
Redox Implications of Extreme Task Performance: The Case in Driver Athletes

College of Health and Human Performance, University of Florida, Gainesville, FL 32611, USA
Cells 2022, 11(5), 899; https://0-doi-org.brum.beds.ac.uk/10.3390/cells11050899
Submission received: 27 January 2022
/
Revised: 26 February 2022
/
Accepted: 3 March 2022
/
Published: 5 March 2022
(This article belongs to the Special Issue Redox Control of Cell Signaling in Cardiac and Skeletal Muscle)
Abstract
:Redox homeostasis and redox-mediated signaling mechanisms are fundamental elements of human biology. Physiological levels of reactive oxygen species (ROS) and reactive nitrogen species (RNS) modulate a range of functional processes at the cellular, tissue, and systemic levels in healthy humans. Conversely, excess ROS or RNS activity can disrupt function, impairing the performance of daily activities. This article analyzes the impact of redox mechanisms on extreme task performance. Such activities (a) require complex motor skills, (b) are physically demanding, (c) are performed in an extreme environment, (d) require high-level executive function, and (e) pose an imminent risk of injury or death. The current analysis utilizes race car driving as a representative example. The physiological challenges of this extreme task include physical exertion, g loading, vibration, heat exposure, dehydration, noise, mental demands, and emotional factors. Each of these challenges stimulates ROS signaling, RNS signaling, or both, alters redox homeostasis, and exerts pro-oxidant effects at either the tissue or systemic levels. These redox mechanisms appear to promote physiological stress during race car driving and impair the performance of driver athletes.
1. Introduction
Over the past half a century, redox stress and redox-mediated signaling mechanisms have emerged as fundamental elements of human biology. Much has been learned about the generation of reactive oxygen species (ROS) and reactive nitrogen species (RNS), their roles in signal transduction and cellular adaptation, and the antioxidant processes that buffer these redox cascades. For obvious reasons, much of this research has focused on redox mechanisms that impair human health in conditions such as aging, acute inflammation, and chronic disease. Less is known about redox mechanisms that modulate physiological processes and can limit the performance of healthy individuals in their daily activities.
Redox limitations to human performance have been demonstrated in laboratory studies of isolated muscle groups, whole-body exercise, and cognitive tasks. By extension, redox mechanisms are thought to also limit the spontaneous performance of more complex activities in a variety of settings. Examples include athletic competition and activities in the workplace. This article considers a special condition that bridges these two fields, i.e., the risks posed by redox stress when performing extreme tasks.
For the purposes of this article, an extreme task is defined by five characteristics: the task requires one or more complex motor skills; the task is physically demanding; it is performed in an extreme environment; successful execution requires high-level executive function; and finally, the task poses an imminent risk of serious injury or death. Extreme tasks are routinely performed by workers in various professions. Examples include war fighters, commercial divers, fighter pilots, astronauts, fire fighters, race car drivers, and alpine rescue teams.
Redox limitations to extreme activities are largely undefined. In part, this reflects the challenges of such research. Collection of primary data is difficult during extreme activities, and laboratory models are imperfect. Accordingly, there are few systematic studies of extreme task performance, and mechanistic research is largely nonexistent. For these reasons, we have little direct knowledge of redox physiology during extreme tasks or the limitations posed by redox mechanisms. Still, a conceptual framework can be used to predict redox limitations by integrating the known traits of a given task with the broader understanding of redox biology.
The present article develops such a framework. For this exercise, race car driving was selected as the representative activity. There are several reasons for this: First, race car driving typifies an extreme activity, meeting each of the criteria stated above. Second, the physiological data needed to define a conceptual framework are documented in the archival literature [1]. Third, racing is reported to alter the redox status of driver athletes [2]. Lastly, race car driving is a widely publicized activity that will be familiar to most readers.
The physiological challenges faced by driver athletes include the physical work of car control (e.g., steering, braking), g-loads, vibration, noise, heat exposure, and mental factors, e.g., cognitive demand, discomfort/pain, emotion, etc. Drivers perceive and adjust to these demands as they race—a classic example of feedback control. An applied model of feedback control is illustrated in Figure 1. This model depicts the basic relationships among the physiological processes that are used in race car driving and are susceptible to driving-related challenges.
Over time, the challenges of racing promote negative outcomes in the driver. These include dehydration, elevated metabolism and cardiovascular demand, fatigue, mental errors, performance loss, and greater risk of accident/injury. The mechanisms that lead to these outcomes appear to have one trait in common: in each case, the challenge stimulates production of ROS or RNS, which mediate negative effects on the driver. Subsequent sections will address the physiological challenges of race car driving, the effects they have on driver athletes, and the redox mechanisms that appear to be involved. Table 1 provides an overview of the pathways thought to regulate these responses.
2. Physical Work
The physical demands of race car driving are substantial. First, car control during competition requires repeated, high-intensity work by specific muscle groups over long periods of time. For example, engineering assessments of driver input illustrate the forces required to drive a high-level race car on a road course during a race that lasted 98 min [3]. Forces developed by the driver during heavy braking averaged 600 N per turn—a one-legged task repeated 225 times. Steering effort averaged 157 N per turn—a task performed 1105 times using arm and shoulder muscles. It is no surprise that professional drivers have identified strength of the limbs and upper body as the major physical demands of their profession [4].
Second, drivers must oppose gravitational (g) forces generated by the race car during changes in direction or velocity. In the horizontal plane, g force along the lateral axis is proportional to the radius of curvature and velocity of a turn. Braking or acceleration develops g force along the longitudinal axis; such forces are proportional to the rate of change in velocity. Modern race cars that are optimized for aerodynamic downforce can exceed 5 g during cornering and braking [5]—physical loads that are applied to the driver.
The physical effort required to resist g loads is among the greatest demands on a race car driver. For example, a driver’s head and race helmet combined weigh approximately 6.4 kg. During heavy braking, the axial loading applied to the head and helmet is effectively 259 N—a force counteracted by the muscles of the neck and upper torso that oppose neck flexion. Moreover, during cornering and braking, drivers use an anti-g straining maneuver developed by fighter pilots [6], in which muscles of the trunk are tensed and the breath may be held [3,7]. This maneuver stabilizes body posture, protects the abdominal viscera, and preserves cerebral blood flow at the expense of muscular effort.
Strenuous exertion places substantial metabolic demand on the driver. This is estimated to range from 5.3 to 13.0 metabolic equivalents, or METs [5,8,9], which is comparable to MET values for traditional sports, e.g., basketball 8.0, soccer 10.0, and boxing 12.8 [10]. Direct measurements of oxygen consumption (VO2) by professional race drivers showed that sustained VO2 averaged 2.76 L·min−1 while driving a typical road course; this was 79% of the maximal VO2 measured in the same drivers under laboratory conditions [11]. Separate measurements of blood chemistry under race conditions have shown robust increases in free fatty acids [12,13], triglycerides [13], and glucose [12,14]—circulating substrates that fuel aerobic metabolism. Longer periods of continuous driving cause an increase in blood lactate levels [12,14,15,16]—further evidence of metabolic demand. Elevated metabolism also increases heat production via aerobic metabolism and blood flow to working muscles; these two factors are discussed in greater detail below.
Fatigue is among the greatest challenges for driver athletes [4]. Over the course of a race, the work required to control the car and resist g forces causes the muscles of the arms, shoulder girdle, neck, trunk, and legs to fatigue [15]. This affects steering, braking, and postural control, impairing the performance of the driver. The degree of fatigue varies between muscles and is time- and task-specific. Fatigue is negligible in events of short duration—e.g., drag or sprint races—but can be a major concern in longer races. The race format also matters. Road races require frequent changes in direction and speed, requiring more work and promoting greater fatigue than speedway races of similar duration. Secondary factors that contribute to driver fatigue include vibration of the car and its controls [17], heat exposure [18], and prolonged mental challenges [19], as discussed in subsequent sections.
In aggregate, drivers perform repetitive, forceful contractions by major muscle groups of the limbs, neck, shoulder girdle, and trunk. This intense, whole-body exercise can continue for up to four hours. During this time, the working muscles exhibit elevated metabolism, greater heat generation, higher rates of perfusion, and increased oxidant production; the latter is of particular interest for this review article.
Oxidants are generated at increased rates by working muscles. Bjugstad et al. [2] have shown that racing induces oxidative changes in driver athletes, shifting the redox potential of mixed venous blood to a more oxidized state. This response was related to pre-race antioxidant capacity. Redox status was less perturbed in drivers who had higher antioxidant capacities and those who took vitamin supplements. In part, the pro-oxidant shift caused by racing reflected an increase in oxidant production by working muscles. Skeletal muscle continually generates ROS, which are detectable within myofibers [20] and in the extracellular space [21]. The parent molecule of the ROS cascade is the superoxide anion radical, which gives rise to hydrogen peroxide, hydroxyl radicals, and other low-molecular-weight derivatives. Sources of ROS in differentiated myofibers include the mitochondrial electron transport chain, nicotinamide adenine dinucleotide (phosphate) oxidase—or NAD(P)H oxidase (NOX)—xanthine oxidase, cyclooxygenases, and lipoxygenases. Among these, ROS from specific oxidases appear to modulate cell signaling via redox-sensitive regulatory proteins [22], whereas ROS that modulate the intracellular redox environment derive from both mitochondria [23] and NOX [24].
In parallel, skeletal muscle continually generates nitric oxide, or NO [25]. This may be produced via either of two constitutively expressed NO synthases: a tissue-specific isoform of neuronal-type nitric oxide synthase—or nNOS [25]—and the endothelial-type NO synthase, or eNOS [26]. NOS-independent synthesis of NO from nitrate may also be possible under certain conditions [27]. NO and its redox derivatives compose the RNS cascade. NO can act on target proteins via cGMP-dependent signaling or via direct redox modification, e.g., S-nitrosylation. By these mechanisms, NO modulates various aspects of skeletal muscle function, including contractile function [25,28], mitochondrial respiration [29], and glucose uptake [30].
Both ROS and RNS activities increase in muscles during repetitive contractions [20,25], such as those that occur in the limb and trunk muscles of driver athletes during competition. Increases in ROS and RNS activities shift the intracellular redox status, promoting oxidation of regulatory proteins and altering myofiber function in a time- and concentration-dependent manner. Increases in oxidant activity can disrupt excitation–contraction coupling [31], calcium sensitivity of myofilament proteins [32,33], mitochondrial energetics [34], and carbohydrate metabolism [30]. These intracellular events combine to depress the specific force of muscle contraction [35] and contribute to muscle fatigue [36]—a near-ubiquitous finding in the muscles of driver athletes following a race [15].
3. Vibration
Race cars vibrate. The sources of vibration include rotation of drivetrain components, wheel-and-brake assemblies, interactions between the tires and the road surface, flexing of aerodynamic surfaces, and harmonics of the chassis, engine, and other components [37,38,39]. The race car structure is stiff, and there is little to dampen the transmission of vibrations to the driver [40,41,42]. Vibrations transmitted to the driver act as both whole-body inputs and localized mechanical stimuli, eliciting distinct physiological responses in each case.
Whole-body vibration has multiple effects on drivers. At high speeds, interactions of the car with variations in the track surface can impose transient vertical loads of up to 3 g on the driver [42]. Vertical loading can cause substantial discomfort and injury in drivers. Indeed, a retrospective study of musculoskeletal injuries in over 130 drivers identified pain in the lumbar back region and upper legs as one of the most common complaints [43]. Whole-body vibration can slightly impair driver vision, although this appears to be a transient phenomenon with no serious consequences. Laboratory studies of healthy adults show that whole-body vibration stimulates reflex increases in heart rate, cardiac output, oxygen consumption, and minute ventilation [44]. These changes mimic the cardiopulmonary responses seen in drivers during competition [1]. Similarly, whole-body vibration causes mental fatigue and diminishes mental alertness in noncompetitive driving [17]. Such issues affect race drivers during long stints, especially in cross-country rallies and endurance racing, and may be caused in part by vibration.
Localized transmission of vibrations to the driver via the steering wheel is a well-defined problem [45]. It predisposes muscles of the hands, arms, and upper body to cramping and fatigue, which can impair car control and distract the driver. Steering wheel vibrations are also linked to symptoms and nerve disorders of the hands and arms [43]. This is consistent with mechanistic research showing that vibration of an appendage affects the associated peripheral nerve. Acute vibration sensitizes nerve fiber populations that carry pain and proprioceptive information—notably Aβ, Aδ, and C fibers [46]. Over longer time periods, repetitive bouts of vibration cause loss of small nerve fibers, increase mitochondrial numbers in fibers, and increase the size of sensory receptors [47]. Adaptations in the afferent pathway are associated with pro-inflammatory changes that include cytokine upregulation and oxidative stress [46].
Vibration also elicits redox responses. Circulating markers of oxidative stress, as documented in driver athletes [2], are among the earliest biochemical indicators of vibration-induced disease in humans [48]. These include changes in the serum activities of superoxide dismutase (SOD) and catalase—enzymes that selectively degrade superoxide anions and hydrogen peroxide, respectively. Mechanistic studies show that selective vibration of a single appendage has widespread effects on redox status. Increased ROS levels have been documented in the appendage skin, arterial walls, and sensory receptors—responses that are sensitive to vibration frequency [47,49]. Appendage vibration also induces oxidative stress in remote tissues, including the dorsal root ganglia that serve the appendage [46] and the heart [50]. Redox adaptation to vibration is suggested by alterations in mRNA levels for nNOS and antioxidant enzymes in the heart, eNOS and oxidative stress genes in the prostate, and pro-inflammatory cytokines in multiple tissues [50].
Experiments have shown that physiological responses stimulated by vibration are sensitized by direct ROS exposure and blunted by selective ROS depletion [51]. This provides proof-of-concept that ROS can play a causal role in the vibration-related problems experienced by race car drivers. Specifically, oxidative stress promotes neuroinflammation [52,53] which mediates lower back pain [54,55]. Such pain is a chronic malady that plagues race car drivers and is caused by whole-body vibration. Similarly, steering wheel vibration causes localized pain, numbness, and cramping in driver forearms. These symptoms are predicted to result from the localized effects of oxidants on peripheral nerves and dorsal root ganglia [56], as well as on muscle spindles [57].
4. Noise
Environmental noise has deleterious effects on human health. Brief periods of noise exposure can interfere with work performance [58] and promote hearing loss that may be irreversible [59]. Repeated noise exposure or continuous long-term exposure can have non-auditory health consequences; these include sleep disturbance [60], impaired cognition [61], circadian dysfunction [62], vascular dysregulation [63], and cardiovascular disease [64]. The severity of such risks has led government agencies to establish standards for occupational noise exposure in numerous countries. For example, United States standards are overseen by both the Occupational Safety and Health Administration (OSHA) and the National Institute of Occupational Safety and Health (NIOSH) [65].
Noise is a prominent feature of auto racing. Driver athletes continuously work near unmuffled race cars and noisy support machinery such as generators, jacks, and compressors. Ambient noise at race tracks can reach peak levels of 130–140 dB [59,66,67], exceeding the exposure limits recommended by OSHA and NIOSH [65,66]. Accordingly, noise has deleterious effects on drivers, interfering with communication [68,69] and potentially impairing performance [58]. Hearing loss is a ubiquitous risk of the profession and a common finding among experienced drivers [66]. Current research predicts that drivers are also at risk of non-auditory health effects of noise on sleep quality, cognitive ability, and cardiovascular regulation. Noise may also promote inner ear balance disorder—a consequence of noise-induced inflammation of the vestibular system. The prevalence and severity of non-auditory noise effects on driver athletes has not been reported.
Noise-induced hearing loss is caused by damage to the cochlea—a delicate structure of the inner ear. Cochlear hair cells transduce mechanical energy to electrical energy, converting sound into afferent neural input that can be interpreted by the brain [70]. Animal experiments show that loud noises damage key components of this sensory pathway, including stereocilia and hair cells [71], hair cell synaptic ribbons [72], spiral ganglia neurons [73], and the stria vascularis [74]. Functional results of this damage include elevated hearing threshold [75], disorders of speech perception and auditory processing [76], and tinnitus [77], all of which are common among driver athletes [66,68,69].
Oxidative stress mediates the damage to auditory structures caused by noise [78]. Noise at damaging levels stimulates ROS activity in the cochlear tissue [79]—a response that precedes cellular injury and persists for a week or more [80]. Evidence of oxidative stress includes increased lipid peroxidation products and protein nitrosylation [81], as well as altered glutathione regulation [82,83]. Importantly, noise-induced cochlear damage is blunted by pretreatment with nonspecific antioxidants, including vitamin E and alpha lipoic acid [84], ebselen [85], glutathione [86], and N-acetylcysteine [87]. Specific evidence for ROS involvement derives from experiments showing that noise-induced damage can be inhibited by ROS-selective probes, such as either exogenous SOD [88] or hydroxylated alpha-phenyl-tert-butylnitrone—a nitrone-based spin trap for hydroxyl and superoxide anion radicals [89]. Mitochondria are the primary source of noise-induced ROS in cochlear tissue [90]. The increase in ROS production is attributed to a rise in aerobic metabolism [90] and loss of mitochondrial membrane potential [75]. These changes drive electron leakage from the electron transport chain, thereby stimulating superoxide anion formation. NOX-derived ROS also contribute to noise-induced cochlear damage. This follows from observations that NOX subunit expression is altered by damaging noise [91], signaling via the NOX3 isoform mediates noise-induced inflammation [92], and NOX inhibition reduces the damage caused by noise exposure [93]. In aggregate, the science indicates that hearing loss, difficulties in speech perception, and tinnitus experienced by driver athletes are the result of cochlear damage caused by repeated bouts of oxidative stress mediated by noise-induced ROS production.
Beyond the acute effects of individual race events, driver athletes live in a professional environment that predisposes them to persistent noise exposure. This is a major environmental stressor that promotes mental stress, disrupts sleep, and is associated with cardiometabolic risk factors [60,64]. Unlike hearing loss, the non-auditory responses to persistent noise are not attributed to a primary unifying mechanism. Rather, the existing research links persistent noise exposure to pathogenic risk factors that include chronic elevation of stress hormone levels and markers of inflammation, e.g., circulating cytokines, pro-inflammatory signaling, nitric oxide synthesis, and ROS activity [64,66,79].
5. Heat
Race cars generate large amounts of thermal energy during competition. Heat from the engine, exhaust system, brakes, and tires combine to warm the cockpit of the car. Cockpit air temperatures of 50–60 °C are common, with solid surfaces reaching 100 °C [5,18,94,95]. In this hot environment, the driver athlete continuously works at 65–85% of maximal VO2 for up to four hours while wearing a helmet, gloves, and multilayered fire-resistant safety clothing. In combination, these factors create the conditions for uncompensable heat stress [96]. Skin temperature rises, core temperature rises faster, and the core-to-skin temperature gradient for heat dissipation falls progressively [97]. Sweating is profuse but ineffective because evaporative cooling is prevented by the safety clothing. As a result, drivers routinely lose 3–4% of body weight due to dehydration, with corresponding increases in hematocrit and urine osmolarity [14,18,98]. Heat stress and dehydration combine to impose thermal and physiological strain on the driver [97,99,100,101] that can increase the perception of exertion and discomfort [101], diminish performance [18,99], impair cognition [102], cause heat stroke [94,95], and induce loss of consciousness [103]. Heat exposure is amplified in closed-cabin cars, hot weather, and prolonged stints of continuous driving [5,18]. Drivers can mitigate the effects of heat through a program of acclimatization [99], and by in-car use of cooling technologies [95,100].
Oxidative stress is associated with environmental heat exposure in various occupations [104,105,106,107,108,109], including athletics [104,110,111,112,113]. This is consistent with data from driver athletes. Drivers exhibited increases in body temperature and oxidative stress after racing under warm conditions (28.3 °C), as compared with baseline measurements under cooler conditions (21.3 °C) [2]. McAnulty et al. [104] first demonstrated that hyperthermia increases oxidative stress during exercise—a robust finding that has been confirmed by others [113,114]. Thus, exercise and environmental heat have additive effects on redox status, amplifying pro-oxidant changes at the systemic level [115].
To define the underlying physiology, researchers have tested the interactions between physical activity, heat, and dehydration in healthy volunteers, measuring plasma biomarkers to assess changes in redox status. Hillman et al. [116] showed that pro-oxidant changes caused by hyperthermic exercise were exaggerated if subjects became dehydrated. This confirms the risk to driver athletes, who routinely become dehydrated during competition. Laitano et al. [117] established that hydration state modulates glutathione regulation by muscles. Glutathione is normally taken up by muscles, whereas dehydration stimulates exercising muscles to release glutathione into the circulation. Consistent with this finding, Georgescu et al. [118] found that dehydration per se alters plasma antioxidant capacity in the absence of heat exposure or exercise. Redox perturbations caused by heat exposure or exercise appear to be mitigated by exercise training [113] and may be acutely reversed by rehydration [119], although the latter finding remains controversial [117,118].
Heat exposure induces oxidative stress at the cellular level by increasing ROS production and decreasing antioxidant protection. Mitochondrial production of superoxide anion radicals is stimulated by damage to the electron transport chain [120] and by a reduction in the synthesis of uncoupling proteins [121]. Generation of superoxide anions by NOX is also stimulated due to upregulation of the NOX1 isoform and an increase in the cytoplasmic ratio between oxidized nicotinamide adenine dinucleotide phosphate (NADP+) and reduced NADP+ or NADPH [122]. In parallel, heat accumulation compromises the oxidant buffering capacity of the cell by inactivating mitochondrial SOD [123] and depleting glutathione levels [124].
King et al. [125] have extended this model to include the effects of exercise and dehydration in vivo. These authors acknowledge the traditional effects of heat on cellular redox status (above), but further observe that evaporative water loss and sweating during hyperthermic exercise leads to hemoconcentration and increased blood viscosity. The rise in blood viscosity increases shear stress on vessel walls. This stimulates endothelial release of nitric oxide [126] and ROS [127] that act on erythrocytes to increase rigidity and impair deformability [128,129]. These biophysical changes alter the rheological properties of blood, promoting hemolysis. The subsequent release of free hemoglobin promotes further ROS generation via Fenton-like reactions, a chemistry catalyzed by the ferrous iron in heme centers.
Redox responses to whole-body heat exposure are well documented. Studies indicate that heat accumulation preferentially affects mitochondria-rich tissues, including the heart, liver, and aerobic skeletal muscles. Thus, heat exposure preferentially targets tissues stressed by the activity of race car driving, imposing an additional physiological load on heavily working skeletal and cardiac muscles. Heat exposure acts on these tissues to disrupt redox homeostasis and mitochondrial function. Malondialdehyde, a marker of lipid peroxidation, is elevated in aerobic muscles following heat accumulation [130,131]. This is accompanied by increased tissue content of SOD mRNA and SOD proteins, as well as elevated SOD enzyme activity [130,132]. Heat stress also alters catalase content and enzyme activity in aerobic muscles [130,132]. Mitochondrial protein analyses showed that cytochrome c, cytochrome c oxidase, voltage-dependent anion channel, pyruvate dehydrogenase, and prohibitin 1 were all elevated [130]. In a separate study [131], biochemical analyses of subsarcolemmal mitochondria confirmed that malondialdehyde levels are elevated following heat exposure and that carbonylation of mitochondrial proteins is widespread—further evidence of oxidative stress in these organelles.
6. Cardiovascular Demand
Cardiovascular regulation is linked to racing-related changes in oxidant activity [2] and is among the best documented aspects of driver physiology. For over half a century, we have known that driver heart rate is elevated before a race begins [133]—an anticipatory response stimulated by increases in circulating epinephrine and norepinephrine [134]. Heart rate remains elevated throughout the event, with sustained heart rates of 65–90% of maximal being common [95,134,135]. Impedance cardiography under race conditions has shown that stroke volume is also increased [136], further contributing to a rise in cardiac output. In general, heart rate increases with the average speed of the car on a given track [11]. However, instantaneous heart rate varies according to driver position on the track, being higher in corners and lower on straight sections [137]. Similarly, average heart rate is greater during races on road courses than races on speedways, despite the latter having higher average speeds [11]. Thus, speed per se is not the sole determinant of heart rate; the physical work of turning and braking is also important. Cardiovascular function is further influenced by positive g loading in the vertical axis, which promotes blood pooling in the lower extremities. Prolonged exposure can limit venous return to the heart, thereby depressing cardiac output and systemic arterial pressure, which can cause cerebral hypoxia. These are real concerns for drivers racing on speedways, where banked corners impose significant vertical g loads. In extreme cases, this can cause visual disturbances and even loss of consciousness in drivers [138]. A final contributor to cardiovascular stress is heat, which elevates sweat rate, blood flow to the skin, heart rate, and cardiac output for a given level of physical work [139]. This thermoregulatory reflex is pervasive in driver athletes across a broad range of racing series [97,101,140]. Prolonged heat exposure promotes dehydration—a common risk for driver athletes [141,142]. Dehydration promotes cardiovascular stress by decreasing the volume of circulating plasma and blunting heart rate variability [143].
The cardiovascular responses of driver athletes are consistent with the acute changes in cardiac function mediated by redox signaling. The contractile function of the healthy heart is modulated by endogenous ROS generated by mitochondria and NOX. Cardiac myocytes produce ROS in proportion to contractile activity and oxidative metabolic rate [144,145]. These are generated by electron leakage from complexes I and III of the mitochondrial electron transport chain [146]. This basal rate of ROS production does not appear to influence contractile function [147,148]; however, endogenous ROS do modulate contraction as second messengers in two parallel pathways.
First, the “fight-or-flight” response seen in driver athletes [149] is mediated in part by sympathetic stimulation of β-adrenergic receptors in the myocardium. This increases the force and frequency of heart contractions, elevating cardiac output. At the cellular level, β-adrenergic stimulation increases endogenous ROS production as well as contractility [150]. Mitochondria and NOX are both implicated as sources of β-adrenergic-stimulated ROS in this response [147]. Potential downstream target(s) of ROS include redox-sensitive kinases, phosphatases, and regulatory proteins of the myofilament lattice, sarcoplasmic reticulum, or sarcolemma [151]. Studies using antioxidant probes indicate that endogenous ROS can either oppose [147,148] or amplify [150] the contractile effect of β-adrenergic stimulation. The net effect may depend on the source of endogenous ROS, the subcellular localization of the source, and the response of the adenylyl cyclase-cAMP-PKA pathway [151].
Second, endothelin-1 (ET-1) is a potent vasoconstrictor that regulates blood pressure under conditions experienced by drivers, including prolonged exercise [152] and altered g loading [153]. ET-1 is constitutively expressed by the cardiac endothelium, vascular smooth muscle, fibroblasts, and myocytes. Among its effects on the heart, ET-1 is an important regulator of myocardial contractility. ET-1 binds to surface receptors, activating a redox-mediated signaling cascade that increases the force of contraction. Cytosolic signaling events in the ET-1 pathway include NOX production of superoxide anion radicals [154]. Superoxide anions activate extracellular signal-regulated kinase 1/2 (ERK1/2), which acts via p90 ribosomal S6 kinase (p90RSK) and sodium/hydrogen exchanger-1 (NHE1) to increase myofilament calcium sensitivity [147], thereby increasing the force of contraction.
Like skeletal muscle, cardiac myocytes constitutively express both nNOS and eNOS, which have distinct subcellular localizations [155]. The nNOS isoform localizes to the sarcoplasmic reticulum (SR) and plays an active role in β-adrenergic receptor signaling [156]; nNOS acts via guanosine 3′,5′-cyclic monophosphate (cGMP) to stimulate phospholamban phosphorylation, which disinhibits calcium uptake by the sarcoplasmic reticulum (SR) calcium ATPase, or SERCA [157]. This increases SR calcium stores, calcium release, and cardiac contractility in response to β-adrenergic stimulation, contributing to the rise in stroke volume and cardiac output in driver athletes under race conditions [136]. In contrast, eNOS localizes to the caveolae of the transverse tubule system, and acts on the L-type calcium channel to limit the inward calcium current. eNOS signaling exerts negative effects on β-adrenergic receptor signaling, blunting calcium release and depressing cardiac contractility [158], as well as limiting arrhythmogenic activity [159].
7. Mental Factors
The physiological basis for elite performance by driver athletes is grounded in motor control and brain neurobiology. Race drivers differ from non-racing drivers in their execution of task-specific motor skills; oculomotor behavior is a notable example. Unlike non-racing drivers, race drivers use head direction to determine gaze angle, which is tightly linked to steering angle and vehicle rotation—a behavior that appears to be controlled at the unconscious level [160]. Moreover, their driving behavior differs from that of non-racing drivers. Race drivers drive a different path when negotiating a racetrack and are more decisive in throttle and brake applications [161]. Furthermore, their gaze strategy is more variable, and there is greater head rotation while turning the vehicle, which led Land and Lee [162] to conclude that race drivers have different perceptual–cognitive skills than non-racing drivers.
Behavioral differences are complemented by differences in brain function and structure. Bernardi et al. [163] used functional magnetic resonance imaging (fMRI) to compare the brain function of race and non-racing drivers while performing standardized mental tasks. For a given task, race drivers were found to recruit smaller volumes of the task-related brain regions and exhibit stronger functional connections between regions. From these results, the authors concluded that that race drivers benefit from greater “neural efficiency”. A second study by the same research team [164] used functional magnetic resonance imaging (fMRI) to measure brain function and structure while subjects watched videos of cars racing on tracks. Race drivers were found to recruit a larger number of brain regions than non-racing drivers, and more consistently recruited the brain regions related to motor control and spatial navigation. Race drivers also had a greater density of gray matter in driving-related brain regions, including the retrosplenial cortex. Among race drivers, gray matter density of the retrosplenial cortex was directly correlated with success in the sport, as reflected by top-three finishes in prior races. The results of these fMRI studies are consistent with the assertion that elite race drivers have unique brains, functionally and structurally different from non-racing drivers [165].
Elite brain notwithstanding, the mental demands of racing are an omnipresent challenge that can affect drivers’ emotions, cognition, and performance [166]. Changes in mental state can be inferred, in part, by activity of the sympathoadrenal axis. Moment-to-moment changes are reflected in driver heart rate. Anticipation causes heart rate to increase in the moments before a race begins [137], imminent danger stimulates heart rate during a crash [167], and competition per se elevates heart rate compared to practice sessions or reconnaissance runs at the same speed [135,140]. Long-term stress responses are evident in measurements of circulating hormones. Total catecholamine levels are elevated during competition [12]. This response surges at the start of a race, slowly declines over time, and is dominated by norepinephrine [13]. Other stress hormones elevated during racing include cortisol [19], aldosterone [16], testosterone [138], and human growth hormone [12].
Environmental and occupational factors can further amplify the problem. Environmental noise interferes with communication [58], promotes annoyance [61], disrupts sleep patterns [168], and activates the sympathoadrenal axis [63]. Sleep deprivation promotes mental stress [169] and is endemic among driver athletes on race weekends. Sleep duration is constrained throughout the weekend by driver commitments to the team, sponsors, race organizers, media, and fans. During available sleep periods, the quality of sleep may be compromised by task-related distractions and pre-performance anxiety. Sleep deprivation is particularly acute during endurance races, which last up to 24 h, or cross-country rallies that continue for days. Finally, international travel in some elite racing series can disrupt circadian rhythm—yet another factor promoting mental stress [170].
What are the redox implications of mental stress? In 1996, an early review by Moller et al. [171] concluded that evidence linking psychological stress to oxidative stress was “rudimentary”, citing only five references. How the field has changed! A PubMed search in January 2022 using the terms “mental stress” and “oxidative stress” returned over 3900 archival reports—the result of exponential growth in published research over the past 35 years. This expansive literature confirms that mental stress impacts redox homeostasis across a broad array of conditions. In humans, circulating biomarkers have been used to screen for oxidative stress under conditions of mental stress, repeatedly demonstrating elevation of circulating oxidation products [172,173,174,175] and decrements in antioxidant status [174,175,176]. Animal models of stress confirm changes in systemic biomarkers [177,178,179]; they further demonstrate oxidative modifications to brain regions that are involved in the stress response, including the hypothalamus [179,180] and brainstem [181]. Driver athletes routinely cope with mental challenges that impact redox status, including strenuous physical activity [174,182,183,184], environmental noise [64], sleep deprivation [169], fatigue [19], and circadian dysregulation [62].
The mechanisms responsible for oxidative stress in acute mental stress are not well defined. Regional effects within the brain likely reflect transient increases in the local activity of neuronal mitochondria and constitutively expressed nNOS or NOX isoforms. At the systemic level, a common denominator across many forms of acute mental stress is the neuroendocrine stress response; this is mediated by central activation of two complementary pathways, both of which are activated during race car driving [12,13,19]. The sympathoadrenal axis is initiated by brain input to sympathetic preganglionic neurons located in the thoracic spinal cord. These neurons respond to excitatory input by stimulating the release of catecholamines—i.e., norepinephrine and epinephrine—via two routes. Preganglionic stimulation of postganglionic neurons causes norepinephrine release from nerve endings in target tissues. Stimulation of chromaffin cells in the adrenal medulla causes the release of norepinephrine and epinephrine into systemic circulation. In contrast, the hypothalamic–pituitary–adrenal (HPA) axis is activated by stress-related glutamatergic input to neuroendocrine neurons in the paraventricular nucleus of the hypothalamus. Once activated, the neuroendocrine neurons release corticotropin-releasing hormone (CRH) and vasopressin—hormones that act on the anterior pituitary to stimulate the release of adrenocorticotropic hormone (ACTH). ACTH is transported by the systemic circulation to the adrenal cortex, where it stimulates the release of glucocorticoids, either cortisol in humans or corticosterone in rodents.
The sympathoadrenal and HPA axes are generally co-activated by conditions that cause mental stress [185], and oxidative stress can be induced by the end products of either axis: catecholamines [186] or glucocorticoids [187]. The mechanisms by which these hormones promote oxidative stress are complex and incompletely understood. However, in response to various mental stressors, there is a strong association between oxidative stress and higher rates of mitochondrial ROS production [188,189,190]. This is consistent with actions of the sympathoadrenal and HPA axes. Catecholamines stimulate oxygen demand in vivo [191], increasing aerobic metabolism via the mitochondrial electron transport chain. Glucocorticoids elevate glucose and free fatty acid concentrations in the circulation, providing additional substrate availability for mitochondrial respiration. Elevated activity of the mitochondrial electron transport chain promotes generation of superoxide anions and superoxide-derived ROS, increasing oxidant activity within minutes-to-hours [192]. Changes in the redox environment of target tissues appear to modulate glucocorticoid receptor signaling. Elevated oxidant levels downregulate expression of the glucocorticoid receptor [193], interfere with the dissociation of heat shock proteins from the receptor in the cytoplasmic compartment [194], and inhibit nuclear internalization of the receptor [194]. This blunts the downstream response of target cells to HPA stimulation at the expense of redox homeostasis, predisposing the target tissue to oxidative stress.
8. Conclusions
Extreme task performance, as illustrated by race car driving, requires an individual to overcome a complex array of physiological challenges. Some are intrinsic to the task; others are imposed by the environment in which the task is performed, while others reflect secondary demands on homeostatic regulation and mental function of the individual. A common property of these challenges is their potential to alter redox homeostasis. Challenges associated with race car driving stimulate the production of ROS or RNS, depress antioxidant capacity, or both [2]. These responses promote oxidative or nitrosative stress that may be localized to specific tissues, including the skeletal muscle, cardiac muscle, brain regions, or the cochlea. Alternatively, whole-body stimuli such as heat, vibration, and sympathoadrenergic activation have pro-oxidant effects that are distributed systemically.
Integrating these individual responses, one may conclude that challenges associated with race car driving disrupt redox homeostasis and promote four types of physiological stress: muscular stress, cardiovascular stress, heat stress, and mental stress. This is illustrated by the integrative model shown in Figure 2, which depicts driving-related challenges that promote each type of physiological stress: First, muscular stress is promoted by the work of steering and braking required for car control, postural work to resist g loading, increased metabolism of working muscles, vibration effects on peripheral nerves and joints, and dehydration effects on muscle blood flow. In combination, these factors can impair driver performance by causing muscle fatigue, muscle cramping, or both. Second, cardiovascular stress is promoted by increased metabolic demand for oxygen and substrate; elevated heart rate, stroke volume, and cardiac output; increased skin blood flow for heat exchange; reflex responses to vibration, noise, anxiety, and discomfort; decreased venous return during vertical g loading; and hemoconcentration and hemolysis secondary to dehydration. If cardiac reserve is insufficient to meet these challenges, perfusion limitation compromises the physical performance, heat tolerance, and mental acuity of the driver. Third, heat stress is caused by muscular heat generation and environmental heat exposure, compounded by the insulating effects of head-to-toe safety clothing. These factors promote heat retention by the driver, increasing core temperature and driver discomfort. In response, thermoregulatory reflexes increase sweat rate; this promotes driver dehydration, which compounds the deleterious effects of heat per se. Finally, mental stress can result from the complex cognitive demands of high-speed driving; the emotional responses to competition, e.g., anxiety, anger, fear, etc.; the discomfort or pain caused by loud noise, sustained vibration, and intense heat; and the loss of mental acuity promoted by physical exhaustion, sleep deprivation, and circadian dysregulation. In aggregate, these factors predispose driver athletes to mental errors of motor control, spatiotemporal judgement, and executive function that worsen performance on track and make accidents more likely.
Driver athletes are affected by all four physiological stresses—muscular, cardiovascular, heat, and mental—any of which may impair performance. Consider muscle cramps that impede steering, or heat stroke, or a mental error that causes a crash—each reflects overt task failure caused by one type of stress. The more common scenario is more nuanced. Drivers complete races but do not win, admitting that they could have done better. Such subpar performance likely reflects multiple stresses acting in parallel. Each exerts pro-oxidant effects on redox homeostasis, promoting oxidative stress that impairs different aspects of the driving task. Thus, redox disruption represents a common mechanism by which physiological stresses oppose optimal task performance.
It follows that nutritional strategies or training regimes that promote redox homeostasis might improve performance. Consistent with this hypothesis, vitamin supplements appear to reduce racing-induced oxidative stress [2], and media reports routinely trumpet the antioxidant-rich diets of professional drivers. Research is needed to determine the effect of such interventions on racing performance. The results could yield data-driven recommendations that would benefit driver athletes and the motorsports industry.
Funding
This work was funded by the University of Florida.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Not applicable.
Acknowledgments
Delainie McNeil for administrative support and Laurel Anderson Reid for editorial assistance.
Conflicts of Interest
The author declares no conflict of interest.
References
- Reid, M.B.; Lightfoot, J.T. The physiology of auto racing. Med. Sci. Sports Exerc. 2019, 51, 2548–2562. [Google Scholar] [CrossRef] [PubMed]
- Bjugstad, K.B.; Gutowski, P.; Pekarek, J.; Bourg, P.; Mains, C.W.; Bar-Or, D. Redox changes in amateur race car drivers before and after racing. Sports Med. Int. Open 2017, 1, E212–E219. [Google Scholar] [CrossRef] [Green Version]
- Pruett, M. You Think Driving an Indy Car Is Easy? Available online: https://www.roadandtrack.com/motorsports/news/a18270/you-think-driving-an-indy-car-is-easy/ (accessed on 25 January 2022).
- Ebben, W.P.; Suchomel, T.J. Physical demands, injuries, and conditioning practices of stock car drivers. J. Strength Cond. Res. 2012, 26, 1188–1198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altmann, D.; Benbow, S.; Bouchard, J. Medicine in Motor Sport; FIA Institute for Motor Sport Safety and Sustainability: London, UK, 2011; pp. 60–69. [Google Scholar]
- Harding, R.M. Human Respiratory Responses during High Performance Flight; Advisory Group for Aerospace Research & Development: Hampshire, UK, 1987. [Google Scholar]
- Norton, C. Formula One Drivers Feel the g-Force. Available online: https://www.telegraph.co.uk/motoring/motorsport/7681665/Formula-One-drivers-feel-the-G-force.html (accessed on 25 January 2022).
- Beaune, B.; Durand, S.; Mariot, J.P. Open-wheel race car driving: Energy cost for pilots. J. Strength Cond. Res. 2010, 24, 2927–2932. [Google Scholar] [CrossRef] [Green Version]
- Lighthall, J.W.; Pierce, J.; Olvey, S. A physiological profile of high-performance race car drivers. SAE Int. 1994, 1, 55–63. [Google Scholar] [CrossRef]
- Ainsworth, B.E.; Haskell, W.L.; Herrmann, S.D.; Meckes, N.; Bassett, D.R., Jr.; Tudor-Locke, C.; Greer, J.L.; Vezina, J.; Whitt-Glover, M.C.; Leon, A.S. 2011 Compendium of physical activities: A second update of codes and MET values. Med. Sci. Sports Exerc. 2011, 43, 1575–1581. [Google Scholar] [CrossRef] [Green Version]
- Jacobs, P.L.; Olvey, S.E.; Johnson, B.M.; Cohn, K. Physiological responses to high-speed, open-wheel racecar driving. Med. Sci. Sports Exerc. 2002, 34, 2085–2090. [Google Scholar] [CrossRef] [PubMed]
- Schwaberger, G. Heart rate, metabolic and hormonal responses to maximal psycho-emotional and physical stress in motor car racing drivers. Int. Arch. Occup. Environ. Health 1987, 59, 579–604. [Google Scholar] [CrossRef]
- Taggart, P.; Carruthers, M. Endogenous hyperlipidaemia induced by emotional stress of racing driving. Lancet 1971, 1, 363–366. [Google Scholar] [CrossRef]
- Collins, V.P. Physiologic Observations on Race Car Drivers. NASA CR-570; NASA Contractor Report; NASA CR: Washington, DC, USA, 1966; pp. 1–114.
- Backman, J.; Hakkinen, K.; Ylinen, J.; Hakkinen, A.; Kyrolainen, H. Neuromuscular performance characteristics of open-wheel and rally drivers. J. Strength Cond. Res. 2005, 19, 777–784. [Google Scholar] [CrossRef]
- Del Rosso, S.; Abreu, L.; Webb, H.E.; Zouhal, H.; Boullosa, D.A. Stress markers during a rally car competition. J. Strength Cond. Res. 2016, 30, 605–614. [Google Scholar] [CrossRef] [PubMed]
- Azizan, M.A.; Fard, M. The influence of vibrations on vehicle occupant fatigue. In Proceedings of the inter.noise 2014. 43rd International Congress on Noise Control Engineering, Melbourne, Australia, 16–19 November 2014; Volume 249, pp. 1767–1777. [Google Scholar]
- Baroody, N.B., Jr.; Thomason, J.M. Blistering the track—and drivers—in stock racing. Phys. Sportsmed. 1975, 3, 36–42. [Google Scholar] [CrossRef] [PubMed]
- Tsopanakis, C.; Tsopanakis, A. Stress hormonal factors, fatigue, and antioxidant responses to prolonged speed driving. Pharmacol. Biochem. Behav. 1998, 60, 747–751. [Google Scholar] [CrossRef]
- Reid, M.B.; Haack, K.E.; Franchek, K.M.; Valberg, P.A.; Kobzik, L.; West, M.S. Reactive oxygen in skeletal muscle. I. Intracellular oxidant kinetics and fatigue in vitro. J. Appl. Physiol. 1992, 73, 1797–1804. [Google Scholar] [CrossRef]
- Reid, M.B.; Shoji, T.; Moody, M.R.; Entman, M.L. Reactive oxygen in skeletal muscle. II. Extracellular release of free radicals. J. Appl. Physiol 1992, 73, 1805–1809. [Google Scholar] [CrossRef]
- Barbieri, E.; Sestili, P. Reactive oxygen species in skeletal muscle signaling. J. Signal Transduct. 2012, 2012, 982794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brand, M.D. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic. Biol. Med. 2016, 100, 14–31. [Google Scholar] [CrossRef]
- Sakellariou, G.K.; Jackson, M.J.; Vasilaki, A. Redefining the major contributors to superoxide production in contracting skeletal muscle. The role of NAD(P)H oxidases. Free Radic. Res. 2014, 48, 12–29. [Google Scholar] [CrossRef]
- Kobzik, L.; Reid, M.B.; Bredt, D.S.; Stamler, J.S. Nitric oxide in skeletal muscle. Nature 1994, 372, 546–548. [Google Scholar] [CrossRef]
- Kobzik, L.; Stringer, B.; Balligand, J.L.; Reid, M.B.; Stamler, J.S. Endothelial type nitric oxide synthase in skeletal muscle fibers: Mitochondrial relationships. Biochem. Biophys. Res. Commun. 1995, 211, 375–381. [Google Scholar] [CrossRef]
- Jones, A.M.; Vanhatalo, A.; Seals, D.R.; Rossman, M.J.; Piknova, B.; Jonvik, K.L. Dietary nitrate and nitric oxide metabolism: Mouth, circulation, skeletal muscle, and exercise performance. Med. Sci. Sports Exerc. 2021, 53, 280–294. [Google Scholar] [CrossRef] [PubMed]
- Morrison, R.J.; Miller, C.C., 3rd; Reid, M.B. Nitric oxide effects on force-velocity characteristics of the rat diaphragm. Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 1998, 119, 203–209. [Google Scholar] [CrossRef]
- Brown, G.C. Nitric oxide regulates mitochondrial respiration and cell functions by inhibiting cytochrome oxidase. FEBS Lett. 1995, 369, 136–139. [Google Scholar] [CrossRef] [Green Version]
- McConell, G.K.; Rattigan, S.; Lee-Young, R.S.; Wadley, G.D.; Merry, T.L. Skeletal muscle nitric oxide signaling and exercise: A focus on glucose metabolism. Am. J. Physiol. Endocrinol. Metab. 2012, 303, E301–E307. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reid, M.B.; Kobzik, L.; Bredt, D.S.; Stamler, J.S. Nitric oxide modulates excitation-contraction coupling in the diaphragm. Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 1998, 119, 211–218. [Google Scholar] [CrossRef]
- Andrade, F.H.; Reid, M.B.; Allen, D.G.; Westerblad, H. Effect of nitric oxide on single skeletal muscle fibres from the mouse. J. Physiol. 1998, 509 Pt 2, 577–586. [Google Scholar] [CrossRef]
- Andrade, F.H.; Reid, M.B.; Allen, D.G.; Westerblad, H. Effect of hydrogen peroxide and dithiothreitol on contractile function of single skeletal muscle fibres from the mouse. J. Physiol. 1998, 509 Pt 2, 565–575. [Google Scholar] [CrossRef]
- Wolin, M.S.; Hintze, T.H.; Shen, W.; Mohazzab, H.K.; Xie, Y.W. Involvement of reactive oxygen and nitrogen species in signalling mechanisms that control tissue respiration in muscle. Biochem. Soc. Trans. 1997, 25, 934–939. [Google Scholar] [CrossRef]
- Reid, M.B. Nitric oxide, reactive oxygen species, and skeletal muscle contraction. Med. Sci. Sports Exerc. 2001, 33, 371–376. [Google Scholar] [CrossRef]
- Ferreira, L.F.; Reid, M.B. Muscle-derived ROS and thiol regulation in muscle fatigue. J. Appl. Physiol. 2008, 104, 853–860. [Google Scholar] [CrossRef] [Green Version]
- Cocco, L.G.P.; Rapuano, S.; Rossi, L. Vibrations measurements of Formula One car electronic devices. roceedings of the 16th IMEKO TC4 Symposium, Exploring New Frontiers of Instrumentation and Methods for Electrical and Electronic Measurements, Florence, Italy, 22–24 September 2008; 2008. [Google Scholar]
- Leslie-Pelecky, D. The Science of Vibration Harmonics. Available online: https://buildingspeed.org/2008/03/06/the_science_of_vibration_harmonics_6/ (accessed on 25 January 2022).
- Sorokin, P.; Khryakov, K.; Gommers, M. Analysis of Dallara T12 race car front wing vibrations. Acta Polytech. 2018, 58, 308. [Google Scholar] [CrossRef]
- Fouladi, M.H.; Namasivayam, S.; Raj, F.R.; Mamtaz, H.; Hon, H.J.; Sivanesan, S. Enhancement of vibration comfort for FSAE race car driver. J. Adv. Res. Dyn. Control Syst. 2018, 13, 1320–1327. [Google Scholar]
- Abrams, R. Formula SAE Race Car Analysis: Simulation & Testing of the Engine as a Structural Member; The University of Western Ontario: London, ON, Canada, 2008. [Google Scholar]
- Gray, W. Racing Vehicle Environment—Forces and Vibrations. Available online: http://atlasf1.autosport.com/99/dec08/gray.html (accessed on 25 January 2022).
- Koutras, C.; Buecking, B.; Jaeger, M.; Ruchholtz, S.; Heep, H. Musculoskeletal injuries in auto racing: A retrospective study of 137 drivers. Phys. Sportsmed. 2014, 42, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Hood, W.B., Jr.; Murray, R.H.; Urschel, C.W.; Bowers, J.A.; Clark, J.G. Cardiopulmonary effects of whole-body vibration in man. J. Appl. Physiol. 1966, 21, 1725–1731. [Google Scholar] [CrossRef]
- Masmejean, E.H.; Chavane, H.; Chantegret, A.; Issermann, J.J.; Alnot, J.Y. The wrist of the Formula One driver. Br. J. Sports Med. 1999, 33, 270–273. [Google Scholar] [CrossRef] [Green Version]
- Pacurari, M.; Waugh, S.; Krajnak, K. Acute vibration induces peripheral nerve sensitization in a rat tail model: Possible role of oxidative stress and inflammation. Neuroscience 2019, 398, 263–272. [Google Scholar] [CrossRef]
- Krajnak, K. Frequency-dependent changes in mitochondrial number and generation of reactive oxygen species in a rat model of vibration-induced injury. J. Toxicol. Environ. Health Part A 2020, 83, 20–35. [Google Scholar] [CrossRef]
- Antoshina, L.I.; Pavlovskaia, N.A. Biochemical markers of vibration effects in workers, and their criteria evaluation. Med. Str. Promyshlennaya Ekol. 2009, 12, 22–27. [Google Scholar]
- Krajnak, K.; Miller, G.R.; Waugh, S.; Johnson, C.; Li, S.; Kashon, M.L. Characterization of frequency-dependent responses of the vascular system to repetitive vibration. J. Occup. Environ. Med. 2010, 52, 584–594. [Google Scholar] [CrossRef] [Green Version]
- Krajnak, K.; Waugh, S. Systemic effects of segmental vibration in an animal model of hand-arm vibration syndrome. J. Occup. Environ. Med. 2018, 60, 886–895. [Google Scholar] [CrossRef]
- Delliaux, S.; Brerro-Saby, C.; Steinberg, J.G.; Jammes, Y. Reactive oxygen species and inflammatory mediators enhance muscle spindles mechanosensitivity in rats. Pflügers Arch.-Eur. J. Physiol. 2009, 457, 877–884. [Google Scholar] [CrossRef] [PubMed]
- Rosales-Corral, S.; Reiter, R.J.; Tan, D.X.; Ortiz, G.G.; Lopez-Armas, G. Functional aspects of redox control during neuroinflammation. Antioxid. Redox Signal. 2010, 13, 193–247. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, H.L.; Yang, C.M. Role of redox signaling in neuroinflammation and neurodegenerative diseases. Biomed. Res. Int. 2013, 2013, 484613. [Google Scholar] [CrossRef] [PubMed]
- Kurutas, E.B.; Gumusalan, Y.; Cetinkaya, A.; Dogan, E. Evaluation of method performance for oxidative stress biomarkers in urine and biological variations in urine of patients with type 2 diabetes mellitus and diabetic nephropathy. Biol. Proced. Online 2015, 17, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Inanir, A.; Sogut, E.; Ayan, M.; Inanir, S. Evaluation of pain intensity and oxidative stress levels in patients with inflammatory and non-inflammatory back pain. Eur. J. Gen. Med. 2013, 10, 185–190. [Google Scholar] [CrossRef]
- Vallbo, A.B.; Hagbarth, K.E.; Torebjork, H.E.; Wallin, B.G. Somatosensory, proprioceptive, and sympathetic activity in human peripheral nerves. Physiol. Rev. 1979, 59, 919–957. [Google Scholar] [CrossRef]
- Bentley, S. Exercise-induced muscle cramp. Proposed mechanisms and management. Sports Med. 1996, 21, 409–420. [Google Scholar] [CrossRef]
- Szalma, J.L.; Hancock, P.A. Noise effects on human performance: A meta-analytic synthesis. Psychol. Bull. 2011, 137, 682–707. [Google Scholar] [CrossRef]
- Lindemann, J.; Brusis, T. Is there a risk of noise-induced hearing loss in automobile drivers and in automobile sport racing? Laryngol. Rhinol. Otol. 1985, 64, 476–480. [Google Scholar] [CrossRef]
- Liu, J.; Ghastine, L.; Um, P.; Rovit, E.; Wu, T. Environmental exposures and sleep outcomes: A review of evidence, potential mechanisms, and implications. Environ. Res. 2021, 196, 110406. [Google Scholar] [CrossRef]
- Jafari, M.J.; Sadeghian, M.; Khavanin, A.; Khodakarim, S.; Jafarpisheh, A.S. Effects of noise on mental performance and annoyance considering task difficulty level and tone components of noise. J. Environ. Health Sci. Eng. 2019, 17, 353–365. [Google Scholar] [CrossRef] [PubMed]
- Wilking, M.; Ndiaye, M.; Mukhtar, H.; Ahmad, N. Circadian rhythm connections to oxidative stress: Implications for human health. Antioxid. Redox Signal. 2013, 19, 192–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munzel, T.; Gori, T.; Babisch, W.; Basner, M. Cardiovascular effects of environmental noise exposure. Eur. Heart J. 2014, 35, 829–836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munzel, T.; Sorensen, M.; Schmidt, F.; Schmidt, E.; Steven, S.; Kroller-Schon, S.; Daiber, A. The adverse effects of environmental noise exposure on oxidative stress and cardiovascular risk. Antioxid. Redox Signal. 2018, 28, 873–908. [Google Scholar] [CrossRef] [PubMed]
- Neiemeier, R.; Dames, B. Recommendations for Occupational Safety and Health: Compendium of Policy Documents and Statements; National Institute for Occupational Safety and Health (NIOSH): Washington, DC, USA, 1992; pp. 1–217. [Google Scholar]
- Gwin, K.K.; Wallingford, K.M.; Morata, T.C.; Van Campen, L.E.; Dallaire, J.; Alvarez, F.J. Ototoxic occupational exposures for a stock car racing team: II. Chemical surveys. J. Occup. Environ. Hyg. 2005, 2, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Rose, A.S.; Ebert, C.S., Jr.; Prazma, J.; Pillsbury, H.C., 3rd. Noise exposure levels in stock car auto racing. Ear Nose Throat J. 2008, 87, 689–692. [Google Scholar] [CrossRef] [Green Version]
- Magara, K.; Jansz, J.; Bertolattia, D. Noise exposure assessment for racing car drivers. World Saf. J. 2016, XXV, 345. [Google Scholar]
- Kardous, F.; Morata, T.C. Occupational and recreational noise exposures at stock car racing circuits: An exploratory survey of three professional race tracks. Noise Control Eng. J. 2010, 58, 54–61. [Google Scholar] [CrossRef]
- Spoendlin, H. Anatomy of cochlear innervation. Am. J. Otolaryngol. 1985, 6, 453–467. [Google Scholar] [CrossRef]
- Gates, G.A.; Mills, J.H. Presbycusis. Lancet 2005, 366, 1111–1120. [Google Scholar] [CrossRef]
- Shi, L.; Guo, X.; Shen, P.; Liu, L.; Tao, S.; Li, X.; Song, Q.; Yu, Z.; Yin, S.; Wang, J. Noise-induced damage to ribbon synapses without permanent threshold shifts in neonatal mice. Neuroscience 2015, 304, 368–377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kujawa, S.G.; Liberman, M.C. Adding insult to injury: Cochlear nerve degeneration after “temporary” noise-induced hearing loss. J. Neurosci. 2009, 29, 14077–14085. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santi, P.A.; Duvall, A.J., 3rd. Stria vascularis pathology and recovery following noise exposure. Otolaryngology 1978, 86, ORL354–ORL361. [Google Scholar] [CrossRef] [PubMed]
- Kurabi, A.; Keithley, E.M.; Housley, G.D.; Ryan, A.F.; Wong, A.C. Cellular mechanisms of noise-induced hearing loss. Hear. Res. 2017, 349, 129–137. [Google Scholar] [CrossRef] [PubMed]
- Kumar, U.A.; Ameenudin, S.; Sangamanatha, A.V. Temporal and speech processing skills in normal hearing individuals exposed to occupational noise. Noise Health 2012, 14, 100–105. [Google Scholar] [CrossRef] [PubMed]
- Shore, S.E.; Roberts, L.E.; Langguth, B. Maladaptive plasticity in tinnitus-triggers, mechanisms and treatment. Nat. Rev. Neurol. 2016, 12, 150–160. [Google Scholar] [CrossRef] [Green Version]
- Henderson, D.; Bielefeld, E.C.; Harris, K.C.; Hu, B.H. The role of oxidative stress in noise-induced hearing loss. Ear Hear. 2006, 27, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Wong, A.C.; Ryan, A.F. Mechanisms of sensorineural cell damage, death and survival in the cochlea. Front. Aging Neurosci. 2015, 7, 58. [Google Scholar] [CrossRef]
- Yamane, H.; Nakai, Y.; Takayama, M.; Iguchi, H.; Nakagawa, T.; Kojima, A. Appearance of free radicals in the guinea pig inner ear after noise-induced acoustic trauma. Eur. Arch. Otorhinolaryngol. 1995, 252, 504–508. [Google Scholar] [CrossRef]
- Wu, F.; Xiong, H.; Sha, S. Noise-induced loss of sensory hair cells is mediated by ROS/AMPKalpha pathway. Redox Biol. 2020, 29, 101406. [Google Scholar] [CrossRef]
- Kil, J.; Pierce, C.; Tran, H.; Gu, R.; Lynch, E.D. Ebselen treatment reduces noise induced hearing loss via the mimicry and induction of glutathione peroxidase. Hear. Res. 2007, 226, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, C.; Chen, G.D.; Hu, B.H.; Chi, L.H.; Li, M.; Zheng, G.; Bielefeld, E.C.; Jamesdaniel, S.; Coling, D.; Henderson, D. The effects of acoustic environment after traumatic noise exposure on hearing and outer hair cells. Hear. Res. 2009, 250, 10–18. [Google Scholar] [CrossRef] [PubMed]
- Xiong, M.; Lai, H.; Yang, C.; Huang, W.; Wang, J.; Fu, X.; He, Q. Comparison of the protective effects of radix astragali, alpha-lipoic acid, and vitamin E on acute acoustic trauma. Clin. Med. Insights Ear Nose Throat 2012, 5, 25–31. [Google Scholar] [CrossRef] [PubMed]
- Pourbakht, A.; Yamasoba, T. Ebselen attenuates cochlear damage caused by acoustic trauma. Hear. Res. 2003, 181, 100–108. [Google Scholar] [CrossRef]
- Ohinata, Y.; Yamasoba, T.; Schacht, J.; Miller, J.M. Glutathione limits noise-induced hearing loss. Hear. Res. 2000, 146, 28–34. [Google Scholar] [CrossRef]
- Kopke, R.D.; Weisskopf, P.A.; Boone, J.L.; Jackson, R.L.; Wester, D.C.; Hoffer, M.E.; Lambert, D.C.; Charon, C.C.; Ding, D.L.; McBride, D. Reduction of noise-induced hearing loss using L-NAC and salicylate in the chinchilla. Hear. Res. 2000, 149, 138–146. [Google Scholar] [CrossRef]
- Seidman, M.D.; Shivapuja, B.G.; Quirk, W.S. The protective effects of allopurinol and superoxide dismutase on noise-induced cochlear damage. Otolaryngol. Head Neck Surg. 1993, 109, 1052–1056. [Google Scholar] [CrossRef]
- Choi, C.H.; Chen, K.; Vasquez-Weldon, A.; Jackson, R.L.; Floyd, R.A.; Kopke, R.D. Effectiveness of 4-hydroxy phenyl N-tert-butylnitrone (4-OHPBN) alone and in combination with other antioxidant drugs in the treatment of acute acoustic trauma in chinchilla. Free Radic. Biol. Med. 2008, 44, 1772–1784. [Google Scholar] [CrossRef]
- Bottger, E.C.; Schacht, J. The mitochondrion: A perpetrator of acquired hearing loss. Hear. Res. 2013, 303, 12–19. [Google Scholar] [CrossRef] [Green Version]
- Vlajkovic, S.M.; Lin, S.C.; Wong, A.C.; Wackrow, B.; Thorne, P.R. Noise-induced changes in expression levels of NADPH oxidases in the cochlea. Hear. Res. 2013, 304, 145–152. [Google Scholar] [CrossRef]
- Mukherjea, D.; Jajoo, S.; Sheehan, K.; Kaur, T.; Sheth, S.; Bunch, J.; Perro, C.; Rybak, L.P.; Ramkumar, V. NOX3 NADPH oxidase couples transient receptor potential vanilloid 1 to signal transducer and activator of transcription 1-mediated inflammation and hearing loss. Antioxid. Redox Signal. 2011, 14, 999–1010. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bielefeld, E.C. Reduction in impulse noise-induced permanent threshold shift with intracochlear application of an NADPH oxidase inhibitor. J. Am. Acad. Audiol. 2013, 24, 461–473. [Google Scholar] [CrossRef] [PubMed]
- Jareño, A.; De La Serna, J.; Cercas, A.; Lobato, A.; Uyá, A. Heat stroke in motor car racing drivers. Br. J. Sports Med. 1987, 21, 48. [Google Scholar] [CrossRef] [Green Version]
- Yanagida, R.; Takahashi, K.; Miura, M.; Nomura, M.; Ogawa, Y.; Aoki, K.; Iwasaki, K.I. Speed ratio but cabin temperature positively correlated with increased heart rates among professional drivers during car races. Environ. Health Prev. Med. 2016, 21, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Cheung, S.S.; McLellan, T.M.; Tenaglia, S. The thermophysiology of uncompensable heat stress. Physiological manipulations and individual characteristics. Sports Med. 2000, 29, 329–359. [Google Scholar] [CrossRef] [PubMed]
- Carlson, L.A.; Ferguson, D.P.; Kenefick, R.W. Physiological strain of stock car drivers during competitive racing. J. Therm. Biol. 2014, 44, 20–26. [Google Scholar] [CrossRef]
- Carlson, L.A.; Lawrence, M.A.; Kenefick, R.W. Hydration status and thermoregulatory responses in drivers during competitive racing. J. Strength Cond. Res. 2018, 32, 2061–2065. [Google Scholar] [CrossRef]
- Walker, S.M.; Dawson, B.; Ackland, T.R. Performance enhancement in rally car drivers via heat acclimation and race simulation. Comp. Biochem. Physiol. Part A Mol. Integr. Physiol. 2001, 128, 701–707. [Google Scholar] [CrossRef]
- Brearley, M.B.; Finn, J.P. Responses of motor-sport athletes to V8 supercar racing in hot conditions. Int. J. Sports Physiol. Perform. 2007, 2, 182–191. [Google Scholar] [CrossRef]
- Potkanowicz, E.S. A real-time case study in driver science: Physiological strain and related variables. Int. J. Sports Physiol. Perform. 2015, 10, 1058–1060. [Google Scholar] [CrossRef]
- Lieberman, H.R. Hydration and cognition: A critical review and recommendations for future research. J. Am. Coll. Nutr. 2007, 26, 555S–561S. [Google Scholar] [CrossRef] [PubMed]
- Carneiro Rodrigues, L.O.; de Castro Megalhães, F. Car racing: In the heat of competition. Rev. Bras. Med. Esporte 2004, 10, 216–219. [Google Scholar]
- McAnulty, S.R.; McAnulty, L.; Pascoe, D.D.; Gropper, S.S.; Keith, R.E.; Morrow, J.D.; Gladden, L.B. Hyperthermia increases exercise-induced oxidative stress. Int. J. Sports Med. 2005, 26, 188–192. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.K.; Lin, C.W.; Chang, C.C.; Chen, P.F.; Wang, C.J.; Hsueh, Y.M.; Chiang, H.C. Heat acclimation decreased oxidative DNA damage resulting from exposure to high heat in an occupational setting. Eur. J. Appl. Physiol. 2012, 112, 4119–4126. [Google Scholar] [CrossRef] [PubMed]
- Mourtakos, S.; Philippou, A.; Papageorgiou, A.; Lembessis, P.; Zaharinova, S.; Hasanova, Y.; Koynova, R.; Bersimis, F.; Tenchov, B.; Geladas, N.; et al. The effect of prolonged intense physical exercise of special forces volunteers on their plasma protein denaturation profile examined by differential scanning calorimetry. J. Therm. Biol. 2021, 96, 102860. [Google Scholar] [CrossRef] [PubMed]
- Barros, B.; Oliveira, M.; Morais, S. Firefighters’ occupational exposure: Contribution from biomarkers of effect to assess health risks. Environ. Int. 2021, 156, 106704. [Google Scholar] [CrossRef]
- Byrne, J.; Ludington-Hoe, S.M.; Voss, J.G. Occupational Heat stress, thermal comfort, and cognitive performance in the OR: An integrative review. AORN J. 2020, 111, 536–545. [Google Scholar] [CrossRef]
- Gharibi, V.; Khanjani, N.; Heidari, H.; Ebrahimi, M.H.; Hosseinabadi, M.B. The effect of heat stress on hematological parameters and oxidative stress among bakery workers. Toxicol. Ind. Health 2020, 36, 1–10. [Google Scholar] [CrossRef]
- Banfi, G.; Malavazos, A.; Iorio, E.; Dolci, A.; Doneda, L.; Verna, R.; Corsi, M.M. Plasma oxidative stress biomarkers, nitric oxide and heat shock protein 70 in trained elite soccer players. Eur. J. Appl. Physiol. 2006, 96, 483–486. [Google Scholar] [CrossRef]
- Knez, W.L.; Periard, J.D. The impact of match-play tennis in a hot environment on indirect markers of oxidative stress and antioxidant status. Br. J. Sports Med. 2014, 48 (Suppl. 1), i59–i63. [Google Scholar] [CrossRef] [Green Version]
- Sureda, A.; Mestre-Alfaro, A.; Banquells, M.; Riera, J.; Drobnic, F.; Camps, J.; Joven, J.; Tur, J.A.; Pons, A. Exercise in a hot environment influences plasma anti-inflammatory and antioxidant status in well-trained athletes. J. Therm. Biol. 2015, 47, 91–98. [Google Scholar] [CrossRef] [PubMed]
- Pilch, W.; Szygula, Z.; Tyka, A.K.; Palka, T.; Tyka, A.; Cison, T.; Pilch, P.; Teleglow, A. Disturbances in pro-oxidant-antioxidant balance after passive body overheating and after exercise in elevated ambient temperatures in athletes and untrained men. PLoS ONE 2014, 9, e85320. [Google Scholar] [CrossRef] [PubMed]
- Laitano, O.; Kalsi, K.K.; Pook, M.; Oliveira, A.R.; Gonzalez-Alonso, J. Separate and combined effects of heat stress and exercise on circulatory markers of oxidative stress in euhydrated humans. Eur. J. Appl. Physiol. 2010, 110, 953–960. [Google Scholar] [CrossRef] [PubMed]
- Quindry, J.; Miller, L.; McGinnis, G.; Kliszczewiscz, B.; Slivka, D.; Dumke, C.; Cuddy, J.; Ruby, B. Environmental temperature and exercise-induced blood oxidative stress. Int. J. Sport Nutr. Exerc. Metab. 2013, 23, 128–136. [Google Scholar] [CrossRef] [PubMed]
- Hillman, A.R.; Vince, R.V.; Taylor, L.; McNaughton, L.; Mitchell, N.; Siegler, J. Exercise-induced dehydration with and without environmental heat stress results in increased oxidative stress. Appl. Physiol. Nutr. Metab. 2011, 36, 698–706. [Google Scholar] [CrossRef]
- Laitano, O.; Kalsi, K.K.; Pearson, J.; Lotlikar, M.; Reischak-Oliveira, A.; Gonzalez-Alonso, J. Effects of graded exercise-induced dehydration and rehydration on circulatory markers of oxidative stress across the resting and exercising human leg. Eur. J. Appl. Physiol. 2012, 112, 1937–1944. [Google Scholar] [CrossRef]
- Georgescu, V.P.; de Souza Junior, T.P.; Behrens, C.; Barros, M.P.; Bueno, C.A.; Utter, A.C.; McAnulty, L.S.; McAnulty, S.R. Effect of exercise-induced dehydration on circulatory markers of oxidative damage and antioxidant capacity. Appl. Physiol. Nutr. Metab. 2017, 42, 694–699. [Google Scholar] [CrossRef] [Green Version]
- McNeely, B.D.; Meade, R.D.; Fujii, N.; Seely, A.J.E.; Sigal, R.J.; Kenny, G.P. Fluid replacement modulates oxidative stress- but not nitric oxide-mediated cutaneous vasodilation and sweating during prolonged exercise in the heat. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2017, 313, R730–R739. [Google Scholar] [CrossRef]
- Zhao, Q.L.; Fujiwara, Y.; Kondo, T. Mechanism of cell death induction by nitroxide and hyperthermia. Free Radic. Biol. Med. 2006, 40, 1131–1143. [Google Scholar] [CrossRef]
- Mujahid, A.; Sato, K.; Akiba, Y.; Toyomizu, M. Acute heat stress stimulates mitochondrial superoxide production in broiler skeletal muscle, possibly via downregulation of uncoupling protein content. Poult. Sci. 2006, 85, 1259–1265. [Google Scholar] [CrossRef]
- Moon, E.J.; Sonveaux, P.; Porporato, P.E.; Danhier, P.; Gallez, B.; Batinic-Haberle, I.; Nien, Y.C.; Schroeder, T.; Dewhirst, M.W. NADPH oxidase-mediated reactive oxygen species production activates hypoxia-inducible factor-1 (HIF-1) via the ERK pathway after hyperthermia treatment. Proc. Natl. Acad. Sci. USA 2010, 107, 20477–20482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, C.Y.; Lin, M.T. Oxidative stress in rats with heatstroke-induced cerebral ischemia. Stroke 2002, 33, 790–794. [Google Scholar] [CrossRef] [PubMed]
- Mitchell, J.B.; Russo, A. Thiols, thiol depletion, and thermosensitivity. Radiat. Res. 1983, 95, 471–485. [Google Scholar] [CrossRef] [PubMed]
- King, M.A.; Clanton, T.L.; Laitano, O. Hyperthermia, dehydration, and osmotic stress: Unconventional sources of exercise-induced reactive oxygen species. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2016, 310, R105–R114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Connes, P.; Simmonds, M.J.; Brun, J.F.; Baskurt, O.K. Exercise hemorheology: Classical data, recent findings and unresolved issues. Clin. Hemorheol. Microcirc. 2013, 53, 187–199. [Google Scholar] [CrossRef] [Green Version]
- Lehoux, S. Redox signalling in vascular responses to shear and stretch. Cardiovasc. Res. 2006, 71, 269–279. [Google Scholar] [CrossRef] [Green Version]
- Ajmani, R.S.; Fleg, J.L.; Demehin, A.A.; Wright, J.G.; O’Connor, F.; Heim, J.M.; Tarien, E.; Rifkind, J.M. Oxidative stress and hemorheological changes induced by acute treadmill exercise. Clin. Hemorheol. Microcirc. 2003, 28, 29–40. [Google Scholar]
- Rifkind, J.M.; Ajmani, R.S.; Heim, J. Impaired hemorheology in the aged associated with oxidative stress. Adv. Exp. Med. Biol. 1997, 428, 7–13. [Google Scholar] [CrossRef]
- Ganesan, S.; Summers, C.M.; Pearce, S.C.; Gabler, N.K.; Valentine, R.J.; Baumgard, L.H.; Rhoads, R.P.; Selsby, J.T. Short-term heat stress causes altered intracellular signaling in oxidative skeletal muscle. J. Anim. Sci. 2017, 95, 2438–2451. [Google Scholar] [CrossRef]
- Mujahid, A.; Pumford, N.; Bottje, W.; Nakagawa, K.; Miyazaqa, T.; Akiba, Y.; Toyomizu, M. Mitochondrial oxidative damage in chicken skeletal muscle induced by acute heat stress. J. Poult. Sci. 2007, 439–445. [Google Scholar] [CrossRef] [Green Version]
- Montilla, S.I.; Johnson, T.P.; Pearce, S.C.; Gardan-Salmon, D.; Gabler, N.K.; Ross, J.W.; Rhoads, R.P.; Baumgard, L.H.; Lonergan, S.M.; Selsby, J.T. Heat stress causes oxidative stress but not inflammatory signaling in porcine skeletal muscle. Temperature 2014, 1, 42–50. [Google Scholar] [CrossRef] [Green Version]
- Taggart, P.; Gibbons, D. Motor-car driving and the heart rate. Br. Med. J. 1967, 1, 411–412. [Google Scholar] [CrossRef] [Green Version]
- Taggart, P.; Gibbons, D.; Somerville, W. Some effects of motor-car driving on the normal and abnormal heart. Br. Med. J. 1969, 4, 130–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsumura, K.; Yamakoshi, T.; Yamakoshi, Y.; Rolfe, P. The effect of competition on heart rate during kart driving: A field study. BMC Res. Notes 2011, 4, 342. [Google Scholar] [CrossRef] [PubMed]
- Mallows, R.J.; Newman, D.G. Cardiovascular data acquisition in a dynamic motion environment. Aviat. Space Environ. Med. 2008, 79, 416–419. [Google Scholar] [CrossRef] [PubMed]
- Watkins, E.S. The physiology and pathology of formula one Grand Prix motor racing. Clin. Neurosurg. 2006, 53, 145–152. [Google Scholar]
- Hawkins, S. Extreme G-Forces Prompt Race Cancellation. Available online: https://abcnews.go.com/US/story?id=93412&page=1 (accessed on 25 January 2022).
- Moran, D.S.; Shitzer, A.; Pandolf, K.B. A physiological strain index to evaluate heat stress. Am. J. Physiol. 1998, 275, R129–R134. [Google Scholar] [CrossRef] [Green Version]
- Turner, A.P.; Richards, H. Physiological and selective attention demands during an international rally motor sport event. Biomed. Res. Int. 2015, 2015, 638659. [Google Scholar] [CrossRef] [Green Version]
- Martinelli, M. Kasey Kahne Opens up about Severe Dehydration, Heat Exhaustion. Available online: https://ftw.usatoday.com/2018/09/kasey-kahne-nascar-health-dehydration-heat-exhaustion-indy (accessed on 25 January 2022).
- Raymond, C. Dehydrated and Hurt: Sergio Perez Is Proof That Formula 1 Drivers Are Super Athletes. Available online: https://www.news24.com/wheels/formulaone/dehydrated-and-hurt-sergio-perez-is-proof-that-formula-1-drivers-are-super-athletes-20211027 (accessed on 25 January 2022).
- Castro-Sepulveda, M.; Cerda-Kohler, H.; Perez-Luco, C.; Monsalves, M.; Andrade, D.C.; Zbinden-Foncea, H.; Baez-San Martin, E.; Ramirez-Campillo, R. Hydration status after exercise affect resting metabolic rate and heart rate variability. Nutr. Hosp. 2014, 31, 1273–1277. [Google Scholar] [CrossRef]
- Heinzel, F.R.; Luo, Y.; Dodoni, G.; Boengler, K.; Petrat, F.; Di Lisa, F.; de Groot, H.; Schulz, R.; Heusch, G. Formation of reactive oxygen species at increased contraction frequency in rat cardiomyocytes. Cardiovasc. Res. 2006, 71, 374–382. [Google Scholar] [CrossRef] [Green Version]
- Saitoh, S.; Zhang, C.; Tune, J.D.; Potter, B.; Kiyooka, T.; Rogers, P.A.; Knudson, J.D.; Dick, G.M.; Swafford, A.; Chilian, W.M. Hydrogen peroxide: A feed-forward dilator that couples myocardial metabolism to coronary blood flow. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 2614–2621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maack, C.; Cortassa, S.; Aon, M.A.; Ganesan, A.N.; Liu, T.; O’Rourke, B. Elevated cytosolic Na+ decreases mitochondrial Ca2+ uptake during excitation-contraction coupling and impairs energetic adaptation in cardiac myocytes. Circ. Res. 2006, 99, 172–182. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kubin, A.M.; Skoumal, R.; Tavi, P.; Konyi, A.; Perjes, A.; Leskinen, H.; Ruskoaho, H.; Szokodi, I. Role of reactive oxygen species in the regulation of cardiac contractility. J. Mol. Cell. Cardiol. 2011, 50, 884–893. [Google Scholar] [CrossRef]
- Mak, S.; Newton, G.E. Vitamin C augments the inotropic response to dobutamine in humans with normal left ventricular function. Circulation 2001, 103, 826–830. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filho, E.; Di Fronso, S.; Mazzoni, C.; Robazza, C.; Bortoli, L.; Bertollo, M. My heart is racing! Psychophysiological dynamics of skilled racecar drivers. J. Sports Sci. 2015, 33, 945–959. [Google Scholar] [CrossRef] [Green Version]
- Andersson, D.C.; Fauconnier, J.; Yamada, T.; Lacampagne, A.; Zhang, S.J.; Katz, A.; Westerblad, H. Mitochondrial production of reactive oxygen species contributes to the beta-adrenergic stimulation of mouse cardiomycytes. J. Physiol. 2011, 589, 1791–1801. [Google Scholar] [CrossRef] [PubMed]
- Perjes, A.; Kubin, A.M.; Konyi, A.; Szabados, S.; Cziraki, A.; Skoumal, R.; Ruskoaho, H.; Szokodi, I. Physiological regulation of cardiac contractility by endogenous reactive oxygen species. Acta Physiol. 2012, 205, 26–40. [Google Scholar] [CrossRef] [PubMed]
- Ahlborg, G.; Weitzberg, E.; Lundberg, J. Metabolic and vascular effects of circulating endothelin-1 during moderately heavy prolonged exercise. J. Appl. Physiol. 1995, 78, 2294–2300. [Google Scholar] [CrossRef] [PubMed]
- Kaufmann, H.; Oribe, E.; Oliver, J.A. Plasma endothelin during upright tilt: Relevance for orthostatic hypotension? Lancet 1991, 338, 1542–1545. [Google Scholar] [CrossRef]
- Sovershaev, M.A.; Egorina, E.M.; Andreasen, T.V.; Jonassen, A.K.; Ytrehus, K. Preconditioning by 17beta-estradiol in isolated rat heart depends on PI3-K/PKB pathway, PKC, and ROS. Am. J. Physiol. Heart Circ. Physiol. 2006, 291, H1554–H1562. [Google Scholar] [CrossRef]
- Barouch, L.A.; Harrison, R.W.; Skaf, M.W.; Rosas, G.O.; Cappola, T.P.; Kobeissi, Z.A.; Hobai, I.A.; Lemmon, C.A.; Burnett, A.L.; O’Rourke, B.; et al. Nitric oxide regulates the heart by spatial confinement of nitric oxide synthase isoforms. Nature 2002, 416, 337–339. [Google Scholar] [CrossRef] [PubMed]
- Frank, K.; Kranias, E.G. Phospholamban and cardiac contractility. Ann. Med. 2000, 32, 572–578. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Kohr, M.J.; Traynham, C.J.; Wheeler, D.G.; Janssen, P.M.; Ziolo, M.T. Neuronal nitric oxide synthase signaling within cardiac myocytes targets phospholamban. Am. J. Physiol.-Cell Physiol. 2008, 294, C1566–C1575. [Google Scholar] [CrossRef] [PubMed]
- Champion, H.C.; Georgakopoulos, D.; Takimoto, E.; Isoda, T.; Wang, Y.; Kass, D.A. Modulation of in vivo cardiac function by myocyte-specific nitric oxide synthase-3. Circ. Res. 2004, 94, 657–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, H.; Kohr, M.J.; Wheeler, D.G.; Ziolo, M.T. Endothelial nitric oxide synthase decreases beta-adrenergic responsiveness via inhibition of the L-type Ca2+ current. Am. J. Physiol. Heart Circ. Physiol. 2008, 294, H1473–H1480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Land, M.F.; Tatler, B.W. Steering with the head. The visual strategy of a racing driver. Curr. Biol. 2001, 11, 1215–1220. [Google Scholar] [CrossRef] [Green Version]
- van Leeuwen, P.M.; de Groot, S.; Happee, R.; de Winter, J.C.F. Differences between racing and non-racing drivers: A simulator study using eye-tracking. PLoS ONE 2017, 12, e0186871. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Land, M.F.; Lee, D.N. Where We Look When We Steer. Nature 1994, 369, 742–744. [Google Scholar] [CrossRef]
- Bernardi, G.; Ricciardi, E.; Sani, L.; Gaglianese, A.; Papasogli, A.; Ceccarelli, R.; Franzoni, F.; Galetta, F.; Santoro, G.; Goebel, R.; et al. How skill expertise shapes the brain functional architecture: An fMRI study of visuo-spatial and motor processing in professional racing-car and naive drivers. PLoS ONE 2013, 8, e77764. [Google Scholar] [CrossRef] [Green Version]
- Bernardi, G.; Cecchetti, L.; Handjaras, G.; Sani, L.; Gaglianese, A.; Ceccarelli, R.; Franzoni, F.; Galetta, F.; Santoro, G.; Goebel, R.; et al. It’s not all in your car: Functional and structural correlates of exceptional driving skills in professional racers. Front. Hum. Neurosci. 2014, 8, 888. [Google Scholar] [CrossRef]
- Lappi, O. The racer’s brain—How domain expertise is reflected in the neural substrates of driving. Front. Hum. Neurosci. 2015, 9, 635. [Google Scholar] [CrossRef] [Green Version]
- Tyrance, S. The Psychology of Auto Racing. Available online: https://nationalpsychologist.com/2012/03/the-psychology-of-auto-racing/101641.html (accessed on 25 January 2022).
- Baroody, N.B.; Thomason, J.M.; O’Bryan, E.C., Jr. The heart of the 500 mile race. Am. Fam. Physician 1973, 8, 184–189. [Google Scholar] [PubMed]
- Basner, M.; McGuire, S. WHO environmental noise guidelines for the European region: A systematic review on environmental noise and effects on sleep. Int. J. Environ. Res. Public Health 2018, 15, 519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nollet, M.; Wisden, W.; Franks, N.P. Sleep deprivation and stress: A reciprocal relationship. Interface Focus 2020, 10, 20190092. [Google Scholar] [CrossRef] [PubMed]
- Koch, C.E.; Leinweber, B.; Drengberg, B.C.; Blaum, C.; Oster, H. Interaction between circadian rhythms and stress. Neurobiol. Stress 2017, 6, 57–67. [Google Scholar] [CrossRef] [Green Version]
- Moller, P.; Wallin, H.; Knudsen, L.E. Oxidative stress associated with exercise, psychological stress and life-style factors. Chem Biol Interact 1996, 102, 17–36. [Google Scholar] [CrossRef]
- Aschbacher, K.; O’Donovan, A.; Wolkowitz, O.M.; Dhabhar, F.S.; Su, Y.; Epel, E. Good stress, bad stress and oxidative stress: Insights from anticipatory cortisol reactivity. NPP 2013, 38, 1698–1708. [Google Scholar] [CrossRef] [Green Version]
- Irie, M.; Asami, S.; Nagata, S.; Miyata, M.; Kasai, H. Relationships between perceived workload, stress and oxidative DNA damage. Int. Arch. Occup. Environ. Health 2001, 74, 153–157. [Google Scholar] [CrossRef]
- McAllister, M.J.; Basham, S.A.; Waldman, H.S.; Smith, J.W.; Mettler, J.A.; Butawan, M.B.; Bloomer, R.J. Effects of psychological stress during exercise on markers of oxidative stress in young healthy, trained men. Physiol. Behav. 2019, 198, 90–95. [Google Scholar] [CrossRef]
- Teixeira, K.R.C.; Dos Santos, C.P.; de Medeiros, L.A.; Mendes, J.A.; Cunha, T.M.; De Angelis, K.; Penha-Silva, N.; de Oliveira, E.P.; Crispim, C.A. Night workers have lower levels of antioxidant defenses and higher levels of oxidative stress damage when compared to day workers. Sci. Rep. 2019, 9, 4455. [Google Scholar] [CrossRef] [Green Version]
- Kim, E.; Zhao, Z.; Rzasa, J.R.; Glassman, M.; Bentley, W.E.; Chen, S.; Kelly, D.L.; Payne, G.F. Association of acute psychosocial stress with oxidative stress: Evidence from serum analysis. Redox Biol. 2021, 47, 102138. [Google Scholar] [CrossRef]
- Said, M.A.; El-Gohary, O.A. Effect of noise stress on cardiovascular system in adult male albino rat: Implication of stress hormones, endothelial dysfunction and oxidative stress. Gen. Physiol. Biophys. 2016, 35, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Solanki, N.; Salvi, A.; Patki, G.; Salim, S. Modulating oxidative stress relieves stress-induced behavioral and cognitive impairments in rats. Int. J. Neuropsychopharmacol. 2017, 20, 550–561. [Google Scholar] [CrossRef] [PubMed]
- Suer, C.; Dolu, N.; Artis, A.S.; Sahin, L.; Yilmaz, A.; Cetin, A. The effects of long-term sleep deprivation on the long-term potentiation in the dentate gyrus and brain oxidation status in rats. Neurosci. Res. 2011, 70, 71–77. [Google Scholar] [CrossRef]
- Lv, H.; Zhu, C.; Wu, R.; Ni, H.; Lian, J.; Xu, Y.; Xia, Y.; Shi, G.; Li, Z.; Caldwell, R.B.; et al. Chronic mild stress induced anxiety-like behaviors can be attenuated by inhibition of NOX2-derived oxidative stress. J. Psychiatr. Res. 2019, 114, 55–66. [Google Scholar] [CrossRef]
- Chan, J.Y.H.; Chan, S.H.H. Differential impacts of brain stem oxidative stress and nitrosative stress on sympathetic vasomotor tone. Pharmacol. Ther. 2019, 201, 120–136. [Google Scholar] [CrossRef]
- Acevedo, E.O.; Webb, H.E.; Weldy, M.L.; Fabianke, E.C.; Orndorff, G.R.; Starks, M.A. Cardiorespiratory responses of Hi Fit and Low Fit subjects to mental challenge during exercise. Int. J. Sports Med. 2006, 27, 1013–1022. [Google Scholar] [CrossRef]
- Aoyagi, Y.; McLellan, T.M.; Shephard, R.J. Effects of endurance training and heat acclimation on psychological strain in exercising men wearing protective clothing. Ergonomics 1998, 41, 328–357. [Google Scholar] [CrossRef]
- Huang, C.J.; Webb, H.E.; Garten, R.S.; Kamimori, G.H.; Evans, R.K.; Acevedo, E.O. Stress hormones and immunological responses to a dual challenge in professional firefighters. Int. J. Psychophysiol. 2010, 75, 312–318. [Google Scholar] [CrossRef]
- Pacak, K.; Palkovits, M. Stressor specificity of central neuroendocrine responses: Implications for stress-related disorders. Endocr. Rev. 2001, 22, 502–548. [Google Scholar] [CrossRef]
- Radakovic, M.; Borozan, S.; Djelic, N.; Ivanovic, S.; Miladinovic, D.C.; Ristanic, M.; Spremo-Potparevic, B.; Stanimirovic, Z. Nitroso-oxidative stress, acute phase response, and cytogenetic damage in Wistar rats treated with adrenaline. Oxidative Med. Cell. Longev. 2018, 2018, 1805354. [Google Scholar] [CrossRef] [PubMed]
- Majer, A.D.; Fasanello, V.J.; Tindle, K.; Frenz, B.J.; Ziur, A.D.; Fischer, C.P.; Fletcher, K.L.; Seecof, O.M.; Gronsky, S.; Vassallo, B.G.; et al. Is there an oxidative cost of acute stress? Characterization, implication of glucocorticoids and modulation by prior stress experience. Proc. Biol. Sci. 2019, 286, 20191698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De, R.; Mazumder, S.; Sarkar, S.; Debsharma, S.; Siddiqui, A.A.; Saha, S.J.; Banerjee, C.; Nag, S.; Saha, D.; Bandyopadhyay, U. Acute mental stress induces mitochondrial bioenergetic crisis and hyper-fission along with aberrant mitophagy in the gut mucosa in rodent model of stress-related mucosal disease. Free Radic. Biol. Med. 2017, 113, 424–438. [Google Scholar] [CrossRef] [PubMed]
- Picard, M.; McManus, M.J.; Gray, J.D.; Nasca, C.; Moffat, C.; Kopinski, P.K.; Seifert, E.L.; McEwen, B.S.; Wallace, D.C. Mitochondrial functions modulate neuroendocrine, metabolic, inflammatory, and transcriptional responses to acute psychological stress. Proc. Natl. Acad. Sci. USA 2015, 112, E6614–E6623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramzan, R.; Vogt, S.; Kadenbach, B. Stress-mediated generation of deleterious ROS in healthy individuals-role of cytochrome c oxidase. J. Mol. Med. 2020, 98, 651–657. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheeren, T.W.; Arndt, J.O. Different response of oxygen consumption and cardiac output to various endogenous and synthetic catecholamines in awake dogs. Crit. Care Med. 2000, 28, 3861–3868. [Google Scholar] [CrossRef]
- Du, J.; Wang, Y.; Hunter, R.; Wei, Y.; Blumenthal, R.; Falke, C.; Khairova, R.; Zhou, R.; Yuan, P.; Machado-Vieira, R.; et al. Dynamic regulation of mitochondrial function by glucocorticoids. Proc. Natl. Acad. Sci. USA 2009, 106, 3543–3548. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.G.; Zhu, L.J.; Chen, C.; Wu, H.Y.; Luo, C.X.; Chang, L.; Zhu, D.Y. Hippocampal neuronal nitric oxide synthase mediates the stress-related depressive behaviors of glucocorticoids by downregulating glucocorticoid receptor. J. Neurosci. 2011, 31, 7579–7590. [Google Scholar] [CrossRef]
- Okamoto, K.; Tanaka, H.; Ogawa, H.; Makino, Y.; Eguchi, H.; Hayashi, S.; Yoshikawa, N.; Poellinger, L.; Umesono, K.; Makino, I. Redox-dependent regulation of nuclear import of the glucocorticoid receptor. J. Biol. Chem. 1999, 274, 10363–10371. [Google Scholar] [CrossRef] [Green Version]
Figure 1.
Development of a negative feedback model for race car driving: (A) Generic model depicting feedback control; the controller incorporates comparator function, error estimation, correction calculation, and output of control signal; the effector transduces the control signal to perform the task; task performance generates a feedback signal to the controller. (B) Biological model under central nervous system (CNS) control stimulates effector organs to perform a psychophysical task monitored by sensory feedback. (C) Driver athlete model depicts CNS regulation of cardiovascular and muscular function during race car driving, with sensory feedback using proprioception, thermoreception, vision, and hearing. Created in BioRender.com; accessed on 26 January 2022.
Figure 1.
Development of a negative feedback model for race car driving: (A) Generic model depicting feedback control; the controller incorporates comparator function, error estimation, correction calculation, and output of control signal; the effector transduces the control signal to perform the task; task performance generates a feedback signal to the controller. (B) Biological model under central nervous system (CNS) control stimulates effector organs to perform a psychophysical task monitored by sensory feedback. (C) Driver athlete model depicts CNS regulation of cardiovascular and muscular function during race car driving, with sensory feedback using proprioception, thermoreception, vision, and hearing. Created in BioRender.com; accessed on 26 January 2022.

Figure 2.
An integrative model of physiological stress caused by race car driving. The model depicts driving-related challenges that act on driver athletes (upper boxes) via redox-mediated pathways (solid lines) to promote distinct categories of physiological stress (lower boxes). Created in BioRender.com; accessed on 27 January 2022.
Figure 2.
An integrative model of physiological stress caused by race car driving. The model depicts driving-related challenges that act on driver athletes (upper boxes) via redox-mediated pathways (solid lines) to promote distinct categories of physiological stress (lower boxes). Created in BioRender.com; accessed on 27 January 2022.


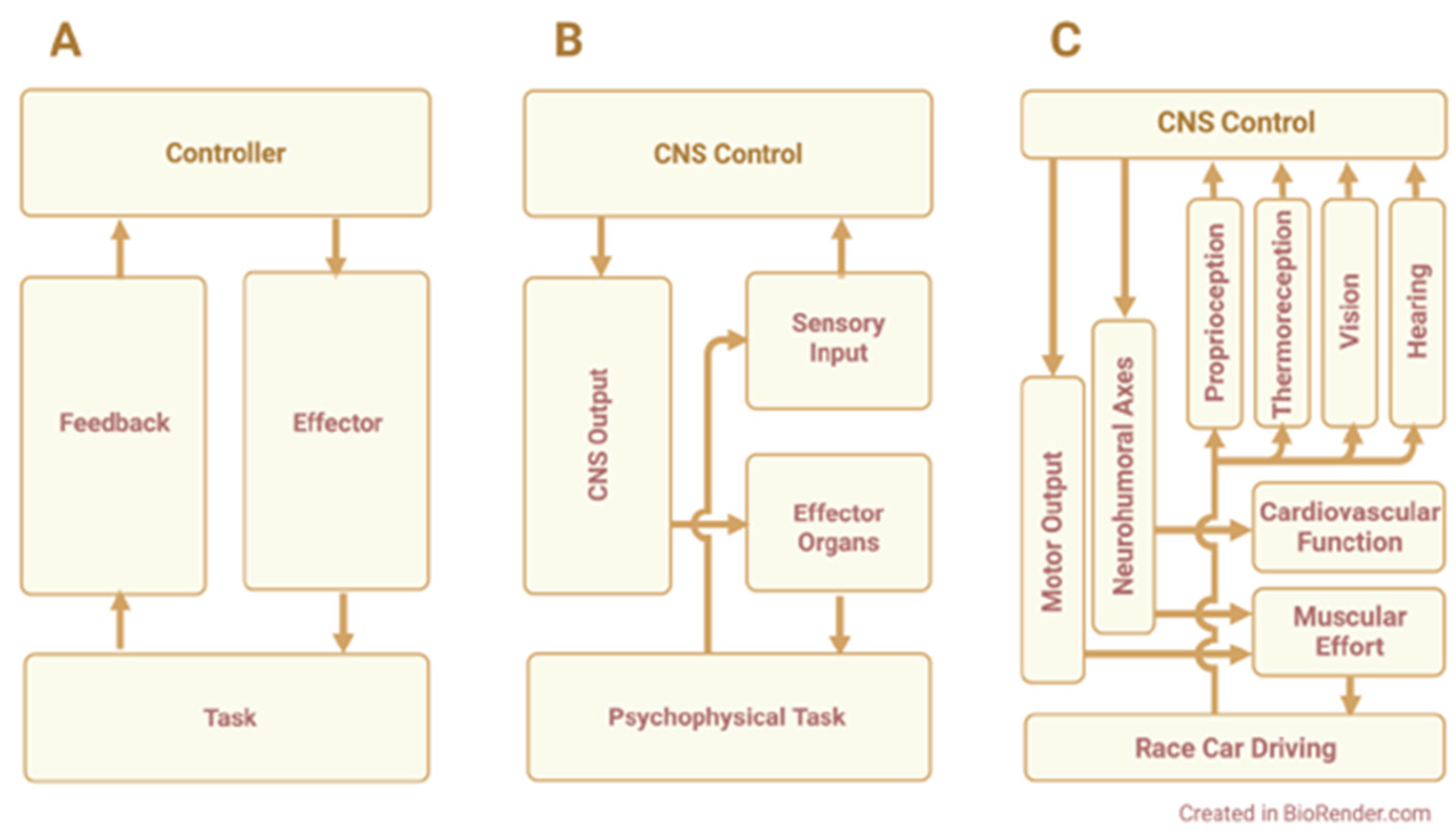{kind=link}
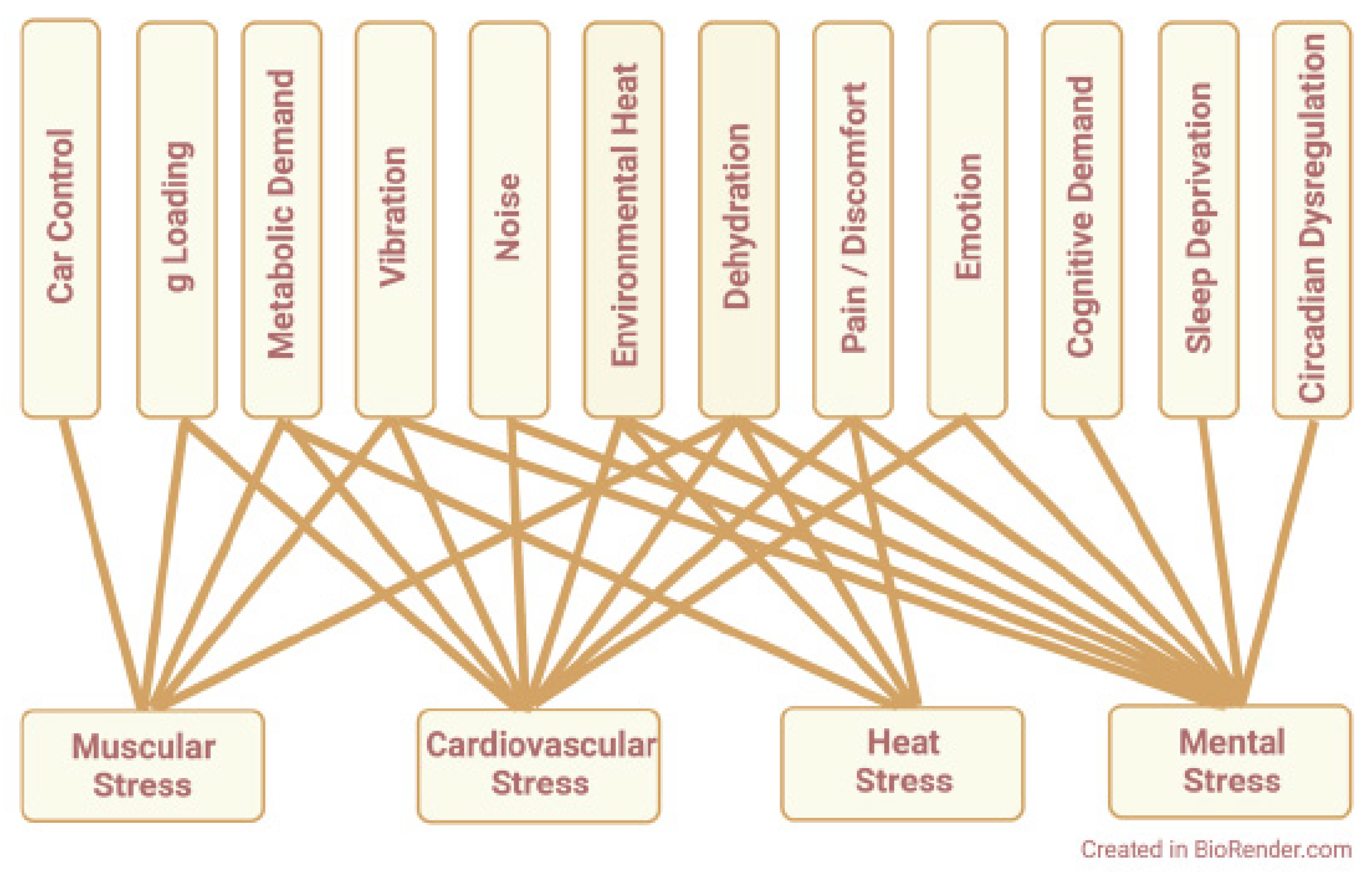{kind=link}
Table 1.
Proposed pathways for redox signaling in race car driving.
| Physiologic Stimulus | Tissue of Origin | Oxidant Cascade | Intracellular Source(s) | Physiologic Outcome | |
|---|---|---|---|---|---|
| Pathways for Redox Signaling | physical work, g loading, metabolic demand | skeletal muscle myofibers | ROS RNS | mitochondria, NOX, nNOS | muscle weakness, fatigue |
| vibration | skin, blood vessels, sensory receptors | ROS | mitochondria | nerve sensitization, muscle cramps, limb and back pain | |
| noise | cochlea | ROS | mitochondria, NOX3 | tinnitus, hearing loss, sleep disruption | |
| heat, dehydration | heart, liver, aerobic skeletal muscle | ROS RNS | mitochondria, NOX1, eNOS | antioxidant depletion, cellular damage, hemolysis | |
| cardiovascular demand | heart, vascular endothelium | ROS RNS | mitochondria, NOX, nNOS, eNOS | altered contractilityand vascular tone | |
| cognitive load, emotion, discomfort, sleeploss, circadian dysregulation | brain, vasculature, stress hormone-sensitive tissues | ROS | mitochondria | systemic oxidative stress, lucocorticoid desensitization |
Details of each pathway with citations are described in Section 2, Section 3, Section 4, Section 5, Section 6 and Section 7; ROS, reactive oxygen species; RNS, reactive nitrogen species; nNOS, neuronal-type nitric oxide synthase; NOX, nicotinamide adenine dinucleotide (phosphate) oxidase or NAD(P)H oxidase; NOX3, NAD(P)H oxidase 3; NOX1, NAD(P)H oxidase 1; eNOS, endothelial-type nitric oxide synthase.
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Reid, M.B. Redox Implications of Extreme Task Performance: The Case in Driver Athletes. Cells 2022, 11, 899. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11050899
AMA Style
Reid MB. Redox Implications of Extreme Task Performance: The Case in Driver Athletes. Cells. 2022; 11(5):899. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11050899
Chicago/Turabian StyleReid, Michael B. 2022. "Redox Implications of Extreme Task Performance: The Case in Driver Athletes" Cells 11, no. 5: 899. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11050899
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.