


JNK Activation Correlates with Cognitive Impairment and Alteration of the Post-Synaptic Element in the 5xFAD AD Mouse Model

 , , ,
, , ,
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Behavioral Test
2.3. Triton Insoluble Fractionation (TIF)
2.4. Western Blot
2.5. ELISA
2.6. Mass Spectrometry
2.7. Statistical Analysis
3. Results
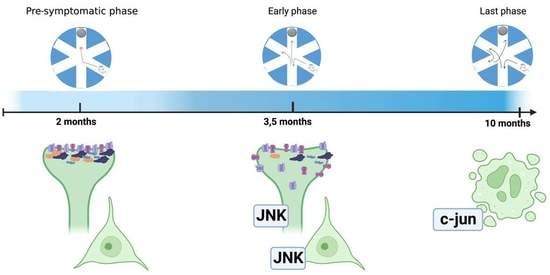
3.1. Pre-Symptomatic Phase: 2-Month-Old 5xFAD Mice Showed an Increase in APP and P-APP Levels but No Cognitive Deficit or JNK Activation
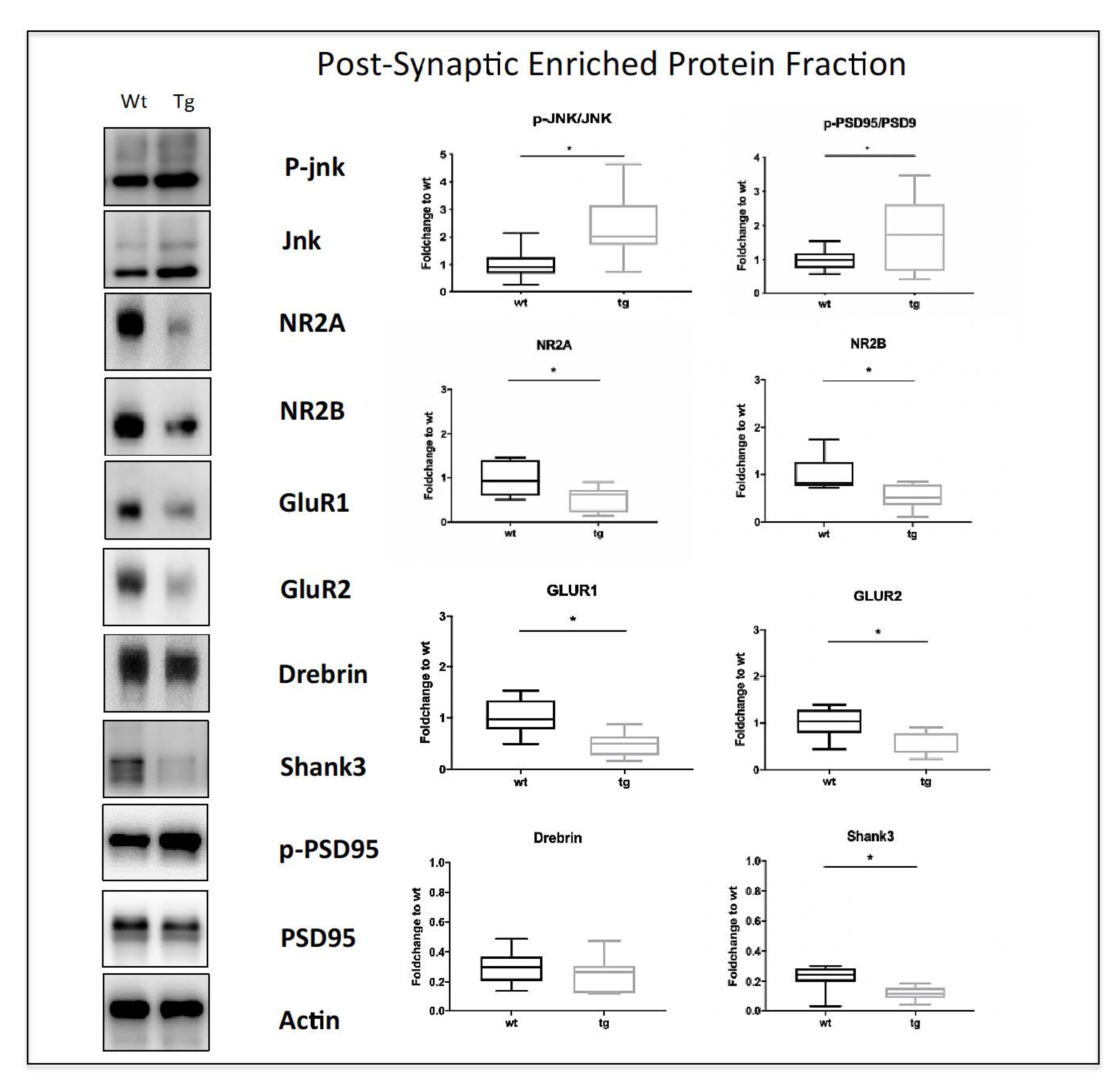
3.2. Early Symptomatic Phase: Cognitive Impairment Correlates with Increased JNK Activation, Aβ Oligomers Deposition, and Synaptic Dysfunction in 3.5-Month-Old 5xFAD Mice
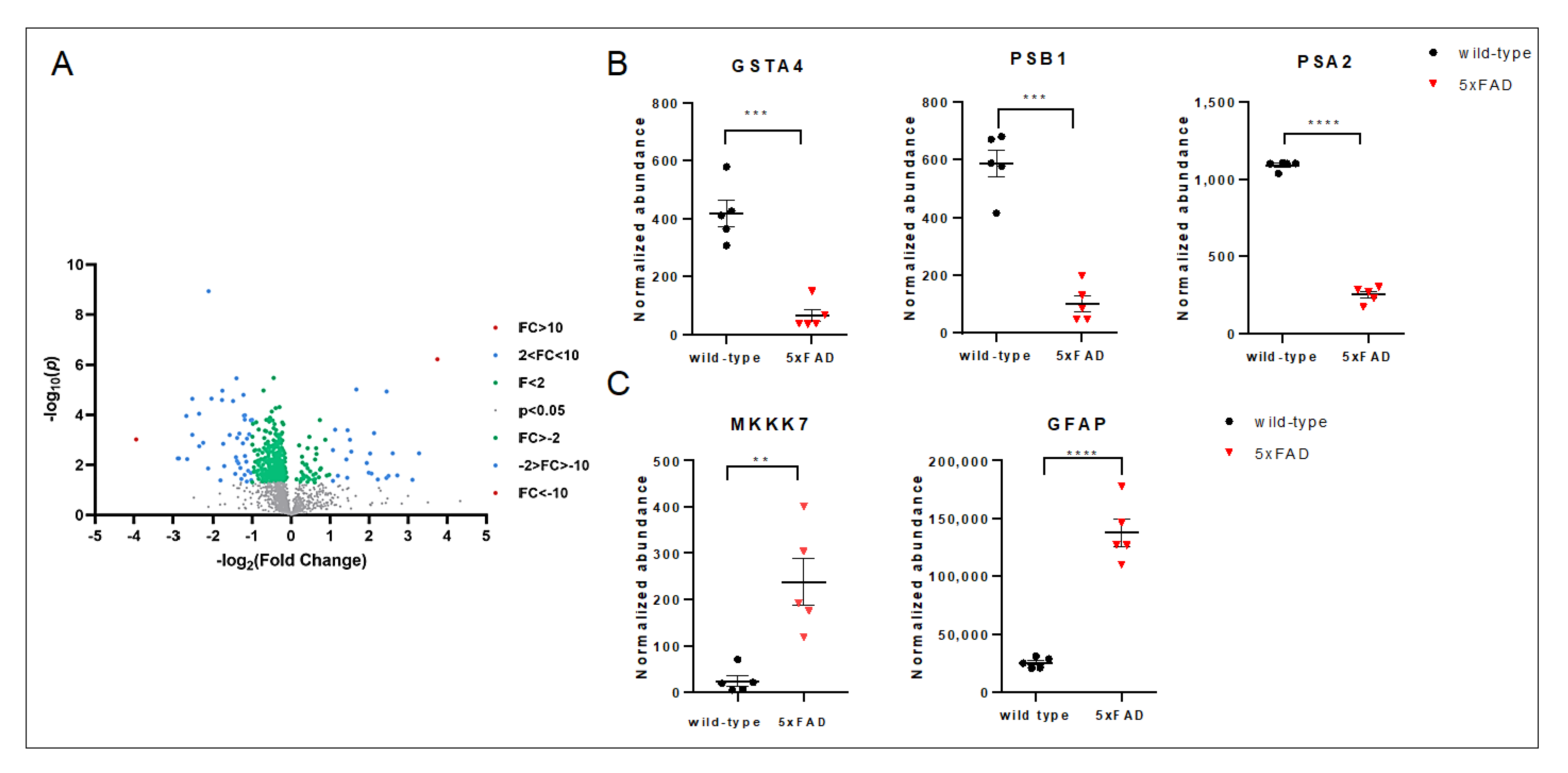
3.3. The Late Phase of the Disease: 10-Month-Old 5xFAD Mice Cortical Lysates Showed Strong Activation of the Cell Death Programs
4. Discussion
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ahmed, T.; Zulfiqar, A.; Arguelles, S.; Rasekhian, M.; Nabavi, S.F.; Silva, A.S.; Nabavi, S.M. Map Kinase Signaling as Therapeutic Target for Neurodegeneration. Pharmacol. Res. 2020, 160, 105090. [Google Scholar] [CrossRef] [PubMed]
- Scheinfeld, M.H.; Roncarati, R.; Vito, P.; Lopez, P.A.; Abdallah, M.; D’Adamio, L. Jun NH2-Terminal Kinase (JNK) Interacting Protein 1 (JIP1) Binds the Cytoplasmic Domain of the Alzheimer’s β-Amyloid Precursor Protein (APP). J. Biol. Chem. 2002, 277, 3767–3775. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sclip, A.; Tozzi, A.; Abaza, A.; Cardinetti, D.; Colombo, I.; Calabresi, P.; Salmona, M.; Welker, E.; Borsello, T. C-Jun N-Terminal Kinase Has a Key Role in Alzheimer Disease Synaptic Dysfunction in Vivo. Cell Death Dis. 2014, 5, e1019. [Google Scholar] [CrossRef] [Green Version]
- Ploia, C.; Antoniou, X.; Sclip, A.; Grande, V.; Cardinetti, D.; Colombo, A.; Canu, N.; Benussi, L.; Ghidoni, R.; Forloni, G.; et al. JNK Plays a Key Role in Tau Hyperphosphorylation in Alzheimer’s Disease Models. J. Alzheimers Dis. JAD 2011, 26, 315–329. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Tatebayashi, Y.; Akagi, T.; Chui, D.-H.; Murayama, M.; Miyasaka, T.; Planel, E.; Tanemura, K.; Sun, X.; Hashikawa, T.; et al. Aberrant Tau Phosphorylation by Glycogen Synthase Kinase-3β and JNK3 Induces Oligomeric Tau Fibrils in COS-7 Cells. J. Biol. Chem. 2002, 277, 42060–42065. [Google Scholar] [CrossRef] [Green Version]
- Goedert, M.; Hasegawa, M.; Jakes, R.; Lawler, S.; Cuenda, A.; Cohen, P. Phosphorylation of Microtubule-Associated Protein Tau by Stress-Activated Protein Kinases. FEBS Lett. 1997, 409, 57–62. [Google Scholar] [CrossRef]
- Coffey, E.T. Nuclear and Cytosolic JNK Signalling in Neurons. Nat. Rev. Neurosci. 2014, 15, 285–299. [Google Scholar] [CrossRef]
- Hollos, P.; John, J.M.; Lehtonen, J.V.; Coffey, E.T. Optogenetic Control of Spine-Head JNK Reveals a Role in Dendritic Spine Regression. eNeuro 2020, 7, ENEURO.0303-19.2019. [Google Scholar] [CrossRef] [Green Version]
- Komulainen, E.; Varidaki, A.; Kulesskaya, N.; Mohammad, H.; Sourander, C.; Rauvala, H.; Coffey, E.T. Impact of JNK and Its Substrates on Dendritic Spine Morphology. Cells 2020, 9, 440. [Google Scholar] [CrossRef] [Green Version]
- Sclip, A.; Antoniou, X.; Colombo, A.; Camici, G.G.; Pozzi, L.; Cardinetti, D.; Feligioni, M.; Veglianese, P.; Bahlmann, F.H.; Cervo, L.; et al. C-Jun N-Terminal Kinase Regulates Soluble Aβ Oligomers and Cognitive Impairment in AD Mouse Model. J. Biol. Chem. 2011, 286, 43871–43880. [Google Scholar] [CrossRef] [Green Version]
- Sclip, A.; Arnaboldi, A.; Colombo, I.; Veglianese, P.; Colombo, L.; Messa, M.; Mancini, S.; Cimini, S.; Morelli, F.; Antoniou, X.; et al. Soluble Aβ Oligomer-Induced Synaptopathy: C-Jun N-Terminal Kinase’s Role. J. Mol. Cell Biol. 2013, 5, 277–279. [Google Scholar] [CrossRef]
- Musi, C.A.; Agrò, G.; Santarella, F.; Iervasi, E.; Borsello, T. JNK3 as Therapeutic Target and Biomarker in Neurodegenerative and Neurodevelopmental Brain Diseases. Cells 2020, 9, 2190. [Google Scholar] [CrossRef] [PubMed]
- Colombo, A.; Bastone, A.; Ploia, C.; Sclip, A.; Salmona, M.; Forloni, G.; Borsello, T. JNK Regulates APP Cleavage and Degradation in a Model of Alzheimer’s Disease. Neurobiol. Dis. 2009, 33, 518–525. [Google Scholar] [CrossRef]
- Busquets, O.; Parcerisas, A.; Verdaguer, E.; Ettcheto, M.; Camins, A.; Beas-Zarate, C.; Castro-Torres, R.D.; Auladell, C. C-Jun N-Terminal Kinases in Alzheimer’s Disease: A Possible Target for the Modulation of the Earliest Alterations. J. Alzheimers Dis. 2021, 82, S127–S139. [Google Scholar] [CrossRef] [PubMed]
- Chishti, M.A.; Yang, D.-S.; Janus, C.; Phinney, A.L.; Horne, P.; Pearson, J.; Strome, R.; Zuker, N.; Loukides, J.; French, J.; et al. Early-Onset Amyloid Deposition and Cognitive Deficits in Transgenic Mice Expressing a Double Mutant Form of Amyloid Precursor Protein 695. J. Biol. Chem. 2001, 276, 21562–21570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Forner, S.; Kawauchi, S.; Balderrama-Gutierrez, G.; Kramár, E.A.; Matheos, D.P.; Phan, J.; Javonillo, D.I.; Tran, K.M.; Hingco, E.; da Cunha, C.; et al. Systematic Phenotyping and Characterization of the 5xFAD Mouse Model of Alzheimer’s Disease. Sci. Data 2021, 8, 270. [Google Scholar] [CrossRef]
- Oakley, H.; Cole, S.L.; Logan, S.; Maus, E.; Shao, P.; Craft, J.; Guillozet-Bongaarts, A.; Ohno, M.; Disterhoft, J.; Van Eldik, L.; et al. Intraneuronal Beta-Amyloid Aggregates, Neurodegeneration, and Neuron Loss in Transgenic Mice with Five Familial Alzheimer’s Disease Mutations: Potential Factors in Amyloid Plaque Formation. J. Neurosci. 2006, 26, 10129–10140. [Google Scholar] [CrossRef] [Green Version]
- Salter, M.W.; Stevens, B. Microglia Emerge as Central Players in Brain Disease. Nat. Med. 2017, 23, 1018–1027. [Google Scholar] [CrossRef]
- Heneka, M.T.; Carson, M.J.; Khoury, J.E.; Landreth, G.E.; Brosseron, F.; Feinstein, D.L.; Jacobs, A.H.; Wyss-Coray, T.; Vitorica, J.; Ransohoff, R.M.; et al. Neuroinflammation in Alzheimer’s Disease. Lancet Neurol. 2015, 14, 388–405. [Google Scholar] [CrossRef] [Green Version]
- Mandrekar-Colucci, S.; Landreth, G.E. Microglia and Inflammation in Alzheimers Disease. CNS Neurol. Disord.-Drug Targets 2010, 9, 156–167. [Google Scholar] [CrossRef]
- Kimura, R.; Ohno, M. Impairments in Remote Memory Stabilization Precede Hippocampal Synaptic and Cognitive Failures in 5xFAD Alzheimer Mouse Model. Neurobiol. Dis. 2009, 33, 229–235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jawhar, S.; Trawicka, A.; Jenneckens, C.; Bayer, T.A.; Wirths, O. Motor Deficits, Neuron Loss, and Reduced Anxiety Coinciding with Axonal Degeneration and Intraneuronal Aβ Aggregation in the 5xFAD Mouse Model of Alzheimer’s Disease. Neurobiol. Aging 2012, 33, 196.e29–196.e40. [Google Scholar] [CrossRef]
- Eimer, W.A.; Vassar, R. Neuron Loss in the 5xFAD Mouse Model of Alzheimer’s Disease Correlates with Intraneuronal Aβ42 Accumulation and Caspase-3 Activation. Mol. Neurodegener. 2013, 8, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gourmaud, S.; Thomas, P.; Thomasseau, S.; Tible, M.; Abadie, C.; Paquet, C.; Hugon, J. Brimapitide Reduced Neuronal Stress Markers and Cognitive Deficits in 5xFAD Transgenic Mice. J. Alzheimers Dis. JAD 2018, 63, 665–674. [Google Scholar] [CrossRef] [PubMed]
- Porte, B.; Marguerit, G.; Thomasseau, S.; Paquet, C.; Hugon, J. Dose-Dependent Neuroprotective Effect of the JNK Inhibitor Brimapitide in 5xFAD Transgenic Mice. Brain Res. 2020, 1727, 146587. [Google Scholar] [CrossRef]
- Musi, C.A.; Castaldo, A.M.; Valsecchi, A.E.; Cimini, S.; Morello, N.; Pizzo, R.; Renieri, A.; Meloni, I.; Bonati, M.; Giustetto, M.; et al. JNK Signaling Provides a Novel Therapeutic Target for Rett Syndrome. BMC Biol. 2021, 19, 256. [Google Scholar] [CrossRef]
- Buccarello, L.; Musi, C.A.; Turati, A.; Borsello, T. The Stress C-Jun N-Terminal Kinase Signaling Pathway Activation Correlates with Synaptic Pathology and Presents A Sex Bias in P301L Mouse Model of Tauopathy. Neuroscience 2018, 393, 196–205. [Google Scholar] [CrossRef]
- Alamed, J.; Wilcock, D.M.; Diamond, D.M.; Gordon, M.N.; Morgan, D. Two-Day Radial-Arm Water Maze Learning and Memory Task; Robust Resolution of Amyloid-Related Memory Deficits in Transgenic Mice. Nat. Protoc. 2006, 1, 1671–1679. [Google Scholar] [CrossRef]
- Musi, C.A.; Agrò, G.; Buccarello, L.; Camuso, S.; Borsello, T. JNK Signaling Activation in the Ube3a Maternal Deficient Mouse Model: Its Specific Inhibition Prevents Post-Synaptic Protein-Enriched Fraction Alterations and Cognitive Deficits in Angelman Syndrome Model. Neurobiol. Dis. 2020, 140, 104812. [Google Scholar] [CrossRef]
- Gardoni, F.; Schrama, L.H.; Kamal, A.; Gispen, W.H.; Cattabeni, F.; Di Luca, M. Hippocampal Synaptic Plasticity Involves Competition between Ca2+/Calmodulin-Dependent Protein Kinase II and Postsynaptic Density 95 for Binding to the NR2A Subunit of the NMDA Receptor. J. Neurosci. Off. J. Soc. Neurosci. 2001, 21, 1501–1509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borsello, T.; Clarke, P.G.H.; Hirt, L.; Vercelli, A.; Repici, M.; Schorderet, D.F.; Bogousslavsky, J.; Bonny, C. A Peptide Inhibitor of C-Jun N-Terminal Kinase Protects against Excitotoxicity and Cerebral Ischemia. Nat. Med. 2003, 9, 1180–1186. [Google Scholar] [CrossRef]
- Yarza, R.; Vela, S.; Solas, M.; Ramirez, M.J. C-Jun N-Terminal Kinase (JNK) Signaling as a Therapeutic Target for Alzheimer’s Disease. Front. Pharmacol. 2016, 6, 321. [Google Scholar] [CrossRef] [Green Version]
- Finder, V.H.; Glockshuber, R. Amyloid-β Aggregation. Neurodegener. Dis. 2007, 4, 13–27. [Google Scholar] [CrossRef]
- Kim, M.J.; Futai, K.; Jo, J.; Hayashi, Y.; Cho, K.; Sheng, M. Synaptic Accumulation of PSD-95 and Synaptic Function Regulated by Phosphorylation of Serine-295 of PSD-95. Neuron 2007, 56, 488–502. [Google Scholar] [CrossRef] [Green Version]
- Kunde, S.-A.; Rademacher, N.; Tzschach, A.; Wiedersberg, E.; Ullmann, R.; Kalscheuer, V.M.; Shoichet, S.A. Characterisation of de Novo MAPK10/JNK3 Truncation Mutations Associated with Cognitive Disorders in Two Unrelated Patients. Hum. Genet. 2013, 132, 461–471. [Google Scholar] [CrossRef] [Green Version]
- Zhu, X.; Raina, A.K.; Rottkamp, C.A.; Aliev, G.; Perry, G.; Boux, H.; Smith, M.A. Activation and Redistribution of C-Jun N-Terminal Kinase/Stress Activated Protein Kinase in Degenerating Neurons in Alzheimer’s Disease. J. Neurochem. 2001, 76, 435–441. [Google Scholar] [CrossRef]
- Standen, C.L.; Brownlees, J.; Grierson, A.J.; Kesavapany, S.; Lau, K.F.; McLoughlin, D.M.; Miller, C.C. Phosphorylation of Thr(668) in the Cytoplasmic Domain of the Alzheimer’s Disease Amyloid Precursor Protein by Stress-Activated Protein Kinase 1b (Jun N-Terminal Kinase-3). J. Neurochem. 2001, 76, 316–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, M.-S.; Kao, S.-C.; Lemere, C.A.; Xia, W.; Tseng, H.-C.; Zhou, Y.; Neve, R.; Ahlijanian, M.K.; Tsai, L.-H. APP Processing Is Regulated by Cytoplasmic Phosphorylation. J. Cell Biol. 2003, 163, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Gourmaud, S.; Paquet, C.; Dumurgier, J.; Pace, C.; Bouras, C.; Gray, F.; Laplanche, J.-L.; Meurs, E.F.; Mouton-Liger, F.; Hugon, J. Increased Levels of Cerebrospinal Fluid JNK3 Associated with Amyloid Pathology: Links to Cognitive Decline. J. Psychiatry Neurosci. JPN 2015, 40, 151–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Musi, C.A.; Bonadonna, C.; Borsello, T. Synaptic Alterations as a Common Phase in Neurological and Neurodevelopmental Diseases: JNK Is a Key Mediator in Synaptic Changes. Neural Regen. Res. 2023, 18, 531–532. [Google Scholar] [CrossRef]
- Mucke, L.; Selkoe, D.J. Neurotoxicity of Amyloid β-Protein: Synaptic and Network Dysfunction. Cold Spring Harb. Perspect. Med. 2012, 2, a006338. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lei, K.; Davis, R.J. JNK Phosphorylation of Bim-Related Members of the Bcl2 Family Induces Bax-Dependent Apoptosis. Proc. Natl. Acad. Sci. USA 2003, 100, 2432–2437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Y.; Spigolon, G.; Bonny, C.; Culman, J.; Vercelli, A.; Herdegen, T. The JNK Inhibitor D-JNKI-1 Blocks Apoptotic JNK Signaling in Brain Mitochondria. Mol. Cell. Neurosci. 2012, 49, 300–310. [Google Scholar] [CrossRef]
- Resende, R.; Moreira, P.I.; Proença, T.; Deshpande, A.; Busciglio, J.; Pereira, C.; Oliveira, C.R. Brain Oxidative Stress in a Triple-Transgenic Mouse Model of Alzheimer Disease. Free Radic. Biol. Med. 2008, 44, 2051–2057. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandal, P.K.; Saharan, S.; Tripathi, M.; Murari, G. Brain Glutathione Levels—A Novel Biomarker for Mild Cognitive Impairment and Alzheimer’s Disease. Biol. Psychiatry 2015, 78, 702–710. [Google Scholar] [CrossRef] [PubMed]
- Ugbode, C.; Garnham, N.; Fort-Aznar, L.; Evans, G.J.O.; Chawla, S.; Sweeney, S.T. JNK Signalling Regulates Antioxidant Responses in Neurons. Redox Biol. 2020, 37, 101712. [Google Scholar] [CrossRef] [PubMed]
- Bonet-Costa, V.; Pomatto, L.C.-D.; Davies, K.J.A. The Proteasome and Oxidative Stress in Alzheimer’s Disease. Antioxid. Redox Signal. 2016, 25, 886–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benzing, W.C.; Wujek, J.R.; Ward, E.K.; Shaffer, D.; Ashe, K.H.; Younkin, S.G.; Brunden, K.R. Evidence for Glial-Mediated Inflammation in Aged APP(SW) Transgenic Mice. Neurobiol. Aging 1999, 20, 581–589. [Google Scholar] [CrossRef]
- Manaye, K.F.; Wang, P.C.; O’Neil, J.N.; Huang, S.Y.; Xu, T.; Lei, D.-L.; Tizabi, Y.; Ottinger, M.A.; Ingram, D.K.; Mouton, P.R. Neuropathological Quantification of Dtg APP/PS1: Neuroimaging, Stereology, and Biochemistry. Age 2007, 29, 87–96. [Google Scholar] [CrossRef] [Green Version]
- Brenner, M.; Messing, A. Regulation of GFAP Expression. ASN Neuro 2021, 13, 1759091420981206. [Google Scholar] [CrossRef]
- Brenner, M.; Messing, A.; Olsen, M.L. AP-1 and the Injury Response of the GFAP Gene. J. Neurosci. Res. 2019, 97, 149–161. [Google Scholar] [CrossRef] [PubMed]






Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Priori, E.C.; Musi, C.A.; Giani, A.; Colnaghi, L.; Milic, I.; Devitt, A.; Borsello, T.; Repici, M. JNK Activation Correlates with Cognitive Impairment and Alteration of the Post-Synaptic Element in the 5xFAD AD Mouse Model. Cells 2023, 12, 904. https://0-doi-org.brum.beds.ac.uk/10.3390/cells12060904
Priori EC, Musi CA, Giani A, Colnaghi L, Milic I, Devitt A, Borsello T, Repici M. JNK Activation Correlates with Cognitive Impairment and Alteration of the Post-Synaptic Element in the 5xFAD AD Mouse Model. Cells. 2023; 12(6):904. https://0-doi-org.brum.beds.ac.uk/10.3390/cells12060904
Chicago/Turabian StylePriori, Erica Cecilia, Clara Alice Musi, Arianna Giani, Luca Colnaghi, Ivana Milic, Andrew Devitt, Tiziana Borsello, and Mariaelena Repici. 2023. "JNK Activation Correlates with Cognitive Impairment and Alteration of the Post-Synaptic Element in the 5xFAD AD Mouse Model" Cells 12, no. 6: 904. https://0-doi-org.brum.beds.ac.uk/10.3390/cells12060904