
Novel AR/AR-V7 and Mnk1/2 Degrader, VNPP433-3β: Molecular Mechanisms of Action and Efficacy in AR-Overexpressing Castration Resistant Prostate Cancer In Vitro and In Vivo Models
 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Culture and Reagents
2.2. Molecular Docking
2.3. Fluorescence Spectroscopy
2.4. Cellular Thermal Shift Assay (CETSA)
2.5. Immunoblotting
2.6. Co-Immunoprecipitation (Co-IP) Assay
2.7. mRNA 5′ Cap (m7GTP) Binding Assay
2.8. RNA-Sequencing and Transcriptome Analyses
2.9. siRNA-Mediated Knockdown of Gene Expression
2.10. In Vivo Tumor Xenograft Studies
2.11. Statistical Analysis
3. Results
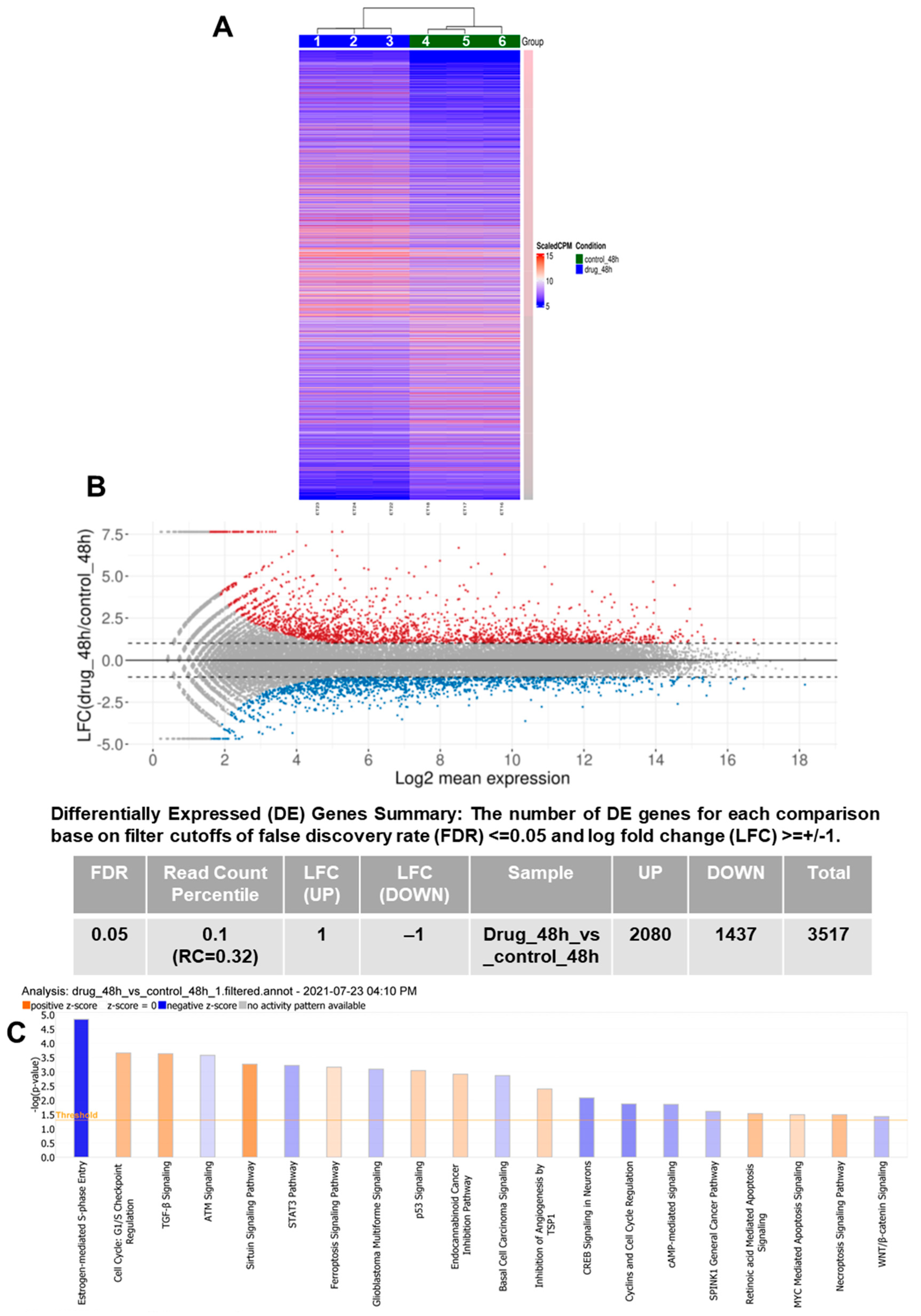
3.1. RNA-Seq Demonstrates Modulation of Multiple Pathways Leading to PCa Inhibition by VNPP433-3β
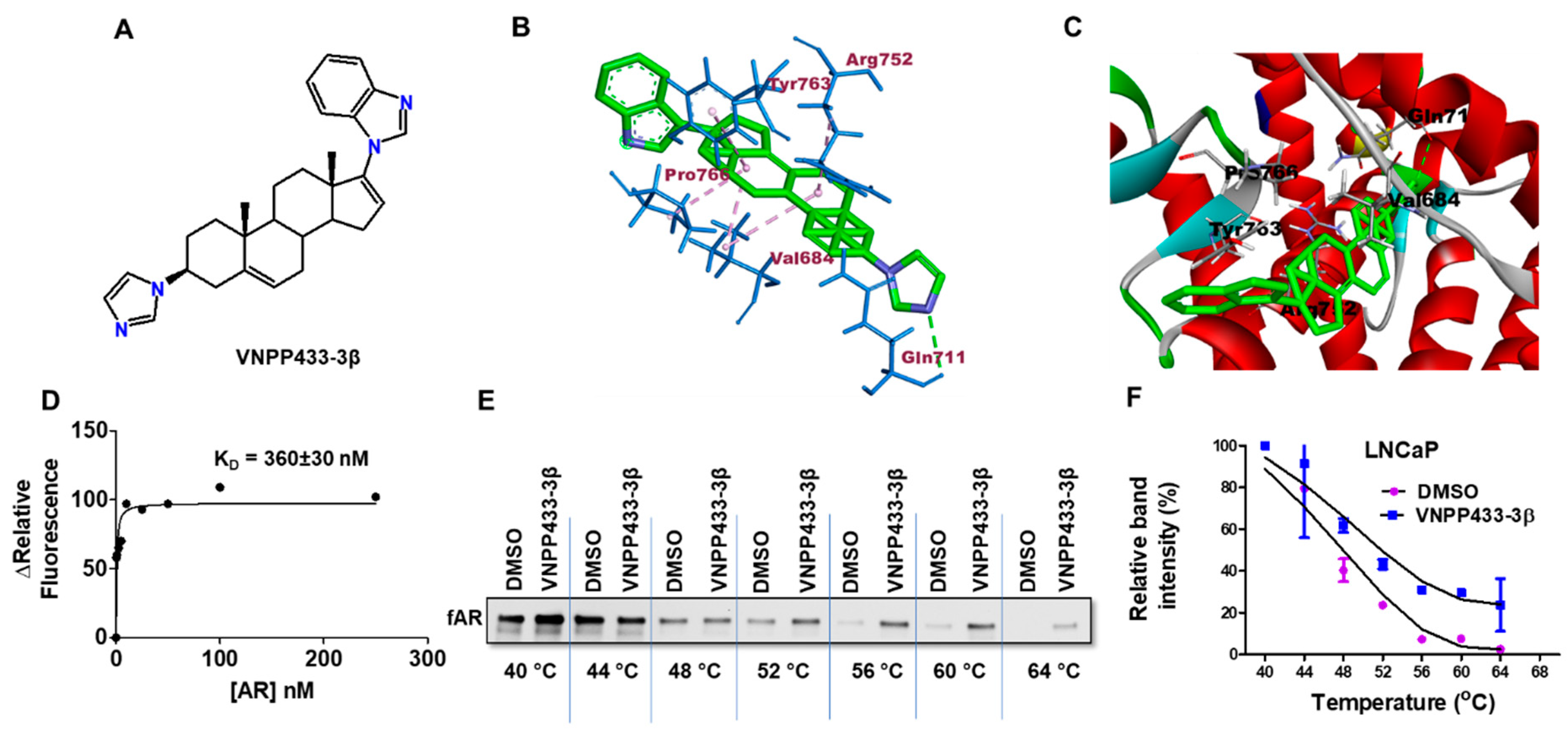
3.2. VNPP433-3β-Induced Degradation of fAR and AR-V7 Is Attributed to the Physical Interaction of VNPP433-3β with fAR
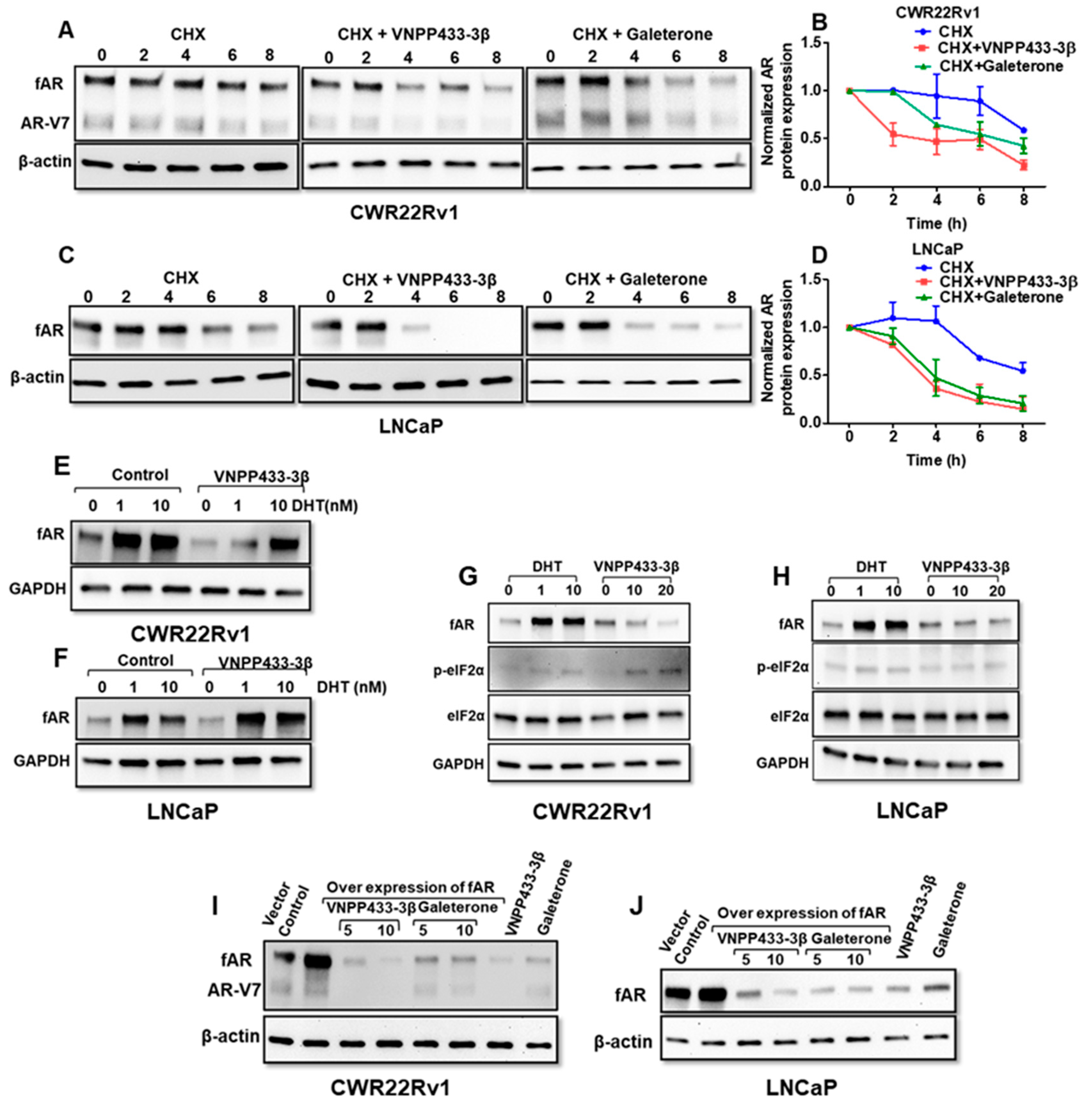
3.3. VNPP433-3β Decreases the Half-Life of fAR and AR-V7 by Enhancing the Rate of Degradation
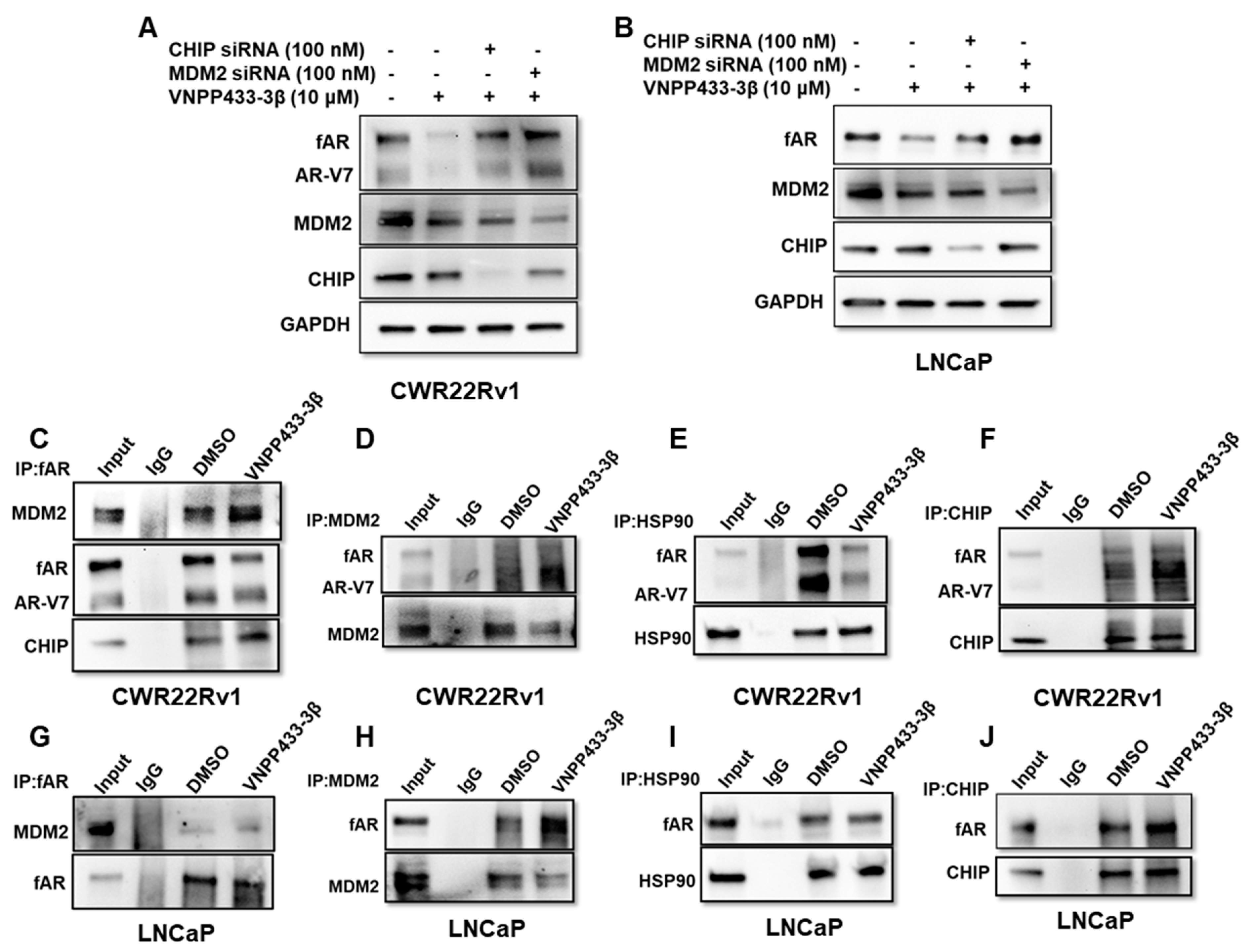
3.4. VNPP433-3β Promotes fAR/AR-V7 Degradation by Enhancing Its Interaction with MDM2/CHIP and Disrupting fAR/AR-V7-HSP90 Interaction
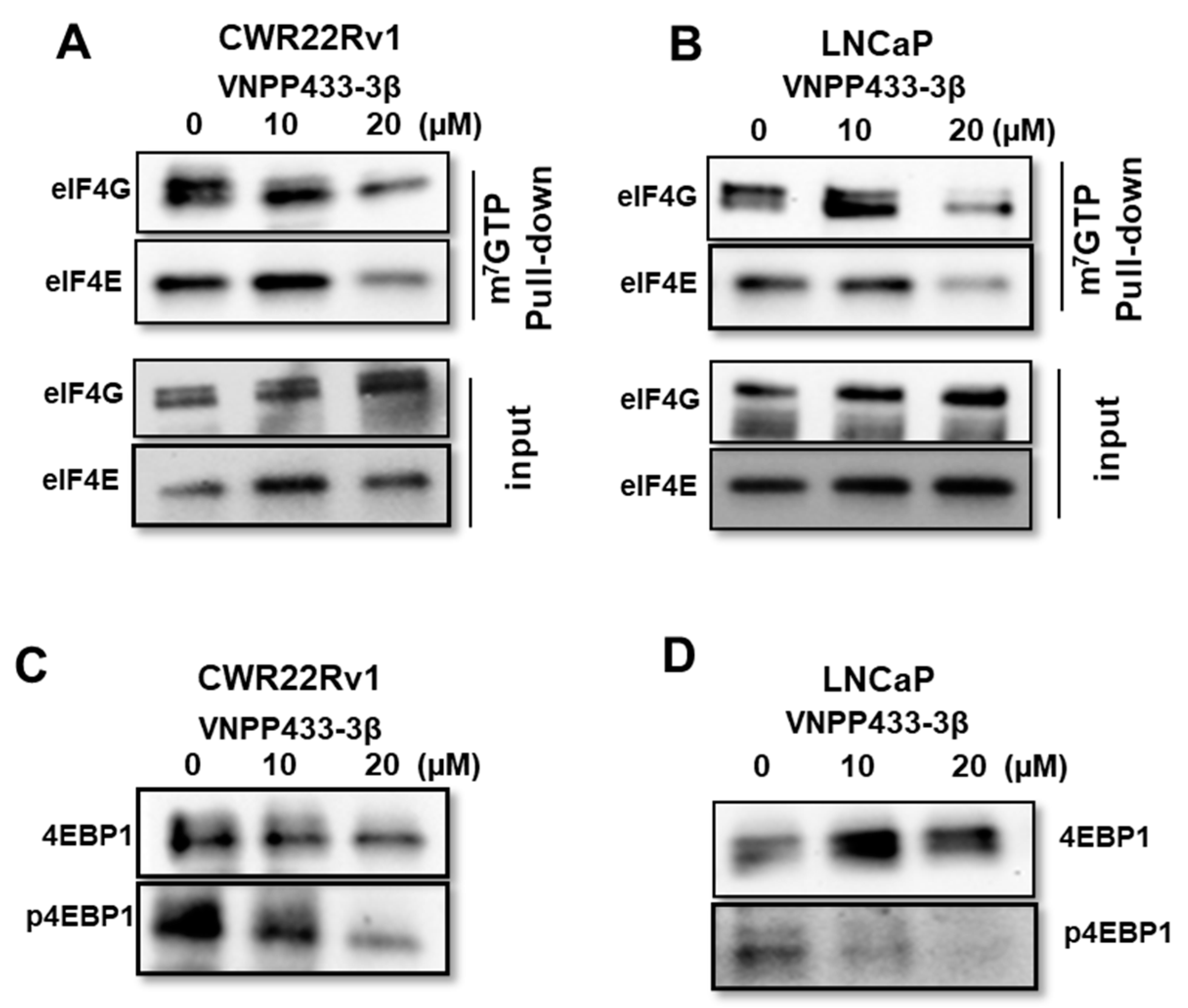
3.5. VNPP433-3β Abates Binding of eIF4E and eIF4G to 5′ Cap of mRNA Thereby Imposing Translational Regulation
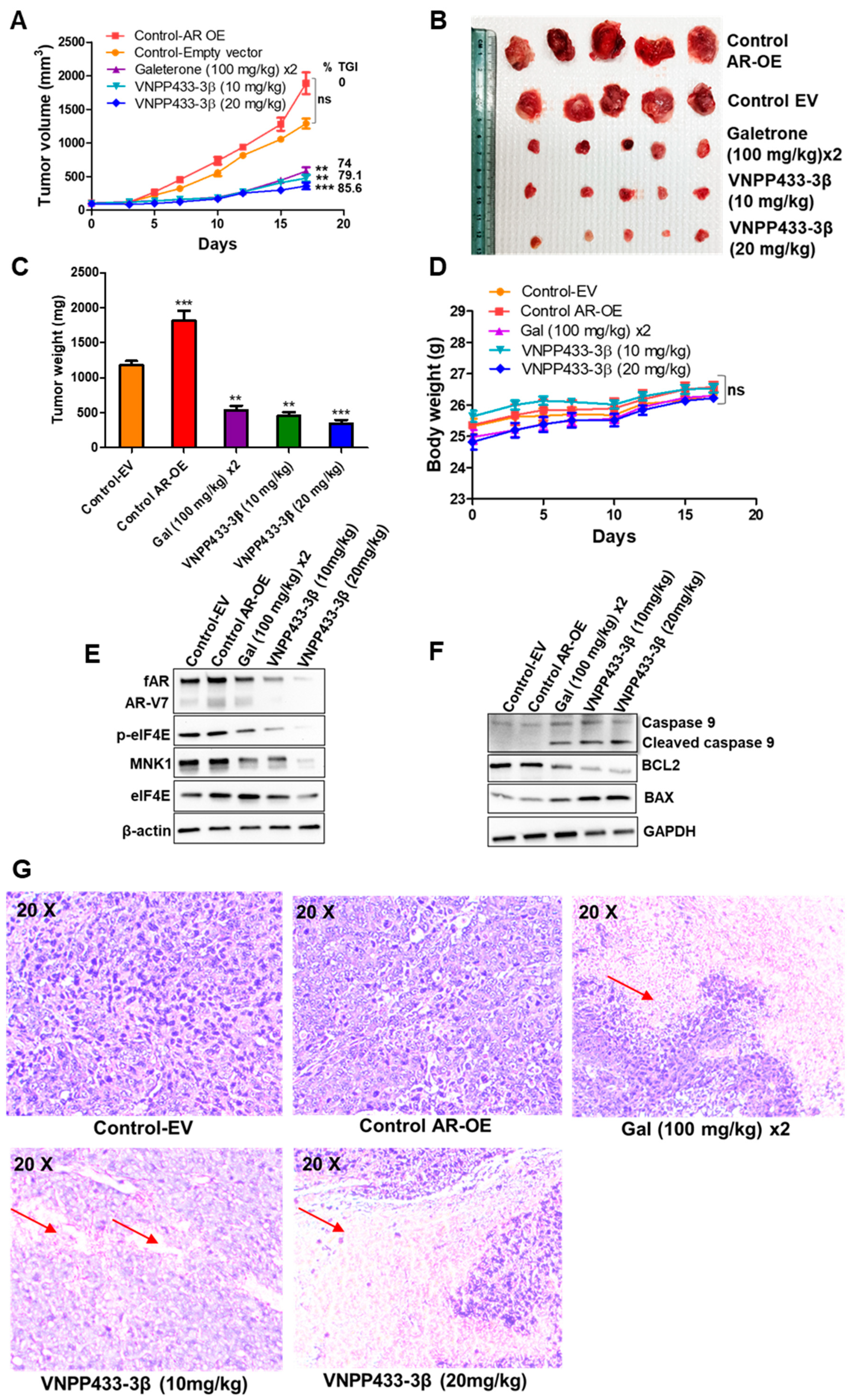
3.6. VNPP433-3β Targets AR In Vivo with Concomitant Tumor Inhibition in CRPC Tumor Xenograft
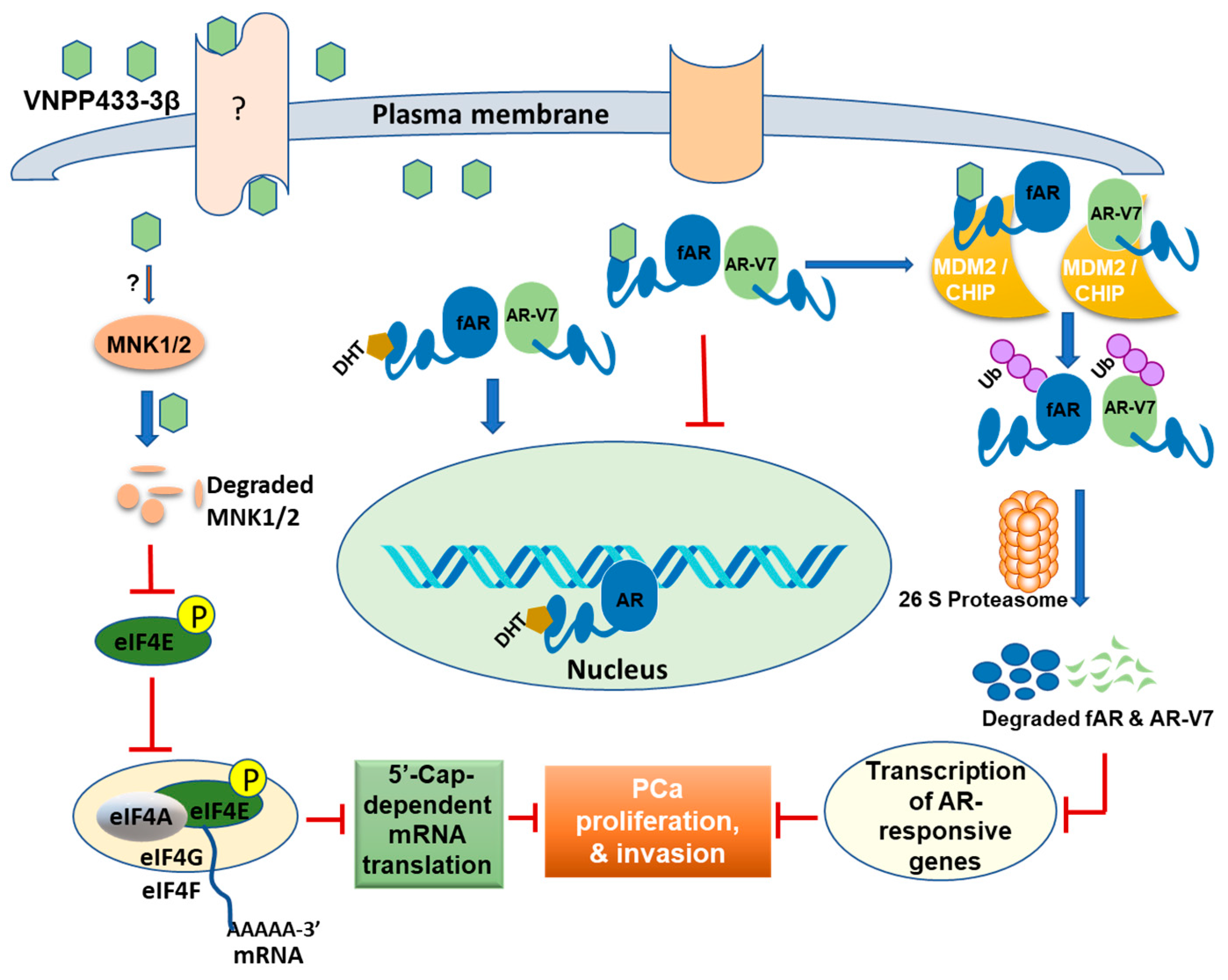
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Torre, L.A.; Bray, F.; Siegel, R.L.; Ferlay, J.; Lortet-Tieulent, J.; Jemal, A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015, 65, 87–108. [Google Scholar] [CrossRef] [PubMed]
- Labriola, M.K.; Atiq, S.; Hirshman, N.; Bitting, R.L. Management of men with metastatic castration-resistant prostate cancer following potent androgen receptor inhibition: A review of novel investigational therapies. Prostate Cancer Prostatic Dis. 2021, 24, 301–309. [Google Scholar] [CrossRef] [PubMed]
- D’Abronzo, L.S.; Ghosh, P.M. eIF4E Phosphorylation in Prostate Cancer. Neoplasia 2018, 20, 563–573. [Google Scholar] [CrossRef] [PubMed]
- Szostak, M.J.; Kyprianou, N. Radiation-induced apoptosis: Predictive and therapeutic significance in radiotherapy of prostate cancer (review). Oncol. Rep. 2000, 7, 699–706. [Google Scholar] [CrossRef] [PubMed]
- Schiewer, M.J.; Augello, M.A.; Knudsen, K.E. The AR dependent cell cycle: Mechanisms and cancer relevance. Mol. Cell Endocrinol. 2012, 352, 34–45. [Google Scholar] [CrossRef] [PubMed]
- Pilling, A.B.; Hwang, C. Targeting prosurvival BCL2 signaling through Akt blockade sensitizes castration-resistant prostate cancer cells to enzalutamide. Prostate 2019, 79, 1347–1359. [Google Scholar] [CrossRef]
- Attard, G.; Parker, C.; Eeles, R.A.; Schröder, F.; Tomlins, S.A.; Tannock, I.; Drake, C.G.; de Bono, J.S. Prostate cancer. Lancet 2016, 387, 70–82. [Google Scholar] [CrossRef]
- Watson, P.A.; Arora, V.K.; Sawyers, C.L. Emerging mechanisms of resistance to androgen receptor inhibitors in prostate cancer. Nat. Rev. Cancer 2015, 15, 701–711. [Google Scholar] [CrossRef]
- Rossi, V.; Di Zazzo, E.; Galasso, G.; De Rosa, C.; Abbondanza, C.; Sinisi, A.A.; Altucci, L.; Migliaccio, A.; Castoria, G. Estrogens Modulate Somatostatin Receptors Expression and Synergize With the Somatostatin Analog Pasireotide in Prostate Cells. Front. Pharmacol. 2019, 10, 28. [Google Scholar] [CrossRef] [Green Version]
- Di Zazzo, E.; Galasso, G.; Giovannelli, P.; Di Donato, M.; Bilancio, A.; Perillo, B.; Sinisi, A.A.; Migliaccio, A.; Castoria, G. Estrogen Receptors in Epithelial-Mesenchymal Transition of Prostate Cancer. Cancers 2019, 11, 1418. [Google Scholar] [CrossRef] [PubMed]
- de Bono, J.S.; Logothetis, C.J.; Molina, A.; Fizazi, K.; North, S.; Chu, L.; Chi, K.N.; Jones, R.J.; Goodman, O.B., Jr.; Saad, F.; et al. Abiraterone and increased survival in metastatic prostate cancer. N. Engl. J. Med. 2011, 364, 1995–2005. [Google Scholar] [CrossRef] [PubMed]
- Ryan, C.J.; Smith, M.R.; De Bono, J.S.; Molina, A.; Logothetis, C.J.; De Souza, P.; Fizazi, K.; Mainwaring, P.; Piulats, J.M.; Ng, S.; et al. Abiraterone in metastatic prostate cancer without previous chemotherapy. N. Engl. J. Med. 2013, 368, 138–148. [Google Scholar] [CrossRef] [PubMed]
- Beer, T.M.; Armstrong, A.J.; Rathkopf, D.E.; Loriot, Y.; Sternberg, C.N.; Higano, C.S.; Iversen, P.; Bhattacharya, S.; Carles, J.; Chowdhury, S.; et al. Enzalutamide in metastatic prostate cancer before chemotherapy. N. Engl. J. Med. 2014, 371, 424–433. [Google Scholar] [CrossRef] [PubMed]
- Scher, H.I.; Fizazi, K.; Saad, F.; Taplin, M.-E.; Sternberg, C.N.; Miller, K.; De Wit, R.; Mulders, P.; Chi, K.N.; Shore, N.D.; et al. Increased survival with enzalutamide in prostate cancer after chemotherapy. N. Engl. J. Med. 2012, 367, 1187–1197. [Google Scholar] [CrossRef]
- Mohler, J.L.; Gregory, C.W.; Ford, O.H.; Kim, D.; Weaver, C.M.; Petrusz, P.; Wilson, E.M.; French, F.S. The androgen axis in recurrent prostate cancer. Clin. Cancer Res. 2004, 10, 440–448. [Google Scholar] [CrossRef]
- Carreira, S.; Romanel, A.; Goodall, J.; Grist, E.; Ferraldeschi, R.; Miranda, S.; Prandi, D.; Lorente, D.; Frenel, J.-S.; Pezaro, C.; et al. Tumor clone dynamics in lethal prostate cancer. Sci. Transl. Med. 2014, 6, 254. [Google Scholar] [CrossRef]
- Visakorpi, T.; Hyytinen, E.; Koivisto, P.; Tanner, M.; Keinänen, R.; Palmberg, C.; Palotie, A.; Tammela, T.; Isola, J.; Kallioniemi, O.P. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat. Genet. 1995, 9, 401–406. [Google Scholar] [CrossRef]
- Huang, Z.Q.; Li, J.; Wong, J. AR possesses an intrinsic hormone-independent transcriptional activity. Mol. Endocrinol. 2002, 16, 924–937. [Google Scholar] [CrossRef]
- Zegarra-Moro, O.L.; Schmidt, L.J.; Huang, H.; Tindall, N.J. Disruption of androgen receptor function inhibits proliferation of androgen-refractory prostate cancer cells. Cancer Res. 2002, 62, 1008–1013. [Google Scholar]
- Balk, S.P. Androgen receptor as a target in androgen-independent prostate cancer. Urology 2002, 60 (Suppl. S1), 132–138; [Google Scholar] [CrossRef]
- Balk, S.P.; Knudsen, K.E. AR, the cell cycle, and prostate cancer. Nucl. Recept. Signal. 2008, 6, e001. [Google Scholar] [CrossRef] [PubMed]
- Njar, V.C.O. Androgen receptor antagonism and impact on inhibitors of androgen synthesis in prostate cancer therapy. Transl. Cancer Res. 2017, 6 (Suppl. S7), S1128–S1131. [Google Scholar] [CrossRef] [PubMed]
- Sarkar, S.; Brautigan, D.L.; Parsons, S.J.; Larner, J.M. Androgen receptor degradation by the E3 ligase CHIP modulates mitotic arrest in prostate cancer cells. Oncogene 2014, 33, 26–33. [Google Scholar] [CrossRef]
- D’Abronzo, L.S.; Bose, S.; Crapuchettes, M.E.; Beggs, R.E.; Vinall, R.L.; Tepper, C.G.; Siddiqui, S.; Mudryj, M.; Melgoza, F.U.; Durbin-Johnson, B.P.; et al. The androgen receptor is a negative regulator of eIF4E phosphorylation at S209: Implications for the use of mTOR inhibitors in advanced prostate cancer. Oncogene 2017, 36, 6359–6373. [Google Scholar] [CrossRef] [PubMed]
- Pyronnet, S.; Imataka, H.; Gingras, A.-C.; Fukunaga, R.; Hunter, T.; Sonenberg, N. Human eukaryotic translation initiation factor 4G (eIF4G) recruits mnk1 to phosphorylate eIF4E. EMBO J. 1999, 18, 270–279. [Google Scholar] [CrossRef]
- Njar, V.C.; Brodie, A.M. Discovery and development of Galeterone (TOK-001 or VN/124-1) for the treatment of all stages of prostate cancer. J. Med. Chem. 2015, 58, 2077–2087. [Google Scholar] [CrossRef]
- Montgomery, B.; Eisenberger, M.A.; Rettig, M.B.; Chu, F.; Pili, R.; Stephenson, J.J.; Vogelzang, N.J.; Koletsky, A.J.; Nordquist, L.T.; Edenfield, W.J.; et al. Androgen Receptor Modulation Optimized for Response (ARMOR) Phase I and II Studies: Galeterone for the Treatment of Castration-Resistant Prostate Cancer. Clin. Cancer Res. 2016, 22, 1356–1363. [Google Scholar] [CrossRef]
- Purushottamachar, P.; Thomas, E.; Thankan, R.S.; Rudchenko, V.; Hualng, G.; Njar, V.C. Large-scale synthesis of galeterone and lead next generation galeterone analog VNPP433-3beta. Steroids 2022, 185, 109062. [Google Scholar] [CrossRef]
- Kwegyir-Afful, A.K.; Ramalingam, S.; Ramamurthy, V.P.; Purushottamachar, P.; Murigi, F.N.; Vasaitis, T.S.; Huang, W.; Kane, M.A.; Zhang, Y.; Ambulos, N.; et al. Galeterone and The Next Generation Galeterone Analogs, VNPP414 and VNPP433-3beta Exert Potent Therapeutic Effects in Castration-/Drug-Resistant Prostate Cancer Preclinical Models In Vitro and In Vivo. Cancers 2019, 11, 637. [Google Scholar] [CrossRef]
- Thomas, E.; Thankan, R.S.; Purushottamachar, P.; Huang, W.; Kane, M.A.; Zhang, Y.; Ambulos, N.; Weber, D.J.; Njar, V.C.O. Transcriptome profiling reveals that VNPP433-3β, the lead next-generation galeterone analog, inhibits prostate cancer stem cells by downregulating EMT and stem cell markers. Mol. Carcinog. 2022, 61, 643–654. [Google Scholar] [CrossRef] [PubMed]
- Purushottamachar, P.; Kwegyir-Afful, A.K.; Martin, M.S.; Ramamurthy, V.P.; Ramalingam, S.; Njar, V.C.O. Identification of Novel Steroidal Androgen Receptor Degrading Agents Inspired by Galeterone 3beta-Imidazole Carbamate. ACS Med. Chem. Lett. 2016, 7, 708–713. [Google Scholar] [CrossRef]
- Matias, P.M.; Donner, P.; Coelho, R.; Thomaz, M.; Peixoto, C.; Macedo, S.; Otto, N.; Joschko, S.; Scholz, P.; Wegg, A.; et al. Structural evidence for ligand specificity in the binding domain of the human androgen receptor. Implications for pathogenic gene mutations. J. Biol. Chem. 2000, 275, 26164–26171. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Thomas, E.; Gopalakrishnan, V.; Hegde, M.; Kumar, S.; Karki, S.S.; Raghavan, S.C.; Choudhary, B. A Novel Resveratrol Based Tubulin Inhibitor Induces Mitotic Arrest and Activates Apoptosis in Cancer Cells. Sci. Rep. 2016, 6, 34653. [Google Scholar] [CrossRef] [PubMed]
- Jafari, R.; Almqvist, H.; Axelsson, H.; Ignatushchenko, M.; Lundbäck, T.; Nordlund, P.; Molina, D.M. The cellular thermal shift assay for evaluating drug target interactions in cells. Nat. Protoc. 2014, 9, 2100–2122. [Google Scholar] [CrossRef]
- Purushottamachar, P.; Thomas, E.; Thankan, R.S.; Njar, V.C. Novel deuterated Mnk1/2 protein degrader VNLG-152R analogs: Synthesis, In vitro Anti-TNBC activities and pharmacokinetics in mice. Eur. J. Med. Chem. 2022, 238, 114441. [Google Scholar] [CrossRef]
- Thomas, E.; Gopalakrishnan, V.; Somasagara, R.R.; Choudhary, B.; Raghavan, S.C. Extract of Vernonia condensata, Inhibits Tumor Progression and Improves Survival of Tumor-allograft Bearing Mouse. Sci. Rep. 2016, 6, 23255. [Google Scholar] [CrossRef] [PubMed]
- Hegde, M.; Karki, S.S.; Thomas, E.; Kumar, S.; Panjamurthy, K.; Ranganatha, S.R.; Rangappa, K.S.; Choudhary, B.; Raghavan, S.C. Novel levamisole derivative induces extrinsic pathway of apoptosis in cancer cells and inhibits tumor progression in mice. PLoS ONE 2012, 7, e43632. [Google Scholar] [CrossRef]
- Liang, J.; Wang, L.; Poluben, L.; Nouri, M.; Arai, S.; Xie, L.; Voznesensky, O.S.; Cato, L.; Yuan, X.; Russo, J.W.; et al. Androgen receptor splice variant 7 functions independently of the full length receptor in prostate cancer cells. Cancer Lett. 2021, 519, 172–184. [Google Scholar] [CrossRef]
- Sheflin, L.; Keegan, B.; Zhang, W.; Spaulding, S.W. Inhibiting proteasomes in human HepG2 and LNCaP cells increases endogenous androgen receptor levels. Biochem. Biophys. Res. Commun. 2000, 276, 144–150. [Google Scholar] [CrossRef] [PubMed]
- Bruno, R.D.; Gover, T.D.; Burger, A.M.; Brodie, A.M.; Njar, V.C. 17alpha-Hydroxylase/17,20 lyase inhibitor VN/124-1 inhibits growth of androgen-independent prostate cancer cells via induction of the endoplasmic reticulum stress response. Mol. Cancer Ther. 2008, 7, 2828–2836. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Cai, C.; Gao, S.; Simon, N.I.; Shen, H.C.; Balk, S.P. Galeterone prevents androgen receptor binding to chromatin and enhances degradation of mutant androgen receptor. Clin. Cancer Res. 2014, 20, 4075–4085. [Google Scholar] [CrossRef]
- Cnop, M.; Toivonen, S.; Igoillo-Esteve, M.; Salpea, P. Endoplasmic reticulum stress and eIF2alpha phosphorylation: The Achilles heel of pancreatic beta cells. Mol. Metab. 2017, 6, 1024–1039. [Google Scholar] [CrossRef] [PubMed]
- Shafi, A.A.; Cox, M.B.; Weigel, N.L. Androgen receptor splice variants are resistant to inhibitors of Hsp90 and FKBP52, which alter androgen receptor activity and expression. Steroids 2013, 78, 548–554. [Google Scholar] [CrossRef] [PubMed]
- Karaki, S.; Andrieu, C.; Ziouziou, H.; Rocchi, P. The Eukaryotic Translation Initiation Factor 4E (eIF4E) as a Therapeutic Target for Cancer. Adv. Protein Chem. Struct. Biol. 2015, 101, 1–26. [Google Scholar] [CrossRef]
- Cinar, B.; de Benedetti, A.; Freeman, M.R. Post-transcriptional regulation of the androgen receptor by Mammalian target of rapamycin. Cancer Res. 2005, 65, 2547–2553. [Google Scholar] [CrossRef]
- Kwegyir-Afful, A.K.; Ramalingam, S.; Purushottamachar, P.; Ramamurthy, V.P.; Njar, V.C.O. Galeterone and VNPT55 induce proteasomal degradation of AR/AR-V7, induce significant apoptosis via cytochrome c release and suppress growth of castration resistant prostate cancer xenografts in vivo. Oncotarget 2015, 6, 27440–27460. [Google Scholar] [CrossRef]
- Kregel, S.; Wang, C.; Han, X.; Xiao, L.; Fernandez-Salas, E.; Bawa, P.; McCollum, B.L.; Wilder-Romans, K.; Apel, I.J.; Cao, X.; et al. Androgen receptor degraders overcome common resistance mechanisms developed during prostate cancer treatment. Neoplasia 2020, 22, 111–119. [Google Scholar] [CrossRef]
- Zhu, Y.; Dalrymple, S.L.; Coleman, I.; Zheng, S.L.; Xu, J.; Hooper, J.E.; Antonarakis, E.S.; De Marzo, A.M.; Meeker, A.K.; Nelson, P.S.; et al. Role of androgen receptor splice variant-7 (AR-V7) in prostate cancer resistance to 2nd-generation androgen receptor signaling inhibitors. Oncogene 2020, 39, 6935–6949. [Google Scholar] [CrossRef]
- Gaughan, L.; Logan, I.R.; Neal, D.E.; Robson, C.N. Regulation of androgen receptor and histone deacetylase 1 by Mdm2-mediated ubiquitylation. Nucleic. Acids Res. 2005, 33, 13–26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Georget, V.; Térouanne, B.; Nicolas, J.-C.; Sultan, C. Mechanism of antiandrogen action: Key role of hsp90 in conformational change and transcriptional activity of the androgen receptor. Biochemistry 2002, 41, 11824–11831. [Google Scholar] [CrossRef]
- Robichaud, N.; del Rincon, S.V.; Huor, B.; Alain, T.; Petruccelli, L.A.; Hearnden, J.; Goncalves, C.; Grotegut, S.; Spruck, C.H.; Furic, L.; et al. Phosphorylation of eIF4E promotes EMT and metastasis via translational control of SNAIL and MMP-3. Oncogene 2015, 34, 2032–2042. [Google Scholar] [CrossRef] [PubMed]
- Cato, L.; de Tribolet-Hardy, J.; Lee, I.; Rottenberg, J.T.; Coleman, I.; Melchers, D.; Houtman, R.; Xiao, T.; Li, W.; Uo, T.; et al. ARv7 Represses Tumor-Suppressor Genes in Castration-Resistant Prostate Cancer. Cancer Cell 2019, 35, 401–413.e6. [Google Scholar] [CrossRef] [PubMed] [Green Version]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thomas, E.; Thankan, R.S.; Purushottamachar, P.; Huang, W.; Kane, M.A.; Zhang, Y.; Ambulos, N.P.; Weber, D.J.; Njar, V.C.O. Novel AR/AR-V7 and Mnk1/2 Degrader, VNPP433-3β: Molecular Mechanisms of Action and Efficacy in AR-Overexpressing Castration Resistant Prostate Cancer In Vitro and In Vivo Models. Cells 2022, 11, 2699. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11172699
Thomas E, Thankan RS, Purushottamachar P, Huang W, Kane MA, Zhang Y, Ambulos NP, Weber DJ, Njar VCO. Novel AR/AR-V7 and Mnk1/2 Degrader, VNPP433-3β: Molecular Mechanisms of Action and Efficacy in AR-Overexpressing Castration Resistant Prostate Cancer In Vitro and In Vivo Models. Cells. 2022; 11(17):2699. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11172699
Chicago/Turabian StyleThomas, Elizabeth, Retheesh S. Thankan, Puranik Purushottamachar, Weiliang Huang, Maureen A. Kane, Yuji Zhang, Nicholas P. Ambulos, David J. Weber, and Vincent C. O. Njar. 2022. "Novel AR/AR-V7 and Mnk1/2 Degrader, VNPP433-3β: Molecular Mechanisms of Action and Efficacy in AR-Overexpressing Castration Resistant Prostate Cancer In Vitro and In Vivo Models" Cells 11, no. 17: 2699. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11172699