

Intranasal Peptide Therapeutics: A Promising Avenue for Overcoming the Challenges of Traditional CNS Drug Development
Abstract
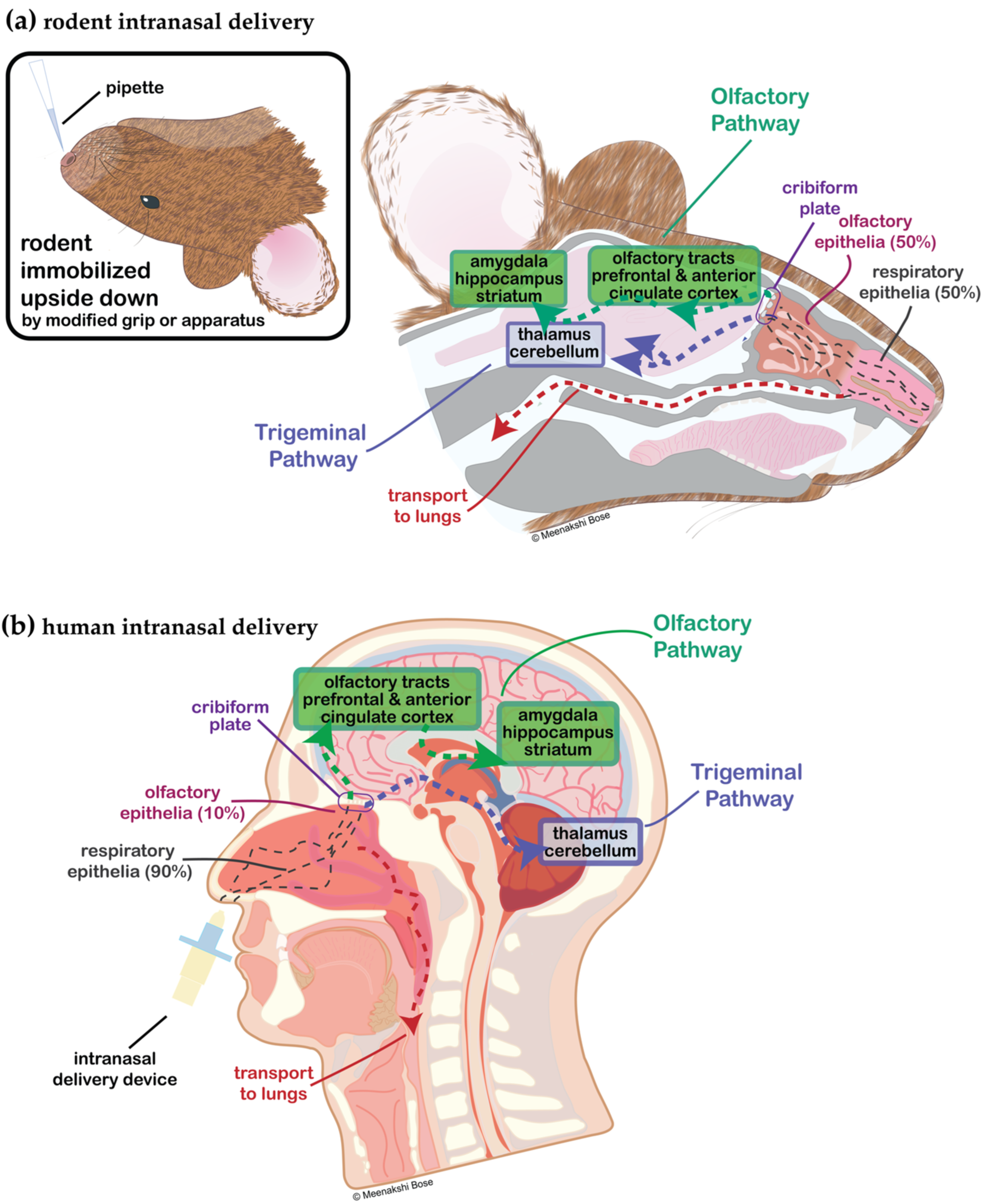
:1. Introduction
2. Brain-Derived Neurotrophic Factor
2.1. IN-BDNF Therapies for Neuropsychiatric Disorders
Major Depressive Disorder
3. EPO
3.1. IN-EPO Therapies for Neurodegenerative Disorders
Alzheimer’s Disease
4. GDNF
4.1. IN-GDNF Therapies for Neurodegenerative Disorders
Parkinson’s Disease
5. GLP-1
5.1. IN-GLP-1 Therapies for Neurodegenerative Disorders
Alzheimer’s Disease
6. Insulin/Insulin-like Growth Factor 1
6.1. IN-Insulin or IN-IGF1 Therapies for Neurodevelopmental Disorders
6.1.1. Autism Spectrum Disorders
6.1.2. Bipolar Disorder
6.1.3. Schizophrenia
6.2. IN-Insulin or IN-IGF1 Therapies for Neuropsychiatric Disorders
6.2.1. Major Depressive Disorder
6.2.2. Generalized/Social Anxiety Disorders
6.3. IN-Insulin or IN-IGF1 Therapies for Neurodegenerative Disorders
6.3.1. Alzheimer’s Disease
6.3.2. Parkinson’s Disease
6.3.3. Huntington’s Disease
7. NAP
7.1. IN-NAP Therapies for Neurodevelopmental Disorders
7.1.1. Autism Spectrum Disorders
7.1.2. Schizophrenia
7.2. IN-NAP Therapies for Neurodegenerative Disorders
7.2.1. Alzheimer’s Disease
7.2.2. Parkinson’s Disease
8. NBD
8.1. IN-NBD Therapies for Neurodegenerative Disorders
Alzheimer’s Disease
9. NGF
9.1. IN-NGF Therapies in Neuropsychiatric Disorders
Major Depressive Disorder
9.2. IN-NGF Therapies in Neurodegenerative Disorders
Frontotemporal Dementia
10. NPY
10.1. IN-NPY Therapies in Neuropsychiatric Disorders
10.1.1. Major Depressive Disorder
10.1.2. Post-Traumatic Stress Disorder
10.2. IN-NPY Therapies in Neurodegenerative Disorders
Huntington’s Disease
11. Oxytocin
11.1. IN-Oxytocin Therapies for Neurodevelopmental Disorders
11.1.1. Autism Spectrum Disorders
11.1.2. Schizophrenia
11.2. IN-Oxytocin Therapies for Neuropsychiatric Disorders
11.2.1. Post-Traumatic Stress Disorder
11.2.2. Generalized/Social Anxiety Disorders
11.3. IN-Oxytocin Therapies for Neurodegenerative Disorders
Frontotemporal Dementia
12. PACAP
12.1. IN-PACAP Therapies for Neurodegenerative Disorders
12.1.1. Alzheimer’s Disease
12.1.2. Huntington’s Disease
13. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gribkoff, V.K.; Kaczmarek, L.K. The need for new approaches in CNS drug discovery: Why drugs have failed, and what can be done to improve outcomes. Neuropharmacology 2017, 120, 11–19. [Google Scholar] [CrossRef] [Green Version]
- Wegener, G.; Rujescu, D. The current development of CNS drug research. Int. J. Neuropsychopharmacol. 2013, 16, 1687–1693. [Google Scholar] [CrossRef] [Green Version]
- Yokley, B.H.; Hartman, M.; Slusher, B.S. Role of Academic Drug Discovery in the Quest for New CNS Therapeutics. ACS Chem. Neurosci. 2017, 8, 429–431. [Google Scholar] [CrossRef] [Green Version]
- Upadhyay, R.K. Drug delivery systems, CNS protection, and the blood brain barrier. BioMed Res. Int. 2014, 2014, 869269. [Google Scholar] [CrossRef] [Green Version]
- Bors, L.A.; Erdő, F. Overcoming the Blood–Brain Barrier. Challenges and Tricks for CNS Drug Delivery. Sci. Pharm. 2019, 87, 6. [Google Scholar] [CrossRef] [Green Version]
- Banks, W.A.; Greig, N.H. Small molecules as central nervous system therapeutics: Old challenges, new directions, and a philosophic divide. Futur. Med. Chem. 2019, 11, 489–493. [Google Scholar] [CrossRef] [Green Version]
- Tsomaia, N. Peptide therapeutics: Targeting the undruggable space. Eur. J. Med. Chem. 2015, 94, 459–470. [Google Scholar] [CrossRef]
- Wetie, A.G.N.; Sokolowska, I.; Woods, A.G.; Roy, U.; Deinhardt, K.; Darie, C.C. Protein–protein interactions: Switch from classical methods to proteomics and bioinformatics-based approaches. Cell. Mol. Life Sci. 2014, 71, 205–228. [Google Scholar] [CrossRef]
- Lu, H.; Zhou, Q.; He, J.; Jiang, Z.; Peng, C.; Tong, R.; Shi, J. Recent advances in the development of protein–protein interactions modulators: Mechanisms and clinical trials. Signal Transduct. Target. Ther. 2020, 5, 1–23. [Google Scholar] [CrossRef]
- Scott, D.E.; Bayly, A.R.; Abell, C.; Skidmore, J. Small molecules, big targets: Drug discovery faces the protein–protein interaction challenge. Nat. Rev. Drug Discov. 2016, 15, 533–550. [Google Scholar] [CrossRef]
- Morimoto, B.H. Therapeutic peptides for CNS indications: Progress and challenges. Bioorganic Med. Chem. 2018, 26, 2859–2862. [Google Scholar] [CrossRef]
- Jiang, Y.; Li, Y.; Liu, X. Intranasal delivery: Circumventing the iron curtain to treat neurological disorders. Expert Opin. Drug Deliv. 2015, 12, 1717–1725. [Google Scholar] [CrossRef]
- Chapman, C.D.; Frey, W.H.; Craft, S.; Danielyan, L.; Hallschmid, M.; Schiöth, H.B.; Benedict, C. Intranasal Treatment of Central Nervous System Dysfunction in Humans. Pharm. Res. 2013, 30, 2475–2484. [Google Scholar] [CrossRef] [Green Version]
- Lochhead, J.J.; Davis, T.P. Perivascular and Perineural Pathways Involved in Brain Delivery and Distribution of Drugs after Intranasal Administration. Pharmaceutics 2019, 11, 598. [Google Scholar] [CrossRef] [Green Version]
- Türker, S.; Onur, E.; Ózer, Y. Nasal route and drug delivery systems. Pharm. World Sci. 2004, 26, 137–142. [Google Scholar] [CrossRef]
- Yadav, S.; Gattacceca, F.; Panicucci, R.; Amiji, M.M. Comparative Biodistribution and Pharmacokinetic Analysis of Cyclosporine-A in the Brain upon Intranasal or Intravenous Administration in an Oil-in-Water Nanoemulsion Formulation. Mol. Pharm. 2015, 12, 1523–1533. [Google Scholar] [CrossRef]
- Al Bakri, W.; Donovan, M.D.; Cueto, M.; Wu, Y.; Orekie, C.; Yang, Z. Overview of intranasally delivered peptides: Key considerations for pharmaceutical development. Expert Opin. Drug Deliv. 2018, 15, 991–1005. [Google Scholar] [CrossRef]
- Rohrer, J.; Lupo, N.; Bernkop-Schnürch, A. Advanced formulations for intranasal delivery of biologics. Int. J. Pharm. 2018, 553, 8–20. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, H.; Ghosh, D.; Williams, R.O., 3rd. Just how prevalent are peptide therapeutic products? A critical review. Int. J. Pharm. 2020, 587, 119491. [Google Scholar] [CrossRef]
- Al-Suhaimi, E.A.; Nawaz, M.; Khan, F.A.; Aljafary, M.A.; Baykal, A.; Homeida, A.M. Emerging trends in the delivery of nanoformulated oxytocin across Blood-Brain barrier. Int. J. Pharm. 2021, 609, 121141. [Google Scholar] [CrossRef]
- Crowe, T.P.; Hsu, W.H. Evaluation of Recent Intranasal Drug Delivery Systems to the Central Nervous System. Pharmaceutics 2022, 14, 629. [Google Scholar] [CrossRef]
- Fortuna, A.; Alves, G.; Serralheiro, A.; Sousa, J.; Falcão, A. Intranasal delivery of systemic-acting drugs: Small-molecules and biomacromolecules. Eur. J. Pharm. Biopharm. 2014, 88, 8–27. [Google Scholar] [CrossRef]
- Sarkar, M.A. Drug Metabolism in the Nasal Mucosa. Pharm. Res. 1992, 9, 1–9. [Google Scholar] [CrossRef]
- Hanson, L.R.; Fine, J.M.; Svitak, A.L.; Faltesek, K.A. Intranasal Administration of CNS Therapeutics to Awake Mice. J. Vis. Exp. 2013, 74, e4440. [Google Scholar] [CrossRef] [Green Version]
- Trevino, J.; Quispe, R.; Khan, F.; Novak, V. Non-Invasive Strategies for Nose-to-Brain Drug Delivery. J. Clin. Trials 2020, 10, 439. [Google Scholar]
- Ma, X.-C.; Liu, P.; Zhang, X.-L.; Jiang, W.-H.; Jia, M.; Wang, C.-X.; Dong, Y.-Y.; Dang, Y.-H.; Gao, C.-G. Intranasal Delivery of Recombinant AAV Containing BDNF Fused with HA2TAT: A Potential Promising Therapy Strategy for Major Depressive Disorder. Sci. Rep. 2016, 6, 22404. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Dong, Y.; Liu, F.; Gao, C.; Ji, C.; Dang, Y.; Ma, X.; Liu, Y. A Study of Antidepressant Effect and Mechanism on Intranasal Delivery of BDNF-HA2TAT/AAV to Rats with Post-Stroke Depression. Neuropsychiatr. Dis. Treat. 2020, 16, 637–649. [Google Scholar] [CrossRef] [Green Version]
- Jelkmann, W. Regulation of erythropoietin production. J. Physiol. 2011, 589, 1251–1258. [Google Scholar] [CrossRef]
- Alnaeeli, M.; Wang, L.; Piknova, B.; Rogers, H.; Li, X.; Noguchi, C.T. Erythropoietin in Brain Development and Beyond. Anat. Res. Int. 2012, 2012, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Maurice, T.; Mustafa, M.H.; Desrumaux, C.; Keller, E.; Naert, G.; Garcia-Barcelo, M.D.; Rodriguez Cruz, Y.; Garcia Rodriguez, J.C. Intranasal formulation of erythropoietin (EPO) showed potent protective activity against amyloid toxicity in the Aβ₂₅₋₃₅ non-transgenic mouse model of Alzheimer’s disease. J. Psychopharmacol. 2013, 27, 1044–1057. [Google Scholar] [CrossRef]
- Cruz, Y.R.; Strehaiano, M.; Obaya, T.R.; Rodríguez, J.C.G.; Maurice, T. An Intranasal Formulation of Erythropoietin (Neuro-EPO) Prevents Memory Deficits and Amyloid Toxicity in the APPSwe Transgenic Mouse Model of Alzheimer’s Disease. J. Alzheimer’s Dis. 2017, 55, 231–248. [Google Scholar] [CrossRef] [PubMed]
- Björklund, A.; Kirik, D.; Rosenblad, C.; Georgievska, B.; Lundberg, C.; Mandel, R. Towards a neuroprotective gene therapy for Parkinson’s disease: Use of adenovirus, AAV and lentivirus vectors for gene transfer of GDNF to the nigrostriatal system in the rat Parkinson model. Brain Res. 2000, 886, 82–98. [Google Scholar] [CrossRef]
- Airaksinen, M.S.; Saarma, M. The GDNF family: Signalling, biological functions and therapeutic value. Nat. Rev. Neurosci. 2002, 3, 383–394. [Google Scholar] [CrossRef]
- Doucet-Beaupré, H.; Gilbert, C.; Profes, M.S.; Chabrat, A.; Pacelli, C.; Giguère, N.; Rioux, V.; Charest, J.; Deng, Q.; Laguna, A.; et al. GDNF-based therapies, GDNF-producing interneurons, and trophic support of the dopaminergic nigrostriatal pathway. Implications for Parkinson’s disease. Front. Neuroanat. 2016, 113, E4387–E4396. [Google Scholar] [CrossRef] [Green Version]
- Lang, A.E.; Gill, S.; Patel, N.K.; Lozano, A.; Nutt, J.G.; Penn, R.; Brooks, D.; Hotton, G.; Moro, E.; Heywood, P.; et al. Randomized controlled trial of intraputamenal glial cell line-derived neurotrophic factor infusion in Parkinson disease. Ann. Neurol. 2006, 59, 459–466. [Google Scholar] [CrossRef]
- Migliore, M.; Ortiz, R.; Dye, S.; Campbell, R.; Amiji, M.; Waszczak, B. Neurotrophic and neuroprotective efficacy of intranasal GDNF in a rat model of Parkinson’s disease. Neuroscience 2014, 274, 11–23. [Google Scholar] [CrossRef]
- Gartziandia, O.; Herrán, E.; Ruiz-Ortega, J.A.; Miguelez, C.; Igartua, M.; Lafuente, J.V.; Pedraz, J.L.; Ugedo, L.; Hernández, R.M. Intranasal Administration of chitosan-Coated Nanostructured Lipid Carriers Loaded with GDNF Improves Behavioral and Histological Recovery in a Partial Lesion Model of Parkinson’s Disease. J. Biomed. Nanotechnol. 2016, 12, 2220–2230. [Google Scholar] [CrossRef]
- Hernando, S.; Herran, E.; Figueiro-Silva, J.; Pedraz, J.L.; Igartua, M.; Carro, E.; Hernandez, R.M. Intranasal Administration of TAT-Conjugated Lipid Nanocarriers Loading GDNF for Parkinson’s Disease. Mol. Neurobiol. 2018, 55, 145–155. [Google Scholar] [CrossRef]
- Holt, M.K.; Trapp, S. The physiological role of the brain GLP-1 system in stress. Cogent Biol. 2016, 2, 1229086. [Google Scholar] [CrossRef]
- Wang, X.; Wang, L.; Xu, Y.; Yu, Q.; Li, L.; Guo, Y. Intranasal administration of Exendin-4 antagonizes Aβ31–35-induced disruption of circadian rhythm and impairment of learning and memory. Aging Clin. Exp. Res. 2016, 28, 1259–1266. [Google Scholar] [CrossRef]
- Kamei, N.; Okada, N.; Ikeda, T.; Choi, H.; Fujiwara, Y.; Okumura, H.; Takeda-Morishita, M. Effective nose-to-brain delivery of exendin-4 via coadministration with cell-penetrating peptides for improving progressive cognitive dysfunction. Sci. Rep. 2018, 8, 17641. [Google Scholar] [CrossRef]
- Wrigley, S.; Arafa, D.; Tropea, D. Insulin-Like Growth Factor 1: At the Crossroads of Brain Development and Aging. Front. Cell. Neurosci. 2017, 11, 14. [Google Scholar] [CrossRef] [Green Version]
- Biessels, G.J.; Reagan, L.P. Hippocampal insulin resistance and cognitive dysfunction. Nat. Rev. Neurosci. 2015, 16, 660–671. [Google Scholar] [CrossRef]
- Dierssen, M.; Fructuoso, M.; de Lagrán, M.M.; Perluigi, M.; Barone, E. Down Syndrome Is a Metabolic Disease: Altered Insulin Signaling Mediates Peripheral and Brain Dysfunctions. Front. Neurosci. 2020, 14, 670. [Google Scholar] [CrossRef]
- Ghasemi, R.; Dargahi, L.; Haeri, A.; Moosavi, M.; Mohamed, Z.; Ahmadiani, A. Brain Insulin Dysregulation: Implication for Neurological and Neuropsychiatric Disorders. Mol. Neurobiol. 2013, 47, 1045–1065. [Google Scholar] [CrossRef]
- Brietzke, E.; Kapczinski, F.; Grassi-Oliveira, R.; Grande, I.; Vieta, E.; McIntyre, R.S. Insulin dysfunction and allostatic load in bipolar disorder. Expert Rev. Neurother. 2011, 11, 1017–1028. [Google Scholar] [CrossRef]
- Delahaye, A.; Toutain, A.; Aboura, A.; Dupont, C.; Tabet, A.; Benzacken, B.; Elion, J.; Verloes, A.; Pipiras, E.; Drunat, S. Chromosome 22q13.3 deletion syndrome with a de novo interstitial 22q13.3 cryptic deletion disrupting SHANK3. Eur. J. Med Genet. 2009, 52, 328–332. [Google Scholar] [CrossRef]
- Cusmano-Ozog, K.; Manning, M.A.; Hoyme, H.E. 22q13.3 deletion syndrome: A recognizable malformation syndrome associated with marked speech and language delay. Am. J. Med Genet. Part C Semin. Med Genet. 2007, 145C, 393–398. [Google Scholar] [CrossRef]
- Kolevzon, A.; Bush, L.; Wang, A.T.; Halpern, D.; Frank, Y.; Grodberg, D.; Rapaport, R.; Tavassoli, T.; Chaplin, W.; Soorya, L.; et al. A pilot controlled trial of insulin-like growth factor-1 in children with Phelan-McDermid syndrome. Mol. Autism 2014, 5, 54. [Google Scholar] [CrossRef] [Green Version]
- Zwanenburg, R.J.; Bocca, G.; Ruiter, S.A.; Dillingh, J.H.; Flapper, B.C.; van den Heuvel, E.R.; van Ravenswaaij-Arts, C. Is there an effect of intranasal insulin on development and behaviour in Phelan-McDermid syndrome? A randomized, double-blind, placebo-controlled trial. Eur. J. Hum. Genet. 2016, 24, 1696–1701. [Google Scholar] [CrossRef] [Green Version]
- Calkin, C.V. Insulin resistance takes center stage: A new paradigm in the progression of bipolar disorder. Ann. Med. 2019, 51, 281–293. [Google Scholar] [CrossRef]
- McIntyre, R.S.; Soczynska, J.; Woldeyohannes, H.O.; Miranda, A.; Vaccarino, A.; MacQueen, G.; Lewis, G.F.; Kennedy, S. A randomized, double-blind, controlled trial evaluating the effect of intranasal insulin on neurocognitive function in euthymic patients with bipolar disorder. Bipolar Disord. 2012, 14, 697–706. [Google Scholar] [CrossRef]
- Fan, X.; Copeland, P.M.; Liu, E.Y.; Chiang, E.; Freudenreich, O.; Goff, D.C.; Henderson, D.C. No Effect of Single-Dose Intranasal Insulin Treatment on Verbal Memory and Sustained Attention in Patients With Schizophrenia. J. Clin. Psychopharmacol. 2011, 31, 231–234. [Google Scholar] [CrossRef]
- Fan, X.; Liu, E.; Freudenreich, O.; Copeland, P.; Hayden, D.; Ghebremichael, M.; Cohen, B.M.; OngurMD, D.; Goff, D.C.; Henderson, D.C. No Effect of Adjunctive, Repeated-Dose Intranasal Insulin Treatment on Psychopathology and Cognition in Patients With Schizophrenia. J. Clin. Psychopharmacol. 2013, 33, 226–230. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Li, X.; Liu, E.; Copeland, P.; Freudenreich, O.; Goff, D.C.; Henderson, D.C.; Song, X.; Fan, X. No effect of adjunctive, repeated dose intranasal insulin treatment on body metabolism in patients with schizophrenia. Schizophr. Res. 2013, 146, 40–45. [Google Scholar] [CrossRef] [Green Version]
- Cha, D.S.; Best, M.W.; Bowie, C.R.; Gallaugher, L.A.; Woldeyohannes, H.O.; Soczynska, J.; Lewis, G.; MacQueen, G.; Sahakian, B.; Kennedy, S.H.; et al. A randomized, double-blind, placebo-controlled, crossover trial evaluating the effect of intranasal insulin on cognition and mood in individuals with treatment-resistant major depressive disorder. J. Affect. Disord. 2017, 210, 57–65. [Google Scholar] [CrossRef]
- Grillo, C.A.; Piroli, G.G.; Kaigler, K.F.; Wilson, S.P.; Wilson, M.A.; Reagan, L.P. Downregulation of hypothalamic insulin receptor expression elicits depressive-like behaviors in rats. Behav. Brain Res. 2011, 222, 230–235. [Google Scholar] [CrossRef] [Green Version]
- Gupta, D.; Radhakrishnan, M.; Kurhe, Y. Insulin reverses anxiety-like behavior evoked by streptozotocin-induced diabetes in mice. Metab. Brain Dis. 2014, 29, 737–746. [Google Scholar] [CrossRef]
- Beirami, E.; Oryan, S.; Tamijani, S.M.S.; Ahmadiani, A.; Dargahi, L. Intranasal insulin treatment alleviates methamphetamine induced anxiety-like behavior and neuroinflammation. Neurosci. Lett. 2017, 660, 122–129. [Google Scholar] [CrossRef]
- Bohringer, A.; Schwabe, L.; Richter, S.; Schachinger, H. Intranasal insulin attenuates the hypothalamic–pituitary–adrenal axis response to psychosocial stress. Psychoneuroendocrinology 2008, 33, 1394–1400. [Google Scholar] [CrossRef]
- De Sá, D.S.F.; Römer, S.; Brückner, A.H.; Issler, T.; Hauck, A.; Michael, T. Effects of intranasal insulin as an enhancer of fear extinction: A randomized, double-blind, placebo-controlled experimental study. Neuropsychopharmacology 2020, 45, 753–760. [Google Scholar] [CrossRef]
- Craft, S.; Peskind, E.; Schwartz, M.W.; Schellenberg, G.D.; Raskind, M.; Porte, D. Cerebrospinal fluid and plasma insulin levels in Alzheimer’s disease: Relationship to severity of dementia and apolipoprotein E genotype. Neurology 1998, 50, 164–168. [Google Scholar] [CrossRef]
- Talbot, K.; Wang, H.Y.; Kazi, H.; Han, L.Y.; Bakshi, K.P.; Stucky, A.; Fuino, R.L.; Kawaguchi, K.R.; Samoyedny, A.J.; Wilson, R.S.; et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J. Clin. Investig. 2012, 122, 1316–1338. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, T.T.; Ta, Q.T.; Nguyen, T.T.; Le, T.T.; Vo, V.G. Role of Insulin Resistance in the Alzheimer’s Disease Progression. Neurochem. Res. 2020, 45, 1481–1491. [Google Scholar] [CrossRef]
- Gabbouj, S.; Natunen, T.; Koivisto, H.; Jokivarsi, K.; Takalo, M.; Marttinen, M.; Wittrahm, R.; Kemppainen, S.; Naderi, R.; Posado-Fernández, A.; et al. Intranasal insulin activates Akt2 signaling pathway in the hippocampus of wild-type but not in APP/PS1 Alzheimer model mice. Neurobiol. Aging 2019, 75, 98–108. [Google Scholar] [CrossRef]
- Mao, Y.F.; Guo, Z.; Zheng, T.; Jiang, Y.; Yan, Y.; Yin, X.; Chen, Y.; Zhang, B. Intranasal insulin alleviates cognitive deficits and amyloid pathology in young adult APPswe/PS1dE9 mice. Aging Cell 2016, 15, 893–902. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Guo, Z.; Mao, Y.F.; Zheng, T.; Zhang, B. Intranasal Insulin Ameliorates Cerebral Hypometabolism, Neuronal Loss, and Astrogliosis in Streptozotocin-Induced Alzheimer’s Rat Model. Neurotox. Res. 2018, 33, 716–724. [Google Scholar] [CrossRef]
- Rajasekar, N.; Nath, C.; Hanif, K.; Shukla, R. Intranasal Insulin Administration Ameliorates Streptozotocin (ICV)-Induced Insulin Receptor Dysfunction, Neuroinflammation, Amyloidogenesis, and Memory Impairment in Rats. Mol. Neurobiol. 2017, 54, 6507–6522. [Google Scholar] [CrossRef]
- Guo, Z.; Chen, Y.; Mao, Y.-F.; Zheng, T.; Jiang, Y.; Yan, Y.; Yin, X.; Zhang, B. Long-term treatment with intranasal insulin ameliorates cognitive impairment, tau hyperphosphorylation, and microglial activation in a streptozotocin-induced Alzheimer’s rat model. Sci. Rep. 2017, 7, 45971. [Google Scholar] [CrossRef] [Green Version]
- Reger, M.; Watson, G.; Frey, W.; Baker, L.; Cholerton, B.; Keeling, M.; Belongia, D.; Fishel, M.; Plymate, S.; Schellenberg, G.; et al. Effects of intranasal insulin on cognition in memory-impaired older adults: Modulation by APOE genotype. Neurobiol. Aging 2006, 27, 451–458. [Google Scholar] [CrossRef]
- Reger, M.A.; Watson, G.S.; Green, P.S.; Wilkinson, C.W.; Baker, L.D.; Cholerton, B.; Fishel, M.A.; Plymate, S.R.; Breitner, J.; DeGroodt, W.; et al. Intranasal insulin improves cognition and modulates -amyloid in early AD. Neurology 2008, 70, 440–448. [Google Scholar] [CrossRef]
- Rosenbloom, M.H.; Barclay, T.R.; Pyle, M.; Owens, B.L.; Cagan, A.B.; Anderson, C.P.; Frey, W.H.; Hanson, L.R. A Single-Dose Pilot Trial of Intranasal Rapid-Acting Insulin in Apolipoprotein E4 Carriers with Mild–Moderate Alzheimer’s Disease. CNS Drugs 2014, 28, 1185–1189. [Google Scholar] [CrossRef]
- Stein, M.S.; Scherer, S.C.; Ladd, K.S.; Harrison, L.C. A Randomized Controlled Trial of High-Dose Vitamin D2 Followed by Intranasal Insulin in Alzheimer’s Disease. J. Alzheimer’s Dis. 2011, 26, 477–484. [Google Scholar] [CrossRef]
- Craft, S.; Baker, L.D.; Montine, T.J.; Minoshima, S.; Watson, G.S.; Claxton, A.; Arbuckle, M.; Callaghan, M.; Tsai, E.; Plymate, S.R.; et al. Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment: A pilot clinical trial. Arch. Neurol. 2012, 69, 29. [Google Scholar] [CrossRef] [Green Version]
- Claxton, A.; Baker, L.D.; Wilkinson, C.W.; Trittschuh, E.H.; Chapman, D.; Watson, G.; Cholerton, B.; Plymate, S.R.; Arbuckle, M.; Craft, S. Sex and ApoE Genotype Differences in Treatment Response to Two Doses of Intranasal Insulin in Adults with Mild Cognitive Impairment or Alzheimer’s Disease. J. Alzheimer’s Dis. 2013, 35, 789–797. [Google Scholar] [CrossRef] [Green Version]
- Claxton, A.; Baker, L.D.; Hanson, A.; Trittschuh, E.H.; Cholerton, B.; Morgan, A.; Callaghan, M.; Arbuckle, M.; Behl, C.; Craft, S. Long-acting intranasal insulin detemir improves cognition for adults with mild cognitive impairment or early-stage Alzheimer’s disease dementia. J. Alzheimer’s Dis. 2015, 44, 897–906. [Google Scholar] [CrossRef] [Green Version]
- Craft, S.; Claxton, A.; Baker, L.D.; Hanson, A.J.; Cholerton, B.; Trittschuh, E.H.; Dahl, D.; Caulder, E.; Neth, B.; Montine, T.J.; et al. Effects of Regular and Long-Acting Insulin on Cognition and Alzheimer’s Disease Biomarkers: A Pilot Clinical Trial. J. Alzheimer’s Dis. 2017, 57, 1325–1334. [Google Scholar] [CrossRef] [Green Version]
- Craft, S.; Raman, R.; Chow, T.W.; Rafii, M.S.; Sun, C.-K.; Rissman, R.A.; Donohue, M.C.; Brewer, J.B.; Jenkins, C.; Harless, K.; et al. Safety, Efficacy, and Feasibility of Intranasal Insulin for the Treatment of Mild Cognitive Impairment and Alzheimer Disease Dementia. JAMA Neurol. 2020, 77, 1099. [Google Scholar] [CrossRef]
- Kellar, D.; Lockhart, S.; Aisen, P.; Raman, R.; Rissman, R.; Brewer, J.; Craft, S. Intranasal Insulin Reduces White Matter Hyperintensity Progression in Association with Improvements in Cognition and CSF Biomarker Profiles in Mild Cognitive Impairment and Alzheimer’s Disease. J. Prev. Alzheimer’s Dis. 2021, 8, 240–248. [Google Scholar] [CrossRef]
- Pang, Y.; Lin, S.; Wright, C.; Shen, J.; Carter, K.; Bhatt, A.; Fan, L.-W. Intranasal insulin protects against substantia nigra dopaminergic neuronal loss and alleviates motor deficits induced by 6-OHDA in rats. Neuroscience 2016, 318, 157–165. [Google Scholar] [CrossRef] [Green Version]
- Fine, J.M.; Stroebel, B.M.; Faltesek, K.A.; Terai, K.; Haase, L.; Knutzen, K.E.; Kosyakovsky, J.; Bowe, T.J.; Fuller, A.K.; Frey, W.H.; et al. Intranasal delivery of low-dose insulin ameliorates motor dysfunction and dopaminergic cell death in a 6-OHDA rat model of Parkinson’s Disease. Neurosci. Lett. 2020, 714, 134567. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Zhang, X.; Li, S.; Wang, H.; Zhang, X.; Liu, L.; Xie, A. Intranasal insulin ameliorates cognitive impairment in a rat model of Parkinson’s disease through Akt/GSK3β signaling pathway. Life Sci. 2020, 259, 118159. [Google Scholar] [CrossRef] [PubMed]
- Novak, P.; Maldonado, D.A.P.; Novak, V. Safety and preliminary efficacy of intranasal insulin for cognitive impairment in Parkinson disease and multiple system atrophy: A double-blinded placebo-controlled pilot study. PLoS ONE 2019, 14, e0214364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopes, C.; Ribeiro, M.; Duarte, A.I.; Humbert, S.; Saudou, F.; Pereira de Almeida, L.; Hayden, M.; Rego, A.C. IGF-1 Intranasal Administration Rescues Huntington’s Disease Phenotypes in YAC128 Mice. Mol. Neurobiol. 2014, 49, 1126–1142. [Google Scholar] [CrossRef] [PubMed]
- Gozes, I.; Stewart, A.; Morimoto, B.; Fox, A.; Sutherland, K.; Schmechel, D. Addressing Alzheimer’s disease tangles: From NAP to AL-108. Curr. Alzheimer. Res. 2009, 6, 455–460. [Google Scholar] [CrossRef] [PubMed]
- Hadar, A.; Kapitansky, O.; Ganaiem, M.; Sragovich, S.; Lobyntseva, A.; Giladi, E.; Yeheskel, A.; Avitan, A.; Vatine, G.D.; Gurwitz, D.; et al. Introducing ADNP and SIRT1 as new partners regulating microtubules and histone methylation. Mol. Psychiatry 2021, 26, 6550–6561. [Google Scholar] [CrossRef]
- Sragovich, S.; Malishkevich, A.; Piontkewitz, Y.; Giladi, E.; Touloumi, O.; Lagoudaki, R.; Grigoriadis, N.; Gozes, I. The autism/neuroprotection-linked ADNP/NAP regulate the excitatory glutamatergic synapse. Transl. Psychiatry 2019, 9, 2. [Google Scholar] [CrossRef] [Green Version]
- Sragovich, S.; Merenlender-Wagner, A.; Gozes, I. ADNP Plays a Key Role in Autophagy: From Autism to Schizophrenia and Alzheimer’s Disease. BioEssays 2017, 39, 1700054. [Google Scholar] [CrossRef]
- Morimoto, B.H.; De Lannoy, I.; Fox, A.W.; Gozes, I.; Stewart, A. Davunetide. Pharmacokinetics and distribution to brain after intravenous or intranasal administration to rat. Chim. Oggi 2009, 27, 16. [Google Scholar]
- Jouroukhin, Y.; Ostritsky, R.; Assaf, Y.; Pelled, G.; Giladi, E.; Gozes, I. NAP (davunetide) modifies disease progression in a mouse model of severe neurodegeneration: Protection against impairments in axonal transport. Neurobiol. Dis. 2013, 56, 79–94. [Google Scholar] [CrossRef]
- Idan-Feldman, A.; Schirer, Y.; Polyzoidou, E.; Touloumi, O.; Lagoudaki, R.; Grigoriadis, N.C.; Gozes, I. Davunetide (NAP) as a preventative treatment for central nervous system complications in a diabetes rat model. Neurobiol. Dis. 2011, 44, 327–339. [Google Scholar] [CrossRef] [PubMed]
- Kapitansky, O.; Giladi, E.; Jaljuli, I.; Bereswill, S.; Heimesaat, M.M.; Gozes, I. Microbiota changes associated with ADNP deficiencies: Rapid indicators for NAP (CP201) treatment of the ADNP syndrome and beyond. J. Neural Transm. 2020, 127, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Ivashko-Pachima, Y.; Ganaiem, M.; Ben-Horin-Hazak, I.; Lobyntseva, A.; Bellaiche, N.; Fischer, I.; Levy, G.; Sragovich, S.; Karmon, G.; Giladi, E.; et al. SH3- and actin-binding domains connect ADNP and SHANK3, revealing a fundamental shared mechanism underlying autism. Mol. Psychiatry, 2022; 1–12, Online ahead of print. [Google Scholar] [CrossRef]
- Karmon, G.; Sragovich, S.; Hacohen-Kleiman, G.; Ben-Horin-Hazak, I.; Kasparek, P.; Schuster, B.; Sedlacek, R.; Pasmanik-Chor, M.; Theotokis, P.; Touloumi, O.; et al. Novel ADNP Syndrome Mice Reveal Dramatic Sex-Specific Peripheral Gene Expression With Brain Synaptic and Tau Pathologies. Biol. Psychiatry 2022, 92, 81–95. [Google Scholar] [CrossRef] [PubMed]
- Merenlender-Wagner, A.; Pikman, R.; Giladi, E.; Andrieux, A.; Gozes, I. NAP (davunetide) enhances cognitive behavior in the STOP heterozygous mouse—A microtubule-deficient model of schizophrenia. Peptides 2010, 31, 1368–1373. [Google Scholar] [CrossRef]
- Merenlender-Wagner, A.; Shemer, Z.; Touloumi, O.; Lagoudaki, R.; Giladi, E.; Andrieux, A.; Grigoriadis, N.C.; Gozes, I. New horizons in schizophrenia treatment: Autophagy protection is coupled with behavioral improvements in a mouse model of schizophrenia. Autophagy 2014, 10, 2324–2332. [Google Scholar] [CrossRef] [Green Version]
- Tropea, D.; Hardingham, N.; Millar, K.; Fox, K. Mechanisms underlying the role of DISC1 in synaptic plasticity. J. Physiol. 2018, 596, 2747–2771. [Google Scholar] [CrossRef] [Green Version]
- Glausier, J.; Lewis, D. Dendritic spine pathology in schizophrenia. Neuroscience 2013, 251, 90–107. [Google Scholar] [CrossRef] [Green Version]
- Vaisburd, S.; Shemer, Z.; Yeheskel, A.; Giladi, E.; Gozes, I. Risperidone and NAP protect cognition and normalize gene expression in a schizophrenia mouse model. Sci. Rep. 2015, 5, 16300. [Google Scholar] [CrossRef] [Green Version]
- Javitt, D.C.; Buchanan, R.W.; Keefe, R.S.; Kern, R.; McMahon, R.P.; Green, M.F.; Lieberman, J.; Goff, D.C.; Csernansky, J.G.; McEvoy, J.P.; et al. Effect of the neuroprotective peptide davunetide (AL-108) on cognition and functional capacity in schizophrenia. Schizophr. Res. 2012, 136, 25–31. [Google Scholar] [CrossRef]
- Gozes, I.; Giladi, E.; Pinhasov, A.; Bardea, A.; Brenneman, D.E. Activity-dependent neurotrophic factor: Intranasal administration of femtomolar-acting peptides improve performance in a water maze. J. Pharmacol. Exp. Ther. 2000, 293, 1091. [Google Scholar]
- Alcalay, R.N.; Giladi, E.; Pick, C.G.; Gozes, I. Intranasal administration of NAP, a neuroprotective peptide, decreases anxiety-like behavior in aging mice in the elevated plus maze. Neurosci. Lett. 2004, 361, 128–131. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, Y.; Gray, A.J.; Hirata-Fukae, C.; Minami, S.S.; Waterhouse, E.G.; Mattson, M.P.; LaFerla, F.M.; Gozes, I.; Aisen, P.S. Intranasal NAP administration reduces accumulation of amyloid peptide and tau hyperphosphorylation in a transgenic mouse model of Alzheimer’s disease at early pathological stage. J. Mol. Neurosci. 2007, 31, 165–170. [Google Scholar] [CrossRef]
- Matsuoka, Y.; Jouroukhin, Y.; Gray, A.J.; Ma, L.; Hirata-Fukae, C.; Li, H.F.; Feng, L.; Lecanu, L.; Walker, B.R.; Planel, E.; et al. A Neuronal Microtubule-Interacting Agent, NAPVSIPQ, Reduces Tau Pathology and Enhances Cognitive Function in a Mouse Model of Alzheimer’s Disease. J. Pharmacol. Exp. Ther. 2008, 325, 146–153. [Google Scholar] [CrossRef] [PubMed]
- Vulih-Shultzman, I.; Pinhasov, A.; Mandel, S.; Grigoriadis, N.; Touloumi, O.; Pittel, Z.; Gozes, I. Activity-Dependent Neuroprotective Protein Snippet NAP Reduces Tau Hyperphosphorylation and Enhances Learning in a Novel Transgenic Mouse Model. J. Pharmacol. Exp. Ther. 2007, 323, 438–449. [Google Scholar] [CrossRef] [Green Version]
- Shiryaev, N.; Jouroukhin, Y.; Giladi, E.; Polyzoidou, E.; Grigoriadis, N.C.; Rosenmann, H.; Gozes, I. NAP protects memory, increases soluble tau and reduces tau hyperphosphorylation in a tauopathy model. Neurobiol. Dis. 2009, 34, 381–388. [Google Scholar] [CrossRef]
- Gozes, I. F1-01-01: Davunetide (AL-108): Targeting tangles. Alzheimer’s Dement. 2009, 5, P74. [Google Scholar] [CrossRef]
- Schmechel, D.E.; Gerard, G.; Vatakis, N.G.; Harper, L.; Ross, J.S.; Bari, M.; Walling, D.; Stedman, M.; Winston, J.L.; Morimoto, B.; et al. P2-377: A phase 2, double-blind, placebo-controlled study to evaluate the safety, tolerability, and effect on cognitive function of AL-108 after 12 weeks of intranasal administration in subjects with mild cognitive impairment. Alzheimer’s Dement. 2008, 4, T483. [Google Scholar] [CrossRef]
- Morimoto, B.H.; Schmechel, D.; Hirman, J.; Blackwell, A.; Keith, J.; Gold, M. A Double-Blind, Placebo-Controlled, Ascending-Dose, Randomized Study to Evaluate the Safety, Tolerability and Effects on Cognition of AL-108 after 12 Weeks of Intranasal Administration in Subjects with Mild Cognitive Impairment. Dement. Geriatr. Cogn. Disord. 2013, 35, 325–339. [Google Scholar] [CrossRef]
- Fleming, S.M.; Mulligan, C.K.; Richter, F.; Mortazavi, F.; Lemesre, V.; Frias, C.; Zhu, C.; Stewart, A.; Gozes, I.; Morimoto, B.; et al. A pilot trial of the microtubule-interacting peptide (NAP) in mice overexpressing alpha-synuclein shows improvement in motor function and reduction of alpha-synuclein inclusions. Mol. Cell. Neurosci. 2011, 46, 597–606. [Google Scholar] [CrossRef] [Green Version]
- Magen, I.; Ostritsky, R.; Richter, F.; Zhu, C.; Fleming, S.M.; Lemesre, V.; Stewart, A.J.; Morimoto, B.H.; Gozes, I.; Chesselet, M. Intranasal NAP (davunetide) decreases tau hyperphosphorylation and moderately improves behavioral deficits in mice overexpressing α-synuclein. Pharmacol. Res. Perspect. 2014, 2, e00065. [Google Scholar] [CrossRef]
- May, M.J.; D’Acquisto, F.; Madge, L.A.; Glockner, J.; Pober, J.S.; Ghosh, S. Selective inhibition of NF-kappaB activation by a peptide that blocks the interaction of NEMO with the IkappaB kinase complex. Science 2000, 289, 1550–1554. [Google Scholar] [CrossRef] [PubMed]
- May, M.J.; Marienfeld, R.B.; Ghosh, S. Characterization of the IκB-kinase NEMO Binding Domain. J. Biol. Chem. 2002, 277, 45992–46000. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.C. NF-κB signaling in inflammation. Signal Transduct. Target. Ther. 2017, 2, 17023. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.; Zhou, M.; Ge, Y.; Wang, X. SIRT1 and aging related signaling pathways. Mech. Ageing Dev. 2020, 187, 111215. [Google Scholar] [CrossRef] [PubMed]
- Jones, S.V.; Kounatidis, I. Nuclear Factor-Kappa B and Alzheimer Disease, Unifying Genetic and Environmental Risk Factors from Cell to Humans. Front. Immunol. 2017, 8, 1805. [Google Scholar] [CrossRef] [Green Version]
- Rangasamy, S.B.; Corbett, G.T.; Roy, A.; Modi, K.K.; Bennett, D.A.; Mufson, E.J.; Ghosh, S.; Pahan, K. Intranasal Delivery of NEMO-Binding Domain Peptide Prevents Memory Loss in a Mouse Model of Alzheimer’s Disease. J. Alzheimer’s Dis. 2015, 47, 385–402. [Google Scholar] [CrossRef] [Green Version]
- Mondal, A.C.; Fatima, M. Direct and indirect evidences of BDNF and NGF as key modulators in depression: Role of antidepressants treatment. Int. J. Neurosci. 2019, 129, 283–296. [Google Scholar] [CrossRef]
- Shi, C.-G.; Wang, L.-M.; Wu, Y.; Wang, P.; Gan, Z.-J.; Lin, K.; Jiang, L.-X.; Xu, Z.-Q.; Fan, M. Intranasal Administration of Nerve Growth Factor Produces Antidepressant-Like Effects in Animals. Neurochem. Res. 2010, 35, 1302–1314. [Google Scholar] [CrossRef]
- de Bellis, A.; de Bellis, M.; Aloe, L. Long-Term Non-Invasive Treatment via Intranasal Administration of Nerve Growth Factor Protects the Human Brain in Frontotemporal Dementia associated with Corticobasal Syndrome: A Pilot Study. J. Alzheimer’s Dis. Rep. 2018, 2, 67–77. [Google Scholar] [CrossRef] [Green Version]
- Benarroch, E.E. Neuropeptide Y: Its multiple effects in the CNS and potential clinical significance. Neurology 2009, 72, 1016–1020. [Google Scholar] [CrossRef]
- Sabban, E.L.; Serova, L.I. Potential of Intranasal Neuropeptide Y (NPY) and/or Melanocortin 4 Receptor (MC4R) Antagonists for Preventing or Treating PTSD. Mil. Med. 2018, 183 (Suppl. 1), 408–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mathé, A.A.; Michaneck, M.; Berg, E.; Charney, D.S.; Murrough, J.W. A Randomized Controlled Trial of Intranasal Neuropeptide Y in Patients With Major Depressive Disorder. Int. J. Neuropsychopharmacol. 2020, 23, 783–790. [Google Scholar] [CrossRef] [PubMed]
- Sabban, E.L.; Serova, L.I.; Alaluf, L.G.; Laukova, M.; Peddu, C. Comparative effects of intranasal neuropeptide Y and HS014 in preventing anxiety and depressive-like behavior elicited by single prolonged stress. Behav. Brain Res. 2015, 295, 9–16. [Google Scholar] [CrossRef] [Green Version]
- Serova, L.; Tillinger, A.; Alaluf, L.; Laukova, M.; Keegan, K.; Sabban, E. Single intranasal neuropeptide Y infusion attenuates development of PTSD-like symptoms to traumatic stress in rats. Neuroscience 2013, 236, 298–312. [Google Scholar] [CrossRef] [PubMed]
- Serova, L.; Laukova, M.; Alaluf, L.; Pucillo, L.; Sabban, E. Intranasal neuropeptide Y reverses anxiety and depressive-like behavior impaired by single prolonged stress PTSD model. Eur. Neuropsychopharmacol. 2014, 24, 142–147. [Google Scholar] [CrossRef]
- Laukova, M.; Alaluf, L.G.; Serova, L.I.; Arango, V.; Sabban, E.L. Early Intervention With Intranasal NPY Prevents Single Prolonged Stress-Triggered Impairments in Hypothalamus and Ventral Hippocampus in Male Rats. Endocrinology 2014, 155, 3920–3933. [Google Scholar] [CrossRef]
- Serova, L.I.; Nwokafor, C.; Van Bockstaele, E.J.; Reyes, B.A.; Lin, X.; Sabban, E.L. Single prolonged stress PTSD model triggers progressive severity of anxiety, altered gene expression in locus coeruleus and hypothalamus and effected sensitivity to NPY. Eur. Neuropsychopharmacol. 2019, 29, 482–492. [Google Scholar] [CrossRef]
- Nwokafor, C.; Serova, L.I.; Sabban, E.L. Preclinical findings on the potential of intranasal neuropeptide Y for treating hyperarousal features of PTSD. Ann. New York Acad. Sci. 2019, 1455, 149–159. [Google Scholar] [CrossRef]
- Nwokafor, C.; Serova, L.I.; Nahvi, R.J.; McCloskey, J.; Sabban, E.L. Activation of NPY receptor subtype 1 by [D-His26]NPY is sufficient to prevent development of anxiety and depressive like effects in the single prolonged stress rodent model of PTSD. Neuropeptides 2020, 80, 102001. [Google Scholar] [CrossRef]
- Sabban, E.L.; Laukova, M.; Alaluf, L.G.; Olsson, E.; Serova, L.I. Locus coeruleus response to single-prolonged stress and early intervention with intranasal neuropeptide Y. J. Neurochem. 2015, 135, 975–986. [Google Scholar] [CrossRef] [Green Version]
- Camp, R.; Stier, C.T.; Serova, L.I.; McCloskey, J.; Edwards, J.G.; Reyes-Zaragoza, M.; Sabban, E.L. Cardiovascular responses to intranasal neuropeptide Y in single prolonged stress rodent model of post-traumatic stress disorder. Neuropeptides 2018, 67, 87–94. [Google Scholar] [CrossRef]
- Serova, L.I.; Hansson, E.; Sabban, E.L. Effect of intranasal administration of neuropeptide Y and single prolonged stress on food consumption and body weight in male rats. Neuropeptides 2020, 82, 102060. [Google Scholar] [CrossRef] [PubMed]
- Nahvi, R.; Tanelian, A.; Nwokafor, C.; Sabban, E. Intranasal Neuropeptide Y as a Potential Therapeutic for Stress-Triggered Disorders in Females (2889). Neurology 2021, 96 (Suppl. S15), 2889. [Google Scholar]
- Sayed, S.; Van Dam, N.; Horn, S.R.; Kautz, M.M.; Parides, M.; Costi, S.; Collins, K.A.; Iacoviello, B.; Iosifescu, D.V.; Mathé, A.A.; et al. A Randomized Dose-Ranging Study of Neuropeptide Y in Patients with Posttraumatic Stress Disorder. Int. J. Neuropsychopharmacol. 2018, 21, 3–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fatoba, O.; Kloster, E.; Saft, C.; Gold, R.; Arning, L.; Ellrichmann, G. L22 Intranasal application of NPY and NPY13–36 ameliorate disease pathology in R6/2 mouse model of huntington’s disease. J. Neurol. Neurosurg. Psychiatry 2016, 87 (Suppl. 1), A97. [Google Scholar] [CrossRef]
- Neumann, I.D.; Landgraf, R. Balance of brain oxytocin and vasopressin: Implications for anxiety, depression, and social behaviors. Trends Neurosci. 2012, 35, 649–659. [Google Scholar] [CrossRef]
- Meyer-Lindenberg, A.; Domes, G.; Kirsch, P.; Heinrichs, M. Oxytocin and vasopressin in the human brain: Social neuropeptides for translational medicine. Nat. Rev. Neurosci. 2011, 12, 524–538. [Google Scholar] [CrossRef]
- Scantamburlo, G.; Hansenne, M.; Fuchs, S.; Pitchot, W.; Maréchal, P.; Pequeux, C.; Ansseau, M.; Legros, J. Plasma oxytocin levels and anxiety in patients with major depression. Psychoneuroendocrinology 2007, 32, 407–410. [Google Scholar] [CrossRef] [Green Version]
- Cyranowski, J.M.; Hofkens, T.L.; Frank, E.; Seltman, H.; Cai, H.-M.; Amico, J.A. Evidence of Dysregulated Peripheral Oxytocin Release Among Depressed Women. Psychosom. Med. 2008, 70, 967–975. [Google Scholar] [CrossRef] [Green Version]
- Yamasue, H.; Domes, G. Oxytocin and Autism Spectrum Disorders; Springer International Publishing: Cham, Switzerland, 2017; pp. 449–465. [Google Scholar]
- Rich, M.E.; Caldwell, H.K. A Role for Oxytocin in the Etiology and Treatment of Schizophrenia. Front. Endocrinol. 2015, 6, 90. [Google Scholar] [CrossRef] [Green Version]
- Huang, H.; Michetti, C.; Busnelli, M.; Managò, F.; Sannino, S.; Scheggia, D.; Giancardo, L.; Sona, D.; Murino, V.; Chini, B.; et al. Chronic and Acute Intranasal Oxytocin Produce Divergent Social Effects in Mice. Neuropsychopharmacology 2014, 39, 1102–1114. [Google Scholar] [CrossRef] [Green Version]
- Hara, Y.; Ago, Y.; Higuchi, M.; Hasebe, S.; Nakazawa, T.; Hashimoto, H.; Matsuda, T.; Takuma, K. Oxytocin attenuates deficits in social interaction but not recognition memory in a prenatal valproic acid-induced mouse model of autism. Horm. Behav. 2017, 96, 130–136. [Google Scholar] [CrossRef] [PubMed]
- Kitagawa, K.; Matsumura, K.; Baba, M.; Kondo, M.; Takemoto, T.; Nagayasu, K.; Ago, Y.; Seiriki, K.; Hayata-Takano, A.; Kasai, A.; et al. Intranasal oxytocin administration ameliorates social behavioral deficits in a POGZWT/Q1038R mouse model of autism spectrum disorder. Mol. Brain 2021, 14, 56. [Google Scholar] [CrossRef]
- Dadds, M.R.; Macdonald, E.; Cauchi, A.; Williams, K.; Levy, F.; Brennan, J. Nasal Oxytocin for Social Deficits in Childhood Autism: A Randomized Controlled Trial. J. Autism Dev. Disord. 2014, 44, 521–531. [Google Scholar] [CrossRef] [PubMed]
- Gordon, I.; Wyk, B.C.V.; Bennett, R.H.; Cordeaux, C.; Lucas, M.V.; Eilbott, J.A.; Zagoory-Sharon, O.; Leckman, J.F.; Feldman, R.; Pelphrey, K.A. Oxytocin enhances brain function in children with autism. Proc. Natl. Acad. Sci. USA 2013, 110, 20953–20958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, I.; Jack, A.; Pretzsch, C.; Wyk, B.V.; Leckman, J.F.; Feldman, R.; Pelphrey, K.A. Intranasal Oxytocin Enhances Connectivity in the Neural Circuitry Supporting Social Motivation and Social Perception in Children with Autism. Sci. Rep. 2016, 6, 35054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greene, R.K.; Spanos, M.; Alderman, C.; Walsh, E.; Bizzell, J.; Mosner, M.G.; Kinard, J.L.; Stuber, G.D.; Chandrasekhar, T.; Politte, L.C.; et al. The effects of intranasal oxytocin on reward circuitry responses in children with autism spectrum disorder. J. Neurodev. Disord. 2018, 10, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Strathearn, L.; Kim, S.; Bastian, D.A.; Jung, J.; Iyengar, U.; Martinez, S.; Kochel, R.; Fonagy, P. Visual systemizing preference in children with autism: A randomized controlled trial of intranasal oxytocin. Dev. Psychopathol. 2018, 30, 511–521. [Google Scholar] [CrossRef] [Green Version]
- Auyeung, B.; Lombardo, M.; Heinrichs, M.; Chakrabarti, B.; Sule, A.; Deakin, J.B.; Bethlehem, R.; Dickens, L.; Mooney, N.; Sipple, J.A.N.; et al. Oxytocin increases eye contact during a real-time, naturalistic social interaction in males with and without autism. Transl. Psychiatry 2015, 5, e507. [Google Scholar] [CrossRef] [Green Version]
- Quintana, D.S.; Westlye, L.T.; Hope, S.; Nærland, T.; Elvsåshagen, T.; Dørum, E.; Rustan, Ø.; Valstad, M.; Rezvaya, L.; Lishaugen, H.; et al. Dose-dependent social-cognitive effects of intranasal oxytocin delivered with novel Breath Powered device in adults with autism spectrum disorder: A randomized placebo-controlled double-blind crossover trial. Transl. Psychiatry 2017, 7, e1136. [Google Scholar] [CrossRef] [Green Version]
- Kruppa, J.A.; Gossen, A.; Weiß, E.O.; Kohls, G.; Großheinrich, N.; Cholemkery, H.; Freitag, C.M.; Karges, W.; Wölfle, E.; Sinzig, J.; et al. Neural modulation of social reinforcement learning by intranasal oxytocin in male adults with high-functioning autism spectrum disorder: A randomized trial. Neuropsychopharmacology 2019, 44, 749–756. [Google Scholar] [CrossRef]
- Le, J.; Kou, J.; Zhao, W.; Fu, M.; Zhang, Y.; Becker, B.; Kendrick, K.M. Oxytocin biases eye-gaze to dynamic and static social images and the eyes of fearful faces: Associations with trait autism. Transl. Psychiatry 2020, 10, 142. [Google Scholar] [CrossRef] [PubMed]
- Mayer, A.V.; Wermter, A.-K.; Stroth, S.; Alter, P.; Haberhausen, M.; Stehr, T.; Paulus, F.M.; Krach, S.; Kamp-Becker, I. Randomized clinical trial shows no substantial modulation of empathy-related neural activation by intranasal oxytocin in autism. Sci. Rep. 2021, 11, 15056. [Google Scholar] [CrossRef] [PubMed]
- Bernaerts, S.; Boets, B.; Bosmans, G.; Steyaert, J.; Alaerts, K. Behavioral effects of multiple-dose oxytocin treatment in autism: A randomized, placebo-controlled trial with long-term follow-up. Mol. Autism 2020, 11, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alaerts, K.; Steyaert, J.; Vanaudenaerde, B.; Wenderoth, N.; Bernaerts, S. Changes in endogenous oxytocin levels after intranasal oxytocin treatment in adult men with autism: An exploratory study with long-term follow-up. Eur. Neuropsychopharmacol. 2021, 43, 147–152. [Google Scholar] [CrossRef] [PubMed]
- Bernaerts, S.; Boets, B.; Steyaert, J.; Wenderoth, N.; Alaerts, K. Oxytocin treatment attenuates amygdala activity in autism: A treatment-mechanism study with long-term follow-up. Transl. Psychiatry 2020, 10, 383. [Google Scholar] [CrossRef] [PubMed]
- Kosaka, H.; Okamoto, Y.; Munesue, T.; Yamasue, H.; Inohara, K.; Fujioka, T.; Anme, T.; Orisaka, M.; Ishitobi, M.; Jung, M.; et al. Oxytocin efficacy is modulated by dosage and oxytocin receptor genotype in young adults with high-functioning autism: A 24-week randomized clinical trial. Transl. Psychiatry 2016, 6, e872. [Google Scholar] [CrossRef] [Green Version]
- Goldman, M.B.; Gomes, A.M.; Carter, C.S.; Lee, R. Divergent effects of two different doses of intranasal oxytocin on facial affect discrimination in schizophrenic patients with and without polydipsia. Psychopharmacology 2011, 216, 101–110. [Google Scholar] [CrossRef]
- Fischer-Shofty, M.; Brüne, M.; Ebert, A.; Shefet, D.; Levkovitz, Y.; Shamay-Tsoory, S. Improving social perception in schizophrenia: The role of oxytocin. Schizophr. Res. 2013, 146, 357–362. [Google Scholar] [CrossRef]
- Davis, M.C.; Lee, J.; Horan, W.P.; Clarke, A.D.; McGee, M.R.; Green, M.F.; Marder, S.R. Effects of single dose intranasal oxytocin on social cognition in schizophrenia. Schizophr. Res. 2013, 147, 393–397. [Google Scholar] [CrossRef]
- Woolley, J.; Chuang, B.; Lam, O.; Lai, W.; O’Donovan, A.; Rankin, K.; Mathalon, D.; Vinogradov, S. Oxytocin administration enhances controlled social cognition in patients with schizophrenia. Psychoneuroendocrinology 2014, 47, 116–125. [Google Scholar] [CrossRef] [Green Version]
- Shin, N.Y.; Park, H.Y.; Jung, W.H.; Park, J.W.; Yun, J.-Y.; Jang, J.H.; Kim, S.N.; Han, H.J.; Kim, S.-Y.; Kang, D.-H.; et al. Effects of Oxytocin on Neural Response to Facial Expressions in Patients with Schizophrenia. Neuropsychopharmacology 2015, 40, 1919–1927. [Google Scholar] [CrossRef] [PubMed]
- Bradley, E.R.; Seitz, A.; Niles, A.N.; Rankin, K.P.; Mathalon, D.H.; O’Donovan, A.; Woolley, J.D. Oxytocin increases eye gaze in schizophrenia. Schizophr. Res. 2019, 212, 177–185. [Google Scholar] [CrossRef] [PubMed]
- de Macedo, L.R.; Zuardi, A.W.; Machado-de-Sousa, J.P.; Chagas, M.H.; Hallak, J.E. Oxytocin does not improve performance of patients with schizophrenia and healthy volunteers in a facial emotion matching task. Psychiatry Res. 2014, 220, 125–128. [Google Scholar] [CrossRef]
- Caravaggio, F.; Gerretsen, P.; Mar, W.; Chung, J.K.; Plitman, E.; Nakajima, S.; Kim, J.; Iwata, Y.; Patel, R.; Chakravarty, M.M.; et al. Intranasal oxytocin does not modulate jumping to conclusions in schizophrenia: Potential interactions with caudate volume and baseline social functioning. Psychoneuroendocrinology 2017, 81, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.C.; Green, M.F.; Lee, J.; Horan, W.P.; Şentürk, D.; Clarke, A.D.; Marder, S.R. Oxytocin-Augmented Social Cognitive Skills Training in Schizophrenia. Neuropsychopharmacology 2014, 39, 2070–2077. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchanan, R.W.; Kelly, D.L.; Strauss, G.P.; Gold, J.M.; Weiner, E.; Zaranski, J.; Chen, S.; Blatt, F.; Holden, J.; Granholm, E. Combined Oxytocin and Cognitive Behavioral Social Skills Training for Social Function in People With Schizophrenia. J. Clin. Psychopharmacol. 2021, 41, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.R.; Wehring, H.J.; McMahon, R.P.; Liu, F.; Linthicum, J.; Buchanan, R.W.; Strauss, G.P.; Rubin, L.H.; Kelly, D.L. The Effect of Intranasal Oxytocin on Measures of Social Cognition in Schizophrenia: A Negative Report. J. Psychiatry Brain Sci. 2019, 4, e190001. [Google Scholar]
- Conley, R.R.; Boggs, D.L.; Kelly, D.L.; McMahon, R.P.; Dickinson, D.; Feldman, S.; Ball, M.P.; Buchanan, R.W. The Effects of Galantamine on Psychopathology in Chronic Stable Schizophrenia. Clin. Neuropharmacol. 2009, 32, 69–74. [Google Scholar] [CrossRef] [Green Version]
- Jarskog, L.F.; Pedersen, C.A.; Johnson, J.L.; Hamer, R.M.; Rau, S.W.; Elliott, T.; Penn, D.L. A 12-week randomized controlled trial of twice-daily intranasal oxytocin for social cognitive deficits in people with schizophrenia. Schizophr. Res. 2017, 185, 88–95. [Google Scholar] [CrossRef]
- Halverson, T.; Jarskog, L.F.; Pedersen, C.; Penn, D. Effects of oxytocin on empathy, introspective accuracy, and social symptoms in schizophrenia: A 12-week twice-daily randomized controlled trial. Schizophr. Res. 2019, 204, 178–182. [Google Scholar] [CrossRef]
- Dagani, J.; Sisti, D.; Abelli, M.; Di Paolo, L.; Pini, S.; Raimondi, S.; Rocchi, M.B.; Saviotti, F.M.; Scocco, P.; Totaro, S.; et al. Do we need oxytocin to treat schizophrenia? A randomized clinical trial. Schizophr. Res. 2016, 172, 158–164. [Google Scholar] [CrossRef] [PubMed]
- Frijling, J.L.; Van Zuiden, M.; Koch, S.B.J.; Nawijn, L.; Veltman, D.J.; Olff, M. Intranasal Oxytocin Affects Amygdala Functional Connectivity after Trauma Script-Driven Imagery in Distressed Recently Trauma-Exposed Individuals. Neuropsychopharmacology 2016, 41, 1286–1296. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frijling, J.L.; van Zuiden, M.; Koch, S.B.J.; Nawijn, L.; Veltman, D.J.; Olff, M. Effects of intranasal oxytocin on amygdala reactivity to emotional faces in recently trauma-exposed individuals. Soc. Cogn. Affect. Neurosci. 2016, 11, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Frijling, J.L. Preventing PTSD with oxytocin: Effects of oxytocin administration on fear neurocircuitry and PTSD symptom development in recently trauma-exposed individuals. Eur. J. Psychotraumatology 2017, 8, 1302652. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Zuiden, M.; Frijling, J.L.; Nawijn, L.; Koch, S.B.; Goslings, J.C.; Luitse, J.S.; Biesheuvel, T.H.; Honig, A.; Veltman, D.J.; Olff, M. Intranasal Oxytocin to Prevent Posttraumatic Stress Disorder Symptoms: A Randomized Controlled Trial in Emergency Department Patients. Biol. Psychiatry 2017, 81, 1030–1040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koch, S.B.; Van Zuiden, M.; Nawijn, L.; Frijling, J.L.; Veltman, D.J.; Olff, M. Intranasal Oxytocin Administration Dampens Amygdala Reactivity towards Emotional Faces in Male and Female PTSD Patients. Neuropsychopharmacology 2016, 41, 1495–1504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nawijn, L.; van Zuiden, M.; Koch, S.B.; Frijling, J.L.; Veltman, D.J.; Olff, M. Intranasal oxytocin increases neural responses to social reward in post-traumatic stress disorder. Soc. Cogn. Affect. Neurosci. 2017, 12, 212–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nawijn, L.; van Zuiden, M.; Koch, S.B.; Frijling, J.L.; Veltman, D.J.; Olff, M. Intranasal oxytocin enhances neural processing of monetary reward and loss in post-traumatic stress disorder and traumatized controls. Psychoneuroendocrinology 2016, 66, 228–237. [Google Scholar] [CrossRef]
- Koch, S.B.; van Zuiden, M.; Nawijn, L.; Frijling, J.L.; Veltman, D.J.; Olff, M. Effects of intranasal oxytocin on distraction as emotion regulation strategy in patients with post-traumatic stress disorder. Eur. Neuropsychopharmacol. 2019, 29, 266–277. [Google Scholar] [CrossRef]
- Sack, M.; Spieler, D.; Wizelman, L.; Epple, G.; Stich, J.; Zaba, M.; Schmidt, U. Intranasal oxytocin reduces provoked symptoms in female patients with posttraumatic stress disorder despite exerting sympathomimetic and positive chronotropic effects in a randomized controlled trial. BMC Med. 2017, 15, 40. [Google Scholar] [CrossRef] [Green Version]
- Flanagan, J.C.; Sippel, L.M.; Wahlquist, A.; Maria, M.M.M.-S.; Back, S.E. Augmenting Prolonged Exposure therapy for PTSD with intranasal oxytocin: A randomized, placebo-controlled pilot trial. J. Psychiatr. Res. 2018, 98, 64–69. [Google Scholar] [CrossRef] [PubMed]
- Sippel, L.M.; Flanagan, J.C.; Holtzheimer, P.E.; Moran-Santa-Maria, M.M.; Brady, K.T.; Joseph, J.E. Effects of intranasal oxytocin on threat- and reward-related functional connectivity in men and women with and without childhood abuse-related PTSD. Psychiatry Res. Neuroimaging 2021, 317, 111368. [Google Scholar] [CrossRef] [PubMed]
- Guastella, A.J.; Howard, A.L.; Dadds, M.; Mitchell, P.; Carson, D.S. A randomized controlled trial of intranasal oxytocin as an adjunct to exposure therapy for social anxiety disorder. Psychoneuroendocrinology 2009, 34, 917–923. [Google Scholar] [CrossRef] [PubMed]
- Fang, A.; Hoge, E.A.; Heinrichs, M.; Hofmann, S.G. Attachment Style Moderates the Effects of Oxytocin on Social Behaviors and Cognitions During Social Rejection. Clin. Psychol. Sci. 2014, 2, 740–747. [Google Scholar] [CrossRef] [PubMed]
- Fang, A.; Treadway, M.T.; Hofmann, S.G. Working hard for oneself or others: Effects of oxytocin on reward motivation in social anxiety disorder. Biol. Psychol. 2017, 127, 157–162. [Google Scholar] [CrossRef] [PubMed]
- Labuschagne, I.; Phan, K.L.; Wood, A.; Angstadt, M.; Chua, P.; Heinrichs, M.; Stout, J.; Nathan, P.J. Oxytocin Attenuates Amygdala Reactivity to Fear in Generalized Social Anxiety Disorder. Neuropsychopharmacology 2010, 35, 2403–2413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Labuschagne, I.; Phan, K.L.; Wood, A.; Angstadt, M.; Chua, P.; Heinrichs, M.; Stout, J.C.; Nathan, P.J. Medial frontal hyperactivity to sad faces in generalized social anxiety disorder and modulation by oxytocin. Int. J. Neuropsychopharmacol. 2012, 15, 883–896. [Google Scholar] [CrossRef] [Green Version]
- Gorka, S.M.; Fitzgerald, D.A.; Labuschagne, I.; Hosanagar, A.; Wood, A.; Nathan, P.J.; Phan, K.L. Oxytocin Modulation of Amygdala Functional Connectivity to Fearful Faces in Generalized Social Anxiety Disorder. Neuropsychopharmacology 2015, 40, 278–286. [Google Scholar] [CrossRef]
- Dodhia, S.; Hosanagar, A.; Fitzgerald, D.A.; Labuschagne, I.; Wood, A.; Nathan, P.J.; Phan, K.L. Modulation of Resting-State Amygdala-Frontal Functional Connectivity by Oxytocin in Generalized Social Anxiety Disorder. Neuropsychopharmacology 2014, 39, 2061–2069. [Google Scholar] [CrossRef] [Green Version]
- Finger, E.C.; MacKinley, J.; Blair, M.; Oliver, L.D.; Jesso, S.; Tartaglia, M.C.; Borrie, M.; Wells, J.; Dziobek, I.; Pasternak, S.; et al. Oxytocin for frontotemporal dementia: A randomized dose-finding study of safety and tolerability. Neurology 2015, 84, 174–181. [Google Scholar] [CrossRef] [Green Version]
- Finger, E.; Jesso, S.; Rankin, K.; Boxer, A.; Kertesz, A. A Dose Finding Study of Intranasal Oxytocin in Behavioural Variant Frontotemporal Dementia (S29.007). Neurology 2012, 78, S29007. [Google Scholar] [CrossRef]
- Oliver, L.D.; Stewart, C.; Coleman, K.; Kryklywy, J.H.; Bartha, R.; Mitchell, D.G.; Finger, E.C. Neural effects of oxytocin and mimicry in frontotemporal dementia. Neurology 2020, 95, e2635–e2647. [Google Scholar] [CrossRef]
- Hirabayashi, T.; Nakamachi, T.; Shioda, S. Discovery of PACAP and its receptors in the brain. J. Headache Pain 2018, 19, 28. [Google Scholar] [CrossRef]
- Nonaka, N.; Farr, S.A.; Nakamachi, T.; Morley, J.E.; Nakamura, M.; Shioda, S.; Banks, W.A. Intranasal administration of PACAP: Uptake by brain and regional brain targeting with cyclodextrins. Peptides 2012, 36, 168–175. [Google Scholar] [CrossRef] [Green Version]
- Reglodi, D.; Kiss, P.; Szabadfi, K.; Atlasz, T.; Gabriel, R.; Horvath, G.; Szakaly, P.; Sandor, B.; Lubics, A.; Laszlo, E.; et al. PACAP is an Endogenous Protective Factor—Insights from PACAP-Deficient Mice. J. Mol. Neurosci. 2012, 48, 482–492. [Google Scholar] [CrossRef]
- Han, P.; Caselli, R.J.; Baxter, L.; Serrano, G.; Yin, J.; Beach, T.G.; Reiman, E.M.; Shi, J. Association of Pituitary Adenylate Cyclase–Activating Polypeptide With Cognitive Decline in Mild Cognitive Impairment Due to Alzheimer Disease. JAMA Neurol. 2015, 72, 333. [Google Scholar] [CrossRef] [Green Version]
- Reglodi, D.; Atlasz, T.; Jungling, A.; Szabo, E.; Kovari, P.; Manavalan, S.; Tamas, A. Alternative Routes of Administration of the Neuroprotective Pituitary Adenylate Cyclase Activating Polypeptide. Curr. Pharm. Des. 2018, 24, 3892–3904. [Google Scholar] [CrossRef]
- Rat, D.; Schmitt, U.; Tippmann, F.; Dewachter, I.; Theunis, C.; Wieczerzak, E.; Postina, R.; Van Leuven, F.; Fahrenholz, F.; Kojro, E. Neuropeptide pituitary adenylate cyclase-activating polypeptide (PACAP) slows down Alzheimer’s disease-like pathology in amyloid precursor protein-transgenic mice. FASEB J. 2011, 25, 3208–3218. [Google Scholar] [CrossRef] [Green Version]
- Cabezas-Llobet, N.; Vidal-Sancho, L.; Masana, M.; Fournier, A.; Alberch, J.; Vaudry, D.; Xifró, X. Pituitary Adenylate Cyclase-Activating Polypeptide (PACAP) Enhances Hippocampal Synaptic Plasticity and Improves Memory Performance in Huntington’s Disease. Mol. Neurobiol. 2018, 55, 8263–8277. [Google Scholar] [CrossRef]
- Solés-Tarrés, I.; Cabezas-Llobet, N.; Lefranc, B.; Leprince, J.; Alberch, J.; Vaudry, D.; Xifró, X. Pituitary Adenylate Cyclase-Activating Polypeptide (PACAP) Protects Striatal Cells and Improves Motor Function in Huntington’s Disease Models: Role of PAC1 Receptor. Front. Pharmacol. 2021, 12, 797541. [Google Scholar] [CrossRef]
- Arnegard, M.E.; Whitten, L.A.; Hunter, C.; Clayton, J.A. Sex as a Biological Variable: A 5-Year Progress Report and Call to Action. J. Women’s Health 2020, 29, 858–864. [Google Scholar] [CrossRef]


{kind=link}
| Peptide | Disorder | Preclinical (Rodents) | Sample Size | References | Clinical | Sample Size | References | |
|---|---|---|---|---|---|---|---|---|
| BDNF | NPD | MDD | ⇑hippocampal BDNF ⇑body weight ⇓anxiety-like behaviors | 80–83 | [26,27] | -- | -- | -- |
| EPO | ND | AD | ⇑locomotor activity, cognition, memory ⇓microglial activation ⇓TNF, Bax/Bcl2-ratio, Fas, IL-1β ∅ hippocampal neurodegeneration | 84–154 | [30,31] | -- | -- | -- |
| GDNF | ND | PD | ⇑locomotor activity ⇓microglial hyperinflammation ⇓lesion severity (40%) ∅6-OHDA induced weight loss ⊕dopaminergic neurons | 16–53 | [36,37,38] | -- | -- | -- |
| GLP-1 | ND | AD | ⇑spatial learning and memory ⇓Aβ plaque formation ∅circadian rhythm disturbances | 3–8 | [40,41] | -- | -- | -- |
| Insulin/IGF-1 | NDD | ASD | -- | -- | -- | ⇑social and cognitive improvement ⇓ restrictive behaviors in children <3yo | 9–25 | [49,50] |
| BD | -- | -- | -- | No significant effects | 62 | [52] | ||
| SZ | -- | -- | -- | ⇑POMC expression | 39–45 | [53,54,55] | ||
| NPD | MDD | -- | -- | -- | No significant effects | 35 | [56] | |
| GSAD | -- | -- | -- | ⇓skin conductance response ⇓saliva and plasma cortisol | 26–123 | [61] | ||
| ND | AD | ⇑ glucose uptake/CNS insulin signaling ⇑cognition ⇑ neurogenesis ⇓ Aβ, tau ⇓microglial activation | 7–44 | [65,66,67,68,69,70] | ⇑ cognition ⇑ working memory/recall ⇓peripheral insulin resistance | 12–289 | [71,72,73,74,75,76,77,78,79] | |
| PD | ⇑motor performance ⇓ODHA neurotoxicity/DAergic neuron loss ⊕ CNS insulin/Akt/GSK3β signaling | 8–39 | [80,81,82] | ⇑ motor performance ⇑ FAS word score ⇓ parkinsonism severity | 8 | [83] | ||
| HD | ⇑locomotor activity ⇑cortical & striatal Akt/mTOR signaling ⇓mHtt phosphorylation | 32 | [84] | -- | -- | -- | ||
| NAP | NDD | ASD | ⇑cognition ⇓ inflammatory cytokines (TNF, IL-6, IL-12) ⊕structural abnormalities ∅irregular immune activation | 12–28 | [87,92,93,94] | -- | -- | -- |
| SZ | ⇑cognitive function ⇓ anxiety ⊕Foxp2 expression | 61 | [95,96,99] | ⇑functionally-significant cognition | 63 | [100] | ||
| ND | AD | ⇑ cognitive performance ⇓Aβ40/Aβ42, tau ⇓anxiety-like behaviors ∅loss of choline acetyltransferase activity | 20–35 | [101,102,103,104,105,106] | No significant effects | 144 | [107,108,109] | |
| PD | ⇑locomotor activity ∅dopaminergic neuron loss | 56 | [110,111] | -- | -- | -- | ||
| NBD | ND | AD | ⇑spatial learning and memory ⇓Aβ plaque formation ⇓neurodegeneration ∅NF-kB activation, neuroinflammation ⊕CREB, mGluR1 expression | 24 | [117] | -- | -- | -- |
| NGF | NPD | MDD | ⇑hippocampal neurogenesis ⇓inflammatory markers ∅stress-induced anhedonia ⊕cortical & hippocampal 5-HT expression | 24 | [119] | -- | -- | -- |
| ND | FTD | -- | -- | -- | ⇑word usage ⇓rigidity | 2 | [120] | |
| NPY | NPD | MDD | -- | -- | -- | ⇓MDD severity (on MARDS) at +5 and +24 h | 30 | [123] |
| PTSD | ⇓depressive and anxiety behaviors ∅stress-induced increases in ACTH, corticosterone, hippocampal glucocorticoids ∅hyperarousal | 18–36 | [122,125,126,127,128,129,130,131,132,133,134] | ⇓anxiety (BAI) | 24 | [135] | ||
| ND | HD | ⇑locomotor activity ⇓mHtt phosphorylation ⇓microglial hyperinflammation | 10 | [136] | -- | -- | -- | |
| Oxytocin | NDD | ASD | ⇑c-Fos in PVN, PFC, and SSC ⇑opposite-sex social behaviors ⇓(selective) OXTr expression ⊕POGZ-reduced OXTr expression | 21–44 | [143,144,145] | ⇑striatal, prefrontal, and motor activity ⇑social-emotional cognition ∅eye fixation anomalies ⊕bilateral amygdala activity | 17–38 | [146,147,148,149,150,151,152,153,154,156,157,158,159] |
| SZ | -- | -- | -- | ⇑controlled cognition ⇑empathetic accuracy ⇑outcomes with combined skills therapy ⊕bilateral amygdala activity | 23–68 | [160,161,162,163,164,165,166,167,168,169,170,171,173,174] | ||
| NPD | PTSD | -- | -- | -- | ⊕multi-regional amygdala connectivity | 34–107 | [175,176,177,178,179,180,181,182,183,184,185] | |
| GSAD | -- | -- | -- | ∅abnormal PFC and ACC activity ⊕amygdala-frontal connectivity | 36 | [186,187,188,189,190,191,192] | ||
| ND | FTD | -- | -- | -- | ⇑frontotemporal and limbic connectivity ⇑empathy ⇓apathy | 25–60 | [193,194,195] | |
| PACAP | ND | AD | ⇑nonamyloidogenic processing ⇓BDNF, Bax/Bcl2-ratio ⊕cognitive function | 14–30 | [201] | -- | -- | -- |
| HD | ⇑locomotor activity ⇑spatial learning and memory ⇑hippocampal VGlut1,PSD95 ⊕BDNF, CREB-binding protein, PAC1r | 24 | [202,203] | -- | -- | -- | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bose, M.; Farias Quipildor, G.; Ehrlich, M.E.; Salton, S.R. Intranasal Peptide Therapeutics: A Promising Avenue for Overcoming the Challenges of Traditional CNS Drug Development. Cells 2022, 11, 3629. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11223629
Bose M, Farias Quipildor G, Ehrlich ME, Salton SR. Intranasal Peptide Therapeutics: A Promising Avenue for Overcoming the Challenges of Traditional CNS Drug Development. Cells. 2022; 11(22):3629. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11223629
Chicago/Turabian StyleBose, Meenakshi, Gabriela Farias Quipildor, Michelle E. Ehrlich, and Stephen R. Salton. 2022. "Intranasal Peptide Therapeutics: A Promising Avenue for Overcoming the Challenges of Traditional CNS Drug Development" Cells 11, no. 22: 3629. https://0-doi-org.brum.beds.ac.uk/10.3390/cells11223629