
Reversible Thiol Oxidation Increases Mitochondrial Electron Transport Complex Enzyme Activity but Not Respiration in Cardiomyocytes from Patients with End-Stage Heart Failure

 , , , ,
, , , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Human Subjects
2.2. Biopsy Collection
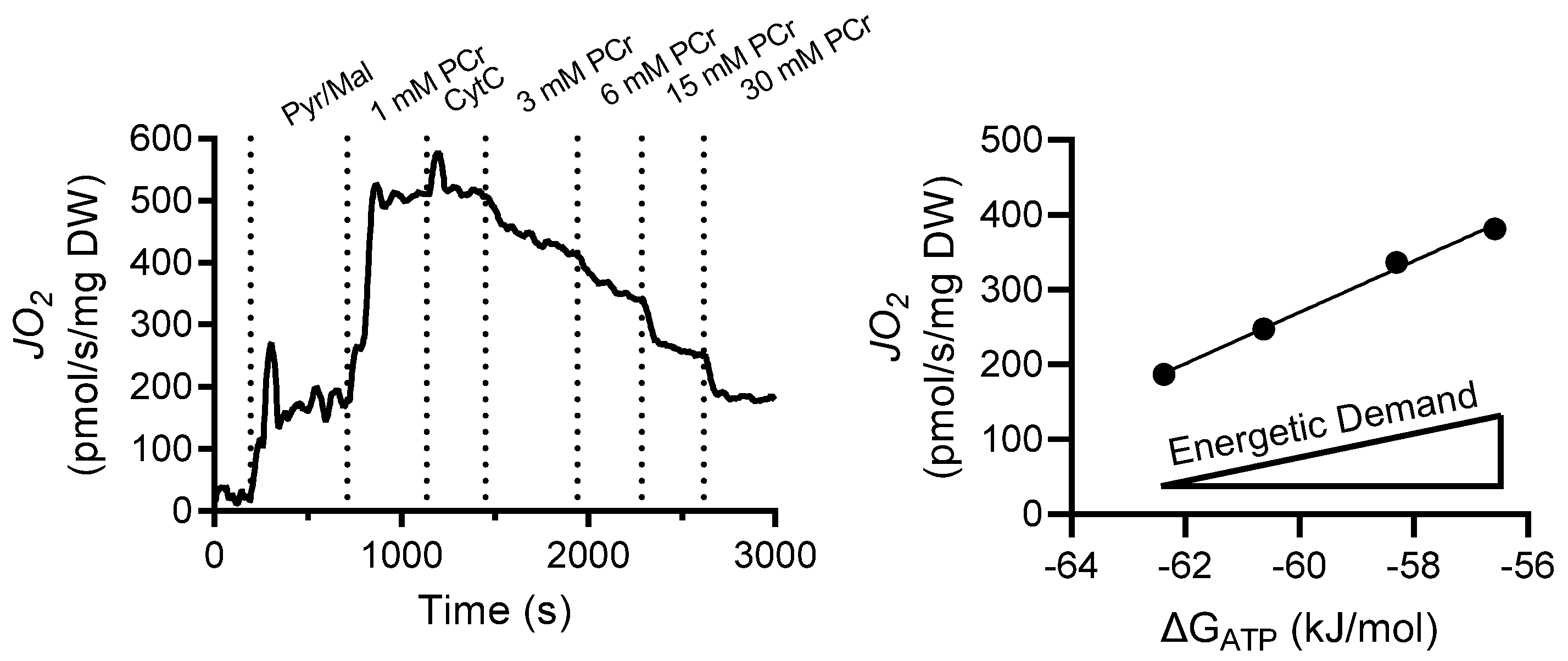
2.3. Preparation of Permeabilized Cardiomyocyte Bundles and Mitochondrial Respiration
2.4. Mitochondrial Isolation
2.5. Mitochondrial Complex Activity Assays
2.6. Statistics
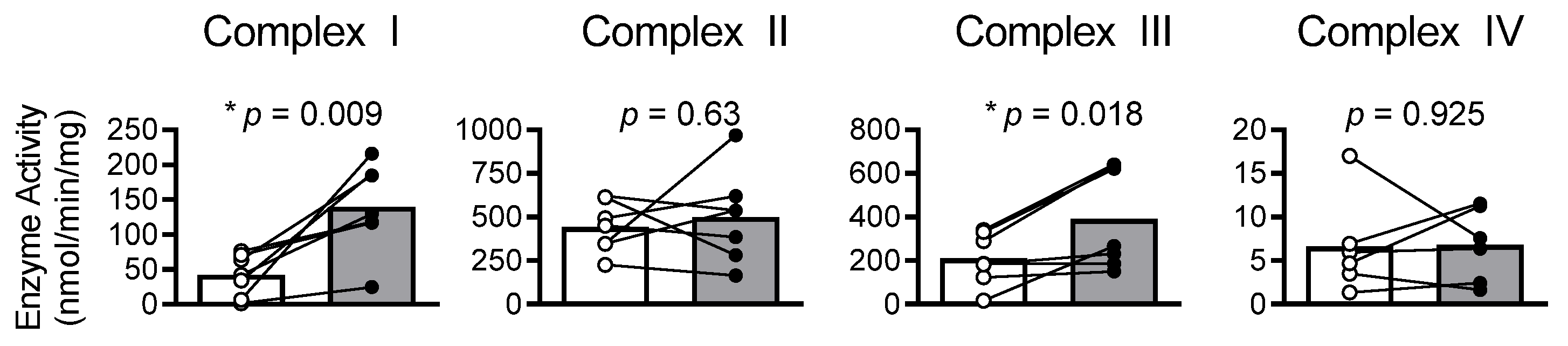
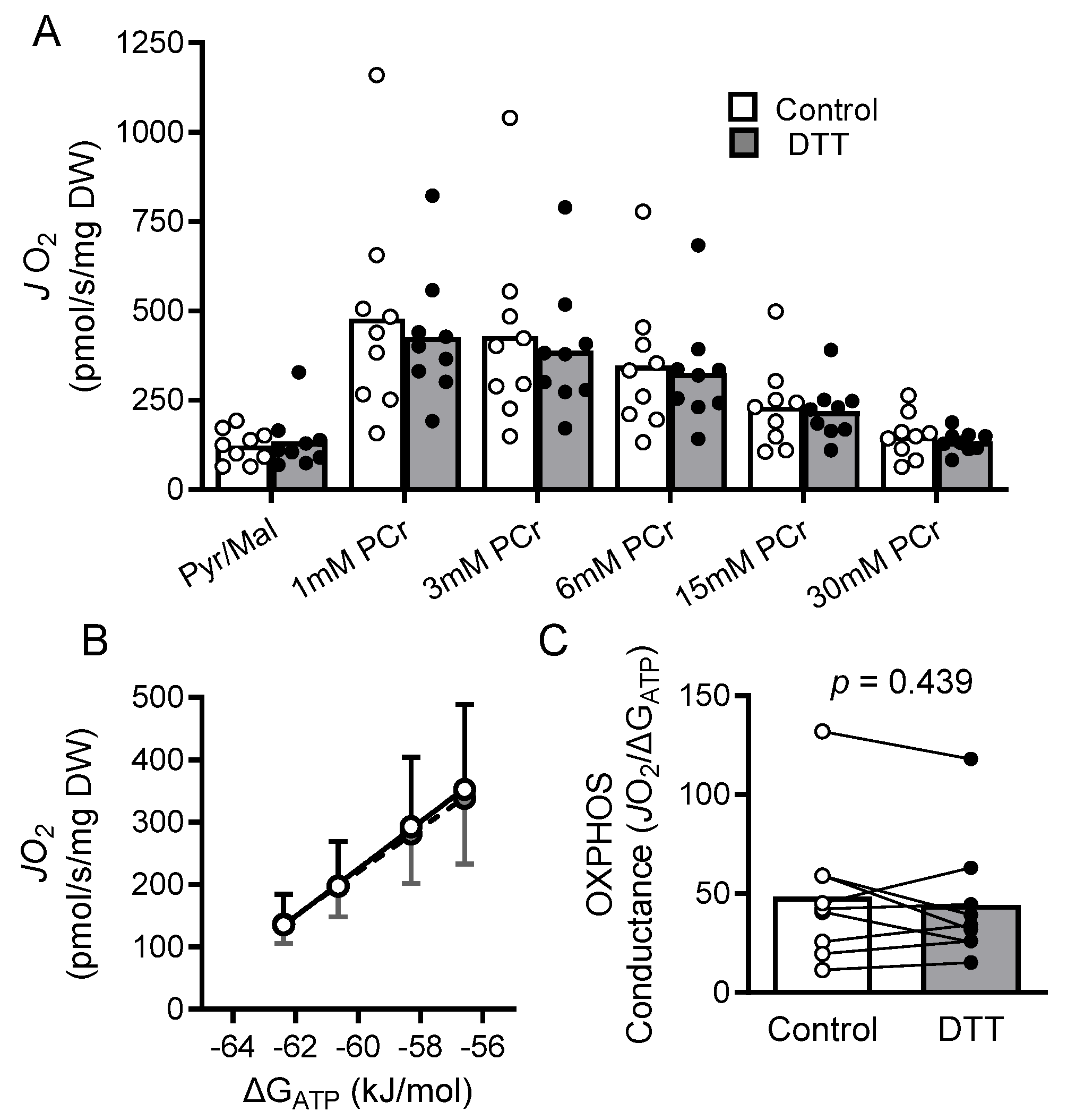
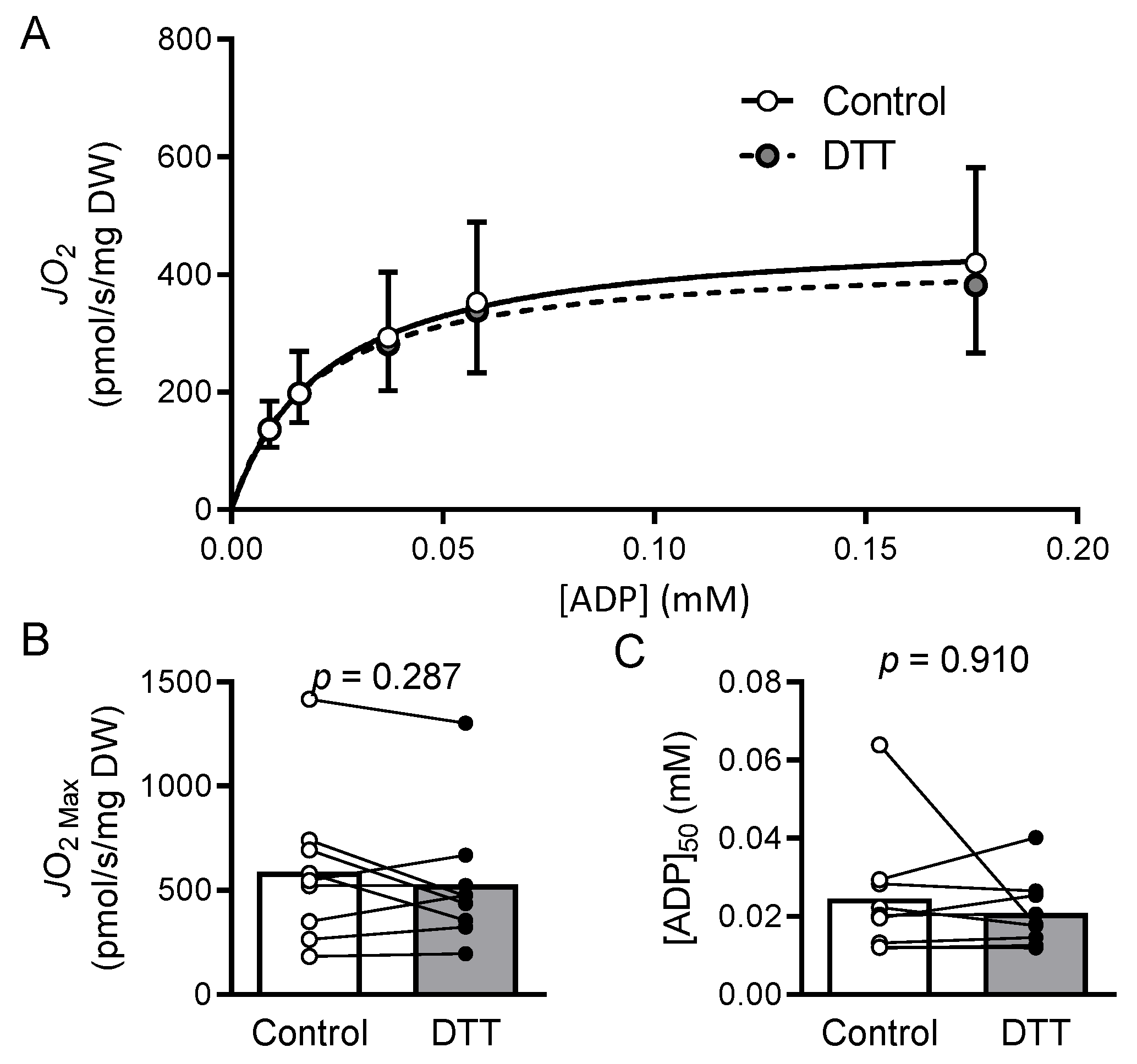
2.7. Results
2.8. Discussion
2.9. Mitochondrial Bioenergetics and Dysfunction in HFrEF
2.10. Myocardial Oxidized Shifts
2.11. Limitations
3. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lemieux, H.; Semsroth, S.; Antretter, H.; Höfer, D.; Gnaiger, E. Mitochondrial respiratory control and early defects of oxidative phosphorylation in the failing human heart. Int. J. Biochem. Cell Biol. 2011, 43, 1729–1738. [Google Scholar] [CrossRef] [PubMed]
- Park, S.Y.; Trinity, J.D.; Gifford, J.R.; Diakos, N.A.; McCreath, L.; Drakos, S.; Richardson, R.S. Mitochondrial function in heart failure: The impact of ischemic and non-ischemic etiology. Int. J. Cardiol. 2016, 220, 711–717. [Google Scholar] [CrossRef] [PubMed]
- Schipper, D.A.; Palsma, R.; Marsh, K.M.; O’Hare, C.; Dicken, D.S.; Lick, S.; Kazui, T.; Johnson, K.; Smolenski, R.T.; Duncker, D.J.; et al. Chronic Myocardial Ischemia Leads to Loss of Maximal Oxygen Consumption and Complex I Dysfunction. Ann. Thorac. Surg. 2017, 104, 1298–1304. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharov, V.G.; Todor, A.V.; Silverman, N.; Goldstein, S.; Sabbah, H.N. Abnormal mitochondrial respiration in failed human myocardium. J. Mol. Cell. Cardiol. 2000, 32, 2361–2367. [Google Scholar] [CrossRef] [PubMed]
- Stride, N.; Larsen, S.; Hey-Mogensen, M.; Sander, K.; Lund, J.T.; Gustafsson, F.; Køber, L.; Dela, F. Decreased mitochondrial oxidative phosphorylation capacity in the human heart with left ventricular systolic dysfunction. Eur. J. Heart Fail. 2013, 15, 150–157. [Google Scholar] [CrossRef] [PubMed]
- Neubauer, S.; Horn, M.; Pabst, T.; Gödde, M.; Lübke, D.; Jilling, B.; Hahn, D.; Ertl, G. Contributions of 31P-magnetic resonance spectroscopy to the understanding of dilated heart muscle disease. Eur. Heart J. 1995, 16 (Suppl. O), 115–118. [Google Scholar] [CrossRef] [PubMed]
- Neubauer, S.; Horn, M.; Cramer, M.; Harre, K.; Newell, J.B.; Peters, W.; Pabst, T.; Ertl, G.; Hahn, D.; Ingwall, J.S.; et al. Myocardial Phosphocreatine-to-ATP Ratio Is a Predictor of Mortality in Patients With Dilated Cardiomyopathy. Circulation 1997, 96, 2190–2196. [Google Scholar] [CrossRef]
- Neubauer, S. The failing heart—an engine out of fuel. N. Engl. J. Med. 2007, 356, 1140–1151. [Google Scholar] [CrossRef] [Green Version]
- Kiyuna, L.A.; Albuquerque, R.P.E.; Chen, C.H.; Mochly-Rosen, D.; Ferreira, J.C.B. Targeting mitochondrial dysfunction and oxidative stress in heart failure: Challenges and opportunities. Free Radic. Biol. Med. 2018, 129, 155–168. [Google Scholar] [CrossRef]
- Brown, D.A.; Perry, J.B.; Allen, M.E.; Sabbah, H.N.; Stauffer, B.L.; Shaikh, S.R.; Cleland, J.G.; Colucci, W.S.; Butler, J.; Voors, A.A.; et al. Expert consensus document: Mitochondrial function as a therapeutic target in heart failure. Nat. Rev. Cardiol. 2017, 14, 238–250. [Google Scholar] [CrossRef]
- Stanley, W.C.; Recchia, F.A.; Lopaschuk, G.D. Myocardial substrate metabolism in the normal and failing heart. Physiol. Rev. 2005, 85, 1093–1129. [Google Scholar] [CrossRef] [PubMed]
- Nietzel, T.; Mostertz, J.; Hochgräfe, F.; Schwarzländer, M. Redox regulation of mitochondrial proteins and proteomes by cysteine thiol switches. Mitochondrion 2017, 33, 72–83. [Google Scholar] [CrossRef] [PubMed]
- Mailloux, R.J.; Jin, X.; Willmore, W.G. Redox regulation of mitochondrial function with emphasis on cysteine oxidation reactions. Redox Biol. 2013, 2, 123–139. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomin, T.; Schittmayer, M.; Sedej, S.; Bugger, H.; Gollmer, J.; Honeder, S.; Darnhofer, B.; Liesinger, L.; Zuckermann, A.; Rainer, P.P.; et al. Mass Spectrometry-Based Redox and Protein Profiling of Failing Human Hearts. Int. J. Mol. Sci. 2021, 22, 1787. [Google Scholar] [CrossRef]
- Sverdlov, A.L.; Elezaby, A.; Behring, J.B.; Bachschmid, M.M.; Luptak, I.; Tu, V.H.; Siwik, D.A.; Miller, E.J.; Liesa, M.; Shirihai, O.S.; et al. High fat, high sucrose diet causes cardiac mitochondrial dysfunction due in part to oxidative post-translational modification of mitochondrial complex II. J. Mol. Cell. Cardiol. 2015, 78, 165–173. [Google Scholar] [CrossRef] [Green Version]
- Oliveira, P.J.; Santos, M.S.; Wallace, K.B. Doxorubicin-induced thiol-dependent alteration of cardiac mitochondrial permeability transition and respiration. Biochemistry 2006, 71, 194–199. [Google Scholar] [CrossRef] [Green Version]
- Thome, T.; Kumar, R.A.; Burke, S.K.; Khattri, R.B.; Salyers, Z.R.; Kelley, R.C.; Coleman, M.D.; Christou, D.D.; Hepple, R.T.; Scali, S.T.; et al. Impaired muscle mitochondrial energetics is associated with uremic metabolite accumulation in chronic kidney disease. JCI Insight. 2020, 6, e139826. [Google Scholar] [CrossRef]
- Fisher-Wellman, K.H.; Davidson, M.T.; Narowski, T.M.; Lin, C.T.; Koves, T.R.; Muoio, D.M. Mitochondrial Diagnostics: A Multiplexed Assay Platform for Comprehensive Assessment of Mitochondrial Energy Fluxes. Cell Rep. 2018, 24, 3593–3606.e10. [Google Scholar] [CrossRef] [Green Version]
- Thome, T.; Salyers, Z.R.; Kumar, R.A.; Hahn, D.; Berru, F.N.; Ferreira, L.F.; Scali, S.T.; Ryan, T.E. Uremic metabolites impair skeletal muscle mitochondrial energetics through disruption of the electron transport system and matrix dehydrogenase activity. Am. J. Physiol. Cell Physiol. 2019, 317, C701–C713. [Google Scholar] [CrossRef]
- Spinazzi, M.; Casarin, A.; Pertegato, V.; Salviati, L.; Angelini, C. Assessment of mitochondrial respiratory chain enzymatic activities on tissues and cultured cells. Nat. Protoc. 2012, 7, 1235–1246. [Google Scholar] [CrossRef]
- Mitter, S.S.; Shah, S.J.; Thomas, J.D. A Test in Context: E/A and E/e′ to Assess Diastolic Dysfunction and LV Filling Pressure. J. Am. Coll. Cardiol. 2017, 69, 1451–1464. [Google Scholar] [CrossRef] [PubMed]
- Dunlay, S.M.; Givertz, M.M.; Aguilar, D.; Allen, L.A.; Chan, M.; Desai, A.S.; Deswal, A.; Dickson, V.V.; Kosiborod, M.N.; Lekavich, C.L.; et al. Type 2 Diabetes Mellitus and Heart Failure: A Scientific Statement From the American Heart Association and the Heart Failure Society of America: This statement does not represent an update of the 2017 ACC/AHA/HFSA heart failure guideline update. Circulation 2019, 140, e294–e324. [Google Scholar] [CrossRef] [PubMed]
- Opie, L.H. Heart Physiology: From Cell to Circulation; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2004. [Google Scholar]
- Barth, E.; Stämmler, G.; Speiser, B.; Schaper, J. Ultrastructural quantitation of mitochondria and myofilaments in cardiac muscle from 10 different animal species including man. J. Mol. Cell. Cardiol. 1992, 24, 669–681. [Google Scholar] [CrossRef]
- Wu, F.; Zhang, J.; Beard, D.A. Experimentally observed phenomena on cardiac energetics in heart failure emerge from simulations of cardiac metabolism. Proc. Natl. Acad. Sci. USA 2009, 106, 7143–7148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheubel, R.J.; Tostlebe, M.; Simm, A.; Rohrbach, S.; Prondzinsky, R.; Gellerich, F.N.; Silber, R.-E.; Holtz, J. Dysfunction of mitochondrial respiratory chain complex I in human failing myocardium is not due to disturbed mitochondrial gene expression. J. Am. Coll. Cardiol. 2002, 40, 2174–2181. [Google Scholar] [CrossRef] [Green Version]
- Sheeran, F.L.; Pepe, S. Posttranslational modifications and dysfunction of mitochondrial enzymes in human heart failure. Am. J. Physiol. Endocrinol. Metab. 2016, 311, E449–E460. [Google Scholar] [CrossRef]
- Jarreta, D.; Orus, J.; Barrientos, A.; Miro, O.; Roig, E.; Heras, M.; Moraes, C.T.; Cardellach, F.; Casademont, J. Mitochondrial function in heart muscle from patients with idiopathic dilated cardiomyopathy. Cardiovasc. Res. 2000, 45, 860–865. [Google Scholar] [CrossRef] [Green Version]
- Chaanine, A.H.; Joyce, L.D.; Stulak, J.M.; Maltais, S.; Joyce, D.L.; Dearani, J.A.; Klaus, K.; Nair, K.S.; Hajjar, R.J.; Redfield, M.M. Mitochondrial Morphology, Dynamics, and Function in Human Pressure Overload or Ischemic Heart Disease with Preserved or Reduced Ejection Fraction. Circ. Heart Fail. 2019, 12, e005131. [Google Scholar] [CrossRef]
- Neufer, P.D. The Bioenergetics of Exercise. Cold Spring Harb. Perspect. Med. 2018, 8, a029678. [Google Scholar] [CrossRef]
- Nicholls, D.G.; Ferguson, S. Bioenergetics, 4th ed.; Elsevier: Amsterdam, The Netherlands, 2013; pp. 1–419. [Google Scholar]
- Willis, W.T.; Jackman, M.R.; Messer, J.I.; Kuzmiak-Glancy, S.; Glancy, B. A Simple Hydraulic Analog Model of Oxidative Phosphorylation. Med. Sci. Sports Exerc. 2016, 48, 990–1000. [Google Scholar] [CrossRef]
- Glancy, B.; Willis, W.T.; Chess, D.J.; Balaban, R.S. Effect of calcium on the oxidative phosphorylation cascade in skeletal muscle mitochondria. Biochemistry 2013, 52, 2793–2809. [Google Scholar] [CrossRef] [PubMed]
- Saks, V.A.; Belikova, Y.O.; Kuznetsov, A.V.; Khuchua, Z.A.; Branishte, T.H.; Semenovsky, M.L.; Naumov, V.G. Phosphocreatine pathway for energy transport: ADP diffusion and cardiomyopathy. Am. J. Physiol.-Heart Circ. Physiol. 1991, 261, 30–38. [Google Scholar] [CrossRef]
- Sheeran, F.L.; Rydström, J.; Shakhparonov, M.I.; Pestov, N.B.; Pepe, S. Diminished NADPH transhydrogenase activity and mitochondrial redox regulation in human failing myocardium. Biochim. Biophys. Acta 2010, 1797, 1138–1148. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mailloux, R.J.; Xuan, J.Y.; Beauchamp, B.; Jui, L.; Lou, M.; Harper, M.E. Glutaredoxin-2 is required to control proton leak through uncoupling protein-3. J. Biol. Chem. 2013, 288, 8365–8379. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, H.; Lin, L.; Giblin, F.; Ho, Y.S.; Lou, M.F. Glutaredoxin 2 knockout increases sensitivity to oxidative stress in mouse lens epithelial cells. Free Radic. Biol. Med. 2011, 51, 2108–2117. [Google Scholar] [CrossRef] [Green Version]
- Hill, B.G.; Higdon, A.N.; Dranka, B.P.; Darley-Usmar, V.M. Regulation of vascular smooth muscle cell bioenergetic function by protein glutathiolation. Biochim. Biophys. Acta 2010, 1797, 285–295. [Google Scholar] [CrossRef] [Green Version]
- Hurd, T.R.; Requejo, R.; Filipovska, A.; Brown, S.; Prime, T.A.; Robinson, A.J.; Fearnley, I.M.; Murphy, M.P. Complex I within oxidatively stressed bovine heart mitochondria is glutathionylated on Cys-531 and Cys-704 of the 75-kDa subunit: Potential role of CYS residues in decreasing oxidative damage. J. Biol. Chem. 2008, 283, 24801–24815. [Google Scholar] [CrossRef] [Green Version]
- Beer, S.M.; Taylor, E.R.; Brown, S.E.; Dahm, C.C.; Costa, N.J.; Runswick, M.J.; Murphy, M.P. Glutaredoxin 2 catalyzes the reversible oxidation and glutathionylation of mitochondrial membrane thiol proteins: Implications for mitochondrial redox regulation and antioxidant DEFENSE. J. Biol. Chem. 2004, 279, 47939–47951. [Google Scholar] [CrossRef] [Green Version]
- Taylor, E.R.; Hurrell, F.; Shannon, R.J.; Lin, T.K.; Hirst, J.; Murphy, M.P. Reversible glutathionylation of complex I increases mitochondrial superoxide formation. J. Biol. Chem. 2003, 278, 19603–19610. [Google Scholar] [CrossRef] [Green Version]
- Cleland, W.W. Dithiothreitol, a New Protective Reagent for SH Groups*. Biochemistry 1964, 3, 480–482. [Google Scholar] [CrossRef]
- Andrade, F.H.; Reid, M.B.; Allen, D.G.; Westerblad, H. Effect of hydrogen peroxide and dithiothreitol on contractile function of single skeletal muscle fibres from the mouse. J. Physiol. 1998, 509, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.B.; Foster, D.B.; Rucker, J.; O’Rourke, B.; Kass, D.A.; Van Eyk, J.E. Redox regulation of mitochondrial ATP synthase: Implications for cardiac resynchronization therapy. Circ. Res. 2011, 109, 750–757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borutaite, V.; Budriunaite, A.; Brown, G.C. Reversal of nitric oxide-, peroxynitrite- and S-nitrosothiol-induced inhibition of mitochondrial respiration or complex I activity by light and thiols. Biochim. Biophys. Acta (BBA)-Bioenerg. 2000, 1459, 405–412. [Google Scholar] [CrossRef] [Green Version]
- Omidi, M.; Niknahad, H.; Mohammadi-Bardbori, A. Dithiothreitol (DTT) rescues mitochondria from nitrofurantoin-induced mitotoxicity in rat. J. Biochem. Mol. Toxicol. 2016, 30, 588–592. [Google Scholar] [CrossRef]
- Sharma, A.; Fonarow, G.C.; Butler, J.; Ezekowitz, J.A.; Felker, G.M. Coenzyme Q10 and Heart Failure: A State-of-the-Art Review. Circ. Heart Fail. 2016, 9, e002639. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| All Patients (n = 9) | |
|---|---|
| Age (years) | 55.9 ± 14.0 |
| Male, % (n) | 77.8 (7) |
| Ischemic CM, % (n) | 11.1 (1) |
| Hypertension, % (n) | 100 (9) |
| Hyperlipidemia, % (n) | 55.6 (5) |
| Atrial fibrillation, % (n) | 77.8 (7) |
| Asthma, % (n) | 11.1 (1) |
| COPD, % (n) | 22.2 (2) |
| CKD, % (n) | 55.6 (5) |
| Diabetes mellitus, % (n) | 44.4 (4) |
| CAD, % (n) | 55.6 (5) |
| All Patients (n = 9) | |
|---|---|
| EF (%) | 17.2 ± 8.3 |
| LVIDd (cm) | 8.0 ± 1.2 |
| LVIDs (cm) | 7.3 ± 1.3 |
| RVH (yes/no) | (4/5) |
| RVSP (mmHg) | 42.4 ± 14.2 |
| E wave (cm/s) | 78.1 ± 25.6 |
| A wave (cm/s) | 61.4 ± 37.5 |
| E’ (cm/s) | 8.19 ± 2.49 |
| Dt (s) | 155.2 ± 65.5 |
| E/A | 1.59 ± 0.78 |
| E/E’ | 9.99 ± 3.21 |
| E/DT (cm/s2) | 0.62 ± 0.38 |
| Diastolic dysfunction Grade I/II/III (n, %) | 1 (11)/2 (22)/1 (11) |
| PCr (mM) | ΔGATP (kJ/mol) | ADP (mM) | ATP (mM) | Cr (mM) |
|---|---|---|---|---|
| 1 | −54.16 | 0.176 | 4.82 | 4.824 |
| 3 | −57.09 | 0.058 | 4.94 | 4.942 |
| 6 | −58.32 | 0.037 | 4.96 | 4.963 |
| 15 | −60.63 | 0.016 | 4.98 | 4.984 |
| 30 | −62.37 | 0.009 | 4.99 | 4.991 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, R.A.; Thome, T.; Sharaf, O.M.; Ryan, T.E.; Arnaoutakis, G.J.; Jeng, E.I.; Ferreira, L.F. Reversible Thiol Oxidation Increases Mitochondrial Electron Transport Complex Enzyme Activity but Not Respiration in Cardiomyocytes from Patients with End-Stage Heart Failure. Cells 2022, 11, 2292. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11152292
Kumar RA, Thome T, Sharaf OM, Ryan TE, Arnaoutakis GJ, Jeng EI, Ferreira LF. Reversible Thiol Oxidation Increases Mitochondrial Electron Transport Complex Enzyme Activity but Not Respiration in Cardiomyocytes from Patients with End-Stage Heart Failure. Cells. 2022; 11(15):2292. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11152292
Chicago/Turabian StyleKumar, Ravi A., Trace Thome, Omar M. Sharaf, Terence E. Ryan, George J. Arnaoutakis, Eric I. Jeng, and Leonardo F. Ferreira. 2022. "Reversible Thiol Oxidation Increases Mitochondrial Electron Transport Complex Enzyme Activity but Not Respiration in Cardiomyocytes from Patients with End-Stage Heart Failure" Cells 11, no. 15: 2292. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11152292