Mitochondrial DNA Integrity: Role in Health and Disease


Department of Nutritional Sciences and New Jersey Institute for Food, Nutrition, and Health, Rutgers University, New Brunswick, NJ 08901, USA
*
Author to whom correspondence should be addressed.
Cells 2019, 8(2), 100; https://0-doi-org.brum.beds.ac.uk/10.3390/cells8020100
Submission received: 31 December 2018
/
Revised: 25 January 2019
/
Accepted: 28 January 2019
/
Published: 29 January 2019
(This article belongs to the Special Issue Mitochondrial Biology in Health and Disease)
Abstract
:As the primary cellular location for respiration and energy production, mitochondria serve in a critical capacity to the cell. Yet, by virtue of this very function of respiration, mitochondria are subject to constant oxidative stress that can damage one of the unique features of this organelle, its distinct genome. Damage to mitochondrial DNA (mtDNA) and loss of mitochondrial genome integrity is increasingly understood to play a role in the development of both severe early-onset maladies and chronic age-related diseases. In this article, we review the processes by which mtDNA integrity is maintained, with an emphasis on the repair of oxidative DNA lesions, and the cellular consequences of diminished mitochondrial genome stability.
1. Introduction
Coming from the Greek words for thread (mitos) and granules (khondros), mitochondria serve the essential function of energy generation, earning them the oft-used moniker of being the “powerhouses” of the cell. Through the coordinated processes of oxidative phosphorylation, mitochondria indeed generate most of the cellular adenosine triphosphate (ATP) and also establish the mitochondrial membrane potential, which is critical to their function. They accomplish these activities through the synergistic actions of over a thousand proteins, the majority of which are encoded in the nuclear genome and imported into the mitochondria [1]. However, mitochondria are a unique organelle in that they house their own genome, distinct from the nuclear genome. While this genome encodes only thirteen peptides that are involved in oxidative phosphorylation, [2] they are a critical component of the cellular energy production machinery. Much of our understanding of mitochondrial organization and function has arisen from the study of mitochondrial diseases. Apart from the better-described severe maladies of mitochondrial origin, it is now becoming clear that chronic accumulation of lower levels of mtDNA damage and decreases in mtDNA copy number are not only associated with the aging process, but may also be causally linked to age-related diseases such as neurodegeneration and diabetes [3,4,5,6]. Given their reparable nature, oxidative lesions represent an important target in our quest to mitigate the age-related disease burden. In this review, we summarize recent progress in our understanding of the maintenance of mtDNA integrity via DNA repair pathways that target oxidative damage and the cellular consequences of a loss of mtDNA integrity.
2. Mitochondrial DNA: Structure
mtDNA encodes for many of the essential components of the oxidative phosphorylation chain, and is thus crucial to ATP generation. Save for a couple of notable anomalies such as budding yeast that can survive anaerobic growing conditions and mtDNA-depleted cells that can subsist on glucose and glycolysis while in culture [7,8,9], mtDNA is essential to life. Interestingly, smaller non-life threatening mutations have also been shown to induce pathologies that may manifest later in the life-cycle or under conditions of metabolic stress [4,10,11,12]. The contribution of such mutations to cellular function is notably harder to decipher, since disease symptoms may exist on a spectrum which is dependent on the number of mitochondria harboring a given mutation. The existence of both healthy and mutated mtDNA within a given mitochondrion is referred to as heteroplasmy (Figure 1), and likely has a profound influence on cellular function [13]. Cellular tolerance of mtDNA heteroplasmy is a topic that is not completely understood, as tolerance is likely to (a) differ based upon the mutation and cell type, (b) be significantly influenced by competing processes such as mitochondrial biogenesis and degradation, and (c) result in a spectrum of phenotypes, many of which may be below clinical detection levels.
Human mitochondrial DNA (mtDNA) is a double-stranded circular molecule of 16,569 base pairs with a molecular mass of 107 daltons [2,14]. It is associated with proteins and organized in nucleoids located within the mitochondrial matrix, in close proximity to the mitochondrial membrane [2,15,16,17,18]. mtDNA encodes 13 polypeptides of the oxidative phosphorylation complex (OXPHOS) that are localized to the mitochondrial inner membrane. These include seven subunits of complex I, one subunit of complex III, three subunits of complex IV, and two of complex V, all of which are required for the generation of ATP by oxidative phosphorylation [2,11,15]. The vast majority of mitochondrial proteins, however, are encoded by the nuclear genome and are imported into the mitochondria by translocation systems localized to the outer and inner mitochondrial membranes [19]. The mitochondrial genome also encodes for 22 transfer RNAs and 2 ribosomal RNAs (12S and 16S rRNAs) for mitochondrial translation [2,20]. Unlike genomic DNA, mtDNA lacks histones and is maternally inherited [11,14,21,22], although evidence for partial paternal inheritance has recently been described [23].
The double-stranded structure of mtDNA consists of a guanine-rich heavy (H) strand and a light (L) strand [24]. The organization of mtDNA across vertebrates is similar to that of humans, with the majority of noncoding DNA being located within a ~1 kb region dubbed the noncoding region (NCR). The NCR is the most polymorphic site within mtDNA with several known polymorphisms within two hypervariable regions (HVR) in the NCR [25,26]. Sequencing of the HVRs of mtDNA can be used to ascribe mitochondrial haplogroups and is useful in tracing genetic lineages of human populations [27]. The noncoding region (NCR) serves a regulatory function within mtDNA, exerting control over transcription and translation. The mtDNA control region contains the replication origin for one strand and the transcription origin for both strands [20,28]. The control region is also the site of the displacement loop (D-loop) of mtDNA. While often used interchangeably, the D-loop only forms part of the control region and is not analogous to the NCR. The D-loop consists of a triple strand of DNA containing the heavy and light strands and a partially replicated heavy strand, hydrogen bonded to the light strand [29,30]. Replication of mtDNA begins in the D-loop, which also contains promoters of transcripts adjacent to the D-loop. While the exact function of the D-loop is unclear, it is noteworthy that this region is subject to high sequence variability [31] and has been shown to be associated with the incidence of particular types of cancers [32,33,34,35,36,37]. Mutations in the mitochondrial control region have also been associated with aging in skeletal muscle [38] and skin fibroblasts [39].
3. Mitochondrial DNA: Transcription and Replication
Apart from the NCR, the remainder of the mtDNA molecule consists of genes without intervening intronic structures and often with overlapping reading frames between adjacent genes [40]. Further, every mitochondrion can contain multiple copies of mtDNA, with cells carrying numerous functional mitochondria per cell. The compact organization of mtDNA, along with the presence of multiple copies of mtDNA within cells, may help explain the relatively high gene density of mtDNA. Despite the minuscule size of the mtDNA molecule relative to the nuclear genome, mRNA of mitochondrial origin can represent a large proportion of total cellular mRNA, ranging from 5% in the lung to up to 30% in the human heart [40,41].
mtDNA transcription is initiated from two promoters on the H-strand (HSP1 and HSP2) and one on the L-strand (LSP) [42,43,44]. Mitochondrial genes are transcribed as polycistrionic precursors that are subsequently processed into individual RNAs [28,42,45]. mtDNA is transcribed into three primary transcripts, the shortest of which originates at HSP1. The second transcript originates at HSP2 and continues for almost the entire length of mtDNA. The third transcript originating at the LSP is close to genomic length [42,46,47,48,49]. These primary transcripts are further processed via sequential processes of cleavage, polyadenylation, and tRNA and rRNA modifications [42]. While mtDNA abundance is clearly correlated with the abundance of mitochondrial transcripts, much of the regulatory effects on mitochondrial transcription are exerted by nuclear-encoded proteins, many of which are still being identified. mtDNA transcription was previously thought to be regulated solely by the mitochondrial RNA polymerase (POLRMT), two transcription factors (TFAM and TF2BM), an elongation factor (TEFM), and a transcription termination factor (MTERF1) [50,51,52]. However, more recent studies have indicated a role for other proteins that have previously been shown to regulate nuclear gene transcription [42,53,54,55,56,57]. Further, mitochondrial transcript abundance is also regulated by cellular ATP levels and rates of transcript turnover [42,58]. There is also some evidence to suggest that similar to genomic DNA, mtDNA methylation may play a role in regulating transcript abundance [42,59,60,61]. These, and other aspects of mitochondrial transcription, including the existence of two origins on the heavy strand, the relative strengths of each of the origins, and mechanisms mediating the relative abundance of transcripts originating from each of the sites are all areas of active investigation, as discussed in greater detail in a recent review [42]. mtDNA replication (Box 1) is a complex process, our understanding of which is ongoing. For a more comprehensive review of this evolving and fascinating topic, we direct the reader to several excellent reviews dedicated to mtDNA replication [25,40]. Briefly, mitochondria house a distinct DNA replication system that includes nuclear-encoded proteins such as DNA polymerase γ, TWINKLE helicase, mitochondrial single-stranded DNA binding protein (mtSSB), and POLRMT [62,63,64,65,66,67], as well as RNase H1, DNA ligase III, and topoisomerase 3α [68,69,70,71,72]. Despite using a similar cast of players, mtDNA replication is distinct from nuclear replication in many ways, including the fact that mtDNA replication occurs not only in dividing cells, but also in post-mitotic tissues and across all stages of the cell cycle. The most widely-accepted model for mtDNA replication involves a strand-displacement mechanism, wherein leading and lagging strand synthesis are uncoupled (Box 1) [73,74,75]. However, in recent years, competing models of mt DNA replication, including the RITOLS model and conventional coupled strand synthesis have also been suggested [76,77,78,79]. The precise mechanisms involved in these alternative mode of replication have been recently reviewed [25]. Defects in mtDNA replication proteins are associated with severe multi-system diseases such as ataxia-neuropathy [80]. Given the importance of accurate replication of its genome to mitochondrial function, the study of mtDNA replication is an active and critical area of inquiry.
While the processes of mitochondrial transcription and replication are well-studied, the impact of oxidative DNA lesions on the rates and fidelity of these two processes is far from clear. This topic is discussed below, following a brief review of mtDNA damage and repair pathways.
Box 1. Mitochondrial DNA Replication—Competing Theories.
In the more classical and widely-accepted model of mtDNA replication, the strand displacement model, single-stranded replication of the heavy (H) strand begins with further expansion of a nascent displacement loop(D-loop). This continues until the light (L) strand origin (OL) is exposed, after about two-thirds of the heavy strand has been replicated, when synthesis of the L-strand begins in the opposite direction. This asymmetrical strand synthesis results in one of two daughter molecules containing a partially-synthesized L-strand, until the replication process is completed. In an alternatively proposed strand-coupled or synchronous model of mtDNA replication, the replication process is initiated within the D-loop, with both strands being synthesized bidirectionally and simultaneously. In the RITOLS model, replication begins at one of two sites of origin. This is followed by leading strand synthesis with simultaneous incorporation of RNA on the lagging strand. Lagging strand synthesis is presumed to begin at OL, following which the lagging strand RNA is either converted to or replaced by DNA.
4. Induction of Mitochondrial DNA Damage
Given the indispensable role of mitochondrially-encoded proteins in regulating energy production in the cell, enzymes responsible for maintaining mtDNA stability and integrity are of critical importance to cellular energetics. The need to maintain mtDNA integrity is complicated by the fact that mtDNA is particularly vulnerable to damage, especially through the generation of oxidative lesions [81,82]. There are several potential reasons for this increased sensitivity of mtDNA to oxidative lesions. First, mtDNA resides in close proximity to the site of reactive oxygen species (ROS) production in the mitochondrial membrane. Second, mtDNA replication proceeds via an asymmetric route (Box 1), resulting in parts of the heavy strand existing as single-stranded structures for extended periods of time; this can lead to the spontaneous deamination of nucleotides [83,84]. Further, factors such as the asymmetric distribution of dNTPs in mitochondria, favoring higher levels of dGTP, can result in decreased fidelity during mtDNA replication, contributing to the higher rate of spontaneous mutagenesis in the mitochondrial genome [85]. By various estimates, mtDNA undergoes a high rate of mutagenesis, potentially 10- to 20-fold higher than that of the nuclear genome [86,87,88,89].
Both the process of respiration, as well as exogenous exposures, can result in damage to DNA bases [90]. Further, mutations in proteins involved in oxidative phosphorylation can also serve to increase cellular ROS level [91,92]. Mitochondrial respiration generates multiple reactive species, including superoxide, hydrogen peroxide, nitric oxide, peroxynitrite, hypochlorous acid, singlet oxygen, and the hydroxyl radical, which react with and damage cellular proteins, lipids, and DNA [81,82,93,94,95]. Cellular ROS levels are regulated by enzymatic antioxidants such as superoxide dismutase (SOD), glutathione peroxidase, and catalase [96,97,98], many of which are activated by the nuclear erythroid-2 like factor-2 transcription factor binding to antioxidant response elements in their promoter regions [99]. Mitochondrial ROS reduction is also achieved via proton leak across the mitochondrial membrane, a process regulated by uncoupling proteins at the expense of ATP generation [100]. Despite the existence of these numerous mechanisms, as well as cellular antioxidants such as ascorbate and tocopherols, ROS quenching is by no means a perfect process. ROS-induced damage results in base substitutions, missense mutations, and deletions within the mitochondrial genome, all of which impact mitochondrial function. mtDNA has been shown to be more prone to damage as a consequence of aging [89,101,102,103,104,105,106,107,108], potentially due to both increased ROS production as well as reduced ROS quenching, and decreased DNA repair capacity with increasing age [81,82,109,110]. mtDNA has also been shown to be damaged at lower ROS levels than genomic DNA. Further, repair of mtDNA is a slower process than genomic repair, especially following longer durations of oxidative stress [89,101,102,103,104,105,106,108].
Mitochondria co-opt nuclear DNA repair factors to repair mtDNA lesions via a number of DNA repair pathways, including mismatch repair and non-homologous end joining, which have been reviewed elsewhere [86,90,108,111]. However, the best characterized and predominant repair pathway in mitochondria is the base excision repair (BER) pathway. BER removes small non-helix distorting lesions such as oxidized and deaminated bases, alkylation damage, and single-strand breaks [112,113,114]. The importance of this pathway for the maintenance of mtDNA integrity is evidenced by the numerous pathological phenotypes described in human cohorts and animal models with defects in BER [81,82,115,116,117,118,119].
5. Base-Excision Repair of Mitochondrial DNA
BER is a multi-step pathway that can act via either short-patch (insertion of 1 nucleotide) or long-patch (insertion of 2–10 nucleotides) mechanisms. The overall steps of this pathway include (i) recognition and excision of the damaged base, (ii) removal of the abasic site, (iii) end processing, (iv) gap filling, and (v) DNA ligation (Figure 2). The structure and biochemistry of each of the enzymes involved in this multi-step pathway has been recently reviewed [108,120,121,122]. BER is initiated by DNA glycosylases that recognize specific DNA lesions and cleave the N-glycosidic bond between the damaged base and the DNA backbone [81,82,121]. DNA glycosylases can be divided into mono- or bifunctional glycosylases. Monofunctional glycosylases such as adenine DNA glycosylase (MUTY) possess only glycosylase activity, necessitating the activity of a downstream enzyme, AP endonuclease (APE1), to generate a single-strand break. Non-oxidative lesions such as deaminated and alkylated bases are primarily excised by monofunctional DNA glycosylases, with MUTY being responsible for excision of mispaired adenines across from oxidized guanines [123]. In contrast, most oxidative lesions are recognized by bifunctional glycosylases such as endonuclease III like-1 (NTH1) that possess both glycosylase and AP lyase activities. Base removal by bifunctional glycosylases requires further processing of DNA ends by APE1 and polynucleotide kinase phosphate (PNKP), followed by nucleotide insertion by POLG (or polymerase beta in the nucleus) [108,121,124,125,126].
DNA glycosylases are encoded in the nucleus with several enzymes or variants containing a mitochondrial targeting sequence (MTS) that allows for mitochondrial localization of the glycosylase (Table 1). Six DNA glycosylases have been described to repair oxidized DNA lesions in humans [81,82,108,121]. These include MUTY homolog (also called MYH), 8-oxoguanine DNA glycosylase (OGG1), endonuclease three homolog 1 (NTH1), and Nei endonuclease VIII-like 1, 2, and 3 (NEIL1, NEIL2, and NEIL3). Of these glycosylases, MUTY, OGG1, NEIL1, NEIL2, and NTH1 have thus far been detected in mitochondria (see Table 1 for references).
As noted above, the enzyme MUTY is responsible for the cleavage of mispaired adenines opposite oxidized guanine (8-oxoG). MUTY is ubiquitously expressed in the human, with the highest expression levels reported in thymus and testis, as well as in embryonic tissues [127]. The NEIL family of glycosylases acts upon oxidized pyrimidines such as FapyA and G, hydroxyuracil, further oxidation products of 8-oxoG such as the hydantoin lesions, and thymine glycol, among other lesions. NEIL1 [128,129,130] is expressed ubiquitously with the highest expression reported in the liver, thymus, and pancreas. NEIL2 [131] has been reported to have the highest expression levels in the testes and skeletal muscle, and NEIL3 [130] has been reported to be selectively expressed in areas of the brain containing stem cells. NTH1 and OGG1 are bifunctional DNA glycosylases that recognize and repair thymine glycol and oxidized guanine (8-oxoG, FapyG) lesions, respectively [81,82]. NTH1 is ubiquitously expressed in rodents, with high levels of expression being reported in the heart and brain [130,132]. Human OGG1 (hOgg1) has been reported to be expressed at the highest levels in the thymus, testis, intestine, brain, and germinal centers of B cells, as well as in differentiated keratinocytes in the upper granular layer of the epidermis [81,82,112,113,133,134,135]. In addition to these DNA glycosylases that act on oxidized lesions, the enzyme MutT homolog 1 (MTH1), while strictly speaking not involved in BER, is also partially mitochondrially-localized (Table 1) [136,137,138] and likely contributes to sanitation of mitochondrial dNTP pools to suppress rates of oxidative lesion incorporation into mtDNA [139,140].
6. Impact of Oxidative Damage on Mitochondrial Replication and Transcription
Despite the high rate of oxidative damage to mtDNA, the significance of ROS-induced mitochondrial lesions to transcription and replication are unclear. POLG is known to be unable to replicate past single strand breaks. However, it can bypass 8-oxoG in an error-prone manner, following some degree of replication stalling [160,161,162]. This stalling at 8-oxoG sites was found to be increased under low cellular dNTP conditions, as might be expected in post-mitotic tissues, where mtDNA replication is still an active process [160]. Replication stalling at 8-oxoG was shown to be mediated by the proofreading function of POLG, pointing to the presumed mutagenicity of 8-oxoG [160]. With regard to mitochondrial transcription, POLRMT has been shown to be able to bypass oxidative lesions such as 8-oxoG and thymine glycol, albeit while generating transcripts containing G:C to T:A transversions [163,164,165,166]. This bypass was shown to be more efficient in the presence of a nuclear elongation factor, TFIIS [166,167]. More recently, it has been demonstrated that lesion bypass by POLRMT is enhanced in the presence of the TEFM, effectively reducing early transcript termination at 8-oxoG sites [163,164]. Thus, while the transcription of mtDNA containing oxidative lesions is likely to occur in vivo, the impact of such lesion bypasses on transcript sequences or the generation of aberrant proteins has, to our knowledge, not been systematically investigated. In particular, while several mitochondrial transcription and replication studies have been performed using reconstituted enzymes and in vitro systems [163,164,165,166,167], there is a marked paucity of data regarding the impact of mtDNA lesions on transcription in vivo. This is an especially important deficit, since differences in in vitro experimental systems, such as those using promoter-independent transcription vs. a more complete transcription system, have yielded divergent results with regard to error bypass [163]. This is a notable shortcoming of in vitro transcription systems, especially given that our understanding of the proteins involved in mitochondrial transcription is still evolving.
In this regard, we recently demonstrated that mice with enhanced expression of mitochondrial OGG1 had an increase in mitochondrial transcript abundance under conditions of oxidative stress [116]. This resulted in an increase in mitochondrial OXPHOS subunits and enhanced oxidative respiration in these mice [116]. Although indirectly, these data argue for a role for OGG1 in potentially regulating the rate and fidelity of mitochondrial transcription. However, it is unclear if this increase in mitochondrial transcript formation is due to a reduction in 8-oxoG content, an overall reduction in oxidative stress, or due to an as yet unidentified role of OGG1 in regulating mitochondrial transcription. Given the importance of mitochondrial replication and transcription to cellular metabolism, further studies in this area, particularly using in vivo models, are warranted.
7. Pathologies Associated with Mitochondrial Repair Defects
While the structure, mode of action, and biochemical regulation of these DNA glycosylases has been extensively studied, we know far less about the physiological relevance of unrepaired mtDNA damage to the development of disease. Given the known correlation between aging and the accumulation of DNA damage, perhaps it is not surprising that many of the pathologies associated with defects in BER can be considered diseases of aging. Through the study of human polymorphic variants and rodent models with engineered defects in glycosylase function, it has become evident that the efficient repair of oxidative lesions in genomic and mtDNA is integral to the prevention of cancers, neurodegenerative diseases, and metabolic pathologies. The role of genomic DNA damage in the progression of chronic and age-related diseases has been reviewed elsewhere by us and others [81,82,168,169,170,171]. The section below will focus on our current, more limited understanding of the role of mtDNA damage and repair in disease progression.
mtDNA mutations can be categorized into four broad classes, namely polypeptide mutations, rRNA and tRNA mutations that result in defects in protein synthesis, rearrangement mutations, and mutations within the control region that can impact mtDNA replication and transcription. Using databases of mtDNA sequence and variation such as the comprehensive MITOMAP initiative [172], a number of associations between specific mitochondrial mutations and proclivity or protection from disease have been reported. For instance, mtDNA mutations and deletions are implicated in many severe diseases (Table 2), including Mitochondrial Encephalopathy, Lactic acidosis, and Stroke-like episodes (MELAS), Kearns-Sayre Syndrome (KSS), Pearson syndrome, and in about 20% of cases of Leigh syndrome [173,174,175,176]. Like many severe maladies of mitochondrial origin, MELAS is typically an early-onset disease, with most patients developing symptoms before age twenty. MELAS is characterized by seizures, headaches, and stroke [173]. KSS is another early-onset neuromuscular disease characterized by pigmentary retinopathy and cardiac conduction defects, among other symptoms [176]. Pearson syndrome is often fatal in infancy and is characterized by exocrine pancreas insufficiency and sideroblastic anemia [174]. Leigh syndrome, a severe neurological disorder, is often diagnosed within the first year of life and generally results in respiratory failure and death within two to three years [175]. Other diseases of known mitochondrial origin include Leber hereditary optic atrophy [177] and a type of epilepsy called MERFF (Myoclonus Epilepsy with Ragged Red Fibers), among others [178].
Apart from these severe maladies, it is becoming increasingly clear that several chronic age-related diseases are also related to impaired mitochondrial function [183,184]. While mitochondrial deficits do not necessarily originate within the mitochondria, mutations within mtDNA and alterations in mtDNA repair enzymes can be a direct cause of age-related pathologies [183,184].
8. Cancer
Defects in mitochondrial function have long been proposed to be involved in the etiology of cancers. Cancerous cells undergo profound alterations in oxidative metabolism, resulting in reduced flux through mitochondrial energy generating pathways [185]. Since mtDNA encodes for several critical proteins of the respiratory chain, it is highly plausible that mutations in mtDNA may impact the respiratory function of cells and thereby contribute, if not to cancer initiation, at least to propagation of the metabolic phenotype of cancer cells. However, unraveling the role of mtDNA mutations and deletions in regulating cellular metabolism is complicated by several factors, including the presence of high levels of heteroplasmy, the use of inappropriate reference sequences for control groups in previously published studies, and the complexity of segregating primary ‘driver’ mutations from secondary ‘passenger’ mutations [186,187]. Further, since many described mitochondrial mutations associated with cancers do not induce amino acid changes, the role of such mutations to carcinogenesis is not clear [10,188].
Despite these caveats, several specific mutations and deletions of mtDNA affecting both the control and coding regions have been reported in various types of cancers [10]. For instance, a 264 bp deletion in the region encoding for NADH:ubiquinone oxidoreductase (complex I) of the electron transport chain has been demonstrated to be associated with renal cell carcinoma [189]. In another notable example, a T8993G mutation in the coding region of the Atp6 mitochondrial gene has been reported in a prostate cancer cell line. This results not only in reduced levels of ATP6 protein, but also in increased ROS production that could presumably serve to perpetuate the oxidative DNA damage cycle [188]. In this particular example, the mutation results in an amino acid change in an integral respiratory chain protein and is associated with increased tumor size. However, to our knowledge, no one has demonstrated why these and other mitochondrial mutations are increased in tumors or cancer cells, and also, whether they are drivers of the cancer phenotype.
9. Neurodegenerative Diseases: Parkinson’s and Alzheimer’s
Oxidative stress and accumulation of oxidative damage is associated with neurodegenerative diseases [82,168,169,190,191,192]. However, a clear understanding of mtDNA damage and repair being mechanistically linked to the onset or progression of neurodegeneration, including Parkinson’s and Alzheimer’s disease, is only beginning to emerge. In this regard, neural stem cells (NSCs) from OGG1-deficient (Ogg1−/−) mice have been shown to accumulate more mtDNA damage, committing them to an astrocytic lineage at the expense of neurogenesis [193]. Treatment of these NSCs with antioxidants, or the introduction of a mitochondrially-targeted OGG1 protein ameliorated mtDNA damage and rescued neurogenesis, indicating the importance of mtDNA repair to NSC fate determination [193].
Studies using mice deficient for OGG1 and MTH1 have demonstrated that double-knockout mice suffer from severe striatal neurodegeneration [194]. Cortical neurons isolated from double-knockout mice demonstrated poor neurite outgrowth, secondary to increased mtDNA damage, especially in the absence of exogenous antioxidants in the culture media [195]. Interestingly, the increase in neuronal stress in the absence of OGG1 and MTH1 is likely a consequence of MUTY-mediated repair of mispaired adenines across 8-oxoG sites [194]. Deletion of MUTY effectively abrogated the neurodegeneration observed in Ogg1−/− mice [194]. Similarly, transgenic overexpression of human MTH1 also reduced mtDNA 8-oxoG accumulation and efficiently prevented neurodegeneration in OGG1-deficient animals. Apart from these studies using rodent models of DNA repair defects, studies of human cohorts have suggested that carriers of the mitochondrial haplogroup H5 may have an increased risk for Alzheimer’s disease [196].
Higher levels of mtDNA deletions have been reported in the striatum of Parkinson’s patients, relative to unaffected age-matched controls [27,197,198]. Mutations reported to be associated with Parkinsonism include those in mitochondrially-encoded subunits of complex I [197]. Interestingly, defects in PolG have also been reported to be associated with Parkinsonism in some families [199]. There have been mixed reports about a role for mitochondrial repair enzymes such as OGG1, APE1, and NEIL1 in modulating risk for Parkinson’s, either as a stand-alone risk factor or in combination with other exposures [137,200,201,202,203,204]. However, more studies are required to determine if specific defects in the mitochondrial forms of these enzymes represent a causative risk for development of Parkinson’s.
10. Metabolic Disease
Chronic consumption of hypercaloric diets, including those high in fat or simple sugars, is causatively linked to the development of obesity and related metabolic derangements, including fatty liver disease, type-2 diabetes, adipose tissue inflammation, and cardiovascular disease. While increased caloric intake is a recognized source of oxidative stress, the role of DNA repair enzymes in regulating metabolic disease had not been explored until recently. In this regard, the first demonstration of a BER enzyme being important to the prevention of metabolic disorders came from mice lacking the BER glycosylase, NEIL1 (Neil1−/−). Neil1−/− mice are prone to obesity and glucose intolerance [118,141]. Chronic high-fat diet further impaired metabolic function in these mice, secondary to an increase in mtDNA deletions and reduced mitochondrial protein content [118,141]. Similarly, engineered deletion of NEIL3 has been recently shown to increase susceptibility to atherosclerosis and increased size of atherosclerotic plaques in hypercholesterolemic mice deficient for the apolipoprotein, APOE3 [205]. The role of mitochondrial NEIL1 vs. the nuclear form of the enzyme in mediating metabolic phenotypes has not yet been determined, and NEIL3 does not have any reported mitochondrial localization.
In a similar, better-characterized paradigm, mice deficient for the BER glycosylase OGG1 are prone to obesity and insulin resistance, due to a specific reduction in fatty acid oxidation [117]. More recently, OGG1-deficient mice were shown to have impaired control over hepatic gluconeogenesis [206]. Our group also reported that OGG1-deficient mice have increased ectopic lipid accumulation in skeletal muscle, associated with increased mitochondrial fission and accelerated muscle function decline [115]. OGG1, like other DNA glycosylases, has been shown to have both nuclear and mitochondrial localization [82]. While the -2a or β isoform of OGG1 was initially presumed to be the main mitochondrial isoform [135], a recent study demonstrated that the endogenous mitochondrial-targeting sequence of the predominant -1a isoform is sufficient to target both the nucleus and mitochondria [151]. In prior experiments using cells in culture, several reports established a protective role for mitochondrial OGG1 in lipid-induced insulin resistance and apoptosis [146,147,148,207]. A recent study from our lab demonstrated that mitochondrial-targeting of OGG1 confers significant protection against diet-induced obesity and insulin resistance, even in the complete absence of nuclear OGG1 [116]. This metabolic protection occurs secondary to increased mitochondrial protein content and increased mitochondrial length, that combine to enhance mitochondrial respiration and whole-body energy expenditure [116]. These data are the first demonstration of mitochondrial repair of oxidative DNA lesions being sufficient to protect animals against the adverse outcomes of chronic high-fat diet consumption. It is notable that in this model of OGG1 mitochondrial overexpression, multiple metabolic parameters were affected primarily in white adipose tissue [116]. This was accompanied by a reduction in 8-oxoG levels in adipose tissue of these transgenic mice, indicating that the DNA repair activity of OGG1 likely mediates the metabolic protection observed in these mice. A previous study that reported on overexpression of the -2a isoform of OGG1 reported no protection against metabolic disease in this model [208]. This isoform of OGG1 has been reported to be devoid of DNA glycosylase activity [208], thereby lending further support to the hypothesis that the DNA-repair functionality of mitochondrial OGG1 is required for its metabolic effects [116].
In human populations, a relatively common OGG1 polymorphism, Ser326Cys, is associated with reduced glycosylase activity, and has been shown to be correlated with increased body mass index and elevated total cholesterol and fasting blood glucose in Japanese individuals homozygous for the mutation [209]. Similarly, in a Mexican cohort, the Ser326Cys polymorphism was independently correlated with increased incidence of type-2 diabetes [119]. Further, in a recent report from the European PREDIMED study, individuals homozygous for the Cys/Cys genotype had increased total mortality, attributable to cardiovascular disease-related deaths, but not to cancer-related mortality [210]. Together, these data in human populations underscore a role for OGG1 in modulating metabolic risk. Given our recent report of mitochondrial OGG1 being protective against obesity and metabolic disease [116], it will be interesting to determine if alterations in mtDNA integrity and mitochondrial function may be observed in carriers of the Ser326Cys mutation.
11. Summary
While mitochondrial function has long been understood to be critical to the health of the cell, the complex topic of mitochondrial origins of disease is only beginning to be explored. In this regard, the study of mtDNA integrity is challenging due to, among other factors, the lower abundance of mtDNA relative to genomic DNA and the existence of varying levels of heteroplasmy that may contribute to disease. However, despite these challenges, our understanding of mitochondrial genome integrity as a driver of health and disease continues to expand. Further, other mitochondrial processes including the regulation of mitochondrial biogenesis, maintenance of mitochondrial networks via fission and fusion pathways, the process of selective degradation of defective mitochondria via mitophagy, and interorganelle communication between mitochondria and organelles such as the endoplasmic reticulum are all increasingly understood to be vital to cellular respiration and energy production. Whether or not these processes are also regulated by underlying changes in mtDNA integrity is likely to be a fascinating novel area of investigation in upcoming years.
Author Contributions
P.S. and H.S. wrote and edited the manuscript.
Funding
This research was funded by NIH DK100640 to H.S.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Fox, T.D. Mitochondrial protein synthesis, import, and assembly. Genetics 2012, 192, 1203–1234. [Google Scholar] [CrossRef] [PubMed]
- Anderson, S.; Bankier, A.T.; Barrell, B.G.; de Bruijn, M.H.; Coulson, A.R.; Drouin, J.; Eperon, I.C.; Nierlich, D.P.; Roe, B.A.; Sanger, F.; et al. Sequence and organization of the human mitochondrial genome. Nature 1981, 290, 457–465. [Google Scholar] [CrossRef]
- Ohkubo, K.; Yamano, A.; Nagashima, M.; Mori, Y.; Anzai, K.; Akehi, Y.; Nomiyama, R.; Asano, T.; Urae, A.; Ono, J. Mitochondrial gene mutations in the tRNA(Leu(UUR)) region and diabetes: Prevalence and clinical phenotypes in Japan. Clin. Chem. 2001, 47, 1641–1648. [Google Scholar] [PubMed]
- Mancuso, M.; Orsucci, D.; Angelini, C.; Bertini, E.; Carelli, V.; Comi, G.P.; Donati, M.A.; Federico, A.; Minetti, C.; Moggio, M.; et al. Redefining phenotypes associated with mitochondrial DNA single deletion. J. Neurol. 2015, 262, 1301–1309. [Google Scholar] [CrossRef]
- Huoponen, K. Leber hereditary optic neuropathy: Clinical and molecular genetic findings. Neurogenetics 2001, 3, 119–125. [Google Scholar] [CrossRef] [PubMed]
- Fukuda, M.; Nakano, S.; Imaizumi, N.; Kitazawa, M.; Nishizawa, M.; Kigoshi, T.; Uchida, K. Mitochondrial DNA mutations are associated with both decreased insulin secretion and advanced microvascular complications in Japanese diabetic subjects. J. Diabetes Its Complicat. 1999, 13, 277–283. [Google Scholar] [CrossRef]
- Goldring, E.S.; Grossman, L.I.; Krupnick, D.; Cryer, D.R.; Marmur, J. The petite mutation in yeast. Loss of mitochondrial deoxyribonucleic acid during induction of petites with ethidium bromide. J. Mol. Biol. 1970, 52, 323–335. [Google Scholar] [CrossRef]
- Desjardins, P.; Frost, E.; Morais, R. Ethidium bromide-induced loss of mitochondrial DNA from primary chicken embryo fibroblasts. Mol. Cell. Biol. 1985, 5, 1163–1169. [Google Scholar] [CrossRef]
- King, M.P.; Attardi, G. Human cells lacking mtDNA: Repopulation with exogenous mitochondria by complementation. Science 1989, 246, 500–503. [Google Scholar] [CrossRef]
- Chatterjee, A.; Mambo, E.; Sidransky, D. Mitochondrial DNA mutations in human cancer. Oncogene 2006, 25, 4663–4674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nunnari, J.; Suomalainen, A. Mitochondria: In sickness and in health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef]
- Taylor, R.W.; Turnbull, D.M. Mitochondrial DNA mutations in human disease. Nat. Rev. Genet. 2005, 6, 389–402. [Google Scholar] [CrossRef] [Green Version]
- Wallace, D.C.; Chalkia, D. Mitochondrial DNA genetics and the heteroplasmy conundrum in evolution and disease. Cold Spring Harb. Perspect. Biol. 2013, 5, a021220. [Google Scholar] [CrossRef] [PubMed]
- Giles, R.E.; Blanc, H.; Cann, H.M.; Wallace, D.C. Maternal inheritance of human mitochondrial DNA. Proc. Natl. Acad. Sci. USA 1980, 77, 6715–6719. [Google Scholar] [CrossRef] [PubMed]
- Garcia, I.; Jones, E.; Ramos, M.; Innis-Whitehouse, W.; Gilkerson, R. The little big genome: The organization of mitochondrial DNA. Front. Biosci. 2017, 22, 710–721. [Google Scholar]
- Hillar, M.; Rangayya, V.; Jafar, B.B.; Chambers, D.; Vitzu, M.; Wyborny, L.E. Membrane-bound mitochondrial DNA: Isolation, transcription and protein. Arch. Int. Physiol. Biochim. 1979, 87, 29–49. [Google Scholar]
- Chen, X.J.; Butow, R.A. The organization and inheritance of the mitochondrial genome. Nat. Rev. Genet. 2005, 6, 815–825. [Google Scholar] [CrossRef]
- Spelbrink, J.N.; Li, F.Y.; Tiranti, V.; Nikali, K.; Yuan, Q.P.; Tariq, M.; Wanrooij, S.; Garrido, N.; Comi, G.; Morandi, L.; et al. Human mitochondrial DNA deletions associated with mutations in the gene encoding Twinkle, a phage T7 gene 4-like protein localized in mitochondria. Nat. Genet. 2001, 28, 223–231. [Google Scholar] [CrossRef]
- Wiedemann, N.; Pfanner, N. Mitochondrial Machineries for Protein Import and Assembly. Annu. Rev. Biochem. 2017, 86, 685–714. [Google Scholar] [CrossRef]
- Moraes, C.T.; Srivastava, S.; Kirkinezos, I.; Oca-Cossio, J.; van Waveren, C.; Woischnick, M.; Diaz, F. Mitochondrial DNA structure and function. Int. Rev. Neurobiol. 2002, 53, 3–23. [Google Scholar]
- Sato, M.; Sato, K. Degradation of paternal mitochondria by fertilization-triggered autophagy in C. elegans embryos. Science 2011, 334, 1141–1144. [Google Scholar] [CrossRef]
- Al Rawi, S.; Louvet-Vallee, S.; Djeddi, A.; Sachse, M.; Culetto, E.; Hajjar, C.; Boyd, L.; Legouis, R.; Galy, V. Postfertilization autophagy of sperm organelles prevents paternal mitochondrial DNA transmission. Science 2011, 334, 1144–1147. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Valencia, C.A.; Zhang, J.; Lee, N.C.; Slone, J.; Gui, B.; Wang, X.; Li, Z.; Dell, S.; Brown, J.; et al. Biparental Inheritance of Mitochondrial DNA in Humans. Proc. Natl. Acad. Sci. USA 2018, 115, 13039–13044. [Google Scholar] [CrossRef] [PubMed]
- Kasamatsu, H.; Vinograd, J. Replication of circular DNA in eukaryotic cells. Annu. Rev. Biochem. 1974, 43, 695–719. [Google Scholar] [CrossRef] [PubMed]
- Holt, I.J.; Reyes, A. Human mitochondrial DNA replication. Cold Spring Harb. Perspect. Biol. 2012, 4, a012971. [Google Scholar] [CrossRef]
- Stoneking, M. Hypervariable sites in the mtDNA control region are mutational hotspots. Am. J. Hum. Genet. 2000, 67, 1029–1032. [Google Scholar] [CrossRef]
- Chocron, E.S.; Munkacsy, E.; Pickering, A.M. Cause or casualty: The role of mitochondrial DNA in aging and age-associated disease. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 9, 285–297. [Google Scholar] [CrossRef]
- Clayton, D.A. Replication and transcription of vertebrate mitochondrial DNA. Annu. Rev. Cell Biol. 1991, 7, 453–478. [Google Scholar] [CrossRef]
- Arnberg, A.; van Bruggen, E.F.; Borst, P. The presence of DNA molecules with a displacement loop in standard mitochondrial DNA preparations. Biochim. Biophys. Acta 1971, 246, 353–357. [Google Scholar] [CrossRef]
- Kasamatsu, H.; Robberson, D.L.; Vinograd, J. A novel closed-circular mitochondrial DNA with properties of a replicating intermediate. Proc. Natl. Acad. Sci. USA 1971, 68, 2252–2257. [Google Scholar] [CrossRef]
- Ingman, M.; Kaessmann, H.; Paabo, S.; Gyllensten, U. Mitochondrial genome variation and the origin of modern humans. Nature 2000, 408, 708–713. [Google Scholar] [CrossRef] [PubMed]
- Yu, M.; Zhou, Y.; Shi, Y.; Ning, L.; Yang, Y.; Wei, X.; Zhang, N.; Hao, X.; Niu, R. Reduced mitochondrial DNA copy number is correlated with tumor progression and prognosis in Chinese breast cancer patients. IUBMB Life 2007, 59, 450–457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, R.T.; Sun, Y.; Bu, L.X.; Jia, M.Y. Gene mutations in the D-loop region of mitochondrial DNA in oral squamous cell carcinoma. Mol. Med. Rep. 2015, 11, 4496–4500. [Google Scholar] [CrossRef] [PubMed]
- Sharma, H.; Singh, A.; Sharma, C.; Jain, S.K.; Singh, N. Mutations in the mitochondrial DNA D-loop region are frequent in cervical cancer. Cancer Cell Int. 2005, 5, 34. [Google Scholar] [CrossRef] [PubMed]
- Miyazono, F.; Schneider, P.M.; Metzger, R.; Warnecke-Eberz, U.; Baldus, S.E.; Dienes, H.P.; Aikou, T.; Hoelscher, A.H. Mutations in the mitochondrial DNA D-Loop region occur frequently in adenocarcinoma in Barrett’s esophagus. Oncogene 2002, 21, 3780–3783. [Google Scholar] [CrossRef] [PubMed]
- Hung, W.Y.; Lin, J.C.; Lee, L.M.; Wu, C.W.; Tseng, L.M.; Yin, P.H.; Chi, C.W.; Lee, H.C. Tandem duplication/triplication correlated with poly-cytosine stretch variation in human mitochondrial DNA D-loop region. Mutagenesis 2008, 23, 137–142. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.G.; Guo, Q.N. Mutations in the mitochondrial DNA D-Loop region occur frequently in human osteosarcoma. Cancer Lett. 2006, 239, 151–155. [Google Scholar] [CrossRef]
- Wang, Y.; Michikawa, Y.; Mallidis, C.; Bai, Y.; Woodhouse, L.; Yarasheski, K.E.; Miller, C.A.; Askanas, V.; Engel, W.K.; Bhasin, S.; et al. Muscle-specific mutations accumulate with aging in critical human mtDNA control sites for replication. Proc. Natl. Acad. Sci. USA 2001, 98, 4022–4027. [Google Scholar] [CrossRef] [Green Version]
- Michikawa, Y.; Mazzucchelli, F.; Bresolin, N.; Scarlato, G.; Attardi, G. Aging-dependent large accumulation of point mutations in the human mtDNA control region for replication. Science 1999, 286, 774–779. [Google Scholar] [CrossRef]
- Yasukawa, T.; Kang, D. An overview of mammalian mitochondrial DNA replication mechanisms. J. Biochem. 2018, 164, 183–193. [Google Scholar] [CrossRef]
- Mercer, T.R.; Neph, S.; Dinger, M.E.; Crawford, J.; Smith, M.A.; Shearwood, A.M.; Haugen, E.; Bracken, C.P.; Rackham, O.; Stamatoyannopoulos, J.A.; et al. The human mitochondrial transcriptome. Cell 2011, 146, 645–658. [Google Scholar] [CrossRef] [PubMed]
- Shokolenko, I.N.; Alexeyev, M.F. Mitochondrial transcription in mammalian cells. Front. Biosci. 2017, 22, 835–853. [Google Scholar] [Green Version]
- Montoya, J.; Christianson, T.; Levens, D.; Rabinowitz, M.; Attardi, G. Identification of initiation sites for heavy-strand and light-strand transcription in human mitochondrial DNA. Proc. Natl. Acad. Sci. USA 1982, 79, 7195–7199. [Google Scholar] [CrossRef]
- Litonin, D.; Sologub, M.; Shi, Y.; Savkina, M.; Anikin, M.; Falkenberg, M.; Gustafsson, C.M.; Temiakov, D. Human mitochondrial transcription revisited: Only TFAM and TFB2M are required for transcription of the mitochondrial genes in vitro. J. Biol. Chem. 2010, 285, 18129–18133. [Google Scholar] [CrossRef] [PubMed]
- Asin-Cayuela, J.; Gustafsson, C.M. Mitochondrial transcription and its regulation in mammalian cells. Trends Biochem. Sci. 2007, 32, 111–117. [Google Scholar] [CrossRef]
- Cantatore, P.; Attardi, G. Mapping of nascent light and heavy strand transcripts on the physical map of HeLa cell mitochondrial DNA. Nucleic Acids Res. 1980, 8, 2605–2625. [Google Scholar] [CrossRef]
- Sanchez, M.I.; Mercer, T.R.; Davies, S.M.; Shearwood, A.M.; Nygard, K.K.; Richman, T.R.; Mattick, J.S.; Rackham, O.; Filipovska, A. RNA processing in human mitochondria. Cell Cycle 2011, 10, 2904–2916. [Google Scholar] [CrossRef] [Green Version]
- Martin, M.; Cho, J.; Cesare, A.J.; Griffith, J.D.; Attardi, G. Termination factor-mediated DNA loop between termination and initiation sites drives mitochondrial rRNA synthesis. Cell 2005, 123, 1227–1240. [Google Scholar] [CrossRef]
- Chang, D.D.; Clayton, D.A. Precise identification of individual promoters for transcription of each strand of human mitochondrial DNA. Cell 1984, 36, 635–643. [Google Scholar] [CrossRef]
- Hillen, H.S.; Temiakov, D.; Cramer, P. Structural basis of mitochondrial transcription. Nat. Struct. Mol. Biol. 2018, 25, 754–765. [Google Scholar] [CrossRef]
- Guja, K.E.; Garcia-Diaz, M. Hitting the brakes: Termination of mitochondrial transcription. Biochim. Biophys. Acta 2012, 1819, 939–947. [Google Scholar] [CrossRef] [Green Version]
- Adan, C.; Matsushima, Y.; Hernandez-Sierra, R.; Marco-Ferreres, R.; Fernandez-Moreno, M.A.; Gonzalez-Vioque, E.; Calleja, M.; Aragon, J.J.; Kaguni, L.S.; Garesse, R. Mitochondrial transcription factor B2 is essential for metabolic function in Drosophila melanogaster development. J. Biol. Chem. 2008, 283, 12333–12342. [Google Scholar] [CrossRef] [PubMed]
- Bestwick, M.L.; Shadel, G.S. Accessorizing the human mitochondrial transcription machinery. Trends Biochem. Sci. 2013, 38, 283–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barshad, G.; Marom, S.; Cohen, T.; Mishmar, D. Mitochondrial DNA Transcription and Its Regulation: An Evolutionary Perspective. Trends Genet. 2018, 34, 682–692. [Google Scholar] [CrossRef] [PubMed]
- Rebelo, A.P.; Dillon, L.M.; Moraes, C.T. Mitochondrial DNA transcription regulation and nucleoid organization. J. Inherit. Metab. Dis. 2011, 34, 941–951. [Google Scholar] [CrossRef]
- Wang, Z.; Cotney, J.; Shadel, G.S. Human mitochondrial ribosomal protein MRPL12 interacts directly with mitochondrial RNA polymerase to modulate mitochondrial gene expression. J. Biol. Chem. 2007, 282, 12610–12618. [Google Scholar] [CrossRef]
- Surovtseva, Y.V.; Shutt, T.E.; Cotney, J.; Cimen, H.; Chen, S.Y.; Koc, E.C.; Shadel, G.S. Mitochondrial ribosomal protein L12 selectively associates with human mitochondrial RNA polymerase to activate transcription. Proc. Natl. Acad. Sci. USA 2011, 108, 17921–17926. [Google Scholar] [CrossRef]
- Enriquez, J.A.; Fernandez-Silva, P.; Perez-Martos, A.; Lopez-Perez, M.J.; Montoya, J. The synthesis of mRNA in isolated mitochondria can be maintained for several hours and is inhibited by high levels of ATP. Eur. J. Biochem. 1996, 237, 601–610. [Google Scholar] [CrossRef]
- Ghosh, S.; Sengupta, S.; Scaria, V. Comparative analysis of human mitochondrial methylomes shows distinct patterns of epigenetic regulation in mitochondria. Mitochondrion 2014, 18, 58–62. [Google Scholar] [CrossRef]
- Van der Wijst, M.G.; Rots, M.G. Mitochondrial epigenetics: An overlooked layer of regulation? Trends Genet. 2015, 31, 353–356. [Google Scholar] [CrossRef]
- Bellizzi, D.; D’Aquila, P.; Scafone, T.; Giordano, M.; Riso, V.; Riccio, A.; Passarino, G. The control region of mitochondrial DNA shows an unusual CpG and non-CpG methylation pattern. DNA Res. 2013, 20, 537–547. [Google Scholar] [CrossRef] [PubMed]
- Fan, L.; Kim, S.; Farr, C.L.; Schaefer, K.T.; Randolph, K.M.; Tainer, J.A.; Kaguni, L.S. A novel processive mechanism for DNA synthesis revealed by structure, modeling and mutagenesis of the accessory subunit of human mitochondrial DNA polymerase. J. Mol. Biol. 2006, 358, 1229–1243. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Kennedy, W.D.; Yin, Y.W. Structural insight into processive human mitochondrial DNA synthesis and disease-related polymerase mutations. Cell 2009, 139, 312–324. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.; Curth, U.; Urbanke, C.; Kang, C. Crystal structure of human mitochondrial single-stranded DNA binding protein at 2.4 A resolution. Nat. Struct. Biol. 1997, 4, 153–157. [Google Scholar] [CrossRef]
- Wanrooij, S.; Fuste, J.M.; Farge, G.; Shi, Y.; Gustafsson, C.M.; Falkenberg, M. Human mitochondrial RNA polymerase primes lagging-strand DNA synthesis in vitro. Proc. Natl. Acad. Sci. USA 2008, 105, 11122–11127. [Google Scholar] [CrossRef] [Green Version]
- Korhonen, J.A.; Gaspari, M.; Falkenberg, M. TWINKLE Has 5′ -> 3′ DNA helicase activity and is specifically stimulated by mitochondrial single-stranded DNA-binding protein. J. Biol. Chem. 2003, 278, 48627–48632. [Google Scholar] [CrossRef] [PubMed]
- Miralles Fuste, J.; Shi, Y.; Wanrooij, S.; Zhu, X.; Jemt, E.; Persson, O.; Sabouri, N.; Gustafsson, C.M.; Falkenberg, M. In vivo occupancy of mitochondrial single-stranded DNA binding protein supports the strand displacement mode of DNA replication. PLoS Genet. 2014, 10, e1004832. [Google Scholar] [CrossRef] [PubMed]
- Nicholls, T.J.; Nadalutti, C.A.; Motori, E.; Sommerville, E.W.; Gorman, G.S.; Basu, S.; Hoberg, E.; Turnbull, D.M.; Chinnery, P.F.; Larsson, N.G.; et al. Topoisomerase 3alpha Is Required for Decatenation and Segregation of Human mtDNA. Mol. Cell 2018, 69, 9–23.e6. [Google Scholar] [CrossRef] [PubMed]
- Cerritelli, S.M.; Frolova, E.G.; Feng, C.; Grinberg, A.; Love, P.E.; Crouch, R.J. Failure to produce mitochondrial DNA results in embryonic lethality in Rnaseh1 null mice. Mol. Cell 2003, 11, 807–815. [Google Scholar] [CrossRef]
- Simsek, D.; Jasin, M. DNA ligase III: A spotty presence in eukaryotes, but an essential function where tested. Cell Cycle 2011, 10, 3636–3644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lakshmipathy, U.; Campbell, C. Antisense-mediated decrease in DNA ligase III expression results in reduced mitochondrial DNA integrity. Nucleic Acids Res. 2001, 29, 668–676. [Google Scholar] [CrossRef] [Green Version]
- Ruhanen, H.; Ushakov, K.; Yasukawa, T. Involvement of DNA ligase III and ribonuclease H1 in mitochondrial DNA replication in cultured human cells. Biochim. Biophys. Acta 2011, 1813, 2000–2007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shadel, G.S.; Clayton, D.A. Mitochondrial DNA maintenance in vertebrates. Annu. Rev. Biochem. 1997, 66, 409–435. [Google Scholar] [CrossRef]
- Phillips, A.F.; Millet, A.R.; Tigano, M.; Dubois, S.M.; Crimmins, H.; Babin, L.; Charpentier, M.; Piganeau, M.; Brunet, E.; Sfeir, A. Single-Molecule Analysis of mtDNA Replication Uncovers the Basis of the Common Deletion. Mol. Cell 2017, 65, 527–538.e526. [Google Scholar] [CrossRef] [PubMed]
- Brown, T.A.; Cecconi, C.; Tkachuk, A.N.; Bustamante, C.; Clayton, D.A. Replication of mitochondrial DNA occurs by strand displacement with alternative light-strand origins, not via a strand-coupled mechanism. Genes Dev. 2005, 19, 2466–2476. [Google Scholar] [CrossRef] [Green Version]
- Yasukawa, T.; Yang, M.Y.; Jacobs, H.T.; Holt, I.J. A bidirectional origin of replication maps to the major noncoding region of human mitochondrial DNA. Mol. Cell 2005, 18, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.Y.; Bowmaker, M.; Reyes, A.; Vergani, L.; Angeli, P.; Gringeri, E.; Jacobs, H.T.; Holt, I.J. Biased incorporation of ribonucleotides on the mitochondrial L-strand accounts for apparent strand-asymmetric DNA replication. Cell 2002, 111, 495–505. [Google Scholar] [CrossRef]
- Holt, I.J.; Lorimer, H.E.; Jacobs, H.T. Coupled leading- and lagging-strand synthesis of mammalian mitochondrial DNA. Cell 2000, 100, 515–524. [Google Scholar] [CrossRef]
- Bowmaker, M.; Yang, M.Y.; Yasukawa, T.; Reyes, A.; Jacobs, H.T.; Huberman, J.A.; Holt, I.J. Mammalian mitochondrial DNA replicates bidirectionally from an initiation zone. J. Biol. Chem. 2003, 278, 50961–50969. [Google Scholar] [CrossRef] [PubMed]
- Copeland, W.C. Defects of mitochondrial DNA replication. J. Child Neurol. 2014, 29, 1216–1224. [Google Scholar] [CrossRef] [PubMed]
- Sampath, H.; McCullough, A.K.; Lloyd, R.S. Regulation of DNA glycosylases and their role in limiting disease. Free Radic. Res. 2012, 46, 460–478. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sampath, H. Oxidative DNA damage in disease–insights gained from base excision repair glycosylase-deficient mouse models. Environ. Mol. Mutagen. 2014, 55, 689–703. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, M.; Ozawa, T. Strand asymmetry in human mitochondrial DNA mutations. Genomics 1994, 22, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Reyes, A.; Gissi, C.; Pesole, G.; Saccone, C. Asymmetrical directional mutation pressure in the mitochondrial genome of mammals. Mol. Biol. Evol. 1998, 15, 957–966. [Google Scholar] [CrossRef] [Green Version]
- Song, S.; Pursell, Z.F.; Copeland, W.C.; Longley, M.J.; Kunkel, T.A.; Mathews, C.K. DNA precursor asymmetries in mammalian tissue mitochondria and possible contribution to mutagenesis through reduced replication fidelity. Proc. Natl. Acad. Sci. USA 2005, 102, 4990–4995. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stein, A.; Sia, E.A. Mitochondrial DNA repair and damage tolerance. Front. Biosci. (Landmark Ed.) 2017, 22, 920–943. [Google Scholar]
- Brown, W.M.; George, M., Jr.; Wilson, A.C. Rapid evolution of animal mitochondrial DNA. Proc. Natl. Acad. Sci. USA 1979, 76, 1967–1971. [Google Scholar] [CrossRef] [PubMed]
- Ballard, J.W.; Whitlock, M.C. The incomplete natural history of mitochondria. Mol. Ecol. 2004, 13, 729–744. [Google Scholar] [CrossRef]
- Richter, C.; Park, J.W.; Ames, B.N. Normal oxidative damage to mitochondrial and nuclear DNA is extensive. Proc. Natl. Acad. Sci. USA 1988, 85, 6465–6467. [Google Scholar] [CrossRef]
- Kazak, L.; Reyes, A.; Holt, I.J. Minimizing the damage: Repair pathways keep mitochondrial DNA intact. Nat. Rev. Mol. Cell Biol. 2012, 13, 659–671. [Google Scholar] [CrossRef]
- Indo, H.P.; Davidson, M.; Yen, H.C.; Suenaga, S.; Tomita, K.; Nishii, T.; Higuchi, M.; Koga, Y.; Ozawa, T.; Majima, H.J. Evidence of ROS generation by mitochondria in cells with impaired electron transport chain and mitochondrial DNA damage. Mitochondrion 2007, 7, 106–118. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Briede, J.J.; Jennen, D.G.; Van Summeren, A.; Saritas-Brauers, K.; Schaart, G.; Kleinjans, J.C.; de Kok, T.M. Increased mitochondrial ROS formation by acetaminophen in human hepatic cells is associated with gene expression changes suggesting disruption of the mitochondrial electron transport chain. Toxicol. Lett. 2015, 234, 139–150. [Google Scholar] [CrossRef]
- Murphy, M.P.; Holmgren, A.; Larsson, N.G.; Halliwell, B.; Chang, C.J.; Kalyanaraman, B.; Rhee, S.G.; Thornalley, P.J.; Partridge, L.; Gems, D.; et al. Unraveling the biological roles of reactive oxygen species. Cell Metab. 2011, 13, 361–366. [Google Scholar] [CrossRef] [PubMed]
- Lambert, A.J.; Brand, M.D. Reactive oxygen species production by mitochondria. Methods Mol. Biol. (Clifton, NJ) 2009, 554, 165–181. [Google Scholar]
- Lambert, A.J.; Brand, M.D. Research on mitochondria and aging, 2006–2007. Aging Cell 2007, 6, 417–420. [Google Scholar] [CrossRef]
- Bresciani, G.; da Cruz, I.B.; Gonzalez-Gallego, J. Manganese superoxide dismutase and oxidative stress modulation. Adv. Clin. Chem. 2015, 68, 87–130. [Google Scholar]
- Couto, N.; Wood, J.; Barber, J. The role of glutathione reductase and related enzymes on cellular redox homoeostasis network. Free Radic. Biol. Med. 2016, 95, 27–42. [Google Scholar] [CrossRef]
- Glorieux, C.; Calderon, P.B. Catalase, a remarkable enzyme: Targeting the oldest antioxidant enzyme to find a new cancer treatment approach. Biol. Chem. 2017, 398, 1095–1108. [Google Scholar] [CrossRef] [PubMed]
- Smith, R.E.; Tran, K.; Smith, C.C.; McDonald, M.; Shejwalkar, P.; Hara, K. The Role of the Nrf2/ARE Antioxidant System in Preventing Cardiovascular Diseases. Diseases 2016, 4, 34. [Google Scholar] [CrossRef]
- Slocinska, M.; Barylski, J.; Jarmuszkiewicz, W. Uncoupling proteins of invertebrates: A review. IUBMB Life 2016, 68, 691–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yakes, F.M.; Van Houten, B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc. Natl. Acad. Sci. USA 1997, 94, 514–519. [Google Scholar] [CrossRef] [Green Version]
- Ames, B.N.; Shigenaga, M.K.; Hagen, T.M. Oxidants, antioxidants, and the degenerative diseases of aging. Proc. Natl. Acad. Sci. USA 1993, 90, 7915–7922. [Google Scholar] [CrossRef] [PubMed]
- Salazar, J.J.; Van Houten, B. Preferential mitochondrial DNA injury caused by glucose oxidase as a steady generator of hydrogen peroxide in human fibroblasts. Mutat. Res. 1997, 385, 139–149. [Google Scholar] [CrossRef]
- Shokolenko, I.N.; Wilson, G.L.; Alexeyev, M.F. Aging: A mitochondrial DNA perspective, critical analysis and an update. World J. Exp. Med. 2014, 4, 46–57. [Google Scholar] [CrossRef] [PubMed]
- Shokolenko, I.; Venediktova, N.; Bochkareva, A.; Wilson, G.L.; Alexeyev, M.F. Oxidative stress induces degradation of mitochondrial DNA. Nucleic Acids Res. 2009, 37, 2539–2548. [Google Scholar] [CrossRef] [Green Version]
- Ballinger, S.W.; Van Houten, B.; Jin, G.F.; Conklin, C.A.; Godley, B.F. Hydrogen peroxide causes significant mitochondrial DNA damage in human RPE cells. Exp. Eye Res. 1999, 68, 765–772. [Google Scholar] [CrossRef]
- Indo, H.P.; Yen, H.C.; Nakanishi, I.; Matsumoto, K.; Tamura, M.; Nagano, Y.; Matsui, H.; Gusev, O.; Cornette, R.; Okuda, T.; et al. A mitochondrial superoxide theory for oxidative stress diseases and aging. J. Clin. Biochem. Nutr. 2015, 56, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexeyev, M.; Shokolenko, I.; Wilson, G.; LeDoux, S. The maintenance of mitochondrial DNA integrity--critical analysis and update. Cold Spring Harb. Perspect. Biol. 2013, 5, a012641. [Google Scholar] [CrossRef]
- Gorbunova, V.; Seluanov, A.; Mao, Z.; Hine, C. Changes in DNA repair during aging. Nucleic Acids Res. 2007, 35, 7466–7474. [Google Scholar] [CrossRef] [Green Version]
- Alexeyev, M.F.; Ledoux, S.P.; Wilson, G.L. Mitochondrial DNA and aging. Clin. Sci. (Lond. Engl.) 2004, 107, 355–364. [Google Scholar] [CrossRef]
- Van Houten, B.; Hunter, S.E.; Meyer, J.N. Mitochondrial DNA damage induced autophagy, cell death, and disease. Front. Biosci. (Landmark Ed.) 2016, 21, 42–54. [Google Scholar] [CrossRef]
- De Souza-Pinto, N.C.; Wilson, D.M.; Stevnsner, T.V.; Bohr, V.A. Mitochondrial DNA, base excision repair and neurodegeneration. DNA Repair 2008, 7, 1098–1109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krokan, H.E.; Bjørås, M. Base Excision Repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012575. [Google Scholar] [CrossRef] [PubMed]
- Sykora, P.; Wilson, D.M., 3rd; Bohr, V.A. Repair of persistent strand breaks in the mitochondrial genome. Mech. Ageing Dev. 2012, 133, 169–175. [Google Scholar] [CrossRef] [Green Version]
- Vartanian, V.; Tumova, J.; Dobrzyn, P.; Dobrzyn, A.; Nakabeppu, Y.; Lloyd, R.S.; Sampath, H. 8-oxoguanine DNA glycosylase (OGG1) deficiency elicits coordinated changes in in lipid and mitochondrial metabolism in muscle. PLoS ONE 2017, 12, 0181687. [Google Scholar] [CrossRef] [PubMed]
- Komakula, S.S.B.; Tumova, J.; Kumaraswamy, D.; Burchat, N.; Vartanian, V.; Ye, H.; Dobrzyn, A.; Lloyd, R.S.; Sampath, H. The DNA Repair Protein OGG1 Protects Against Obesity by Altering Mitochondrial Energetics in White Adipose Tissue. Sci. Rep. 2018, 8, 14886. [Google Scholar] [CrossRef] [PubMed]
- Sampath, H.; Vartanian, V.; Rollins, M.R.; Sakumi, K.; Nakabeppu, Y.; Lloyd, R.S. 8-Oxoguanine DNA glycosylase (OGG1) deficiency increases susceptibility to obesity and metabolic dysfunction. PLoS ONE 2012, 7, 17. [Google Scholar] [CrossRef] [PubMed]
- Sampath, H.; Batra, A.K.; Vartanian, V.; Carmical, J.R.; Prusak, D.; King, I.B.; Lowell, B.; Earley, L.F.; Wood, T.G.; Marks, D.L.; et al. Variable penetrance of metabolic phenotypes and development of high-fat diet-induced adiposity in NEIL-1 deficient mice. Am. J. Physiol. Endocrinol. Metab. 2011, 300, E724–E734. [Google Scholar] [CrossRef]
- Thameem, F.; Puppala, S.; Lehman, D.M.; Stern, M.P.; Blangero, J.; Abboud, H.E.; Duggirala, R.; Habib, S.L. The Ser(326)Cys polymorphism of 8-oxoguanine glycosylase 1 (OGG1) is associated with Type 2 diabetes in Mexican Americans. Hum Hered 2010, 70, 97–101. [Google Scholar] [CrossRef]
- Zinovkina, L.A. Mechanisms of Mitochondrial DNA Repair in Mammals. Biochemistry 2018, 83, 233–249. [Google Scholar] [CrossRef] [PubMed]
- Prakash, A.; Doublie, S. Base Excision Repair in the Mitochondria. J. Cell Biochem. 2015, 116, 1490–1499. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.J.; Wilson, D.M., 3rd. Overview of base excision repair biochemistry. Curr. Mol. Pharmacol. 2012, 5, 3–13. [Google Scholar] [CrossRef]
- Parker, A.R.; Eshleman, J.R. Human MutY: Gene structure, protein functions and interactions, and role in carcinogenesis. Cell. Mol. Life Sci. 2003, 60, 2064–2083. [Google Scholar] [CrossRef] [PubMed]
- Caldecott, K.W. Polynucleotide kinase: A versatile molecule makes a clean break. Structure (Lond. Engl.) 2002, 10, 1151–1152. [Google Scholar] [CrossRef]
- Wiederhold, L.; Leppard, J.B.; Kedar, P.; Karimi-Busheri, F.; Rasouli-Nia, A.; Weinfeld, M.; Tomkinson, A.E.; Izumi, T.; Prasad, R.; Wilson, S.H.; et al. AP endonuclease-independent DNA base excision repair in human cells. Mol. Cell 2004, 15, 209–220. [Google Scholar] [CrossRef] [PubMed]
- Das, A.; Wiederhold, L.; Leppard, J.B.; Kedar, P.; Prasad, R.; Wang, H.; Boldogh, I.; Karimi-Busheri, F.; Weinfeld, M.; Tomkinson, A.E.; et al. NEIL2-initiated, APE-independent repair of oxidized bases in DNA: Evidence for a repair complex in human cells. DNA Repair 2006, 5, 1439–1448. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oda, H.; Nakabeppu, Y.; Furuichi, M.; Sekiguchi, M. Regulation of expression of the human MTH1 gene encoding 8-oxo-dGTPase. Alternative splicing of transcription products. J. Biol. Chem. 1997, 272, 17843–17850. [Google Scholar] [CrossRef]
- Hazra, T.K.; Izumi, T.; Boldogh, I.; Imhoff, B.; Kow, Y.W.; Jaruga, P.; Dizdaroglu, M.; Mitra, S. Identification and characterization of a human DNA glycosylase for repair of modified bases in oxidatively damaged DNA. Proc. Natl. Acad. Sci. USA 2002, 99, 3523–3528. [Google Scholar] [CrossRef] [Green Version]
- Morland, I.; Rolseth, V.; Luna, L.; Rognes, T.; Bjoras, M.; Seeberg, E. Human DNA glycosylases of the bacterial Fpg/MutM superfamily: An alternative pathway for the repair of 8-oxoguanine and other oxidation products in DNA. Nucleic Acids Res. 2002, 30, 4926–4936. [Google Scholar] [CrossRef]
- Rolseth, V.; Runden-Pran, E.; Luna, L.; McMurray, C.; Bjoras, M.; Ottersen, O.P. Widespread distribution of DNA glycosylases removing oxidative DNA lesions in human and rodent brains. DNA Repair 2008, 7, 1578–1588. [Google Scholar] [CrossRef] [Green Version]
- Hazra, T.K.; Kow, Y.W.; Hatahet, Z.; Imhoff, B.; Boldogh, I.; Mokkapati, S.K.; Mitra, S.; Izumi, T. Identification and characterization of a novel human DNA glycosylase for repair of cytosine-derived lesions. J. Biol. Chem. 2002, 277, 30417–30420. [Google Scholar] [CrossRef] [PubMed]
- Karahalil, B.; Hogue, B.A.; de Souza-Pinto, N.C.; Bohr, V.A. Base excision repair capacity in mitochondria and nuclei: Tissue-specific variations. FASEB J. Off. Publ. Fed. Am. Soc. Exp. Biol. 2002, 16, 1895–1902. [Google Scholar] [CrossRef] [PubMed]
- Kuo, F.C.; Sklar, J. Augmented expression of a human gene for 8-oxoguanine DNA glycosylase (MutM) in B lymphocytes of the dark zone in lymph node germinal centers. J. Exp. Med. 1997, 186, 1547–1556. [Google Scholar] [CrossRef] [PubMed]
- Gredilla, R.; Garm, C.; Holm, R.; Bohr, V.A.; Stevnsner, T. Differential age-related changes in mitochondrial DNA repair activities in mouse brain regions. Neurobiol. Aging 2010, 31, 993–1002. [Google Scholar] [CrossRef]
- Nishioka, K.; Ohtsubo, T.; Oda, H.; Fujiwara, T.; Kang, D.; Sugimachi, K.; Nakabeppu, Y. Expression and differential intracellular localization of two major forms of human 8-oxoguanine DNA glycosylase encoded by alternatively spliced OGG1 mRNAs. Mol. Biol. Cell 1999, 10, 1637–1652. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, H.; Kajitani, K.; Dan, Y.; Furuichi, M.; Ohno, M.; Sakumi, K.; Kang, D.; Nakabeppu, Y. MTH1, an oxidized purine nucleoside triphosphatase, protects the dopamine neurons from oxidative damage in nucleic acids caused by 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. Cell Death Differ. 2006, 13, 551–563. [Google Scholar] [CrossRef]
- Nakabeppu, Y.; Tsuchimoto, D.; Yamaguchi, H.; Sakumi, K. Oxidative damage in nucleic acids and Parkinson’s disease. J. Neurosci. Res. 2007, 85, 919–934. [Google Scholar] [CrossRef]
- Kajitani, K.; Yamaguchi, H.; Dan, Y.; Furuichi, M.; Kang, D.; Nakabeppu, Y. MTH1, an oxidized purine nucleoside triphosphatase, suppresses the accumulation of oxidative damage of nucleic acids in the hippocampal microglia during kainate-induced excitotoxicity. J. Neurosci. 2006, 26, 1688–1698. [Google Scholar] [CrossRef]
- Nakabeppu, Y.; Ohta, E.; Abolhassani, N. MTH1 as a nucleotide pool sanitizing enzyme: Friend or foe? Free Radic. Biol. Med. 2017, 107, 151–158. [Google Scholar] [CrossRef]
- Nakabeppu, Y. Molecular genetics and structural biology of human MutT homolog, MTH1. Mutat. Res. 2001, 477, 59–70. [Google Scholar] [CrossRef]
- Vartanian, V.; Lowell, B.; Minko, I.G.; Wood, T.G.; Ceci, J.D.; George, S.; Ballinger, S.W.; Corless, C.L.; McCullough, A.K.; Lloyd, R.S. The metabolic syndrome resulting from a knockout of the NEIL1 DNA glycosylase. Proc. Natl. Acad. Sci. USA 2006, 103, 1864–1869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, N.; Chakravarthy, S.; Longley, M.J.; Copeland, W.C.; Prakash, A. The C-terminal tail of the NEIL1 DNA glycosylase interacts with the human mitochondrial single-stranded DNA binding protein. DNA Repair 2018, 65, 11–19. [Google Scholar]
- Takao, M.; Kanno, S.; Shiromoto, T.; Hasegawa, R.; Ide, H.; Ikeda, S.; Sarker, A.H.; Seki, S.; Xing, J.Z.; Le, X.C.; et al. Novel nuclear and mitochondrial glycosylases revealed by disruption of the mouse Nth1 gene encoding an endonuclease III homolog for repair of thymine glycols. EMBO J. 2002, 21, 3486–3493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mandal, S.M.; Hegde, M.L.; Chatterjee, A.; Hegde, P.M.; Szczesny, B.; Banerjee, D.; Boldogh, I.; Gao, R.; Falkenberg, M.; Gustafsson, C.M.; et al. Role of human DNA glycosylase Nei-like 2 (NEIL2) and single strand break repair protein polynucleotide kinase 3′-phosphatase in maintenance of mitochondrial genome. J. Biol. Chem. 2012, 287, 2819–2829. [Google Scholar] [CrossRef] [PubMed]
- Radicella, J.P.; Dherin, C.; Desmaze, C.; Fox, M.S.; Boiteux, S. Cloning and characterization of hOGG1, a human homolog of the OGG1 gene of Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 1997, 94, 8010–8015. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rachek, L.I.; Thornley, N.P.; Grishko, V.I.; LeDoux, S.P.; Wilson, G.L. Protection of INS-1 cells from free fatty acid-induced apoptosis by targeting hOGG1 to mitochondria. Diabetes 2006, 55, 1022–1028. [Google Scholar] [CrossRef] [PubMed]
- Yuzefovych, L.V.; Solodushko, V.A.; Wilson, G.L.; Rachek, L.I. Protection from palmitate-induced mitochondrial DNA damage prevents from mitochondrial oxidative stress, mitochondrial dysfunction, apoptosis, and impaired insulin signaling in rat L6 skeletal muscle cells. Endocrinology 2012, 153, 92–100. [Google Scholar] [CrossRef]
- Yuzefovych, L.V.; Schuler, A.M.; Chen, J.; Alvarez, D.F.; Eide, L.; Ledoux, S.P.; Wilson, G.L.; Rachek, L.I. Alteration of mitochondrial function and insulin sensitivity in primary mouse skeletal muscle cells isolated from transgenic and knockout mice: Role of ogg1. Endocrinology 2013, 154, 2640–2649. [Google Scholar] [CrossRef]
- Rachek, L.I.; Grishko, V.I.; Musiyenko, S.I.; Kelley, M.R.; LeDoux, S.P.; Wilson, G.L. Conditional targeting of the DNA repair enzyme hOGG1 into mitochondria. J. Biol. Chem. 2002, 277, 44932–44937. [Google Scholar] [CrossRef] [PubMed]
- Souza-Pinto, N.C.; Croteau, D.L.; Hudson, E.K.; Hansford, R.G.; Bohr, V.A. Age-associated increase in 8-oxo-deoxyguanosine glycosylase/AP lyase activity in rat mitochondria. Nucleic Acids Res. 1999, 27, 1935–1942. [Google Scholar] [CrossRef] [Green Version]
- Lia, D.; Reyes, A.; de Melo Campos, J.T.A.; Piolot, T.; Baijer, J.; Radicella, J.P.; Campalans, A. Mitochondrial maintenance under oxidative stress depends on mitochondrially localised alpha-OGG1. J. Cell Sci. 2018, 131, jcs213538. [Google Scholar] [CrossRef]
- De Souza-Pinto, N.C.; Hogue, B.A.; Bohr, V.A. DNA repair and aging in mouse liver: 8-oxodG glycosylase activity increase in mitochondrial but not in nuclear extracts. Free Radic. Biol. Med. 2001, 30, 916–923. [Google Scholar] [CrossRef]
- Imai, K.; Sarker, A.H.; Akiyama, K.; Ikeda, S.; Yao, M.; Tsutsui, K.; Shohmori, T.; Seki, S. Genomic structure and sequence of a human homologue (NTHL1/NTH1) of Escherichia coli endonuclease III with those of the adjacent parts of TSC2 and SLC9A3R2 genes. Gene 1998, 222, 287–295. [Google Scholar] [CrossRef]
- Luna, L.; Bjoras, M.; Hoff, E.; Rognes, T.; Seeberg, E. Cell-cycle regulation, intracellular sorting and induced overexpression of the human NTH1 DNA glycosylase involved in removal of formamidopyrimidine residues from DNA. Mutat. Res. 2000, 460, 95–104. [Google Scholar] [CrossRef]
- Ikeda, S.; Kohmoto, T.; Tabata, R.; Seki, Y. Differential intracellular localization of the human and mouse endonuclease III homologs and analysis of the sorting signals. DNA Repair 2002, 1, 847–854. [Google Scholar] [CrossRef]
- Ichinoe, A.; Behmanesh, M.; Tominaga, Y.; Ushijima, Y.; Hirano, S.; Sakai, Y.; Tsuchimoto, D.; Sakumi, K.; Wake, N.; Nakabeppu, Y. Identification and characterization of two forms of mouse MUTYH proteins encoded by alternatively spliced transcripts. Nucleic Acids Res. 2004, 32, 477–487. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.M.; Wang, C.; Hu, Z.; Greeley, G.H.; Makalowski, W.; Hellmich, H.L.; Englander, E.W. Hypoxia induces mitochondrial DNA damage and stimulates expression of a DNA repair enzyme, the Escherichia coli MutY DNA glycosylase homolog (MYH), in vivo, in the rat brain. J. Neurochem. 2002, 80, 928–937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ohtsubo, T.; Nishioka, K.; Imaiso, Y.; Iwai, S.; Shimokawa, H.; Oda, H.; Fujiwara, T.; Nakabeppu, Y. Identification of human MutY homolog (hMYH) as a repair enzyme for 2-hydroxyadenine in DNA and detection of multiple forms of hMYH located in nuclei and mitochondria. Nucleic Acids Res. 2000, 28, 1355–1364. [Google Scholar] [CrossRef] [Green Version]
- Yoshimura, D.; Sakumi, K.; Ohno, M.; Sakai, Y.; Furuichi, M.; Iwai, S.; Nakabeppu, Y. An oxidized purine nucleoside triphosphatase, MTH1, suppresses cell death caused by oxidative stress. J. Biol. Chem. 2003, 278, 37965–37973. [Google Scholar] [CrossRef]
- Stojkovic, G.; Makarova, A.V.; Wanrooij, P.H.; Forslund, J.; Burgers, P.M.; Wanrooij, S. Oxidative DNA damage stalls the human mitochondrial replisome. Sci. Rep. 2016, 6, 28942. [Google Scholar] [CrossRef] [Green Version]
- Hanes, J.W.; Thal, D.M.; Johnson, K.A. Incorporation and replication of 8-oxo-deoxyguanosine by the human mitochondrial DNA polymerase. J. Biol. Chem. 2006, 281, 36241–36248. [Google Scholar] [CrossRef]
- Garcia-Gomez, S.; Reyes, A.; Martinez-Jimenez, M.I.; Chocron, E.S.; Mouron, S.; Terrados, G.; Powell, C.; Salido, E.; Mendez, J.; Holt, I.J.; et al. PrimPol, an archaic primase/polymerase operating in human cells. Mol. Cell 2013, 52, 541–553. [Google Scholar] [CrossRef] [PubMed]
- Posse, V.; Shahzad, S.; Falkenberg, M.; Hallberg, B.M.; Gustafsson, C.M. TEFM is a potent stimulator of mitochondrial transcription elongation in vitro. Nucleic Acids Res. 2015, 43, 2615–2624. [Google Scholar] [CrossRef] [Green Version]
- Nakanishi, N.; Fukuoh, A.; Kang, D.; Iwai, S.; Kuraoka, I. Effects of DNA lesions on the transcription reaction of mitochondrial RNA polymerase: Implications for bypass RNA synthesis on oxidative DNA lesions. Mutagenesis 2013, 28, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Tornaletti, S.; Maeda, L.S.; Kolodner, R.D.; Hanawalt, P.C. Effect of 8-oxoguanine on transcription elongation by T7 RNA polymerase and mammalian RNA polymerase II. DNA Repair 2004, 3, 483–494. [Google Scholar] [CrossRef]
- Charlet-Berguerand, N.; Feuerhahn, S.; Kong, S.E.; Ziserman, H.; Conaway, J.W.; Conaway, R.; Egly, J.M. RNA polymerase II bypass of oxidative DNA damage is regulated by transcription elongation factors. EMBO J. 2006, 25, 5481–5491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuraoka, I.; Suzuki, K.; Ito, S.; Hayashida, M.; Kwei, J.S.; Ikegami, T.; Handa, H.; Nakabeppu, Y.; Tanaka, K. RNA polymerase II bypasses 8-oxoguanine in the presence of transcription elongation factor TFIIS. DNA Repair 2007, 6, 841–851. [Google Scholar] [CrossRef] [PubMed]
- Maynard, S.; Fang, E.F.; Scheibye-Knudsen, M.; Croteau, D.L.; Bohr, V.A. DNA Damage, DNA Repair, Aging, and Neurodegeneration. Cold Spring Harb. Perspect. Med. 2015, 5, a025130. [Google Scholar] [CrossRef] [PubMed]
- Madabhushi, R.; Pan, L.; Tsai, L.H. DNA damage and its links to neurodegeneration. Neuron 2014, 83, 266–282. [Google Scholar] [CrossRef]
- Bautista-Nino, P.K.; Portilla-Fernandez, E.; Vaughan, D.E.; Danser, A.H.; Roks, A.J. DNA Damage: A Main Determinant of Vascular Aging. Int. J. Mol. Sci. 2016, 17, 748. [Google Scholar] [CrossRef] [PubMed]
- Moehrle, B.M.; Geiger, H. Aging of hematopoietic stem cells: DNA damage and mutations? Exp. Hematol. 2016, 44, 895–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brandon, M.C.; Lott, M.T.; Nguyen, K.C.; Spolim, S.; Navathe, S.B.; Baldi, P.; Wallace, D.C. MITOMAP: A human mitochondrial genome database—2004 update. Nucleic Acids Res. 2005, 33, D611–D613. [Google Scholar] [CrossRef]
- El-Hattab, A.W.; Adesina, A.M.; Jones, J.; Scaglia, F. MELAS syndrome: Clinical manifestations, pathogenesis, and treatment options. Mol. Genet. Metab. 2015, 116, 4–12. [Google Scholar] [CrossRef]
- Crippa, B.L.; Leon, E.; Calhoun, A.; Lowichik, A.; Pasquali, M.; Longo, N. Biochemical abnormalities in Pearson syndrome. Am. J. Med. Genet. Part A 2015, 3, 621–628. [Google Scholar] [CrossRef] [PubMed]
- Lake, N.J.; Compton, A.G.; Rahman, S.; Thorburn, D.R. Leigh syndrome: One disorder, more than 75 monogenic causes. Ann. Neurol. 2016, 79, 190–203. [Google Scholar] [CrossRef]
- Saldana-Martinez, A.; Munoz, M.L.; Perez-Ramirez, G.; Montiel-Sosa, J.F.; Montoya, J.; Emperador, S.; Ruiz-Pesini, E.; Cuevas-Covarrubias, S.; Lopez-Valdez, J.; Ramirez, R.G. Whole sequence of the mitochondrial DNA genome of Kearns Sayre Syndrome patients: Identification of deletions and variants. Gene 2018, 688, 171–181. [Google Scholar] [CrossRef]
- Kim, U.S.; Jurkute, N.; Yu-Wai-Man, P. Leber Hereditary Optic Neuropathy-Light at the End of the Tunnel? Asia Pac. J. Ophthalmol. 2018, 7, 242–245. [Google Scholar] [CrossRef]
- Lamperti, C.; Zeviani, M. Myoclonus epilepsy in mitochondrial disorders. Epileptic Disord. 2016, 18, 94–102. [Google Scholar]
- Yu-Wai-Man, P.; Votruba, M.; Moore, A.T.; Chinnery, P.F. Treatment strategies for inherited optic neuropathies: Past, present and future. Eye (Lond. Engl.) 2014, 28, 521–537. [Google Scholar] [CrossRef] [PubMed]
- Yu-Wai-Man, P.; Chinnery, P.F. Leber Hereditary Optic Neuropathy—Therapeutic Challenges and Early Promise. Taiwan J. Ophthalmol. 2011, 1, 12–15. [Google Scholar] [CrossRef] [PubMed]
- Szuhai, K.; Ouweland, J.; Dirks, R.; Lemaitre, M.; Truffert, J.; Janssen, G.; Tanke, H.; Holme, E.; Maassen, J.; Raap, A. Simultaneous A8344G heteroplasmy and mitochondrial DNA copy number quantification in myoclonus epilepsy and ragged-red fibers (MERRF) syndrome by a multiplex molecular beacon based real-time fluorescence PCR. Nucleic Acids Res. 2001, 29, E13. [Google Scholar] [CrossRef]
- Pearson, H.A.; Lobel, J.S.; Kocoshis, S.A.; Naiman, J.L.; Windmiller, J.; Lammi, A.T.; Hoffman, R.; Marsh, J.C. A new syndrome of refractory sideroblastic anemia with vacuolization of marrow precursors and exocrine pancreatic dysfunction. J. Pediatr. 1979, 95, 976–984. [Google Scholar] [CrossRef]
- Srivastava, S. The Mitochondrial Basis of Aging and Age-Related Disorders. Genes 2017, 8, 398. [Google Scholar] [CrossRef]
- Lane, R.K.; Hilsabeck, T.; Rea, S.L. The role of mitochondrial dysfunction in age-related diseases. Biochim. Biophys. Acta 2015, 1847, 1387–1400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liberti, M.V.; Locasale, J.W. The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem. Sci. 2016, 41, 211–218. [Google Scholar] [CrossRef] [Green Version]
- Salas, A.; Yao, Y.G.; Macaulay, V.; Vega, A.; Carracedo, A.; Bandelt, H.J. A critical reassessment of the role of mitochondria in tumorigenesis. PLoS Med. 2005, 2, e296. [Google Scholar] [CrossRef] [PubMed]
- Stratton, M.R.; Campbell, P.J.; Futreal, P.A. The cancer genome. Nature 2009, 458, 719–724. [Google Scholar] [CrossRef] [PubMed]
- Petros, J.A.; Baumann, A.K.; Ruiz-Pesini, E.; Amin, M.B.; Sun, C.Q.; Hall, J.; Lim, S.; Issa, M.M.; Flanders, W.D.; Hosseini, S.H.; et al. mtDNA mutations increase tumorigenicity in prostate cancer. Proc. Natl. Acad. Sci. USA 2005, 102, 719–724. [Google Scholar] [CrossRef] [Green Version]
- Horton, T.M.; Petros, J.A.; Heddi, A.; Shoffner, J.; Kaufman, A.E.; Graham, S.D., Jr.; Gramlich, T.; Wallace, D.C. Novel mitochondrial DNA deletion found in a renal cell carcinoma. Genes Chromosom. Cancer 1996, 15, 95–101. [Google Scholar] [CrossRef]
- Ahmad, W.; Ijaz, B.; Shabbiri, K.; Ahmed, F.; Rehman, S. Oxidative toxicity in diabetes and Alzheimer’s disease: Mechanisms behind ROS/RNS generation. J. Biomed. Sci. 2017, 24, 76. [Google Scholar] [CrossRef]
- Tse, K.H.; Herrup, K. Re-imagining Alzheimer’s disease—The diminishing importance of amyloid and a glimpse of what lies ahead. J. Neurochem. 2017, 143, 432–444. [Google Scholar] [CrossRef] [PubMed]
- Kwiatkowski, D.; Czarny, P.; Toma, M.; Jurkowska, N.; Sliwinska, A.; Drzewoski, J.; Bachurska, A.; Szemraj, J.; Maes, M.; Berk, M.; et al. Associations between DNA Damage, DNA Base Excision Repair Gene Variability and Alzheimer’s Disease Risk. Dement. Geriatr. Cogn. Disord. 2016, 41, 152–171. [Google Scholar] [CrossRef] [PubMed]
- Wang, W.; Esbensen, Y.; Kunke, D.; Suganthan, R.; Rachek, L.; Bjoras, M.; Eide, L. Mitochondrial DNA damage level determines neural stem cell differentiation fate. J. Neurosci. 2011, 31, 9746–9751. [Google Scholar] [CrossRef]
- Sheng, Z.; Oka, S.; Tsuchimoto, D.; Abolhassani, N.; Nomaru, H.; Sakumi, K.; Yamada, H.; Nakabeppu, Y. 8-Oxoguanine causes neurodegeneration during MUTYH-mediated DNA base excision repair. J. Clin. Investig. 2012, 122, 4344–4361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leon, J.; Sakumi, K.; Castillo, E.; Sheng, Z.; Oka, S.; Nakabeppu, Y. 8-Oxoguanine accumulation in mitochondrial DNA causes mitochondrial dysfunction and impairs neuritogenesis in cultured adult mouse cortical neurons under oxidative conditions. Sci. Rep. 2016, 6, 22086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Santoro, A.; Balbi, V.; Balducci, E.; Pirazzini, C.; Rosini, F.; Tavano, F.; Achilli, A.; Siviero, P.; Minicuci, N.; Bellavista, E.; et al. Evidence for sub-haplogroup h5 of mitochondrial DNA as a risk factor for late onset Alzheimer’s disease. PLoS ONE 2010, 5, 0012037. [Google Scholar] [CrossRef]
- Ikebe, S.; Tanaka, M.; Ohno, K.; Sato, W.; Hattori, K.; Kondo, T.; Mizuno, Y.; Ozawa, T. Increase of deleted mitochondrial DNA in the striatum in Parkinson’s disease and senescence. Biochem. Biophys. Res. Commun. 1990, 170, 1044–1048. [Google Scholar] [CrossRef]
- Simon, D.K.; Pulst, S.M.; Sutton, J.P.; Browne, S.E.; Beal, M.F.; Johns, D.R. Familial multisystem degeneration with parkinsonism associated with the 11778 mitochondrial DNA mutation. Neurology 1999, 53, 1787–1793. [Google Scholar] [CrossRef]
- Luoma, P.; Melberg, A.; Rinne, J.O.; Kaukonen, J.A.; Nupponen, N.N.; Chalmers, R.M.; Oldfors, A.; Rautakorpi, I.; Peltonen, L.; Majamaa, K.; et al. Parkinsonism, premature menopause, and mitochondrial DNA polymerase gamma mutations: Clinical and molecular genetic study. Lancet 2004, 364, 875–882. [Google Scholar] [CrossRef]
- Sanders, L.H.; Paul, K.C.; Howlett, E.H.; Lawal, H.; Boppana, S.; Bronstein, J.M.; Ritz, B.; Greenamyre, J.T. Editor’s Highlight: Base Excision Repair Variants and Pesticide Exposure Increase Parkinson’s Disease Risk. Toxicol. Sci. 2017, 158, 188–198. [Google Scholar] [CrossRef]
- Cornetta, T.; Patrono, C.; Terrenato, I.; De Nigris, F.; Bentivoglio, A.R.; Testa, A.; Palma, V.; Poggioli, T.; Padua, L.; Cozzi, R. Epidemiological, clinical, and molecular study of a cohort of Italian Parkinson disease patients: Association with glutathione-S-transferase and DNA repair gene polymorphisms. Cell. Mol. Neurobiol. 2013, 33, 673–680. [Google Scholar] [CrossRef] [PubMed]
- Fukae, J.; Mizuno, Y.; Hattori, N. Mitochondrial dysfunction in Parkinson’s disease. Mitochondrion 2007, 7, 58–62. [Google Scholar] [CrossRef]
- Fukae, J.; Takanashi, M.; Kubo, S.; Nishioka, K.; Nakabeppu, Y.; Mori, H.; Mizuno, Y.; Hattori, N. Expression of 8-oxoguanine DNA glycosylase (OGG1) in Parkinson’s disease and related neurodegenerative disorders. Acta Neuropathol. 2005, 109, 256–262. [Google Scholar] [CrossRef]
- Coppede, F.; Ceravolo, R.; Migheli, F.; Fanucchi, F.; Frosini, D.; Siciliano, G.; Bonuccelli, U.; Migliore, L. The hOGG1 Ser326Cys polymorphism is not associated with sporadic Parkinson’s disease. Neurosci. Lett. 2010, 473, 248–251. [Google Scholar] [CrossRef]
- Skarpengland, T.; Holm, S.; Scheffler, K.; Gregersen, I.; Dahl, T.B.; Suganthan, R.; Segers, F.M.; Ostlie, I.; Otten, J.J.; Luna, L.; et al. Neil3-dependent base excision repair regulates lipid metabolism and prevents atheroslerosis in Apoe-deficient mice. Sci. Rep. 2016, 6, 28337. [Google Scholar] [CrossRef] [PubMed]
- Scheffler, K.; Rachek, L.; You, P.; Rowe, A.D.; Wang, W.; Kusnierczyk, A.; Kittelsen, L.; Bjoras, M.; Eide, L. 8-oxoguanine DNA glycosylase (Ogg1) controls hepatic gluconeogenesis. DNA Repair 2018, 61, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Yuzefovych, L.V.; Musiyenko, S.I.; Wilson, G.L.; Rachek, L.I. Mitochondrial DNA damage and dysfunction, and oxidative stress are associated with endoplasmic reticulum stress, protein degradation and apoptosis in high fat diet-induced insulin resistance mice. PLoS ONE 2013, 8, e54059. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Xie, C.; Spencer, H.J.; Zuo, C.; Higuchi, M.; Ranganathan, G.; Kern, P.A.; Chou, M.W.; Huang, Q.; Szczesny, B.; et al. Obesity and hepatosteatosis in mice with enhanced oxidative DNA damage processing in mitochondria. Am. J. Pathol. 2011, 178, 1715–1727. [Google Scholar] [CrossRef]
- Hara, M.; Nakamura, K.; Nanri, H.; Nishida, Y.; Hishida, A.; Kawai, S.; Hamajima, N.; Kita, Y.; Suzuki, S.; Mantjoro, E.M.; et al. Associations between hOGG1 Ser326Cys polymorphism and increased body mass index and fasting glucose level in the Japanese general population. J. Epidemiol. 2014, 24, 379–384. [Google Scholar] [CrossRef]
- Corella, D.; Ramirez-Sabio, J.B.; Coltell, O.; Ortega-Azorin, C.; Estruch, R.; Martinez-Gonzalez, M.A.; Salas-Salvado, J.; Sorli, J.V.; Castaner, O.; Aros, F.; et al. Effects of the Ser326Cys Polymorphism in the DNA Repair OGG1 Gene on Cancer. J. Acad. Nutr. Diet. 2018, 118, 589–605. [Google Scholar] [CrossRef]
Figure 1.
Mechanisms of inheritance of mtDNA mutations. Each mitochondrion consists of multiple copies of mtDNA, some of which may harbor harmful mutations. Upon mitochondrial fission, fusion, or mtDNA replication, these mtDNA molecules may be randomly segregated to daughter mitochondria, resulting in either reduced or increased levels of heteroplasmy. The contribution of heteroplasmy to disease development is difficult to study, as the disease threshold for each mutation may be different and may lead to a range of clinical and sub-clinical phenotypes. (Green ovals = functional mitochondria; red = dysfunctional; blue = suboptimal function).
Figure 1.
Mechanisms of inheritance of mtDNA mutations. Each mitochondrion consists of multiple copies of mtDNA, some of which may harbor harmful mutations. Upon mitochondrial fission, fusion, or mtDNA replication, these mtDNA molecules may be randomly segregated to daughter mitochondria, resulting in either reduced or increased levels of heteroplasmy. The contribution of heteroplasmy to disease development is difficult to study, as the disease threshold for each mutation may be different and may lead to a range of clinical and sub-clinical phenotypes. (Green ovals = functional mitochondria; red = dysfunctional; blue = suboptimal function).

Figure 2.
Base Excision Repair (BER) in mitochondria. Generation of free radicals induces DNA lesions in the form of oxidized bases, affecting base-pairing properties. Oxidative lesions are repaired via the BER pathway. BER is initiated by the activity of DNA glycosylases such as monofunctional glycosylases which recognize and cleave the N-glycosidic bond between the modified base and sugar, creating an abasic site and bifunctional glycosylases which have an additional intrinsic Apurinic/Apyrimidinic (AP)lyase activity. The incision of the AP site occurs via β elimination or β-δ elimination which is further processed by APE1 or PNKP, followed by gap-filling by DNA pol gamma (PolG). Once the AP site has been processed and the correct nucleotide recruited by PolG, the free DNA ends are ligated by DNA ligase III (LIG3).
Figure 2.
Base Excision Repair (BER) in mitochondria. Generation of free radicals induces DNA lesions in the form of oxidized bases, affecting base-pairing properties. Oxidative lesions are repaired via the BER pathway. BER is initiated by the activity of DNA glycosylases such as monofunctional glycosylases which recognize and cleave the N-glycosidic bond between the modified base and sugar, creating an abasic site and bifunctional glycosylases which have an additional intrinsic Apurinic/Apyrimidinic (AP)lyase activity. The incision of the AP site occurs via β elimination or β-δ elimination which is further processed by APE1 or PNKP, followed by gap-filling by DNA pol gamma (PolG). Once the AP site has been processed and the correct nucleotide recruited by PolG, the free DNA ends are ligated by DNA ligase III (LIG3).


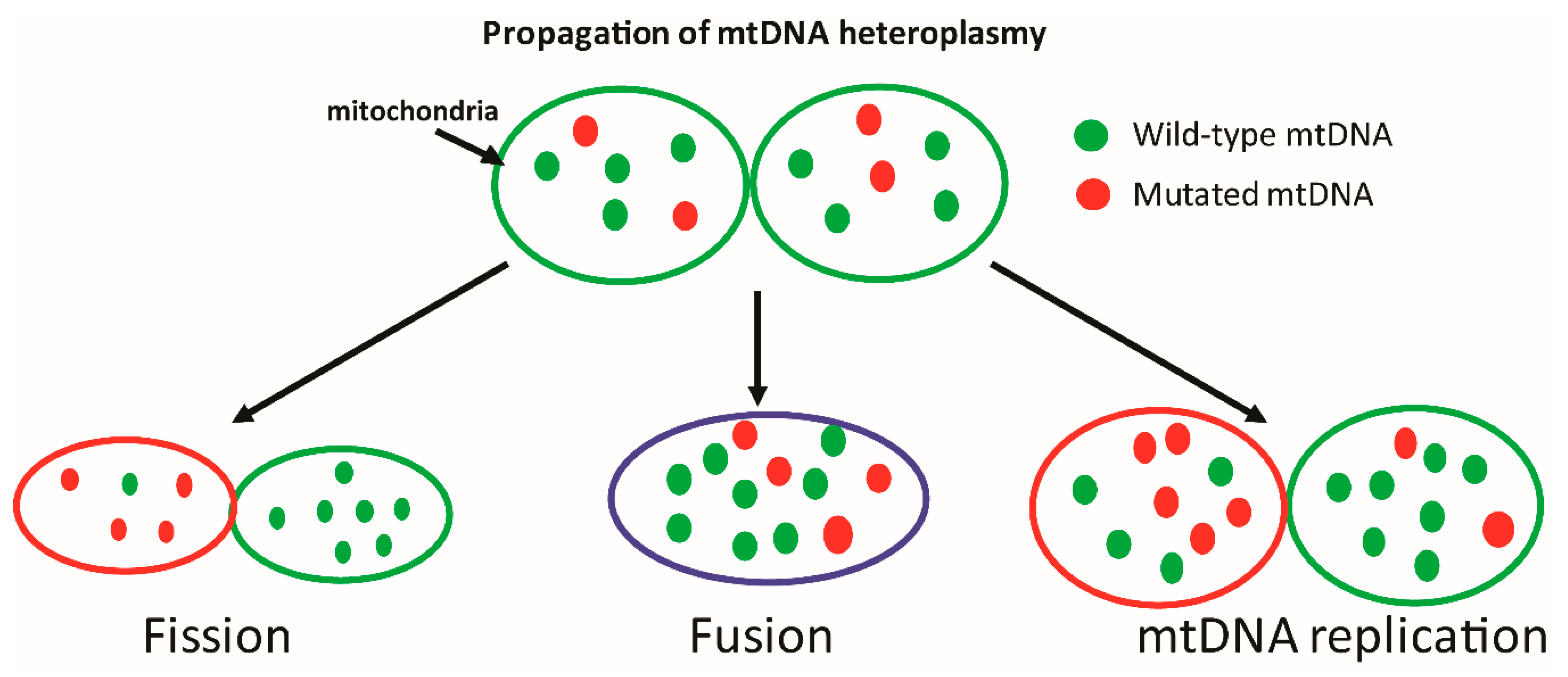{kind=link}
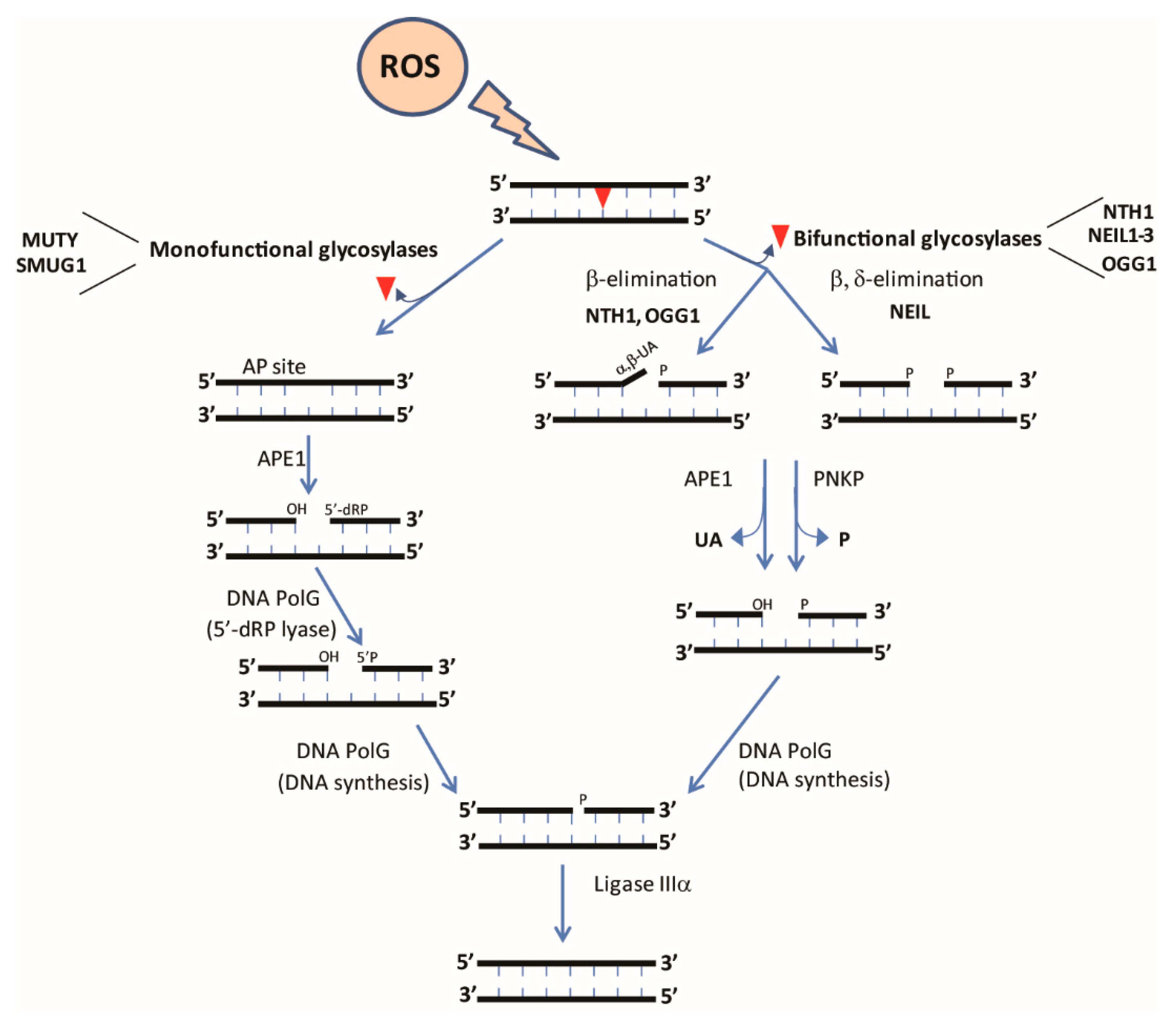{kind=link}
Table 1.
Tissue specificity, cellular localization, and known or inferred functions of mitochondrial isoforms of selected BER glycosylases and MTH1.
Table 1.
Tissue specificity, cellular localization, and known or inferred functions of mitochondrial isoforms of selected BER glycosylases and MTH1.
| Enzyme | Glycosylase Family | Tissue Specificity | Inferred or Confirmed Function(s) of Mitochondrial Isoform | Mitochondrial Localization Described |
|---|---|---|---|---|
| NEIL1 | Fpg/Nei Helix-two turns-helix | Liver, thymus, pancreas, brain [128,129,130] | Potential role in mediating metabolic syndrome in Neil1−/− mice [141]; Binding partner for mtSSB [142] | [142,143] |
| NEIL2 | Fpg/Nei Helix-two turns-helix | Testes and skeletal muscle [131] | Removal of oxidized bases from mitochondrial genome [144] | [144] |
| OGG1 (1a) | Helix-hairpin- helix | Thymus, testis, intestine, brain, and germinal center of B cells [130,133,135,145] | Role in protecting against oxidative stress [146,147,148,149]; Prevention of metabolic syndrome [116] | [150,151,152] |
| NTH1 | Helix-hairpin- helix | Heart, brain [130,132,153] | Unknown; potentially compensated for by NEIL1 activity | Mouse isoform is exclusively mitochondrial; human isoform is thought to be exclusively nuclear [154,155] |
| MUTY | Helix-hairpin- helix | Thymus, intestine, heart, lung [156] | Potentially involved in repair of hypoxia induced damage in brain [157] | [156,158] |
| MTH1 | Oxidized purine nucleoside triphosphatase | Thymus, testis, embryonic tissues [127] | Protection from oxidative damage in models of Parkinson’s disease [137] | [136,138,159] |
Table 2.
Mitochondrial diseases and their genomic localization.
| Disease | Clinical Manifestations | Location |
|---|---|---|
| Leber Hereditary Optic Neuropathy (LHON) | Bilateral, painless subacute visual failure | Mutations in MT-ND1, MT-ND4 or MT-ND6 gene [179,180] |
| Myoclonic Epilepsy with Ragged Red Fibers (MERRF) | Myoclonus epilepsy, ataxia, muscle weakness and dementia | Mutations in MT-TK encoding tRNA lysine (tRNALys). A-to-G transition at nucleotide 8344 [178,181] |
| Pearson’s syndrome | Sideroblastic anemia and exocrine pancreas dysfunction | Mitochondrial DNA deletion [174,182] |
| Kearns-Sayre Syndrome (KSS) | Pigmentary retinopathy | Mitochondrial DNA deletion [176] |
| Mitochondrial encephalopathy, lactic acidosis and strokelike episodes (MELAS) | Mitochondrial encephalomyopathy, lactic acidosis and strokelike episodes. Other features include headache, seizures, muscle weakness | Point mutation in the tRNA leucineUUR gene of mitochondrial DNA. A to G transition at nucleotide 3243 [173] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Sharma, P.; Sampath, H. Mitochondrial DNA Integrity: Role in Health and Disease. Cells 2019, 8, 100. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8020100
AMA Style
Sharma P, Sampath H. Mitochondrial DNA Integrity: Role in Health and Disease. Cells. 2019; 8(2):100. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8020100
Chicago/Turabian StyleSharma, Priyanka, and Harini Sampath. 2019. "Mitochondrial DNA Integrity: Role in Health and Disease" Cells 8, no. 2: 100. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8020100
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.