
Unleashed Actin Assembly in Capping Protein-Deficient B16-F1 Cells Enables Identification of Multiple Factors Contributing to Filopodium Formation

Abstract
:1. Introduction
2. Materials and Methods
2.1. Constructs
2.2. Cell Culture and Transfection
2.3. Genome Editing by CRISPR/Cas9
2.4. Protein Purification
2.5. Antibodies
2.6. Immunoblotting
2.7. Immunofluorescence and Imaging
2.8. Quantification of F-Actin Content by Flow Cytometry
2.9. Quantification of the F- to G-Actin Ratio and of Global Actin Levels
2.10. Quantification of Peripheral Filopodia
2.11. Analysis of Focal Adhesions
2.12. Statistical Analyses
3. Results
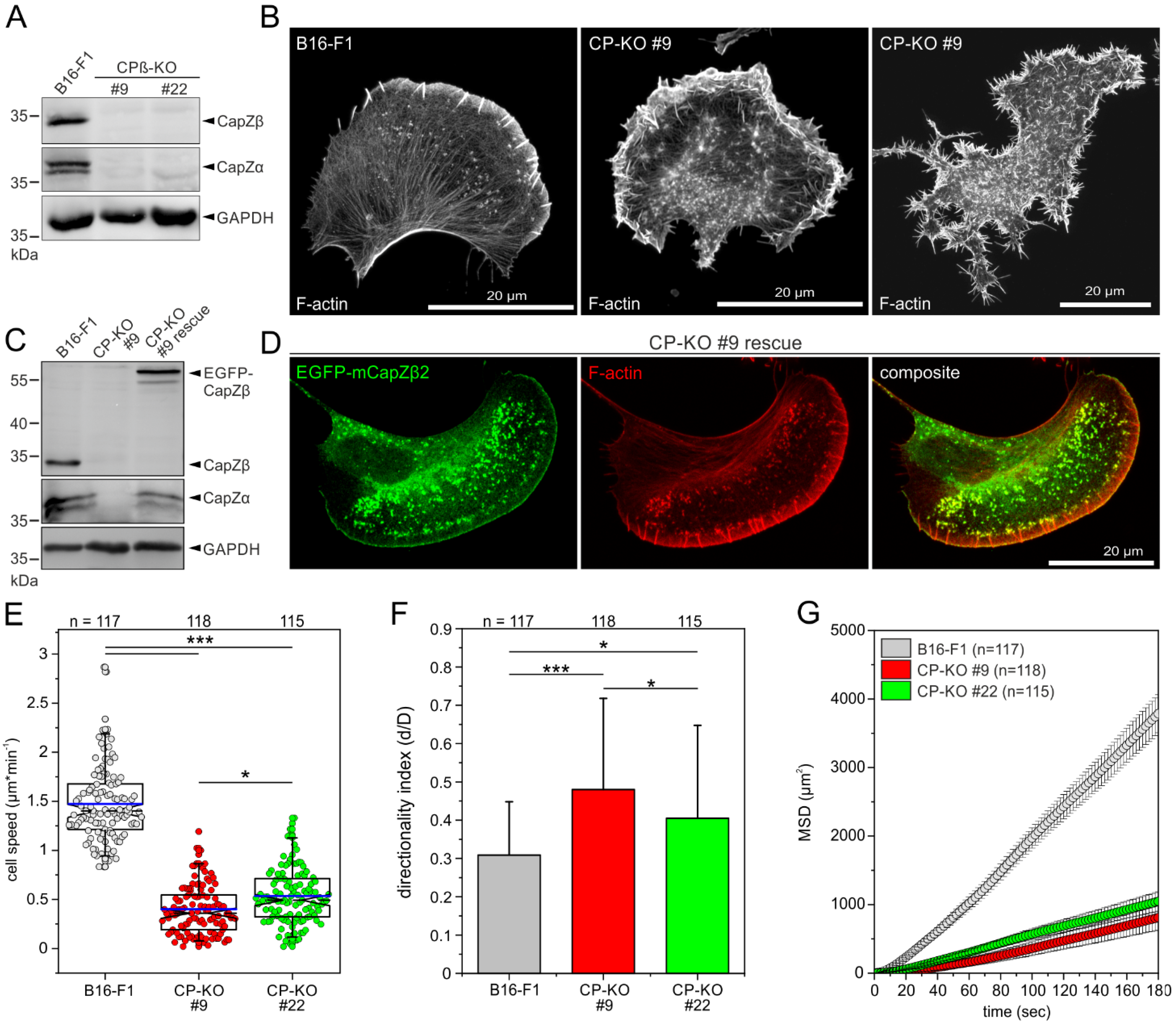
3.1. Loss of CP in B16-F1 Cells Triggers the Massive Formation of Filopodia and Impairs 2D-Cell Migration
3.2. Loss of CP in B16-F1 Cells Increases Global Levels of Filamentous and Total Actin
3.3. Loss of CP in NIH 3T3 Fibroblasts Induces the Massive Formation FAs and SFs Instead of Filopodia on Fibronectin
3.4. CP-Deficent NIH 3T3 Cells form Numerous Filopodia on Poorly Adhesive Substrates
3.5. Loss of CP in Ena/VASP-Deficient B16-F1 Cells Does Not Abrogate Filopodium Formation
3.6. Endogenous MyoX, FMNL2 and -3, but Not mDia2, Reside at the Tips of Filopodia in CP-KO Cells
3.7. Loss of MyoX in EVM/CP-KO Cells Leads to an Additional Reduction of Filopodia Length
3.8. Combined Inactivation of Ena/VASP, MyoX and FMNL2 and -3 Is Required to Drastically Impair Filopodium Formation in CP-Deficient Cells
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Blanchoin, L.; Boujemaa-Paterski, R.; Sykes, C.; Plastino, J. Actin Dynamics, Architecture, and Mechanics in Cell Motility. Physiol. Rev. 2014, 94, 235–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rottner, K.; Faix, J.; Bogdan, S.; Linder, S.; Kerkhoff, E. Actin assembly mechanisms at a glance. J. Cell Sci. 2017, 130, 3427–3435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rottner, K.; Schaks, M. Assembling actin filaments for protrusion. Curr. Opin. Cell Biol. 2019, 56, 53–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krause, M.; Gautreau, A. Steering cell migration: Lamellipodium dynamics and the regulation of directional persistence. Nat. Rev. Mol. Cell Biol. 2014, 15, 577–590. [Google Scholar] [CrossRef]
- Le Clainche, C.; Carlier, M.-F.; Eelen, G.; de Zeeuw, P.; Treps, L.; Harjes, U.; Wong, B.W.; Carmeliet, P.; Schwab, A.; Fabian, A.; et al. Regulation of Actin Assembly Associated with Protrusion and Adhesion in Cell Migration. Physiol. Rev. 2008, 88, 489–513. [Google Scholar] [CrossRef] [Green Version]
- Lappalainen, P.; Kotila, T.; Jégou, A.; Romet-Lemonne, G. Biochemical and mechanical regulation of actin dynamics. Nat. Rev. Mol. Cell Biol. 2022, 23, 836–852. [Google Scholar] [CrossRef]
- Pollard, T.D.; Borisy, G.G. Cellular Motility Driven by Assembly and Disassembly of Actin Filaments. Cell 2003, 112, 453–465. [Google Scholar] [CrossRef] [Green Version]
- Svitkina, T.M. Ultrastructure of the actin cytoskeleton. Curr. Opin. Cell Biol. 2018, 54, 1–8. [Google Scholar] [CrossRef]
- Rottner, K.; Stradal, T.E. Actin dynamics and turnover in cell motility. Curr. Opin. Cell Biol. 2011, 23, 569–578. [Google Scholar] [CrossRef]
- Faix, J.; Rottner, K. Ena/VASP proteins in cell edge protrusion, migration and adhesion. J. Cell Sci. 2022, 135, jcs259226. [Google Scholar] [CrossRef]
- Eden, S.; Rohatgi, R.; Podtelejnikov, A.V.; Mann, M.; Kirschner, M.W. Mechanism of regulation of WAVE1-induced actin nucleation by Rac1 and Nck. Nature 2002, 418, 790–793. [Google Scholar] [CrossRef] [PubMed]
- Ismail, A.M.; Padrick, S.B.; Chen, B.; Umetani, J.; Rosen, M.K. The WAVE regulatory complex is inhibited. Nat. Struct. Mol. Biol. 2009, 16, 561–563. [Google Scholar] [CrossRef] [PubMed]
- Molinie, N.; Gautreau, A. The Arp2/3 Regulatory System and Its Deregulation in Cancer. Physiol. Rev. 2018, 98, 215–238. [Google Scholar] [CrossRef]
- Steffen, A.; Ladwein, M.; Dimchev, G.A.; Hein, A.; Schwenkmezger, L.; Arens, S.; Ladwein, K.I.; Holleboom, J.M.; Schur, F.; Small, J.V.; et al. Rac function is critical for cell migration but not required for spreading and focal adhesion formation. J. Cell Sci. 2013, 126, 4572–4588. [Google Scholar] [CrossRef] [Green Version]
- Pollard, T.D. Regulation of Actin Filament Assembly by Arp2/3 Complex and Formins. Annu. Rev. Biophys. Biomol. Struct. 2007, 36, 451–477. [Google Scholar] [CrossRef]
- Xu, Y.; Moseley, J.B.; Sagot, I.; Poy, F.; Pellman, D.; Goode, B.L.; Eck, M.J. Crystal Structures of a Formin Homology-2 Domain Reveal a Tethered Dimer Architecture. Cell 2004, 116, 711–723. [Google Scholar] [CrossRef] [Green Version]
- Paul, A.S.; Pollard, T.D. Energetic Requirements for Processive Elongation of Actin Filaments by FH1FH2-formins. J. Biol. Chem. 2009, 284, 12533–12540. [Google Scholar] [CrossRef] [Green Version]
- Sagot, I.; Rodal, A.A.; Moseley, J.; Goode, B.L.; Pellman, D. An actin nucleation mechanism mediated by Bni1 and Profilin. Nat. Cell Biol. 2002, 4, 626–631. [Google Scholar] [CrossRef]
- Kühn, S.; Geyer, M. Formins as effector proteins of Rho GTPases. Small GTPases 2014, 5, e29513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Block, J.; Stradal, T.; Hänisch, J.; Geffers, R.; Köstler, S.; Urban, E.; Small, J.; Rottner, K.; Faix, J. Filopodia formation induced by active mDia2/Drf3. J. Microsc. 2008, 231, 506–517. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Czech, L.; Gerboth, S.; Kojima, S.-I.; Scita, G.; Svitkina, T. Novel Roles of Formin mDia2 in Lamellipodia and Filopodia Formation in Motile Cells. PLoS Biol. 2007, 5, e317. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Block, J.; Breitsprecher, D.; Kühn, S.; Winterhoff, M.; Kage, F.; Geffers, R.; Duwe, P.; Rohn, J.L.; Baum, B.; Brakebusch, C.; et al. FMNL2 Drives Actin-Based Protrusion and Migration Downstream of Cdc42. Curr. Biol. 2012, 22, 1005–1012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kage, F.; Winterhoff, M.; Dimchev, V.; Mueller, J.; Thalheim, T.; Freise, A.; Brühmann, S.; Kollasser, J.; Block, J.; Dimchev, G.; et al. FMNL formins boost lamellipodial force generation. Nat. Commun. 2017, 8, 14832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, E.S.; Gauvin, T.J.; Heimsath, E.G.; Higgs, H.N. Assembly of filopodia by the formin FRL2 (FMNL3). Cytoskeleton 2010, 67, 755–772. [Google Scholar] [CrossRef] [Green Version]
- Breitsprecher, D.; Kiesewetter, A.K.; Linkner, J.; Urbanke, C.; Resch, G.P.; Small, J.V.; Faix, J. Clustering of VASP actively drives processive, WH2 domain-mediated actin filament elongation. EMBO J. 2008, 27, 2943–2954. [Google Scholar] [CrossRef]
- Hansen, S.D.; Mullins, R.D. VASP is a processive actin polymerase that requires monomeric Actin for barbed end association. J. Cell Biol. 2010, 191, 571–584. [Google Scholar] [CrossRef] [Green Version]
- Breitsprecher, D.; Kiesewetter, A.K.; Linkner, J.; Vinzenz, M.; Stradal, T.E.B.; Small, J.V.; Curth, U.; Dickinson, R.B.; Faix, J. Molecular mechanism of Ena/VASP-mediated Actin-filament elongation. EMBO J. 2011, 30, 456–467. [Google Scholar] [CrossRef]
- Brühmann, S.; Ushakov, D.S.; Winterhoff, M.; Dickinson, R.B.; Curth, U.; Faix, J. Distinct VASP tetramers synergize in the processive elongation of individual actin filaments from clustered arrays. Proc. Natl. Acad. Sci. USA 2017, 114, E5815–E5824. [Google Scholar] [CrossRef] [Green Version]
- Disanza, A.; Bisi, S.; Winterhoff, M.; Milanesi, F.; Ushakov, D.S.; Kast, D.; Marighetti, P.; Romet-Lemonne, G.; Müller, H.-M.; Nickel, W.; et al. CDC42 switches IRSp53 from inhibition of actin growth to elongation by clustering of VASP. EMBO J. 2013, 32, 2735–2750. [Google Scholar] [CrossRef] [Green Version]
- Hansen, S.D.; Mullins, R.D. Lamellipodin promotes actin assembly by clustering Ena/VASP proteins and tethering them to actin filaments. Elife 2015, 4, e06585. [Google Scholar] [CrossRef]
- Pokrant, T.; Hein, J.I.; Körber, S.; Disanza, A.; Pich, A.; Scita, G.; Rottner, K.; Faix, J. Ena/VASP clustering at microspike tips involves lamellipodin but not I-BAR proteins, and absolutely requires unconventional myosin-X. Proc. Natl. Acad. Sci. USA 2023, 120, e2217437120. [Google Scholar] [CrossRef] [PubMed]
- Dent, E.; Kwiatkowski, A.V.; Mebane, L.M.; Philippar, U.; Barzik, M.; Rubinson, D.A.; Gupton, S.; van Veen, J.E.; Furman, C.; Zhang, J.; et al. Filopodia are required for cortical neurite initiation. Nat. Cell Biol. 2007, 9, 1347–1359. [Google Scholar] [CrossRef]
- Kwiatkowski, A.V.; Rubinson, D.A.; Dent, E.W.; van Veen, J.E.; Leslie, J.D.; Zhang, J.; Mebane, L.M.; Philippar, U.; Pinheiro, E.M.; Burds, A.A.; et al. Ena/VASP Is Required for Neuritogenesis in the Developing Cortex. Neuron 2007, 56, 441–455. [Google Scholar] [CrossRef] [Green Version]
- Damiano-Guercio, J.; Kurzawa, L.; Mueller, J.; Dimchev, G.; Schaks, M.; Nemethova, M.; Pokrant, T.; Brühmann, S.; Linkner, J.; Blanchoin, L.; et al. Loss of Ena/VASP interferes with lamellipodium architecture, motility and integrin-dependent adhesion. Elife 2020, 9, e55351. [Google Scholar] [CrossRef] [PubMed]
- Dimchev, G.; Amiri, B.; Fäßler, F.; Falcke, M.; Schur, F.K. Computational toolbox for ultrastructural quantitative analysis of filament networks in cryo-ET data. J. Struct. Biol. 2021, 213, 107808. [Google Scholar] [CrossRef]
- Small, J.; Stradal, T.; Vignal, E.; Rottner, K. The lamellipodium: Where motility begins. Trends Cell Biol. 2002, 12, 112–120. [Google Scholar] [CrossRef]
- Mellor, H. The role of formins in filopodia formation. Biochim. Biophys. Acta 2010, 1803, 191–200. [Google Scholar] [CrossRef] [Green Version]
- Heimsath, E.G., Jr.; Yim, Y.-I.; Mustapha, M.; Hammer, J.A.; Cheney, R.E. Myosin-X knockout is semi-lethal and demonstrates that myosin-X functions in neural tube closure, pigmentation, hyaloid vasculature regression, and filopodia formation. Sci. Rep. 2017, 7, 17354. [Google Scholar] [CrossRef] [PubMed]
- Mejillano, M.R.; Kojima, S.-I.; Applewhite, D.A.; Gertler, F.B.; Svitkina, T.M.; Borisy, G.G. Lamellipodial Versus Filopodial Mode of the Actin Nanomachinery: Pivotal Role of the Filament Barbed End. Cell 2004, 118, 363–373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riedl, J.; Crevenna, A.; Kessenbrock, K.; Yu, J.H.; Neukirchen, D.; Bista, M.; Bradke, F.; Jenne, D.; Holak, T.A.; Werb, Z.; et al. Lifeact: A versatile marker to visualize F-Actin. Nat. Methods 2008, 5, 605–607. [Google Scholar] [CrossRef]
- Ran, F.A.; Hsu, P.D.; Wright, J.; Agarwala, V.; Scott, D.A.; Zhang, F. Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 2013, 8, 2281–2308. [Google Scholar] [CrossRef] [Green Version]
- Brinkman, E.K.; Chen, T.; Amendola, M.; Van Steensel, B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 2014, 42, e168. [Google Scholar] [CrossRef] [PubMed]
- Platonova, E.; Winterflood, C.M.; Junemann, A.; Albrecht, D.; Faix, J.; Ewers, H. Single-molecule microscopy of molecules tagged with GFP or RFP derivatives in mammalian cells using nanobody binders. Methods 2015, 88, 89–97. [Google Scholar] [CrossRef]
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. NIH Image to ImageJ: 25 Years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Meijering, E.; Dzyubachyk, O.; Smal, I. Methods for Cell and Particle Tracking. Methods Enzymol. 2012, 504, 183–200. [Google Scholar] [CrossRef] [PubMed]
- Litschko, C.; Damiano-Guercio, J.; Brühmann, S.; Faix, J. Analysis of Random Migration of Dictyostelium Amoeba in Confined and Unconfined Environments. Methods Mol. Biol. 2018, 1749, 341–350. [Google Scholar] [CrossRef]
- Otsu, N. Threshold selection method from gray-level histograms. IEEE Trans. Syst. Man Cybern. 1979, 9, 62–66. [Google Scholar] [CrossRef] [Green Version]
- Funk, J.; Merino, F.; Schaks, M.; Rottner, K.; Raunser, S.; Bieling, P. A barbed end interference mechanism reveals how capping protein promotes nucleation in branched actin networks. Nat. Commun. 2021, 12, 5329. [Google Scholar] [CrossRef]
- Sinnar, S.A.; Antoku, S.; Saffin, J.-M.; Cooper, J.A.; Halpain, S. Capping protein is essential for cell migration in vivo and for filopodial morphology and dynamics. Mol. Biol. Cell 2014, 25, 2152–2160. [Google Scholar] [CrossRef] [PubMed]
- Hug, C.; Jay, P.Y.; Reddy, I.; McNally, J.G.; Bridgman, P.C.; Elson, E.L.; Cooper, J. Capping protein levels influence actin assembly and cell motility in dictyostelium. Cell 1995, 81, 591–600. [Google Scholar] [CrossRef] [Green Version]
- Hogg, K.; Thomas, J.; Ashford, D.; Cartwright, J.; Coldwell, R.; Weston, D.J.; Pillmoor, J.; Surry, D.; O’Toole, P. Quantification of proteins by flow cytometry: Quantification of human hepatic transporter P-Gp and OATP1B1 using flow cytometry and mass spectrometry. Methods 2015, 82, 38–46. [Google Scholar] [CrossRef] [PubMed]
- Burridge, K.; Guilluy, C. Focal adhesions, stress fibers and mechanical tension. Exp. Cell Res. 2016, 343, 14–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mueller, J.; Szep, G.; Nemethova, M.; de Vries, I.; Lieber, A.D.; Winkler, C.; Kruse, K.; Small, J.V.; Schmeiser, C.; Keren, K.; et al. Load Adaptation of Lamellipodial Actin Networks. Cell 2017, 171, 188–200.e16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colom, A.; Derivery, E.; Soleimanpour, S.; Tomba, C.; Molin, M.D.; Sakai, N.; González-Gaitán, M.; Matile, S.; Roux, A. A fluorescent membrane tension probe. Nat. Chem. 2018, 10, 1118–1125. [Google Scholar] [CrossRef]
- Tokuo, H.; Mabuchi, K.; Ikebe, M. The motor activity of myosin-X promotes actin fiber convergence at the cell periphery to initiate filopodia formation. J. Cell Biol. 2007, 179, 229–238. [Google Scholar] [CrossRef] [PubMed]
- Berg, J.S.; Cheney, R.E. Myosin-X is an unconventional myosin that undergoes intrafilopodial motility. Nat. Cell Biol. 2002, 4, 246–250. [Google Scholar] [CrossRef] [PubMed]
- Bohil, A.B.; Robertson, B.W.; Cheney, R.E. Myosin-X is a molecular motor that functions in filopodia formation. Proc. Natl. Acad. Sci. USA 2006, 103, 12411–12416. [Google Scholar] [CrossRef] [Green Version]
- Dimchev, G.; Steffen, A.; Kage, F.; Dimchev, V.; Pernier, J.; Carlier, M.-F.; Rottner, K. Efficiency of lamellipodia protrusion is determined by the extent of cytosolic actin assembly. Mol. Biol. Cell 2017, 28, 1311–1325. [Google Scholar] [CrossRef]
- Dieluweit, S.; Csiszár, A.; Rubner, W.; Fleischhauer, J.; Houben, S.; Merkel, R. Mechanical Properties of Bare and Protein-Coated Giant Unilamellar Phospholipid Vesicles. A Comparative Study of Micropipet Aspiration and Atomic Force Microscopy. Langmuir 2010, 26, 11041–11049. [Google Scholar] [CrossRef]
- Zhang, H.; Berg, J.; Li, Z.; Wang, Y.; Lång, P.; Sousa, A.; Bhaskar, A.; Cheney, R.; Strömblad, S. Myosin-X provides a motor-based link between integrins and the cytoskeleton. Nat. Cell Biol. 2004, 6, 523–531. [Google Scholar] [CrossRef]
- Rothkegel, M.; Mayboroda, O.; Rohde, M.; Wucherpfennig, C.; Valenta, R.; Jockusch, B. Plant and animal profilins are functionally equivalent and stabilize microfilaments in living animal cells. J. Cell Sci. 1996, 109, 83–90. [Google Scholar] [CrossRef] [PubMed]
- Carlsson, L.; Blikstad, I. Colchicine treatment of HeLa cells alters the G/F actin ratio. FEBS Lett. 1981, 124, 282–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carlier, M.-F.; Pantaloni, D. Control of Actin Assembly Dynamics in Cell Motility. J. Biol. Chem. 2007, 282, 23005–23009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cramer, L.P. Role of Actin-filament disassembly in lamellipodium protrusion in motile cells revealed using the drug jasplakinolide. Curr. Biol. 1999, 9, 1095–1105. [Google Scholar] [CrossRef] [Green Version]
- Westermann, L.; Li, Y.; Göcmen, B.; Niedermoser, M.; Rhein, K.; Jahn, J.; Cascante, I.; Schöler, F.; Moser, N.; Neubauer, B.; et al. Wildtype heterogeneity contributes to clonal variability in genome edited cells. Sci. Rep. 2022, 12, 18211. [Google Scholar] [CrossRef]
- Miyoshi, T.; Tsuji, T.; Higashida, C.; Hertzog, M.; Fujita, A.; Narumiya, S.; Scita, G.; Watanabe, N. Actin turnover–dependent fast dissociation of capping protein in the dendritic nucleation actin network: Evidence of frequent filament severing. J. Cell Biol. 2006, 175, 947–955. [Google Scholar] [CrossRef]
- Caldwell, J.E.; Heiss, S.G.; Mermall, V.; Cooper, J.A. Effects of CapZ, an Actin-capping protein of muscle, on the polymerization of Actin. Biochemistry 1989, 28, 8506–8514. [Google Scholar] [CrossRef]
- Pocaterra, A.; Santinon, G.; Romani, P.; Brian, I.; Dimitracopoulos, A.; Ghisleni, A.; Carnicer-Lombarte, A.; Forcato, M.; Braghetta, P.; Montagner, M.; et al. F-actin dynamics regulates mammalian organ growth and cell fate maintenance. J. Hepatol. 2019, 71, 130–142. [Google Scholar] [CrossRef] [Green Version]
- Faix, J.; Breitsprecher, D.; Stradal, T.E.; Rottner, K. Filopodia: Complex models for simple rods. Int. J. Biochem. Cell Biol. 2009, 41, 1656–1664. [Google Scholar] [CrossRef]
- Young, L.E.; Heimsath, E.G.; Higgs, H.N. Cell type–dependent mechanisms for formin-mediated assembly of filopodia. Mol. Biol. Cell 2015, 26, 4646–4659. [Google Scholar] [CrossRef]
- Harris, E.S.; Li, F.; Higgs, H.N. The Mouse Formin, FRLα, Slows Actin Filament Barbed End Elongation, Competes with Capping Protein, Accelerates Polymerization from Monomers, and Severs Filaments. J. Biol. Chem. 2004, 279, 20076–20087. [Google Scholar] [CrossRef] [Green Version]
- Seth, A.; Otomo, C.; Rosen, M.K. Autoinhibition regulates cellular localization and actin assembly activity of the diaphanous-related formins FRLα and mDia1. J. Cell Biol. 2006, 174, 701–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Junemann, A.; Filić, V.; Winterhoff, M.; Nordholz, B.; Litschko, C.; Schwellenbach, H.; Stephan, T.; Weber, I.; Faix, J. A Diaphanous-related formin links Ras signaling directly to actin assembly in macropinocytosis and phagocytosis. Proc. Natl. Acad. Sci. USA 2016, 113, E7464–E7473. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramalingam, N.; Zhao, H.; Breitsprecher, D.; Lappalainen, P.; Faix, J.; Schleicher, M. Phospholipids regulate localization and activity of mDia1 formin. Eur. J. Cell Biol. 2010, 89, 723–732. [Google Scholar] [CrossRef]
- Watanabe, S.; Okawa, K.; Miki, T.; Sakamoto, S.; Morinaga, T.; Segawa, K.; Arakawa, T.; Kinoshita, M.; Ishizaki, T.; Narumiya, S. Rho and Anillin-dependent Control of mDia2 Localization and Function in Cytokinesis. Mol. Biol. Cell 2010, 21, 3193–3204. [Google Scholar] [CrossRef] [Green Version]
- Lau, E.O.-C.; Damiani, D.; Chehade, G.; Ruiz-Reig, N.; Saade, R.; Jossin, Y.; Aittaleb, M.; Schakman, O.; Tajeddine, N.; Gailly, P.; et al. DIAPH3 deficiency links microtubules to mitotic errors, defective neurogenesis, and brain dysfunction. Elife 2021, 10, e61974. [Google Scholar] [CrossRef]
- Watanabe, S.; De Zan, T.; Ishizaki, T.; Yasuda, S.; Kamijo, H.; Yamada, D.; Aoki, T.; Kiyonari, H.; Kaneko, H.; Shimizu, R.; et al. Loss of a Rho-Regulated Actin Nucleator, mDia2, Impairs Cytokinesis during Mouse Fetal Erythropoiesis. Cell Rep. 2013, 5, 926–932. [Google Scholar] [CrossRef] [Green Version]
- Watanabe, S.; Ando, Y.; Yasuda, S.; Hosoya, H.; Watanabe, N.; Ishizaki, T.; Narumiya, S. mDia2 Induces the Actin Scaffold for the Contractile Ring and Stabilizes Its Position during Cytokinesis in NIH 3T3 Cells. Mol. Biol. Cell 2008, 19, 2328–2338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Houdusse, A.; Titus, M.A. The many roles of myosins in filopodia, microvilli and stereocilia. Curr. Biol. 2021, 31, R586–R602. [Google Scholar] [CrossRef] [PubMed]
- Salles, F.T.; Jr, R.C.M.; Manor, U.; Dougherty, G.W.; Sousa, A.D.; Moore, J.E.; Yengo, C.M.; Dosé, A.C.; Kachar, B. Myosin IIIa boosts elongation of stereocilia by transporting espin 1 to the plus ends of actin filaments. Nat. Cell Biol. 2009, 11, 443–450. [Google Scholar] [CrossRef] [Green Version]
- Liu, R.; Billington, N.; Yang, Y.; Bond, C.; Hong, A.; Siththanandan, V.; Takagi, Y.; Sellers, J.R. A binding protein regulates myosin-7a dimerization and actin bundle assembly. Nat. Commun. 2021, 12, 563. [Google Scholar] [CrossRef] [PubMed]
- Belyantseva, I.A.; Boger, E.T.; Friedman, T.B. Myosin XVa Localizes to the Tips of Inner Ear Sensory Cell Stereocilia and Is Essential for Staircase Formation of the Hair Bundle. Proc. Natl. Acad. Sci. USA 2003, 100, 13958–13963. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goh, W.I.; Lim, K.B.; Sudhaharan, T.; Sem, K.P.; Bu, W.; Chou, A.M.; Ahmed, S. mDia1 and WAVE2 Proteins Interact Directly with IRSp53 in Filopodia and Are Involved in Filopodium Formation. J. Biol. Chem. 2012, 287, 4702–4714. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Litschko, C.; Brühmann, S.; Csiszár, A.; Stephan, T.; Dimchev, V.; Damiano-Guercio, J.; Junemann, A.; Körber, S.; Winterhoff, M.; Nordholz, B.; et al. Functional integrity of the contractile actin cortex is safeguarded by multiple Diaphanous-related formins. Proc. Natl. Acad. Sci. USA 2019, 116, 3594–3603. [Google Scholar] [CrossRef] [Green Version]
- Jaiswal, R.; Breitsprecher, D.; Collins, A.; Correa, I.; Xu, M.-Q.; Goode, B.L. The Formin Daam1 and Fascin Directly Collaborate to Promote Filopodia Formation. Curr. Biol. 2013, 23, 1373–1379. [Google Scholar] [CrossRef] [Green Version]
- Hoffmann, A.; Naj, X.; Linder, S. Daam1 is a regulator of filopodia formation and phagocytic uptake of Borrelia burgdorferi by primary human macrophages. FASEB J. 2014, 28, 3075–3089. [Google Scholar] [CrossRef]
- Svitkina, T.M.; Bulanova, E.A.; Chaga, O.Y.; Vignjevic, D.M.; Kojima, S.-I.; Vasiliev, J.M.; Borisy, G.G. Mechanism of filopodia initiation by reorganization of a dendritic network. J. Cell Biol. 2003, 160, 409–421. [Google Scholar] [CrossRef]
- Bieling, P.; Hansen, S.D.; Akin, O.; Li, T.; Hayden, C.C.; Fletcher, D.A.; Mullins, R.D. WH2 and proline-rich domains of WASP-family proteins collaborate to accelerate actin filament elongation. EMBO J. 2017, 37, 102–121. [Google Scholar] [CrossRef] [PubMed]
- Vignjevic, D.; Kojima, S.-I.; Aratyn, Y.; Danciu, O.; Svitkina, T.; Borisy, G.G. Role of fascin in filopodial protrusion. J. Cell Biol. 2006, 174, 863–875. [Google Scholar] [CrossRef] [Green Version]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Mutant Cell Lines | Filopodia Length (µm) | Normalized to CP-KO (%) | n 1 | Number of Filopodia/Cell | Normalized to CP-KO (%) | n 2 |
|---|---|---|---|---|---|---|
| CP-KO | 2.8 ± 0.7 | 100.0 ± 25.6 | 208 | 89.9 ± 14.3 | 100.0 ± 15.9 | 59 |
| EVM/CP-KO | 2.2 ± 0.5 | 78.6 ± 24.5 | 204 | 90.1 ± 14.2 | 100.1 ± 15.7 | 54 |
| EVM/CP/MyoX-KO | 1.6 ± 0.3 | 57.7 ± 20.6 | 206 | 89.0 ± 11.4 | 99.0 ± 12.8 | 60 |
| EVM/CP/FMNL3-KO | 1.1 ± 0.2 | 39.7 ± 20.7 | 198 | 64.2 ± 11.7 | 71.4 ± 18.3 | 60 |
| EVM/CP/MyoX/FMNL2/3-KO | 1.1 ± 0.9 | 40.0 ± 16.8 | 194 | 62.0 ± 10.8 | 69.0 ± 17.3 | 60 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hein, J.I.; Scholz, J.; Körber, S.; Kaufmann, T.; Faix, J. Unleashed Actin Assembly in Capping Protein-Deficient B16-F1 Cells Enables Identification of Multiple Factors Contributing to Filopodium Formation. Cells 2023, 12, 890. https://0-doi-org.brum.beds.ac.uk/10.3390/cells12060890
Hein JI, Scholz J, Körber S, Kaufmann T, Faix J. Unleashed Actin Assembly in Capping Protein-Deficient B16-F1 Cells Enables Identification of Multiple Factors Contributing to Filopodium Formation. Cells. 2023; 12(6):890. https://0-doi-org.brum.beds.ac.uk/10.3390/cells12060890
Chicago/Turabian StyleHein, Jens Ingo, Jonas Scholz, Sarah Körber, Thomas Kaufmann, and Jan Faix. 2023. "Unleashed Actin Assembly in Capping Protein-Deficient B16-F1 Cells Enables Identification of Multiple Factors Contributing to Filopodium Formation" Cells 12, no. 6: 890. https://0-doi-org.brum.beds.ac.uk/10.3390/cells12060890