
The Src-Family Kinases SRC and BLK Contribute to the CLDN6-Adhesion Signaling

Department of Basic Pathology, Fukushima Medical University School of Medicine, Fukushima 960-1295, Japan
*
Author to whom correspondence should be addressed.
†
These authors contributed equally to this work.
‡
Present address: Department of Pharmacology, Kansai Medical University, Osaka 573-1010, Japan.
§
Present address: Department of Pathology, Dokkyo Medical University School of Medicine, Mibu 321-0293, Japan.
Cells 2023, 12(13), 1696; https://0-doi-org.brum.beds.ac.uk/10.3390/cells12131696
Submission received: 15 May 2023
/
Revised: 16 June 2023
/
Accepted: 21 June 2023
/
Published: 23 June 2023
(This article belongs to the Special Issue Structure and Function of Tight Junctions)
Abstract
:Cell adhesion molecules, including integrins, cadherins, and claudins (CLDNs), are known to activate Src-family kinases (SFKs) that organize a variety of physiological and pathological processes; however, the underlying molecular basis remains unclear. Here, we identify the SFK members that are coupled with the CLDN6-adhesion signaling. Among SFK subtypes, BLK, FGR, HCK, and SRC were highly expressed in F9 cells and concentrated with CLDN6 along cell borders during epithelial differentiation. Immunoprecipitation assay showed that BLK and SRC, but not FGR or HCK, form a complex with CLDN6 via the C-terminal cytoplasmic domain. We also demonstrated, by pull-down assay, that recombinant BLK and SRC proteins directly bind to the C-terminal cytoplasmic domain of CLDN6 (CLDN6C). Unexpectedly, both recombinant SFK proteins recognized the CLDN6C peptide in a phosphotyrosine-independent manner. Furthermore, by comparing phenotypes of F9:Cldn6:Blk−/− and F9:Cldn6:Src−/− cells with those of wild-type F9 and F9:Cldn6 cells, we revealed that BLK and SRC are essential for CLDN6-triggered cellular events, namely epithelial differentiation and the expression of retinoid acid receptor target genes. These results indicate that selective SFK members appear to participate in the CLDN-adhesion signaling.
1. Introduction
The members belonging to the Src-family kinases (SFKs), including BLK, FGR, FRK, FYN, HCK, LCK, LYN, SRC, and YES, are non-receptor tyrosine kinases [1,2,3,4]. They consist of the N-terminal Src-homology 4 (SH4) domain, which contains a myristoylation site for membrane localization; a unique domain; the SH3 domain, which binds proline-rich sequences; the SH2 domain, which recognizes phosphotyrosine (pY) motifs; and the kinase (SH1) domain [5,6,7,8]. SFKs are activated by the binding of respective ligands to various transmembrane-receptor-associated tyrosine kinases (RTKs), such as the epidermal growth factor receptor (EGFR), human epidermal growth factor receptor 2 (HER2), and insulin-like growth factor receptor 1 (IGF-1R). In more detail, the liganded RTKs, adaptors or effectors, are known to interact with the SH2 and/or SH3 domains of SFKs, leading to the conformational change from the inactive closed form to the active open one [7,8,9,10,11,12]. The activated SFKs phosphorylate substrates, including phosphoinositide 3-kinase (PI3K) and focal adhesion kinase (FAK), which sequentially initiate the intracellular signaling cascades and cellular responses. On the other hand, SFKs are also activated by the engagement of cell–cell or cell–matrix adhesion molecules lacking intrinsic kinase activity, such as cadherins and integrins [13,14,15,16,17]. However, information on the cell-adhesion-triggered SFK signaling is still fragmentary.
The claudin (CLDN) family members are the major transmembrane proteins of tight junctions, the apical-most apparatus of apical junctional complexes in vertebrate epithelial cell sheets [18]. They are tetra-spanning membrane proteins with a short cytoplasmic N-terminus, two extracellular loops (EC1 and EC2), and a C-terminal cytoplasmic domain. The CLDN family is composed of more than 20 members in mammals and shows distinct expression patterns in tissue- and cell-type-specific manners [19,20,21]. The fundamental function of CLDNs is to create paracellular barriers and pores for ions and substances in the corresponding EC1-dependent fashion [19,20,21,22,23,24,25,26].
CLDNs are also thought to possess signaling properties, but the underlying molecular basis has yet to be established [27]. We previously showed that the engagement of CLDN6 recruits and activates SFKs in the EC2-dependent and the C-terminal cytoplasmic-domain-dependent manners, resulting in epithelial differentiation from F9 stem cells [28,29]. We also demonstrated that both Y196 and Y200 in the first half of the C-terminal cytoplasmic domain of CLDN6 are absolutely required to activate SFKs, which in turn phosphorylate CLDN6 at Y196/200. Moreover, we found that CLDN6/SFK signal propagates the PI3K/AKT cascade, which targets the retinoic acid receptor γ (RARγ) and the estrogen receptor α (ERα) and stimulates their activities independently of the ligands. Additionally, we reported that aberrant CLDN6/SFK signaling promotes endometrial cancer progression in vitro and in vivo by hijacking the CLDN6–ERα axis [29,30,31]. Nevertheless, it remains unknown which SFK members cooperate with CLDN6 to initiate these cellular processes.
Here, we report that BLK and SRC directly bind to the C-terminal cytoplasmic domain of CLDN6 (CLDN6C). Unexpectedly, both SFK subtypes recognized the CLDN6C peptide in a pY-independent manner. We also demonstrate that BLK and SRC are indispensable for the CLDN6-adhesion signaling to induce epithelial differentiation and the expression of RAR target genes.
2. Materials and Methods
2.1. Antibodies
The antibodies used in this study are listed in Table 1.
2.2. Expression Vectors
The coding region of mouse Cldn6 (full length, amino acids [AA] 1-219; Δ1/2C, AA 1-201; ΔC, AA 1-183) was amplified by RT-PCR using total RNA from F9 cells. A 3 × HA tag was added to the 5′ end of the Cldn6 sequence using a long-tail primer. The fragments were cloned into the XbaI/KpnI site of pD402 plasmid, in which the gene expression is driven by the PGK promoter. Mouse SFKs (Blk, Fgr, Frk, Fyn, Hck, Lck, Lyn, Src, and Yes1) were similarly amplified by PCR using cDNA of F9 cells or Blk Mouse Tagged ORF Clone (MR208004, OriGene, Rockville, MD, USA), and cloned into the EcoRI/SalI site of pFLAG-CMV-5.1 (E6908, Merck, Darmstadt, Germany). To produce the recombinant proteins, Blk-FLAG and Src-FLAG were subcloned into the KpnI/SacI site and the KpnI/HindIII site of plEx/Bac-4 (71726, Merck), while the C-terminus of Cldn6 (C185-V219) was inserted into the KpnI/HindIII site of plEx/Bac-4, which contains glutathione S-transferase (GST).
2.3. Cell Culture
F9 L32T2 cells, in which both doxycycline (Dox)-inducible gene expression and tamoxifen-dependent Cre-mediated recombination are allowed, F9:iCldn6 (hereafter, “i” refers to Dox-inducible expression of a given gene), and F9:Cldn6 cells were generated as described previously [32,33]. The cells were plated on a gelatin-coated dish and maintained in DMEM with 10% FBS. F9:iCldn6 cells were treated with 1 µg/mL Dox (D3447, Merck) 12 h after seeding and harvested 72–96 h after treatment when epithelial differentiation was microscopically evident.
The human endometrial carcinoma cell line Ishikawa was obtained from Dr. Yamada (Wakayama Medical University), and Ishikawa:CLDN6 cells were established as reported previously [31]. These cells were grown in DMEM with 10% FBS.
For transient expression of the target genes (HA-Cldn6, HA-Cldn6Δ1/2C, HA-Cldn6ΔC, Blk-FLAG, Fgr-FLAG, Frk-FLAG, Fyn-FLAG, Hck-FLAG, Lck-FLAG, Lyn-FLAG, Src-FLAG, and Yes1-FLAG), 1.0 × 106 cells were transfected with 1 µg of the plasmids and pHRL-Puro [32] by Lipofectamine 3000 (15292465, Thermo Fisher Scientific, Waltham, MA, USA) in a 6-well plate. Twenty-four hours after transfection, the cells were treated with 0.1 µg/mL of puromycin. After 2–3 days, when the cells were grown to 70–80% confluency, the whole cell extracts were collected for immunoprecipitation.
For gene expression analysis, the cells were maintained in charcoal-treated FBS and treated for 12 h with 1 nM of all-trans retinoic acid (RA; R2625, Merck) 24 h after seeding.
2.4. Genome Editing
sgRNA/Cas9 all-in-one expression clones targeting mouse Blk and Src genes were obtained from GeneCopoeia (Rockville, MD, USA; MCP226646-CG01-1, MCP226646-CG01-1). The plasmids were transfected by electroporation. The cells were treated with 0.1 µg/mL of puromycin and cloned by limiting dilution. Knockout of Blk and Src genes was verified by genomic PCR and Sanger sequencing, as well as by Western blotting. The knockout clones were then transfected with pD402-Cldn6 by electroporation as previously described [33].
2.5. Immunoprecipitation and Immunoblot
Total cell extracts were collected by using CellLytic MT Cell Lysis Reagent (C3228, Merck) and sonicated with three or four bursts of 5–10 s. Immunoprecipitation was performed using Immunoprecipitation Kit Protein G (11719386001, Merck) according to the manufacturer’s protocol. One microgram of ChromPure Mouse IgG (015-000-003, Jackson ImmunoResearch, West Grove, PA, USA) was used as a negative control. Whole-cell lysates or immunoprecipitated samples were mixed with sample loading buffer containing 2-mercaptoethanol and incubated for 10 min at 95 °C. They were resolved by one-dimensional SDS-PAGE and transferred onto a polyvinylidene difluoride membrane. The membranes were saturated with PVDF Blocking Reagent for Can Get Signal (NYPBR01, TOYOBO, Osaka, Japan) for 30 min. After rinsing in TBS containing 0.1% Tween 20, they were incubated with a primary antibody diluted in PBS or Can Get Signal Solution 1 (NKB-101, TOYOBO) for 1 h at room temperature or overnight at 4 °C, followed by 1 h incubation with horseradish peroxidase (HRP)-conjugated secondary antibodies diluted in PBS or Can Get Signal Solution 2 (NKB-101, TOYOBO). They were rinsed again and exposed to EzWestLumi One (WSE-7110, ATTO, Tokyo, Japan). After rinsing with 10% H2O2 to inactivate HRP, each membrane was hybridized with HRP-conjugated anti-beta actin antibody as loading controls. Whole-cell extracts from a breast cancer cell line MDA-MB-231 and 293T cells transiently overexpressing SFKs or CLDN6 were used as positive controls.
2.6. Immunofluorescence and Imaging
Cells were grown on a glass-based dish (3910-035, IWAKI, Shizuoka, Japan) coated with Cellmatrix Type I-A (Nitta gelatin, Osaka, Japan). The samples were fixed in 4% paraformaldehyde and 0.2% Triton-X for 10 min at room temperature. After washing with PBS, they were preincubated in PBS containing 5% skimmed milk. They were subsequently incubated overnight at 4 °C with primary antibodies diluted in Signal Booster Immunostain F (BCL-ISF, Beacle, Kyoto, Japan) and rinsed with PBS, followed by a reaction for 1 h at room temperature with appropriate secondary antibodies. All samples were examined using a laser-scanning confocal microscope (FV1000, Olympus, Tokyo, Japan). Photographs were processed with Photoshop CC (Adobe, San Jose, CA, USA) and ImageJ software (Wayne Rasband National Institutes of Health, Bethesda, MD, USA).
2.7. Pull-Down Assay
plEx/Bac-4, plEx/Bac-4-Blk-FLAG, and plEx/Bac-4-Src-FLAG were transfected into Sf9 insect cells using Insect GeneJuice reagent (71259, Merck). Recombinant BLK (recBLK) and recSRC proteins were obtained from these cell lysates by GST (glutathione S-transferase)-tagged protein purification kit (635619, Clontech, Kusatsu, Japan) according to the manufacturer’s instructions. For the GST-pull-down assay, 10 µg of the recGST, recBLK-FLAG, and recSRC proteins was bound to Glutathione-Superflow Resin (635607, Clontech) and incubated with 3 µg of recCLDN6C protein for 1 h at room temperature. The resins were washed five times in wash buffer (140 mM NaCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, pH 7.5). The proteins were then eluted with SDS-PAGE sample buffer and subjected to immunoblotting under reducing conditions.
2.8. ELISA
A 96-well ELISA plate was coated with 500 ng of CLDN6C peptide synthesized by Eurofins Scientific, dissolved in 100 µL of TBS, and incubated overnight at 4 °C. After washing with TBS-T and blocking with 2% bovine serum albumin (BSA) at 37 °C for 30 min, recGST, recBLK (ab60876, abcam, Cambridge, UK), or recSRC (ab51424, abcam) was dissolved in TBS-T and incubated at 37 °C for 1 h. The wells were then washed three times with TBS-T and probed with horseradish peroxidase (HRP)-conjugated anti-GST antibody (sc-138 HRP, Santa Cruz Biotechnology, Dallas, TX, USA) for 1 h at room temperature. After five more washes with TBS-T, the bound HRP was detected using 3,3′,5,5′-tetramethylbenzidine (TMB) substrate (421101, BioLegend, San Diego, CA, USA). The relative absorbance to the mean value of CLDN6C/GST was plotted as a bar graph.
2.9. RT-qPCR
For gene expression analysis, total RNA was isolated from cells using TRIzol RNA Isolation Reagents (15596026, Thermo Fisher Scientific), and reverse transcription was performed using SentiFAST cDNA Synthesis Kit (BIO-65063, meridian BIOSCIENCE, Cincinnati, OH, USA). Quantitative PCR (qPCR) was performed using THUNDERBIRD SYBR qPCR Mix (TOYOBO) and Step One Real-Time PCR System (Applied Biosystems, Waltham, MA, USA) using the primers listed in the previous paper [28]. The expression levels of the target genes were normalized to the corresponding Gapdh expression.
3. Results
3.1. BLK, FGR, HCK, and SRC Are Colocalized with CLDN6 on Epithelial Cell Borders of F9 Cells
We first determined the expression profile of SFK proteins in F9:iCldn6 cells in which CLDN6 expression is induced by Dox treatment [33]. Western blot analysis revealed that four members of SFKs (BLK, FGR HCK, and SRC) were strongly expressed in undifferentiated F9:iCldn6 cells (Figure 1A). Although SFKs were activated in Dox-treated F9:iCldn6 cells as expected and described previously [28], the expression levels of these SFK members were similar to those in undifferentiated F9:iCldn6 cells. On the other hand, FRK, FYN, LCK, LYN, and YES were hardly detected in undifferentiated and differentiated F9:iCldn6 cells. Note that we used 293T cells transfected with the corresponding mouse SFK and Cldn6 genes as positive controls, supporting the validity of our Western blot analysis.
Double immunofluorescence staining showed that pSFK was concentrated with CLDN6 along cell boundaries of Dox-exposed F9:iCldn6 cells (Figure 1B), as reported previously [28]. Furthermore, BLK, FGR HCK, and SRC were colocalized with CLDN6 at mature cell–cell junctions of Dox-treated F9:iCldn6 cells, though their positive signals were also detected in the cytoplasm and nucleus (Figure 1B). By contrast, these SFK members were scarcely concentrated along cell borders in undifferentiated F9:iCldn6 cells (Figure 1C). It should be noted that epithelial differentiation is not induced in the entire cells because F9 cells are stem cells and therefore divide both symmetrically and asymmetrically. Colocalization of BLK and SRC with CLDN6 was also observed along cell borders in Ishikawa:CLDN6 but not in Ishikawa cells (Supplementary Figure S1).
3.2. BLK and SRC Are Associated with CLDN6 through the C-Terminal Cytoplasmic Domain
We subsequently performed an immunoprecipitation assay using F9 L32T2 cells that transiently transfected with either the Blk-FLAG, Fgr-FLAG, Hck-FLAG, or Src-FLAG expression vector along with the HA-Cldn6 expression vector. Among these SFK members, BLK and SRC, but not FGR or HCK, formed a complex with CLDN6 (Figure 2A). We previously showed that pSFKs are associated with CLDN6C [28]. Therefore, the HA-Cldn6, HA-Cldn6ΔC1/2, or HA-Cldn6ΔC expression vectors, in the latter two of which the second half of or the whole C-terminal cytoplasmic domain of CLDN6 is deleted, were introduced with the Blk-FLAG or Src-FLAG expression vector to F9 L32T2 cells and subjected to immunoprecipitation assay. As shown in Figure 2B, an association between CLDN6 and either BLK or SRC was evident even in the absence of the second half of the C-terminal cytoplasmic domain of CLDN6 but disappeared without the whole C-terminal cytoplasmic domain.
3.3. BLK and SRC Directly Bind to the C-Terminal Cytoplasmic Domain of CLDN6
We next verified, by GST pull-down assay, whether full-length recombinant BLK and SRC proteins (recGST-BLK and recGST-SRC) were able to bind directly to recCLDN6C (Figure 3A). As shown in Figure 3B, both recGST-BLK and recGST-SRC appeared to interact with recCLDN6C. Since CLDN6 recruits and activates SFKs in a Y196/200-dependent manner, as previously reported [28], we subsequently determined whether recBLK and recSRC interact with the non-pY196/200- or pY196/200-containing CLDN6C peptides that correspond to the first half of the C-terminal cytoplasmic domain of CLDN6 (Figure 4A).
Unexpectedly, however, both recGST-BLK and recGST-SRC dose-dependently bound not only to the pY196/200-CLDN6C peptide but also to the non-pY196/200-CLDN6C peptide (Figure 4B). In addition, the binding efficiency of the pY196/200-CLDN6C peptide was significantly lower than that of the non-pY196/200-CLDN6C peptide.
3.4. BLK and SRC Are Essential for the CLDN6-Adhesion Signaling
To validate whether BLK and SRC are indispensable for the CLDN6-adhesion signaling, we then generated F9:Cldn6:Blk−/− and F9:Cldn6:Src−/− cells using CRISPR/Cas9-based genome editing and compared their phenotypes with those of wild-type (WT) F9 and F9:Cldn6 cells. The knockout of Blk and Src genes was verified by DNA sequencing, and the absence of the corresponding protein was confirmed by Western blot (Figure 5A,B). The absence of either BLK or SRC led to a decrease in pSFK levels. The CLDN6-initiated epithelial differentiation was hindered in F9:Cldn6:Blk−/− and F9:Cldn6:Src−/− cells compared with F9:Cldn6 cells (Figure 5C). In addition, the induced expression of epithelial markers (ZO-1+ variant and ezrin/radixin/moesin-binding phosphoprotein [EBP50]) was inhibited in F9:Cldn6:Blk−/− and F9:Cldn6:Src−/− cells (Figure 5D). Moreover, the CLDN6-triggered expression of RAR target genes Cyp26a and Cldn6 (endogenous) was completely reversed in F9:Cldn6:Blk−/− and F9:Cldn6:Src−/− cells (Figure 5E). Taken together, these results indicate that BLK and SRC are indispensable for mediating the CLDN6-adhesion signaling.
4. Discussion
We previously reported that pSFK is colocalized with CLDN6 along cell borders in F9:Cldn6 cells, and both signals disappeared upon the SFK inhibitor PP2 treatment [28]. Colocalization of pSFK and CLDN6 was also evident at the cell–cell junctions of epithelia in embryoid bodies [28]. Moreover, we characterized the human endometrial carcinoma cell line Ishikawa expressing CLDN6, further confirming that pSFK is concentrated to cell boundaries together with CLDN6 [31]. In the present study, we demonstrated that at least four members of SFKs (BLK, FGR HCK, and SRC) are highly expressed in undifferentiated F9:iCldn6 cells and Dox-treated F9:iCldn6 cells. On the other hand, our double immunofluorescence staining showed that these SFK subtypes were colocalized with CLDN6 along cell borders in differentiated F9:iCldn6 cells but not in undifferentiated F9:iCldn6 cells. These results indicated that CLDN6 adhesion activates BLK, FGR HCK, and SRC, leading to the alteration of their subcellular localization.
We also identified, via immunoprecipitation assay, the SFK members associated with CLDN6 and obtained evidence showing that BLK and SRC form a complex with CLDN6. By contrast, neither the CLDN6-FGR nor CLDN6-HCK complexes were detected, suggesting that FGR and HCK are concentrated on cell boundaries in a CLDN6-independent manner. We subsequently demonstrated that BLK and SRC are associated with CLDN6 via the first half of the C-terminal cytoplasmic domain, in good agreement with our previous results on the CLDN6-pSFK complex in F9 cells and Ishikawa cells expressing WT and mutant CLDN6 [28]. Furthermore, GST pull-down assay revealed that BLK and SRC directly bind to the C-terminal cytoplasmic domain of CLDN6.
Another finding of the present study was that BLK and SRC bind to both non-pY196/200-CLDN6C and pY196/200-CLDN6C peptides. The binding efficiency of the pY196/200-CLDN6C peptide was lower than that of the non-pY196/200-CLDN6C peptide. These results were unexpected because most of the SH2 domains are known to recognize the pY-containing peptide better than the corresponding non-phosphorylated ones [34,35,36]. However, there are several exceptions. For instance, SH2 domains of Cten (C-terminal tensin-like), Grb7 (growth factor receptor bound 7), SAP (SLAM-associated protein), Shc (Src homologous and collagen), and Tensin2 exhibit pY-independent binding to their ligands [37,38,39,40,41,42,43,44,45,46]. Similar to these SH2 proteins, BLK and SRC seem to recognize the Y196/200-CLDN6C peptide independently of tyrosine phosphorylation. In addition, human and mouse CLDN6Y196 and CLDN6Y200 are conserved in CLDN2/3/4/7/8/10/12/14/16 and CLDN1/2/4/5/9/12/17/18, respectively [29]; therefore, it should be determined whether the C-terminal cytoplasmic domain of these CLDN subtypes could recruit specific SFK members.
Using several SFK inhibitors (i.e., PP1, PP2, SU6656, and aminogenistein), we previously revealed that SFKs are involved in CLDN6-triggered epithelial differentiation in F9 cells and embryoid bodies [28]. In the present study, we compared phenotypes of F9:Cldn6:Blk−/− and F9:Cldn6:Src−/− cells with those of WT F9 and F9:Cldn6 cells and showed that BLK and SRC are essential for CLDN6-initiated cellular events such as epithelial differentiation and the expression of Cyp26a and Cldn6 (endogenous) genes. Since RARγ activates Cldn6 gene expression [28,47], the positive loop of the CLDN6/BLK–RARγ and CLDN6/SRC–RARγ pathways most likely participates not only in provoking but also in the maintenance of CLDN6-initiated cellular processes.
SFKs, including SRC, are frequently activated in many types of cancers and promote cancer progression [4,8,48,49]. We previously reported that abnormal CLDN6/SFK signaling accelerates malignant phenotypes of endometrial cancer cells via hijacking the CLDN6–ERα axis [29,31]. Excessive CLDN6/SFK signaling stimulated cell proliferation and migration, as well as tumor growth and invasion into fibrous capsules in xenografts. Furthermore, we have very recently demonstrated that aberrant CLDN4/SFK signaling drives breast cancer metabolism and progression via Liver X receptor β [50]. Hence, the appropriate and extreme CLDN/SFK signaling likely contributes to physiological and pathological processes, respectively.
5. Conclusions
Here, we identified that BLK and SRC are directly associated with CLDN6 through the C-terminal cytoplasmic domain. We also demonstrated that these SFKs bind to CLDN6C in a pY-independent manner. Moreover, we showed that BLK and SRC are critical for the CLDN6 signaling to trigger epithelial differentiation and the expression of RAR target genes.
Supplementary Materials
The following supporting information can be downloaded at https://0-www-mdpi-com.brum.beds.ac.uk/article/10.3390/cells12131696/s1, Figure S1: BLK and SRC are at least in part colocalized with CLDN6 along cell boundaries in Ishikawa:CLDN6 cells but not in Ishikawa cells. Confocal images of indicated proteins in the revealed cell lines.
Author Contributions
N.I.-T., K.S. and H.C. designed the research; N.I.-T., K.S. and K.K. performed the experiments; N.I.-T., K.S., K.K. and H.C. analyzed data; N.I.-T., K.S. and H.C. wrote the paper. All authors have read and agreed to the published version of the manuscript.
Funding
This work was supported by JSPS KAKENHI (Grant Numbers 17K08699, 17K17978, 23M02703, and 24790390), the Uehara Memorial Foundation, and the Takeda Science Foundation.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available on request from the corresponding author.
Acknowledgments
We thank J. Kai, S. Watanabe, and K. Watari for their technical assistance and the Scientific English Editing Section of Fukushima Medical University for their help with the manuscript.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Rous, P. A Transmissible avian neoplasm. (Sarcoma of the common fowl) by Peyton Rous, M.D., Experimental Medicine for Sept. 1, 1910, vol. 12, pp. 696–705. J. Exp. Med. 1979, 150, 738–753. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.T.; Cooper, J.A. Regulation, Substrates and Functions of Src. Biochim. Biophys. Acta 1996, 1287, 121–149. [Google Scholar] [CrossRef] [PubMed]
- Parsons, S.J.; Parsons, J.T. Src Family Kinases, Key Regulators of Signal Transduction. Oncogene 2004, 23, 7906–7909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeatman, T.J. A Renaissance for SRC. Nat. Rev. Cancer 2004, 4, 470–480. [Google Scholar] [CrossRef]
- Boggon, T.J.; Eck, M.J. Structure and Regulation of Src Family Kinases. Oncogene 2004, 23, 7918–7927. [Google Scholar] [CrossRef] [Green Version]
- Kim, L.C.; Song, L.; Haura, E.B. Src Kinases as Therapeutic Targets for Cancer. Nat. Rev. Clin. Oncol. 2009, 6, 587–595. [Google Scholar] [CrossRef]
- Aleshin, A.; Finn, R.S. SRC: A Century of Science Brought to the Clinic. Neoplasia 2010, 12, 599–607. [Google Scholar] [CrossRef] [Green Version]
- Pelaz, S.G.; Tabernero, A. Src: Coordinating Metabolism in Cancer. Oncogene 2022, 41, 4917–4928. [Google Scholar] [CrossRef]
- Martin, G.S. The Hunting of the Src. Nat. Rev. Mol. Cell Biol. 2001, 2, 467–475. [Google Scholar] [CrossRef]
- Harrison, S.C. Variation on an Src-like Theme. Cell 2003, 112, 737–740. [Google Scholar] [CrossRef] [Green Version]
- Shah, N.H.; Amacher, J.F.; Nocka, L.M.; Kuriyan, J. The Src Module: An Ancient Scaffold in the Evolution of Cytoplasmic Tyrosine Kinases. Crit. Rev. Biochem. Mol. Biol. 2018, 53, 535–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lipsick, J. A History of Cancer Research: Tyrosine Kinases. Cold Spring Harb. Perspect. Biol. 2019, 11, a035592. [Google Scholar] [CrossRef] [Green Version]
- McLachlan, R.W.; Kraemer, A.; Helwani, F.M.; Kovacs, E.M.; Yap, A.S. E-Cadherin Adhesion Activates c-Src Signaling at Cell-Cell Contacts. Mol. Biol. Cell 2007, 18, 3214–3223. [Google Scholar] [CrossRef] [Green Version]
- Canel, M.; Serrels, A.; Frame, M.C.; Brunton, V.G. E-Cadherin-Integrin Crosstalk in Cancer Invasion and Metastasis. J. Cell Sci. 2013, 126, 393–401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiao, R.; Xi, X.-D.; Chen, Z.; Chen, S.-J.; Meng, G. Structural Framework of C-Src Activation by Integrin B3. Blood 2013, 121, 700–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ochs, K.; Málaga-Trillo, E. Common Themes in PrP Signaling: The Src Remains the Same. Front. Cell Dev. Biol. 2014, 2, 63. [Google Scholar] [CrossRef] [Green Version]
- Hamidi, H.; Ivaska, J. Every Step of the Way: Integrins in Cancer Progression and Metastasis. Nat. Rev. Cancer 2018, 18, 533–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furuse, M.; Fujita, K.; Hiiragi, T.; Fujimoto, K.; Tsukita, S. Claudin-1 and -2: Novel Integral Membrane Proteins Localizing at Tight Junctions with No Sequence Similarity to Occludin. J. Cell Biol. 1998, 141, 1539–1550. [Google Scholar] [CrossRef] [Green Version]
- Furuse, M.; Tsukita, S. Claudins in Occluding Junctions of Humans and Flies. Trends Cell Biol. 2006, 16, 181–188. [Google Scholar] [CrossRef]
- Chiba, H.; Osanai, M.; Murata, M.; Kojima, T.; Sawada, N. Transmembrane Proteins of Tight Junctions. Biochim. Biophys. Acta 2008, 1778, 588–600. [Google Scholar] [CrossRef] [Green Version]
- Günzel, D.; Yu, A.S.L. Claudins and the Modulation of Tight Junction Permeability. Physiol. Rev. 2013, 93, 525–569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Itallie, C.M.; Anderson, J.M. Claudins and Epithelial Paracellular Transport. Annu. Rev. Physiol. 2006, 68, 403–429. [Google Scholar] [CrossRef] [PubMed]
- Krug, S.M.; Schulzke, J.D.; Fromm, M. Tight Junction, Selective Permeability, and Related Diseases. Semin. Cell Dev. Biol. 2014, 36, 166–176. [Google Scholar] [CrossRef]
- Turner, J.R.; Buschmann, M.M.; Romero-Calvo, I.; Sailer, A.; Shen, L. The Role of Molecular Remodeling in Differential Regulation of Tight Junction Permeability. Semin. Cell Dev. Biol. 2014, 36, 204–212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zihni, C.; Mills, C.; Matter, K.; Balda, M.S. Tight Junctions: From Simple Barriers to Multifunctional Molecular Gates. Nat. Rev. Mol. Cell Biol. 2016, 17, 564–580. [Google Scholar] [CrossRef]
- Tsukita, S.; Tanaka, H.; Tamura, A. The Claudins: From Tight Junctions to Biological Systems. Trends Biochem. Sci. 2019, 44, 141–152. [Google Scholar]
- Cavallaro, U.; Dejana, E. Adhesion Molecule Signalling: Not Always a Sticky Business. Nat. Rev. Mol. Cell Biol. 2011, 12, 189–197. [Google Scholar] [CrossRef]
- Sugimoto, K.; Ichikawa-Tomikawa, N.; Kashiwagi, K.; Endo, C.; Tanaka, S.; Sawada, N.; Watabe, T.; Higashi, T.; Chiba, H. Cell Adhesion Signals Regulate the Nuclear Receptor Activity. Proc. Natl. Acad. Sci. USA 2019, 116, 24600–24609. [Google Scholar] [CrossRef]
- Sugimoto, K.; Chiba, H. The Claudin-Transcription Factor Signaling Pathway. Tissue Barriers 2021, 9, 1908109. [Google Scholar] [CrossRef]
- Kojima, M.; Sugimoto, K.; Tanaka, M.; Endo, Y.; Kato, H.; Honda, T.; Furukawa, S.; Nishiyama, H.; Watanabe, T.; Soeda, S.; et al. Prognostic Significance of Aberrant Claudin-6 Expression in Endometrial Cancer. Cancers 2020, 12, 2748. [Google Scholar] [CrossRef]
- Kojima, M.; Sugimoto, K.; Kobayashi, M.; Ichikawa-Tomikawa, N.; Kashiwagi, K.; Watanabe, T.; Soeda, S.; Fujimori, K.; Chiba, H. Aberrant Claudin-6-Adhesion Signaling Promotes Endometrial Cancer Progression via Estrogen Receptor α. Mol. Cancer Res. 2021, 19, 1208–1220. [Google Scholar] [CrossRef] [PubMed]
- Chiba, H.; Chambon, P.; Metzger, D. F9 Embryonal Carcinoma Cells Engineered for Tamoxifen-Dependent Cre-Mediated Site-Directed Mutagenesis and Doxycycline-Inducible Gene Expression. Exp. Cell Res. 2000, 260, 334–339. [Google Scholar] [CrossRef]
- Sugimoto, K.; Ichikawa-Tomikawa, N.; Satohisa, S.; Akashi, Y.; Kanai, R.; Saito, T.; Sawada, N.; Chiba, H. The Tight-Junction Protein Claudin-6 Induces Epithelial Differentiation from Mouse F9 and Embryonic Stem Cells. PLoS ONE 2013, 8, e75106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadowski, I.; Stone, J.C.; Pawson, T. A Noncatalytic Domain Conserved among Cytoplasmic Protein-Tyrosine Kinases Modifies the Kinase Function and Transforming Activity of Fujinami Sarcoma Virus P130gag-Fps. Mol. Cell. Biol. 1986, 6, 4396–4408. [Google Scholar] [PubMed] [Green Version]
- Olsen, J.V.; Blagoev, B.; Gnad, F.; Macek, B.; Kumar, C.; Mortensen, P.; Mann, M. Global, In Vivo, and Site-Specific Phosphorylation Dynamics in Signaling Networks. Cell 2006, 127, 635–648. [Google Scholar] [CrossRef] [Green Version]
- Huttlin, E.L.; Jedrychowski, M.P.; Elias, J.E.; Goswami, T.; Rad, R.; Beausoleil, S.A.; Villén, J.; Haas, W.; Sowa, M.E.; Gygi, S.P. A Tissue-Specific Atlas of Mouse Protein Phosphorylation and Expression. Cell 2010, 143, 1174–1189. [Google Scholar] [CrossRef] [Green Version]
- Charest, A.; Wagner, J.; Jacob, S.; McGlade, C.J.; Tremblay, M.L. Phosphotyrosine-Independent Binding of SHC to the NPLH Sequence of Murine Protein-Tyrosine Phosphatase-PEST. Evidence for Extended Phosphotyrosine Binding/Phosphotyrosine Interaction Domain Recognition Specificity. J. Biol. Chem. 1996, 271, 8424–8429. [Google Scholar] [CrossRef] [Green Version]
- Raffel, G.D.; Parmar, K.; Rosenberg, N. In Vivo Association of V-Abl with Shc Mediated by a Non-Phosphotyrosine-Dependent SH2 Interaction. J. Biol. Chem. 1996, 271, 4640–4645. [Google Scholar] [CrossRef] [Green Version]
- Sayos, J.; Wu, C.; Morra, M.; Wang, N.; Zhang, X.; Allen, D.; van Schaik, S.; Notarangelo, L.; Geha, R.; Roncarolo, M.G.; et al. The X-Linked Lymphoproliferative-Disease Gene Product SAP Regulates Signals Induced through the Co-Receptor SLAM. Nature 1998, 395, 462–469. [Google Scholar] [CrossRef]
- Li, S.C.; Gish, G.; Yang, D.; Coffey, A.J.; Forman-Kay, J.D.; Ernberg, I.; Kay, L.E.; Pawson, T. Novel Mode of Ligand Binding by the SH2 Domain of the Human XLP Disease Gene Product SAP/SH2D1A. Curr. Biol. 1999, 9, 1355–1362. [Google Scholar] [CrossRef] [Green Version]
- Poy, F.; Yaffe, M.B.; Sayos, J.; Saxena, K.; Morra, M.; Sumegi, J.; Cantley, L.C.; Terhorst, C.; Eck, M.J. Crystal Structures of the XLP Protein SAP Reveal a Class of SH2 Domains with Extended, Phosphotyrosine-Independent Sequence Recognition. Mol. Cell 1999, 4, 555–561. [Google Scholar] [CrossRef] [PubMed]
- Pero, S.C.; Oligino, L.; Daly, R.J.; Soden, A.L.; Liu, C.; Roller, P.P.; Li, P.; Krag, D.N. Identification of Novel Non-Phosphorylated Ligands, Which Bind Selectively to the SH2 Domain of Grb7. J. Biol. Chem. 2002, 277, 11918–11926. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spuches, A.M.; Argiros, H.J.; Lee, K.H.; Haas, L.L.; Pero, S.C.; Krag, D.N.; Roller, P.P.; Wilcox, D.E.; Lyons, B.A. Calorimetric Investigation of Phosphorylated and Non-Phosphorylated Peptide Ligand Binding to the Human Grb7-SH2 Domain. J. Mol. Recognit. 2007, 20, 245–252. [Google Scholar] [CrossRef] [PubMed]
- Liao, Y.-C.; Si, L.; deVere White, R.W.; Lo, S.H. The Phosphotyrosine-Independent Interaction of DLC-1 and the SH2 Domain of Cten Regulates Focal Adhesion Localization and Growth Suppression Activity of DLC-1. J. Cell Biol. 2007, 176, 43–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dai, K.; Liao, S.; Zhang, J.; Zhang, X.; Tu, X. Solution Structure of Tensin2 SH2 Domain and Its Phosphotyrosine-Independent Interaction with DLC-1. PLoS ONE 2011, 6, e21965. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.A.; Engelmann, B.W.; Nash, P.D. The Language of SH2 Domain Interactions Defines Phosphotyrosine-Mediated Signal Transduction. FEBS Lett. 2012, 586, 2597–2605. [Google Scholar] [CrossRef] [Green Version]
- Kubota, H.; Chiba, H.; Takakuwa, Y.; Osanai, M.; Tobioka, H.; Kohama, G.; Mori, M.; Sawada, N. Retinoid X Receptor Alpha and Retinoic Acid Receptor Gamma Mediate Expression of Genes Encoding Tight-Junction Proteins and Barrier Function in F9 Cells during Visceral Endodermal Differentiation. Exp. Cell Res. 2001, 263, 163–172. [Google Scholar] [CrossRef]
- Irby, R.B.; Yeatman, T.J. Role of Src Expression and Activation in Human Cancer. Oncogene 2000, 19, 5636–5642. [Google Scholar] [CrossRef] [Green Version]
- Alvarez, R.H.; Kantarjian, H.M.; Cortes, J.E. The Role of Src in Solid and Hematologic Malignancies: Development of New-Generation Src Inhibitors. Cancer 2006, 107, 1918–1929. [Google Scholar] [CrossRef]
- Murakami-Nishimagi, Y.; Sugimoto, K.; Kobayashi, M.; Tachibana, K.; Kojima, M.; Okano, M.; Hashimoto, Y.; Saji, S.; Ohtake, T.; Chiba, H. Claudin-4-Adhesion Signaling Drives Breast Cancer Metabolism and Progression via Liver X Receptor β. Breast Cancer Res. 2023, 25, 41. [Google Scholar] [CrossRef]
Figure 1.
BLK, FGR, HCK, and SRC are expressed in F9 cells. (A) Western blot analysis showing the expression profile of SFKs in F9:iCldn6 cells grown for 96 h in the presence or absence of 1 µg/mL doxycycline (Dox). 293T cells over-expressing the corresponding mouse SFK and Cldn6 genes were used as positive controls (P/C). A breast cancer cell line MDA-MB-231 was used as the positive control for pSFK. (B,C) Confocal immunofluorescent images of the indicated proteins in Dox-exposed F9:iCldin6 cells. Areas consisting of epithelial cells (B) and the boundary regions between undifferentiated and epithelial cells (C) are indicated. Bars, 20 µm.
Figure 1.
BLK, FGR, HCK, and SRC are expressed in F9 cells. (A) Western blot analysis showing the expression profile of SFKs in F9:iCldn6 cells grown for 96 h in the presence or absence of 1 µg/mL doxycycline (Dox). 293T cells over-expressing the corresponding mouse SFK and Cldn6 genes were used as positive controls (P/C). A breast cancer cell line MDA-MB-231 was used as the positive control for pSFK. (B,C) Confocal immunofluorescent images of the indicated proteins in Dox-exposed F9:iCldin6 cells. Areas consisting of epithelial cells (B) and the boundary regions between undifferentiated and epithelial cells (C) are indicated. Bars, 20 µm.

Figure 2.
BLK and SRC form a complex with CLDN6 via the C-terminal cytoplasmic domain. (A) Association between CLDN6 and the indicated SFK members. (B) Association between CLDN6C and either BLK or SRC. F9 L32T2 cells were transiently transfected with the indicated HA-Cldn6 expression vector along with the revealed SFK-FLAG expression vectors. In the input lanes, 1% of HA of the input protein samples was loaded. The schematic representation of HA-Cldn6 (WT) and the deletion mutants is revealed in the top panel. The conserved tyrosine residues of the C-terminal cytoplasmic domain of human CLDN6 are indicated in red. IP, immunoprecipitation; WT, wild-type.
Figure 2.
BLK and SRC form a complex with CLDN6 via the C-terminal cytoplasmic domain. (A) Association between CLDN6 and the indicated SFK members. (B) Association between CLDN6C and either BLK or SRC. F9 L32T2 cells were transiently transfected with the indicated HA-Cldn6 expression vector along with the revealed SFK-FLAG expression vectors. In the input lanes, 1% of HA of the input protein samples was loaded. The schematic representation of HA-Cldn6 (WT) and the deletion mutants is revealed in the top panel. The conserved tyrosine residues of the C-terminal cytoplasmic domain of human CLDN6 are indicated in red. IP, immunoprecipitation; WT, wild-type.

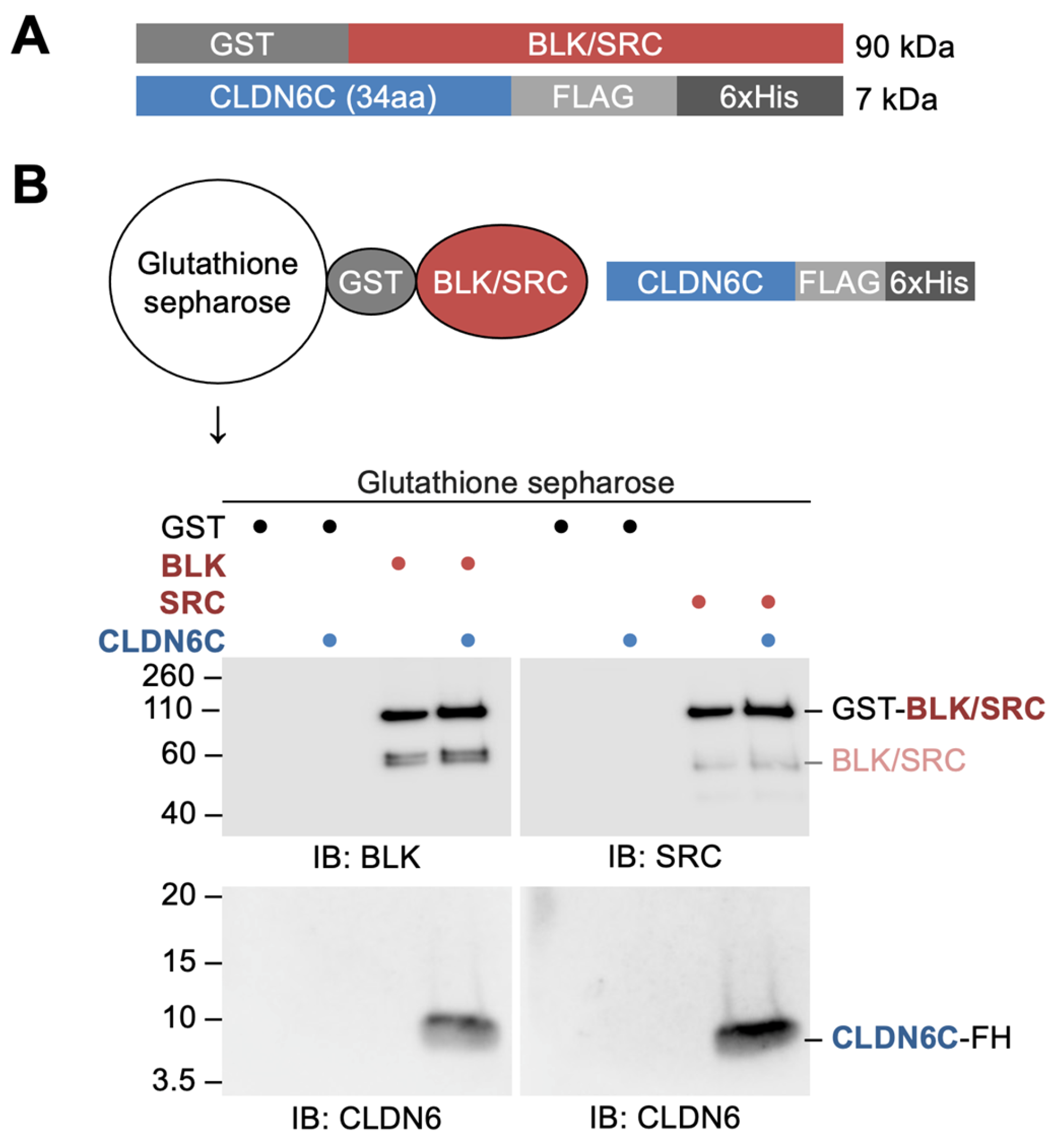
Figure 3.
BLK and SRC directly bind to the C-terminal cytoplasmic domain of CLDN6. (A) The structure of recombinant BLK, SRC, and CLDN6C proteins. (B) GST pull-down assay showing direct binding of recombinant BLK and SRC to recombinant CLDN6C. The mobility of molecular mass markers (kilodaltons) is indicated on the left. GST, glutathione S-transferase; FL, FLAG; IB, immunoblot.
Figure 3.
BLK and SRC directly bind to the C-terminal cytoplasmic domain of CLDN6. (A) The structure of recombinant BLK, SRC, and CLDN6C proteins. (B) GST pull-down assay showing direct binding of recombinant BLK and SRC to recombinant CLDN6C. The mobility of molecular mass markers (kilodaltons) is indicated on the left. GST, glutathione S-transferase; FL, FLAG; IB, immunoblot.

Figure 4.
Recombinant BLK and SRC bind to the CLDN6C peptide in a pY-independent manner. (A) Amino acid sequences of the non-pY196/200-CLDN6C and pY196/200-CLDN6C peptides. The phosphorylation of Y residues is indicated in red. (B) Schema for ELISA assay. Ninety-six-well ELISA plates were coated with CLDN6C peptides and used for subsequent analysis. (C) ELISA analysis showing that recombinant BLK and SRC dose-dependently bind not only to the pY196/200-CLDN6C peptide but also to the non-pY196/200-CLDN6C peptide. Relative absorbance is shown in the histograms (mean ± SD; n = 4).
Figure 4.
Recombinant BLK and SRC bind to the CLDN6C peptide in a pY-independent manner. (A) Amino acid sequences of the non-pY196/200-CLDN6C and pY196/200-CLDN6C peptides. The phosphorylation of Y residues is indicated in red. (B) Schema for ELISA assay. Ninety-six-well ELISA plates were coated with CLDN6C peptides and used for subsequent analysis. (C) ELISA analysis showing that recombinant BLK and SRC dose-dependently bind not only to the pY196/200-CLDN6C peptide but also to the non-pY196/200-CLDN6C peptide. Relative absorbance is shown in the histograms (mean ± SD; n = 4).

Figure 5.
BLK and SRC are indispensable for the CLDN6-initiated cellular events. (A) Knockout (KO) of the Blk and Src genes in F9 cells using the CRISPR/Cas9 method. KO of these genes is verified by DNA sequencing. (B,D) Western blot for the indicated proteins in the revealed F9 cells. (C) Morphological appearance of the indicated F9 cells. The boundaries between undifferentiated and epithelial cells are shown by the dashed yellow lines. (E) RT-qPCR analysis for the indicated molecules in the revealed F9 cells. Relative expression levels are shown in the histograms (mean ± SD; n = 4). Bar, 50 µm.
Figure 5.
BLK and SRC are indispensable for the CLDN6-initiated cellular events. (A) Knockout (KO) of the Blk and Src genes in F9 cells using the CRISPR/Cas9 method. KO of these genes is verified by DNA sequencing. (B,D) Western blot for the indicated proteins in the revealed F9 cells. (C) Morphological appearance of the indicated F9 cells. The boundaries between undifferentiated and epithelial cells are shown by the dashed yellow lines. (E) RT-qPCR analysis for the indicated molecules in the revealed F9 cells. Relative expression levels are shown in the histograms (mean ± SD; n = 4). Bar, 50 µm.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Table 1.
Antibodies.
| Antigen | Type | Host | IF | WB | IP | Source | Identifier |
|---|---|---|---|---|---|---|---|
| BLK | P | Rabbit | 1/100 | 1/1000 | Cell Signaling Technology | #3262 | |
| FGR | P | Rabbit | 1/100 | 1/1000 | Santa Cruz Biotechnology | sc-50338 | |
| FRK | M | Rat | 1/1000 | R&D Systems | MAB3766 | ||
| FYN | P | Rabbit | 1/1000 | GeneTex | GTX109428 | ||
| HCK | P | Rabbit | 1/100 | 1/1000 | Santa Cruz Biotechnology | sc-72 | |
| LCK | M | Mouse | 1/1000 | Santa Cruz Biotechnology | sc-433 | ||
| LYN | P | Rabbit | 1/1000 | Santa Cruz Biotechnology | sc-15 | ||
| SRC | M | Rabbit | 1/100 | 1/1000 | Cell Signaling Technology | #2123 | |
| YES1 | M | Mouse | 1/1000 | Santa Cruz Biotechnology | sc-8403 | ||
| pSFK | P | Rabbit | 1/100 | 1/1000 | Cell Signaling Technology | #2101 | |
| CLDN6 | P | Rabbit | 1/1000 | Immuno-Biological Laboratories | #18865 | ||
| CLDN6 | M | Rat | 1/4 | Kojima et al. [30] | |||
| EBP50 | P | Rabbit | 1/1000 | Affinity Bio Reagents | PA-1-090 | ||
| ZO-1 (α+ variant) | M | Rat | 1/1000 | Santa Cruz Biotechnology | sc33725 | ||
| FLAG | M | Mouse | 1 µg | MBL | M185-3L | ||
| FLAG (HRP) | P | Rabbit | 1/8000 | Merck | SAB4301135 | ||
| HA | M | Mouse | 1 µg | Proteintech | 66006-2-Ig | ||
| HA (HRP) | M | Rat | 1/4000 | Roche | 12013819001 | ||
| βActin (HRP) | M | Mouse | 1/4000 | Santa Cruz Biotechnology | sc-47778 HRP | ||
| Mouse IgG (HRP) | P | Sheep | 1/5000 | GE Health Care | NA934V | ||
| Rabbit IgG (HRP) | P | Goat | 1/2000 | Cell Signaling Technology | #7074 | ||
| Rat IgG (HRP) | P | Goat | 1/2000 | GE Health Care | NA935V | ||
| Rabbit IgG (Alexa 488) | P | Donkey | 1/400 | Jackson ImmunoResearch | 711-165-152 | ||
| Rabbit IgG (Alexa647) | P | Donkey | 1/400 | Jackson ImmunoResearch | 712-605-153 | ||
| Rat IgG (Cy3) | P | Donkey | 1/400 | Jackson ImmunoResearch | 712-165-153 |
IF, immunofluorescence; IHC, immunohistochemistry; WB, Western blotting; IP, immunoprecipitation; M, monoclonal; P, polyclonal.
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Ichikawa-Tomikawa, N.; Sugimoto, K.; Kashiwagi, K.; Chiba, H. The Src-Family Kinases SRC and BLK Contribute to the CLDN6-Adhesion Signaling. Cells 2023, 12, 1696. https://0-doi-org.brum.beds.ac.uk/10.3390/cells12131696
AMA Style
Ichikawa-Tomikawa N, Sugimoto K, Kashiwagi K, Chiba H. The Src-Family Kinases SRC and BLK Contribute to the CLDN6-Adhesion Signaling. Cells. 2023; 12(13):1696. https://0-doi-org.brum.beds.ac.uk/10.3390/cells12131696
Chicago/Turabian StyleIchikawa-Tomikawa, Naoki, Kotaro Sugimoto, Korehito Kashiwagi, and Hideki Chiba. 2023. "The Src-Family Kinases SRC and BLK Contribute to the CLDN6-Adhesion Signaling" Cells 12, no. 13: 1696. https://0-doi-org.brum.beds.ac.uk/10.3390/cells12131696
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.