Targeting the Hedgehog Pathway in Cancer: Current Evidence and Future Perspectives


Division of Medical Oncology, Hospital Sírio-Libanês, Brasilia 70200-730, Brazil
*
Author to whom correspondence should be addressed.
Cells 2019, 8(2), 153; https://0-doi-org.brum.beds.ac.uk/10.3390/cells8020153
Submission received: 27 December 2018
/
Revised: 10 February 2019
/
Accepted: 11 February 2019
/
Published: 12 February 2019
(This article belongs to the Collection Hedgehog Signal Transduction in Physiology and Disease)
Abstract
:The Hedgehog pathway (HhP) plays an important role in normal embryonic development and its abnormal function has been linked to a variety of neoplasms. Recently, the complex mechanisms involved in this pathway have been deciphered and the cross talks with other important pathways involved in carcinogenesis have been characterized. This knowledge has led to the development of targeted therapies against key components of HhP, which culminated in the approval of vismodegib for the treatment of advanced basal cell carcinoma in 2012. Since then, other compounds have been developed and evaluated in preclinical and clinical studies with interesting results. Today, several medications against components of the HhP have demonstrated clinical activity as monotherapies and in combination with cytotoxic treatment or other targeted therapies against mitogenic pathways that are linked to the HhP. This review aims to clarify the mechanism of the HhP and the complex crosstalk with others pathways involved in carcinogenesis and to discuss both the evidence associated with the growing number of medications and combined therapies addressing this pathway and future perspectives.
1. Introduction
The Hedgehog pathway (HhP) plays a fundamental role in embryonic development, tissue patterning, and wound healing [1]. Aberrant functioning of this pathway is related to several congenital abnormalities and the development of cancer in several organs [2,3].
The idea that alterations in the HhP are linked to carcinogenicity came from the discovery of activating mutations in this pathway in patients with basal cell carcinoma (BCC), medulloblastoma and rhabdomyosarcoma [4,5,6,7]. These findings led to a better understanding of the pathway and the development of targeted therapies directed against their effectors [8,9].
In this review, we focus on the characterization of the HhP, its relation with other pathways related to the development of cancer and especially on treatment strategies as a way to fight cancer.
2. Characterization of the HhP and Its Relation with Carcinogenesis
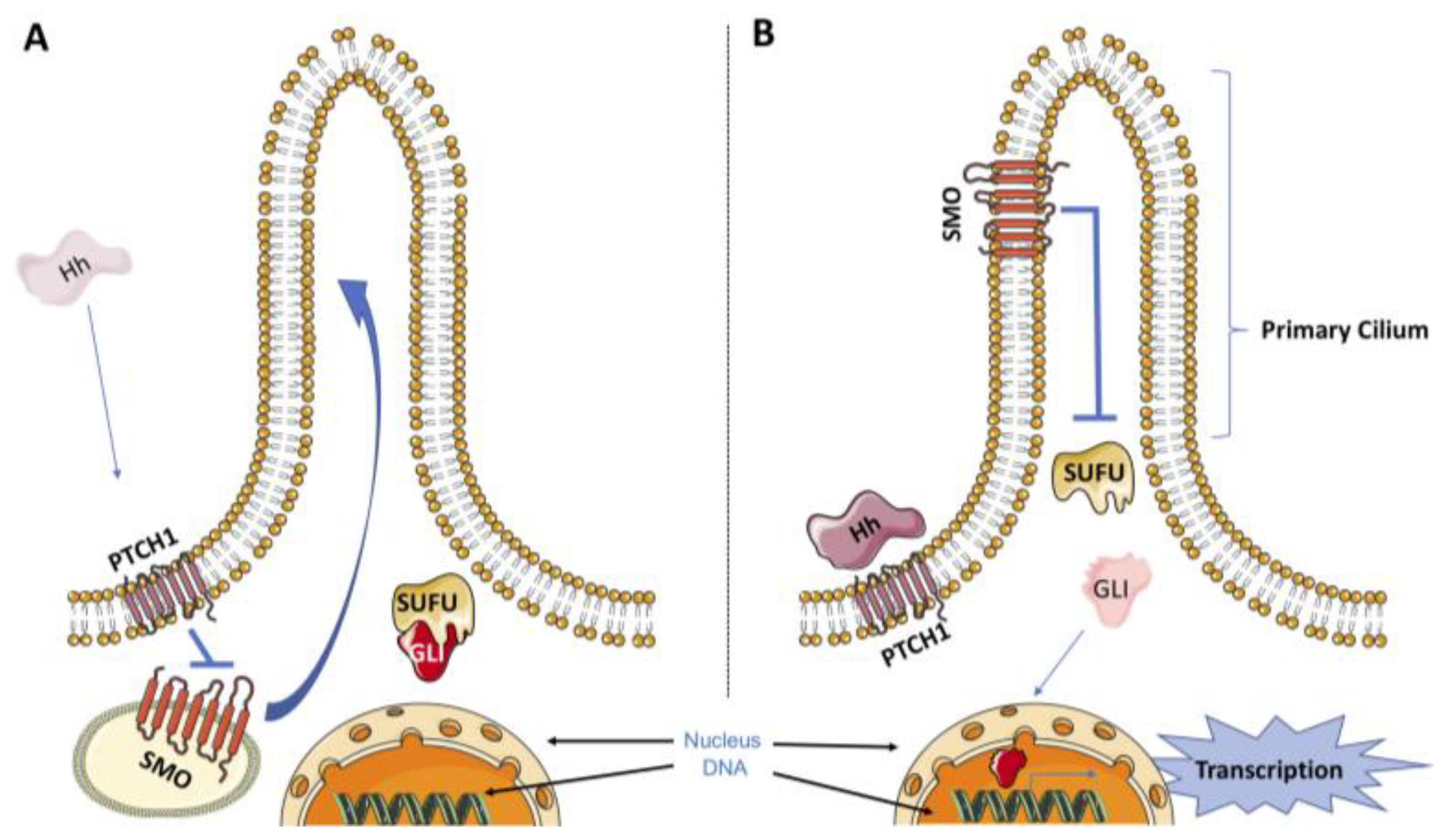
The HhP is very complex and can be divided into two different pathways: canonical and noncanonical. The canonical HhP is initiated by the release of three ligands named Sonic Hedgehog (SHH), Desert Hedgehog (DHH), and Indian Hedgehog (IHH) [10]. In the absence of these ligands, the 12-pass transmembrane receptor Patched1 (PTCH1) exerts an inhibitory effect on the transmembrane transducer smoothened (SMO) [11]. Binding of Hedgehog ligands to PTCH1 relieves the repression of SMO by PTCH1, which results in the translocation of SMO to the primary cilium. The primary cilium is a membrane-encased protrusion usually described as a single nonmotile cilium present in a variety of vertebrate cells. In humans, for instance, virtually all other cells have a primary cilium, with the exception of sperm, epithelia cells in the bronchi and oviducts, and ependymal cells that line the brain vesicles. The translocation of SMO to the primary cilium, in turn, initiates an intracellular signal cascade that promotes the activation of glioma-associated oncogene (GLI) transcription factors [12]. There are three members of the GLI transcription factor family (GLI1, GLI2 and GLI3) that share a similar DNA-binding domain [13]. Once activated in the primary cilium, GLIs dissociate from the suppressor of fused (SUFU), which is a key cytoplasmic negative regulator of the HhP, and translocate into the nucleus to initiate the transcription program related to the HhP (Figure 1) [14]. In the absence of HH ligands, PTCH1 exerts a repressive effect on SMO by preventing its accumulation in the primary cilium. In this state, SMO is not capable of activating GLI transcription factors, which are bound to SUFU and retained in the cytoplasm. In the cytoplasm, the degradation of GLI proteins by the proteasome can occur through phosphorylation by protein kinase A (PKA), casein kinase 1 (CK1) and glycogen synthase kinase 3β (GSK3β) [15,16].
In the noncanonical HhP, activation of the GLI transcription factors can occur independently of the upstream components of the HhP by cross talk with other signaling cascades [16]. Multiple cross talk and synergistic interactions between HhP components and other important oncogenic pathways have been shown to activate HhP and have been implicated in several types of cancer. For example, cross talk between HhP and the mechanistic target of rapamycin (mTOR) pathway has been described in esophageal carcinoma in which GLI1 appears to be activated by ribosomal protein S6 kinase 1 (S6K1) through direct phosphorylation at SEr84 [17]. Conversely, another study demonstrated that V-akt murine thymoma viral oncogene homolog 1 (AKT1) is a direct transcriptional target of GLI1 [18].
Cytotoxic agents, such as radiotherapy and cytokines, can also activate and upregulate the expression of GLI1 independent of canonical pathway activation. For example, tumor necrosis factor alpha (TNF-α) and interleukin-1β (IL-1β) can upregulate the expression of GLI1 through the nuclear factor kappa light chain enhancer of activated B cells (NF-κB) pathway in pancreatic and breast cancer cells [19,20]. Interestingly, evidence now shows that NF-κB subunit p65 can bind directly to the GLI1 promoter region, and inhibition of NF-κB decreased GLI1 activity in breast cancer cells [20]. Transforming growth factor-beta (TGF-β) has also been demonstrated to upregulate GLI1 and GLI2 via the SMAD3 pathway and may also lead to GLI protein accumulation in cancer cells [21,22]. In pancreatic cancer cell lines resistant to Hedgehog inhibitors, the pharmacologic blockade of TGF-β has been shown to inhibit cell proliferation [21]. Similarly, inhibition of TGF-β also contributed to reduced tumor volume in preclinical models of SMO-induced BCC and reduced tumor cell invasion in gastric cancer cell models [22,23].
The RAS/RAF/MEK/ERK pathway can also upregulate and activate GLI transcriptional activity. An interesting study demonstrated that oncogenic KRAS was able to increase GLI activity in pancreatic adenocarcinoma cell lines, and this effect was inhibited by MEK inhibitors [24]. Another study demonstrated that EGFR synergizes with GLI in the carcinogenesis process via the RAS/RAF/MEK/ERK axis and that in vitro dual inhibition of epidermal growth factor receptor (EGFR) and GLI led to a more potent reduction in cell proliferation in BCC cells than either inhibitor alone [25]. Finally, a study shows that c-MYC can directly regulate GLI1 by interacting with the 5′-regulatory region of GLI1 and that inhibition of c-MYC can decrease GLI1 mRNA [26]. This study also provides evidence that the use of GLI1 inhibitors was able to increase tumor control of Burkitt lymphoma cells [26].
3. Other Implications of the HhP and Cancer
As mentioned above, upregulation and overexpression of the HhP are related to several cancer types, such as lung, head and neck, esophagus, colon, pancreas, glioma, breast, ovarian and cervical [1]. Recently, some clinical implications of this upregulation and overexpression have been elucidated. For example, GLI1 activation was associated with distant metastasis and poor outcomes in patients with head and neck squamous cell carcinoma (HNSSC) [27]. Similar findings were described in esophageal squamous cell carcinoma in which GLI1 overexpression was associated with node metastasis and poor prognosis [28]. Upregulation of GLI1 expression in colon cancer was correlated with node metastasis, T-stage and postoperative live metastasis-free survival periods [29]. GLI2 expression was also correlated with poor outcomes in acute myeloid leukemia. A patient cohort indicated that patients with GLI2 overexpression had significantly worse outcomes in terms of event-free survival (EFS), relapse-free survival (RFS), and overall survival (OS) [30].
The HhP is also implicated in resistance to cancer therapies. For example, a study demonstrated the role of the HhP in chemoresistant ovarian cancer cell lines, and the use of SMO antagonists was able to sensitize chemotherapy-resistant cell lines to paclitaxel [31]. One of the reasons why tumors can become resistant to treatment is the epithelial-to-mesenchymal transition (EMT), which has been associated with the HhP. Multiple genes and proteins have been implicated in this mechanism, including those of the HhP, the NOTCH and WNT pathways [32,33] and the transcription factors ZEB1, ZEB2, SNAI1, SLUG and TWIST1 [34,35,36]. EMT is a process that occurs both in normal cells (during wound repairs and embryonic development) and in cancer cells, where it is implicated in resistance to chemotherapy and radiotherapy and increased migratory and invasive properties [34,37,38,39,40]. During the EMT process, cancer cells appear to acquire a stem cell-like phenotype that portents the capacity of self-renewal, resistance to cancer therapies and repopulation after a cytotoxic treatment [41,42,43,44]. The GLI transcriptional factors mediate the stem cell-like phenotype by regulating the transcription of genes involved in this signature, such as NANOG, octamer binding transcription factor 4 (OCT4), SOX2, BMI1, WNT-2, and Kruppel-like factor 4 (KLF4) [45,46].
Preclinical data have shown that in HNSSC cells, the expression of GLI transcription factors is increased in the population of cells that were resistant to EGFR inhibitors and radiotherapy [47,48]. These cell lines expressed higher levels of HhP genes and a stem cell-like phenotype [1]. This process was also described in other cancer types, such as lung, esophagus, gastric and colorectal cancers, in which transcriptional activation of genes related to EMT and stem cell-like phenotype were mediated by the HhP through GLI [49,50,51,52]. In a lung cancer model, HhP inhibition was able to reverse EGFR resistance and the stem cell-like phenotype [49].
4. SMO Inhibitors
A great deal of effort has been focused on targeting SMO in particular [53]. To date, two SMO inhibitors (sonidegib and vismodegib) have received US Food and Drug Administration (FDA) approval for treating BCC, while many clinical trials are being conducted to evaluate the efficacy of this exciting class of targeted therapies in a variety of cancers. Table 1 summarizes the clinical trials that evaluated SMO inhibitors against a variety of cancer types.
4.1. Cyclopamine
Cyclopamine (11-deoxojervine), a name derived from the word cyclopia, is a naturally occurring chemical isolated from the corn lily (Veratrum californicum) and belongs to the family of steroidal alkaloids.
In the 1950s, a group of ranchers in Idaho were surprised when their sheep gave birth to lambs with only one eye. After decades of research, they discovered that the cause of the deformity was the corn lily ingested by the pregnant sheep. Four decades later, the relationship of the SHH gene with cyclopamine was discovered. Upon experimentation, they recreated cyclopia by silencing the SHH gene and then connected their results to the cycloptic sheep noted four decades earlier.
In 1998, the first studies showing that cyclopamine inhibits SHH signal transduction were published [91,92], but the potential effects of cyclopamine or synthetic derivatives on cancer were reported in 2000 [93].
Cyclopamine has a high affinity for SMO, and upon binding, it inhibits the signal. It was the first compound found to inhibit HhP signaling and has been very valuable for understanding the function of Hedgehog signaling. Cyclopamine is widely used as a Hedgehog inhibitor in cell and murine models of various tumors [94,95,96,97]. However, the poor solubility and the low potency of cyclopamine prevent its clinical usage. A cyclopamine synthetic analog with greater solubility, cyclopamine tartrate (CycT), was reported for its activities in HhP signaling-mediated cancer in vitro and in vivo (mice) but had uncertain results and is still not used in clinical practice [98,99].
4.2. Vismodegib (GDC-0449)
Vismodegib is the first FDA-approved SMO inhibitor for the treatment of advanced and metastatic BCC. Currently, vismodegib and many other SMO inhibitors are being investigated in clinical trials in a range of advanced cancers [73,100]. This medication binds to and directly inhibits SMO, blocking the signal transduction in the HhP.
The international, open-label STEVIE trial is the largest study of safety and efficacy of vismodegib in BCC [55]. This trial allocated 1215 patients with locally advanced and metastatic BCC from 36 countries. The response rates were 68.5% in patients with locally advanced BCC and 36.9% in patients with metastatic BCC, but serious side effects occurred in 289 patients (23.8%), with death in 46 patients (3.8%).
It is not clear whether treatment with vismodegib increases the risk of cutaneous squamous cell carcinoma. A small case control study suggested this association and showed that in the vismodegib group, the risk of new cases of squamous cell carcinoma increased eightfold [101]. Controversially, another case control study did not demonstrate this association [102]. Even with these controversial data, continuous skin surveillance should be performed after the initiation of this therapy.
Acquired resistance was identified in 21% of the patients treated while undergoing continuous vismodegib treatment, with a mean time to detected regrowth by clinical examination of 56.4 weeks [103].
4.3. Sonidegib (LDE-225)
Sonidegib is a direct inhibitor of SMO, blocking the signal transduction in the HhP. In the phase I study, sonidegib had an acceptable safety profile in patients with advanced solid tumors and exhibited antitumor activity in advanced BCC and relapsed medulloblastoma [104]. In a phase 2 study, it was shown that 800 mg daily is not more efficacious than 200 mg and causes more adverse effects (grade 3/4 effects adverse of 43.0% vs. 64.0%). As BCC is a slow growing tumor, even after 30 months, the median OS had not been reached. The estimated 2-year OS rates in patients taking 200 mg were 93.2% for advanced disease and 69.3% for those with metastases, and few patients had a complete response [74].
In the comparison of sonidegib with vismodegib, a recent meta-analysis has shown that in locally advanced BCC, overall response rates (ORRs) were similar for vismodegib and sonidegib (69% vs. 57%), but complete response rates were not (31% vs. 3%). In metastatic disease, the ORR of vismodegib was 2.7-fold higher than the ORR of sonidegib (39% vs. 15%). Side effects were similar in both groups, except for upper gastrointestinal symptoms, which were higher in patients receiving sonidegib [105].
Regarding the tumors resistant to Hedgehog inhibitors, a study showed that patients with advanced BCCs who were previously resistant to treatment with vismodegib similarly demonstrated the same treatment resistance with sonidegib [106].
4.4. Saridegib (IPI-926)
Saridegib is a potent and specific inhibitor of SMO derived from cyclopamine. In vitro and in vivo studies have shown the activity of this drug in many types of cancer, such as ovarian, medulloblastoma and head and neck [1]. Furthermore, evidence from a genetically engineered mouse model of pancreatic cancer demonstrated that saridegib can deplete tumor-associated stromal tissue and increase intratumoral mean vessel density [110]. These changes resulted in enhanced delivery of concurrently administered systemic chemotherapy, leading to a decreased tumor burden and prolonged survival in this mouse model.
A multicenter phase Ib study evaluated saridegib in combination with FOLFIRINOX in patients with advanced pancreatic cancer [79]. The objective response rate was high (67%), and patients receiving saridegib maintenance showed further declines in CA19-9 levels even after FOLFIRINOX discontinuation. However, the study closed early when a separate phase II trial of saridegib plus gemcitabine indicated detrimental effects of this combination.
4.5. Taladegib (LY2940680)
Taladegib is an antagonist of the Hedgehog ligand cell surface receptor SMO with potential antineoplastic activity. Recently, a study demonstrated the in vivo efficacy of taladegib in a mouse medulloblastoma allograft model [111].
Taladegib has been studied in several trials involving solid tumors, such as colon cancer, breast cancer and rhabdomyosarcoma. However, a phase 1 trial suggests that taladegib is effective only in BCC. This trial evaluated several advanced solid tumors, and collectively, the clinical responses in BCC patients had an estimated ORR of 46.8%. This study also suggested potential benefit of taladegib not only in Hedgehog treatment-naïve patients but also in patients who were previously treated with Hedgehog inhibitor therapy [87].
4.6. Glasdegib (PF-04449913)
Glasdegib is an oral, potent, selective inhibitor of the HhP that functions through binding to the SMO receptor [112]. This molecule prevents the translocation of SMO into primary cilia and prevents SMO-mediated activation of downstream Hedgehog targets [113]. In preclinical studies, glasdegib inhibited SMO in vitro and induced significant antitumor activity in vivo.
Its use seems to be attractive in myeloid malignancies since it has demonstrated that glasdegib inhibition of SMO reduces the expression of key intracellular leukemia stem cell regulators [114]. In addition, the administration of glasdegib also resulted in a significant reduction in leukemic stem cell (LSC) burden in xenograft models, inhibition of HhP signaling, and a reduction in cell populations expressing LSC markers [115]. A phase 1 study suggested that glasdegib had no effect on solid tumors. In this study, eight patients (34.8%) achieved stable disease, and none had a complete or partial response. Three patients with disease progression at enrollment had prolonged disease stabilization (≥6 months) [90].
4.7. Itraconazole
Itraconazole, an FDA-approved antifungal drug, appears to act on the essential HhP component SMO by a mechanism distinct from that of cyclopamine and other known SMO antagonists [116].
In clinical trials, itraconazole was tested in an open-label, phase II trial for the treatment of BCC in 29 patients. Itraconazole reduced cell proliferation by 45%, HhP activity by 65%, and tumor area by 24% [84]. In another randomized phase II clinical trial of metastatic castration-resistant prostate cancer, 46 chemotherapy-naïve patients were enrolled, of whom 29 received high-dose itraconazole treatment (600 mg/day), and 17 received low-dose (200 mg/day) itraconazole treatment. Prostate-specific antigen progression-free survival (PFS) rates at 24 weeks were 48% and 11.8% with median PFS of 11.9 and 35.9 weeks in the high- and low-dose arms, respectively [86].
5. GLI Inhibitors
As demonstrated above, the HhP can be activated downstream of SMO, and multiple cross talk between the HhP and other mitogenic pathways converges to amplify and activate GLI transcriptional factors. This cross talk is also one of the causes of resistance to SMO inhibitors, along with mutations in SMO [121,122,123]. Strategies to address this mechanism and overcome resistance to SMO inhibitors are targeted therapies against GLI and combination with targeted agents against other oncogenic pathways involved in GLI activation [15].
GANT58 and GANT61 are GLI antagonists that can interfere with GLI translocation into the nucleus and can prevent DNA binding [124]. GANT61 is the most studied and efficient antagonist that can bind to the zinc finger regions 2 and 3 of GLI1 and GLI2 [1]. Several preclinical studies demonstrated antitumor activity in many cancer types, such as lung, acute myeloid leukemia, rhabdomyosarcoma, neuroblastoma, breast cancer, colon, prostate, melanoma, and pancreatic cancer [15,124,125,126,127,128,129]. The main effects of GANT61 are the interference with the cell cycle by induction of G1 arrest and expression of p21 [127,130,131]; the cytotoxic activity through activation of Fas signaling and decreased levels of Bcl2 (an antiapoptotic protein) [15,125,132,133]; the attenuation of the EMT process slowing down cell migration [131,132,134,135]; the decrease in the transcription of genes related to stem cell phenotype, such as NANOG, SOX2, OCT4 and c-MYC [15,136]; and the increase in the production of inflammatory cytokines such as IL8 and MCP1, which increases monocyte recruitment [137].
Arsenic trioxide (ATO) is known for its use in acute promyelocytic leukemia; however, it is also a GLI inhibitor [138]. The mechanism of action is the inhibition of GLI2 by blocking the trafficking in and out the primary cilium, which is necessary for GLI2 activation [139]. In preclinical studies, ATO was able to reduce the growth of medulloblastoma allografts [139]. A similar mechanism of action was described for pirfenidone, which is a drug approved for the treatment of idiopathic pulmonary fibrosis [140]. This drug is also capable of destabilizing GLI2 and decreasing the expression of TGF-β [141]. In fact, some recent preclinical studies have shown promising antitumor results in several cancer types, such as lung, breast, pancreas, glioma and hepatocellular carcinoma [142,143,144,145,146,147].
Four Hedgehog pathway inhibitors (HPIs) with unique mechanisms of action downstream of SMO that are capable of modulating GLI activity have been recently described [15]. HPI-1 likely increases GLI repression through PKA phosphorylation, HPI-2 and HPI-3 modulate the activity of GLI2, and HPI-4 interferes with ciliogenesis and therefore with GLI activation [148]. Pyrvinium, an antihelminthic, is another drug that has been shown to have inhibitory effects on the HhP. The proposed mechanism of action is the activation of CK1, which facilitates the phosphorylation of GLI proteins and consequent degradation [149]. Imiquimod, an agonist of toll-like receptor (TLR) 7 and TLR8, can also modulate GLI activity by activating PKA and subsequent degradation of GLI proteins [150]. Nanoquinacrine, a spherical nanoparticle form of quinacrine, also interferes with the HhP by increasing the expression of GSK3β and interfering with the binding of GLI1 and DNA [151]. Recently, the bromodomain and extra terminal (BET) proteins, specifically the bromodomain-containing protein 4 (BRD4), have been described and associated with HhP inhibition. These molecules can bind directly to GLI promoter regions on DNA and epigenetically regulate its transcription. Two preclinical studies have demonstrated the antitumor properties of these protein inhibitors [152,153].
6. Combination Therapy
The characterization of the noncanonical pathway elucidated cross talk between the HhP and other oncogenic pathways, such as mTOR, EGFR, MAPK, NF-κB and TGF-β. Consequently, several studies evaluated the combination therapy between agents targeting HhP components and agents addressing components of these other pathways. A promising combination is dual inhibition of the HhP and mTOR pathways. Two preclinical studies have demonstrated the efficacy of GANT61 and mTOR inhibitor combination in myeloid leukemia cells and in rhabdomyosarcoma cells [127,154]. In vitro studies have also demonstrated that blockage of mTOR pathways improves the effect of Hedgehog inhibitors in esophageal and head and neck cancers [17,47]. The combination of an SMO inhibitor (sonidegib) with a PI3K inhibitor (NVP-BKM120 or NVP-BEZ235) delayed the development of resistance to SMO inhibitor in medulloblastoma animal models [123]. Similar findings have been described in pancreatic cancer, demonstrating the in vitro benefit of combining mTOR and SMO inhibitors [36], and in biliary tract cancer cells [155]. Another combination that shows promising results in preclinical studies involves EGFR and the HhP. As mentioned above, dual inhibition of EGFR and GLI showed efficient antitumor activity in BCC cell lines of mice with an activated HhP [25].
The combination of targeted therapy against the HhP and cytotoxic agents has also demonstrated promising results in preclinical trials. GANT61 was able to increase the radiosensitivity of renal cell carcinoma cells, and this was also noted in the combination therapy involving GANT61 and hypoxia-inducible factor 2α (HIF2α) inhibitor [15,156]. Similar findings were observed in the HNSCC model, in which an HhP blockade with cyclopamine in chronically irradiated or EGFR-resistant cell lines increased sensitivity to radiotherapy [47] and in non-small cell lung cancer preclinical models, in which vismodegib increase in vivo radiation efficacy [157].
Preclinical evidence that Hedgehog inhibition may sensitize acute myeloid leukemia cells to cytarabine or azacytidine provided rationale for evaluating glasdegib in combination with chemotherapeutic agents. A recent phase 2 trial evaluated the combination of glasdegib with cytarabine and daunorubicin in patients with acute myeloid leukemia or high-risk myelodysplastic syndromes. This trial demonstrated that the combination is feasible and safe with clinical activity. An ongoing phase 3 trial will help clarify the activity of this combination [89]. Itraconazole associated with pemetrexed has been studied in patients with recurrent non-small cell lung cancer. A phase 2 trial was designed to evaluate this combination. The study was stopped early because of the increased use of pemetrexed in the first-line setting, but preliminary results of the 23 enrolled patients had already shown benefit with this combination. At 3 months, 67% of the patients on itraconazole plus pemetrexed were progression-free versus 29% on the control arm of pemetrexed alone. The median PFS was 5.5 months (itraconazole) versus 2.8 months (control). OS was longer in patients receiving itraconazole (median 32 months) versus control (8 months) [85].
However, in some clinical trials, the results have been disappointing. Two phase II trials failed to show the benefit of the combination of HhP inhibitor and chemotherapy. One study failed to show improvement in PFS with vismodegib in combination with standard treatment (FOLFOX or FOLFIRI and bevacizumab) versus standard treatment alone (hazard ratio (HR): 125, p=0.28) in patients with previously untreated metastatic colorectal cancer [70]. The other study evaluated vismodegib as a maintenance treatment in patients with ovarian cancer after second or third complete remission. The study fails to show a significant difference in PFS between vismodegib and placebo (median PFS 7.5 versus 5.8 months, respectively, HR 0.79; 95% confidence interval (CI), 0.46-1.35) [72]. A phase 1b trial evaluated the combination of cetuximab and saridegib for HNSCC patients. One patient experienced a partial response, three stable disease and four disease progression [81]. A pilot trial evaluated the combination of ATO and itraconazole in five patients with metastatic BCC who experienced relapse after SMO inhibitor treatment. This combination was able to reduce GLI1 messenger RNA levels, but the best clinical response was stable disease [158].
7. Conclusion and Future Perspectives
The HhP is essential for embryogenesis and several other physiologic processes, such as wound healing. Recently, this pathway was also related to carcinogenesis in several cancer types. Targeted therapy against the HhP has shown impressive results for BCC, meduloblastoma and rhabdomyosarcoma but disappointing results in other histologies. In addition, resistance to SMO inhibitors in BCC is a reality that needs to be better comprehended. Recently, the elucidation and better understanding of this pathway have led to important discoveries. Multiple cross talk with other important oncogenic pathways opened new avenues to explore targeted therapy and overcome resistance. Several preclinical and animal models have revealed the efficacy and antitumor activity of targeted therapy against HhP components or combination therapy with other targeted agents, which have become promising in the fight against cancer.
Future clinical trials have the potential to add more knowledge to this scenario. Table 2 summarizes the clinical trials that are evaluating HhP inhibitors. This table comprehends single-agent treatments and combination treatments in many types of cancer. For example, a phase II trial is evaluating the combination of sonidegib (SMO inhibitor) with buparlisib (PI3K inhibitor) in patients with locally advanced or metastatic BCC (NCT02303041). Another interesting trial is evaluating the addition of vismodegib to neoadjuvant chemotherapy treatment in breast cancer patients (NCT02694224). Future research will certainly add more insight to the clinical role of this important pathway.
Author Contributions
Conceptualization, D.G. and A.B.; methodology, D.G.; writing—original draft preparation, D.G. and A.B.; writing—review and editing, A.P and G.F.; supervision, G.F. and A.P.
Acknowledgments
The Oncology Center of Hospital Sírio-Libanês supported this work.
Conflicts of Interest
Gustavo dos Santos Fernandes received grants as a consultant or an advisor from Roche. Additionally, he received speaker honorarium and funding for travel, accommodations and other expenses from Roche. He also provided expert testimony on behalf of Novartis. The rest of the authors declare that they have no conflicts of interest. The rest of the authors declare no conflict of interest.
Abbreviations
The following abbreviations are used in this manuscript:
| AKT1 | V-akt murine thymoma viral oncogene homolog 1 |
| ATO | Arsenic trioxide |
| BCC | Basal cell carcinoma |
| BET | Bromodomain and extra terminal |
| BRD4 | Bromodomain-containing protein 4 |
| CK1 | Casein kinase 1 |
| DHH | Desert Hedgehog |
| EFS | Event-free survival |
| EGFR | Epidermal growth factor receptor |
| EMT | Epithelial-to-mesenchymal transition |
| GLI | Glioma-associated oncogene |
| GSK3β | Glycogen synthase kinase 3β |
| HhP | Hedgehog pathway |
| HIF2α | Hypoxia-inducible factor 2α |
| HNSCC | Head and neck squamous cell carcinoma |
| HPI | Hedgehog pathway inhibitors |
| HR | Hazard ratio |
| IHH | Indian Hedgehog |
| IL-1β | Interleukin-1β |
| KLF4 | Kruppel-like factor 4 |
| LSC | Leukemic stem cell |
| mTOR | Mechanistic target of rapamycin |
| NF-κB | Nuclear factor kappa light chain enhancer of activated B cells |
| OCT4 | Octamer binding transcription factor 4 |
| ORR | Overall response rate |
| OS | Overall survival |
| PFS | Progression-free survival |
| PKA | Protein kinase A |
| PTCH1 | Patched1 |
| RFS | Relapse-free survival |
| S6K1 | Ribosomal protein S6 kinase 1 |
| SHH | Sonic Hedgehog |
| SMO | Smoothened |
| SUFU | Suppressor of fused |
| TGF-β | Transforming growth factor-beta |
| TLR | Toll-like receptor |
| TNF-α | Tumor necrosis factor-α |
References
- Gan, G.N.; Jimeno, A. Emerging from their burrow: Hedgehog pathway inhibitors for cancer. Expert. Opin. Investig. Drugs 2016, 25, 1153–1166. [Google Scholar] [CrossRef]
- Ramalho-Santos, M.; Melton, D.A.; McMahon, A.P. Hedgehog signals regulate multiple aspects of gastrointestinal development. Development 2000, 127, 2763–2772. [Google Scholar] [PubMed]
- Jiang, J.; Hui, C.C. Hedgehog signaling in development and cancer. Dev. Cell 2008, 15, 801–812. [Google Scholar] [CrossRef] [PubMed]
- Raffel, C.; Jenkins, R.B.; Frederick, L.; Hebrink, D.; Alderete, B.; Fults, D.W.; James, C.D. Sporadic medulloblastomas contain PTCH mutations. Cancer Res. 1997, 57, 842–845. [Google Scholar] [PubMed]
- Reifenberger, J.; Wolter, M.; Weber, R.G.; Megahed, M.; Ruzicka, T.; Lichter, P.; Reifenberger, G. Missense mutations in SMOH in sporadic basal cell carcinomas of the skin and primitive neuroectodermal tumors of the central nervous system. Cancer Res. 1998, 58, 1798–1803. [Google Scholar] [PubMed]
- Tostar, U.; Malm, C.J.; Meis-Kindblom, J.M.; Kindblom, L.G.; Toftgard, R.; Unden, A.B. Deregulation of the hedgehog signalling pathway: A possible role for the PTCH and SUFU genes in human rhabdomyoma and rhabdomyosarcoma development. J. Pathol. 2006, 208, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Murone, M.; Luoh, S.M.; Ryan, A.; Gu, Q.; Zhang, C.; Bonifas, J.M.; Lam, C.W.; Hynes, M.; Goddard, A.; et al. Activating Smoothened mutations in sporadic basal-cell carcinoma. Nature 1998, 391, 90–92. [Google Scholar] [CrossRef]
- Basset-Seguin, N.; Hauschild, A.; Grob, J.J.; Kunstfeld, R.; Dreno, B.; Mortier, L.; Ascierto, P.A.; Licitra, L.; Dutriaux, C.; Thomas, L.; et al. Vismodegib in patients with advanced basal cell carcinoma (STEVIE): A pre-planned interim analysis of an international, open-label trial. Lancet Oncol. 2015, 16, 729–736. [Google Scholar] [CrossRef]
- Sekulic, A.; Migden, M.R.; Oro, A.E.; Dirix, L.; Lewis, K.D.; Hainsworth, J.D.; Solomon, J.A.; Yoo, S.; Arron, S.T.; Friedlander, P.A.; et al. Efficacy and safety of vismodegib in advanced basal-cell carcinoma. N. Engl. J. Med. 2012, 366, 2171–2179. [Google Scholar] [CrossRef]
- Echelard, Y.; Epstein, D.J.; St-Jacques, B.; Shen, L.; Mohler, J.; McMahon, J.A.; McMahon, A.P. Sonic hedgehog, a member of a family of putative signaling molecules, is implicated in the regulation of CNS polarity. Cell 1993, 75, 1417–1430. [Google Scholar] [CrossRef]
- Chen, M.H.; Wilson, C.W.; Li, Y.J.; Law, K.K.; Lu, C.S.; Gacayan, R.; Zhang, X.; Hui, C.C.; Chuang, P.T. Cilium-independent regulation of Gli protein function by Sufu in Hedgehog signaling is evolutionarily conserved. Genes Dev. 2009, 23, 1910–1928. [Google Scholar] [CrossRef] [PubMed]
- Robbins, D.J.; Fei, D.L.; Riobo, N.A. The Hedgehog signal transduction network. Sci. Signal. 2012, 5, re6. [Google Scholar] [CrossRef] [PubMed]
- Aza-Blanc, P.; Lin, H.Y.; Ruiz i Altaba, A.; Kornberg, T.B. Expression of the vertebrate Gli proteins in Drosophila reveals a distribution of activator and repressor activities. Development 2000, 127, 4293–4301. [Google Scholar]
- Kogerman, P.; Grimm, T.; Kogerman, L.; Krause, D.; Undén, A.B.; Sandstedt, B.; Toftgård, R.; Zaphiropoulos, P.G. Mammalian Suppressor-of-Fused modulates nuclear–cytoplasmic shuttling of GLI-1. Nat. Cell Biol. 1999, 1, 312. [Google Scholar] [CrossRef] [PubMed]
- Gonnissen, A.; Isebaert, S.; Haustermans, K. Targeting the Hedgehog signaling pathway in cancer: Beyond Smoothened. Oncotarget 2015, 6, 13899–13913. [Google Scholar] [CrossRef] [PubMed]
- Teperino, R.; Aberger, F.; Esterbauer, H.; Riobo, N.; Pospisilik, J.A. Canonical and non-canonical Hedgehog signalling and the control of metabolism. Semin. Cell Dev. Biol. 2014, 33, 81–92. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Ding, Q.; Yen, C.J.; Xia, W.; Izzo, J.G.; Lang, J.Y.; Li, C.W.; Hsu, J.L.; Miller, S.A.; Wang, X.; et al. The crosstalk of mTOR/S6K1 and Hedgehog pathways. Cancer Cell 2012, 21, 374–387. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, N.K.; Qu, C.; Kunkalla, K.; Liu, Y.; Vega, F. Transcriptional regulation of serine/threonine protein kinase (AKT) genes by glioma-associated oncogene homolog 1. J. Biol. Chem. 2013, 288, 15390–15401. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Jin, G.; Li, Q.; Wang, Z.; Hu, W.; Li, P.; Li, S.; Wu, H.; Kong, X.; Gao, J.; et al. Hedgehog Signaling Non-Canonical Activated by Pro-Inflammatory Cytokines in Pancreatic Ductal Adenocarcinoma. J. Cancer 2016, 7, 2067–2076. [Google Scholar] [CrossRef] [PubMed]
- Colavito, S.A.; Zou, M.R.; Yan, Q.; Nguyen, D.X.; Stern, D.F. Significance of glioma-associated oncogene homolog 1 (GLI1) expression in claudin-low breast cancer and crosstalk with the nuclear factor kappa-light-chain-enhancer of activated B cells (NFkappaB) pathway. Breast Cancer Res. 2014, 16, 444. [Google Scholar] [CrossRef] [PubMed]
- Dennler, S.; Andre, J.; Alexaki, I.; Li, A.; Magnaldo, T.; ten Dijke, P.; Wang, X.J.; Verrecchia, F.; Mauviel, A. Induction of sonic hedgehog mediators by transforming growth factor-beta: Smad3-dependent activation of Gli2 and Gli1 expression in vitro and in vivo. Cancer Res. 2007, 67, 6981–6986. [Google Scholar] [CrossRef] [PubMed]
- Yoo, Y.A.; Kang, M.H.; Kim, J.S.; Oh, S.C. Sonic hedgehog signaling promotes motility and invasiveness of gastric cancer cells through TGF-beta-mediated activation of the ALK5-Smad 3 pathway. Carcinogenesis 2008, 29, 480–490. [Google Scholar] [CrossRef] [PubMed]
- Fan, Q.; He, M.; Sheng, T.; Zhang, X.; Sinha, M.; Luxon, B.; Zhao, X.; Xie, J. Requirement of TGFbeta signaling for SMO-mediated carcinogenesis. J. Biol. Chem. 2010, 285, 36570–36576. [Google Scholar] [CrossRef]
- Ji, Z.; Mei, F.C.; Xie, J.; Cheng, X. Oncogenic KRAS activates hedgehog signaling pathway in pancreatic cancer cells. J. Biol. Chem. 2007, 282, 14048–14055. [Google Scholar] [CrossRef] [PubMed]
- Schnidar, H.; Eberl, M.; Klingler, S.; Mangelberger, D.; Kasper, M.; Hauser-Kronberger, C.; Regl, G.; Kroismayr, R.; Moriggl, R.; Sibilia, M.; et al. Epidermal growth factor receptor signaling synergizes with Hedgehog/GLI in oncogenic transformation via activation of the MEK/ERK/JUN pathway. Cancer Res. 2009, 69, 1284–1292. [Google Scholar] [CrossRef] [PubMed]
- Yoon, J.W.; Gallant, M.; Lamm, M.L.; Iannaccone, S.; Vieux, K.F.; Proytcheva, M.; Hyjek, E.; Iannaccone, P.; Walterhouse, D. Noncanonical regulation of the Hedgehog mediator GLI1 by c-MYC in Burkitt lymphoma. Mol. Cancer Res. 2013, 11, 604–615. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.H.; Dignam, J.J.; Elizabeth Hammond, M.; Klimowicz, A.C.; Petrillo, S.K.; Magliocco, A.; Jordan, R.; Trotti, A.; Spencer, S.; Cooper, J.S.; et al. Glioma-Associated Oncogene Family Zinc Finger 1 Expression and Metastasis in Patients With Head and Neck Squamous Cell Carcinoma Treated With Radiation Therapy (RTOG 9003). J. Clin. Oncol. 2011, 29, 1326–1334. [Google Scholar] [CrossRef]
- Mori, Y.; Okumura, T.; Tsunoda, S.; Sakai, Y.; Shimada, Y. Gli-1 expression is associated with lymph node metastasis and tumor progression in esophageal squamous cell carcinoma. Oncology 2006, 70, 378–389. [Google Scholar] [CrossRef]
- Ding, Y.-l.; Zhou, Y.; Xiang, L.; Ji, Z.-p.; Luo, Z.-h. Expression of Glioma-associated Oncogene Homolog 1 is Associated with Invasion and Postoperative Liver Metastasis in Colon Cancer. Int. J. Med. Sci. 2012, 9, 334–338. [Google Scholar] [CrossRef]
- Wellbrock, J.; Latuske, E.; Kohler, J.; Wagner, K.; Stamm, H.; Vettorazzi, E.; Vohwinkel, G.; Klokow, M.; Uibeleisen, R.; Ehm, P.; et al. Expression of Hedgehog Pathway Mediator GLI Represents a Negative Prognostic Marker in Human Acute Myeloid Leukemia and Its Inhibition Exerts Antileukemic Effects. Clin. Cancer Res. 2015, 21, 2388–2398. [Google Scholar] [CrossRef]
- Steg, A.D.; Katre, A.A.; Bevis, K.S.; Ziebarth, A.; Dobbin, Z.C.; Shah, M.M.; Alvarez, R.D.; Landen, C.N. Smoothened antagonists reverse taxane resistance in ovarian cancer. Mol. Cancer Ther. 2012, 11, 1587–1597. [Google Scholar] [CrossRef] [PubMed]
- Katoh, Y.; Katoh, M. Hedgehog signaling, epithelial-to-mesenchymal transition and miRNA. Int. J. Mol. Med. 2008, 22, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Ginnebaugh, K.R.; Ahmad, A.; Sarkar, F.H. The therapeutic potential of targeting the epithelial-mesenchymal transition in cancer. Expert Opin. Ther. Targets 2014, 18, 731–745. [Google Scholar] [CrossRef] [PubMed]
- Kurrey, N.K.; Jalgaonkar, S.P.; Joglekar, A.V.; Ghanate, A.D.; Chaskar, P.D.; Doiphode, R.Y.; Bapat, S.A. Snail and slug mediate radioresistance and chemoresistance by antagonizing p53-mediated apoptosis and acquiring a stem-like phenotype in ovarian cancer cells. Stem Cells 2009, 27, 2059–2068. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Zhou, B.P. Snail: More than EMT. Cell Adh. Migr. 2010, 4, 199–203. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.; Nanta, R.; Sharma, J.; Gunewardena, S.; Singh, K.P.; Shankar, S.; Srivastava, R.K. PI3K/AKT/mTOR and sonic hedgehog pathways cooperate together to inhibit human pancreatic cancer stem cell characteristics and tumor growth. Oncotarget 2015, 6, 32039–32060. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Invest. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed]
- Thiery, J.P.; Acloque, H.; Huang, R.Y.; Nieto, M.A. Epithelial-mesenchymal transitions in development and disease. Cell 2009, 139, 871–890. [Google Scholar] [CrossRef] [PubMed]
- Qian, L.W.; Mizumoto, K.; Urashima, T.; Nagai, E.; Maehara, N.; Sato, N.; Nakajima, M.; Tanaka, M. Radiation-induced increase in invasive potential of human pancreatic cancer cells and its blockade by a matrix metalloproteinase inhibitor, CGS27023. Clin. Cancer Res. 2002, 8, 1223–1227. [Google Scholar]
- Wild-Bode, C.; Weller, M.; Rimner, A.; Dichgans, J.; Wick, W. Sublethal irradiation promotes migration and invasiveness of glioma cells: Implications for radiotherapy of human glioblastoma. Cancer Res. 2001, 61, 2744–2750. [Google Scholar]
- Steg, A.D.; Bevis, K.S.; Katre, A.A.; Ziebarth, A.; Dobbin, Z.C.; Alvarez, R.D.; Zhang, K.; Conner, M.; Landen, C.N. Stem cell pathways contribute to clinical chemoresistance in ovarian cancer. Clin. Cancer Res. 2012, 18, 869–881. [Google Scholar] [CrossRef]
- De Boeck, A.; Narine, K.; De Neve, W.; Mareel, M.; Bracke, M.; De Wever, O. Resident and bone marrow-derived mesenchymal stem cells in head and neck squamous cell carcinoma. Oral Oncol. 2010, 46, 336–342. [Google Scholar] [CrossRef] [PubMed]
- Prince, M.E.; Ailles, L.E. Cancer stem cells in head and neck squamous cell cancer. J. Clin. Oncol. 2008, 26, 2871–2875. [Google Scholar] [CrossRef] [PubMed]
- Peacock, C.D.; Wang, Q.; Gesell, G.S.; Corcoran-Schwartz, I.M.; Jones, E.; Kim, J.; Devereux, W.L.; Rhodes, J.T.; Huff, C.A.; Beachy, P.A.; et al. Hedgehog signaling maintains a tumor stem cell compartment in multiple myeloma. Proc. Natl. Acad. Sci. USA 2007, 104, 4048–4053. [Google Scholar] [CrossRef] [PubMed]
- Varnat, F.; Siegl-Cachedenier, I.; Malerba, M.; Gervaz, P.; Ruiz i Altaba, A. Loss of WNT-TCF addiction and enhancement of HH-GLI1 signalling define the metastatic transition of human colon carcinomas. EMBO Mol. Med. 2010, 2, 440–457. [Google Scholar] [CrossRef] [PubMed]
- Clement, V.; Sanchez, P.; de Tribolet, N.; Radovanovic, I.; Altaba, A.R.i. HEDGEHOG-GLI1 signaling regulates human glioma growth, cancer stem cell self-renewal and tumorigenicity. Curr. Biol. 2007, 17, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Gan, G.N.; Eagles, J.; Keysar, S.B.; Wang, G.; Glogowska, M.J.; Altunbas, C.; Anderson, R.T.; Le, P.N.; Morton, J.J.; Frederick, B.; et al. Hedgehog signaling drives radioresistance and stroma-driven tumor repopulation in head and neck squamous cancers. Cancer Res. 2014, 74, 7024–7036. [Google Scholar] [CrossRef]
- Keysar, S.B.; Le, P.N.; Anderson, R.T.; Morton, J.J.; Bowles, D.W.; Paylor, J.J.; Vogler, B.W.; Thorburn, J.; Fernandez, P.; Glogowska, M.J.; et al. Hedgehog signaling alters reliance on EGF receptor signaling and mediates anti-EGFR therapeutic resistance in head and neck cancer. Cancer Res. 2013, 73, 3381–3392. [Google Scholar] [CrossRef]
- Bora-Singhal, N.; Perumal, D.; Nguyen, J.; Chellappan, S. Gli1-Mediated Regulation of Sox2 Facilitates Self-Renewal of Stem-Like Cells and Confers Resistance to EGFR Inhibitors in Non–Small Cell Lung Cancer. Neoplasia 2015, 17, 538–551. [Google Scholar] [CrossRef]
- Ohta, H.; Aoyagi, K.; Fukaya, M.; Danjoh, I.; Ohta, A.; Isohata, N.; Saeki, N.; Taniguchi, H.; Sakamoto, H.; Shimoda, T.; et al. Cross talk between hedgehog and epithelial–mesenchymal transition pathways in gastric pit cells and in diffuse-type gastric cancers. Br. J. Cancer 2009, 100, 389–398. [Google Scholar] [CrossRef]
- Isohata, N.; Aoyagi, K.; Mabuchi, T.; Daiko, H.; Fukaya, M.; Ohta, H.; Ogawa, K.; Yoshida, T.; Sasaki, H. Hedgehog and epithelial-mesenchymal transition signaling in normal and malignant epithelial cells of the esophagus. Int. J. Cancer 2009, 125, 1212–1221. [Google Scholar] [CrossRef] [PubMed]
- Kawamoto, A.; Yokoe, T.; Tanaka, K.; Saigusa, S.; Toiyama, Y.; Yasuda, H.; Inoue, Y.; Miki, C.; Kusunoki, M. Radiation induces epithelial-mesenchymal transition in colorectal cancer cells. Oncol. Rep. 2012, 27, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Amakye, D.; Jagani, Z.; Dorsch, M. Unraveling the therapeutic potential of the Hedgehog pathway in cancer. Nat. Med. 2013, 19, 1410–1422. [Google Scholar] [CrossRef] [PubMed]
- Rizzo, J.M.; Segal, R.J.; Zeitouni, N.C. Combination vismodegib and photodynamic therapy for multiple basal cell carcinomas. Photodiagnosis Photodyn. Ther. 2018, 21, 58–62. [Google Scholar] [CrossRef] [PubMed]
- Basset-Seguin, N.; Hauschild, A.; Kunstfeld, R.; Grob, J.; Dreno, B.; Mortier, L.; Ascierto, P.A.; Licitra, L.; Dutriaux, C.; Thomas, L.; et al. Vismodegib in patients with advanced basal cell carcinoma: Primary analysis of STEVIE, an international, open-label trial. Eur. J. Cancer 2017, 86, 334–348. [Google Scholar] [CrossRef] [PubMed]
- Abou-Alfa, G.K.; Lewis, L.D.; LoRusso, P.; Maitland, M.; Chandra, P.; Cheeti, S.; Colburn, D.; Williams, S.; Simmons, B.; Graham, R.A. Pharmacokinetics and safety of vismodegib in patients with advanced solid malignancies and hepatic impairment. Cancer Chemother. Pharmacol. 2017, 80, 29–36. [Google Scholar] [CrossRef]
- Sekulic, A.; Migden, M.R.; Basset-Seguin, N.; Garbe, C.; Gesierich, A.; Lao, C.D.; Miller, C.; Mortier, L.; Murrell, D.F.; Hamid, O.; et al. Long-term safety and efficacy of vismodegib in patients with advanced basal cell carcinoma: Final update of the pivotal ERIVANCE BCC study. BMC Cancer 2017, 17, 332. [Google Scholar] [CrossRef]
- Dreno, B.; Kunstfeld, R.; Hauschild, A.; Fosko, S.; Zloty, D.; Labeille, B.; Grob, J.J.; Puig, S.; Gilberg, F.; Bergstrom, D.; et al. Two intermittent vismodegib dosing regimens in patients with multiple basal-cell carcinomas (MIKIE): A randomised, regimen-controlled, double-blind, phase 2 trial. Lancet Oncol. 2017, 18, 404–412. [Google Scholar] [CrossRef]
- Tang, J.Y.; Ally, M.S.; Chanana, A.M.; Mackay-Wiggan, J.M.; Aszterbaum, M.; Lindgren, J.A.; Ulerio, G.; Rezaee, M.R.; Gildengorin, G.; Marji, J.; et al. Inhibition of the hedgehog pathway in patients with basal-cell nevus syndrome: Final results from the multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet Oncol. 2016, 17, 1720–1731. [Google Scholar] [CrossRef]
- Maughan, B.L.; Suzman, D.L.; Luber, B.; Wang, H.; Glavaris, S.; Hughes, R.; Sullivan, R.; Harb, R.; Boudadi, K.; Paller, C.; et al. Pharmacodynamic study of the oral hedgehog pathway inhibitor, vismodegib, in patients with metastatic castration-resistant prostate cancer. Cancer Chemother. Pharmacol. 2016, 78, 1297–1304. [Google Scholar] [CrossRef]
- Kwon, G.P.; Ally, M.S.; Bailey-Healy, I.; Oro, A.E.; Kim, J.; Chang, A.L.; Aasi, S.; Tang, J.Y. Update to an open-label clinical trial of vismodegib as neoadjuvant before surgery for high-risk basal cell carcinoma (BCC). J. Am. Acad. Dermatol. 2016, 75, 213–215. [Google Scholar] [CrossRef]
- Belani, C.P.; Dahlberg, S.E.; Rudin, C.M.; Fleisher, M.; Chen, H.X.; Takebe, N.; Velasco, M.R., Jr.; Tester, W.J.; Sturtz, K.; Hann, C.L.; et al. Vismodegib or cixutumumab in combination with standard chemotherapy for patients with extensive-stage small cell lung cancer: A trial of the ECOG-ACRIN Cancer Research Group (E1508). Cancer 2016, 122, 2371–2378. [Google Scholar] [CrossRef]
- Houot, R.; Soussain, C.; Tilly, H.; Haioun, C.; Thieblemont, C.; Casasnovas, O.; Bouabdallah, K.; Morschhauser, F.; Le Gouill, S.; Salles, G.A.; et al. Inhibition of Hedgehog signaling for the treatment of lymphoma and CLL: A phase II study from the LYSA. Ann. Oncol. 2016, 27, 1349–1350. [Google Scholar] [CrossRef] [PubMed]
- Catenacci, D.V.; Junttila, M.R.; Karrison, T.; Bahary, N.; Horiba, M.N.; Nattam, S.R.; Marsh, R.; Wallace, J.; Kozloff, M.; Rajdev, L.; et al. Randomized phase Ib/II study of gemcitabine plus placebo or vismodegib, a hedgehog pathway inhibitor, in patients with metastatic pancreatic cancer. J. Clin. Oncol. 2015, 33, 4284–4292. [Google Scholar] [CrossRef] [PubMed]
- Sofen, H.; Gross, K.G.; Goldberg, L.H.; Sharata, H.; Hamilton, T.K.; Egbert, B.; Lyons, B.; Hou, J.; Caro, I. A phase II, multicenter, open-label, 3-cohort trial evaluating the efficacy and safety of vismodegib in operable basal cell carcinoma. J. Am. Acad. Dermatol. 2015, 73, 99–105. [Google Scholar] [CrossRef]
- Kim, E.J.; Sahai, V.; Abel, E.V.; Griffith, K.A.; Greenson, J.K.; Takebe, N.; Khan, G.N.; Blau, J.L.; Craig, R.; Balis, U.G.; et al. Pilot clinical trial of hedgehog pathway inhibitor GDC-0449 (vismodegib) in combination with gemcitabine in patients with metastatic pancreatic adenocarcinoma. Clin. Cancer Res. 2014, 20, 5937–5945. [Google Scholar] [CrossRef]
- Italiano, A.; Le Cesne, A.; Bellera, C.; Piperno-Neumann, S.; Duffaud, F.; Penel, N.; Cassier, P.; Domont, J.; Takebe, N.; Kind, M.; et al. GDC-0449 in patients with advanced chondrosarcomas: A French sarcoma group/US and French national cancer institute single-arm phase II collaborative study. Ann. Oncol. 2013, 24, 2922–2926. [Google Scholar] [CrossRef]
- Gajjar, A.; Stewart, C.F.; Ellison, D.W.; Kaste, S.; Kun, L.E.; Packer, R.J.; Goldman, S.; Chintagumpala, M.; Wallace, D.; Takebe, N.; et al. Phase I study of vismodegib in children with recurrent or refractory medulloblastoma: A pediatric brain tumor consortium study. Clin. Cancer Res. 2013, 19, 6305–6312. [Google Scholar] [CrossRef]
- Thacker, C.A.; Weiss, G.J.; Tibes, R.; Blaydorn, L.; Downhour, M.; White, E.; Baldwin, J.; Hoff, D.D.; Korn, R.L. 18-FDG PET/CT assessment of basal cell carcinoma with vismodegib. Cancer Med. 2012, 1, 230–236. [Google Scholar] [CrossRef]
- Berlin, J.; Bendell, J.C.; Hart, L.L.; Firdaus, I.; Gore, I.; Hermann, R.C.; Mulcahy, M.F.; Zalupski, M.M.; Mackey, H.M.; Yauch, R.L.; et al. A randomized phase II trial of vismodegib versus placebo with FOLFOX or FOLFIRI and bevacizumab in patients with previously untreated metastatic colorectal cancer. Clin. Cancer Res. 2013, 19, 258–267. [Google Scholar] [CrossRef]
- LoRusso, P.M.; Piha-Paul, S.A.; Mita, M.; Colevas, A.D.; Malhi, V.; Colburn, D.; Yin, M.; Low, J.A.; Graham, R.A. Co-administration of vismodegib with rosiglitazone or combined oral contraceptive in patients with locally advanced or metastatic solid tumors: A pharmacokinetic assessment of drug-drug interaction potential. Cancer Chemother. Pharmacol. 2013, 71, 193–202. [Google Scholar] [CrossRef] [PubMed]
- Kaye, S.B.; Fehrenbacher, L.; Holloway, R.; Amit, A.; Karlan, B.; Slomovitz, B.; Sabbatini, P.; Fu, L.; Yauch, R.L.; Chang, I.; et al. A phase II, randomized, placebo-controlled study of vismodegib as maintenance therapy in patients with ovarian cancer in second or third complete remission. Clin. Cancer Res. 2012, 18, 6509–6518. [Google Scholar] [CrossRef] [PubMed]
- Von Hoff, D.D.; LoRusso, P.M.; Rudin, C.M.; Reddy, J.C.; Yauch, R.L.; Tibes, R.; Weiss, G.J.; Borad, M.J.; Hann, C.L.; Brahmer, J.R.; et al. Inhibition of the hedgehog pathway in advanced basal-cell carcinoma. N. Engl. J. Med. 2009, 361, 1164–1172. [Google Scholar] [CrossRef] [PubMed]
- Lear, J.T.; Migden, M.R.; Lewis, K.D.; Chang, A.L.S.; Guminski, A.; Gutzmer, R.; Dirix, L.; Combemale, P.; Stratigos, A.; Plummer, R.; et al. Long-term efficacy and safety of sonidegib in patients with locally advanced and metastatic basal cell carcinoma: 30-month analysis of the randomized phase 2 BOLT study. J. Eur. Acad. Dermatol. Venereol. 2018, 32, 372–381. [Google Scholar] [CrossRef] [PubMed]
- Kieran, M.W.; Chisholm, J.; Casanova, M.; Brandes, A.A.; Aerts, I.; Bouffet, E.; Bailey, S.; Leary, S.; MacDonald, T.J.; Mechinaud, F.; et al. Phase I study of oral sonidegib (LDE225) in pediatric brain and solid tumors and a phase II study in children and adults with relapsed medulloblastoma. Neuro Oncol. 2017, 19, 1542–1552. [Google Scholar] [CrossRef] [PubMed]
- Stathis, A.; Hess, D.; von Moos, R.; Homicsko, K.; Griguolo, G.; Joerger, M.; Mark, M.; Ackermann, C.J.; Allegrini, S.; Catapano, C.V.; et al. Phase I trial of the oral smoothened inhibitor sonidegib in combination with paclitaxel in patients with advanced solid tumors. Invest. New Drugs 2017, 35, 766–772. [Google Scholar] [CrossRef] [PubMed]
- Minami, H.; Ando, Y.; Ma, B.B.; Hsiang Lee, J.; Momota, H.; Fujiwara, Y.; Li, L.; Fukino, K.; Ito, K.; Tajima, T.; et al. Phase I, multicenter, open-label, dose-escalation study of sonidegib in Asian patients with advanced solid tumors. Cancer Sci. 2016, 107, 1477–1483. [Google Scholar] [CrossRef] [PubMed]
- Pietanza, M.C.; Litvak, A.M.; Varghese, A.M.; Krug, L.M.; Fleisher, M.; Teitcher, J.B.; Holodny, A.I.; Sima, C.S.; Woo, K.M.; Ng, K.K.; et al. A phase I trial of the Hedgehog inhibitor, sonidegib (LDE225), in combination with etoposide and cisplatin for the initial treatment of extensive stage small cell lung cancer. Lung Cancer 2016, 99, 23–30. [Google Scholar] [CrossRef]
- Ko, A.H.; LoConte, N.; Tempero, M.A.; Walker, E.J.; Kate Kelley, R.; Lewis, S.; Chang, W.C.; Kantoff, E.; Vannier, M.W.; Catenacci, D.V.; et al. A Phase I Study of FOLFIRINOX Plus IPI-926, a Hedgehog Pathway Inhibitor, for Advanced Pancreatic Adenocarcinoma. Pancreas 2016, 45, 370–375. [Google Scholar] [CrossRef]
- Sasaki, K.; Gotlib, J.R.; Mesa, R.A.; Newberry, K.J.; Ravandi, F.; Cortes, J.E.; Kelly, P.; Kutok, J.L.; Kantarjian, H.M.; Verstovsek, S. Phase II evaluation of IPI-926, an oral Hedgehog inhibitor, in patients with myelofibrosis. Leuk. Lymphoma 2015, 56, 2092–2097. [Google Scholar] [CrossRef]
- Bowles, D.W.; Keysar, S.B.; Eagles, J.R.; Wang, G.; Glogowska, M.J.; McDermott, J.D.; Le, P.N.; Gao, D.; Ray, C.E.; Rochon, P.J.; et al. A pilot study of cetuximab and the hedgehog inhibitor IPI-926 in recurrent/metastatic head and neck squamous cell carcinoma. Oral Oncol. 2016, 53, 74–79. [Google Scholar] [CrossRef] [PubMed]
- Jimeno, A.; Weiss, G.J.; Miller, W.H., Jr.; Gettinger, S.; Eigl, B.J.; Chang, A.L.; Dunbar, J.; Devens, S.; Faia, K.; Skliris, G.; et al. Phase I study of the Hedgehog pathway inhibitor IPI-926 in adult patients with solid tumors. Clin. Cancer Res. 2013, 19, 2766–2774. [Google Scholar] [CrossRef] [PubMed]
- Lee, M.; Hong, H.; Kim, W.; Zhang, L.; Friedlander, T.W.; Fong, L.; Lin, A.M.; Small, E.J.; Wei, X.X.; Rodvelt, T.J.; et al. Itraconazole as a Noncastrating Treatment for Biochemically Recurrent Prostate Cancer: A Phase 2 Study. Clin. Genitourin. Cancer 2018, 7, e92–e96. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.J.; Kim, J.; Spaunhurst, K.; Montoya, J.; Khodosh, R.; Chandra, K.; Fu, T.; Gilliam, A.; Molgo, M.; Beachy, P.A.; et al. Open-label, exploratory phase II trial of oral itraconazole for the treatment of basal cell carcinoma. J. Clin. Oncol. 2014, 32, 745–751. [Google Scholar] [CrossRef] [PubMed]
- Rudin, C.M.; Brahmer, J.R.; Juergens, R.A.; Hann, C.L.; Ettinger, D.S.; Sebree, R.; Smith, R.; Aftab, B.T.; Huang, P.; Liu, J.O. Phase 2 study of pemetrexed and itraconazole as second-line therapy for metastatic nonsquamous non-small-cell lung cancer. J. Thorac. Oncol. 2013, 8, 619–623. [Google Scholar] [CrossRef] [PubMed]
- Antonarakis, E.S.; Heath, E.I.; Smith, D.C.; Rathkopf, D.; Blackford, A.L.; Danila, D.C.; King, S.; Frost, A.; Ajiboye, A.S.; Zhao, M.; et al. Repurposing itraconazole as a treatment for advanced prostate cancer: A noncomparative randomized phase II trial in men with metastatic castration-resistant prostate cancer. Oncologist 2013, 18, 163–173. [Google Scholar] [CrossRef] [PubMed]
- Bendell, J.; Andre, V.; Ho, A.; Kudchadkar, R.; Migden, M.; Infante, J.; Tiu, R.V.; Pitou, C.; Tucker, T.; Brail, L.; et al. Phase I Study of LY2940680, a Smo Antagonist, in Patients with Advanced Cancer Including Treatment-Naive and Previously Treated Basal Cell Carcinoma. Clin. Cancer Res. 2018, 24, 2082–2091. [Google Scholar] [CrossRef]
- Savona, M.R.; Pollyea, D.A.; Stock, W.; Oehler, V.G.; Schroeder, M.A.; Lancet, J.; McCloskey, J.; Kantarjian, H.M.; Ma, W.W.; Shaik, M.N.; et al. Phase Ib Study of Glasdegib, a Hedgehog Pathway Inhibitor, in Combination with Standard Chemotherapy in Patients with AML or High-Risk MDS. Clin. Cancer Res. 2018, 24, 2294–2303. [Google Scholar] [CrossRef]
- Cortes, J.E.; Douglas Smith, B.; Wang, E.S.; Merchant, A.; Oehler, V.G.; Arellano, M.; DeAngelo, D.J.; Pollyea, D.A.; Sekeres, M.A.; Robak, T.; et al. Glasdegib in combination with cytarabine and daunorubicin in patients with AML or high-risk MDS: Phase 2 study results. Am. J. Hematol. 2018, 93, 1301–1310. [Google Scholar] [CrossRef]
- Wagner, A.J.; Messersmith, W.A.; Shaik, M.N.; Li, S.; Zheng, X.; McLachlan, K.R.; Cesari, R.; Courtney, R.; Levin, W.J.; El-Khoueiry, A.B. A phase I study of PF-04449913, an oral hedgehog inhibitor, in patients with advanced solid tumors. Clin. Cancer Res. 2015, 21, 1044–1051. [Google Scholar] [CrossRef]
- Incardona, J.P.; Gaffield, W.; Kapur, R.P.; Roelink, H. The teratogenic Veratrum alkaloid cyclopamine inhibits sonic hedgehog signal transduction. Development 1998, 125, 3553–3562. [Google Scholar] [PubMed]
- Cooper, M.K.; Porter, J.A.; Young, K.E.; Beachy, P.A. Teratogen-mediated inhibition of target tissue response to Shh signaling. Science 1998, 280, 1603–1607. [Google Scholar] [CrossRef] [PubMed]
- Taipale, J.; Chen, J.K.; Cooper, M.K.; Wang, B.; Mann, R.K.; Milenkovic, L.; Scott, M.P.; Beachy, P.A. Effects of oncogenic mutations in Smoothened and Patched can be reversed by cyclopamine. Nature 2000, 406, 1005–1009. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, P.; Ruiz i Altaba, A. In vivo inhibition of endogenous brain tumors through systemic interference of Hedgehog signaling in mice. Mech. Dev. 2005, 122, 223–230. [Google Scholar] [CrossRef] [PubMed]
- Karhadkar, S.S.; Bova, G.S.; Abdallah, N.; Dhara, S.; Gardner, D.; Maitra, A.; Isaacs, J.T.; Berman, D.M.; Beachy, P.A. Hedgehog signalling in prostate regeneration, neoplasia and metastasis. Nature 2004, 431, 707–712. [Google Scholar] [CrossRef] [PubMed]
- Berman, D.M.; Karhadkar, S.S.; Maitra, A.; Montes De Oca, R.; Gerstenblith, M.R.; Briggs, K.; Parker, A.R.; Shimada, Y.; Eshleman, J.R.; Watkins, D.N.; et al. Widespread requirement for Hedgehog ligand stimulation in growth of digestive tract tumours. Nature 2003, 425, 846–851. [Google Scholar] [CrossRef] [PubMed]
- Watkins, D.N.; Berman, D.M.; Burkholder, S.G.; Wang, B.; Beachy, P.A.; Baylin, S.B. Hedgehog signalling within airway epithelial progenitors and in small-cell lung cancer. Nature 2003, 422, 313–317. [Google Scholar] [CrossRef]
- Fan, Q.; Gu, D.; He, M.; Liu, H.; Sheng, T.; Xie, G.; Li, C.X.; Zhang, X.; Wainwright, B.; Garrossian, A.; et al. Tumor shrinkage by cyclopamine tartrate through inhibiting hedgehog signaling. Chin. J. Cancer 2011, 30, 472–481. [Google Scholar] [CrossRef]
- Alam, M.M.; Sohoni, S.; Kalainayakan, S.P.; Garrossian, M.; Zhang, L. Cyclopamine tartrate, an inhibitor of Hedgehog signaling, strongly interferes with mitochondrial function and suppresses aerobic respiration in lung cancer cells. BMC Cancer 2016, 16, 150. [Google Scholar] [CrossRef]
- Di Magno, L.; Coni, S.; Di Marcotullio, L.; Canettieri, G. Digging a hole under Hedgehog: Downstream inhibition as an emerging anticancer strategy. Biochim. Biophys. Acta 2015, 1856, 62–72. [Google Scholar] [CrossRef]
- Mohan, S.V.; Chang, J.; Li, S.; Henry, A.S.; Wood, D.J.; Chang, A.L. Increased Risk of Cutaneous Squamous Cell Carcinoma After Vismodegib Therapy for Basal Cell Carcinoma. JAMA Dermatol. 2016, 152, 527–532. [Google Scholar] [CrossRef] [PubMed]
- Bhutani, T.; Abrouk, M.; Sima, C.S.; Sadetsky, N.; Hou, J.; Caro, I.; Chren, M.M.; Arron, S.T. Risk of cutaneous squamous cell carcinoma after treatment of basal cell carcinoma with vismodegib. J. Am. Acad. Dermatol. 2017, 77, 713–718. [Google Scholar] [CrossRef] [PubMed]
- Chang, A.L.; Oro, A.E. Initial assessment of tumor regrowth after vismodegib in advanced Basal cell carcinoma. Arch. Dermatol. 2012, 148, 1324–1325. [Google Scholar] [CrossRef] [PubMed]
- Rodon, J.; Tawbi, H.A.; Thomas, A.L.; Stoller, R.G.; Turtschi, C.P.; Baselga, J.; Sarantopoulos, J.; Mahalingam, D.; Shou, Y.; Moles, M.A.; et al. A phase I, multicenter, open-label, first-in-human, dose-escalation study of the oral smoothened inhibitor Sonidegib (LDE225) in patients with advanced solid tumors. Clin. Cancer Res. 2014, 20, 1900–1909. [Google Scholar] [CrossRef] [PubMed]
- Xie, P.; Lefrancois, P. Efficacy, safety, and comparison of sonic hedgehog inhibitors in basal cell carcinomas: A systematic review and meta-analysis. J. Am. Acad. Dermatol. 2018, 79, 1089–1100. [Google Scholar] [CrossRef]
- Danial, C.; Sarin, K.Y.; Oro, A.E.; Chang, A.L. An Investigator-Initiated Open-Label Trial of Sonidegib in Advanced Basal Cell Carcinoma Patients Resistant to Vismodegib. Clin. Cancer Res. 2016, 22, 1325–1329. [Google Scholar] [CrossRef] [PubMed]
- Della Corte, C.M.; Bellevicine, C.; Vicidomini, G.; Vitagliano, D.; Malapelle, U.; Accardo, M.; Fabozzi, A.; Fiorelli, A.; Fasano, M.; Papaccio, F.; et al. SMO gene amplification and activation of the hedgehog pathway as novel mechanisms of resistance to anti-epidermal growth factor receptor drugs in human lung cancer. Clin. Cancer Res. 2015, 21, 4686–4697. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Rodova, M.; Nanta, R.; Meeker, D.; Van Veldhuizen, P.J.; Srivastava, R.K.; Shankar, S. NPV-LDE-225 (Erismodegib) inhibits epithelial mesenchymal transition and self-renewal of glioblastoma initiating cells by regulating miR-21, miR-128, and miR-200. Neuro Oncol. 2013, 15, 691–706. [Google Scholar] [CrossRef]
- D’Amato, C.; Rosa, R.; Marciano, R.; D’Amato, V.; Formisano, L.; Nappi, L.; Raimondo, L.; Di Mauro, C.; Servetto, A.; Fulciniti, F.; et al. Inhibition of Hedgehog signalling by NVP-LDE225 (Erismodegib) interferes with growth and invasion of human renal cell carcinoma cells. Br. J. Cancer 2014, 111, 1168–1179. [Google Scholar] [CrossRef]
- Olive, K.P.; Jacobetz, M.A.; Davidson, C.J.; Gopinathan, A.; McIntyre, D.; Honess, D.; Madhu, B.; Goldgraben, M.A.; Caldwell, M.E.; Allard, D.; et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science 2009, 324, 1457–1461. [Google Scholar] [CrossRef]
- Wang, C.; Zhu, M.; Lu, X.; Wang, H.; Zhao, W.; Zhang, X.; Dong, X. Synthesis and evaluation of novel dimethylpyridazine derivatives as hedgehog signaling pathway inhibitors. Bioorg. Med. Chem. 2018, 26, 3308–3320. [Google Scholar] [CrossRef] [PubMed]
- Munchhof, M.J.; Li, Q.; Shavnya, A.; Borzillo, G.V.; Boyden, T.L.; Jones, C.S.; LaGreca, S.D.; Martinez-Alsina, L.; Patel, N.; Pelletier, K.; et al. Discovery of PF-04449913, a Potent and Orally Bioavailable Inhibitor of Smoothened. ACS Med. Chem. Lett. 2012, 3, 106–111. [Google Scholar] [CrossRef] [PubMed]
- Singh, M.; Chaudhry, P.; Merchant, A.A. Primary cilia are present on human blood and bone marrow cells and mediate Hedgehog signaling. Exp. Hematol. 2016, 44, 1181–1187 e1182. [Google Scholar] [CrossRef] [PubMed]
- Sadarangani, A.; Pineda, G.; Lennon, K.M.; Chun, H.J.; Shih, A.; Schairer, A.E.; Court, A.C.; Goff, D.J.; Prashad, S.L.; Geron, I.; et al. GLI2 inhibition abrogates human leukemia stem cell dormancy. J. Transl. Med. 2015, 13, 98. [Google Scholar] [CrossRef] [PubMed]
- Fukushima, N.; Minami, Y.; Kakiuchi, S.; Kuwatsuka, Y.; Hayakawa, F.; Jamieson, C.; Kiyoi, H.; Naoe, T. Small-molecule Hedgehog inhibitor attenuates the leukemia-initiation potential of acute myeloid leukemia cells. Cancer Sci. 2016, 107, 1422–1429. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Tang, J.Y.; Gong, R.; Kim, J.; Lee, J.J.; Clemons, K.V.; Chong, C.R.; Chang, K.S.; Fereshteh, M.; Gardner, D.; et al. Itraconazole, a commonly used antifungal that inhibits Hedgehog pathway activity and cancer growth. Cancer Cell 2010, 17, 388–399. [Google Scholar] [CrossRef] [PubMed]
- Tsubamoto, H.; Sonoda, T.; Yamasaki, M.; Inoue, K. Impact of combination chemotherapy with itraconazole on survival for patients with recurrent or persistent ovarian clear cell carcinoma. Anticancer Res. 2014, 34, 2007–2014. [Google Scholar] [PubMed]
- Tsubamoto, H.; Sonoda, T.; Inoue, K. Impact of itraconazole on the survival of heavily pre-treated patients with triple-negative breast cancer. Anticancer Res. 2014, 34, 3839–3844. [Google Scholar]
- Tsubamoto, H.; Sonoda, T.; Ikuta, S.; Tani, S.; Inoue, K.; Yamanaka, N. Combination chemotherapy with itraconazole for treating metastatic pancreatic cancer in the second-line or additional setting. Anticancer Res. 2015, 35, 4191–4196. [Google Scholar]
- Tsubamoto, H.; Sonoda, T.; Ikuta, S.; Tani, S.; Inoue, K.; Yamanaka, N. impact of itraconazole after first-line chemotherapy on survival of patients with metastatic biliary tract cancer. Anticancer Res. 2015, 35, 4923–4927. [Google Scholar]
- Yauch, R.L.; Dijkgraaf, G.J.; Alicke, B.; Januario, T.; Ahn, C.P.; Holcomb, T.; Pujara, K.; Stinson, J.; Callahan, C.A.; Tang, T.; et al. Smoothened mutation confers resistance to a Hedgehog pathway inhibitor in medulloblastoma. Science 2009, 326, 572–574. [Google Scholar] [CrossRef] [PubMed]
- Dijkgraaf, G.J.; Alicke, B.; Weinmann, L.; Januario, T.; West, K.; Modrusan, Z.; Burdick, D.; Goldsmith, R.; Robarge, K.; Sutherlin, D.; et al. Small molecule inhibition of GDC-0449 refractory smoothened mutants and downstream mechanisms of drug resistance. Cancer Res. 2011, 71, 435–444. [Google Scholar] [CrossRef] [PubMed]
- Buonamici, S.; Williams, J.; Morrissey, M.; Wang, A.; Guo, R.; Vattay, A.; Hsiao, K.; Yuan, J.; Green, J.; Ospina, B.; et al. Interfering with resistance to smoothened antagonists by inhibition of the PI3K pathway in medulloblastoma. Sci. Transl. Med. 2010, 2, 51ra70. [Google Scholar] [CrossRef] [PubMed]
- Lauth, M.; Bergstrom, A.; Shimokawa, T.; Toftgard, R. Inhibition of GLI-mediated transcription and tumor cell growth by small-molecule antagonists. Proc. Natl. Acad. Sci. USA 2007, 104, 8455–8460. [Google Scholar] [CrossRef] [PubMed]
- Huang, L.; Walter, V.; Hayes, D.N.; Onaitis, M. Hedgehog-GLI signaling inhibition suppresses tumor growth in squamous lung cancer. Clin. Cancer Res. 2014, 20, 1566–1575. [Google Scholar] [CrossRef] [PubMed]
- Latuske, E.M.; Stamm, H.; Klokow, M.; Vohwinkel, G.; Muschhammer, J.; Bokemeyer, C.; Jucker, M.; Kebenko, M.; Fiedler, W.; Wellbrock, J. Combined inhibition of GLI and FLT3 signaling leads to effective anti-leukemic effects in human acute myeloid leukemia. Oncotarget 2017, 8, 29187–29201. [Google Scholar] [CrossRef]
- Srivastava, R.K.; Kaylani, S.Z.; Edrees, N.; Li, C.; Talwelkar, S.S.; Xu, J.; Palle, K.; Pressey, J.G.; Athar, M. GLI inhibitor GANT-61 diminishes embryonal and alveolar rhabdomyosarcoma growth by inhibiting Shh/AKT-mTOR axis. Oncotarget 2014, 5, 12151–12165. [Google Scholar] [CrossRef]
- Wickstrom, M.; Dyberg, C.; Shimokawa, T.; Milosevic, J.; Baryawno, N.; Fuskevag, O.M.; Larsson, R.; Kogner, P.; Zaphiropoulos, P.G.; Johnsen, J.I. Targeting the hedgehog signal transduction pathway at the level of GLI inhibits neuroblastoma cell growth in vitro and in vivo. Int. J. Cancer 2013, 132, 1516–1524. [Google Scholar] [CrossRef]
- Benvenuto, M.; Masuelli, L.; De Smaele, E.; Fantini, M.; Mattera, R.; Cucchi, D.; Bonanno, E.; Di Stefano, E.; Frajese, G.V.; Orlandi, A.; et al. In vitro and in vivo inhibition of breast cancer cell growth by targeting the Hedgehog/GLI pathway with SMO (GDC-0449) or GLI (GANT-61) inhibitors. Oncotarget 2016, 7, 9250–9270. [Google Scholar] [CrossRef]
- Mazumdar, T.; Sandhu, R.; Qadan, M.; DeVecchio, J.; Magloire, V.; Agyeman, A.; Li, B.; Houghton, J.A. Hedgehog signaling regulates telomerase reverse transcriptase in human cancer cells. PLoS ONE 2013, 8, e75253. [Google Scholar] [CrossRef]
- Yan, M.; Wang, L.; Zuo, H.; Zhang, Z.; Chen, W.; Mao, L.; Zhang, P. HH/GLI signalling as a new therapeutic target for patients with oral squamous cell carcinoma. Oral Oncol. 2011, 47, 504–509. [Google Scholar] [CrossRef] [PubMed]
- Yan, R.; Peng, X.; Yuan, X.; Huang, D.; Chen, J.; Lu, Q.; Lv, N.; Luo, S. Suppression of growth and migration by blocking the Hedgehog signaling pathway in gastric cancer cells. Cell. Oncol. 2013, 36, 421–435. [Google Scholar] [CrossRef] [PubMed]
- Mazumdar, T.; DeVecchio, J.; Shi, T.; Jones, J.; Agyeman, A.; Houghton, J.A. Hedgehog signaling drives cellular survival in human colon carcinoma cells. Cancer Res. 2011, 71, 1092–1102. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Rodova, M.; Roy, S.K.; Sharma, J.; Singh, K.P.; Srivastava, R.K.; Shankar, S. GANT-61 inhibits pancreatic cancer stem cell growth in vitro and in NOD/SCID/IL2R gamma null mice xenograft. Cancer Lett. 2013, 330, 22–32. [Google Scholar] [CrossRef] [PubMed]
- Samarzija, I.; Beard, P. Hedgehog pathway regulators influence cervical cancer cell proliferation, survival and migration. Biochem. Biophys. Res. Commun. 2012, 425, 64–69. [Google Scholar] [CrossRef]
- Santini, R.; Pietrobono, S.; Pandolfi, S.; Montagnani, V.; D’Amico, M.; Penachioni, J.Y.; Vinci, M.C.; Borgognoni, L.; Stecca, B. SOX2 regulates self-renewal and tumorigenicity of human melanoma-initiating cells. Oncogene 2014, 33, 4697–4708. [Google Scholar] [CrossRef] [PubMed]
- Yoshimoto, A.N.; Bernardazzi, C.; Carneiro, A.J.; Elia, C.C.; Martinusso, C.A.; Ventura, G.M.; Castelo-Branco, M.T.; de Souza, H.S. Hedgehog pathway signaling regulates human colon carcinoma HT-29 epithelial cell line apoptosis and cytokine secretion. PLoS ONE 2012, 7, e45332. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.Y.; Chen, Z. Differentiation and apoptosis induction therapy in acute promyelocytic leukaemia. Lancet Oncol. 2000, 1, 101–106. [Google Scholar] [CrossRef]
- Kim, J.; Lee, J.J.; Kim, J.; Gardner, D.; Beachy, P.A. Arsenic antagonizes the Hedgehog pathway by preventing ciliary accumulation and reducing stability of the Gli2 transcriptional effector. Proc. Natl. Acad. Sci. USA 2010, 107, 13432–13437. [Google Scholar] [CrossRef]
- Noble, P.W.; Albera, C.; Bradford, W.Z.; Costabel, U.; Glassberg, M.K.; Kardatzke, D.; King, T.E., Jr.; Lancaster, L.; Sahn, S.A.; Szwarcberg, J.; et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): Two randomised trials. Lancet 2011, 377, 1760–1769. [Google Scholar] [CrossRef]
- Didiasova, M.; Singh, R.; Wilhelm, J.; Kwapiszewska, G.; Wujak, L.; Zakrzewicz, D.; Schaefer, L.; Markart, P.; Seeger, W.; Lauth, M.; et al. Pirfenidone exerts antifibrotic effects through inhibition of GLI transcription factors. FASEB J. 2017, 31, 1916–1928. [Google Scholar] [CrossRef] [PubMed]
- Polydorou, C.; Mpekris, F.; Papageorgis, P.; Voutouri, C.; Stylianopoulos, T. Pirfenidone normalizes the tumor microenvironment to improve chemotherapy. Oncotarget 2017, 8, 24506–24517. [Google Scholar] [CrossRef]
- Iwata, T.; Yoshida, S.; Fujiwara, T.; Wada, H.; Nakajima, T.; Suzuki, H.; Yoshino, I. Effect of Perioperative Pirfenidone Treatment in Lung Cancer Patients With Idiopathic Pulmonary Fibrosis. Ann. Thorac. Surg. 2016, 102, 1905–1910. [Google Scholar] [CrossRef]
- Kozono, S.; Ohuchida, K.; Eguchi, D.; Ikenaga, N.; Fujiwara, K.; Cui, L.; Mizumoto, K.; Tanaka, M. Pirfenidone inhibits pancreatic cancer desmoplasia by regulating stellate cells. Cancer Res. 2013, 73, 2345–2356. [Google Scholar] [CrossRef] [PubMed]
- Mediavilla-Varela, M.; Boateng, K.; Noyes, D.; Antonia, S.J. The anti-fibrotic agent pirfenidone synergizes with cisplatin in killing tumor cells and cancer-associated fibroblasts. BMC Cancer 2016, 16, 176. [Google Scholar] [CrossRef] [PubMed]
- Burghardt, I.; Tritschler, F.; Opitz, C.A.; Frank, B.; Weller, M.; Wick, W. Pirfenidone inhibits TGF-beta expression in malignant glioma cells. Biochem. Biophys. Res. Commun. 2007, 354, 542–547. [Google Scholar] [CrossRef]
- Zou, W.J.; Huang, Z.; Jiang, T.P.; Shen, Y.P.; Zhao, A.S.; Zhou, S.; Zhang, S. Pirfenidone Inhibits Proliferation and Promotes Apoptosis of Hepatocellular Carcinoma Cells by Inhibiting the Wnt/beta-Catenin Signaling Pathway. Med. Sci. Monit. 2017, 23, 6107–6113. [Google Scholar] [CrossRef]
- Hyman, J.M.; Firestone, A.J.; Heine, V.M.; Zhao, Y.; Ocasio, C.A.; Han, K.; Sun, M.; Rack, P.G.; Sinha, S.; Wu, J.J.; et al. Small-molecule inhibitors reveal multiple strategies for Hedgehog pathway blockade. Proc. Natl. Acad. Sci. USA 2009, 106, 14132–14137. [Google Scholar] [CrossRef]
- Li, B.; Fei, D.L.; Flaveny, C.A.; Dahmane, N.; Baubet, V.; Wang, Z.; Bai, F.; Pei, X.H.; Rodriguez-Blanco, J.; Hang, B.; et al. Pyrvinium attenuates Hedgehog signaling downstream of smoothened. Cancer Res. 2014, 74, 4811–4821. [Google Scholar] [CrossRef]
- Wolff, F.; Loipetzberger, A.; Gruber, W.; Esterbauer, H.; Aberger, F.; Frischauf, A.M. Imiquimod directly inhibits Hedgehog signalling by stimulating adenosine receptor/protein kinase A-mediated GLI phosphorylation. Oncogene 2013, 32, 5574–5581. [Google Scholar] [CrossRef] [PubMed]
- Nayak, A.; Satapathy, S.R.; Das, D.; Siddharth, S.; Tripathi, N.; Bharatam, P.V.; Kundu, C. Nanoquinacrine induced apoptosis in cervical cancer stem cells through the inhibition of hedgehog-GLI1 cascade: Role of GLI-1. Sci. Rep. 2016, 6, 20600. [Google Scholar] [CrossRef]
- Long, J.; Li, B.; Rodriguez-Blanco, J.; Pastori, C.; Volmar, C.-H.; Wahlestedt, C.; Capobianco, A.; Bai, F.; Pei, X.-H.; Ayad, N.G.; et al. The BET Bromodomain Inhibitor I-BET151 Acts Downstream of Smoothened Protein to Abrogate the Growth of Hedgehog Protein-driven Cancers. J. Biol. Chem. 2014, 289, 35494–35502. [Google Scholar] [CrossRef]
- Tang, Y.; Gholamin, S.; Schubert, S.; Willardson, M.I.; Lee, A.; Bandopadhayay, P.; Bergthold, G.; Masoud, S.; Nguyen, B.; Vue, N.; et al. Epigenetic targeting of Hedgehog pathway transcriptional output through BET bromodomain inhibition. Nat. Med. 2014, 20, 732–740. [Google Scholar] [CrossRef]
- Pan, D.; Li, Y.; Li, Z.; Wang, Y.; Wang, P.; Liang, Y. Gli inhibitor GANT61 causes apoptosis in myeloid leukemia cells and acts in synergy with rapamycin. Leuk. Res. 2012, 36, 742–748. [Google Scholar] [CrossRef]
- Zuo, M.; Rashid, A.; Churi, C.; Vauthey, J.N.; Chang, P.; Li, Y.; Hung, M.C.; Li, D.; Javle, M. Novel therapeutic strategy targeting the Hedgehog signalling and mTOR pathways in biliary tract cancer. Br. J. Cancer 2015, 112, 1042–1051. [Google Scholar] [CrossRef]
- Zhou, J.; Wu, K.; Gao, D.; Zhu, G.; Wu, D.; Wang, X.; Chen, Y.; Du, Y.; Song, W.; Ma, Z.; et al. Reciprocal regulation of hypoxia-inducible factor 2alpha and GLI1 expression associated with the radioresistance of renal cell carcinoma. Int. J. Radiat. Oncol. Biol. Phys. 2014, 90, 942–951. [Google Scholar] [CrossRef]
- Zeng, J.; Aziz, K.; Chettiar, S.T.; Aftab, B.T.; Armour, M.; Gajula, R.; Gandhi, N.; Salih, T.; Herman, J.M.; Wong, J.; et al. Hedgehog pathway inhibition radiosensitizes non-small cell lung cancers. Int. J. Radiat. Oncol. Biol. Phys. 2013, 86, 143–149. [Google Scholar] [CrossRef]
- Ally, M.S.; Ransohoff, K.; Sarin, K.; Atwood, S.X.; Rezaee, M.; Bailey-Healy, I.; Kim, J.; Beachy, P.A.; Chang, A.L.S.; Oro, A.; et al. Effects of combined treatment with arsenic trioxide and itraconazole in patients with refractory metastatic basal cell carcinoma. JAMA Dermatol. 2016, 152, 452–456. [Google Scholar] [CrossRef]
Figure 1.
A simplified model for canonical HhP. (A) The Hedgehog receptor PTCH1 inhibits SMO signaling in the absence of the Hedgehog ligand, which turns off the Hedgehog signaling pathway. (B) In the presence of Hedgehog ligand, PTCH1 stops inhibiting SMO, resulting in the translocation of SMO to the primary cilium. Once activated in the primary cilium, SMO promotes the release of GLI from the SUFU, which allows GLI to enter the nucleus to initiate the transcription program related to HhP. Dysregulation of the HhP has been associated with carcinogenesis.
Figure 1.
A simplified model for canonical HhP. (A) The Hedgehog receptor PTCH1 inhibits SMO signaling in the absence of the Hedgehog ligand, which turns off the Hedgehog signaling pathway. (B) In the presence of Hedgehog ligand, PTCH1 stops inhibiting SMO, resulting in the translocation of SMO to the primary cilium. Once activated in the primary cilium, SMO promotes the release of GLI from the SUFU, which allows GLI to enter the nucleus to initiate the transcription program related to HhP. Dysregulation of the HhP has been associated with carcinogenesis.


{kind=link}
Table 1.
SMO inhibitors in malignant tumors tested in clinical trials completed by October 2018.
| Study | Phase | Type of Cancer | Experimental Arm | Control Arm | Results of Primary EP |
|---|---|---|---|---|---|
| NCT02639117 | Phase 1 | Multiple BCC | Vismodegib + photodynamic therapy sessions + topical application of 20% 5-aminolevulinic acid (ALA) | Combination PDT-vismodegib therapy was overall well tolerated (50% dysgeusia, 50% myalgia, 75% flu-like symptoms) [54]. | |
| STEVIE NCT01367665 | Phase 2 | Locally advanced and metastatic BCC | Vismodegib | Serious side effects (grade ≥ 3) in 289 patients (23.8%) and death in 46 patients (3.8%) [55]. | |
| NCT01546519 | Phase 1b | Advanced solid malignancies and hepatic impairment | Vismodegib | 96.8% in all groups, experienced at least one AE. 67.7% of all AEs reported were grade 3 or 4 [56]. | |
| ERIVANCE BCC NCT00833417 | Phase 2 | Locally advanced and metastatic BCC | Vismodegib | ORR of 60.3% in patients with locally advanced BCC and 48.5% metastatic BCC [57]. | |
| MIKIE NCT01815840 | Phase 2 | Multiple BCC | A. Vismodegib 12 w - placebo 8 w - vismodegib 12 w B. Vismodegib 24 w - placebo 8 w - vismodegib 8 w | The mean number of BCC lesions at week 73 was reduced from baseline by 62.7% in group A and 54% in group B [58]. | |
| NCT00957229 | Phase 2 | Basal cell nevus syndrome (BCNS) | Vismodegib | Placebo | Reduced rate of new surgically eligible BCC (2 vs 34 per patient per year) [59]. |
| NCT02115828 | Phase 2 | Metastatic castration-resistant prostate cancer | Vismodegib | Gli1 mRNA was significantly suppressed by vismodegib in both tumor tissue (57%) and benign skin biopsies (75%) [60]. | |
| NCT01631331 | Phase 1 | BCC | Neoadjuvant vismodegib | Reduction of the final surgical defect size by 34.8% compared with baseline [61]. | |
| E1508 NCT00887159 | Phase 2 | Extensive stage small cell lung carcinoma | A. Cisplatin + etoposide B. Vismodegib C. Cixutumumab | The median PFS times in arms A, B, and C were 4.4, 4.4, and 4.6 months, respectively [62]. | |
| VISMOLY NCT01944943 | Phase 2 | Refractory or relapsed B-cell lymphoma or chronic lymphocytic leukemia | Vismodegib | The best overall response: DLBCL: 0 (0%), iNHL: 1 (16.7%), PCNSL: 0 (0%), CLL: (0%), all: 1 (3.2%) [63]. | |
| NCT01064622 | Phase 1b/2 | Metastatic pancreatic cancer | Gemcitabine + vismodegib | Gemcitabine plus Placebo | Median PFS was 4.0 and 2.5 months for GV and GP arms, respectively [64] |
| NCT01201915 | Phase 2 | BCC | Neoadjuvant vismodegib
| Complete histologic clearance was achieved by 42%, 16%, and 44% of patients in cohorts 1, 2, and 3, respectively [65]. | |
| NCT01195415 | Phase 2 | Metastatic pancreatic adenocarcinoma | Vismodegib plus gemcitabine | GLI1 and PTCH1 decreased in 95.6% and 82.6%, respectively [66]. | |
| NCT01267955 | Phase 2 | Advanced chondrosarcoma | Vismodegib | The 6-month clinical benefit rate was 25.6% [67]. | |
| NCT00822458 | Phase 1 | Medulloblastoma | Vismodegib | 3 dose-limiting toxicities but no drug-related bone toxicity. The median vismodegib penetration in the CSF was 0.53 (ratio of the concentration of vismodegib in the CSF to that of the unbound drug in plasma) [68]. | |
| NCT00607724 | Phase 1 | BCC | Vismodegib | SUVmax decreased (median 33%, SD ± 45%) with metabolic activity normalizing or disappearing in 42% of lesions [69] | |
| NCT00636610 | Phase 2 | Metastatic colorectal cancer | Vismodegib + FOLFOX or FOLFIRI + bevacizumab | Placebo + FOLFOX or FOLFIRI + bevacizumab | Median PFS hazard ratio (HR) was 1.25 [70]. |
| NCT01209143 | Phase 1b | Solid cancers |
| Systemic exposure of rosiglitazone or oral contraceptive is not altered with concomitant vismodegib [71]. | |
| NCT00739661 | Phase 2 | Ovarian cancer | Vismodegib | Placebo | Median PFS in vismodegib and placebo groups were 7.5 months and 5.8 months, respectively [72]. |
| NCT00607724 | Phase 1 | Solid tumor | Vismodegib | 8 grade 3 adverse events in 6 patients. 1 patient withdrew from the study because of adverse events [73] | |
| BOLT NCT01327053 | Phase 2 | BCC | Sonidegib 200 mg and 800 mg | Sonidegib 200 mg (approved dose), objective response rates were 56.1% (central) and 71.2% (investigator) in laBCC and 7.7% (central) and 23.1% (investigator) in mBCC [74]. | |
| NCT01125800 | Phase 1/2 | Medulloblastoma, rhabdomyosarcoma, neuroblastoma, hepatoblastoma, glioma, astrocytoma | Sonidegib 233 mg/m2 daily; 372 mg/m2 daily; 425 mg/m2 daily; 680 mg/m2 daily; 800 mg/m2 daily | The recommended phase II dose in pediatric patients was 680 mg/m2 once daily. The results were 4 complete responders (2 pediatric and 2 adult) and 1 partial response (adult) [75]. | |
| NCT01954355 | Phase 1 | Solid tumorovarian cancer | Sonidegib 400, 600 and 800 mg + paclitaxel | The recommended phase II dose was 800 mg in combination with paclitaxel [76]. | |
| NCT01208831 | Phase 1 | Advanced solid tumor cancers, medulloblastoma, BCC | Sonidegib | The recommended dose in East Asian patients (400 mg) was lower than in patients from Europe and the USA (800 mg and 250 mg, respectively, twice daily) [77]. | |
| NCT01579929 | Phase 1 | Lung cancer | Sonidegib + etoposide + cisplatin | Sonidegib 800 mg daily was the maximum tolerated dose when administered with EP [78]. | |
| NCT01383538 | Phase 1 | Advanced pancreatic adenocarcinoma | Saridegib + FOLFIRINOX | The combination was active and safe [79]. | |
| NCT01371617 | Phase 2 | Primary myelofibrosis | Saridegib | Nine out of fourteen patients (79%) did not respond. Saridegib is not active in myelofibrosis as monotherapy [80]. | |
| NCT01255800 | Phase 1 | Recurrent head and neck cancer | Saridegib + cetuximab | The recommended phase 2 dose was 160 mg, the same as the single-agent saridegib maximum tolerated dose [81]. | |
| NCT00761696 | Phase 1 | Solid tumor | Saridegib | The maximum tolerated dose (MTD) of saridegib was 160 mg QD within 28-day cycles [82]. | |
| NCT01787331 | Phase 2 | Biochemically relapsed prostate cancer | Itraconazole | One patient (5%) had a > 50% PSA decline [83]. | |
| NCT01108094 | Phase 2 | BCC | Itraconazole | Itraconazole reduced cell proliferation by 45%, HH pathway activity by 65% and reduced tumor area by 24% [84]. | |
| NCT00769600 | Phase 2 | Recurrent non-small cell lung cancer | Itraconazole + pemetrexed | Pemetrexed | Median PFS was 5.5 months (itraconazole) versus 2.8 months (control). There were no evident differences in toxicity between the study arms [85]. |
| NCT00887458 | Phase 2 | Metastatic castration-resistant prostate cancer | Itraconazole 200 mg daily and 600 mg daily | The PSA PFS rates at 24 weeks were 11.8% in the low-dose arm and 48.0% in the high-dose arm [86]. | |
| NCT01919398 | Phase 1 | Metastatic solid tumor | Taladegib | No dose-limiting toxicities were observed at doses of 100 mg or 200 mg; 3 of the 9 patients evaluable for DLTs at the 400 mg dose level experienced DLTs [83]. | |
| NCT01226485 | Phase 1 | Advanced cancer | Taladegib | The maximum tolerable dose was 400 mg [87]. | |
| NCT01546038 | Phase 1/2 | Acute myeloid leukemia High-risk myelodysplastic syndromes | A: Glasdegib + low-dose ARA-C B: Glasdegib + decitabine C: Glasdegib + daunorubicin + cytarabine | No dose-limiting toxicities (DLT) were observed in arms A and B; 1 DLT (grade 4 neuropathy) occurred in arm C. 46.4% of patient achieved investigator-reported. Among patients ≥55 years old (n = 60), 40.0% achieved complete remission [88,89]. | |
| NCT01286467 | Phase 1 | Solid tumors | Glasdegib | The first-cycle DLT rate at the 640 mg dose level was 33.3%, and the O maximum tolerable dose was estimated to be 320 mg once daily [90]. |
EP: end point; BCC: basal cell carcinoma; AE: adverse events; DLBCL: diffuse large B-cell lymphoma; iNHL: indolent lymphoma; PCNSL: primary central nervous system lymphoma; CLL: chronic lymphocytic leukemia; laBCC: locally advanced basal cell carcinoma; mBCC: metastatic basal cell carcinoma.
Table 2.
HhP inhibitors that are being evaluated in clinical trials as of October 2018. Data from www.clinicaltrials.gov.
Table 2.
HhP inhibitors that are being evaluated in clinical trials as of October 2018. Data from www.clinicaltrials.gov.
| Study | Phase | Type of Cancer | Experimental Arm | Control Arm | Status |
|---|---|---|---|---|---|
| NCT02138929 | Phase 1 | Advanced gastroesophageal cancer | Sonidegib + everolimus | Active, not recruiting | |
| NCT01485744 | Phase 1 | Advanced pancreatic adenocarcinoma | Sonidegib + FOLFIRINOX | Active, not recruiting | |
| NCT01431794 | Phase 1/2 | Pancreatic | Neoadjuvant gemcitabine, nab-paclitaxel and sonidegib | Neoadjuvant gemcitabine, nab-paclitaxel | Active, not recruiting |
| NCT02151864 | Phase 1 | Hepatocellular carcinoma | Sonidegib in patients intolerant to sorafenib | Active, not recruiting | |
| NCT02303041 | Phase 2 | Metastatic basal cell cancer | Sonidegib + buparlisib | Completed | |
| NCT01576666 | Phase 1 | Advanced solid tumors | Sonidegib + buparlisib | Completed | |
| NCT03434262 | Phase 1 | Recurrent brain tumors | Sonidegib among others | Recruiting | |
| NCT01787331 | Phase 2 | Biochemically relapsed prostate cancer | Itraconazole | Active, not recruiting | |
| NCT02735356 | Early phase 1 | Basal cell cancer | Topical itraconazole | Active, not recruiting | |
| NCT02357836 | Early phase 1 | Non-small cell lung cancer | Neoadjuvant itraconazole | Recruiting | |
| NCT02749513 | Early phase 1 | Esophagus | Itraconazole | Recruiting | |
| NCT01835626 | Phase 2 | Basal cell cancer | Vismodegib + radiotherapy | Recruiting | |
| NCT03052478 | Phase 2 | Advanced gastric cancer | Vismodegib | Recruiting | |
| NCT01878617 | Phase 2 | Medulloblastoma | Vismodegib among others | Recruiting | |
| NCT03035188 | Phase 2 | Basal cell cancer | Neoadjuvant vismodegib | Recruiting | |
| NCT02694224 | Phase 2 | Triple-negative breast cancer | Neoadjuvant vismodegib + paclitaxel, epirubicin and cyclophosphamide | Neoadjuvant paclitaxel, epirubicin and cyclophosphamide | Recruiting |
| NCT01791894 | Phase 1/2 | Basal cell cancer | Arsenic trioxide | Completed | |
| NCT01470248 | Phase 2 | Advanced small cell lung cancer | Arsenic trioxide | Completed | |
| NCT03503864 | Phase 2 | Advanced neuroblastoma | Arsenic trioxide + conventional induction chemotherapy | Conventional induction chemotherapy | Recruiting |
| NCT03466450 | Phase 1/2 | Glioblastoma | Glasdegib + temozolomide | Recruiting | |
| NCT02530437 | Phase 1/2 | Localized esophageal or gastroesophageal junction cancer | Taladegib + carboplatin, paclitaxel and radiation | Active, not recruiting | |
| NCT01130142 | Phase 1/2 | Metastatic pancreatic adenocarcinoma | Saridegib + gemcitabine | Gemcitabine | Completed |
| NCT01383538 | Phase 1 | Metastatic pancreatic adenocarcinoma | Saridegib + FOLFIRINOX | Completed | |
| NCT01310816 | Phase 2 | Metastatic or locally advanced chondrosarcoma | Saridegib | Placebo | Completed |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Girardi, D.; Barrichello, A.; Fernandes, G.; Pereira, A. Targeting the Hedgehog Pathway in Cancer: Current Evidence and Future Perspectives. Cells 2019, 8, 153. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8020153
AMA Style
Girardi D, Barrichello A, Fernandes G, Pereira A. Targeting the Hedgehog Pathway in Cancer: Current Evidence and Future Perspectives. Cells. 2019; 8(2):153. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8020153
Chicago/Turabian StyleGirardi, Daniel, Adriana Barrichello, Gustavo Fernandes, and Allan Pereira. 2019. "Targeting the Hedgehog Pathway in Cancer: Current Evidence and Future Perspectives" Cells 8, no. 2: 153. https://0-doi-org.brum.beds.ac.uk/10.3390/cells8020153
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.