
Molecular Mechanisms in Pentanucleotide Repeat Diseases


1
Genetics of Cognitive Dysfunction Laboratory, i3S- Instituto de Investigação e Inovação em Saúde, Universidade do Porto, 4200-135 Porto, Portugal
2
Institute for Molecular and Cell Biology, Universidade do Porto, 4200-135 Porto, Portugal
3
Instituto de Ciências Biomédicas Abel Salazar, Universidade do Porto, 4050-313 Porto, Portugal
*
Author to whom correspondence should be addressed.
Cells 2022, 11(2), 205; https://0-doi-org.brum.beds.ac.uk/10.3390/cells11020205
Submission received: 10 November 2021
/
Revised: 4 January 2022
/
Accepted: 5 January 2022
/
Published: 8 January 2022
(This article belongs to the Collection Spinocerebellar Ataxia (SCA): Molecular Mechanisms and Novel Treatment Strategies)
Abstract
:The number of neurodegenerative diseases resulting from repeat expansion has increased extraordinarily in recent years. In several of these pathologies, the repeat can be transcribed in RNA from both DNA strands producing, at least, one toxic RNA repeat that causes neurodegeneration by a complex mechanism. Recently, seven diseases have been found caused by a novel intronic pentanucleotide repeat in distinct genes encoding proteins highly expressed in the cerebellum. These disorders are clinically heterogeneous being characterized by impaired motor function, resulting from ataxia or epilepsy. The role that apparently normal proteins from these mutant genes play in these pathologies is not known. However, recent advances in previously known spinocerebellar ataxias originated by abnormal non-coding pentanucleotide repeats point to a gain of a toxic function by the pathogenic repeat-containing RNA that abnormally forms nuclear foci with RNA-binding proteins. In cells, RNA foci have been shown to be formed by phase separation. Moreover, the field of repeat expansions has lately achieved an extraordinary progress with the discovery that RNA repeats, polyglutamine, and polyalanine proteins are crucial for the formation of nuclear membraneless organelles by phase separation, which is perturbed when they are expanded. This review will cover the amazing advances on repeat diseases.
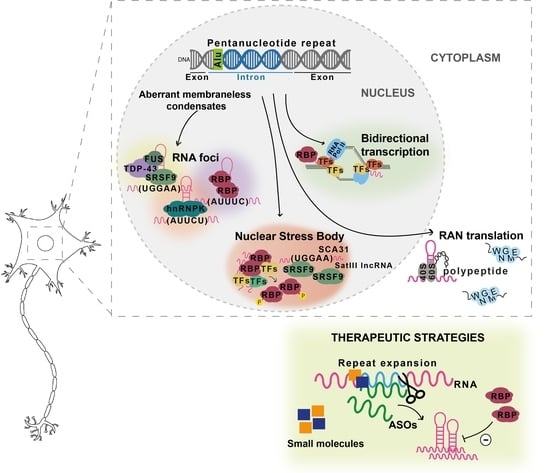
1. Introduction
The number of hereditary disorders originated by unstable microsatellite repeat tracts has been rapidly increasing. Most of them are neurological, neuromuscular, or neurodegenerative diseases. Recent work has shown that a growing group of these conditions is caused by global RNA dysregulation [1,2,3]. The identified RNA alterations modify transcription, generation of antisense RNA, RNA processing, RNA translation, and RNA nuclear export [4,5,6,7]. The fact that most of the genes harboring repeat tracts have a role in transcription or are transcription factors (TFs) also points for a crucial role of RNA processing dysregulation in repeat diseases.
These repeat tracts can be located in coding or non-coding gene regions. In protein-coding gene regions, only trinucleotide repeat expansions have been found, whereas within introns, promoters, 3’ and 5’ UTRs not only trinucleotide but also tetra-, penta-, hexa-, and dodecanucleotide repeats expanded above a given repeat size threshold have been identified in pathological chromosomes (Figure 1).
There is evidence that expanded repeats in protein-coding gene regions could cause pathology by gain-of-function of mislocalized and aggregated polyglutamine [67] or polyalanine [68,69] proteins, many of them with both transcriptional and DNA repair functions, which could lead to their loss-of-function with disruption of DNA damage and repair processes [67,70] and/or interfere with the formation of condensates [3]. The abnormal DNA repair could explain the unstable nature of expanded repeat sequences that are characterized by changing their repeat size in parent-offspring transmissions and within a subject among tissues. The polyglutamine and polyalanine tracts in several proteins are in intrinsically disordered regions (IDR) involved in the formation of cellular membraneless condensates through a mechanism named liquid-liquid phase separation [3] (Figure 2). This is a spontaneous thermodynamically triggered mechanism in which two simultaneously existing liquid phases separate from one homogeneous solution. Several polyglutamine and polyalanine proteins have been demonstrated to form cellular condensates, by liquid-liquid phase separation, which have their composition and/or dynamics altered when these proteins are expanded [1,3,4]. Polyglutamine protein TBP binds to the TATA-box of gene promoters to trigger transcription, but when the polyglutamine tract is expanded, in spinocerebellar ataxia type 17 (SCA17), changes its phase separation capacity and probably its ability to co-condense with transcription co-activators (Figure 2), leading to transcriptional dysregulation, a mechanism implicated in many polyglutamine diseases [1,3,67,70]. Remarkably, Ataxin-2, an RNA-binding protein (RBP) harboring a polyglutamine sequence that when is expanded causes SCA2, has been implicated in the assembly of P-bodies and stress granules (Figure 2), two important cytoplasmic membraneless condensates involved in RNA processing [4]. Stress granule dynamics are perturbed by abnormal interaction of expanded Ataxin-2 with other proteins, which consequently aggregates [1,4]. Interestingly, in cytoplasmic aggregates, Ataxin-2 intermediate-length polyglutamine has been shown to interact with TDP-43, a nuclear RBP mislocalized in the cytoplasm in amyotrophic lateral sclerosis and other neurodegenerative diseases [1,5].
In non-coding gene regions, repeat expansions can originate epigenetic changes with transcriptional repression and protein loss-of-function such as in fragile X syndrome [73], due to CpG methylation of the (CGG)n, and Friedreich ataxia [74], caused by histone hypermethylation of the FXN. In Unverricht-Lundborg progressive myoclonus epilepsy (EPM1), a repeat expansion impairs transcription by altering the spacing of promoter elements. On the other hand, transcribed repeat expansions from non-coding gene sequences cause disease by a complex mechanism involving a toxic RNA gain-of-function through the production of an expanded RNA repeat that leads to the formation of nuclear RNA foci recruiting RBPs and originating splicing misregulation [7,75,76]. In cells, RNA foci have been shown to be formed by phase separation of the repeat-containing RNA [77]. RNAs transcribed from many repeat expansion loci, namely with AT-rich repeat motifs, have common properties of being able to form an RNA hairpin and due to their nucleotide repetitive nature drive multivalent intermolecular interactions [1,77]. The multivalent base-pairing causes expanded RNA repeats to undergo an abrupt sol–gel transition in vitro [77]. In cells, repeat-containing RNAs form aberrant nuclear RNA foci that display liquid-like properties [77]. The splicing misregulation is originated by loss-of-function of aberrantly recruited RBPs, for the RNA foci, most of them are transcription or splicing factors, resulting in the production of abnormal protein isoforms crucial for biological processes in the affected tissue [1], being able to interfere with phase separation capacity, as they have other amino acids with different physical or chemical properties [72]. These expanded non-coding RNA repeats abnormally recruit RBPs with a role in normal physiology [2], disturbing the biological function of these RNAs in the guidance of RBPs for the formation of nuclear compartments [71], consequently impacting genome regulation [78] and possibly the instability in non-coding repeat diseases.
The high complexity of mechanisms involved in repeat diseases is demonstrated by the fact that an expanded RNA repeat transcribed from both protein-coding and non-coding gene regions can lead to the formation of toxic peptides by repeat-associated non-AUG initiated (RAN) translation [6,79]. These RAN peptides have already been found in affected tissue of tri-, tetra-, penta-, and hexanucleotide repeat diseases [6].
To further increase the complexity of repeat expansion loci, they can be transcribed in sense and antisense orientations, originating neurotoxic sense and antisense expanded RNA repeats that, in addition, can be translated by RAN in all reading frames, producing toxic peptides [47,79,80,81,82,83]. The first antisense RNA repeat has been found in myotonic dystrophy type 1 (DM1) [81] followed, soon, by spinocerebellar ataxia type 8 (SCA8) [47], but currently, they are known for at least 14 diseases [84].
Interestingly, many repeat expansion pathologies are caused by expansions that have arisen in genes with TF activity as can be observed in Figure 1. Remarkably, TFs, transcriptional co-activators, and several RBPs composing the transcriptional machinery have recently been shown to have IDRs, leading to the formation of transcriptional condensates important for the recruitment of RNA polymerase II [85]. The presence of repeat expansions in TF IDRs may alter their phase separation capacity, which is necessary for the correct assembly of these transcriptional condensates. Disruption of the phase separation capacity of TFs encoded by TF genes involved in repeat expansion diseases has recently been demonstrated by Basu and colleagues for HOXA13, HOXD13, RUNX2, and TBP [3]. This evidence, combined with the high number of TFs with expanded repeats, suggests that the disruption of transcriptional condensates could be a common mechanism underlying many repeat expansion diseases.
During the last decade, a new type of repeat expansion disease has emerged (Figure 3). These diseases have arisen due to a nucleotide substitution in a non-pathogenic pentanucleotide repeat tract, originating a novel and pathogenic pentanucleotide repeat insertion with adjacent or flanking non-pathogenic pentanucleotide repeats [34,35,86,87]. This type of repeat insertion was first identified in SCA31, in 2009 [34]. Later, in 2017, the second repeat insertion of a new pentanucleotide repeat motif was found causing SCA37 [35]. More recently, ATTTC pentanucleotide repeat insertions have been discovered in six genes causing familial adult myoclonic epilepsies (FAME) type 1, 2, 3, 4, 6, and 7 [36,37,38,39].
This review focuses on pentanucleotide repeat insertions and their contribution to the current understanding of how repeat expansions cause neurodegenerative diseases.
2. Pentanucleotide Repeats in Spinocerebellar Ataxias
The SCAs are a genetically heterogeneous group of usually adult onset autosomal-dominant neurodegenerative diseases characterized by progressive loss of balance and coordination, accompanied by slurred speech. The most common SCAs are caused by repeat expansions in coding or non-coding gene regions, but an unknown proportion of these disorders result from non-repeat mutations. Both groups of SCAs have already been reviewed [84,88,89,90], we will cover the SCAs originated by pentanucleotide repeat inserted mutations.
2.1. ATXN10 ATTCT Repeat Expansions and Inserted Interruptions in SCA10
SCA10 is characterized by cerebellar atrophy, ataxia, and seizures [91]. This disease is one of the most common SCAs in Mexico and Brazil [27,92,93,94,95] and has been reported in diverse North, Central, and South American populations [96,97,98,99] and Asia [100,101]. SCA10 is caused by an (ATTCT)n expansion in intron 9 of ATXN10 [27], a gene required for the survival of cerebellar neurons inducing neuritogenesis [102]. Non-pathogenic alleles range from 10 to 32 repeats, whereas expanded alleles in affected individuals vary from 800 to 4500 repeats (Table 1). Intermediate-size alleles with reduced penetrance have also been reported [93,99,103]. Remarkably, in subjects with epileptic seizures, the repeat expansion is interrupted by inserted pentanucleotide motifs such as (ATTCC)n, (ATCCC)n, and (ATCCT)n that modify disease presentation [103,104,105,106]. Interestingly, the pathogenic repeat is in the poly-A tail of an Alu DNA element [107] similar to other pentanucleotide repeat mutations (Figure 3A).
In SCA10, the expression of ATXN10 and its neighboring genes remains unchanged and the pre-mRNA with the expanded AUUCU is spliced normally in affected subjects-derived cells [108]. ATXN10-null mice have shown embryonic lethality, whereas heterozygous mutants have not developed SCA10 phenotype, ruling out a simple loss-of-function of ATXN10 as the major pathogenic mechanism in SCA10 [108]. The spliced AUUCU expanded RNA repeat forms RNA-protein complexes that aggregate in RNA foci in the cytosol and nuclei of SCA10 human cells and transgenic mouse brains [109,110]. The RNA-binding protein heterogeneous nuclear ribonucleoprotein K (hnRNP K) has been shown co-localizing with nuclear AUUCU foci in SCA10 human cells and transgenic mouse brain. The sequestration of hnRNP K in these RNA foci, likely formed by phase separation [77], changes the splicing regulation of β-tropomyosin and releases protein kinase Cδ that translocates to the mitochondria, leading to apoptosis [109]. Notably, overexpression of hnRNP K in cells expressing expanded AUUCU RNA repeats has been demonstrated to rescue them from apoptosis, showing that the spliced expanded AUUCU RNA repeat triggers neuronal death in SCA10 through loss-of-function of hnRNP K. In interrupted alleles, the inserted pentanucleotide motifs present in affected individuals with epileptic seizures likely recruit additional RBPs relevant for phase separation of condensates with a function in gene regulation of genes associated with epilepsy.
2.2. TGGAA Repeat Insertion in BEAN1 and TK2 in SCA31
SCA31, one of the most common types of SCA in Japan, is caused by an insertion of complex pentanucleotide repeats containing (TGGAA)n in an intron belonging to brain expressed associated with NEDD4-1 (BEAN1) and Thymidine kinase 2 (TK2) genes, in chromosome 16q22.1 [34]. The (TGGAA)n in the BEAN1 strand orientation and the (TTCCA)n in the TK2 orientation are the only repeats exclusively found in affected individuals among the complex repeat tract seen at this location [34,111].
Of note, the complex repeat is located in the poly-A tail of an AluSx element [34] (Figure 3A). Most of the Japanese subjects have a short (TAAAA)n containing between 8 and 20 repeats (Table 1), whereas sequencing of large insertions in control chromosomes detected a long pure stretch of (TAAAA)n or a complex repeat containing (TAAAA)n, (TAGAA)n and (TAAAATAGAA)n [34,86]. SCA31 individuals share the same ancestral haplotype and the 5′-end of pathogenic alleles has a configuration that may vary from TAAAATAGAA(TGGAA)n to TAAAA(TAGAA)4(TGGAA)n [112]. This may suggest that pathogenic alleles have arisen by nucleotide A > G [86], or in the opposite strand T > C, substitution similar to what has been proposed for the mutational mechanism of the (ATTTC)n insertion in SCA37 [87].
In SCA31, there is an inverse correlation between the size of the total complex repeat with the (TGGAA)n insertion and the age of disease onset. Furthermore, the total complex repeat with this insertion has a tendency for expansion upon transmission to the youngest generations [34,113]. Apart from Japan, SCA31 is very rare worldwide [86,111,114], but an affected subject has been described in China with the complex (TGGAA)n and the same C > T change in puratrophin-1 gene found in most Japanese SCA31 individuals [115], showing a strong founder effect.
Clinically, SCA31 is characterized by progressive cerebellar ataxia with a pure cerebellar syndrome [86]. Affected cerebellar tissue on neuropathological examination displayed predominant cerebellar Purkinje cell loss with the remaining Purkinje cells presenting shrinkage of its cell body and amorphous structures with calbindin-positive sprouts surrounding the cell body [86]. RNA foci, likely an aberrant condensate, have been identified in the nuclei of Purkinje cells from brain tissue of affected subjects using probes to target the complex repeat or specifically to the (TGGAA)n, in BEAN1 orientation [34,116]. Expression of the (TGGAA)n in Drosophila also causes accumulation of nuclear RNA foci in the fly eye accompanied by eye degeneration [117]. These observations indicate that the (UGGAA)n forms potentially toxic nuclear RNA foci [86].
An in vitro RNA pulldown assay has identified TDP-43, fused in sarcoma (FUS) and hnRNPs as UGGAA-binding proteins, and TDP-43 has further been confirmed to co-localize with nuclear RNA foci in human SCA31 cerebellar Purkinje cells [117]. Co-expression of TDP-43 suppresses fly eye degeneration and decreases the formation of nuclear RNA foci in (UGGAA)exp-expressing flies, suggesting that TDP-43 prevents RNA foci formation, resulting in the rescue of (UGGAA)exp RNA toxicity via their direct interactions [117]. Further investigation has demonstrated that the binding of TDP-43 to UGGAA RNA repeat changes its structure and prevents its aggregation, functioning as an RNA chaperone for this RNA repeat. Similar investigations with FUS and hnRNPA2B1 have shown dramatic suppression of eye degeneration and attenuated the formation of RNA foci in (UGGAA)exp-expressing flies, confirming their function as RNA chaperones [86,117].
Remarkably the (TGGAA)n is present in Satellite III (SatIII) DNA located in pericentromeric heterochromatin, which is transcribed in a SatIII non-coding RNA. Recent findings suggest that the SCA31 (UGGAA)n RNA competes with long non-coding RNA transcribed from SatIII for SRSF9 splicing factor binding [118], which is abnormally sequestered in SCA31 RNA foci [34]. This SRSF9 is normally recruited to nuclear stress bodies (nSB) with other RBPs, chromatin-remodeling factors, and TFs, resulting in the assembly of nSB, nuclear condensates formed by phase separation, that during thermal stress recovery promote the rapid adaptation of gene expression by target intron retention [119].
The small molecule NCD inhibits the assembly of RNA foci, namely releasing TDP-43 from the foci [118]. Although this NCD molecule interferes with the normal assembly of nSB with SRSF9, it is able to suppress eye degeneration in a SCA31 Drosophila model, being a therapeutic candidate for the disease [118].
Interestingly, in the Drosophila model and in human cerebellar tissues, pentapeptide repeat proteins, originated from the (UGGAA)exp, have likely been produced by repeat-associated RAN translation (Figure 2) [117]. These pentapeptide repeat proteins have been detected in the eye imaginal discs of flies and in the cell bodies and dendrites of SCA31 cerebellar Purkinje cells. However, additional investigations are required to understand the role of these pentapeptides in SCA31.
2.3. DAB1 ATTTC Repeat Insertion in SCA37
In 2017, we found a pentanucleotide ATTTC repeat insertion in a polymorphic ATTTT repeat as the cause of SCA37 [35]. This ATTTC repeat is in intron 1 or 3 of the 5′ UTR of the DAB1, reelin adaptor protein (DAB1) gene, depending on promoter usage [35]. In the reference genome, there is only an (ATTTT)n at this position. Non-pathogenic alleles are very polymorphic, with sizes varying from 7 to 400 ATTTTs (Table 1). On the other hand, in affected subjects, pathogenic alleles have the (ATTTT)60–79(ATTTC)31–75(ATTTT)58–90 configuration (Figure 3B) [120]. The disease is characterized by pure cerebellar ataxia and distinctively onset of dysarthria in late adolescence to adulthood [35,121]. Fifty percent of this variability in age of onset is explained by the size of the (ATTTC)n that shows an inverse correlation with age of disease onset [35]. Both ATTTT and ATTTC repeats are very unstable upon transmission to the next generation. For the (ATTTC)n, this intergenerational instability is biased towards expansion as no contraction has been reported [35,122].
In the DAB1 antisense strand, the (ATTTC)n is in the middle A-rich region of an AluJb element (Figure 3A), which may explain the high instability of this repeat region. Like in other repeat diseases [123,124,125], a small fraction of non-pathogenic repeat alleles are interrupted by other nucleotide motifs, while no interruptions were seen in pathogenic alleles. Interruptions of the (ATTTT)n are AT-rich, with a single A or A(T)n, varying from a di- to an octanucleotide interruption [87]. In non-pathogenic alleles, the existence of one shared haplotype for short pure, interrupted, and large pure alleles shows that they may become unstable at both repeat size and sequence [87].
There is a haplotype shared by all Portuguese subjects with SCA37 [35]. This haplotype has been found in two healthy individuals carrying one non-pathogenic (ATTTT)200 allele, which, together with the observed instability of normal alleles, suggest that the ATTTC repeat has arisen, first, by lengthening of the ATTTT repeat, followed by one or more nucleotide T > C substitutions, in the last T of the pentanucleotide repeat motif [87]. This mutational mechanism is plausible also for the other ATTTC repeat diseases recently identified [36,37,38,39].
Like in other repeat expansions located in non-coding gene regions, the repeat insertion RNA in SCA37 forms nuclear RNA aggregates in human cells [35]. This has been shown by overexpression of the transfected ATTTC repeat in a human cell line followed by fluorescent in situ hybridization (FISH) with a probe predicted to hybridize to the (AUUUC)n RNA, which detected the widespread formation of nuclear RNA aggregates, 48 h after transfection, not present in cells transfected with the corresponding normal (ATTTT)7 or (ATTTT)139 alleles. These RNA aggregates are similar to RNA foci, but larger, probably sequester RBPs, forming these aberrant condensates that compete with nuclear membraneless condensates for RBP-binding, disrupting their normal function as happens in SCA31, DM1, DM2, SCA8, and other repeat diseases [1,5,7,75,126].
In vivo, the AUUUC-containing RNA is toxic and impairs early embryonic development in zebrafish embryos [35]. Zebrafish embryos injected with the (AUUUC)58 pathological insertion have presented a significantly higher lethality rate compared with embryos injected with control RNA. Strikingly, the number of embryos that developed normally following (AUUUC)58 injection has also been significantly lower, demonstrating the high toxicity of this repeat [35]. Sequestration of RBPs by the (AUUUC)n could affect the splicing of several genes crucial for zebrafish development.
DAB1 encodes an adaptor protein of the reelin signaling pathway that controls accurate positioning of neurons and maturation of synaptic connections in the brain during development [127]. Neuropathological findings in two Spanish subjects with SCA37 have shown severe loss of cerebellar Purkinje cells with abundant astrogliosis [122]. Immunostaining with anti-DAB1 suggested specific overexpression in the SCA37 cerebellum compared with age-matched controls. In control cerebellar tissue, DAB1 was present in soma and dendrites, while SCA37 tissue displayed perisomatic and perinuclear punctate staining in the remaining cerebellar Purkinje cells [122]. Moreover, some Purkinje cells presented abnormal arborization and mispositioning within the granular layer, suggesting that the normal function of the DAB1 gene is disrupted in SCA37.
3. Biallelic RFC1 AAGGG Expansions in CANVAS
Cerebellar ataxia with neuropathy and vestibular areflexia (CANVAS) is a late-onset slowly progressive recessive disorder characterized by imbalance, sensory neuropathy, bilateral vestibulopathy, and chronic cough [128]. Recently, a biallelic AAGGG repeat expansion in intron 2 of the Replication factor C subunit 1 (RFC1) gene has been identified as the cause of this disease [32,33], estimated to have arisen in Europe more than 25,000 years ago [33].
In the reference genome, there is an (AAAAG)11 allele, whereas in affected individuals the expansion size of the novel (AAGGG)exp ranges from 400 to 2,000 repeats (Table 1). In control chromosomes, 75% of the alleles are (AAAAG)11, whereas 13% are very polymorphic with large (AAAAG)n sizes that can reach the expansion range; a small fraction (0.8%), however, have (AAGGG)n in the expansion range. The size of the repeat is much larger in (AAGGG)exp alleles, which can reach more than 1,000 units than in (AAAGG)n alleles in the expansion range, that are below 1,000 repeats; most of the (AAAAG)n large alleles have less than 200 units [32]. Three additional repeat structures, AGAGG, AAGAG, and ACAGG have been observed in adult-onset ataxia cases [129,130], as well as a new allele with an (AAAGG)10-25 tract preceding the (AAGGG)n [131] further showing the unstable nature of this repeat, being advisable to give careful attention to its sequence and size before releasing a molecular diagnosis.
Of note, in the RFC1 antisense strand, the novel pentanucleotide repeat is in the poly-A tail of an AluSx3 element (Figure 3A) [32,33]. The role of this Alu element in the high repeat instability seen in RFC1 alleles is unknown. The large (AAAGG)n alleles in the expansion range often show interruptions and nucleotide changes in the sequence, while 3% of alleles in healthy individuals could not be amplified by standard PCR and have given negative for RP-PCR targeting AAAAG, AAAGG, or AAGGG repeats [32], suggesting the presence of other repeat motifs. In fact, this high instability resembles that observed for other repeat expansions in poly-A regions of Alu elements such as SCA10, DM2, and SCA37 [87,103,107,132].
The disease-associated haplotype has been observed in carriers of two large (AAAGG)n alleles and of (AAGGG)exp/large (AAAGG)n chromosomes [32]. This seems to indicate that the pathogenic (AAGGG)exp allele has arisen by nucleotide A > G substitution, or T > C in the complementary strand, in a large (AAAGG)n chromosome, resembling the mutational mechanism in the origin of SCA37 chromosomes [87] and possibly SCA31, based on analysis of the published data [86,112].
Given the pattern of inheritance and the finding of biallelic expansions, a recessive mode of transmission is likely for CANVAS [32,33]. In Friedrich ataxia, a recessive disease caused by biallelic (GAA)n expansions in the FXN gene, there is a decrease in mRNA expression in brain tissue from affected subjects [24,133]. In CANVAS, contrary to what would be expected in a loss-of-function scenario, RFC1 mRNA assessment in lymphoblasts, muscle, and cerebellar vermis has not shown any significant decrease and no RFC1 protein reduction was detected [32]. Moreover, no formation of abnormal RNA foci in affected brain tissue has been reported [32]. Thus, the mechanism by which such a large pentanucleotide repeat in RFC1 causes CANVAS remains intriguing.
4. Pentanucleotide Repeats in Familial Adult Myoclonic Epilepsy
Familial adult myoclonic epilepsy (FAME), also named familial cortical myoclonic tremor and epilepsy (FCMTE), benign adult familial myoclonic epilepsy (BAFME), and several other designations, has first been described in Japan [134]. The disease is characterized by non-progressive adult-onset of cortical myoclonus and generalized seizures inherited in an autosomal-dominant mode [135]. Usually, myoclonus is the first symptom that manifests between the age of 10 and 60 years by tremulous finger movements and myoclonus of the extremities aggravated by action and posture. Additional manifestations include cerebellar ataxia, dysarthria, cognitive decline, and migraine [134,136]. The diagnosis of FAME is supported by electroencephalographic changes. Interestingly, pathology [137], imaging [138] and clinical presentation suggest also the existence of cerebellar dysfunction [134,139]. There are six known genetic types of FAME and they are all caused by (ATTTC)n insertions [140].
4.1. SAMD12 Repeat Insertion in FAME1
In Japanese families with FAME that mapped to the previously known FAME1 locus, on chromosome 8q24 [141,142], an unstable ATTTC repeat with adjacent or flanked ATTTT repeat tracts of variable lengths has been identified in intron 4 of the Sterile α-motif domain-containing 12 (SAMD12) gene [36]. In the reference genome, in the position of this mutation there is ATTTT, but not ATTTC repeats. A total of 82 affected subjects from 48 families have initially shown the ATTTC repeat with adjacent ATTTTs, whereas in 3 affected individuals from one pedigree the ATTTT repeat is flanking the ATTTCs such as in SCA37 (Figure 3B). In fact, long-read sequencing with the Oxford Nanopore Technology identified each of the configurations (ATTTT)598(ATTTC)458 or (ATTTT)221(ATTTC)225(ATTTT)81 in two affected individuals (Figure 3B). In control subjects, most of the alleles have about 20 ATTTT repeats, while a small fraction has allele sizes larger than 100 repeats (Table 1) [36]. Like in SCA37, no ATTTC repeats have been detected in control subjects [36,143]. Of note, the total length of ATTTT and ATTTC repeat tracts together, determined by Southern blot analysis, correlates with age of onset of epilepsy and age of onset of myoclonic tremor [36].
Interestingly, the repeat is located in the poly-A tail of an AluSq2 element in the SAMD12 antisense strand (Figure 3A), which may not only contribute to its high instability but be relevant for the FAME1 pathogenesis. In fact, Southern blot analysis of DNA from affected individual autopsy tissues, including brain, liver, and kidney, showed a broad smear, indicative of high somatic instability [36]. Moreover, intergenerational instability has been detected in DNA from peripheral blood leukocytes by Southern blot analysis. The identification by RNA-seq analysis of short reads filled with the repeat motifs ACUUC or GAAGU and AGUUC or GAACU only in the brain and not in liver or LCL of affected subjects, not present in control brain or LCL, is intriguing [36]. The high instability of this repeat is further demonstrated by the recent finding of a Chinese family with a different nucleotide change in the pentanucleotide repeat, a G instead of a C, originating an (ATTTG)n insertion in SAMD12 [144]. This also confirms that repeat-primed PCR alone cannot guarantee the detection of pentanucleotide repeat insertions [87,120].
The levels of SAMD12 transcripts in autopsied brains are similar to that of control subjects, but SAMD12 protein is significantly reduced. Nevertheless, transcription of the ATTTC repeat originates an RNA that forms RNA foci in neurons. In autopsy brain tissue, FISH using a probe predicted to hybridize with the AUUUC repeat has allowed the detection of nuclear RNA foci in cortical neurons and in Purkinje cells from affected subjects, but not in neurons from control brains [36]. These nuclear RNA foci in FAME1 brain tissue and the formation of nuclear (AUUUC)n RNA aggregates in human embryonic transfected cells with in vivo toxicity [35] show that the (AUUUC)n likely recruits RBPs originating aberrant nuclear condensates that impair the formation of nuclear membraneless organelles crucial for neurons.
Notably, haplotype analysis has detected a shared disease haplotype among Japanese affected subjects with the two known repeat sequence configurations, suggesting a common ancestral founder. Chinese families with FAME1, presenting the configuration of adjacent ATTTT repeats, have also exhibited a shared core haplotype with Japanese pedigrees [143]. The ATTTC repeat insertion in SAMD12 has been identified in 50 Japanese and 23 Chinese families and, more recently, two pedigrees of Sri Lankan and Indian origin, and one Thai family have been reported [143,145,146,147,148,149]. All the affected Asian subjects share a core ancestral haplotype and mutation dating based on the length of this haplotype has estimated that it has arisen 12,000–17,000 years ago [148,149]. The old age of this mutation could account for a larger spread than the currently reported, being important to carry out genetic screens for more than one repeat motif. The high instability observed is likely related to the pathogenic RNA that disturbs the physiological function of RBPs recruited for aberrant condensates.
4.2. STARD7 ATTTC Repeat Insertion in FAME2
The second locus for FAME has been mapped to chromosome 2p11 in an Italian family [150]. Many previous efforts to identify the gene mutation had been unsuccessful, but the finding of additional pedigrees has allowed to narrow down the candidate region [151,152]. Following the finding of three genes harboring ATTTC repeat insertions causing FAME [36], the effort made to identify the FAME2 gene mutation by analysis of whole-genome sequencing data with ExpansionHunter [153] and exSTRa [154] repeat expansion detection methods has finally been successful. This strategy culminated in the finding of an ATTTC repeat insertion in the first intron of the Start domain-containing protein 7 (STARD7) gene [37]. Oxford Nanopore Technology sequencing of the DNA from two affected individuals identified the (ATTTT)390(ATTTC)345 and (ATTTT)340(ATTTC)588 configurations (Figure 3B). The ATTTC repeat insertion has been identified in a total of 22 families with 266 affected individuals. The families are from different origins, but almost all have European ancestry.
The STARD7 gene encodes a protein involved in lipid transport and metabolism broadly expressed throughout the brain, LCL and fibroblasts. Analysis of the expression levels of STARD7 transcript and protein assessed by quantitative RT-PCR and Western blot, respectively, in affected subject-derived fibroblasts have shown, however, no significant differences compared with controls [37]. Thus, an RNA gain-of-function, such as in SCA37 and FAME1 [35,36], is the most plausible mechanism for this disease.
4.3. MARCH6 Repeat Insertion in FAME3
In 2010, Depienne and colleagues mapped the third locus of FAME to chromosome 5p15, in a large French family [135]. Genomic and RNA expression analysis failed to detect pathogenic variants. Later, the identification of (ATTTC)n insertions causing FAME [36] prompt the search of these repeats within the candidate region, in whole-genome sequencing reads, using ExpansionHunter [153] and other similar bioinformatics tools such as exSTRa [154], STRetch [155] and TRhist [156] programs. This investigation has shown an (ATTTC)n in intron 1 of the Membrane-associated ring-ch finger protein (MARCH6) gene segregating with the disease in affected individuals with FAME3 [38]. Long-read Oxford Nanopore sequencing has allowed the identification of (ATTTT)exp(ATTTC)exp tracts (Figure 3) with between 791 and 1035 repeats, in total (Table 1) [38]. The repeat insertion has been identified in five families of diverse European origins, including French, Dutch, and German.
MARCH6 encodes an E3 ubiquitin ligase ubiquitously expressed that mediates degradation of misfolded proteins in the endoplasmic reticulum. The (ATTTC)n insertion in MARCH6 causes no significant changes in gene expression in LCL or fibroblasts of affected individuals when compared with controls [38].
The size of the (ATTTC)n in MARCH6 inversely correlates with the age of seizures onset. Moreover, the individuals with the largest expansions present the most severe symptoms. Remarkably, two of these individuals have shown micro-rearrangements of the (ATTTC)n and adjacent ATTTTs, resulting in variable repeat configurations [38]. These rearrangements seem to indicate that this large repeat breaks during replication, transcription, or both, but their role in the disease process is still unknown.
4.4. Repeat Insertions in Other Types of FAME
The gene for FAME4 has been mapped to chromosome 3q in a large Thai pedigree [157]. More recently, the same methodology of whole-genome long-read sequencing followed by searching repeat motifs in the sequencing data allowed the finding of an ATTTC repeat with adjacent ATTTTs in intron 1 of Yeats domain-containing protein 2 (YEATS2) gene causing FAME4 [39]. At this genomic location, most control subjects have from 4 to 119 ATTTT repeats, while very few have approximately 1000 ATTTTs (Table 1). In an affected subject, the complete sequencing of the pathogenic allele has shown the configuration (ATTTT)819(ATTTC)221 (Figure 3B).
In Japanese FAME families without the ATTTC insertion in SAMD12 the search for ATTTC repeats in ATTTT repeat regions of the reference genome has allowed FAME6 gene identification. Expansion of an ATTTC repeat in a 5′ UTR intron of Trinucleotide repeat-containing 6A (TNRC6A) gene, on chromosome 16p, has been identified in one family by searching whole-genome sequencing data followed by Southern blot analysis [36]. In the reference genome, there is only an (ATTTT)18 allele in this repeat location. The ATTTC repeat is flanked by ATTTT repeat tracts (Figure 3B) originating the configuration (ATTTT)22(ATTTC)exp(ATTTT)exp. In control subjects, there are no ATTTC repeat tracts (Table 1).
The strategy used to identify the ATTTC repeat insertion in FAME6 has successfully enabled the finding of another ATTTC repeat insertion in intron 14 of Rap guanine nucleotide-exchange factor 2 (RAPGEF2) gene, on chromosome 4q32.1, in a Japanese family with FAME7 [36]. Again, in the reference genome, there is only an ATTTT repeat in this position. In affected subjects, the ATTTC is also flanked by ATTTT repeats (Figure 3B). The control subjects analyzed have shown no ATTTC repeats. This disease has also been identified in a Chinese family, showing the same structure of (ATTTC)n flanked by ATTTT repeats [145].
5. Alu Repeat Expansions and Insertions
Analysis of pentanucleotide repeat flanking regions shows that they are all, except in FAME7 RAPGEF2, located in Alu elements (Figure 3A) [34,35,36,37,38,39,87,107]. Alu elements are primate-specific retrotransposons that successfully colonized these genomes by the accumulation of new insertions. These elements, with approximately 300pb, compose about 11% of the human genome [158]. They are composed of two monomers and two poly-As, one in the middle and the other in the 3′-end of the element (Figure 3A). Alu elements are divided into three subfamilies according to their age, AluJ is the oldest, AluS, the intermediate, and AluY, the most recent [159]. The oldest Alu elements accumulated mutations in their sequence, including in their poly-As, being previously associated with the birth and expansion of microsatellite repeats [132,160,161]. These elements were more active in retrotransposition during earlier primate evolution, but they continue to be transcribed and inserted into human genomes with an impact on biology and pathology [159]. Besides the consequences of their retrotransposition, Alu RNAs not only can regulate gene expression [162], but they can drive nuclear localization and fate of transcripts, including long non-coding RNAs and mRNAs, with integrated Alu elements [163]. This has further been shown to be achieved by interaction with an RBP, hnRNP K [163]. The mechanism by which hnRNP K affects nuclear enrichment is not known, but the recent discoveries [71,72] allow us to propose that Alu-containing RNAs could have a say in the formation of nuclear condensates. In Alu orientation, the Alu pentanucleotide repeat motifs differ only by one nucleotide in all pathogenic repeats except CANVAS RFC1, which suggests that they could drive neurodegeneration by a similar mechanism of RNA gain-of-function and likely by disruption of the function of the Alu element, compromising neuronal gene expression of specific genes, a hypothesis that needs to be tested.
6. Therapeutic Strategies
One of the most promising therapeutic strategies for diseases caused by transcribed repeat expansion relies on antisense oligonucleotides (ASOs) [164,165,166,167]. ASOs are short nucleotide sequences chemically modified that bind to the target RNA repeat by Watson–Crick base-pairing, modulating splicing, inhibiting translation, preventing RBP aggregation, or promoting RNA degradation mediated by RNase-H [164,168]. ASOs delivery to the nervous system as well as their stability has led to two critical challenges for therapeutic intervention. Therefore, several delivery carriers and chemical modifications have been studied to increase ASOs uptake and nuclease-resistance [164,167,169,170].
ASOs have shown encouraging results for several neurodegenerative and neuromuscular disorders [171,172,173,174,175,176,177,178,179,180,181,182,183]. In preclinical studies, non-allele specific ASOs acting by RNase-H degradation have been successful for SCA1 [171], SCA3 [173,174], SCA7 [177] and DM1 [178]. In addition, ASOs leading to polyQ exon skipping [175,176], namely combining two ASOs for ATXN3 exon 9 and 10 skipping, have shown therapeutic value in SCA3 [176].
The most promising ASOs reached clinical trials phase I, being the case of ASOs BIIB105 (NCT04494256) for ALS, and both WVE-004 (NCT04931862) and BIIB078 (NCT03626012) for C9ORF72 FTD/ALS [172,179,180]. Interestingly, the ASO BIIB105 has been designed against polyQ-ATXN2 for the treatment of ALS subjects carrying an ATXN2 intermediate allele (30-33 CAG/CAA repeats) [172]. Although several nonspecific-allele ASOs targeting both pathogenic and non-pathogenic RNA repeats have been successful with minor effects in wild-type transcript function [167], ASOs targeting single nucleotide polymorphisms in linkage disequilibrium with expanded alleles have also been tested in preclinical studies for SCA3 [181] and in clinical trials for HD (WVE-003, NCT05032196) [182], lowering the levels of mutant RNAs.
To this date, no studies have been reported with ASOs for the treatment of pentanucleotide repeat diseases. Instead, small molecule compounds have successfully targeting the SCA10 RNA repeat [183] and SCA31 RNA repeat insertion [118], inhibiting RNA foci formation in vitro. An increasing number of studies have shown that both sense and antisense transcripts are involved in pathogenesis, therefore, the development of ASOs targeting both sense and antisense RNAs may be required for more efficient rescue of disease phenotype [84].
7. Conclusions and Future Perspectives
Pathogenic pentanucleotide repeats have been identified in three different types of neurological diseases, SCAs, CANVAS, and FAMEs. These pathogenic repeats are all in non-coding gene regions, thus being transcribed in the same orientation of their host gene, but not translated as part of the gene protein. In the host gene antisense strand, they are located in Alu elements, but these Alu transcripts are difficult to grasp and have not been reported. In SCA31 and FAME1 the pathogenic repeat forms RNA foci in the nuclei of cerebellar Purkinje cells that, in SCA31, interact with the ALS protein TDP-43. In SCA10, the nuclear RNA foci sequester hnRNP K causing its loss of function and leading to apoptosis. The AUUUC RNA repeat forms intranuclear RNA foci in transfected human embryonic cells and causes lethal zebrafish developmental malformations in SCA37, whereas FAME1 triggers the formation of nuclear RNA foci in the nucleus of Purkinje and cortical neurons in affected brain tissue. RNA foci are formed by multivalent intermolecular base-pairing of RNAs with repeated nucleotide motifs that recruit RBPs and phase separate in cellular condensates. As all but one pathogenic pentanucleotide repeat is located in Alu elements, differ by only one nucleotide, and, at least, four of them originate the formation of nuclear RNA foci with RBPs that drive nuclear localization, we hypothesized that each one could be toxic through the formation of aberrant nuclear condensates. Thus, in pentanucleotide SCAs and FAME, the data gathered support a role for the pathogenic RNA in neuronal dysfunction by an RNA-gain-of function mechanism. The specificity of the disease alterations likely results from temporal and spatial differences in cellular and tissue gene expression, namely the levels of expression of the affected gene together with free RBP levels for interaction. Furthermore, the abnormal RAN pentapeptides in cell bodies and dendrites of Purkinje cells from SCA31 cerebellar tissue suggest that this mechanism deserves further investigation in the other pentanucleotide repeat disorders. Moreover, since these pentanucleotide repeat diseases present some clinical, genetic, or neuropathological overlap, shared mechanisms underlying the neuronal pathology are expected. The new opportunities for research created by the discovery of pathogenic pentanucleotide repeats will advance knowledge regarding the complexity of mysterious non-coding gene regions in the human genome and their essential role in neurons.
Author Contributions
J.R.L., A.F.C., A.S.F. and I.S. writing of original draft, reviewing and editing; I.S. conceptualization and supervision. All authors have read and agreed to the published version of the manuscript.
Funding
This work was financed by Fundo Europeu de Desenvolvimento Regional (FEDER), through the COMPETE 2020 Operational Program for Competitiveness and Internationalization (POCI) of Portugal 2020, and by the Fundação para a Ciência e a Tecnologia (FCT) and Ministério da Ciência, Tecnologia e Ensino Superior (Portugal), in the framework of the project with Grant/Award Number: POCI-01-0145-FEDER-029255 (PTDC/MED-GEN/29255/2017) to I.S. A.F.C is recipient of scholarship 2020.00528.BD. A.S.F. is recipient of scholarship 2021.05757.BD.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Conflicts of Interest
On behalf of all authors, the corresponding author states that there is no conflict of interest.
References
- Malik, I.; Kelley, C.P.; Wang, E.T.; Todd, P.K. Author Correction: Molecular mechanisms underlying nucleotide repeat expansion disorders. Nat. Rev. Mol. Cell Biol. 2021, 22, 644. [Google Scholar] [CrossRef] [PubMed]
- Morriss, G.R.; Cooper, T.A. Protein sequestration as a normal function of long noncoding RNAs and a pathogenic mechanism of RNAs containing nucleotide repeat expansions. Hum. Genet. 2017, 136, 1247–1263. [Google Scholar] [CrossRef] [PubMed]
- Basu, S.; Mackowiak, S.D.; Niskanen, H.; Knezevic, D.; Asimi, V.; Grosswendt, S.; Geertsema, H.; Ali, S.; Jerkovic, I.; Ewers, H.; et al. Unblending of Transcriptional Condensates in Human Repeat Expansion Disease. Cell 2020, 181, 1062–1079.e1030. [Google Scholar] [CrossRef]
- Paul, S.; Dansithong, W.; Figueroa, K.P.; Scoles, D.R.; Pulst, S.M. Staufen1 links RNA stress granules and autophagy in a model of neurodegeneration. Nat. Commun. 2018, 9, 3648. [Google Scholar] [CrossRef]
- Loureiro, J.R.; Oliveira, C.L.; Silveira, I. Unstable repeat expansions in neurodegenerative diseases: Nucleocytoplasmic transport emerges on the scene. Neurobiol. Aging 2016, 39, 174–183. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, L.; Cleary, J.D.; Ranum, L.P.W. Repeat-Associated Non-ATG Translation: Molecular Mechanisms and Contribution to Neurological Disease. Annu. Rev. Neurosci. 2019, 42, 227–247. [Google Scholar] [CrossRef]
- Sznajder, L.J.; Swanson, M.S. Short Tandem Repeat Expansions and RNA-Mediated Pathogenesis in Myotonic Dystrophy. Int. J. Mol. Sci. 2019, 20, 3365. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lalioti, M.D.; Scott, H.S.; Buresi, C.; Rossier, C.; Bottani, A.; Morris, M.A.; Malafosse, A.; Antonarakis, S.E. Dodecamer repeat expansion in cystatin B gene in progressive myoclonus epilepsy. Nature 1997, 386, 847–851. [Google Scholar] [CrossRef]
- LaCroix, A.J.; Stabley, D.; Sahraoui, R.; Adam, M.P.; Mehaffey, M.; Kernan, K.; Myers, C.T.; Fagerstrom, C.; Anadiotis, G.; Akkari, Y.M.; et al. GGC Repeat Expansion and Exon 1 Methylation of XYLT1 Is a Common Pathogenic Variant in Baratela-Scott Syndrome. Am. J. Hum. Genet. 2019, 104, 35–44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmes, S.E.; O’Hearn, E.E.; McInnis, M.G.; Gorelick-Feldman, D.A.; Kleiderlein, J.J.; Callahan, C.; Kwak, N.G.; Ingersoll-Ashworth, R.G.; Sherr, M.; Sumner, A.J.; et al. Expansion of a novel CAG trinucleotide repeat in the 5′ region of PPP2R2B is associated with SCA12. Nat. Genet. 1999, 23, 391–392. [Google Scholar] [CrossRef]
- O’Hearn, E.E.; Hwang, H.S.; Holmes, S.E.; Rudnicki, D.D.; Chung, D.W.; Seixas, A.I.; Cohen, R.L.; Ross, C.A.; Trojanowski, J.Q.; Pletnikova, O.; et al. Neuropathology and Cellular Pathogenesis of Spinocerebellar Ataxia Type 12. Mov. Disord. 2015, 30, 1813–1824. [Google Scholar] [CrossRef] [Green Version]
- Verkerk, A.J.; Pieretti, M.; Sutcliffe, J.S.; Fu, Y.H.; Kuhl, D.P.; Pizzuti, A.; Reiner, O.; Richards, S.; Victoria, M.F.; Zhang, F.P.; et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell 1991, 65, 905–914. [Google Scholar] [CrossRef]
- Knight, S.J.; Flannery, A.V.; Hirst, M.C.; Campbell, L.; Christodoulou, Z.; Phelps, S.R.; Pointon, J.; Middleton-Price, H.R.; Barnicoat, A.; Pembrey, M.E.; et al. Trinucleotide repeat amplification and hypermethylation of a CpG island in FRAXE mental retardation. Cell 1993, 74, 127–134. [Google Scholar] [CrossRef]
- Metsu, S.; Rooms, L.; Rainger, J.; Taylor, M.S.; Bengani, H.; Wilson, D.I.; Chilamakuri, C.S.; Morrison, H.; Vandeweyer, G.; Reyniers, E.; et al. FRA2A is a CGG repeat expansion associated with silencing of AFF3. PLoS Genet. 2014, 10, e1004242. [Google Scholar] [CrossRef]
- Metsu, S.; Rainger, J.K.; Debacker, K.; Bernhard, B.; Rooms, L.; Grafodatskaya, D.; Weksberg, R.; Fombonne, E.; Taylor, M.S.; Scherer, S.W.; et al. A CGG-repeat expansion mutation in ZNF713 causes FRA7A: Association with autistic spectrum disorder in two families. Hum. Mutat. 2014, 35, 1295–1300. [Google Scholar] [CrossRef]
- Debacker, K.; Winnepenninckx, B.; Longman, C.; Colgan, J.; Tolmie, J.; Murray, R.; van Luijk, R.; Scheers, S.; Fitzpatrick, D.; Kooy, F. The molecular basis of the folate-sensitive fragile site FRA11A at 11q13. Cytogenet. Genome Res. 2007, 119, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Winnepenninckx, B.; Debacker, K.; Ramsay, J.; Smeets, D.; Smits, A.; FitzPatrick, D.R.; Kooy, R.F. CGG-repeat expansion in the DIP2B gene is associated with the fragile site FRA12A on chromosome 12q13.1. Am. J. Hum. Genet. 2007, 80, 221–231. [Google Scholar] [CrossRef] [Green Version]
- Van Kuilenburg, A.B.P.; Tarailo-Graovac, M.; Richmond, P.A.; Drogemoller, B.I.; Pouladi, M.A.; Leen, R.; Brand-Arzamendi, K.; Dobritzsch, D.; Dolzhenko, E.; Eberle, M.A.; et al. Glutaminase Deficiency Caused by Short Tandem Repeat Expansion in GLS. N. Engl. J. Med. 2019, 380, 1433–1441. [Google Scholar] [CrossRef]
- Hagerman, R.J.; Leehey, M.; Heinrichs, W.; Tassone, F.; Wilson, R.; Hills, J.; Grigsby, J.; Gage, B.; Hagerman, P.J. Intention tremor, parkinsonism, and generalized brain atrophy in male carriers of fragile X. Neurology 2001, 57, 127–130. [Google Scholar] [CrossRef]
- Cronister, A.; Schreiner, R.; Wittenberger, M.; Amiri, K.; Harris, K.; Hagerman, R.J. Heterozygous fragile X female: Historical, physical, cognitive, and cytogenetic features. Am. J. Med. Genet. 1991, 38, 269–274. [Google Scholar] [CrossRef]
- Tian, Y.; Wang, J.L.; Huang, W.; Zeng, S.; Jiao, B.; Liu, Z.; Chen, Z.; Li, Y.; Wang, Y.; Min, H.X.; et al. Expansion of Human-Specific GGC Repeat in Neuronal Intranuclear Inclusion Disease-Related Disorders. Am. J. Hum. Genet. 2019, 105, 166–176. [Google Scholar] [CrossRef] [Green Version]
- Sone, J.; Mitsuhashi, S.; Fujita, A.; Mizuguchi, T.; Hamanaka, K.; Mori, K.; Koike, H.; Hashiguchi, A.; Takashima, H.; Sugiyama, H.; et al. Long-read sequencing identifies GGC repeat expansions in NOTCH2NLC associated with neuronal intranuclear inclusion disease. Nat. Genet. 2019, 51, 1215–1221. [Google Scholar] [CrossRef]
- Ishiura, H.; Shibata, S.; Yoshimura, J.; Suzuki, Y.; Qu, W.; Doi, K.; Almansour, M.A.; Kikuchi, J.K.; Taira, M.; Mitsui, J.; et al. Noncoding CGG repeat expansions in neuronal intranuclear inclusion disease, oculopharyngodistal myopathy and an overlapping disease. Nat. Genet. 2019, 51, 1222–1232. [Google Scholar] [CrossRef]
- Campuzano, V.; Montermini, L.; Molto, M.D.; Pianese, L.; Cossee, M.; Cavalcanti, F.; Monros, E.; Rodius, F.; Duclos, F.; Monticelli, A.; et al. Friedreich’s ataxia: Autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 1996, 271, 1423–1427. [Google Scholar] [CrossRef]
- DeJesus-Hernandez, M.; Mackenzie, I.R.; Boeve, B.F.; Boxer, A.L.; Baker, M.; Rutherford, N.J.; Nicholson, A.M.; Finch, N.A.; Flynn, H.; Adamson, J.; et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011, 72, 245–256. [Google Scholar] [CrossRef] [Green Version]
- Renton, A.E.; Majounie, E.; Waite, A.; Simon-Sanchez, J.; Rollinson, S.; Gibbs, J.R.; Schymick, J.C.; Laaksovirta, H.; van Swieten, J.C.; Myllykangas, L.; et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011, 72, 257–268. [Google Scholar] [CrossRef] [Green Version]
- Matsuura, T.; Yamagata, T.; Burgess, D.L.; Rasmussen, A.; Grewal, R.P.; Watase, K.; Khajavi, M.; McCall, A.E.; Davis, C.F.; Zu, L.; et al. Large expansion of the ATTCT pentanucleotide repeat in spinocerebellar ataxia type 10. Nat. Genet. 2000, 26, 191–194. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Murias, M.; Quintans, B.; Arias, M.; Seixas, A.I.; Cacheiro, P.; Tarrio, R.; Pardo, J.; Millan, M.J.; Arias-Rivas, S.; Blanco-Arias, P.; et al. ‘Costa da Morte’ ataxia is spinocerebellar ataxia 36: Clinical and genetic characterization. Brain 2012, 135, 1423–1435. [Google Scholar] [CrossRef] [Green Version]
- Kobayashi, H.; Abe, K.; Matsuura, T.; Ikeda, Y.; Hitomi, T.; Akechi, Y.; Habu, T.; Liu, W.; Okuda, H.; Koizumi, A. Expansion of intronic GGCCTG hexanucleotide repeat in NOP56 causes SCA36, a type of spinocerebellar ataxia accompanied by motor neuron involvement. Am. J. Hum. Genet. 2011, 89, 121–130. [Google Scholar] [CrossRef] [Green Version]
- Liquori, C.L.; Ricker, K.; Moseley, M.L.; Jacobsen, J.F.; Kress, W.; Naylor, S.L.; Day, J.W.; Ranum, L.P. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science 2001, 293, 864–867. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wieben, E.D.; Aleff, R.A.; Tosakulwong, N.; Butz, M.L.; Highsmith, W.E.; Edwards, A.O.; Baratz, K.H. A common trinucleotide repeat expansion within the transcription factor 4 (TCF4, E2-2) gene predicts Fuchs corneal dystrophy. PLoS ONE 2012, 7, e49083. [Google Scholar] [CrossRef]
- Cortese, A.; Simone, R.; Sullivan, R.; Vandrovcova, J.; Tariq, H.; Yau, W.Y.; Humphrey, J.; Jaunmuktane, Z.; Sivakumar, P.; Polke, J.; et al. Biallelic expansion of an intronic repeat in RFC1 is a common cause of late-onset ataxia. Nat. Genet. 2019, 51, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Rafehi, H.; Szmulewicz, D.J.; Bennett, M.F.; Sobreira, N.L.M.; Pope, K.; Smith, K.R.; Gillies, G.; Diakumis, P.; Dolzhenko, E.; Eberle, M.A.; et al. Bioinformatics-Based Identification of Expanded Repeats: A Non-reference Intronic Pentamer Expansion in RFC1 Causes CANVAS. Am. J. Hum. Genet. 2019, 105, 151–165. [Google Scholar] [CrossRef] [Green Version]
- Sato, N.; Amino, T.; Kobayashi, K.; Asakawa, S.; Ishiguro, T.; Tsunemi, T.; Takahashi, M.; Matsuura, T.; Flanigan, K.M.; Iwasaki, S.; et al. Spinocerebellar ataxia type 31 is associated with “inserted” penta-nucleotide repeats containing (TGGAA)n. Am. J. Hum. Genet. 2009, 85, 544–557. [Google Scholar] [CrossRef] [Green Version]
- Seixas, A.I.; Loureiro, J.R.; Costa, C.; Ordonez-Ugalde, A.; Marcelino, H.; Oliveira, C.L.; Loureiro, J.L.; Dhingra, A.; Brandao, E.; Cruz, V.T.; et al. A Pentanucleotide ATTTC Repeat Insertion in the Non-coding Region of DAB1, Mapping to SCA37, Causes Spinocerebellar Ataxia. Am. J. Hum. Genet. 2017, 101, 87–103. [Google Scholar] [CrossRef] [Green Version]
- Ishiura, H.; Doi, K.; Mitsui, J.; Yoshimura, J.; Matsukawa, M.K.; Fujiyama, A.; Toyoshima, Y.; Kakita, A.; Takahashi, H.; Suzuki, Y.; et al. Expansions of intronic TTTCA and TTTTA repeats in benign adult familial myoclonic epilepsy. Nat. Genet. 2018, 50, 581–590. [Google Scholar] [CrossRef]
- Corbett, M.A.; Kroes, T.; Veneziano, L.; Bennett, M.F.; Florian, R.; Schneider, A.L.; Coppola, A.; Licchetta, L.; Franceschetti, S.; Suppa, A.; et al. Intronic ATTTC repeat expansions in STARD7 in familial adult myoclonic epilepsy linked to chromosome 2. Nat. Commun. 2019, 10, 4920. [Google Scholar] [CrossRef] [Green Version]
- Florian, R.T.; Kraft, F.; Leitao, E.; Kaya, S.; Klebe, S.; Magnin, E.; van Rootselaar, A.F.; Buratti, J.; Kuhnel, T.; Schroder, C.; et al. Unstable TTTTA/TTTCA expansions in MARCH6 are associated with Familial Adult Myoclonic Epilepsy type 3. Nat. Commun. 2019, 10, 4919. [Google Scholar] [CrossRef]
- Yeetong, P.; Pongpanich, M.; Srichomthong, C.; Assawapitaksakul, A.; Shotelersuk, V.; Tantirukdham, N.; Chunharas, C.; Suphapeetiporn, K.; Shotelersuk, V. TTTCA repeat insertions in an intron of YEATS2 in benign adult familial myoclonic epilepsy type 4. Brain 2019, 142, 3360–3366. [Google Scholar] [CrossRef]
- Bragg, D.C.; Mangkalaphiban, K.; Vaine, C.A.; Kulkarni, N.J.; Shin, D.; Yadav, R.; Dhakal, J.; Ton, M.L.; Cheng, A.; Russo, C.T.; et al. Disease onset in X-linked dystonia-parkinsonism correlates with expansion of a hexameric repeat within an SVA retrotransposon in TAF1. Proc. Natl. Acad. Sci. USA 2017, 114, E11020–E11028. [Google Scholar] [CrossRef] [Green Version]
- Mahadevan, M.; Tsilfidis, C.; Sabourin, L.; Shutler, G.; Amemiya, C.; Jansen, G.; Neville, C.; Narang, M.; Barcelo, J.; O’Hoy, K.; et al. Myotonic dystrophy mutation: An unstable CTG repeat in the 3′ untranslated region of the gene. Science 1992, 255, 1253–1255. [Google Scholar] [CrossRef]
- Brook, J.D.; McCurrach, M.E.; Harley, H.G.; Buckler, A.J.; Church, D.; Aburatani, H.; Hunter, K.; Stanton, V.P.; Thirion, J.P.; Hudson, T.; et al. Molecular basis of myotonic dystrophy: Expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell 1992, 68, 799–808. [Google Scholar] [CrossRef]
- Seixas, A.I.; Holmes, S.E.; Takeshima, H.; Pavlovich, A.; Sachs, N.; Pruitt, J.L.; Silveira, I.; Ross, C.A.; Margolis, R.L.; Rudnicki, D.D. Loss of junctophilin-3 contributes to huntington disease-like 2 pathogenesis. Ann. Neurol. 2012, 71, 245–257. [Google Scholar] [CrossRef]
- Wilburn, B.; Rudnicki, D.D.; Zhao, J.; Weitz, T.M.; Cheng, Y.; Gu, X.; Greiner, E.; Park, C.S.; Wang, N.; Sopher, B.L.; et al. An antisense CAG repeat transcript at JPH3 locus med.diates expanded polyglutamine protein toxicity in Huntington’s disease-like 2 mice. Neuron 2011, 70, 427–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holmes, S.E.; O’Hearn, E.; Rosenblatt, A.; Callahan, C.; Hwang, H.S.; Ingersoll-Ashworth, R.G.; Fleisher, A.; Stevanin, G.; Brice, A.; Potter, N.T.; et al. A repeat expansion in the gene encoding junctophilin-3 is associated with Huntington disease-like 2. Nat. Genet. 2001, 29, 377–378. [Google Scholar] [CrossRef]
- Koob, M.D.; Moseley, M.L.; Schut, L.J.; Benzow, K.A.; Bird, T.D.; Day, J.W.; Ranum, L.P. An untranslated CTG expansion causes a novel form of spinocerebellar ataxia (SCA8). Nat. Genet. 1999, 21, 379–384. [Google Scholar] [CrossRef]
- Moseley, M.L.; Zu, T.; Ikeda, Y.; Gao, W.; Mosemiller, A.K.; Daughters, R.S.; Chen, G.; Weatherspoon, M.R.; Clark, H.B.; Ebner, T.J.; et al. Bidirectional expression of CUG and CAG expansion transcripts and intranuclear polyglutamine inclusions in spinocerebellar ataxia type 8. Nat. Genet. 2006, 38, 758–769. [Google Scholar] [CrossRef]
- Koide, R.; Ikeuchi, T.; Onodera, O.; Tanaka, H.; Igarashi, S.; Endo, K.; Takahashi, H.; Kondo, R.; Ishikawa, A.; Hayashi, T.; et al. Unstable expansion of CAG repeat in hereditary dentatorubral-pallidoluysian atrophy (DRPLA). Nat. Genet. 1994, 6, 9–13. [Google Scholar] [CrossRef]
- The Huntington’s Disease Collaborative Research Group. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 1993, 72, 971–983. [Google Scholar] [CrossRef]
- Orr, H.T.; Chung, M.Y.; Banfi, S.; Kwiatkowski, T.J.; Servadio, A.; Beaudet, A.L.; McCall, A.E.; Duvick, L.A.; Ranum, L.P.; Zoghbi, H.Y. Expansion of an unstable trinucleotide CAG repeat in spinocerebellar ataxia type 1. Nat. Genet. 1993, 4, 221–226. [Google Scholar] [CrossRef]
- La Spada, A.R.; Wilson, E.M.; Lubahn, D.B.; Harding, A.E.; Fischbeck, K.H. Androgen receptor gene mutations in X-linked spinal and bulbar muscular atrophy. Nature 1991, 352, 77–79. [Google Scholar] [CrossRef] [PubMed]
- Imbert, G.; Saudou, F.; Yvert, G.; Devys, D.; Trottier, Y.; Garnier, J.M.; Weber, C.; Mandel, J.L.; Cancel, G.; Abbas, N.; et al. Cloning of the gene for spinocerebellar ataxia 2 reveals a locus with high sensitivity to expanded CAG/glutamine repeats. Nat. Genet. 1996, 14, 285–291. [Google Scholar] [CrossRef]
- Kawaguchi, Y.; Okamoto, T.; Taniwaki, M.; Aizawa, M.; Inoue, M.; Katayama, S.; Kawakami, H.; Nakamura, S.; Nishimura, M.; Akiguchi, I. CAG expansions in a novel gene for Machado-Joseph disease at chromosome 14q32.1. Nat. Genet. 1994, 8, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Zhuchenko, O.; Bailey, J.; Bonnen, P.; Ashizawa, T.; Stockton, D.W.; Amos, C.; Dobyns, W.B.; Subramony, S.H.; Zoghbi, H.Y.; Lee, C.C. Autosomal dominant cerebellar ataxia (SCA6) associated with small polyglutamine expansions in the alpha 1A-voltage-dependent calcium channel. Nat. Genet. 1997, 15, 62–69. [Google Scholar] [CrossRef] [PubMed]
- Lindblad, K.; Savontaus, M.L.; Stevanin, G.; Holmberg, M.; Digre, K.; Zander, C.; Ehrsson, H.; David, G.; Benomar, A.; Nikoskelainen, E.; et al. An expanded CAG repeat sequence in spinocerebellar ataxia type 7. Genome Res. 1996, 6, 965–971. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koide, R.; Kobayashi, S.; Shimohata, T.; Ikeuchi, T.; Maruyama, M.; Saito, M.; Yamada, M.; Takahashi, H.; Tsuji, S. A neurological disease caused by an expanded CAG trinucleotide repeat in the TATA-binding protein gene: A new polyglutamine disease? Hum. Mol. Genet. 1999, 8, 2047–2053. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brais, B.; Bouchard, J.P.; Xie, Y.G.; Rochefort, D.L.; Chretien, N.; Tome, F.M.; Lafreniere, R.G.; Rommens, J.M.; Uyama, E.; Nohira, O.; et al. Short GCG expansions in the PABP2 gene cause oculopharyngeal muscular dystrophy. Nat. Genet. 1998, 18, 164–167. [Google Scholar] [CrossRef] [PubMed]
- Stromme, P.; Mangelsdorf, M.E.; Shaw, M.A.; Lower, K.M.; Lewis, S.M.; Bruyere, H.; Lutcherath, V.; Gedeon, A.K.; Wallace, R.H.; Scheffer, I.E.; et al. Mutations in the human ortholog of Aristaless cause X-linked mental retardation and epilepsy. Nat. Genet. 2002, 30, 441–445. [Google Scholar] [CrossRef]
- De Baere, E.; Beysen, D.; Oley, C.; Lorenz, B.; Cocquet, J.; De Sutter, P.; Devriendt, K.; Dixon, M.; Fellous, M.; Fryns, J.P.; et al. FOXL2 and BPES: Mutational hotspots, phenotypic variability, and revision of the genotype-phenotype correlation. Am. J. Hum. Genet. 2003, 72, 478–487. [Google Scholar] [CrossRef] [Green Version]
- Mundlos, S.; Otto, F.; Mundlos, C.; Mulliken, J.B.; Aylsworth, A.S.; Albright, S.; Lindhout, D.; Cole, W.G.; Henn, W.; Knoll, J.H.; et al. Mutations involving the transcription factor CBFA1 cause cleidocranial dysplasia. Cell 1997, 89, 773–779. [Google Scholar] [CrossRef] [Green Version]
- Amiel, J.; Laudier, B.; Attie-Bitach, T.; Trang, H.; de Pontual, L.; Gener, B.; Trochet, D.; Etchevers, H.; Ray, P.; Simonneau, M.; et al. Polyalanine expansion and frameshift mutations of the paired-like homeobox gene PHOX2B in congenital central hypoventilation syndrome. Nat. Genet. 2003, 33, 459–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goodman, F.R.; Bacchelli, C.; Brady, A.F.; Brueton, L.A.; Fryns, J.P.; Mortlock, D.P.; Innis, J.W.; Holmes, L.B.; Donnenfeld, A.E.; Feingold, M.; et al. Novel HOXA13 mutations and the phenotypic spectrum of hand-foot-genital syndrome. Am. J. Hum. Genet. 2000, 67, 197–202. [Google Scholar] [CrossRef] [Green Version]
- Brown, S.A.; Warburton, D.; Brown, L.Y.; Yu, C.Y.; Roeder, E.R.; Stengel-Rutkowski, S.; Hennekam, R.C.; Muenke, M. Holoprosencephaly due to mutations in ZIC2, a homologue of Drosophila odd-paired. Nat. Genet. 1998, 20, 180–183. [Google Scholar] [CrossRef]
- Muragaki, Y.; Mundlos, S.; Upton, J.; Olsen, B.R. Altered growth and branching patterns in synpolydactyly caused by mutations in HOXD13. Science 1996, 272, 548–551. [Google Scholar] [CrossRef]
- Kato, M.; Das, S.; Petras, K.; Kitamura, K.; Morohashi, K.; Abuelo, D.N.; Barr, M.; Bonneau, D.; Brady, A.F.; Carpenter, N.J.; et al. Mutations of ARX are associated with striking pleiotropy and consistent genotype-phenotype correlation. Hum. Mutat. 2004, 23, 147–159. [Google Scholar] [CrossRef]
- Laumonnier, F.; Ronce, N.; Hamel, B.C.; Thomas, P.; Lespinasse, J.; Raynaud, M.; Paringaux, C.; Van Bokhoven, H.; Kalscheuer, V.; Fryns, J.P.; et al. Transcription factor SOX3 is involved in X-linked mental retardation with growth hormone deficiency. Am. J. Hum. Genet. 2002, 71, 1450–1455. [Google Scholar] [CrossRef] [Green Version]
- McLoughlin, H.S.; Moore, L.R.; Paulson, H.L. Pathogenesis of SCA3 and implications for other polyglutamine diseases. Neurobiol. Dis. 2019, 134, 104635. [Google Scholar] [CrossRef]
- Abu-Baker, A.; Rouleau, G.A. Oculopharyngeal muscular dystrophy: Recent advances in the understanding of the molecular pathogenic mechanisms and treatment strategies. Biochim. Biophys. Acta 2007, 1772, 173–185. [Google Scholar] [CrossRef] [Green Version]
- Hughes, J.N.; Thomas, P.Q. Molecular pathology of polyalanine expansion disorders: New perspectives from mouse models. Methods Mol. Biol. 2013, 1017, 135–151. [Google Scholar] [CrossRef] [PubMed]
- Massey, T.H.; Jones, L. The central role of DNA damage and repair in CAG repeat diseases. Dis. Model. Mech. 2018, 11, dmm031930. [Google Scholar] [CrossRef] [Green Version]
- Quinodoz, S.A.; Jachowicz, J.W.; Bhat, P.; Ollikainen, N.; Banerjee, A.K.; Goronzy, I.N.; Blanco, M.R.; Chovanec, P.; Chow, A.; Markaki, Y.; et al. RNA promotes the formation of spatial compartments in the nucleus. Cell 2021, 184, 5775–5790.e30. [Google Scholar] [CrossRef]
- Wiedner, H.J.; Giudice, J. It’s not just a phase: Function and characteristics of RNA-binding proteins in phase separation. Nat. Struct. Mol. Biol. 2021, 28, 465–473. [Google Scholar] [CrossRef]
- Nelson, D.L.; Orr, H.T.; Warren, S.T. The unstable repeats—Three evolving faces of neurological disease. Neuron 2013, 77, 825–843. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Lu, Y.; Polak, U.; Lin, K.; Shen, J.; Farmer, J.; Seyer, L.; Bhalla, A.D.; Rozwadowska, N.; Lynch, D.R.; et al. Expanded GAA repeats impede transcription elongation through the FXN gene and induce transcriptional silencing that is restricted to the FXN locus. Hum. Mol. Genet. 2015, 24, 6932–6943. [Google Scholar] [CrossRef] [Green Version]
- Mankodi, A.; Logigian, E.; Callahan, L.; McClain, C.; White, R.; Henderson, D.; Krym, M.; Thornton, C.A. Myotonic dystrophy in transgenic mice expressing an expanded CUG repeat. Science 2000, 289, 1769–1773. [Google Scholar] [CrossRef] [Green Version]
- Lee, K.Y.; Li, M.; Manchanda, M.; Batra, R.; Charizanis, K.; Mohan, A.; Warren, S.A.; Chamberlain, C.M.; Finn, D.; Hong, H.; et al. Compound loss of muscleblind-like function in myotonic dystrophy. EMBO Mol. Med. 2013, 5, 1887–1900. [Google Scholar] [CrossRef]
- Jain, A.; Vale, R.D. RNA phase transitions in repeat expansion disorders. Nature 2017, 546, 243–247. [Google Scholar] [CrossRef] [Green Version]
- Hall, L.L.; Lawrence, J.B. RNA as a fundamental component of interphase chromosomes: Could repeats prove key? Curr. Opin. Genet. Dev. 2016, 37, 137–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zu, T.; Gibbens, B.; Doty, N.S.; Gomes-Pereira, M.; Huguet, A.; Stone, M.D.; Margolis, J.; Peterson, M.; Markowski, T.W.; Ingram, M.A.; et al. Non-ATG-initiated translation directed by microsatellite expansions. Proc. Natl. Acad. Sci. USA 2011, 108, 260–265. [Google Scholar] [CrossRef] [Green Version]
- Cleary, J.D.; Pattamatta, A.; Ranum, L.P.W. Repeat-associated non-ATG (RAN) translation. J. Biol. Chem. 2018, 293, 16127–16141. [Google Scholar] [CrossRef] [Green Version]
- Cho, D.H.; Thienes, C.P.; Mahoney, S.E.; Analau, E.; Filippova, G.N.; Tapscott, S.J. Antisense transcription and heterochromatin at the DM1 CTG repeats are constrained by CTCF. Mol. Cell 2005, 20, 483–489. [Google Scholar] [CrossRef] [PubMed]
- Ladd, P.D.; Smith, L.E.; Rabaia, N.A.; Moore, J.M.; Georges, S.A.; Hansen, R.S.; Hagerman, R.J.; Tassone, F.; Tapscott, S.J.; Filippova, G.N. An antisense transcript spanning the CGG repeat region of FMR1 is upregulated in premutation carriers but silenced in full mutation individuals. Hum. Mol. Genet. 2007, 16, 3174–3187. [Google Scholar] [CrossRef] [Green Version]
- Zu, T.; Liu, Y.; Banez-Coronel, M.; Reid, T.; Pletnikova, O.; Lewis, J.; Miller, T.M.; Harms, M.B.; Falchook, A.E.; Subramony, S.H.; et al. RAN proteins and RNA foci from antisense transcripts in C9ORF72 ALS and frontotemporal dementia. Proc. Natl. Acad. Sci. USA 2013, 110, E4968–E4977. [Google Scholar] [CrossRef] [Green Version]
- Castro, A.F.; Loureiro, J.R.; Bessa, J.; Silveira, I. Antisense Transcription across Nucleotide Repeat Expansions in Neurodegenerative and Neuromuscular Diseases: Progress and Mysteries. Genes 2020, 11, 1418. [Google Scholar] [CrossRef]
- Plys, A.J.; Kingston, R.E. Dynamic condensates activate transcription. Science 2018, 361, 329–330. [Google Scholar] [CrossRef]
- Ishikawa, K.; Nagai, Y. Molecular Mechanisms and Future Therapeutics for Spinocerebellar Ataxia Type 31 (SCA31). Neurotherapeutics 2019, 16, 1106–1114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loureiro, J.R.; Oliveira, C.L.; Mota, C.; Castro, A.F.; Costa, C.; Loureiro, J.L.; Coutinho, P.; Martins, S.; Sequeiros, J.; Silveira, I. Mutational mechanism for DAB1 (ATTTC)n insertion in SCA37: ATTTT repeat lengthening and nucleotide substitution. Hum. Mutat. 2019, 40, 404–412. [Google Scholar] [CrossRef]
- Klockgether, T.; Mariotti, C.; Paulson, H.L. Spinocerebellar ataxia. Nat. Rev. Dis. Primers 2019, 5, 24. [Google Scholar] [CrossRef] [PubMed]
- Huang, M.; Verbeek, D.S. Why do so many genetic insults lead to Purkinje Cell degeneration and spinocerebellar ataxia? Neurosci. Lett. 2019, 688, 49–57. [Google Scholar] [CrossRef] [Green Version]
- Buijsen, R.A.M.; Toonen, L.J.A.; Gardiner, S.L.; van Roon-Mom, W.M.C. Genetics, Mechanisms, and Therapeutic Progress in Polyglutamine Spinocerebellar Ataxias. Neurotherapeutics 2019, 16, 263–286. [Google Scholar] [CrossRef] [Green Version]
- Lin, X.; Ashizawa, T. SCA10 and ATTCT repeat expansion: Clinical features and molecular aspects. Cytogenet. Genome Res. 2003, 100, 184–188. [Google Scholar] [CrossRef]
- Teive, H.A.; Roa, B.B.; Raskin, S.; Fang, P.; Arruda, W.O.; Neto, Y.C.; Gao, R.; Werneck, L.C.; Ashizawa, T. Clinical phenotype of Brazilian families with spinocerebellar ataxia 10. Neurology 2004, 63, 1509–1512. [Google Scholar] [CrossRef]
- Alonso, I.; Jardim, L.; Artigalas, O.; Saraiva-Pereira, M.; Matsuura, T.; Ashizawa, T.; Sequeiros, J.; Silveira, I. Reduced penetrance of intermediate size alleles in spinocerebellar ataxia type 10. Neurology 2006, 66, 1602–1604. [Google Scholar] [CrossRef]
- Rasmussen, A.; Matsuura, T.; Ruano, L.; Yescas, P.; Ochoa, A.; Ashizawa, T.; Alonso, E. Clinical and genetic analysis of four Mexican families with spinocerebellar ataxia type 10. Ann. Neurol. 2001, 50, 234–239. [Google Scholar] [CrossRef]
- Almeida, T.; Alonso, I.; Martins, S.; Ramos, E.M.; Azevedo, L.; Ohno, K.; Amorim, A.; Saraiva-Pereira, M.L.; Jardim, L.B.; Matsuura, T.; et al. Ancestral origin of the ATTCT repeat expansion in spinocerebellar ataxia type 10 (SCA10). PLoS ONE 2009, 4, e4553. [Google Scholar] [CrossRef] [Green Version]
- Bushara, K.; Bower, M.; Liu, J.; McFarland, K.N.; Landrian, I.; Hutter, D.; Teive, H.A.; Rasmussen, A.; Mulligan, C.J.; Ashizawa, T. Expansion of the Spinocerebellar ataxia type 10 (SCA10) repeat in a patient with Sioux Native American ancestry. PLoS ONE 2013, 8, e81342. [Google Scholar] [CrossRef] [Green Version]
- Leonardi, L.; Marcotulli, C.; McFarland, K.N.; Tessa, A.; DiFabio, R.; Santorelli, F.M.; Pierelli, F.; Ashizawa, T.; Casali, C. Spinocerebellar ataxia type 10 in Peru: The missing link in the Amerindian origin of the disease. J. Neurol. 2014, 261, 1691–1694. [Google Scholar] [CrossRef] [Green Version]
- Baizabal-Carvallo, J.F.; Xia, G.; Botros, P.; Laguna, J.; Ashizawa, T.; Jankovic, J. Bolivian kindred with combined spinocerebellar ataxia types 2 and 10. Acta Neurol. Scand. 2015, 132, 139–142. [Google Scholar] [CrossRef] [Green Version]
- Cornejo-Olivas, M.; Inca-Martinez, M.; Castilhos, R.M.; Furtado, G.V.; Mattos, E.P.; Bampi, G.B.; Leistner-Segal, S.; Marca, V.; Mazzetti, P.; Saraiva-Pereira, M.L.; et al. Genetic Analysis of Hereditary Ataxias in Peru Identifies SCA10 Families with Incomplete Penetrance. Cerebellum 2020, 19, 208–215. [Google Scholar] [CrossRef]
- Wang, K.; McFarland, K.N.; Liu, J.; Zeng, D.; Landrian, I.; Xia, G.; Hao, Y.; Jin, M.; Mulligan, C.J.; Gu, W.; et al. Spinocerebellar ataxia type 10 in Chinese Han. Neurol. Genet. 2015, 1, e26. [Google Scholar] [CrossRef]
- Naito, H.; Takahashi, T.; Kamada, M.; Morino, H.; Yoshino, H.; Hattori, N.; Maruyama, H.; Kawakami, H.; Matsumoto, M. First report of a Japanese family with spinocerebellar ataxia type 10: The second report from Asia after a report from China. PLoS ONE 2017, 12, e0177955. [Google Scholar] [CrossRef] [PubMed]
- Waragai, M.; Nagamitsu, S.; Xu, W.; Li, Y.J.; Lin, X.; Ashizawa, T. Ataxin 10 induces neuritogenesis via interaction with G-protein beta2 subunit. J. Neurosci. Res. 2006, 83, 1170–1178. [Google Scholar] [CrossRef]
- Matsuura, T.; Fang, P.; Pearson, C.E.; Jayakar, P.; Ashizawa, T.; Roa, B.B.; Nelson, D.L. Interruptions in the expanded ATTCT repeat of spinocerebellar ataxia type 10: Repeat purity as a disease modifier? Am. J. Hum. Genet. 2006, 78, 125–129. [Google Scholar] [CrossRef] [Green Version]
- McFarland, K.N.; Liu, J.; Landrian, I.; Godiska, R.; Shanker, S.; Yu, F.; Farmerie, W.G.; Ashizawa, T. SMRT Sequencing of Long Tandem Nucleotide Repeats in SCA10 Reveals Unique Insight of Repeat Expansion Structure. PLoS ONE 2015, 10, e0135906. [Google Scholar] [CrossRef]
- McFarland, K.N.; Liu, J.; Landrian, I.; Zeng, D.; Raskin, S.; Moscovich, M.; Gatto, E.M.; Ochoa, A.; Teive, H.A.; Rasmussen, A.; et al. Repeat interruptions in spinocerebellar ataxia type 10 expansions are strongly associated with epileptic seizures. Neurogenetics 2014, 15, 59–64. [Google Scholar] [CrossRef] [Green Version]
- Hashem, V.; Tiwari, A.; Bewick, B.; Teive, H.A.G.; Moscovich, M.; Schuele, B.; Bushara, K.; Bower, M.; Rasmussen, A.; Tsai, Y.C.; et al. Pulse-Field capillary electrophoresis of repeat-primed PCR amplicons for analysis of large repeats in Spinocerebellar Ataxia Type 10. PLoS ONE 2020, 15, e0228789. [Google Scholar] [CrossRef]
- Kurosaki, T.; Matsuura, T.; Ohno, K.; Ueda, S. Alu-mediated acquisition of unstable ATTCT pentanucleotide repeats in the human ATXN10 gene. Mol. Biol. Evol. 2009, 26, 2573–2579. [Google Scholar] [CrossRef]
- Wakamiya, M.; Matsuura, T.; Liu, Y.; Schuster, G.C.; Gao, R.; Xu, W.; Sarkar, P.S.; Lin, X.; Ashizawa, T. The role of ataxin 10 in the pathogenesis of spinocerebellar ataxia type 10. Neurology 2006, 67, 607–613. [Google Scholar] [CrossRef]
- White, M.C.; Gao, R.; Xu, W.; Mandal, S.M.; Lim, J.G.; Hazra, T.K.; Wakamiya, M.; Edwards, S.F.; Raskin, S.; Teive, H.A.; et al. Inactivation of hnRNP K by expanded intronic AUUCU repeat induces apoptosis via translocation of PKCdelta to mitochondria in spinocerebellar ataxia 10. PLoS Genet. 2010, 6, e1000984. [Google Scholar] [CrossRef]
- White, M.; Xia, G.; Gao, R.; Wakamiya, M.; Sarkar, P.S.; McFarland, K.; Ashizawa, T. Transgenic mice with SCA10 pentanucleotide repeats show motor phenotype and susceptibility to seizure: A toxic RNA gain-of-function model. J. Neurosci. Res. 2012, 90, 706–714. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, K.; Durr, A.; Klopstock, T.; Muller, S.; De Toffol, B.; Vidailhet, M.; Vighetto, A.; Marelli, C.; Wichmann, H.E.; Illig, T.; et al. Pentanucleotide repeats at the spinocerebellar ataxia type 31 (SCA31) locus in Caucasians. Neurology 2011, 77, 1853–1855. [Google Scholar] [CrossRef]
- Sakai, H.; Yoshida, K.; Shimizu, Y.; Morita, H.; Ikeda, S.; Matsumoto, N. Analysis of an insertion mutation in a cohort of 94 patients with spinocerebellar ataxia type 31 from Nagano, Japan. Neurogenetics 2010, 11, 409–415. [Google Scholar] [CrossRef] [Green Version]
- Yoshida, K.; Matsushima, A.; Nakamura, K. Inter-generational instability of inserted repeats during transmission in spinocerebellar ataxia type 31. J. Hum. Genet. 2017, 62, 923–925. [Google Scholar] [CrossRef]
- Lee, Y.C.; Liu, C.S.; Lee, T.Y.; Lo, Y.C.; Lu, Y.C.; Soong, B.W. SCA31 is rare in the Chinese population on Taiwan. Neurobiol. Aging 2012, 33, 426.e23–426.e24. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, Y.; He, Z.; Li, L.; Qin, X.; Zhao, Y.; Yuan, L. Spinocerebellar ataxia type 31 exists in northeast China. J. Neurol. Sci. 2012, 316, 164–167. [Google Scholar] [CrossRef]
- Niimi, Y.; Takahashi, M.; Sugawara, E.; Umeda, S.; Obayashi, M.; Sato, N.; Ishiguro, T.; Higashi, M.; Eishi, Y.; Mizusawa, H.; et al. Abnormal RNA structures (RNA foci) containing a penta-nucleotide repeat (UGGAA)n in the Purkinje cell nucleus is associated with spinocerebellar ataxia type 31 pathogenesis. Neuropathology 2013, 33, 600–611. [Google Scholar] [CrossRef]
- Ishiguro, T.; Sato, N.; Ueyama, M.; Fujikake, N.; Sellier, C.; Kanegami, A.; Tokuda, E.; Zamiri, B.; Gall-Duncan, T.; Mirceta, M.; et al. Regulatory Role of RNA Chaperone TDP-43 for RNA Misfolding and Repeat-Associated Translation in SCA31. Neuron 2017, 94, 108–124.e7. [Google Scholar] [CrossRef] [Green Version]
- Shibata, T.; Nagano, K.; Ueyama, M.; Ninomiya, K.; Hirose, T.; Nagai, Y.; Ishikawa, K.; Kawai, G.; Nakatani, K. Small molecule targeting r(UGGAA)n disrupts RNA foci and alleviates disease phenotype in Drosophila model. Nat. Commun. 2021, 12, 236. [Google Scholar] [CrossRef] [PubMed]
- Ninomiya, K.; Adachi, S.; Natsume, T.; Iwakiri, J.; Terai, G.; Asai, K.; Hirose, T. LncRNA-dependent nuclear stress bodies promote intron retention through SR protein phosphorylation. EMBO J. 2020, 39, e102729. [Google Scholar] [CrossRef]
- Loureiro, J.R.; Oliveira, C.L.; Sequeiros, J.; Silveira, I. A repeat-primed PCR assay for pentanucleotide repeat alleles in spinocerebellar ataxia type 37. J. Hum. Genet. 2018, 63, 981–987. [Google Scholar] [CrossRef]
- Serrano-Munuera, C.; Corral-Juan, M.; Stevanin, G.; San Nicolas, H.; Roig, C.; Corral, J.; Campos, B.; de Jorge, L.; Morcillo-Suarez, C.; Navarro, A.; et al. New subtype of spinocerebellar ataxia with altered vertical eye movements mapping to chromosome 1p32. JAMA Neurol. 2013, 70, 764–771. [Google Scholar] [CrossRef] [Green Version]
- Corral-Juan, M.; Serrano-Munuera, C.; Rabano, A.; Cota-Gonzalez, D.; Segarra-Roca, A.; Ispierto, L.; Cano-Orgaz, A.T.; Adarmes, A.D.; Mendez-Del-Barrio, C.; Jesus, S.; et al. Clinical, genetic and neuropathological characterization of spinocerebellar ataxia type 37. Brain 2018, 141, 1981–1997. [Google Scholar] [CrossRef]
- Chung, M.Y.; Ranum, L.P.; Duvick, L.A.; Servadio, A.; Zoghbi, H.Y.; Orr, H.T. Evidence for a mechanism predisposing to intergenerational CAG repeat instability in spinocerebellar ataxia type I. Nat. Genet. 1993, 5, 254–258. [Google Scholar] [CrossRef] [PubMed]
- Ramos, E.M.; Martins, S.; Alonso, I.; Emmel, V.E.; Saraiva-Pereira, M.L.; Jardim, L.B.; Coutinho, P.; Sequeiros, J.; Silveira, I. Common Origin of Pure and Interrupted Repeat Expansions in Spinocerebellar Ataxia Type 2 (SCA2). Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2010, 153B, 524–531. [Google Scholar] [CrossRef] [PubMed]
- Liquori, C.L.; Ikeda, Y.; Weatherspoon, M.; Ricker, K.; Schoser, B.G.; Dalton, J.C.; Day, J.W.; Ranum, L.P. Myotonic dystrophy type 2: Human founder haplotype and evolutionary conservation of the repeat tract. Am. J. Hum. Genet. 2003, 73, 849–862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daughters, R.S.; Tuttle, D.L.; Gao, W.; Ikeda, Y.; Moseley, M.L.; Ebner, T.J.; Swanson, M.S.; Ranum, L.P. RNA gain-of-function in spinocerebellar ataxia type 8. PLoS Genet. 2009, 5, e1000600. [Google Scholar] [CrossRef] [Green Version]
- Sheldon, M.; Rice, D.S.; D’Arcangelo, G.; Yoneshima, H.; Nakajima, K.; Mikoshiba, K.; Howell, B.W.; Cooper, J.A.; Goldowitz, D.; Curran, T. Scrambler and yotari disrupt the disabled gene and produce a reeler-like phenotype in mice. Nature 1997, 389, 730–733. [Google Scholar] [CrossRef] [PubMed]
- Szmulewicz, D.J.; Roberts, L.; McLean, C.A.; MacDougall, H.G.; Halmagyi, G.M.; Storey, E. Proposed diagnostic criteria for cerebellar ataxia with neuropathy and vestibular areflexia syndrome (CANVAS). Neurol. Clin. Pract. 2016, 6, 61–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akcimen, F.; Ross, J.P.; Bourassa, C.V.; Liao, C.; Rochefort, D.; Gama, M.T.D.; Dicaire, M.J.; Barsottini, O.G.; Brais, B.; Pedroso, J.L.; et al. Investigation of the RFC1 Repeat Expansion in a Canadian and a Brazilian Ataxia Cohort: Identification of Novel Conformations. Front. Genet. 2019, 10, 1219. [Google Scholar] [CrossRef] [Green Version]
- Scriba, C.K.; Beecroft, S.J.; Clayton, J.S.; Cortese, A.; Sullivan, R.; Yau, W.Y.; Dominik, N.; Rodrigues, M.; Walker, E.; Dyer, Z.; et al. A novel RFC1 repeat motif (ACAGG) in two Asia-Pacific CANVAS families. Brain 2020, 143, 2904–2910. [Google Scholar] [CrossRef] [PubMed]
- Beecroft, S.J.; Cortese, A.; Sullivan, R.; Yau, W.Y.; Dyer, Z.; Wu, T.Y.; Mulroy, E.; Pelosi, L.; Rodrigues, M.; Taylor, R.; et al. A Maori specific RFC1 pathogenic repeat configuration in CANVAS, likely due to a founder allele. Brain 2020, 143, 2673–2680. [Google Scholar] [CrossRef] [PubMed]
- Kurosaki, T.; Ueda, S.; Ishida, T.; Abe, K.; Ohno, K.; Matsuura, T. The unstable CCTG repeat responsible for myotonic dystrophy type 2 originates from an AluSx element insertion into an early primate genome. PLoS ONE 2012, 7, e38379. [Google Scholar] [CrossRef] [PubMed]
- Pastore, A.; Puccio, H. Frataxin: A protein in search for a function. J. Neurochem. 2013, 126 (Suppl. S1), 43–52. [Google Scholar] [CrossRef]
- Van den Ende, T.; Sharifi, S.; van der Salm, S.M.A.; van Rootselaar, A.F. Familial Cortical Myoclonic Tremor and Epilepsy, an Enigmatic Disorder: From Phenotypes to Pathophysiology and Genetics. A Systematic Review. Tremor Other Hyperkinet. Mov. 2018, 8, 503. [Google Scholar] [CrossRef]
- Depienne, C.; Magnin, E.; Bouteiller, D.; Stevanin, G.; Saint-Martin, C.; Vidailhet, M.; Apartis, E.; Hirsch, E.; LeGuern, E.; Labauge, P.; et al. Familial cortical myoclonic tremor with epilepsy: The third locus (FCMTE3) maps to 5p. Neurology 2010, 74, 2000–2003. [Google Scholar] [CrossRef] [PubMed]
- Striano, P.; Caranci, F.; Di Benedetto, R.; Tortora, F.; Zara, F.; Striano, S. 1H-MR spectroscopy indicates prominent cerebellar dysfunction in benign adult familial myoclonic epilepsy. Epilepsia 2009, 50, 1491–1497. [Google Scholar] [CrossRef]
- Van Rootselaar, A.F.; van der Salm, S.M.; Bour, L.J.; Edwards, M.J.; Brown, P.; Aronica, E.; Rozemuller-Kwakkel, J.M.; Koehler, P.J.; Koelman, J.H.; Rothwell, J.C.; et al. Decreased cortical inhibition and yet cerebellar pathology in ‘familial cortical myoclonic tremor with epilepsy’. Mov. Disord. 2007, 22, 2378–2385. [Google Scholar] [CrossRef]
- Buijink, A.W.; Broersma, M.; van der Stouwe, A.M.; Sharifi, S.; Tijssen, M.A.; Speelman, J.D.; Maurits, N.M.; van Rootselaar, A.F. Cerebellar Atrophy in Cortical Myoclonic Tremor and Not in Hereditary Essential Tremor-a Voxel-Based Morphometry Study. Cerebellum 2016, 15, 696–704. [Google Scholar] [CrossRef]
- Striano, P.; Louis, E.D.; Manto, M. Autosomal dominant cortical tremor, myoclonus, and epilepsy: Is the origin in the cerebellum? Editorial. Cerebellum 2013, 12, 145–146. [Google Scholar] [CrossRef] [Green Version]
- Depienne, C.; Mandel, J.L. 30 years of repeat expansion disorders: What have we learned and what are the remaining challenges? Am. J. Hum. Genet. 2021, 108, 764–785. [Google Scholar] [CrossRef] [PubMed]
- Mikami, M.; Yasuda, T.; Terao, A.; Nakamura, M.; Ueno, S.; Tanabe, H.; Tanaka, T.; Onuma, T.; Goto, Y.; Kaneko, S.; et al. Localization of a gene for benign adult familial myoclonic epilepsy to chromosome 8q23.3-q24.1. Am. J. Hum. Genet. 1999, 65, 745–751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plaster, N.M.; Uyama, E.; Uchino, M.; Ikeda, T.; Flanigan, K.M.; Kondo, I.; Ptacek, L.J. Genetic localization of the familial adult myoclonic epilepsy (FAME) gene to chromosome 8q24. Neurology 1999, 53, 1180–1183. [Google Scholar] [CrossRef] [PubMed]
- Cen, Z.; Jiang, Z.; Chen, Y.; Zheng, X.; Xie, F.; Yang, X.; Lu, X.; Ouyang, Z.; Wu, H.; Chen, S.; et al. Intronic pentanucleotide TTTCA repeat insertion in the SAMD12 gene causes familial cortical myoclonic tremor with epilepsy type 1. Brain 2018, 141, 2280–2288. [Google Scholar] [CrossRef] [PubMed]
- Cen, Z.; Chen, Y.; Yang, D.; Zhu, Q.; Chen, S.; Chen, X.; Wang, B.; Xie, F.; Ouyang, Z.; Jiang, Z.; et al. Intronic (TTTGA)n insertion in SAMD12 also causes familial cortical myoclonic tremor with epilepsy. Mov. Disord. 2019, 34, 1571–1576. [Google Scholar] [CrossRef] [PubMed]
- Lei, X.X.; Liu, Q.; Lu, Q.; Huang, Y.; Zhou, X.Q.; Sun, H.Y.; Wu, L.W.; Cui, L.Y.; Zhang, X. TTTCA repeat expansion causes familial cortical myoclonic tremor with epilepsy. Eur. J. Neurol. 2019, 26, 513–518. [Google Scholar] [CrossRef] [PubMed]
- Zeng, S.; Zhang, M.Y.; Wang, X.J.; Hu, Z.M.; Li, J.C.; Li, N.; Wang, J.L.; Liang, F.; Yang, Q.; Liu, Q.; et al. Long-read sequencing identified intronic repeat expansions in SAMD12 from Chinese pedigrees affected with familial cortical myoclonic tremor with epilepsy. J. Med. Genet. 2019, 56, 265–270. [Google Scholar] [CrossRef]
- Mizuguchi, T.; Toyota, T.; Adachi, H.; Miyake, N.; Matsumoto, N.; Miyatake, S. Detecting a long insertion variant in SAMD12 by SMRT sequencing: Implications of long-read whole-genome sequencing for repeat expansion diseases. J. Hum. Genet. 2019, 64, 191–197. [Google Scholar] [CrossRef]
- Bennett, M.F.; Oliver, K.L.; Regan, B.M.; Bellows, S.T.; Schneider, A.L.; Rafehi, H.; Sikta, N.; Crompton, D.E.; Coleman, M.; Hildebrand, M.S.; et al. Familial adult myoclonic epilepsy type 1 SAMD12 TTTCA repeat expansion arose 17,000 years ago and is present in Sri Lankan and Indian families. Eur. J. Hum. Genet. 2020, 28, 973–978. [Google Scholar] [CrossRef]
- Yeetong, P.; Chunharas, C.; Pongpanich, M.; Bennett, M.F.; Srichomthong, C.; Pasutharnchat, N.; Suphapeetiporn, K.; Bahlo, M.; Shotelersuk, V. Founder effect of the TTTCA repeat insertions in SAMD12 causing BAFME1. Eur. J. Hum. Genet. 2021, 29, 343–348. [Google Scholar] [CrossRef] [PubMed]
- Guerrini, R.; Bonanni, P.; Patrignani, A.; Brown, P.; Parmeggiani, L.; Grosse, P.; Brovedani, P.; Moro, F.; Aridon, P.; Carrozzo, R.; et al. Autosomal dominant cortical myoclonus and epilepsy (ADCME) with complex partial and generalized seizures: A newly recognized epilepsy syndrome with linkage to chromosome 2p11.1-q12.2. Brain 2001, 124, 2459–2475. [Google Scholar] [CrossRef] [Green Version]
- Henden, L.; Freytag, S.; Afawi, Z.; Baldassari, S.; Berkovic, S.F.; Bisulli, F.; Canafoglia, L.; Casari, G.; Crompton, D.E.; Depienne, C.; et al. Identity by descent fine mapping of familial adult myoclonus epilepsy (FAME) to 2p11.2-2q11.2. Hum. Genet. 2016, 135, 1117–1125. [Google Scholar] [CrossRef] [PubMed]
- Saint-Martin, C.; Bouteiller, D.; Stevanin, G.; Popescu, C.; Charon, C.; Ruberg, M.; Baulac, S.; LeGuern, E.; Labauge, P.; Depienne, C. Refinement of the 2p11.1-q12.2 locus responsible for cortical tremor associated with epilepsy and exclusion of candidate genes. Neurogenetics 2008, 9, 69–71. [Google Scholar] [CrossRef] [PubMed]
- Dolzhenko, E.; van Vugt, J.; Shaw, R.J.; Bekritsky, M.A.; van Blitterswijk, M.; Narzisi, G.; Ajay, S.S.; Rajan, V.; Lajoie, B.R.; Johnson, N.H.; et al. Detection of long repeat expansions from PCR-free whole-genome sequence data. Genome Res. 2017, 27, 1895–1903. [Google Scholar] [CrossRef] [Green Version]
- Tankard, R.M.; Bennett, M.F.; Degorski, P.; Delatycki, M.B.; Lockhart, P.J.; Bahlo, M. Detecting Expansions of Tandem Repeats in Cohorts Sequenced with Short-Read Sequencing Data. Am. J. Hum. Genet. 2018, 103, 858–873. [Google Scholar] [CrossRef] [Green Version]
- Dashnow, H.; Lek, M.; Phipson, B.; Halman, A.; Sadedin, S.; Lonsdale, A.; Davis, M.; Lamont, P.; Clayton, J.S.; Laing, N.G.; et al. STRetch: Detecting and discovering pathogenic short tandem repeat expansions. Genome Biol. 2018, 19, 121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Doi, K.; Monjo, T.; Hoang, P.H.; Yoshimura, J.; Yurino, H.; Mitsui, J.; Ishiura, H.; Takahashi, Y.; Ichikawa, Y.; Goto, J.; et al. Rapid detection of expanded short tandem repeats in personal genomics using hybrid sequencing. Bioinformatics 2014, 30, 815–822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeetong, P.; Ausavarat, S.; Bhidayasiri, R.; Piravej, K.; Pasutharnchat, N.; Desudchit, T.; Chunharas, C.; Loplumlert, J.; Limotai, C.; Suphapeetiporn, K.; et al. A newly identified locus for benign adult familial myoclonic epilepsy on chromosome 3q26.32-3q28. Eur. J. Hum. Genet. 2013, 21, 225–228. [Google Scholar] [CrossRef] [Green Version]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deininger, P. Alu elements: Know the SINEs. Genome Biol. 2011, 12, 236. [Google Scholar] [CrossRef] [Green Version]
- Kelkar, Y.D.; Eckert, K.A.; Chiaromonte, F.; Makova, K.D. A matter of life or death: How microsatellites emerge in and vanish from the human genome. Genome Res. 2011, 21, 2038–2048. [Google Scholar] [CrossRef] [Green Version]
- Clark, R.M.; Dalgliesh, G.L.; Endres, D.; Gomez, M.; Taylor, J.; Bidichandani, S.I. Expansion of GAA triplet repeats in the human genome: Unique origin of the FRDA mutation at the center of an Alu. Genomics 2004, 83, 373–383. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.L.; Yang, L. ALUternative Regulation for Gene Expression. Trends Cell Biol. 2017, 27, 480–490. [Google Scholar] [CrossRef] [Green Version]
- Lubelsky, Y.; Ulitsky, I. Sequences enriched in Alu repeats drive nuclear localization of long RNAs in human cells. Nature 2018, 555, 107–111. [Google Scholar] [CrossRef]
- Hammond, S.M.; Aartsma-Rus, A.; Alves, S.; Borgos, S.E.; Buijsen, R.A.M.; Collin, R.W.J.; Covello, G.; Denti, M.A.; Desviat, L.R.; Echevarria, L.; et al. Delivery of oligonucleotide-based therapeutics: Challenges and opportunities. EMBO Mol. Med. 2021, 13, e13243. [Google Scholar] [CrossRef]
- Crooke, S.T.; Liang, X.H.; Baker, B.F.; Crooke, R.M. Antisense technology: A review. J. Biol. Chem. 2021, 296, 100416. [Google Scholar] [CrossRef]
- Scoles, D.R.; Minikel, E.V.; Pulst, S.M. Antisense oligonucleotides: A primer. Neurol. Genet. 2019, 5, e323. [Google Scholar] [CrossRef] [Green Version]
- Silva, A.C.; Lobo, D.D.; Martins, I.M.; Lopes, S.M.; Henriques, C.; Duarte, S.P.; Dodart, J.C.; Nobre, R.J.; Pereira de Almeida, L. Antisense oligonucleotide therapeutics in neurodegenerative diseases: The case of polyglutamine disorders. Brain 2020, 143, 407–429. [Google Scholar] [CrossRef]
- Kuijper, E.C.; Bergsma, A.J.; Pijnappel, W.; Aartsma-Rus, A. Opportunities and challenges for antisense oligonucleotide therapies. J. Inherit. Metab. Dis. 2021, 44, 72–87. [Google Scholar] [CrossRef]
- Oliveira, C.; Silveira, I.; Veiga, F.; Ribeiro, A.J. Recent advances in characterization of nonviral vectors for delivery of nucleic acids: Impact on their biological performance. Expert Opin. Drug Deliv. 2015, 12, 27–39. [Google Scholar] [CrossRef]
- Evers, M.M.; Toonen, L.J.; van Roon-Mom, W.M. Antisense oligonucleotides in therapy for neurodegenerative disorders. Adv. Drug Deliv. Rev. 2015, 87, 90–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedrich, J.; Kordasiewicz, H.B.; O’Callaghan, B.; Handler, H.P.; Wagener, C.; Duvick, L.; Swayze, E.E.; Rainwater, O.; Hofstra, B.; Benneyworth, M.; et al. Antisense oligonucleotide-mediated ataxin-1 reduction prolongs survival in SCA1 mice and reveals disease-associated transcriptome profiles. JCI Insight 2018, 3, e123193. [Google Scholar] [CrossRef] [PubMed]
- Scoles, D.R.; Meera, P.; Schneider, M.D.; Paul, S.; Dansithong, W.; Figueroa, K.P.; Hung, G.; Rigo, F.; Bennett, C.F.; Otis, T.S.; et al. Antisense oligonucleotide therapy for spinocerebellar ataxia type 2. Nature 2017, 544, 362–366. [Google Scholar] [CrossRef]
- McLoughlin, H.S.; Moore, L.R.; Chopra, R.; Komlo, R.; McKenzie, M.; Blumenstein, K.G.; Zhao, H.; Kordasiewicz, H.B.; Shakkottai, V.G.; Paulson, H.L. Oligonucleotide therapy mitigates disease in spinocerebellar ataxia type 3 mice. Ann. Neurol. 2018, 84, 64–77. [Google Scholar] [CrossRef]
- Moore, L.R.; Rajpal, G.; Dillingham, I.T.; Qutob, M.; Blumenstein, K.G.; Gattis, D.; Hung, G.; Kordasiewicz, H.B.; Paulson, H.L.; McLoughlin, H.S. Evaluation of Antisense Oligonucleotides Targeting ATXN3 in SCA3 Mouse Models. Mol. Ther. Nucleic Acids 2017, 7, 200–210. [Google Scholar] [CrossRef] [Green Version]
- Toonen, L.J.; Schmidt, I.; Luijsterburg, M.S.; van Attikum, H.; van Roon-Mom, W.M. Antisense oligonucleotide-mediated exon skipping as a strategy to reduce proteolytic cleavage of ataxin-3. Sci. Rep. 2016, 6, 35200. [Google Scholar] [CrossRef] [Green Version]
- Evers, M.M.; Tran, H.D.; Zalachoras, I.; Pepers, B.A.; Meijer, O.C.; den Dunnen, J.T.; van Ommen, G.J.; Aartsma-Rus, A.; van Roon-Mom, W.M. Ataxin-3 protein modification as a treatment strategy for spinocerebellar ataxia type 3: Removal of the CAG containing exon. Neurobiol. Dis. 2013, 58, 49–56. [Google Scholar] [CrossRef] [Green Version]
- Niu, C.; Prakash, T.P.; Kim, A.; Quach, J.L.; Huryn, L.A.; Yang, Y.; Lopez, E.; Jazayeri, A.; Hung, G.; Sopher, B.L.; et al. Antisense oligonucleotides targeting mutant Ataxin-7 restore visual function in a mouse model of spinocerebellar ataxia type 7. Sci. Transl. Med. 2018, 10, eaap8677. [Google Scholar] [CrossRef] [Green Version]
- Yadava, R.S.; Mandal, M.; Giese, J.M.; Rigo, F.; Bennett, C.F.; Mahadevan, M.S. Modeling muscle regeneration in RNA toxicity mice. Hum. Mol. Genet. 2021, 30, 1111–1130. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Dodart, J.C.; Tran, H.; Berkovitch, S.; Braun, M.; Byrne, M.; Durbin, A.F.; Hu, X.S.; Iwamoto, N.; Jang, H.G.; et al. Variant-selective stereopure oligonucleotides protect against pathologies associated with C9orf72-repeat expansion in preclinical models. Nat. Commun. 2021, 12, 847. [Google Scholar] [CrossRef]
- Jiang, J.; Zhu, Q.; Gendron, T.F.; Saberi, S.; McAlonis-Downes, M.; Seelman, A.; Stauffer, J.E.; Jafar-Nejad, P.; Drenner, K.; Schulte, D.; et al. Gain of Toxicity from ALS/FTD-Linked Repeat Expansions in C9ORF72 Is Alleviated by Antisense Oligonucleotides Targeting GGGGCC-Containing RNAs. Neuron 2016, 90, 535–550. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toonen, L.J.A.; Rigo, F.; van Attikum, H.; van Roon-Mom, W.M.C. Antisense Oligonucleotide-Mediated Removal of the Polyglutamine Repeat in Spinocerebellar Ataxia Type 3 Mice. Mol. Ther. Nucleic Acids 2017, 8, 232–242. [Google Scholar] [CrossRef] [Green Version]
- Kingwell, K. Double setback for ASO trials in Huntington disease. Nat. Rev. Drug Discov. 2021, 20, 412–413. [Google Scholar] [CrossRef]
- Yang, W.Y.; Gao, R.; Southern, M.; Sarkar, P.S.; Disney, M.D. Design of a bioactive small molecule that targets r(AUUCU) repeats in spinocerebellar ataxia 10. Nat. Commun. 2016, 7, 11647. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Genomic localization of pathogenic repeat expansions. Pathogenic repeats are found in protein-coding genes or non-coding RNAs; in gene-coding regions, these repeats are found in promoters, 5′ UTRs, exons, introns and 3′ UTRs; in red boxes are represented expanded repeats that trigger gene silencing or downregulation (gene loss-of-function); in blue boxes the non-coding transcribed repeat expansions (RNA-mediated toxicity); in green boxes are expanded repeats in protein coding regions or that lead to upregulation (protein gain-of-function). The pentanucleotide repeats are highlighted in blue. In promoters are located the pathogenic repeats causing progressive myoclonus epilepsy of the Unverricht-Lundborg (EPM1) [8] and Baratela-Scott Syndrome (BSS) [9]; the repeat expansion leading to spinocerebellar ataxia type 12 (SCA12) is in the promoter or the 5’ UTR, depending on the transcript [10,11]. In 5′ UTRs are found the expanded repeats associated with fragile X syndrome (FXS or FRAXA) [12], FRAXE [13], FRA2A [14], FRA7A [15], FRA11A [16], FRA12A [17], glutaminase deficiency (GAD) [18], fragile-X-associated tremor ataxia syndrome (FXTAS) [19], fragile X-associated primary ovarian insufficiency (FXPOI) [20], neuronal intranuclear inclusion disease (NIID) [21,22,23], and oculopharyngodistal myopathy (OPDM1) [23]. In introns, abnormal repeat tracts may lead to Friedrich ataxia (FRDA) [24], frontotemporal dementia/amyotrophic lateral sclerosis (FTD/ALS) [25,26], SCA10 [27], SCA36 [28,29], myotonic dystrophy type 2 (DM2) [30], and Fuchs endothelial corneal dystrophy (FECD) [31]. The pathogenic pentanucleotide repeats causing cerebellar ataxia with neuropathy and vestibular areflexia (CANVAS) [32,33], SCA31 [34], SCA37 [35], familial adult myoclonic epilepsy type 1 (FAME1) [36], FAME2 [37], FAME3 [38], FAME4 [39], FAME6, and FAME7 [36] are also intronic; a repeat-containing retrotransposon insertion potentially interferes with transcription of surrounding genomic elements in X-linked dystonia-parkinsonism (XDP) [40]. In 3′ UTR, expanded trinucleotide repeats originate DM1 [41,42]. The Huntington’s disease-like 2 (HDL2) repeat expansion is located both in the 3′ UTR and in an exon, depending on the transcript, being bidirectional transcribed [43,44,45] as in SCA8 [46,47]. In exons, trinucleotide repeat expansions encode abnormal polyglutamine (polyQ) tracts in dentatorubral-pallidoluysian atrophy (DRPLA) [48], HD [49], SCA1 [50], spinobulbar muscular atrophy (SBMA) [51], SCA2 [52], Machado-Joseph disease/SCA3 [53], SCA6 [54], SCA7 [55], and SCA17 [56] or polyalanine (polyA) stretches in oculopharyngeal muscular dystrophy (OPMD) [57], X-linked mental retardation (XLMR) [58], blepharophimosis syndrome (BPES) [59], cleidocranial dysplasia (CCD) [60], congenital central hypoventilation syndrome (CCHS) [61], hand-foot-genital syndrome (HFGS) [62], holoprosencephaly (HPE) [63], synpolydactyly (SPD) [64], X-linked mental retardation and abnormal genitalia (XLAG) [65], or XLMR and growth hormone deficit (XLMRGHD) [66]. An expanded repeat in opposite DNA strands of non-coding RNAs LOC642361/ NUTM2B-AS1 cause oculopharyngeal myopathy with leukoencephalopathy (OPML1) [23]. * The pathogenic repeat threshold is not clear in FTD/ALS, some studies consider >30 repeats.
Figure 1.
Genomic localization of pathogenic repeat expansions. Pathogenic repeats are found in protein-coding genes or non-coding RNAs; in gene-coding regions, these repeats are found in promoters, 5′ UTRs, exons, introns and 3′ UTRs; in red boxes are represented expanded repeats that trigger gene silencing or downregulation (gene loss-of-function); in blue boxes the non-coding transcribed repeat expansions (RNA-mediated toxicity); in green boxes are expanded repeats in protein coding regions or that lead to upregulation (protein gain-of-function). The pentanucleotide repeats are highlighted in blue. In promoters are located the pathogenic repeats causing progressive myoclonus epilepsy of the Unverricht-Lundborg (EPM1) [8] and Baratela-Scott Syndrome (BSS) [9]; the repeat expansion leading to spinocerebellar ataxia type 12 (SCA12) is in the promoter or the 5’ UTR, depending on the transcript [10,11]. In 5′ UTRs are found the expanded repeats associated with fragile X syndrome (FXS or FRAXA) [12], FRAXE [13], FRA2A [14], FRA7A [15], FRA11A [16], FRA12A [17], glutaminase deficiency (GAD) [18], fragile-X-associated tremor ataxia syndrome (FXTAS) [19], fragile X-associated primary ovarian insufficiency (FXPOI) [20], neuronal intranuclear inclusion disease (NIID) [21,22,23], and oculopharyngodistal myopathy (OPDM1) [23]. In introns, abnormal repeat tracts may lead to Friedrich ataxia (FRDA) [24], frontotemporal dementia/amyotrophic lateral sclerosis (FTD/ALS) [25,26], SCA10 [27], SCA36 [28,29], myotonic dystrophy type 2 (DM2) [30], and Fuchs endothelial corneal dystrophy (FECD) [31]. The pathogenic pentanucleotide repeats causing cerebellar ataxia with neuropathy and vestibular areflexia (CANVAS) [32,33], SCA31 [34], SCA37 [35], familial adult myoclonic epilepsy type 1 (FAME1) [36], FAME2 [37], FAME3 [38], FAME4 [39], FAME6, and FAME7 [36] are also intronic; a repeat-containing retrotransposon insertion potentially interferes with transcription of surrounding genomic elements in X-linked dystonia-parkinsonism (XDP) [40]. In 3′ UTR, expanded trinucleotide repeats originate DM1 [41,42]. The Huntington’s disease-like 2 (HDL2) repeat expansion is located both in the 3′ UTR and in an exon, depending on the transcript, being bidirectional transcribed [43,44,45] as in SCA8 [46,47]. In exons, trinucleotide repeat expansions encode abnormal polyglutamine (polyQ) tracts in dentatorubral-pallidoluysian atrophy (DRPLA) [48], HD [49], SCA1 [50], spinobulbar muscular atrophy (SBMA) [51], SCA2 [52], Machado-Joseph disease/SCA3 [53], SCA6 [54], SCA7 [55], and SCA17 [56] or polyalanine (polyA) stretches in oculopharyngeal muscular dystrophy (OPMD) [57], X-linked mental retardation (XLMR) [58], blepharophimosis syndrome (BPES) [59], cleidocranial dysplasia (CCD) [60], congenital central hypoventilation syndrome (CCHS) [61], hand-foot-genital syndrome (HFGS) [62], holoprosencephaly (HPE) [63], synpolydactyly (SPD) [64], X-linked mental retardation and abnormal genitalia (XLAG) [65], or XLMR and growth hormone deficit (XLMRGHD) [66]. An expanded repeat in opposite DNA strands of non-coding RNAs LOC642361/ NUTM2B-AS1 cause oculopharyngeal myopathy with leukoencephalopathy (OPML1) [23]. * The pathogenic repeat threshold is not clear in FTD/ALS, some studies consider >30 repeats.

Figure 2.
Representation of cellular condensates and location of RNA and proteins with expansions. Transcriptional condensates, formed by liquid–liquid phase separation, concentrate TFs, mediators, and other crucial players in transcription initiation. They are closely located to nuclear speckles, small reservoirs of splicing factors acting in mRNA splicing, and to paraspeckles, that assemble TFs and RBPs, thus regulating gene expression. Histone loci bodies drive histone synthesis and storage [71], while RNA transport granules carry mRNAs from and to the nucleus. The nucleolus is the place of rRNA synthesis and early-stage ribosomal subunit assembly. Under stress conditions (1) in nuclear stress bodies, the phosphorylation of RBPs leads to alterations in mRNA splicing of their target genes; in SCA31, the toxic (UGGAA)n competes with SatIII (UGGAA)n non-coding RNA for SRSF9 binding; (2) in the cytoplasm, the stress granules sequester mRNA, arresting translational initiation; Ataxin-2 protein has been implicated in the assembly of stress granules when expanded interacts with TDP-43 and recruits Staufen1 (STAU1) protein, leading to alterations in stress granule dynamics [4]. Perinucleolar compartment is thought to be the place where RNA Pol III transcripts are processed; additionally, recent work has demonstrated that the recruitment of PTBP1 by PNCTR long non-coding RNA controls the splicing patterns of RNA Pol II transcripts [72]; cleavage bodies are regions of mRNA processing. P bodies stall untranslated mRNAs, mediating its decay. U bodies assemble and store small nuclear ribonucleoproteins (snRNPs), previously biosynthesized in Cajal bodies. There are several mechanisms contributing to pathology in repeat expansion diseases (e.g., bidirectional transcription, RNA foci formation and RAN translation; most of them disturb phase-separation capacity, interfering with the correct assembly of condensates. Polyalanine—polyA; polyglutamine—polyQ.
Figure 2.
Representation of cellular condensates and location of RNA and proteins with expansions. Transcriptional condensates, formed by liquid–liquid phase separation, concentrate TFs, mediators, and other crucial players in transcription initiation. They are closely located to nuclear speckles, small reservoirs of splicing factors acting in mRNA splicing, and to paraspeckles, that assemble TFs and RBPs, thus regulating gene expression. Histone loci bodies drive histone synthesis and storage [71], while RNA transport granules carry mRNAs from and to the nucleus. The nucleolus is the place of rRNA synthesis and early-stage ribosomal subunit assembly. Under stress conditions (1) in nuclear stress bodies, the phosphorylation of RBPs leads to alterations in mRNA splicing of their target genes; in SCA31, the toxic (UGGAA)n competes with SatIII (UGGAA)n non-coding RNA for SRSF9 binding; (2) in the cytoplasm, the stress granules sequester mRNA, arresting translational initiation; Ataxin-2 protein has been implicated in the assembly of stress granules when expanded interacts with TDP-43 and recruits Staufen1 (STAU1) protein, leading to alterations in stress granule dynamics [4]. Perinucleolar compartment is thought to be the place where RNA Pol III transcripts are processed; additionally, recent work has demonstrated that the recruitment of PTBP1 by PNCTR long non-coding RNA controls the splicing patterns of RNA Pol II transcripts [72]; cleavage bodies are regions of mRNA processing. P bodies stall untranslated mRNAs, mediating its decay. U bodies assemble and store small nuclear ribonucleoproteins (snRNPs), previously biosynthesized in Cajal bodies. There are several mechanisms contributing to pathology in repeat expansion diseases (e.g., bidirectional transcription, RNA foci formation and RAN translation; most of them disturb phase-separation capacity, interfering with the correct assembly of condensates. Polyalanine—polyA; polyglutamine—polyQ.

Figure 3.
Pathogenic pentanucleotide repeats. (A) Schematic representation of pathogenic pentanucleotide repeats located in poly-A regions of ancient Alu families. Repeats are represented in Alu orientation; orange for repeats in genes with the Alu in antisense DNA strand; blue for repeats with Alu in shared introns of genes transcribed in opposite directions. The SCA37 repeat is located in the middle poly-A of an AluJb and the remaining are in 3′ poly-A tails of AluS elements. LM—left Alu monomer; RM—right Alu monomer; boxes A and B—internal regions of an RNA pol III promoter degenerated in ancient Alu families. (B) Schematic representation of non-pathogenic and pathogenic alleles with ATTTC repeat insertions, in intronic gene regions. Orange represents repeat composition of non-pathogenic alleles, whereas red is the pathogenic repeat insertion. Most non-pathogenic chromosomes have less than 30 repeats, but some can be larger than the pathogenic tract.
Figure 3.
Pathogenic pentanucleotide repeats. (A) Schematic representation of pathogenic pentanucleotide repeats located in poly-A regions of ancient Alu families. Repeats are represented in Alu orientation; orange for repeats in genes with the Alu in antisense DNA strand; blue for repeats with Alu in shared introns of genes transcribed in opposite directions. The SCA37 repeat is located in the middle poly-A of an AluJb and the remaining are in 3′ poly-A tails of AluS elements. LM—left Alu monomer; RM—right Alu monomer; boxes A and B—internal regions of an RNA pol III promoter degenerated in ancient Alu families. (B) Schematic representation of non-pathogenic and pathogenic alleles with ATTTC repeat insertions, in intronic gene regions. Orange represents repeat composition of non-pathogenic alleles, whereas red is the pathogenic repeat insertion. Most non-pathogenic chromosomes have less than 30 repeats, but some can be larger than the pathogenic tract.


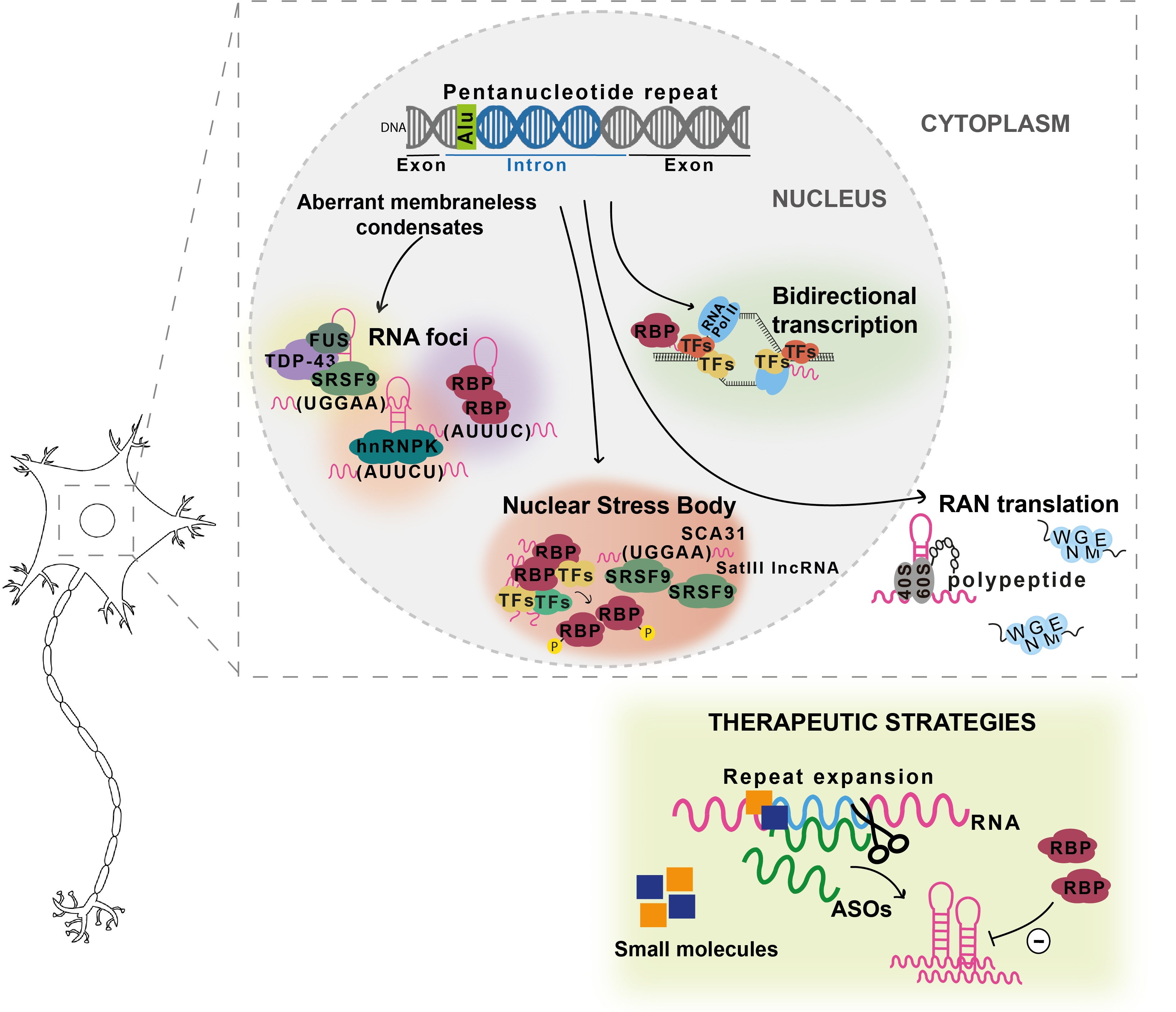{kind=link}
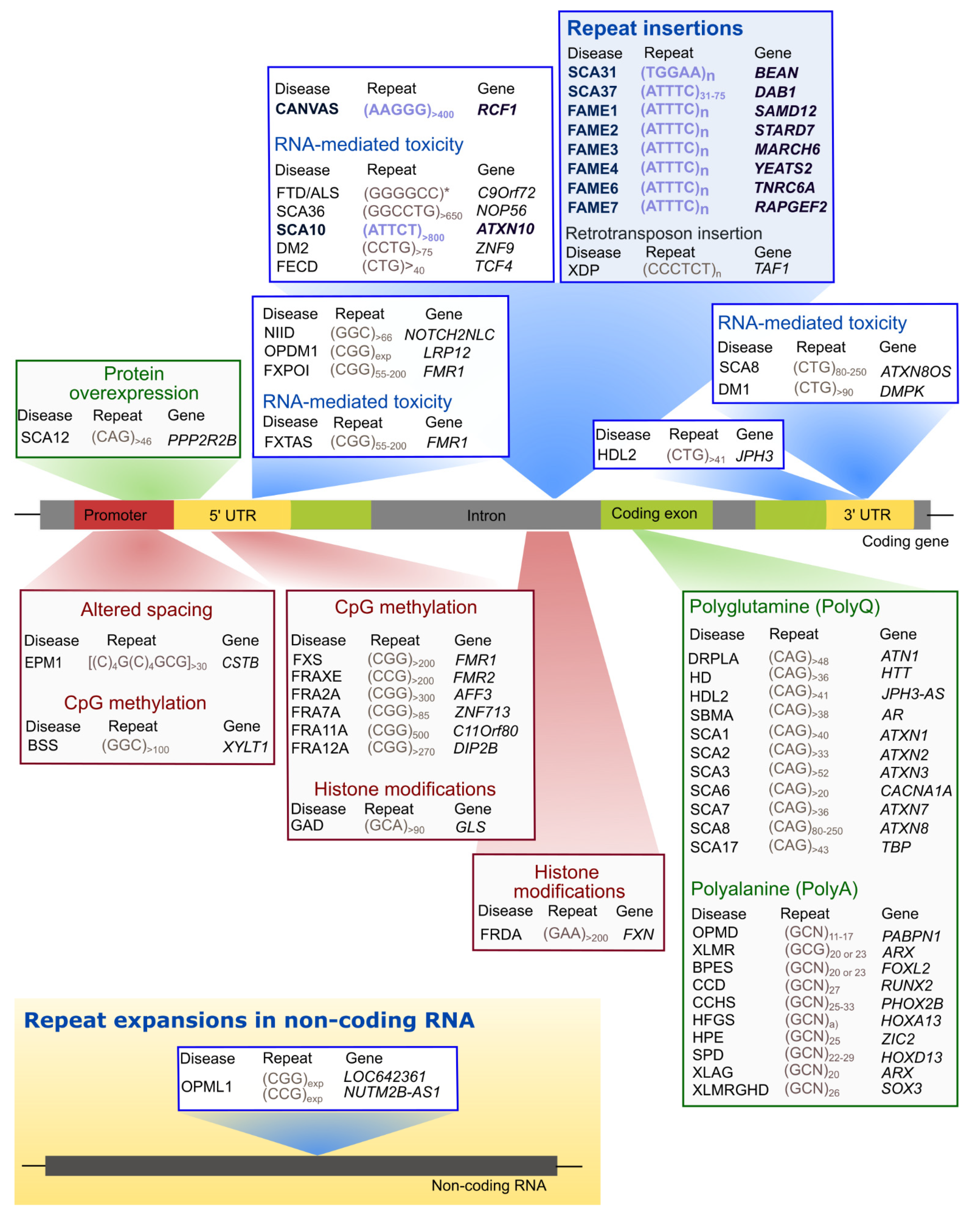{kind=link}
{kind=link}
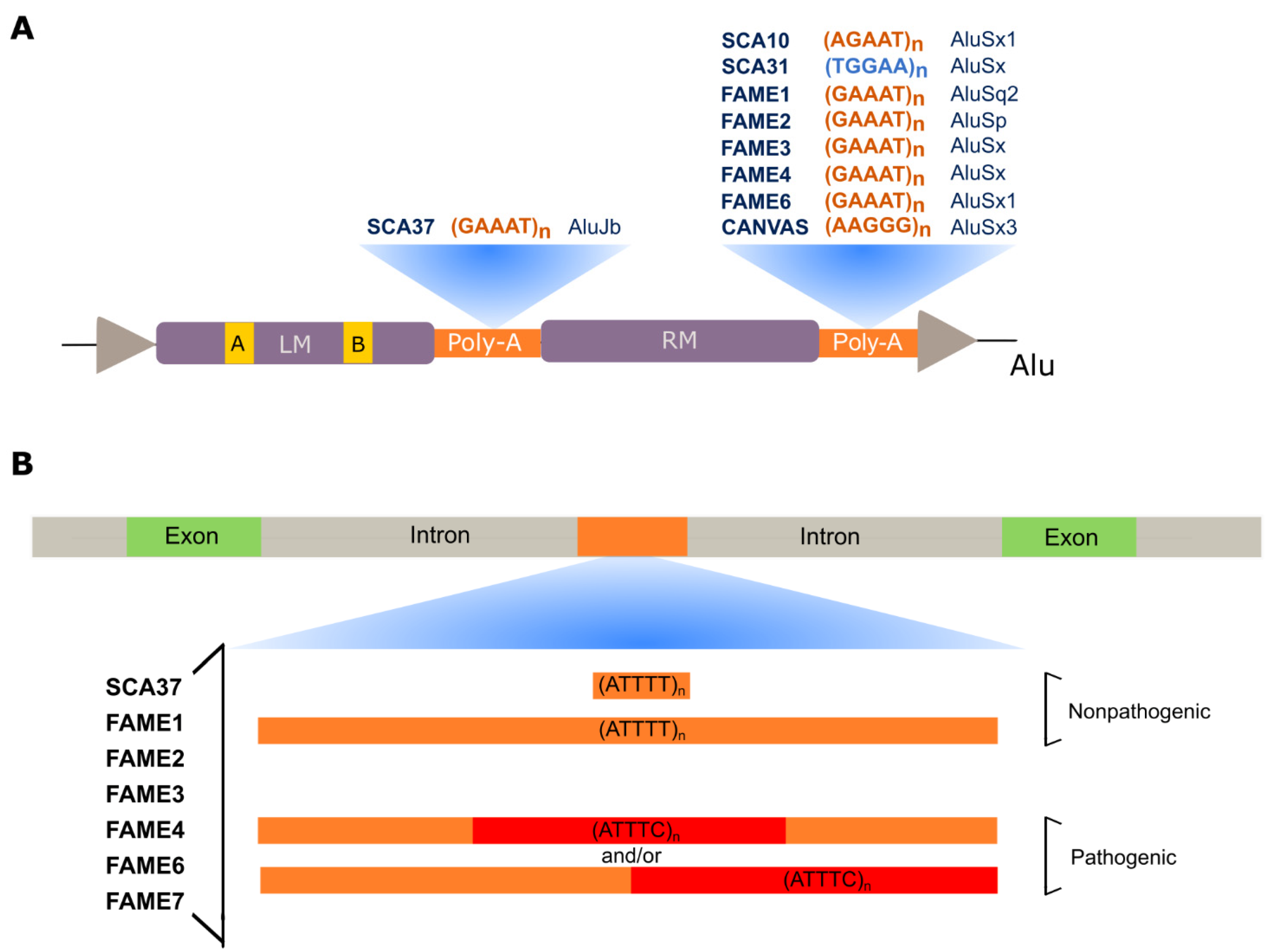{kind=link}
Table 1.
Neurological diseases caused by pentanucleotide repeat expansions.
| Disease | Type | Gene | Pathogenic Alleles | Non-Pathogenic Alleles | |
|---|---|---|---|---|---|
| Common | Less Common | ||||
| SCA | 27SCA10 | ATXN10 | (ATTCT)800–4500 | (ATTCT)10–16 | (ATTCT)17–32 |
| 34SCA31 | BEAN1 | TAAAA(TAGAA)1–4(TGGAA)n | (TAAAA)8–20 | (TAAAA)exp | |
| (TAAAA)n(TAGAA)n and (TAAAATAGAA)n | |||||
| 35SCA37 | DAB1 | (ATTTT)60–79(ATTTC)31–75(ATTTT)58–90 | (ATTTT)7–30 | (ATTTT)31–400 | |
| CANVAS | 32CANVAS | RFC1 | (AAGGG)400-2000 | (AAAAG)11 | (AAAAG)15–200 |
| (AAAGG)40–1000 | |||||
| 130(ACAGG)~1000 | |||||
| 131(AAAGG)10–25(AAGGG)exp | |||||
| FAME | 36FAME1 | SAMD12 | (ATTTT)exp(ATTTC)exp | (ATTTT)<100 | (ATTTT)>100 |
| [(ATTTT)exp(ATTTC)exp(ATTTT)exp]440–3680 | |||||
| 143(ATTTT)n(ATTTG)n | |||||
| 37FAME2 | STARD7 | (ATTTT)340–390(ATTTC)345–588 | (ATTTT)n | n.d. | |
| 38FAME3 | MARCH6 | [(ATTTT)exp(ATTTC)exp]791–1035 | (ATTTT)9–20 | n.d. | |
| 39FAME4 | YEATS2 | (ATTTT)819(ATTTC)221 | (ATTTT)4–120 | (ATTTT)121–1219 | |
| 36FAME6 | TNRC6A | (ATTTT)22(ATTTC)exp(ATTTT)exp | (ATTTT)<200 bp * | (ATTTT)>200 bp * | |
| 36FAME7 | RAPGEF2 | (ATTTT)exp(ATTTC)exp(ATTTT)n | (ATTTT)<300 bp * | (ATTTT)300–3000 bp * | |
n.d.—not determined; * Repeat size is in base pairs.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Loureiro, J.R.; Castro, A.F.; Figueiredo, A.S.; Silveira, I. Molecular Mechanisms in Pentanucleotide Repeat Diseases. Cells 2022, 11, 205. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11020205
AMA Style
Loureiro JR, Castro AF, Figueiredo AS, Silveira I. Molecular Mechanisms in Pentanucleotide Repeat Diseases. Cells. 2022; 11(2):205. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11020205
Chicago/Turabian StyleLoureiro, Joana R., Ana F. Castro, Ana S. Figueiredo, and Isabel Silveira. 2022. "Molecular Mechanisms in Pentanucleotide Repeat Diseases" Cells 11, no. 2: 205. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11020205
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.