

Patient-Derived Pancreatic Cancer Cells Induce C2C12 Myotube Atrophy by Releasing Hsp70 and Hsp90
 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
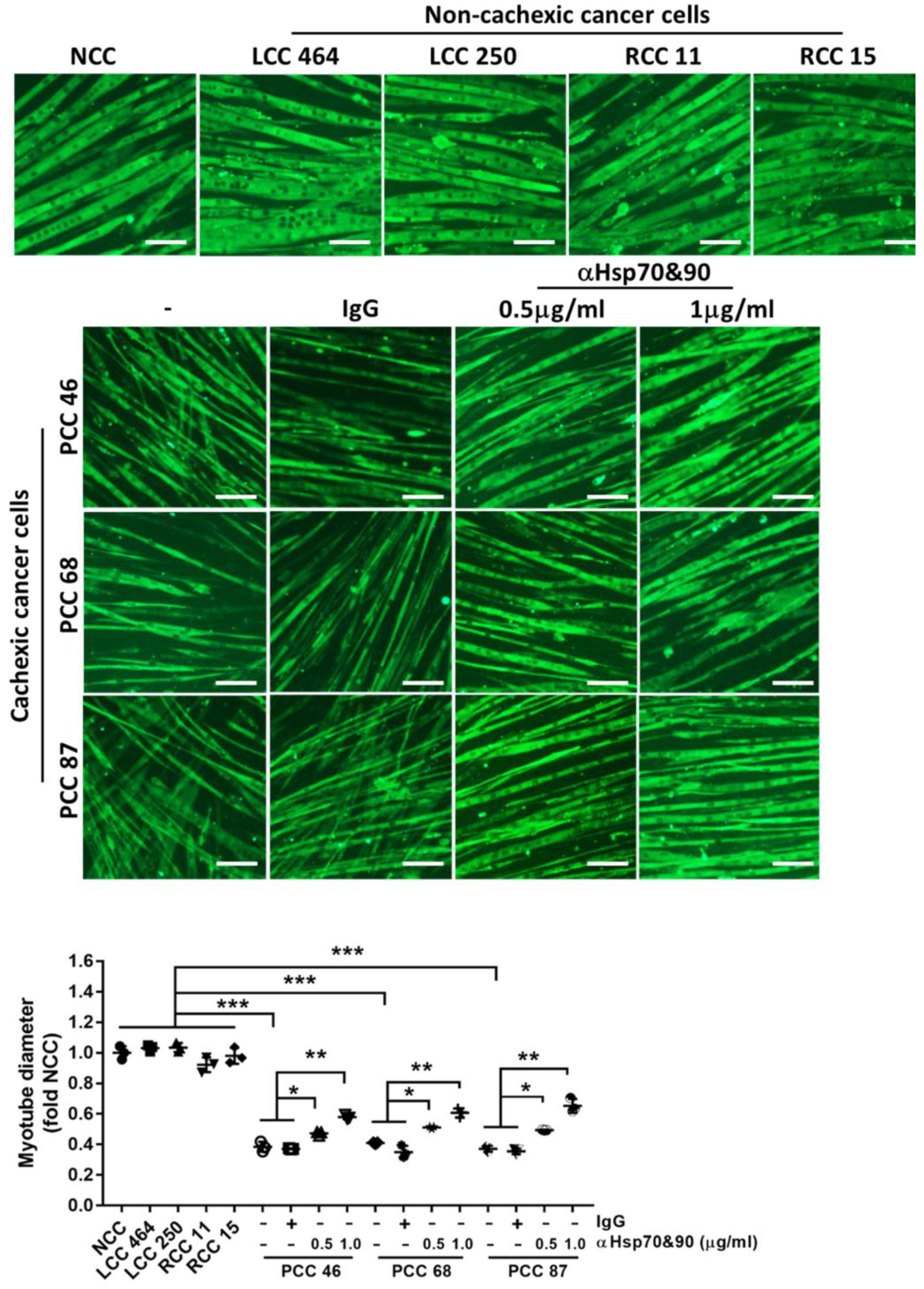{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Cultures
2.2. Quantification of Extracellular Hsp70 and Hsp90
2.3. Extracellular Vesicle (EV) Isolation and Quantitation
2.4. Western Blot Analysis
2.5. Immunoprecipitation
2.6. Fluorescence Microscopy
2.7. Statistical Analyses
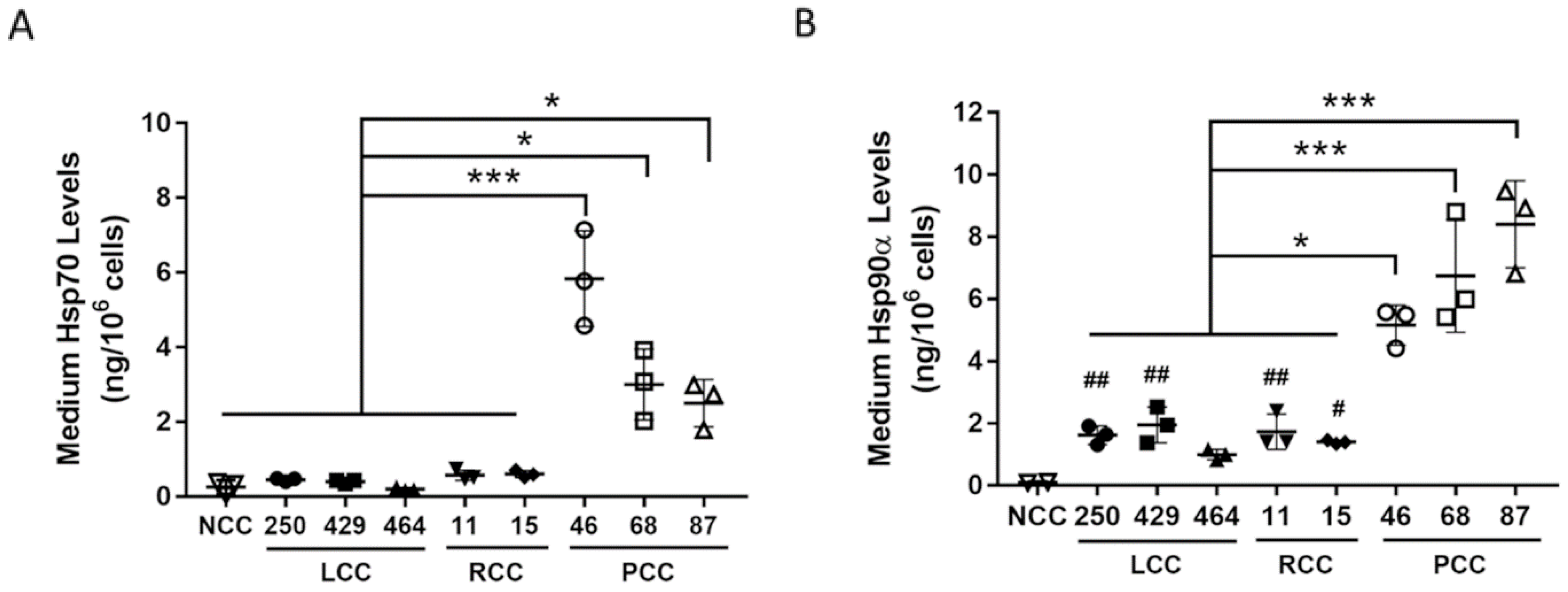
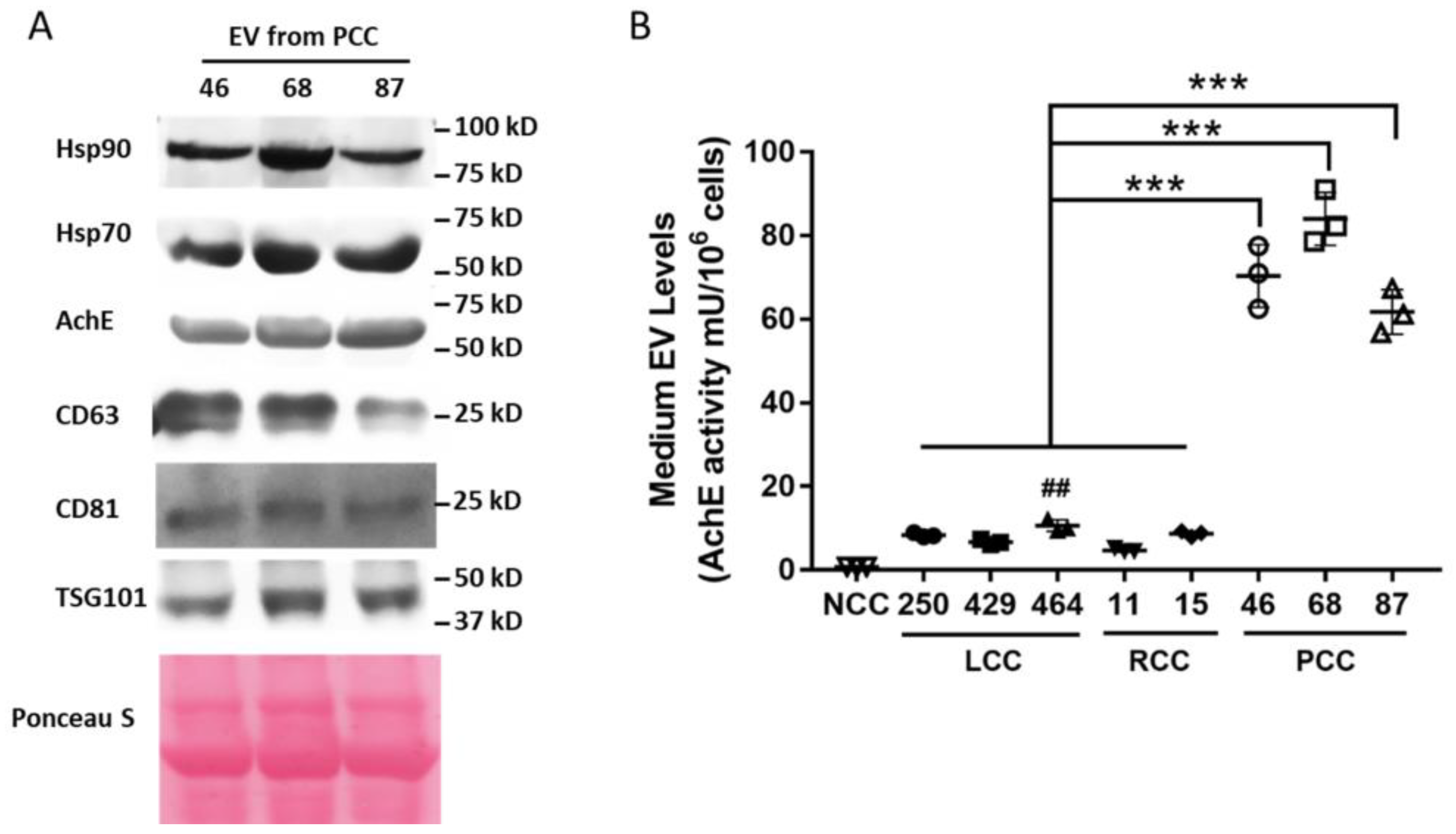
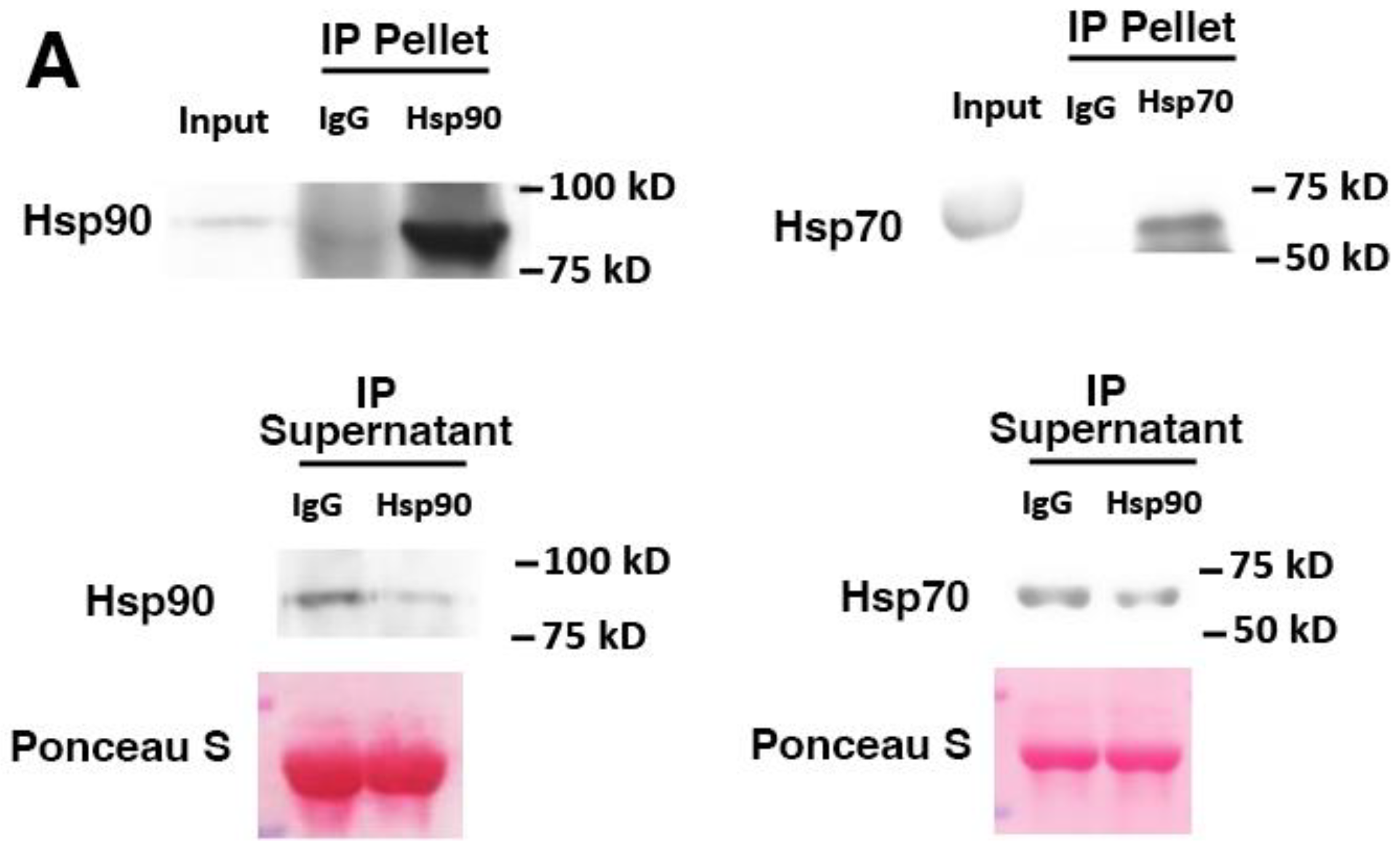
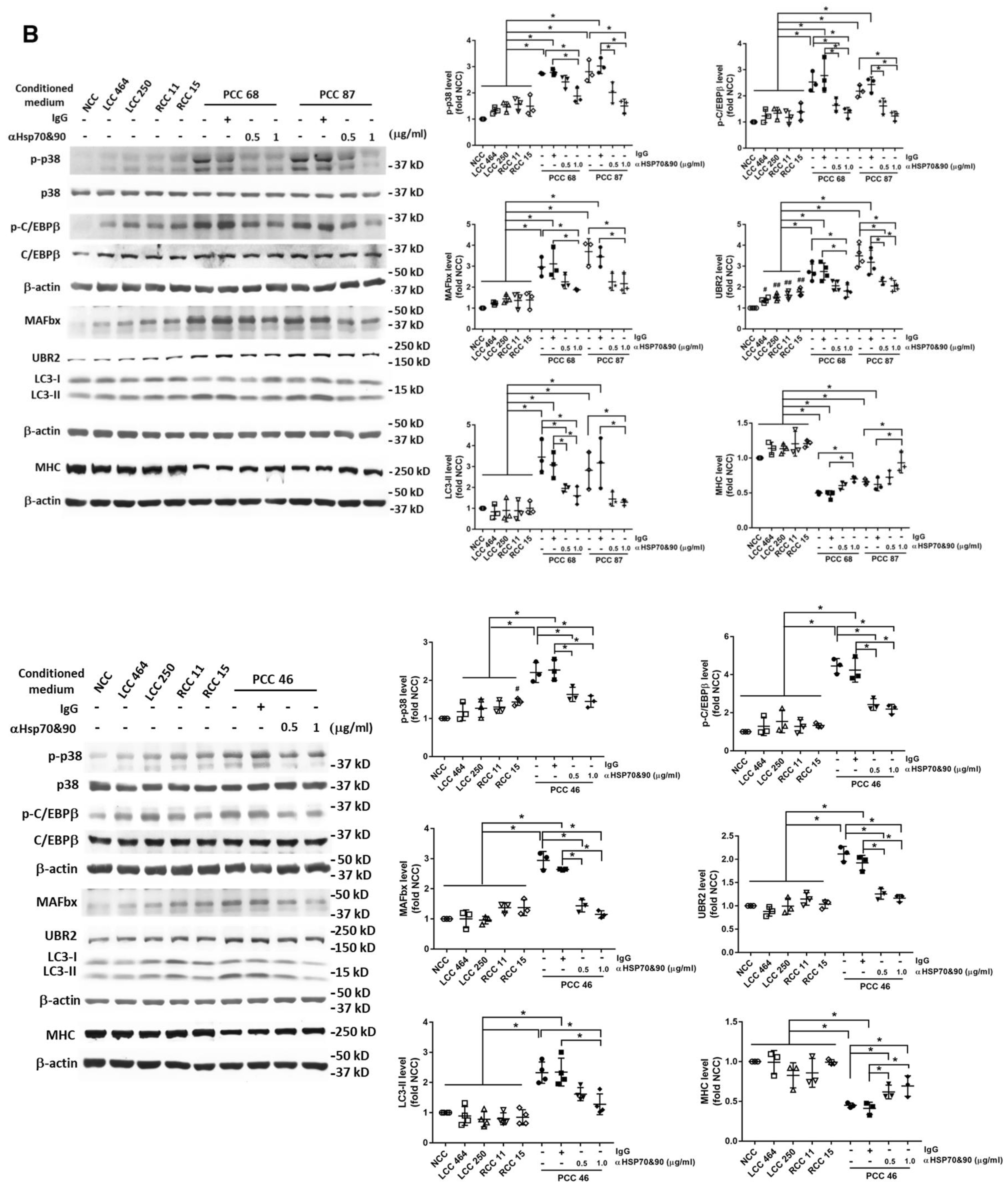
3. Results
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Fuchs, H.E.; Jemal, A. Cancer statistics, 2022. CA Cancer J. Clin. 2022, 72, 7–33. [Google Scholar] [CrossRef] [PubMed]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef]
- Palesty, J.A.; Dudrick, S.J. What we have learned about cachexia in gastrointestinal cancer. Dig. Dis. 2003, 21, 198–213. [Google Scholar] [CrossRef] [PubMed]
- Martignoni, M.E.; Kunze, P.; Friess, H. Cancer cachexia. Mol. Cancer 2003, 2, 36. [Google Scholar] [CrossRef]
- Baracos, V.E.; Martin, L.; Korc, M.; Guttridge, D.C.; Fearon, K.C.H. Cancer-associated cachexia. Nat. Rev. Dis. Primers 2018, 4, 17105. [Google Scholar] [CrossRef] [PubMed]
- Martin, L.; Birdsell, L.; Macdonald, N.; Reiman, T.; Clandinin, M.T.; McCargar, L.J.; Murphy, R.; Ghosh, S.; Sawyer, M.B.; Baracos, V.E. Cancer cachexia in the age of obesity: Skeletal muscle depletion is a powerful prognostic factor, independent of body mass index. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2013, 31, 1539–1547. [Google Scholar] [CrossRef]
- Lee, S.J.; Glass, D.J. Treating cancer cachexia to treat cancer. Skelet Muscle 2011, 1, 2. [Google Scholar] [CrossRef]
- Fearon, K.C.; Glass, D.J.; Guttridge, D.C. Cancer cachexia: Mediators, signaling, and metabolic pathways. Cell Metab. 2012, 16, 153–166. [Google Scholar] [CrossRef]
- Rupert, J.E.; Narasimhan, A.; Jengelley, D.H.A.; Jiang, Y.; Liu, J.; Au, E.; Silverman, L.M.; Sandusky, G.; Bonetto, A.; Cao, S.; et al. Tumor-derived IL-6 and trans-signaling among tumor, fat, and muscle mediate pancreatic cancer cachexia. J. Exp. Med. 2021, 218, e20190450. [Google Scholar] [CrossRef]
- Zhong, X.; Pons, M.; Poirier, C.; Jiang, Y.; Liu, J.; Sandusky, G.E.; Shahda, S.; Nakeeb, A.; Schmidt, C.M.; House, M.G.; et al. The systemic activin response to pancreatic cancer: Implications for effective cancer cachexia therapy. J. Cachexia Sarcopenia Muscle 2019, 10, 1083–1101. [Google Scholar] [CrossRef] [Green Version]
- Talbert, E.E.; Lewis, H.L.; Farren, M.R.; Ramsey, M.L.; Chakedis, J.M.; Rajasekera, P.; Haverick, E.; Sarna, A.; Bloomston, M.; Pawlik, T.M.; et al. Circulating monocyte chemoattractant protein-1 (MCP-1) is associated with cachexia in treatment-naive pancreatic cancer patients. J. Cachexia Sarcopenia Muscle 2018, 9, 358–368. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Anderson, L.J.; Gao, S.; Sin, T.K.; Zhang, Z.; Wu, H.; Jafri, S.H.; Graf, S.A.; Wu, P.C.; Dash, A.; et al. Weight Loss in Cancer Patients Correlates With p38beta MAPK Activation in Skeletal Muscle. Front. Cell Dev. Biol. 2021, 9, 784424. [Google Scholar] [CrossRef] [PubMed]
- Dutta, S.K.; Girotra, M.; Singla, M.; Dutta, A.; Otis Stephen, F.; Nair, P.P.; Merchant, N.B. Serum HSP70: A novel biomarker for early detection of pancreatic cancer. Pancreas 2012, 41, 530–534. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, K.; Ito, Y.; Wakai, K.; Kawado, M.; Hashimoto, S.; Seki, N.; Ando, M.; Nishino, Y.; Kondo, T.; Watanabe, Y.; et al. Serum heat shock protein 70 levels and lung cancer risk: A case-control study nested in a large cohort study. Cancer Epidemiol. Biomark. Prev. 2006, 15, 1733–1737. [Google Scholar] [CrossRef]
- Shi, Y.; Liu, X.; Lou, J.; Han, X.; Zhang, L.; Wang, Q.; Li, B.; Dong, M.; Zhang, Y. Plasma levels of heat shock protein 90 alpha associated with lung cancer development and treatment responses. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2014, 20, 6016–6022. [Google Scholar] [CrossRef]
- Bianchi, M.E. DAMPs, PAMPs and alarmins: All we need to know about danger. J. Leukoc. Biol. 2007, 81, 1–5. [Google Scholar] [CrossRef]
- Zhang, G.; Liu, Z.; Ding, H.; Zhou, Y.; Doan, H.A.; Sin, K.W.T.; Zhu, Z.J.; Flores, R.; Wen, Y.; Gong, X.; et al. Tumor induces muscle wasting in mice through releasing extracellular Hsp70 and Hsp90. Nat. Commun. 2017, 8, 589. [Google Scholar] [CrossRef]
- Liu, Z.; Sin, K.W.T.; Ding, H.; Doan, H.A.; Gao, S.; Miao, H.; Wei, Y.; Wang, Y.; Zhang, G.; Li, Y.-P. p38beta MAPK mediates ULK1-dependent induction of autophagy in skeletal muscle of tumor-bearing mice. Cell Stress 2018, 2, 311–324. [Google Scholar] [CrossRef]
- Zhang, G.; Liu, Z.; Ding, H.; Miao, H.; Garcia, J.M.; Li, Y.P. Toll-like receptor 4 mediates Lewis lung carcinoma-induced muscle wasting via coordinate activation of protein degradation pathways. Sci. Rep. 2017, 7, 2273. [Google Scholar] [CrossRef]
- Ding, H.; Zhang, G.; Sin, K.W.; Liu, Z.; Lin, R.K.; Li, M.; Li, Y.P. Activin A induces skeletal muscle catabolism via p38beta mitogen-activated protein kinase. J. Cachexia Sarcopenia Muscle 2017, 8, 202–212. [Google Scholar] [CrossRef]
- Yang, J.; Zhang, Z.; Zhang, Y.; Ni, X.; Zhang, G.; Cui, X.; Liu, M.; Xu, C.; Zhang, Q.; Zhu, H.; et al. ZIP4 Promotes Muscle Wasting and Cachexia in Mice With Orthotopic Pancreatic Tumors by Stimulating RAB27B-Regulated Release of Extracellular Vesicles From Cancer Cells. Gastroenterology 2019, 156, 722–734 e726. [Google Scholar] [CrossRef] [PubMed]
- Hidalgo, M.; Amant, F.; Biankin, A.V.; Budinska, E.; Byrne, A.T.; Caldas, C.; Clarke, R.B.; de Jong, S.; Jonkers, J.; Maelandsmo, G.M.; et al. Patient-derived xenograft models: An emerging platform for translational cancer research. Cancer Discov. 2014, 4, 998–1013. [Google Scholar] [CrossRef] [PubMed]
- Pham, K.; Delitto, D.; Knowlton, A.E.; Hartlage, E.R.; Madhavan, R.; Gonzalo, D.H.; Thomas, R.M.; Behrns, K.E.; George, T.J., Jr.; Hughes, S.J.; et al. Isolation of Pancreatic Cancer Cells from a Patient-Derived Xenograft Model Allows for Practical Expansion and Preserved Heterogeneity in Culture. Am. J. Pathol. 2016, 186, 1537–1546. [Google Scholar] [CrossRef] [PubMed]
- Delitto, D.; Judge, S.M.; Delitto, A.E.; Nosacka, R.L.; Rocha, F.G.; DiVita, B.B.; Gerber, M.H.; George, T.J., Jr.; Behrns, K.E.; Hughes, S.J.; et al. Human pancreatic cancer xenografts recapitulate key aspects of cancer cachexia. Oncotarget 2017, 8, 1177–1189. [Google Scholar] [CrossRef]
- Valente, M.J.; Henrique, R.; Costa, V.L.; Jeronimo, C.; Carvalho, F.; Bastos, M.L.; de Pinho, P.G.; Carvalho, M. A rapid and simple procedure for the establishment of human normal and cancer renal primary cell cultures from surgical specimens. PLoS ONE 2011, 6, e19337. [Google Scholar] [CrossRef]
- Chen, Y.; Zhang, R.; Wang, L.; Correa, A.M.; Pataer, A.; Xu, Y.; Zhang, X.; Ren, C.; Wu, S.; Meng, Q.H.; et al. Tumor characteristics associated with engraftment of patient-derived non-small cell lung cancer xenografts in immunocompromised mice. Cancer 2019, 125, 3738–3748. [Google Scholar] [CrossRef]
- Zhang, X.; Zhang, R.; Chen, H.; Wang, L.; Ren, C.; Pataer, A.; Wu, S.; Meng, Q.H.; Ha, M.J.; Morris, J.; et al. KRT-232 and navitoclax enhance trametinib’s anti-Cancer activity in non-small cell lung cancer patient-derived xenografts with KRAS mutations. Am. J. Cancer Res. 2020, 10, 4464–4475. [Google Scholar]
- Taylor, D.D.; Zacharias, W.; Gercel-Taylor, C. Exosome isolation for proteomic analyses and RNA profiling. Methods Mol. Biol. 2011, 728, 235–246. [Google Scholar] [CrossRef]
- Zhang, G.; Jin, B.; Li, Y.P. C/EBPbeta mediates tumour-induced ubiquitin ligase atrogin1/MAFbx upregulation and muscle wasting. EMBO J. 2011, 30, 4323–4335. [Google Scholar] [CrossRef]
- Doyle, A.; Zhang, G.; Abdel Fattah, E.A.; Eissa, N.T.; Li, Y.P. Toll-like receptor 4 mediates lipopolysaccharide-induced muscle catabolism via coordinate activation of ubiquitin-proteasome and autophagy-lysosome pathways. FASEB J. 2011, 25, 99–110. [Google Scholar] [CrossRef]
- Pu, X.; Zhang, R.; Wang, L.; Chen, Y.; Xu, Y.; Pataer, A.; Meraz, I.M.; Zhang, X.; Wu, S.; Wu, L.; et al. Patient-derived tumor immune microenvironments in patient-derived xenografts of lung cancer. J. Transl. Med. 2018, 16, 328. [Google Scholar] [CrossRef] [PubMed]
- Chalmin, F.; Ladoire, S.; Mignot, G.; Vincent, J.; Bruchard, M.; Remy-Martin, J.P.; Boireau, W.; Rouleau, A.; Simon, B.; Lanneau, D.; et al. Membrane-associated Hsp72 from tumor-derived exosomes mediates STAT3-dependent immunosuppressive function of mouse and human myeloid-derived suppressor cells. J. Clin. Investig. 2010, 120, 457–471. [Google Scholar] [CrossRef] [PubMed]
- McCready, J.; Sims, J.D.; Chan, D.; Jay, D.G. Secretion of extracellular hsp90alpha via exosomes increases cancer cell motility: A role for plasminogen activation. BMC Cancer 2010, 10, 294. [Google Scholar] [CrossRef]
- Lotvall, J.; Hill, A.F.; Hochberg, F.; Buzas, E.I.; Di Vizio, D.; Gardiner, C.; Gho, Y.S.; Kurochkin, I.V.; Mathivanan, S.; Quesenberry, P.; et al. Minimal experimental requirements for definition of extracellular vesicles and their functions: A position statement from the International Society for Extracellular Vesicles. J. Extracell. Vesicles 2014, 3, 26913. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Li, Y.P. p38beta MAPK upregulates atrogin1/MAFbx by specific phosphorylation of C/EBPbeta. Skelet Muscle 2012, 2, 20. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Lin, R.K.; Kwon, Y.T.; Li, Y.P. Signaling mechanism of tumor cell-induced up-regulation of E3 ubiquitin ligase UBR2. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2013, 27, 2893–2901. [Google Scholar] [CrossRef] [PubMed]
- Rong, B.; Zhao, C.; Liu, H.; Ming, Z.; Cai, X.; Gao, W.; Yang, S. Identification and verification of Hsp90-beta as a potential serum biomarker for lung cancer. Am. J. Cancer Res. 2014, 4, 874–885. [Google Scholar]
- Balazs, M.; Zsolt, H.; Laszlo, G.; Gabriella, G.; Lilla, T.; Gyula, O.; Balazs, D.; Eva, M.; Zoltan, B.; Zoltan, P.; et al. Serum Heat Shock Protein 70, as a Potential Biomarker for Small Cell Lung Cancer. Pathol. Oncol. Res. 2016, 23, 377–383. [Google Scholar] [CrossRef]
- Puppa, M.J.; Gao, S.; Narsale, A.A.; Carson, J.A. Skeletal muscle glycoprotein 130’s role in Lewis lung carcinoma-induced cachexia. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2014, 28, 998–1009. [Google Scholar] [CrossRef]
- Li, Y.P.; Chen, Y.; John, J.; Moylan, J.; Jin, B.; Mann, D.L.; Reid, M.B. TNF-alpha acts via p38 MAPK to stimulate expression of the ubiquitin ligase atrogin1/MAFbx in skeletal muscle. FASEB J. 2005, 19, 362–370. [Google Scholar] [CrossRef]
- Li, W.; Moylan, J.S.; Chambers, M.A.; Smith, J.; Reid, M.B. Interleukin-1 stimulates catabolism in C2C12 myotubes. Am. J. Physiol. Cell Physiol. 2009, 297, C706–C714. [Google Scholar] [CrossRef] [PubMed]
- Lucas, K.; Maes, M. Role of the Toll Like receptor (TLR) radical cycle in chronic inflammation: Possible treatments targeting the TLR4 pathway. Mol. Neurobiol. 2013, 48, 190–204. [Google Scholar] [CrossRef] [PubMed]
- Carson, J.A.; Hardee, J.P.; VanderVeen, B.N. The emerging role of skeletal muscle oxidative metabolism as a biological target and cellular regulator of cancer-induced muscle wasting. Semin. Cell Dev. Biol. 2016, 54, 53–67. [Google Scholar] [CrossRef] [PubMed]
- Jin, B.; Li, Y.P. Curcumin prevents lipopolysaccharide-induced atrogin-1/MAFbx upregulation and muscle mass loss. J. Cell. Biochem. 2007, 100, 960–969. [Google Scholar] [CrossRef]
- McClung, J.M.; Judge, A.R.; Powers, S.K.; Yan, Z. p38 MAPK links oxidative stress to autophagy-related gene expression in cachectic muscle wasting. Am. J. Physiol. Cell Physiol. 2010, 298, C542–C549. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, H.-Y.; Trevino, J.G.; Fang, B.-L.; Riner, A.N.; Vudatha, V.; Zhang, G.-H.; Li, Y.-P. Patient-Derived Pancreatic Cancer Cells Induce C2C12 Myotube Atrophy by Releasing Hsp70 and Hsp90. Cells 2022, 11, 2756. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11172756
Wu H-Y, Trevino JG, Fang B-L, Riner AN, Vudatha V, Zhang G-H, Li Y-P. Patient-Derived Pancreatic Cancer Cells Induce C2C12 Myotube Atrophy by Releasing Hsp70 and Hsp90. Cells. 2022; 11(17):2756. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11172756
Chicago/Turabian StyleWu, Hong-Yu, Jose G. Trevino, Bing-Liang Fang, Andrea N. Riner, Vignesh Vudatha, Guo-Hua Zhang, and Yi-Ping Li. 2022. "Patient-Derived Pancreatic Cancer Cells Induce C2C12 Myotube Atrophy by Releasing Hsp70 and Hsp90" Cells 11, no. 17: 2756. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11172756