
β-Glucocerebrosidase Deficiency Activates an Aberrant Lysosome-Plasma Membrane Axis Responsible for the Onset of Neurodegeneration

 ,
,  , ,
, ,  , ,
, ,  , , ,
, , ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Generation of Murine Cerebellar Granule Neurons
2.3. iPSC Culture
2.4. Differentiation of iPSC into Dopaminergic Neurons
2.5. Cell Sphingolipid Labelling with [1-3H]-Sphingosine
2.6. CBE Treatment
2.7. Protein Determination
2.8. Cell Surface Protein Biotinylation and Isolation of Plasma Membrane Proteins by Streptavidin Pulldown Assay
2.9. Cell Surface Protein Biotinylation and Isolation of Detergent-Resistant Membrane Fractions
2.10. Immunoblotting
2.11. Evaluation of Enzymatic Activities in Cell Lysates
2.12. Evaluation of Plasma Membrane Enzymatic Activities in Living Cells
2.13. Lipid Analysis
2.14. Evaluation of GlcCer Release
2.15. Proteomic Analysis
2.15.1. Protein Digestion
2.15.2. LC-MS/MS Analysis
2.15.3. Peptide Chromatography
2.15.4. SWATH Acquisition
2.15.5. Data-Dependent Acquisition
2.16. Data Analysis
2.16.1. SWATH—Data Analysis
2.16.2. DDA—Data Analysis
2.17. Statistical Analyses
3. Results
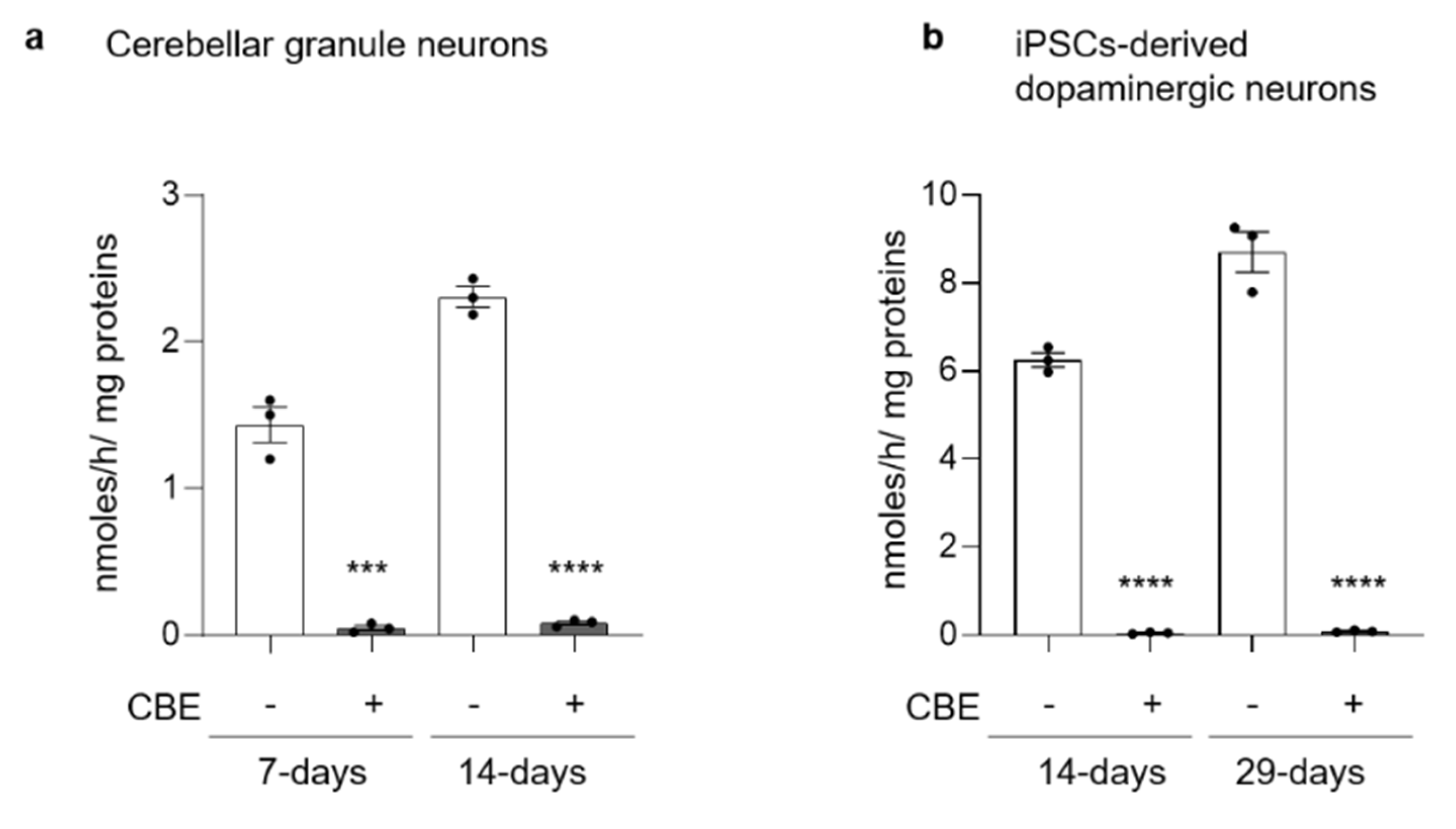
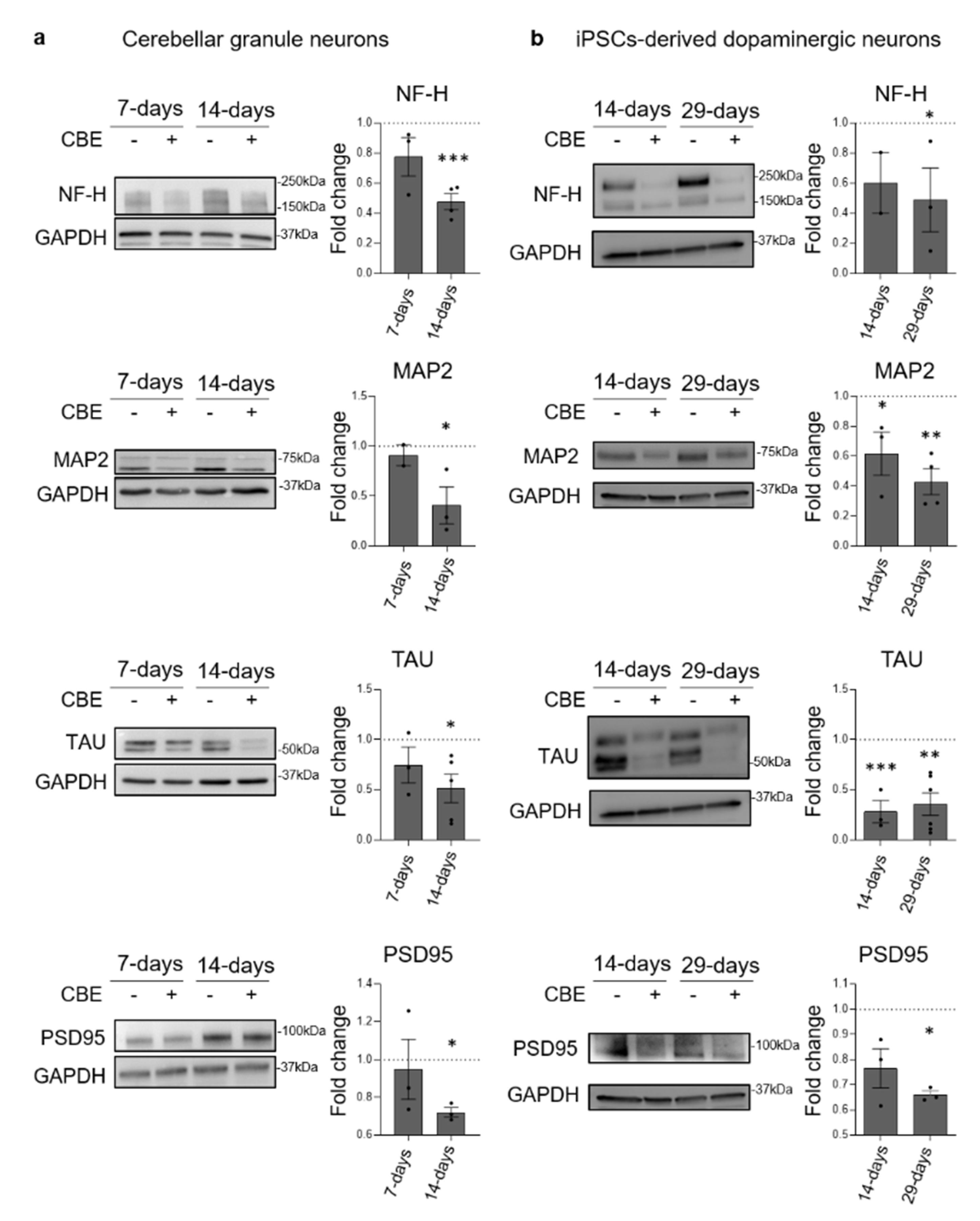
3.1. GCase Inhibition Induces a Neurodegenerative Phenotype
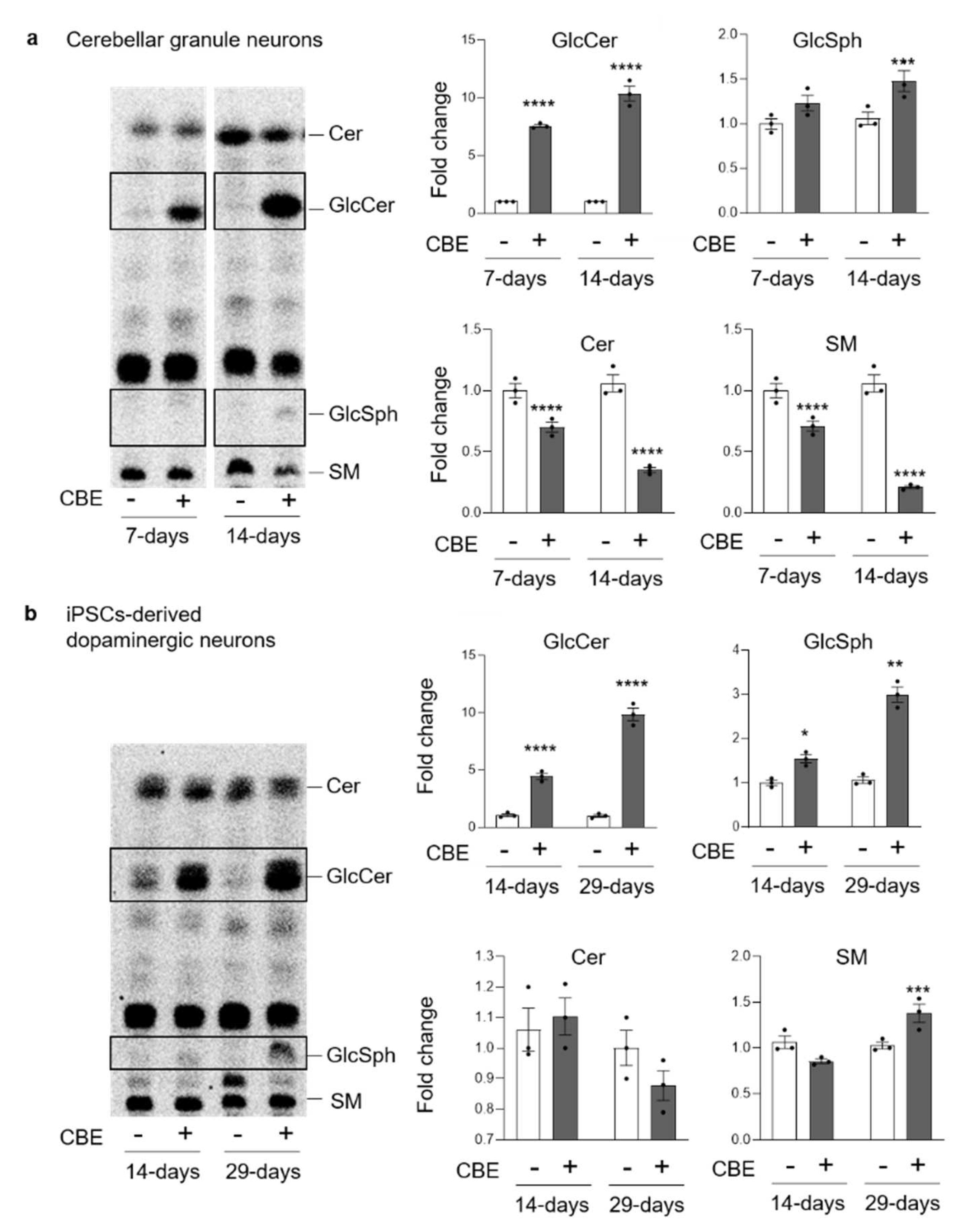
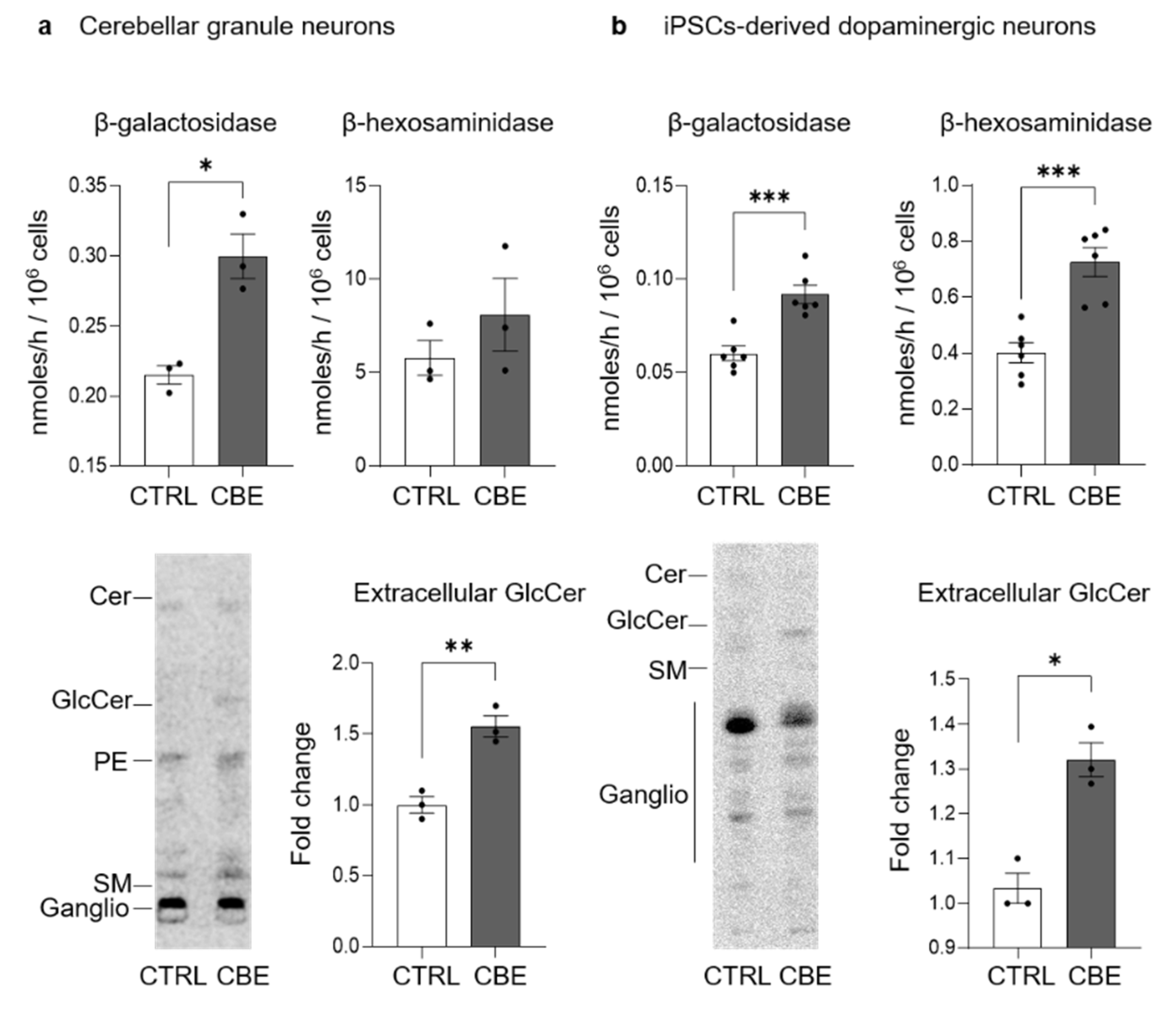
3.2. GCase Inhibition Alters the SL Pattern
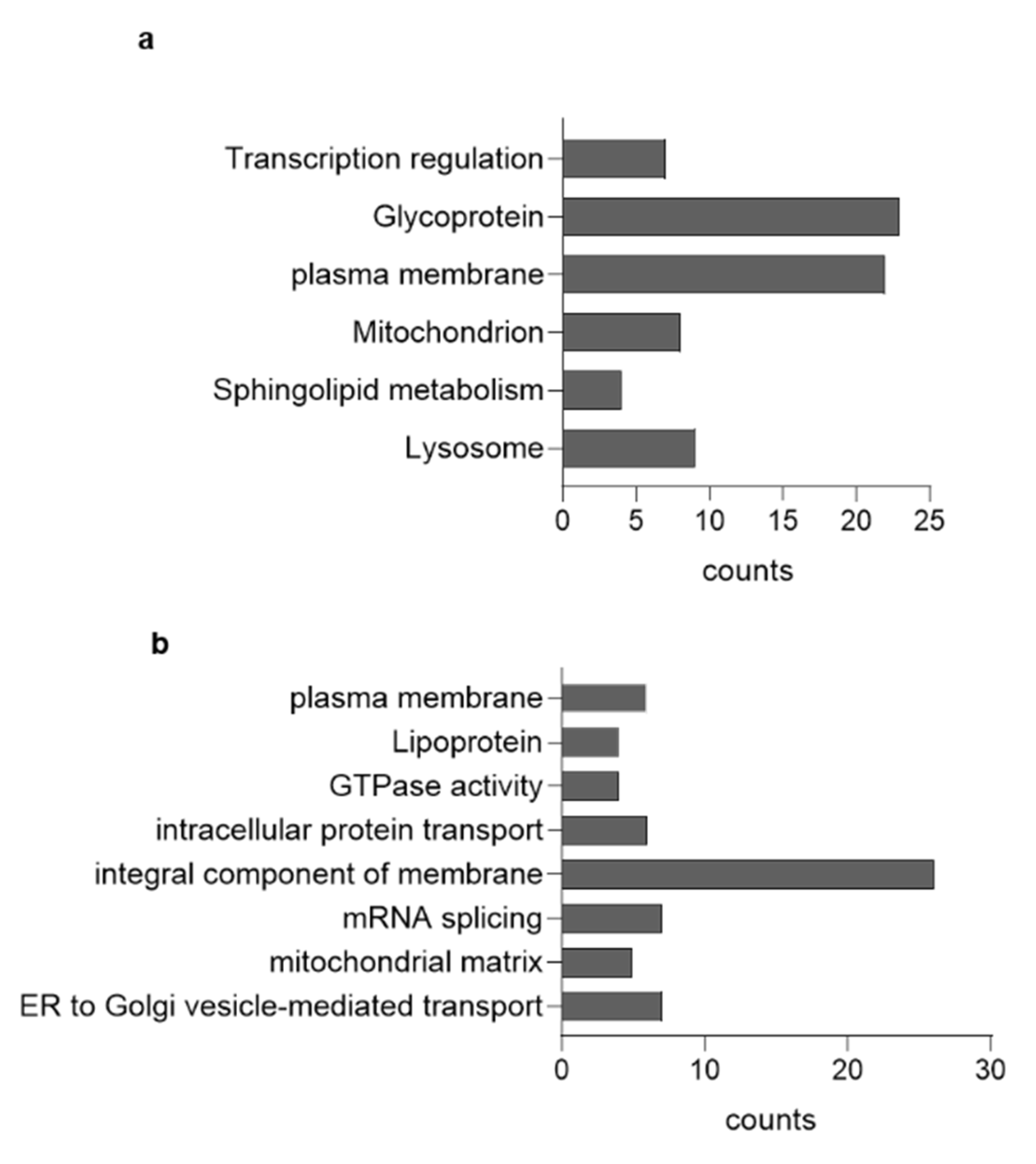
3.3. GCase Inhibition Alters the Proteomic Profile
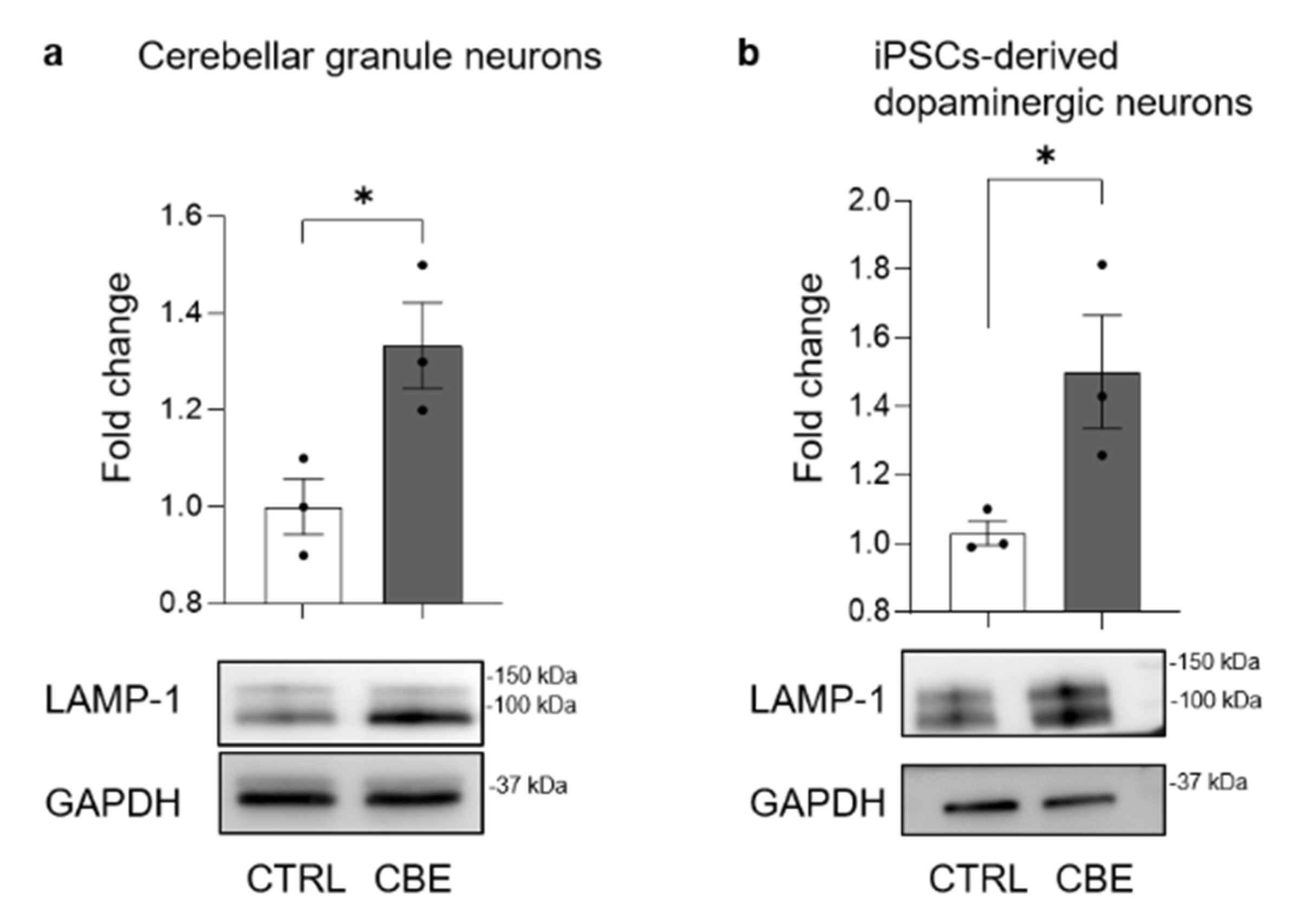
3.4. GlcCer Accumulation Affects the Lysosomal Compartment
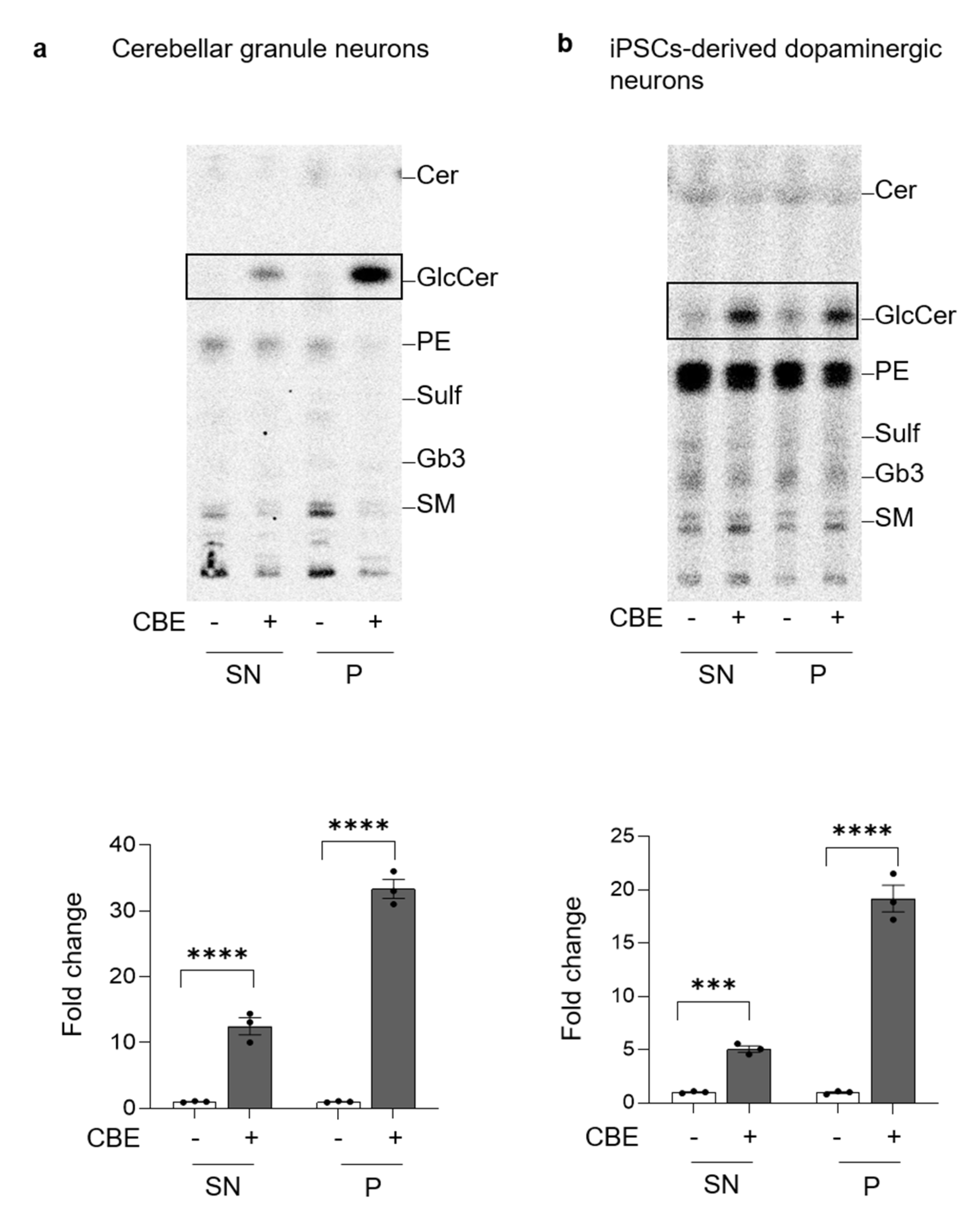
3.5. GCase-Inhibited Neurons Are Characterized by Aberrant Lysosomal Exocytosis
3.6. GlcCer Accumulation Is Not Confined to Lysosomes
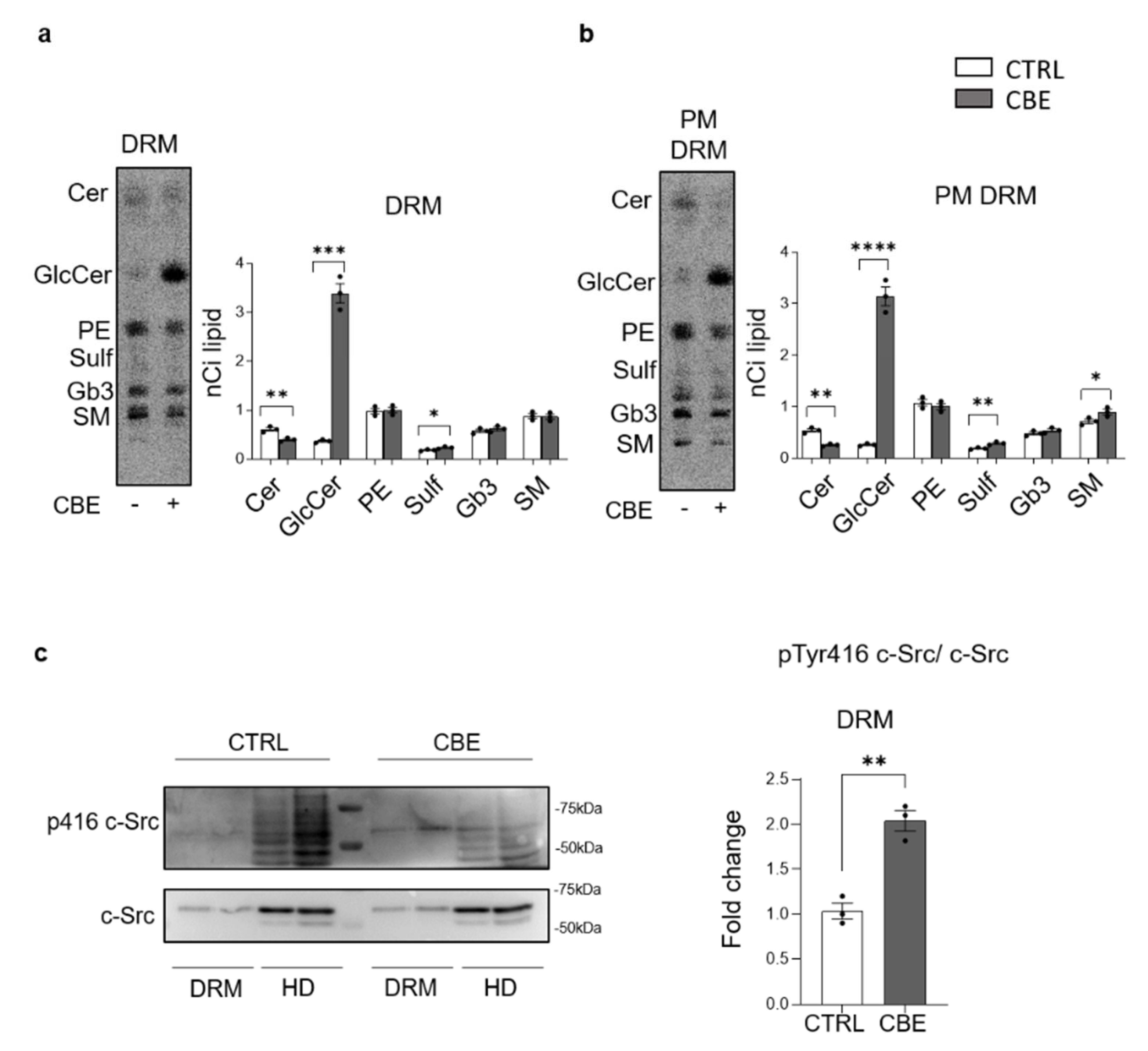
3.7. GCase Inhibition Induces Changes in PM Microdomains
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| GCase | β-glucocerebrosidase; |
| GSL | glycosphingolipid; |
| GlcCer | glucosylceramide; |
| GD | Gaucher disease; |
| LSD | lysosomal storage disorder; |
| PD | Parkinson’s disease; |
| CGN | cerebellar granule neurons; |
| iPSC | induced pluripotent stem cell; |
| DA | dopaminergic; |
| CBE | conduritol B epoxide; |
| PM | plasma membrane; |
| SL | sphingolipid; |
| DIC | days in culture; |
| TH | tyrosine hydroxylase; |
| GlcSph | glucosylsphingosine; |
| P | precipitate; |
| SN | supernatant; |
| HD | high-density fractions; |
| DRM | detergent resistant membrane. |
References
- Stirnemann, J.; Belmatoug, N.; Camou, F.; Serratrice, C.; Froissart, R.; Caillaud, C.; Levade, T.; Astudillo, L.; Serratrice, J.; Brassier, A.; et al. A Review of Gaucher Disease Pathophysiology, Clinical Presentation and Treatments. Int. J. Mol. Sci. 2017, 18, 441. [Google Scholar] [CrossRef] [PubMed]
- Chiricozzi, E.; Aureli, M.; Mauri, L.; Di Biase, E.; Lunghi, G.; Fazzari, M.; Valsecchi, M.; Carsana, E.V.; Loberto, N.; Prinetti, A.; et al. Glycosphingolipids. Adv. Exp. Med. Biol. 2021, 1325, 61–102. [Google Scholar] [CrossRef] [PubMed]
- Lwin, A.; Orvisky, E.; Goker-Alpan, O.; LaMarca, M.E.; Sidransky, E. Glucocerebrosidase mutations in subjects with parkinsonism. Mol. Genet. Metab. 2004, 81, 70–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goker-Alpan, O.; Schiffmann, R.; LaMarca, M.E.; Nussbaum, R.L.; McInerney-Leo, A.; Sidransky, E. Parkinsonism among Gaucher disease carriers. J. Med. Genet. 2004, 41, 937–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eblan, M.J.; Walker, J.M.; Sidransky, E. The glucocerebrosidase gene and Parkinson’s disease in Ashkenazi Jews. N. Engl. J. Med. 2005, 352, 728–731, author reply 728–731. [Google Scholar] [CrossRef]
- Sidransky, E. Heterozygosity for a Mendelian disorder as a risk factor for complex disease. Clin. Genet. 2006, 70, 275–282. [Google Scholar] [CrossRef]
- Straniero, L.; Asselta, R.; Bonvegna, S.; Rimoldi, V.; Melistaccio, G.; Solda, G.; Aureli, M.; Della Porta, M.; Lucca, U.; Di Fonzo, A.; et al. The SPID-GBA study: Sex distribution, Penetrance, Incidence, and Dementia in GBA-PD. Neurol. Genet. 2020, 6, e523. [Google Scholar] [CrossRef]
- Gegg, M.E.; Burke, D.; Heales, S.J.; Cooper, J.M.; Hardy, J.; Wood, N.W.; Schapira, A.H. Glucocerebrosidase deficiency in substantia nigra of parkinson disease brains. Ann. Neurol. 2012, 72, 455–463. [Google Scholar] [CrossRef] [Green Version]
- Seo, B.A.; Kim, D.; Hwang, H.; Kim, M.S.; Ma, S.X.; Kwon, S.H.; Kweon, S.H.; Wang, H.; Yoo, J.M.; Choi, S.; et al. TRIP12 ubiquitination of glucocerebrosidase contributes to neurodegeneration in Parkinson’s disease. Neuron 2021, 109, 3758–3774.e3711. [Google Scholar] [CrossRef]
- Dilsizoglu Senol, A.; Samarani, M.; Syan, S.; Guardia, C.M.; Nonaka, T.; Liv, N.; Latour-Lambert, P.; Hasegawa, M.; Klumperman, J.; Bonifacino, J.S.; et al. alpha-Synuclein fibrils subvert lysosome structure and function for the propagation of protein misfolding between cells through tunneling nanotubes. PLoS Biol. 2021, 19, e3001287. [Google Scholar] [CrossRef]
- Murphy, K.E.; Gysbers, A.M.; Abbott, S.K.; Tayebi, N.; Kim, W.S.; Sidransky, E.; Cooper, A.; Garner, B.; Halliday, G.M. Reduced glucocerebrosidase is associated with increased alpha-synuclein in sporadic Parkinson’s disease. Brain 2014, 137, 834–848. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goker-Alpan, O.; Hruska, K.S.; Orvisky, E.; Kishnani, P.S.; Stubblefield, B.K.; Schiffmann, R.; Sidransky, E. Divergent phenotypes in Gaucher disease implicate the role of modifiers. J. Med. Genet. 2005, 42, e37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazzulli, J.R.; Xu, Y.H.; Sun, Y.; Knight, A.L.; McLean, P.J.; Caldwell, G.A.; Sidransky, E.; Grabowski, G.A.; Krainc, D. Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 2011, 146, 37–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richter, F.; Fleming, S.M.; Watson, M.; Lemesre, V.; Pellegrino, L.; Ranes, B.; Zhu, C.; Mortazavi, F.; Mulligan, C.K.; Sioshansi, P.C.; et al. A GCase chaperone improves motor function in a mouse model of synucleinopathy. Neurotherapeutics 2014, 11, 840–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.; Yun, S.P.; Lee, S.; Umanah, G.E.; Bandaru, V.V.R.; Yin, X.; Rhee, P.; Karuppagounder, S.S.; Kwon, S.H.; Lee, H.; et al. GBA1 deficiency negatively affects physiological alpha-synuclein tetramers and related multimers. Proc. Natl. Acad. Sci. USA 2018, 115, 798–803. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prence, E.M.; Chaturvedi, P.; Newburg, D.S. In vitro accumulation of glucocerebroside in neuroblastoma cells: A model for study of Gaucher disease pathobiology. J. Neurosci. Res. 1996, 43, 365–371. [Google Scholar] [CrossRef]
- Di Biase, E.; Lunghi, G.; Fazzari, M.; Maggioni, M.; Pome, D.Y.; Valsecchi, M.; Samarani, M.; Fato, P.; Ciampa, M.G.; Prioni, S.; et al. Gangliosides in the differentiation process of primary neurons: The specific role of GM1-oligosaccharide. Glycocon. J. 2020, 37, 329–343. [Google Scholar] [CrossRef]
- Zhang, P.; Xia, N.; Reijo Pera, R.A. Directed dopaminergic neuron differentiation from human pluripotent stem cells. J. Vis. Exp. 2014, 51737. [Google Scholar] [CrossRef]
- Schiumarini, D.; Loberto, N.; Mancini, G.; Bassi, R.; Giussani, P.; Chiricozzi, E.; Samarani, M.; Munari, S.; Tamanini, A.; Cabrini, G.; et al. Evidence for the Involvement of Lipid Rafts and Plasma Membrane Sphingolipid Hydrolases in Pseudomonas aeruginosa Infection of Cystic Fibrosis Bronchial Epithelial Cells. Mediat. Inflamm. 2017, 2017, 1730245. [Google Scholar] [CrossRef] [Green Version]
- Schondorf, D.C.; Aureli, M.; McAllister, F.E.; Hindley, C.J.; Mayer, F.; Schmid, B.; Sardi, S.P.; Valsecchi, M.; Hoffmann, S.; Schwarz, L.K.; et al. iPSC-derived neurons from GBA1-associated Parkinson’s disease patients show autophagic defects and impaired calcium homeostasis. Nat. Commun. 2014, 5, 4028. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lunghi, G.; Fazzari, M.; Di Biase, E.; Mauri, L.; Sonnino, S.; Chiricozzi, E. Modulation of calcium signaling depends on the oligosaccharide of GM1 in Neuro2a mouse neuroblastoma cells. Glycocon. J. 2020, 37, 713–727. [Google Scholar] [CrossRef] [PubMed]
- Aureli, M.; Loberto, N.; Lanteri, P.; Chigorno, V.; Prinetti, A.; Sonnino, S. Cell surface sphingolipid glycohydrolases in neuronal differentiation and aging in culture. J. Neurochem. 2011, 116, 891–899. [Google Scholar] [CrossRef] [PubMed]
- Loberto, N.; Lunghi, G.; Schiumarini, D.; Samarani, M.; Chiricozzi, E.; Aureli, M. Methods for Assay of Ganglioside Catabolic Enzymes. Methods Mol. Biol. 2018, 1804, 383–400. [Google Scholar] [CrossRef]
- Schueler, U.H.; Kolter, T.; Kaneski, C.R.; Blusztajn, J.K.; Herkenham, M.; Sandhoff, K.; Brady, R.O. Toxicity of glucosylsphingosine (glucopsychosine) to cultured neuronal cells: A model system for assessing neuronal damage in Gaucher disease type 2 and 3. NeuroBiol. Dis 2003, 14, 595–601. [Google Scholar] [CrossRef]
- Sueyoshi, N.; Maehara, T.; Ito, M. Apoptosis of Neuro2a cells induced by lysosphingolipids with naturally occurring stereochemical configurations. J. Lipid Res. 2001, 42, 1197–1202. [Google Scholar] [CrossRef]
- Liu, J.; Halene, S.; Yang, M.; Iqbal, J.; Yang, R.; Mehal, W.Z.; Chuang, W.L.; Jain, D.; Yuen, T.; Sun, L.; et al. Gaucher disease gene GBA functions in immune regulation. Proc. Natl. Acad. Sci. USA 2012, 109, 10018–10023. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dennis, G., Jr.; Sherman, B.T.; Hosack, D.A.; Yang, J.; Gao, W.; Lane, H.C.; Lempicki, R.A. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003, 4, P3. [Google Scholar] [CrossRef] [PubMed]
- Szklarczyk, D.; Gable, A.L.; Nastou, K.C.; Lyon, D.; Kirsch, R.; Pyysalo, S.; Doncheva, N.T.; Legeay, M.; Fang, T.; Bork, P.; et al. The STRING database in 2021: Customizable protein-protein networks, and functional characterization of user-uploaded gene/measurement sets. Nucleic Acids Res. 2021, 49, D605–D612. [Google Scholar] [CrossRef] [PubMed]
- Samarani, M.; Loberto, N.; Solda, G.; Straniero, L.; Asselta, R.; Duga, S.; Lunghi, G.; Zucca, F.A.; Mauri, L.; Ciampa, M.G.; et al. A lysosome-plasma membrane-sphingolipid axis linking lysosomal storage to cell growth arrest. FASEB J. 2018, 32, 5685–5702. [Google Scholar] [CrossRef]
- Sardiello, M.; Palmieri, M.; di Ronza, A.; Medina, D.L.; Valenza, M.; Gennarino, V.A.; Di Malta, C.; Donaudy, F.; Embrione, V.; Polishchuk, R.S.; et al. A gene network regulating lysosomal biogenesis and function. Science 2009, 325, 473–477. [Google Scholar] [CrossRef] [Green Version]
- Aureli, M.; Prioni, S.; Mauri, L.; Loberto, N.; Casellato, R.; Ciampa, M.G.; Chigorno, V.; Prinetti, A.; Sonnino, S. Photoactivable sphingosine as a tool to study membrane microenvironments in cultured cells. J. Lipid Res. 2010, 51, 798–808. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lingwood, D.; Simons, K. Lipid rafts as a membrane-organizing principle. Science 2010, 327, 46–50. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonnino, S.; Aureli, M.; Grassi, S.; Mauri, L.; Prioni, S.; Prinetti, A. Lipid rafts in neurodegeneration and neuroprotection. Mol. NeuroBiol. 2014, 50, 130–148. [Google Scholar] [CrossRef]
- Riboldi, G.M.; Di Fonzo, A.B. GBA, Gaucher Disease, and Parkinson’s Disease: From Genetic to Clinic to New Therapeutic Approaches. Cells 2019, 8, 364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cilia, R.; Tunesi, S.; Marotta, G.; Cereda, E.; Siri, C.; Tesei, S.; Zecchinelli, A.L.; Canesi, M.; Mariani, C.B.; Meucci, N.; et al. Survival and dementia in GBA-associated Parkinson’s disease: The mutation matters. Ann. Neurol. 2016, 80, 662–673. [Google Scholar] [CrossRef]
- Sidransky, E.; Nalls, M.A.; Aasly, J.O.; Aharon-Peretz, J.; Annesi, G.; Barbosa, E.R.; Bar-Shira, A.; Berg, D.; Bras, J.; Brice, A.; et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N. Engl. J. Med. 2009, 361, 1651–1661. [Google Scholar] [CrossRef] [Green Version]
- McNeill, A.; Duran, R.; Hughes, D.A.; Mehta, A.; Schapira, A.H. A clinical and family history study of Parkinson’s disease in heterozygous glucocerebrosidase mutation carriers. J. Neurol. Neurosurg. Psychiatry 2012, 83, 853–854. [Google Scholar] [CrossRef] [Green Version]
- Nalls, M.A.; Duran, R.; Lopez, G.; Kurzawa-Akanbi, M.; McKeith, I.G.; Chinnery, P.F.; Morris, C.M.; Theuns, J.; Crosiers, D.; Cras, P.; et al. A multicenter study of glucocerebrosidase mutations in dementia with Lewy bodies. JAMA Neurol. 2013, 70, 727–735. [Google Scholar] [CrossRef]
- Johnson, P.H.; Weinreb, N.J.; Cloyd, J.C.; Tuite, P.J.; Kartha, R.V. GBA1 mutations: Prospects for exosomal biomarkers in alpha-synuclein pathologies. Mol. Genet. Metab. 2020, 129, 35–46. [Google Scholar] [CrossRef] [PubMed]
- Farfel-Becker, T.; Vitner, E.B.; Futerman, A.H. Animal models for Gaucher disease research. Dis. Model. Mech. 2011, 4, 746–752. [Google Scholar] [CrossRef] [Green Version]
- Enquist, I.B.; Lo Bianco, C.; Ooka, A.; Nilsson, E.; Mansson, J.E.; Ehinger, M.; Richter, J.; Brady, R.O.; Kirik, D.; Karlsson, S. Murine models of acute neuronopathic Gaucher disease. Proc. Natl. Acad. Sci. USA 2007, 104, 17483–17488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sillence, D.J.; Puri, V.; Marks, D.L.; Butters, T.D.; Dwek, R.A.; Pagano, R.E.; Platt, F.M. Glucosylceramide modulates membrane traffic along the endocytic pathway. J. Lipid Res. 2002, 43, 1837–1845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orvisky, E.; Park, J.K.; LaMarca, M.E.; Ginns, E.I.; Martin, B.M.; Tayebi, N.; Sidransky, E. Glucosylsphingosine accumulation in tissues from patients with Gaucher disease: Correlation with phenotype and genotype. Mol. Genet. Metab. 2002, 76, 262–270. [Google Scholar] [CrossRef]
- Hein, L.K.; Meikle, P.J.; Hopwood, J.J.; Fuller, M. Secondary sphingolipid accumulation in a macrophage model of Gaucher disease. Mol. Genet. Metab. 2007, 92, 336–345. [Google Scholar] [CrossRef]
- Walkley, S.U.; Vanier, M.T. Secondary lipid accumulation in lysosomal disease. Biochim. Biophys. Acta 2009, 1793, 726–736. [Google Scholar] [CrossRef] [Green Version]
- Medina, D.L.; Fraldi, A.; Bouche, V.; Annunziata, F.; Mansueto, G.; Spampanato, C.; Puri, C.; Pignata, A.; Martina, J.A.; Sardiello, M.; et al. Transcriptional activation of lysosomal exocytosis promotes cellular clearance. Dev. Cell 2011, 21, 421–430. [Google Scholar] [CrossRef]
- Kim, O.S.; Park, E.J.; Joe, E.H.; Jou, I. JAK-STAT signaling mediates gangliosides-induced inflammatory responses in brain microglial cells. J. Biol. Chem. 2002, 277, 40594–40601. [Google Scholar] [CrossRef] [Green Version]
- Sonnino, S.; Prinetti, A.; Mauri, L.; Chigorno, V.; Tettamanti, G. Dynamic and structural properties of sphingolipids as driving forces for the formation of membrane domains. Chem. Rev. 2006, 106, 2111–2125. [Google Scholar] [CrossRef]
- Chiricozzi, E.; Di Biase, E.; Lunghi, G.; Fazzari, M.; Loberto, N.; Aureli, M.; Mauri, L.; Sonnino, S. Turning the spotlight on the oligosaccharide chain of GM1 ganglioside. Glycoconj. J. 2021, 38, 101–117. [Google Scholar] [CrossRef]
- Lunghi, G.; Fazzari, M.; Di Biase, E.; Mauri, L.; Chiricozzi, E.; Sonnino, S. The structure of gangliosides hides a code for determining neuronal functions. FEBS Open Bio. 2021, 11, 3193–3200. [Google Scholar] [CrossRef]
- Aureli, M.; Masilamani, A.P.; Illuzzi, G.; Loberto, N.; Scandroglio, F.; Prinetti, A.; Chigorno, V.; Sonnino, S. Activity of plasma membrane beta-galactosidase and beta-glucosidase. FEBS Lett. 2009, 583, 2469–2473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Svennerholm, L.; Bostrom, K.; Fredman, P.; Mansson, J.E.; Rosengren, B.; Rynmark, B.M. Human brain gangliosides: Developmental changes from early fetal stage to advanced age. Biochim. Biophys. Acta 1989, 1005, 109–117. [Google Scholar] [CrossRef]
- Batta, G.; Soltesz, L.; Kovacs, T.; Bozo, T.; Meszar, Z.; Kellermayer, M.; Szollosi, J.; Nagy, P. Alterations in the properties of the cell membrane due to glycosphingolipid accumulation in a model of Gaucher disease. Sci. Rep. 2018, 8, 157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hein, L.K.; Duplock, S.; Hopwood, J.J.; Fuller, M. Lipid composition of microdomains is altered in a cell model of Gaucher disease. J. Lipid Res. 2008, 49, 1725–1734. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piccinini, M.; Scandroglio, F.; Prioni, S.; Buccinna, B.; Loberto, N.; Aureli, M.; Chigorno, V.; Lupino, E.; DeMarco, G.; Lomartire, A.; et al. Deregulated sphingolipid metabolism and membrane organization in neurodegenerative disorders. Mol. NeuroBiol. 2010, 41, 314–340. [Google Scholar] [CrossRef] [PubMed]
- Roskoski, R., Jr. Src protein-tyrosine kinase structure, mechanism, and small molecule inhibitors. Pharmacol. Res. 2015, 94, 9–25. [Google Scholar] [CrossRef] [PubMed]
- Holzer, R.G.; Park, E.J.; Li, N.; Tran, H.; Chen, M.; Choi, C.; Solinas, G.; Karin, M. Saturated fatty acids induce c-Src clustering within membrane subdomains, leading to JNK activation. Cell 2011, 147, 173–184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kajiwara, K.; Yamada, T.; Bamba, T.; Fukusaki, E.; Imamoto, F.; Okada, M.; Oneyama, C. c-Src-induced activation of ceramide metabolism impairs membrane microdomains and promotes malignant progression by facilitating the translocation of c-Src to focal adhesions. Biochem. J. 2014, 458, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.W.; Wang, S.G.; Lee, C.H.; Chan, W.L.; Lin, M.L.; Chen, S.S. Enforced C-Src Activation Causes Compartmental Dysregulation of PI3K and PTEN Molecules in Lipid Rafts of Tongue Squamous Carcinoma Cells by Attenuating Rac1-Akt-GLUT-1-Mediated Sphingolipid Synthesis. Int. J. Mol. Sci. 2020, 21, 5812. [Google Scholar] [CrossRef]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lunghi, G.; Carsana, E.V.; Loberto, N.; Cioccarelli, L.; Prioni, S.; Mauri, L.; Bassi, R.; Duga, S.; Straniero, L.; Asselta, R.; et al. β-Glucocerebrosidase Deficiency Activates an Aberrant Lysosome-Plasma Membrane Axis Responsible for the Onset of Neurodegeneration. Cells 2022, 11, 2343. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11152343
Lunghi G, Carsana EV, Loberto N, Cioccarelli L, Prioni S, Mauri L, Bassi R, Duga S, Straniero L, Asselta R, et al. β-Glucocerebrosidase Deficiency Activates an Aberrant Lysosome-Plasma Membrane Axis Responsible for the Onset of Neurodegeneration. Cells. 2022; 11(15):2343. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11152343
Chicago/Turabian StyleLunghi, Giulia, Emma Veronica Carsana, Nicoletta Loberto, Laura Cioccarelli, Simona Prioni, Laura Mauri, Rosaria Bassi, Stefano Duga, Letizia Straniero, Rosanna Asselta, and et al. 2022. "β-Glucocerebrosidase Deficiency Activates an Aberrant Lysosome-Plasma Membrane Axis Responsible for the Onset of Neurodegeneration" Cells 11, no. 15: 2343. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11152343