgs, B.W.; Zhu, W.; Brown, P.; Yao, Y.; Fiorentino, D. Interferon and biologic signatures in dermatomyositis skin: Specificity and heterogeneity across diseases. PLoS ONE 2012, 7, e29161. [Google Scholar] [CrossRef] [Green Version]
- Arndt, V.; Dick, N.; Tawo, R.; Dreiseidler, M.; Wenzel, D.; Hesse, M.; Fürst, D.O.; Saftig, P.; Saint, R.; Fleischmann, B.K.; et al. Chaperone-assisted selective autophagy is essential for muscle maintenance. Curr. Biol. 2010, 20, 143–148. [Google Scholar] [CrossRef]
- Milisenda, J.C.; Pinal-Fernandez, I.; Lloyd, T.E.; Grau, J.M.; Miller, F.W.; Selva-O’Callaghan, A.; Christopher-Stine, L.; Stenzel, W.; Mammen, A.L.; Corse, A.M. Accumulation of autophagosome cargo protein p62 is common in idiopathic inflammatory myopathies. Clin. Exp. Rheumatol. 2021, 39, 351–356. [Google Scholar]
- Fischer, N.; Preusse, C.; Radke, J.; Pehl, D.; Allenbach, Y.; Schneider, U.; Feist, E.; von Casteleyn, V.; Hahn, K.; Ruck, T.; et al. Sequestosome-1 (p62) expression reveals chaperone-assisted selective autophagy in immune-mediated necrotizing myopathies. Brain Pathol. 2020, 30, 261–271. [Google Scholar] [CrossRef] [Green Version]









| Patients with jIIM | |
|---|---|
| Number of patients | 15 |
| Age at biopsy (median) | 8 years |
| range | (3–17 years) |
| female | 11 (73%) |
| Skeletal muscle symptoms | 14 (93%) |
| Myalgia | 11 (73%) |
| Exercise induced myalgia | 6 (40%) |
| Proximal weakness | 14 (93%) |
| Skin symptoms | 15/15 (100%) |
| Dry skin | 6 (40%) |
| Exanthema | 3 (20%) |
| Erythema | 4 (27%) |
| Butterfly rash | 5 (33%) |
| Redness, livid coloration | 8 (53%) |
| Raynaud’s phenomenon | 1 (7%) |
| Gottron’s papules | 8 (53%) |
| Nail fold changes | 1 (7%) |
| Calcinosis | 4 (27%) |
| Accompanying symptoms | 15/15 (100%) |
| Fever | 3 (20%) |
| Difficulty swallowing | 1 (7%) |
| Morning stiffness | 1 (7%) |
| Arthritis | 1 (7%) |
| Weight gain | 2 (13%) |
| Weight loss | 1 (7%) |
| Night sweat | 1 (7%) |
| Oedema | 3 (20%) |
| Lymphadenopathy | 1 (7%) |
| Fatigue, concentration difficulties | 7 (47%) |
| Loss of appetite/rejection to drink | 1 (7%) |
| Sadness/mood slump/mood swings | 5 (33%) |
| Social withdrawal | 3 (20%) |
| Extramuscular symptoms | 5/15 (33%) |
| Pulmonary restriction | 2 (13%) |
| Pneumonia | 1 (7%) |
| Cardial involvement | 4 (27%) |
| Comorbidities | 5/15 (33%) |
| Cystic fibrosis | 1 (7%) |
| Factor-V-Leiden mutation | 1 (7%) |
| Hypothyroidism | 1 (7%) |
| Steatosis hepatis | 1 (7%) |
| HLA-B27 enthesitis | 1 (7%) |
| Laboratory results | 15/15 (100%) |
| Creatine kinase (CK) ↑ | 14 (93%) |
| Lactatdehydrogenase (LDH) ↑ | 11 (73%) |
| GOT ↑ | 11 (73%) |
| GPT ↑ | 7 (47%) |
| Serum antibodies | |
| Myositis-specific-antibodies (MSA) | 6/12 (50%) |
| Anti-NXP-2 | 2 (17%) |
| Anti-PL-7 | 2 (17%) |
| Anti–Jo-1 | 2 (17%) |
| Myositis-associated-antibodies (MAA) | 2/12 (17%) |
| Anti-Ro52 | 1 (8%) |
| PM75 | 1 (8%) |
| Antinuclear antibodies (ANA) | 9/13 (69%) |
| Systemic Lupus erythematodes (SLE) | |
| Anti-double-stranded-DNA-antibodies (Anti-dsDNA) | 1 (8%) |
| Smith-Antibodies (Anti-Sm/Sm-AK) | 1 (8%) |
| Anti RNP/Sm-AK | 2 (17%) |
| MRI skeletal muscle | 11/15 (73%) |
| Normal | 2 (18%) |
| Oedema | 6 (54%) |
| Enhancement of contrast medium/signal alterations | 4 (36%) |
| Compatible with myositis | 8 (73%) |
| Musculoskeletal Ultrasound | 9/15 (60%) |
| Oedema | 3 (33%) |
| Enhancement of echogenicity/signal alterations | 6 (67%) |
| Compatible with myositis | 6 (67%) |
| Electromyography (EMG) | 11/15 (73%) |
| Myopathic, compatible with myositis | 8 (72%) |
| Unspecific sign | 3 (27%) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schänzer, A.; Rager, L.; Dahlhaus, I.; Dittmayer, C.; Preusse, C.; Della Marina, A.; Goebel, H.-H.; Hahn, A.; Stenzel, W. Morphological Characteristics of Idiopathic Inflammatory Myopathies in Juvenile Patients. Cells 2022, 11, 109. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11010109
Schänzer A, Rager L, Dahlhaus I, Dittmayer C, Preusse C, Della Marina A, Goebel H-H, Hahn A, Stenzel W. Morphological Characteristics of Idiopathic Inflammatory Myopathies in Juvenile Patients. Cells. 2022; 11(1):109. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11010109
Chicago/Turabian StyleSchänzer, Anne, Leonie Rager, Iris Dahlhaus, Carsten Dittmayer, Corinna Preusse, Adela Della Marina, Hans-Hilmar Goebel, Andreas Hahn, and Werner Stenzel. 2022. "Morphological Characteristics of Idiopathic Inflammatory Myopathies in Juvenile Patients" Cells 11, no. 1: 109. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11010109