

HIV-Associated Insults Modulate ADAM10 and Its Regulator Sirtuin1 in an NMDA Receptor-Dependent Manner
 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Dissection and Maintenance Primary Rat Neuronal Cultures
2.2. HIV/MDM Generation
2.3. Neuronal Culture Treatments
2.4. Immunoblotting
2.5. Immunohistochemistry
2.6. Statistical Analysis
3. Results
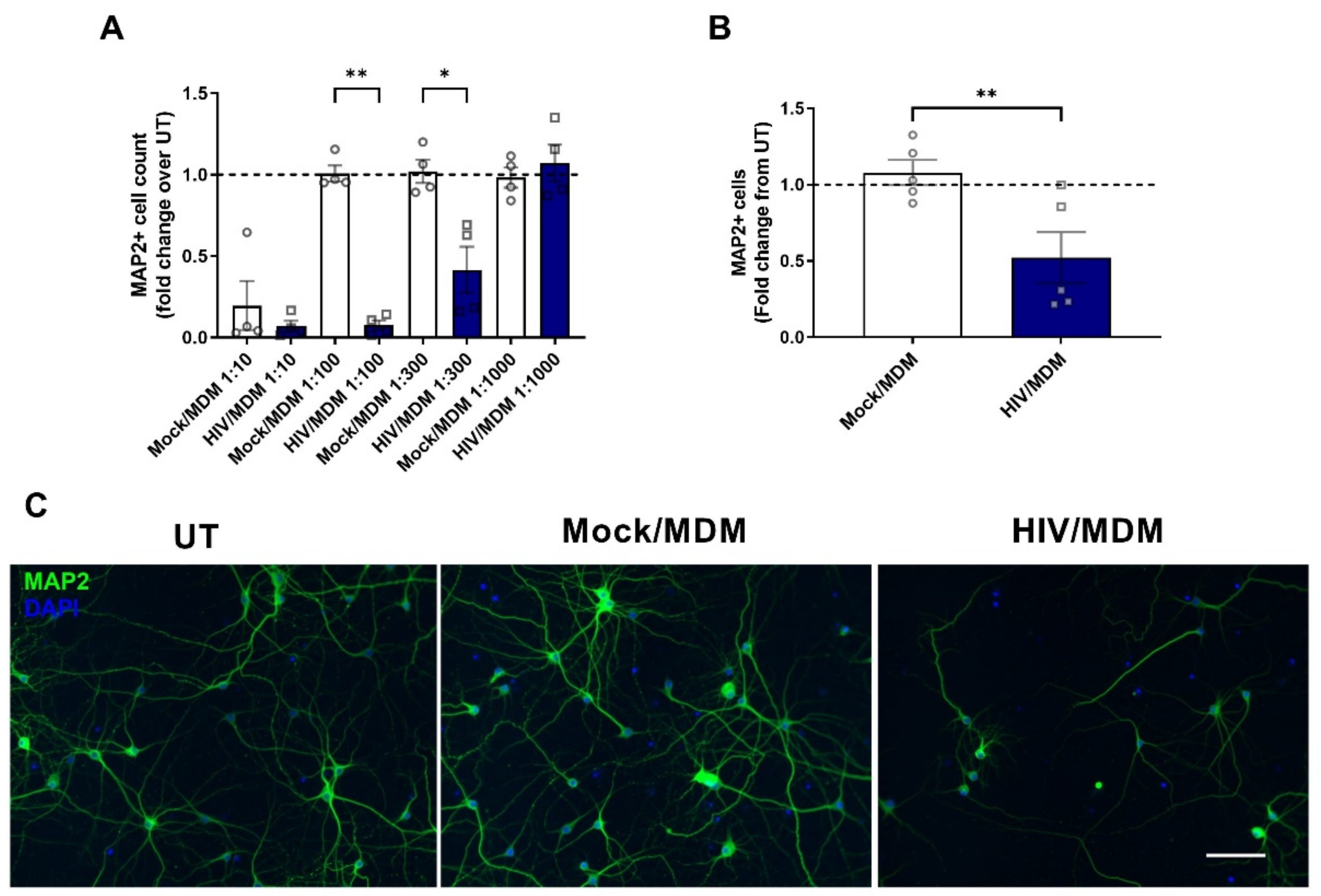
3.1. HIV/MDM Supernatants Lead to Neurotoxicity in Rat Neuronal Cultures
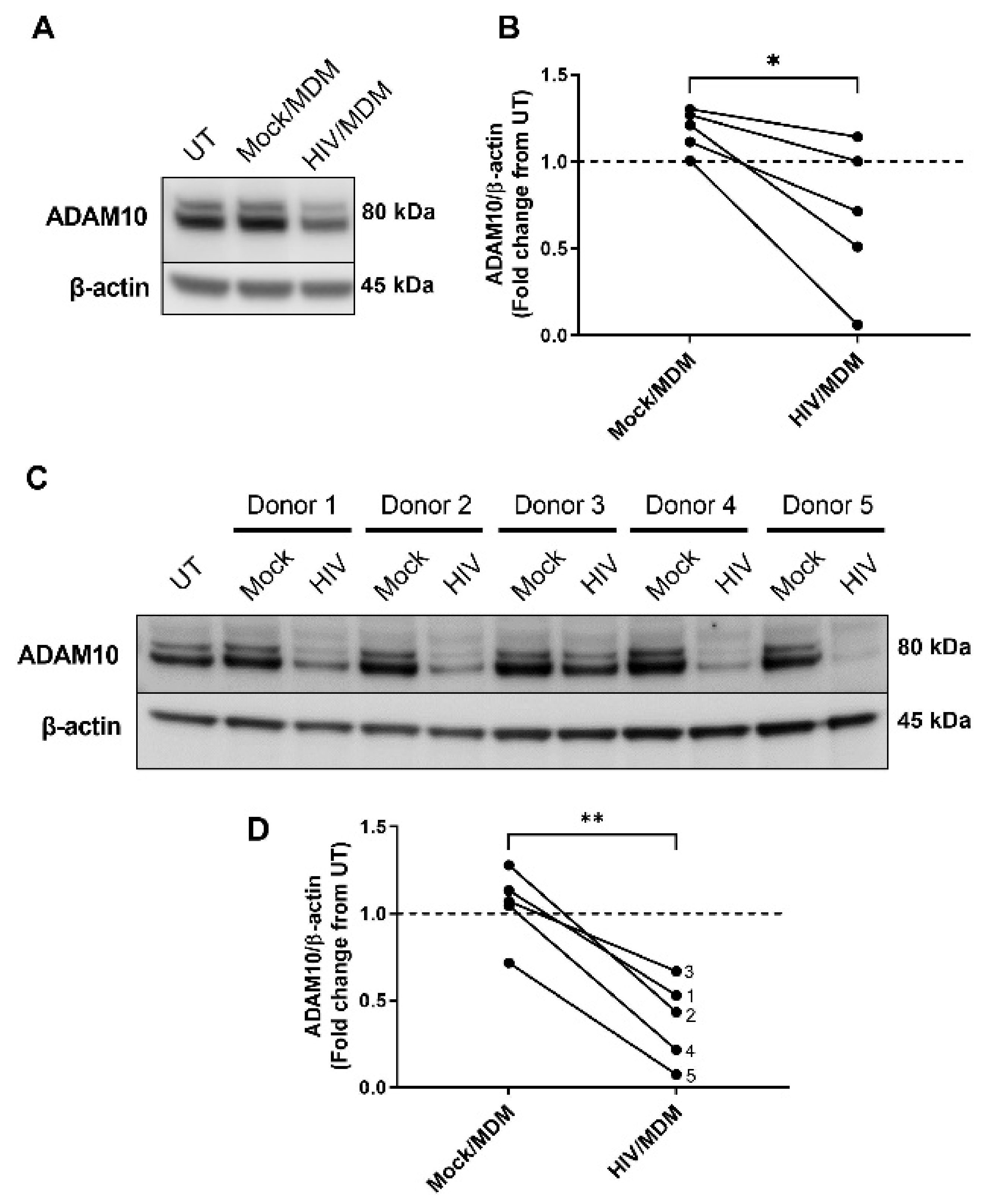
3.2. HIV/MDMs Reduce Levels of ADAM10 Protein
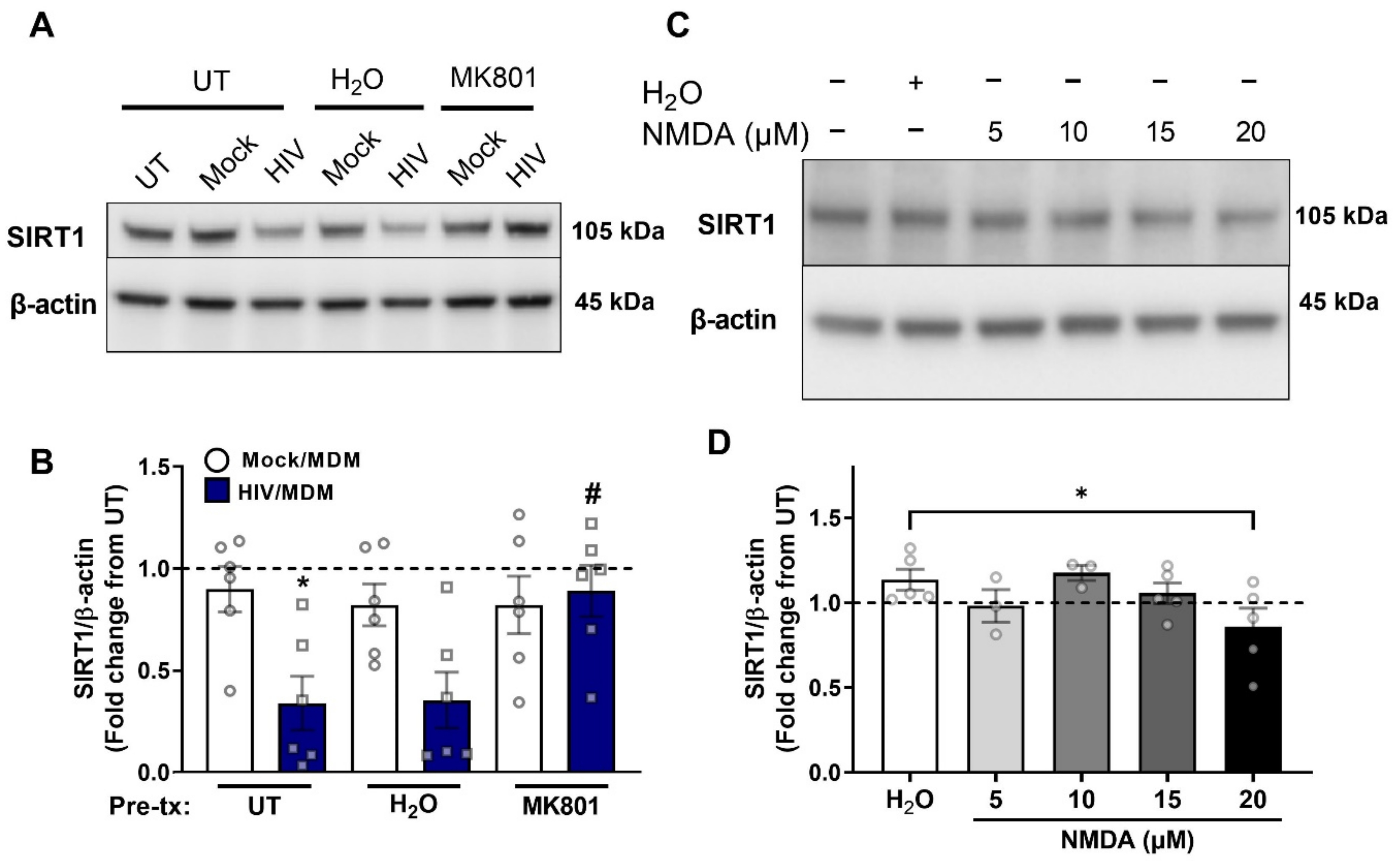
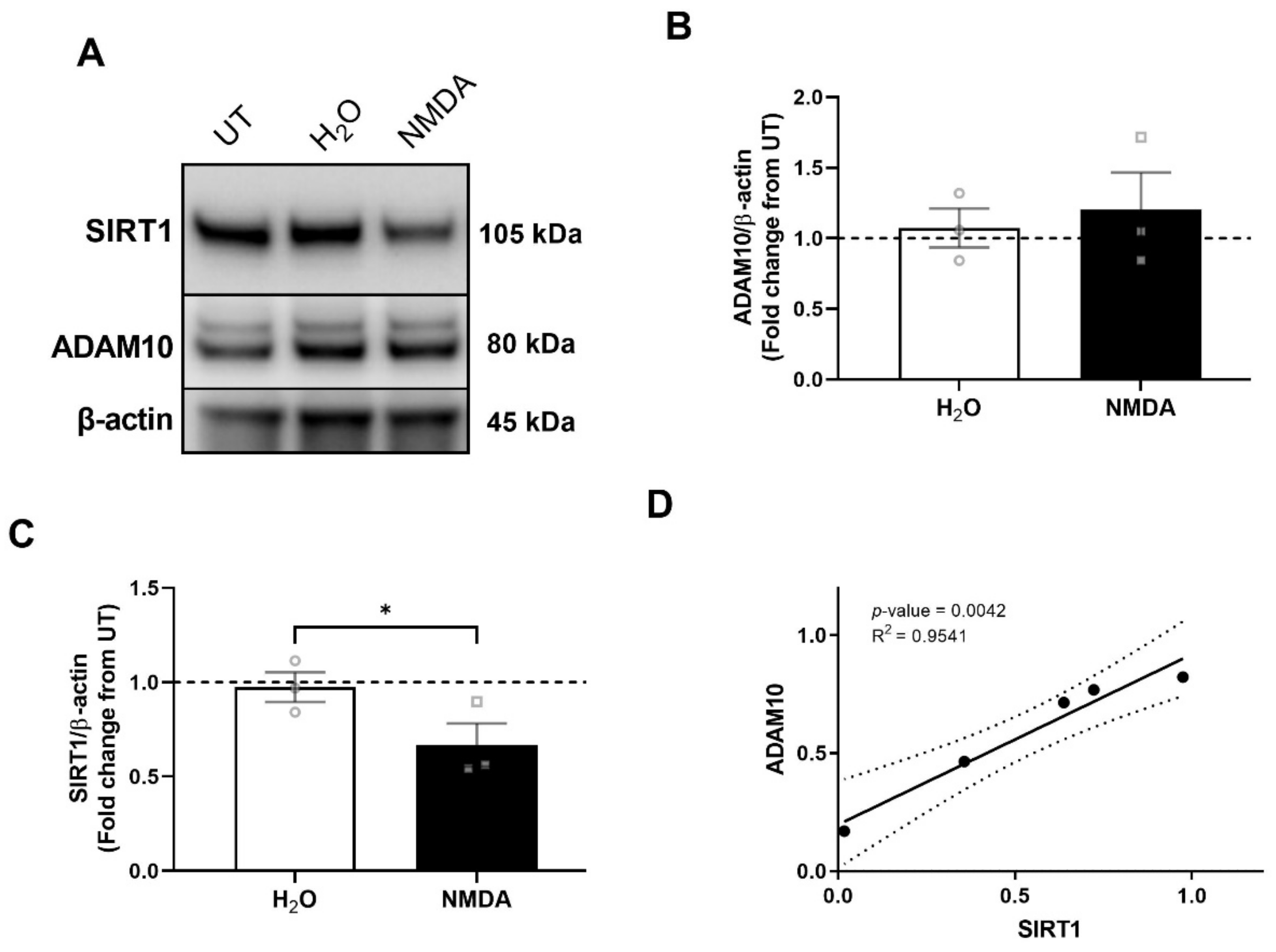
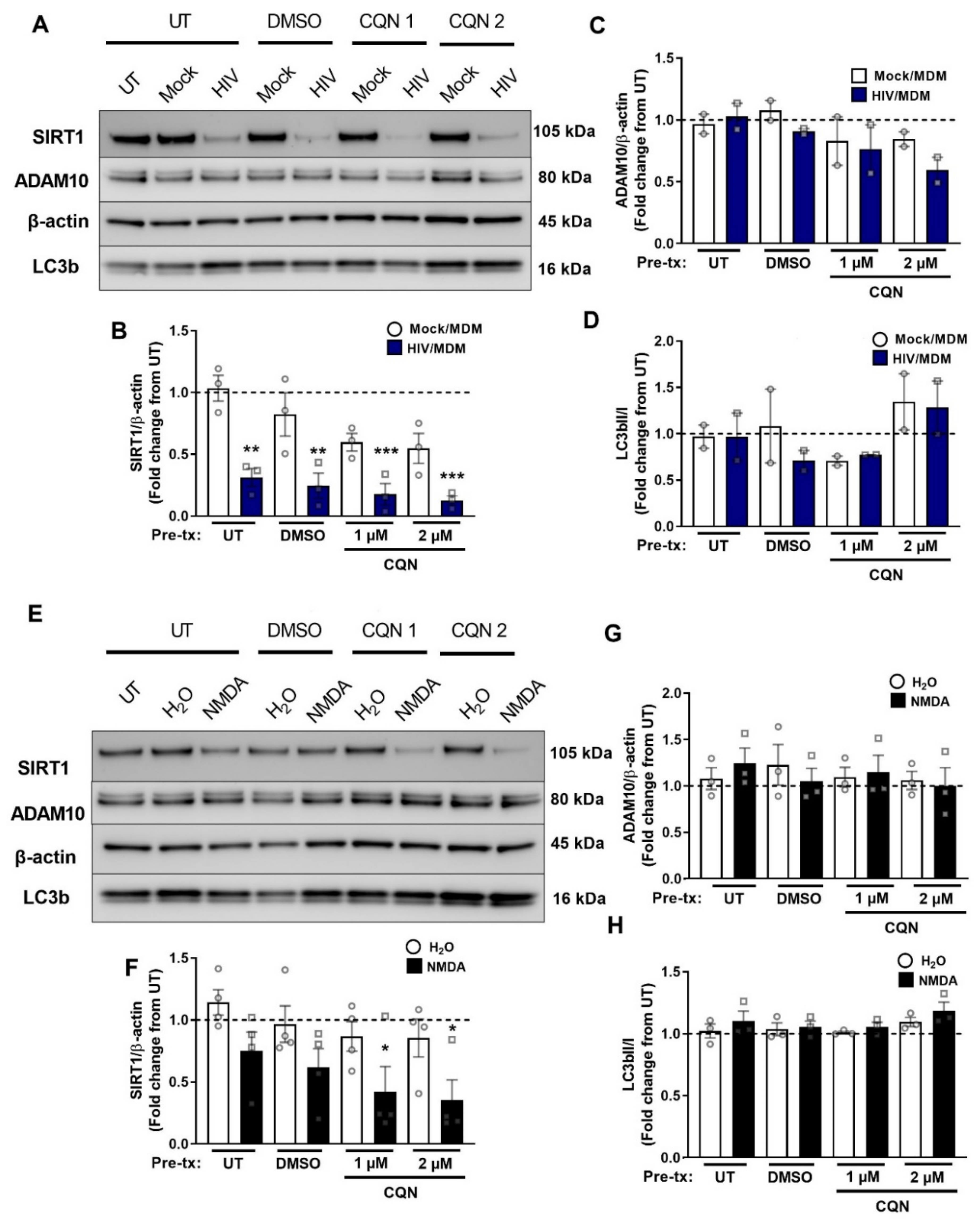
3.3. NMDA Receptor Activation Is Necessary and Sufficient for ADAM10 Decrease by HIV/MDMs
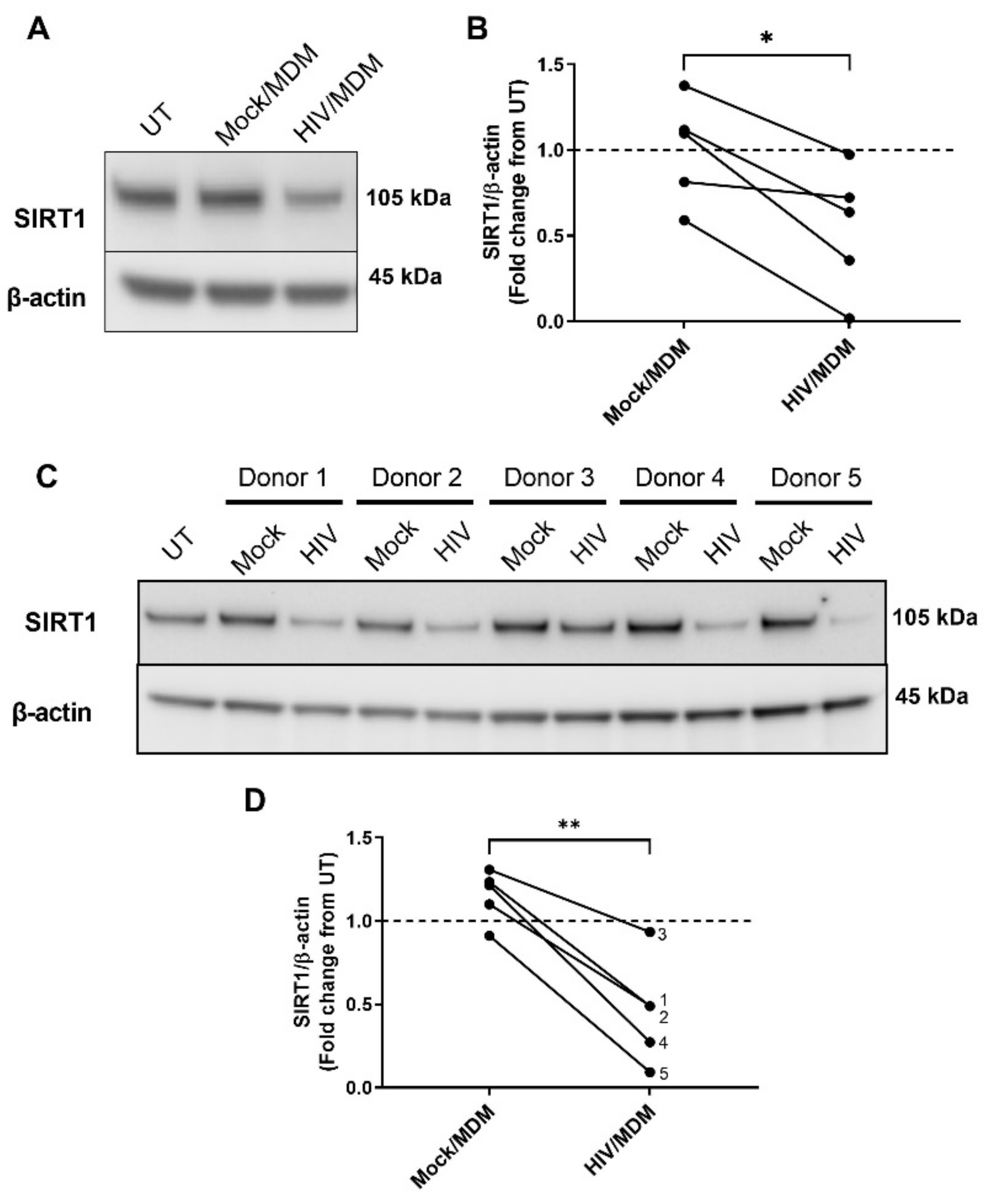
3.4. HIV/MDMs Reduce Levels of the ADAM10 Regulator SIRT1
3.5. NMDA Receptor Activation Is Necessary and Sufficient for SIRT1 Decrease by HIV/MDMs
3.6. SIRT1 Decrease Precedes ADAM10 Decrease
3.7. Autophagy Inhibitors Do Not Block Early SIRT1 Decrease by HIV/MDMs
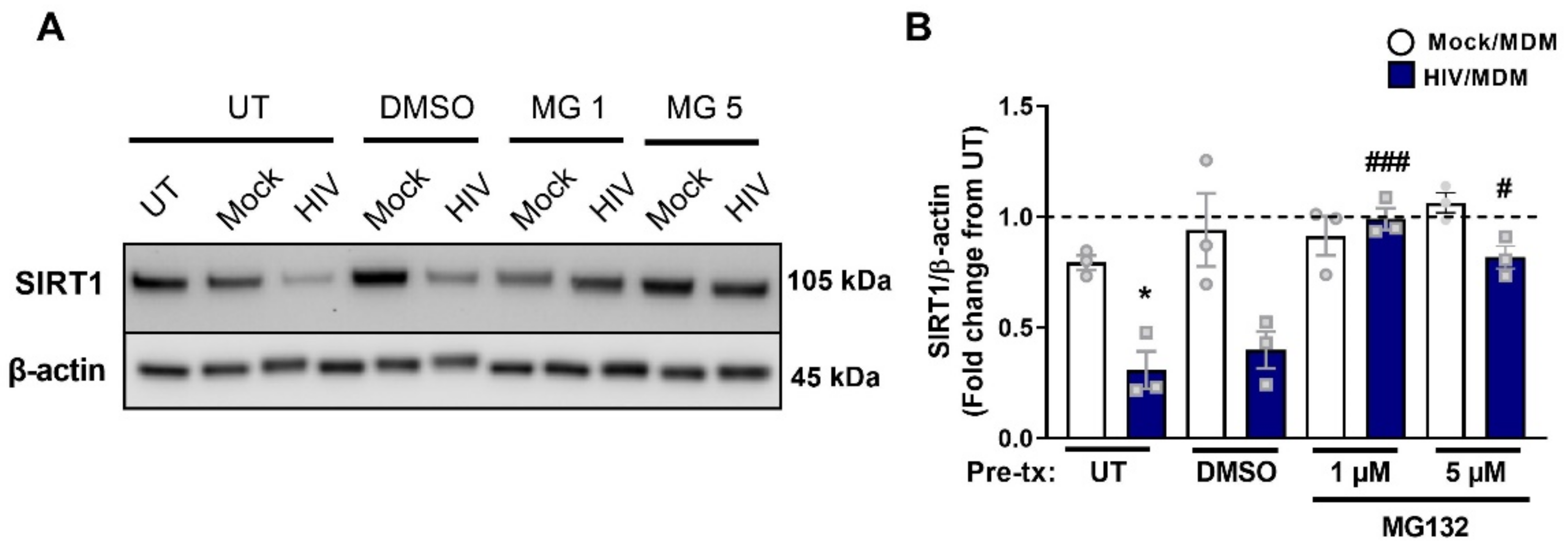
3.8. Early SIRT1 Decrease in Response to HIV/MDM Supernatant Treatment Occurs via Proteasomal Degradation
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Heaton, R.K.; Clifford, D.B.; Franklin, D.R., Jr.; Woods, S.P.; Ake, C.; Vaida, F.; Ellis, R.J.; Letendre, S.L.; Marcotte, T.D.; Atkinson, J.H.; et al. HIV-associated neurocognitive disorders persist in the era of potent antiretroviral therapy: CHARTER Study. Neurology 2010, 75, 2087–2096. [Google Scholar] [CrossRef] [PubMed]
- Heaton, R.K.; Franklin, D.R., Jr.; Deutsch, R.; Letendre, S.; Ellis, R.J.; Casaletto, K.; Marquine, M.J.; Woods, S.P.; Vaida, F.; Atkinson, J.H.; et al. Neurocognitive change in the era of HIV combination antiretroviral therapy: The longitudinal CHARTER study. Clin. Infect. Dis. 2015, 60, 473–480. [Google Scholar] [CrossRef] [PubMed]
- Heaton, R.K.; Franklin, D.R.; Ellis, R.J.; McCutchan, J.A.; Letendre, S.L.; Leblanc, S.; Corkran, S.H.; Duarte, N.A.; Clifford, D.B.; Woods, S.P.; et al. HIV-associated neurocognitive disorders before and during the era of combination antiretroviral therapy: Differences in rates, nature, and predictors. J. Neurovirol. 2011, 17, 3–16. [Google Scholar] [CrossRef] [PubMed]
- Ferrarese, C.; Aliprandi, A.; Tremolizzo, L.; Stanzani, L.; De Micheli, A.; Dolara, A.; Frattola, L. Increased glutamate in CSF and plasma of patients with HIV dementia. Neurology 2001, 57, 671–675. [Google Scholar] [CrossRef] [PubMed]
- Kaul, M.; Garden, G.A.; Lipton, S.A. Pathways to neuronal injury and apoptosis in HIV-associated dementia. Nature. 2001, 410, 988–994. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, L.; Cho, H.J.; Toborek, M. Blood-brain barrier pericytes as a target for HIV-1 infection. Brain 2019, 142, 502–511. [Google Scholar] [CrossRef]
- Nakagawa, S.; Castro, V.; Toborek, M. Infection of human pericytes by HIV-1 disrupts the integrity of the blood-brain barrier. J. Cell. Mol. Med. 2012, 16, 2950–2957. [Google Scholar] [CrossRef]
- Lindl, K.A.; Marks, D.R.; Kolson, D.L.; Jordan-Sciutto, K.L. HIV-associated neurocognitive disorder: Pathogenesis and therapeutic opportunities. J. Neuroimmune Pharmacol. 2010, 5, 294–309. [Google Scholar] [CrossRef]
- Kaul, M.; Lipton, S.A. Mechanisms of neuroimmunity and neurodegeneration associated with HIV-1 infection and AIDS. J. Neuroimmune Pharmacol. 2006, 1, 138–151. [Google Scholar] [CrossRef]
- Kaul, M.; Zheng, J.; Okamoto, S.; Gendelman, H.E.; Lipton, S.A. HIV-1 infection and AIDS: Consequences for the central nervous system. Cell Death Differ. 2005, 12 (Suppl. S1), 878–892. [Google Scholar] [CrossRef] [Green Version]
- Akay, C.; Cooper, M.; Odeleye, A.; Jensen, B.K.; White, M.G.; Vassoler, F.; Gannon, P.J.; Mankowski, J.; Dorsey, J.L.; Buch, A.M.; et al. Antiretroviral drugs induce oxidative stress and neuronal damage in the central nervous system. J. Neurovirol. 2014, 20, 39–53. [Google Scholar] [CrossRef]
- Stern, A.L.; Lee, R.N.; Panvelker, N.; Li, J.; Harowitz, J.; Jordan-Sciutto, K.L.; Akay-Espinoza, C. Differential Effects of Antiretroviral Drugs on Neurons In Vitro: Roles for Oxidative Stress and Integrated Stress Response. J. Neuroimmune Pharmacol. 2017, 13, 64–76. [Google Scholar] [CrossRef]
- Canet, G.; Dias, C.; Gabelle, A.; Simonin, Y.; Gosselet, F.; Marchi, N.; Makinson, A.; Tuaillon, E.; Van de Perre, P.; Givalois, L.; et al. HIV Neuroinfection and Alzheimer’s Disease: Similarities and Potential Links? Front. Cell Neurosci. 2018, 12, 307. [Google Scholar] [CrossRef]
- Brew, B.J.; Pemberton, L.; Blennow, K.; Wallin, A.; Hagberg, L. CSF amyloid beta42 and tau levels correlate with AIDS dementia complex. Neurology 2005, 65, 1490–1492. [Google Scholar] [CrossRef]
- Green, D.A.; Masliah, E.; Vinters, H.V.; Beizai, P.; Moore, D.J.; Achim, C.L. Brain deposition of beta-amyloid is a common pathologic feature in HIV positive patients. AIDS 2005, 19, 407–411. [Google Scholar] [CrossRef]
- Clifford, D.B.; Fagan, A.M.; Holtzman, D.M.; Morris, J.C.; Teshome, M.; Shah, A.R.; Kauwe, J.S. CSF biomarkers of Alzheimer disease in HIV-associated neurologic disease. Neurology 2009, 73, 1982–1987. [Google Scholar] [CrossRef]
- Soontornniyomkij, V.; Moore, D.J.; Gouaux, B.; Soontornniyomkij, B.; Tatro, E.T.; Umlauf, A.; Masliah, E.; Levine, A.J.; Singer, E.J.; Vinters, H.V.; et al. Cerebral beta-amyloid deposition predicts HIV-associated neurocognitive disorders in APOE epsilon4 carriers. AIDS 2012, 26, 2327–2335. [Google Scholar] [CrossRef]
- Stern, A.L.; Ghura, S.; Gannon, P.J.; Akay-Espinoza, C.; Phan, J.M.; Yee, A.C.; Vassar, R.; Gelman, B.B.; Kolson, D.L.; Jordan-Sciutto, K.L. BACE1 Mediates HIV-Associated and Excitotoxic Neuronal Damage Through an APP-Dependent Mechanism. J. Neurosci. 2018, 38, 4288–4300. [Google Scholar] [CrossRef]
- Umlauf, A.; Soontornniyomkij, B.; Sundermann, E.E.; Gouaux, B.; Ellis, R.J.; Levine, A.J.; Moore, D.J.; Soontornniyomkij, V. Risk of developing cerebral beta-amyloid plaques with posttranslational modification among HIV-infected adults. AIDS 2019, 33, 2157–2166. [Google Scholar] [CrossRef]
- Ortega, M.; Ances, B.M. Role of HIV in amyloid metabolism. J. Neuroimmune Pharmacol. 2014, 9, 483–491. [Google Scholar] [CrossRef]
- Soontornniyomkij, V.; Moore, D.J.; Gouaux, B.; Soontornniyomkij, B.; Sinsheimer, J.S.; Levine, A.J. Associations of regional amyloid-beta plaque and phospho-tau pathology with biological factors and neuropsychological functioning among HIV-infected adults. J. Neurovirol. 2019, 25, 741–753. [Google Scholar] [CrossRef]
- Kodidela, S.; Gerth, K.; Haque, S.; Gong, Y.; Ismael, S.; Singh, A.; Tauheed, I.; Kumar, S. Extracellular Vesicles: A Possible Link between HIV and Alzheimer’s Disease-Like Pathology in HIV Subjects? Cells 2019, 8, 968. [Google Scholar] [CrossRef]
- Cho, Y.E.; Lee, M.H.; Song, B.J. Neuronal Cell Death and Degeneration through Increased Nitroxidative Stress and Tau Phosphorylation in HIV-1 Transgenic Rats. PLoS ONE 2017, 12, e0169945. [Google Scholar] [CrossRef]
- Gannon, P.J.; Akay-Espinoza, C.; Yee, A.C.; Briand, L.A.; Erickson, M.A.; Gelman, B.B.; Gao, Y.; Haughey, N.J.; Zink, M.C.; Clements, J.E.; et al. HIV Protease Inhibitors Alter Amyloid Precursor Protein Processing via beta-Site Amyloid Precursor Protein Cleaving Enzyme-1 Translational Up-Regulation. Am. J. Pathol. 2017, 187, 91–109. [Google Scholar] [CrossRef]
- Sil, S.; Hu, G.; Liao, K.; Niu, F.; Callen, S.; Periyasamy, P.; Fox, H.S.; Buch, S. HIV-1 Tat-mediated astrocytic amyloidosis involves the HIF-1alpha/lncRNA BACE1-AS axis. PLoS Biol. 2020, 18, e3000660. [Google Scholar] [CrossRef]
- Rempel, H.C.; Pulliam, L. HIV-1 Tat inhibits neprilysin and elevates amyloid beta. AIDS 2005, 19, 127–135. [Google Scholar] [CrossRef]
- Kuhn, P.H.; Wang, H.; Dislich, B.; Colombo, A.; Zeitschel, U.; Ellwart, J.W.; Kremmer, E.; Rossner, S.; Lichtenthaler, S.F. ADAM10 is the physiologically relevant, constitutive alpha-secretase of the amyloid precursor protein in primary neurons. EMBO J. 2010, 29, 3020–3032. [Google Scholar] [CrossRef]
- Obregon, D.; Hou, H.; Deng, J.; Giunta, B.; Tian, J.; Darlington, D.; Shahaduzzaman, M.; Zhu, Y.; Mori, T.; Mattson, M.P.; et al. Soluble amyloid precursor protein-alpha modulates beta-secretase activity and amyloid-beta generation. Nat. Commun. 2012, 3, 777. [Google Scholar] [CrossRef]
- Dar, N.J.; Glazner, G.W. Deciphering the neuroprotective and neurogenic potential of soluble amyloid precursor protein alpha (sAPPalpha). Cell Mol. Life Sci. 2020, 77, 2315–2330. [Google Scholar] [CrossRef]
- Fukumoto, H.; Cheung, B.S.; Hyman, B.T.; Irizarry, M.C. Beta-secretase protein and activity are increased in the neocortex in Alzheimer disease. Arch. Neurol. 2002, 59, 1381–1389. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.B.; Lindholm, K.; Yan, R.; Citron, M.; Xia, W.; Yang, X.L.; Beach, T.; Sue, L.; Wong, P.; Price, D.; et al. Elevated beta-secretase expression and enzymatic activity detected in sporadic Alzheimer disease. Nat. Med. 2003, 9, 3–4. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Hui, L.; Geiger, N.H.; Haughey, N.J.; Geiger, J.D. Endolysosome involvement in HIV-1 transactivator protein-induced neuronal amyloid beta production. Neurobiol. Aging 2013, 34, 2370–2378. [Google Scholar] [CrossRef] [PubMed]
- Hui, L.; Ye, Y.; Soliman, M.L.; Lakpa, K.L.; Miller, N.M.; Afghah, Z.; Geiger, J.D.; Chen, X. Antiretroviral Drugs Promote Amyloidogenesis by De-Acidifying Endolysosomes. J. Neuroimmune Pharmacol. 2021, 16, 159–168. [Google Scholar] [CrossRef] [PubMed]
- Colciaghi, F.; Borroni, B.; Pastorino, L.; Marcello, E.; Zimmermann, M.; Cattabeni, F.; Padovani, A.; Di Luca, M. alpha-Secretase ADAM10 as well as alpha-APPs is reduced in platelets and CSF of Alzheimer disease patients. Mol. Med. 2002, 8, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.T.; Cao, K.; Xiang, J.; Shan, L.; Guan, Z.Z. Silent Mating-Type Information Regulation 2 Homolog 1 Attenuates the Neurotoxicity Associated with Alzheimer Disease via a Mechanism Which May Involve Regulation of Peroxisome Proliferator-Activated Receptor Gamma Coactivator 1-alpha. Am. J. Pathol. 2020, 190, 1545–1564. [Google Scholar] [CrossRef] [PubMed]
- Sennvik, K.; Fastbom, J.; Blomberg, M.; Wahlund, L.-O.; Winblad, B.; Benedikz, E. Levels of alpha- and beta-secretase cleaved amyloid precursor protein in the cerebrospinal fuid of Alzheimer’s disease patients. Neurosci. Lett. 2000, 278, 169–172. [Google Scholar] [CrossRef]
- Lesne, S.; Ali, C.; Gabriel, C.; Croci, N.; MacKenzie, E.T.; Glabe, C.G.; Plotkine, M.; Marchand-Verrecchia, C.; Vivien, D.; Buisson, A. NMDA receptor activation inhibits alpha-secretase and promotes neuronal amyloid-beta production. J. Neurosci. 2005, 25, 9367–9377. [Google Scholar] [CrossRef]
- Bordji, K.; Becerril-Ortega, J.; Nicole, O.; Buisson, A. Activation of extrasynaptic, but not synaptic, NMDA receptors modifies amyloid precursor protein expression pattern and increases amyloid-ss production. J. Neurosci. 2010, 30, 15927–15942. [Google Scholar] [CrossRef]
- Min, S.W.; Sohn, P.D.; Cho, S.H.; Swanson, R.A.; Gan, L. Sirtuins in neurodegenerative diseases: An update on potential mechanisms. Front. Aging Neurosci. 2013, 5, 53. [Google Scholar] [CrossRef]
- Herskovits, A.Z.; Guarente, L. SIRT1 in neurodevelopment and brain senescence. Neuron 2014, 81, 471–483. [Google Scholar] [CrossRef] [Green Version]
- Qin, W.; Yang, T.; Ho, L.; Zhao, Z.; Wang, J.; Chen, L.; Zhao, W.; Thiyagarajan, M.; MacGrogan, D.; Rodgers, J.T.; et al. Neuronal SIRT1 activation as a novel mechanism underlying the prevention of Alzheimer disease amyloid neuropathology by calorie restriction. J. Biol. Chem. 2006, 281, 21745–21754. [Google Scholar] [CrossRef]
- Qin, W.; Chachich, M.; Lane, M.; Roth, G.; Bryant, M.; de Cabo, R.; Ottinger, M.A.; Mattison, J.; Ingram, D.; Gandy, S.; et al. Calorie restriction attenuates Alzheimer’s disease type brain amyloidosis in Squirrel monkeys (Saimiri sciureus). J. Alzheimers Dis. 2006, 10, 417–422. [Google Scholar] [CrossRef]
- Thonda, S.; Puttapaka, S.N.; Kona, S.V.; Kalivendi, S.V. Extracellular-Signal-Regulated Kinase Inhibition Switches APP Processing from beta- to alpha-Secretase under Oxidative Stress: Modulation of ADAM10 by SIRT1/NF-kappaB Signaling. ACS Chem. Neurosci. 2021, 12, 4175–4186. [Google Scholar] [CrossRef]
- Quintas, A.; de Solis, A.J.; Diez-Guerra, F.J.; Carrascosa, J.M.; Bogonez, E. Age-associated decrease of SIRT1 expression in rat hippocampus: Prevention by late onset caloric restriction. Exp. Gerontol. 2012, 47, 198–201. [Google Scholar] [CrossRef]
- Julien, C.; Tremblay, C.; Émond, V.; Lebbadi, M.; Norman Salem, J.; Bennett, D.A.; Calon, F. Sirtuin 1 Reduction Parallels the Accumulation of Tau in Alzheimer Disease. J. Neuropathol. Exp. Neurol. 2009, 68, 48–58. [Google Scholar] [CrossRef]
- Jurkowska, K.; Szymanska, B.; Knysz, B.; Kuzniarski, A.; Piwowar, A. Sirtuins as Interesting Players in the Course of HIV Infection and Comorbidities. Cells 2021, 10, 2739. [Google Scholar] [CrossRef]
- Chaudhuri, A.D.; Yelamanchili, S.V.; Marcondes, M.C.; Fox, H.S. Up-regulation of microRNA-142 in simian immunodeficiency virus encephalitis leads to repression of sirtuin1. FASEB J. 2013, 27, 3720–3729. [Google Scholar] [CrossRef]
- Hu, G.; Liao, K.; Yang, L.; Pendyala, G.; Kook, Y.; Fox, H.S.; Buch, S. Tat-Mediated Induction of miRs-34a & -138 Promotes Astrocytic Activation via Downregulation of SIRT1: Implications for Aging in HAND. J. Neuroimmune Pharmacol. 2017, 12, 420–432. [Google Scholar] [CrossRef]
- Li, H.; McLaurin, K.A.; Mactutus, C.F.; Likins, B.; Huang, W.; Chang, S.L.; Booze, R.M. Intraneuronal beta-Amyloid Accumulation: Aging HIV-1 Human and HIV-1 Transgenic Rat Brain. Viruses 2022, 14, 1268. [Google Scholar] [CrossRef]
- Ryan, S.K.; Gonzalez, M.V.; Garifallou, J.P.; Bennett, F.C.; Williams, K.S.; Sotuyo, N.P.; Mironets, E.; Cook, K.; Hakonarson, H.; Anderson, S.A.; et al. Neuroinflammation and EIF2 Signaling Persist despite Antiretroviral Treatment in an hiPSC Tri-culture Model of HIV Infection. Stem Cell Rep. 2020, 14, 703–716. [Google Scholar] [CrossRef]
- Lacey, D.C.; Achuthan, A.; Fleetwood, A.J.; Dinh, H.; Roiniotis, J.; Scholz, G.M.; Chang, M.W.; Beckman, S.K.; Cook, A.D.; Hamilton, J.A. Defining GM-CSF- and macrophage-CSF-dependent macrophage responses by in vitro models. J. Immunol. 2012, 188, 5752–5765. [Google Scholar] [CrossRef]
- Chen, W.; Sulcove, J.; Frank, I.; Jaffer, S.; Ozdener, H.; Kolson, D.L. Development of a human neuronal cell model for human immunodeficiency virus (HIV)-infected macrophage-induced neurotoxicity: Apoptosis induced by HIV type 1 primary isolates and evidence for involvement of the Bcl-2/Bcl-xL-sensitive intrinsic apoptosis pathway. J. Virol. 2002, 76, 9407–9419. [Google Scholar] [CrossRef]
- O’Donnell, L.A.; Agrawal, A.; Jordan-Sciutto, K.L.; Dichter, M.A.; Lynch, D.R.; Kolson, D.L. Human immunodeficiency virus (HIV)-induced neurotoxicity: Roles for the NMDA receptor subtypes. J. Neurosci. 2006, 26, 981–990. [Google Scholar] [CrossRef]
- Wang, Y.; White, M.G.; Akay, C.; Chodroff, R.A.; Robinson, J.; Lindl, K.A.; Dichter, M.A.; Qian, Y.; Mao, Z.; Kolson, D.L.; et al. Activation of cyclin-dependent kinase 5 by calpains contributes to human immunodeficiency virus-induced neurotoxicity. J. Neurochem. 2007, 103, 439–455. [Google Scholar] [CrossRef]
- Akay, C.; Lindl, K.A.; Wang, Y.; White, M.G.; Isaacman-Beck, J.; Kolson, D.L.; Jordan-Sciutto, K.L. Site-specific hyperphosphorylation of pRb in HIV-induced neurotoxicity. Mol. Cell Neurosci. 2011, 47, 154–165. [Google Scholar] [CrossRef]
- Zhang, Z.; Shen, Q.; Wu, X.; Zhang, D.; Xing, D. Activation of PKA/SIRT1 signaling pathway by photobiomodulation therapy reduces Abeta levels in Alzheimer’s disease models. Aging Cell 2020, 19, e13054. [Google Scholar] [CrossRef]
- Xu, C.; Wang, L.; Fozouni, P.; Evjen, G.; Chandra, V.; Jiang, J.; Lu, C.; Nicastri, M.; Bretz, C.; Winkler, J.D.; et al. SIRT1 is downregulated by autophagy in senescence and ageing. Nat. Cell Biol. 2020, 22, 1170–1179. [Google Scholar] [CrossRef]
- Zhang, Q.; Zhang, P.; Qi, G.J.; Zhang, Z.; He, F.; Lv, Z.X.; Peng, X.; Cai, H.W.; Li, T.X.; Wang, X.M.; et al. Cdk5 suppression blocks SIRT1 degradation via the ubiquitin-proteasome pathway in Parkinson’s disease models. Biochim. Biophys. Acta Gen. Subj. 2018, 1862, 1443–1451. [Google Scholar] [CrossRef] [PubMed]
- Hoey, S.E.; Williams, R.J.; Perkinton, M.S. Synaptic NMDA receptor activation stimulates alpha-secretase amyloid precursor protein processing and inhibits amyloid-beta production. J. Neurosci. 2009, 29, 4442–4460. [Google Scholar] [CrossRef]
- Wan, X.Z.; Li, B.; Li, Y.C.; Yang, X.L.; Zhang, W.; Zhong, L.; Tang, S.J. Activation of NMDA receptors upregulates a disintegrin and metalloproteinase 10 via a Wnt/MAPK signaling pathway. J. Neurosci. 2012, 32, 3910–3916. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, X.; Hollern, D.; Liao, J.; Andrechek, E.; Wang, H. NMDA receptor-mediated excitotoxicity depends on the coactivation of synaptic and extrasynaptic receptors. Cell Death Dis. 2013, 4, e560. [Google Scholar] [CrossRef] [PubMed]
- Xia, P.; Chen, H.S.; Zhang, D.; Lipton, S.A. Memantine preferentially blocks extrasynaptic over synaptic NMDA receptor currents in hippocampal autapses. J. Neurosci. 2010, 30, 11246–11250. [Google Scholar] [CrossRef] [PubMed]
- Alley, G.M.; Bailey, J.A.; Chen, D.; Ray, B.; Puli, L.K.; Tanila, H.; Banerjee, P.K.; Lahiri, D.K. Memantine lowers amyloid-beta peptide levels in neuronal cultures and in APP/PS1 transgenic mice. J. Neurosci. Res. 2010, 88, 143–154. [Google Scholar] [CrossRef]
- Companys-Alemany, J.; Turcu, A.L.; Bellver-Sanchis, A.; Loza, M.I.; Brea, J.M.; Canudas, A.M.; Leiva, R.; Vazquez, S.; Pallas, M.; Grinan-Ferre, C. A Novel NMDA Receptor Antagonist Protects against Cognitive Decline Presented by Senescent Mice. Pharmaceutics 2020, 12, 284. [Google Scholar] [CrossRef]
- Companys-Alemany, J.; Turcu, A.L.; Schneider, M.; Muller, C.E.; Vazquez, S.; Grinan-Ferre, C.; Pallas, M. NMDA receptor antagonists reduce amyloid-beta deposition by modulating calpain-1 signaling and autophagy, rescuing cognitive impairment in 5XFAD mice. Cell Mol Life Sci. 2022, 79, 408. [Google Scholar] [CrossRef]
- Reiss, K.; Maretzky, T.; Ludwig, A.; Tousseyn, T.; de Strooper, B.; Hartmann, D.; Saftig, P. ADAM10 cleavage of N-cadherin and regulation of cell-cell adhesion and beta-catenin nuclear signalling. EMBO J. 2005, 24, 742–752. [Google Scholar] [CrossRef]
- Kuhn, P.H.; Colombo, A.V.; Schusser, B.; Dreymueller, D.; Wetzel, S.; Schepers, U.; Herber, J.; Ludwig, A.; Kremmer, E.; Montag, D.; et al. Systematic substrate identification indicates a central role for the metalloprotease ADAM10 in axon targeting and synapse function. Elife 2016, 5, e12748. [Google Scholar] [CrossRef]
- Cao, L.; Liu, C.; Wang, F.; Wang, H. SIRT1 negatively regulates amyloid-beta-induced inflammation via the NF-kappaB pathway. Braz. J. Med. Biol. Res. 2013, 46, 659–669. [Google Scholar] [CrossRef]
- Bortell, N.; Basova, L.; Najera, J.A.; Morsey, B.; Fox, H.S.; Marcondes, M.C.G. Sirtuin 1-Chromatin-Binding Dynamics Points to a Common Mechanism Regulating Inflammatory Targets in SIV Infection and in the Aging Brain. J. Neuroimmune Pharmacol. 2018, 13, 163–178. [Google Scholar] [CrossRef]
- Basova, L.V.; Kesby, J.P.; Kaul, M.; Semenova, S.; Marcondes, M.C.G. Systems Biology Analysis of the Antagonizing Effects of HIV-1 Tat Expression in the Brain over Transcriptional Changes Caused by Methamphetamine Sensitization. Viruses 2020, 12, 426. [Google Scholar] [CrossRef] [Green Version]
- Pallas, M.; Pizarro, J.G.; Gutierrez-Cuesta, J.; Crespo-Biel, N.; Alvira, D.; Tajes, M.; Yeste-Velasco, M.; Folch, J.; Canudas, A.M.; Sureda, F.X.; et al. Modulation of SIRT1 expression in different neurodegenerative models and human pathologies. Neuroscience 2008, 154, 1388–1397. [Google Scholar] [CrossRef]
- Yang, X.; Sun, X.; Wu, J.; Ma, J.; Si, P.; Yin, L.; Zhang, Y.; Yan, L.J.; Zhang, C. Regulation of the SIRT1 signaling pathway in NMDA-induced Excitotoxicity. Toxicol. Lett. 2020, 322, 66–76. [Google Scholar] [CrossRef]
- Chen, J.; Zhou, Y.; Mueller-Steiner, S.; Chen, L.F.; Kwon, H.; Yi, S.; Mucke, L.; Gan, L. SIRT1 protects against microglia-dependent amyloid-beta toxicity through inhibiting NF-kappaB signaling. J. Biol. Chem. 2005, 280, 40364–40374. [Google Scholar] [CrossRef]
- Min, S.W.; Sohn, P.D.; Li, Y.; Devidze, N.; Johnson, J.R.; Krogan, N.J.; Masliah, E.; Mok, S.A.; Gestwicki, J.E.; Gan, L. SIRT1 Deacetylates Tau and Reduces Pathogenic Tau Spread in a Mouse Model of Tauopathy. J. Neurosci. 2018, 38, 3680–3688. [Google Scholar] [CrossRef]
- Redmann, M.; Benavides, G.A.; Berryhill, T.F.; Wani, W.Y.; Ouyang, X.; Johnson, M.S.; Ravi, S.; Barnes, S.; Darley-Usmar, V.M.; Zhang, J. Inhibition of autophagy with bafilomycin and chloroquine decreases mitochondrial quality and bioenergetic function in primary neurons. Redox Biol. 2017, 11, 73–81. [Google Scholar] [CrossRef]
- Liu, P.Y.; Chen, C.C.; Chin, C.Y.; Liu, T.J.; Tsai, W.C.; Chou, J.L.; Huang, C.Y.; Chen, Y.G.; Chen, Y.C. E3 ubiquitin ligase Grail promotes hepatic steatosis through Sirt1 inhibition. Cell Death Dis. 2021, 12, 323. [Google Scholar] [CrossRef]
- Caldeira, M.V.; Curcio, M.; Leal, G.; Salazar, I.L.; Mele, M.; Santos, A.R.; Melo, C.V.; Pereira, P.; Canzoniero, L.M.; Duarte, C.B. Excitotoxic stimulation downregulates the ubiquitin-proteasome system through activation of NMDA receptors in cultured hippocampal neurons. Biochim. Biophys. Acta 2013, 1832, 263–274. [Google Scholar] [CrossRef]
- Ferreira, J.S.; Kellermayer, B.; Carvalho, A.L.; Groc, L. Interplay between NMDA receptor dynamics and the synaptic proteasome. Eur. J. Neurosci. 2021, 54, 6000–6011. [Google Scholar] [CrossRef]
- Nguyen, T.P.; Soukup, V.M.; Gelman, B.B. Persistent hijacking of brain proteasomes in HIV-associated dementia. Am. J. Pathol. 2010, 176, 893–902. [Google Scholar] [CrossRef]
- Gruenewald, A.L.; Garcia-Mesa, Y.; Gill, A.J.; Garza, R.; Gelman, B.B.; Kolson, D.L. Neuroinflammation associates with antioxidant heme oxygenase-1 response throughout the brain in persons living with HIV. J. Neurovirol. 2020, 26, 846–862. [Google Scholar] [CrossRef]
- Lee, H.R.; Shin, H.K.; Park, S.Y.; Kim, H.Y.; Lee, W.S.; Rhim, B.Y.; Hong, K.W.; Kim, C.D. Cilostazol suppresses beta-amyloid production by activating a disintegrin and metalloproteinase 10 via the upregulation of SIRT1-coupled retinoic acid receptor-beta. J. Neurosci. Res. 2014, 92, 1581–1590. [Google Scholar] [CrossRef] [PubMed]
- Tippmann, F.; Hundt, J.; Schneider, A.; Endres, K.; Fahrenholz, F. Up-regulation of the alpha-secretase ADAM10 by retinoic acid receptors and acitretin. FASEB J. 2009, 23, 1643–1654. [Google Scholar] [CrossRef] [PubMed]
- Deng, H.; Mi, M.T. Resveratrol Attenuates Abeta25-35 Caused Neurotoxicity by Inducing Autophagy Through the TyrRS-PARP1-SIRT1 Signaling Pathway. Neurochem. Res. 2016, 41, 2367–2379. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Si, P.; Qin, H.; Yin, L.; Yan, L.J.; Zhang, C. The Neuroprotective Effects of SIRT1 on NMDA-Induced Excitotoxicity. Oxid. Med. Cell Longev. 2017, 2017, 2823454. [Google Scholar] [CrossRef]
- Ibi, D.; Kondo, S.; Ohmi, A.; Kojima, Y.; Nakasai, G.; Takaba, R.; Hiramatsu, M. Preventive Effect of Betaine Against Cognitive Impairments in Amyloid beta Peptide-Injected Mice Through Sirtuin1 in Hippocampus. Neurochem. Res. 2022, 47, 2333–2344. [Google Scholar] [CrossRef]
- Gomes, B.A.Q.; Silva, J.P.B.; Romeiro, C.F.R.; Dos Santos, S.M.; Rodrigues, C.A.; Goncalves, P.R.; Sakai, J.T.; Mendes, P.F.S.; Varela, E.L.P.; Monteiro, M.C. Neuroprotective Mechanisms of Resveratrol in Alzheimer’s Disease: Role of SIRT1. Oxid. Med. Cell Longev. 2018, 2018, 8152373. [Google Scholar] [CrossRef]
- Tosatti, J.A.G. Effects of Resveratrol Supplementation on the Cognitive Function of Patients with Alzheimer’s Disease: A Systematic Review of Randomized Controlled Trials. Drugs Aging 2022, 39, 285–295. [Google Scholar] [CrossRef]
- Moussa, C.; Hebron, M.; Huang, X.; Ahn, J.; Rissman, R.A.; Aisen, P.S.; Turner, R.S. Resveratrol regulates neuro-inflammation and induces adaptive immunity in Alzheimer’s disease. J. Neuroinflamm. 2017, 14, 1. [Google Scholar] [CrossRef]
- Sawda, C.; Moussa, C.; Turner, R.S. Resveratrol for Alzheimer’s disease. Ann. N. Y. Acad. Sci. 2017, 1403, 142–149. [Google Scholar] [CrossRef]
- Donmez, G.; Outeiro, T.F. SIRT1 and SIRT2: Emerging targets in neurodegeneration. EMBO Mol. Med. 2013, 5, 344–352. [Google Scholar] [CrossRef]
- Manzine, P.R.; Ettcheto, M.; Cano, A.; Busquets, O.; Marcello, E.; Pelucchi, S.; Di Luca, M.; Endres, K.; Olloquequi, J.; Camins, A.; et al. ADAM10 in Alzheimer’s disease: Pharmacological modulation by natural compounds and its role as a peripheral marker. Biomed. Pharmacother. 2019, 113, 108661. [Google Scholar] [CrossRef]
- Peron, R.; Vatanabe, I.P.; Manzine, P.R.; Camins, A.; Cominetti, M.R. Alpha-Secretase ADAM10 Regulation: Insights into Alzheimer’s Disease Treatment. Pharmaceuticals 2018, 11, 12. [Google Scholar] [CrossRef] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lopez Lloreda, C.; Chowdhury, S.; Ghura, S.; Alvarez-Periel, E.; Jordan-Sciutto, K. HIV-Associated Insults Modulate ADAM10 and Its Regulator Sirtuin1 in an NMDA Receptor-Dependent Manner. Cells 2022, 11, 2962. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11192962
Lopez Lloreda C, Chowdhury S, Ghura S, Alvarez-Periel E, Jordan-Sciutto K. HIV-Associated Insults Modulate ADAM10 and Its Regulator Sirtuin1 in an NMDA Receptor-Dependent Manner. Cells. 2022; 11(19):2962. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11192962
Chicago/Turabian StyleLopez Lloreda, Claudia, Sarah Chowdhury, Shivesh Ghura, Elena Alvarez-Periel, and Kelly Jordan-Sciutto. 2022. "HIV-Associated Insults Modulate ADAM10 and Its Regulator Sirtuin1 in an NMDA Receptor-Dependent Manner" Cells 11, no. 19: 2962. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11192962